
- Department of Aging Neurobiology, Center for Development of Advanced Medicine for Dementia, National Center for Geriatrics and Gerontology, Aichi, Japan
In Alzheimer’s disease (AD), tau hyperphosphorylation and neurofibrillary tangle (NFT) formation are strongly associated with dementia, a characteristic and early feature of this disease. Glycogen synthase kinase 3β (GSK-3β) is a pivotal kinase in both the normal and pathological phosphorylation of tau. In the diseased state, hyperphosphorylated tau is deposited in NFTs, the formation of which, drive the disease process. GSK-3β which is also involved in long-term depression induction, interacts with tau to inhibit synaptic long-term potentiation. Strong lines of evidence suggest that the activation of GSK-3β is responsible for the memory deficits seen in both advanced age and AD. In this review, we will focus on the role of GSK-3β in brain function, particularly in memory maintenance. We will examine human and mouse studies which suggest a role for GSK-3β in memory maintenance and the eventual development of memory deficits.
Alzheimer’s Disease and Memory
Memory impairment in old age is a hallmark of the initial stage of Alzheimer’s disease (AD), with dementia developing in the final stages (Poissonnet et al., 2012). AD is characterized by the extensive deposition of amyloid β (Aβ), outside of neurons, and the formation of neurofibrillary tangles (NFTs) consisting of hyperphosphorylated tau, as intraneuronal inclusions (Selkoe, 1986). The relationship between the clinical course of AD and the observed pathological changes is not yet fully understood. Genetic studies of familial AD identified three causative genes, APP, PSEN 1, and PSEN 2 (Tandon et al., 2000). Since these genes form part of a cascade that results in Aβ generation, the Aβ hypothesis emerged as a mechanism for AD pathophysiology (Hardy and Selkoe, 2002). This theory states that Aβ deposition directly affects neurons, inducing NFTs and neuronal death, leading to dementia. Inheritance of the APP mutation leads to AD with 100% penetrance (Goate and Hardy, 2011). Mice engineered to overexpress mutant human APP, show memory impairment along with Aβ deposition (Gotz and Ittner, 2008), supporting the Aβ hypothesis. Electrophysiological analyses indicate an inverse correlation between Aβ levels and the amplitude of hippocampal long-term potentiation (LTP; Walsh et al., 2002; Westerman et al., 2002), an underlying mechanism of memory. A recent study found that reducing tau alleviated Aβ-induced memory impairment in APP transgenic (Tg) mice (Roberson et al., 2007), suggesting that tau contributes to memory impairment in APP Tg mice. However, contrary to these results, recent clinical trials show that reducing Aβ generation, or removing Aβ deposits fail to halt the progression of dementia (Holmes et al., 2008).
NFT Formation Promotes Memory Impairment and Dementia
The number of NFTs, unlike the extent of Aβ deposition, correlates strongly with the degree of dementia (Gomez-Isla et al., 1997). In diseased brains, synaptic and neuronal loss are prominent in regions with detectable NFTs, implicating NFT formation in AD associated memory impairment and dementia (Masliah et al., 1992). Based on the observations of Braak and Braak (1990), as AD progresses, NFTs are observed first in the entorhinal cortex, a region integral to memory formation and maintenance, later spreading into the limbic cortex and neocortex, regions associated with emotions, and higher functioning such as thought, respectively. Considering the role of these regions in normal brain function, this sequential formation of NFTs could go some way to explaining the clinical progression of AD. Before NFT formation, tau is hyperphosphorylated by glycogen synthase kinase 3β (GSK-3β) activation and forms granular tau oligomers. This hyperphosphorylated tau is associated with synapse loss (Kimura et al., 2007), while granular tau oligomers are involved in neuronal death. These data imply that the neuronal dysfunction resulting from synaptic and neuronal loss (Kimura et al., 2010), occurs when NFTs are formed.
NFT Formation Promotes Neuronal Dysfunction
Mice that overexpress P301L, a mutant form of tau, display age-related NFTs, neuronal death, and memory deficits (Ramsden et al., 2005; Santacruz et al., 2005). Although inhibiting mutant tau overexpression in these mice blocks neuronal death and improves memory, NFTs continue to form (Ramsden et al., 2005; Spires et al., 2006). This suggests that NFTs in themselves are not toxic, but instead, the processes of NFT formation, neuronal death and neuronal dysfunction underly the pathogenic mechanism.
The formation of tau fibrils follows three sequential steps (Maeda et al., 2007; Kimura et al., 2008; Takashima, 2008), and has been studied using atomic force microscopy (AFM). AFM allows direct observation of tau aggregation in experimental solutions, with no special pretreatments, in contrast to scanning electron microscopy which requires several pretreatment steps. First, hyperphosphorylated monomeric tau binds together to form soluble oligomers. The structure of these oligomers however, is not discernible under AFM. Second, the soluble tau oligomers take on a β-sheet structure, forming insoluble tau aggregates. These aggregates become granular-shaped oligomers consisting of approximately 40 tau molecules, which are detectable under AFM. Third and finally, the increased concentration of granular tau causes these oligomers to fuse, forming tau fibrils (Maeda et al., 2007).
As a major tau kinase, GSK-3β induces tau hyperphosphorylation, as one of the earliest events in NFT formation (Ishiguro et al., 1988, 1993). Hyperphosphorylated tau or soluble tau oligomers are associated with loss of synapses in wild type tau Tg mice (Kimura et al., 2007), while granular tau oligomers are associated with loss of neurons in P301L tau Tg mice (Kimura et al., 2010). Thus, the intermediary, soluble and granular tau oligomers can promote synaptic and neuronal loss before NFT formation. This suggests that rather than being the cause of cell death, NFTs represent a biological tombstone, marking the sites of neuron death. Therefore, memory impairment probably occurs when NFTs are seen in the entorhinal cortex and hippocampus, since synaptic and neuronal loss occur before the formation of NFTs in these regions.
Tau Phosphorylation by GSK-3β
Tau protein kinase I (TPKI; Ishiguro et al., 1988), is encoded by a nucleotide sequence identical to that of GSK-3β (Ishiguro et al., 1993), but not GSK-3α. This kinase is activated by aggregated Aβ and induces tau hyperphosphorylation as seen in NFTs and neuron death, in hippocampal cultures (Takashima et al., 1993, 1996). Phosphorylation of the tau Ser422 residue, a site not phosphorylated by GSK-3β, is specifically seen in NFTs (Morishima-Kawashima et al., 1995) indicating the involvement of additional kinases in this process. While the Ser422 residue can be phosphorylated by c-Jun amino-terminal kinase (JNK), this is not enough to promote tau aggregation. In cultured cells at least, both JNK and GSK-3β activation are needed to generate tau aggregation (Sato et al., 2002). These results point to GSK-3β activation as a requirement for AD pathogenesis.
Mice overexpressing GSK-3β show an accumulation of hyperphosphorylated tau, neuronal death in the hippocampus, and memory impairment in object recognition tests (Lucas et al., 2001; Hernandez et al., 2002). These mice also exhibit reduced hippocampal LTP (Hooper et al., 2007), and this memory deficit is reversed when tau expression stops (de Barreda et al., 2010). Reducing tau levels (Roberson et al., 2007) and inhibiting GSK-3 (Sereno et al., 2009) can each rescue memory impairment in APP Tg mice. Aβ activates GSK-3β, inducing tau hyperphosphorylation in hippocampal neurons, and it is this GSK-3β activation that leads to reduced LTP and eventual memory impairment in APP Tg mice. Again, evidence shows that activation of GSK-3β is a key factor in AD associated memory impairment, promoting the idea that inhibitors of GSK-3β, may be potential therapeutic agents for this disease.
GSK-3 Inhibitors
Peineau et al. (2007) showed that GSK-3β localizes to postsynaptic regions and that GSK-3 inhibitors block NMDA-dependent long-term depression (LTD) induction. Our own data (unpublished) shows a blockade of LTD induction in GSK-3β heterozygote knockout mice. Although several companies have developed GSK-3 inhibitors, there are currently no successful candidates in Phase III trials. Lithium, a longstanding therapeutic drug used in bipolar disorder (Gould et al., 2006), is a specific inhibitor for GSK-3 (Klein and Melton, 1996). Lithium inhibits GSK-3 directly by competing with magnesium binding sites. It also acts indirectly, by enhancing serine phosphorylation of GSK-3, as well as through β-arrestin complex formation (reviewed in this Research Topic series: Eldar-Finkelman and Martinez, 2011; Freland and Beaulieu, 2012). Lithium treatment inhibits tau hyperphosphorylation, and NFT formation (Engel et al., 2006; Leroy et al., 2010), alleviating memory deficits not only in mice overexpressing tau, but also mice expressing both APP and PS1 (Zhang et al.,2011). Therefore hypothetically, lithium inhibition of GSK-3β should halt the clinical progression of AD in humans. While short-term lithium treatment failed to improve cognitive function, a biomarker for AD (Hampel et al., 2009), long-term treatment significantly reduced phosphorylated tau levels in cerebrospinal fluid, a potential biomarker for AD, and improved cognitive function (Forlenza et al., 2011). Interestingly, a retrospective study of bipolar and unipolar-depression patients with a history of lithium treatment, found that these patients had a higher risk of developing dementia (Dunn et al., 2005). It therefore appears that GSK-3 performs a dual role. In patients without dementia, GSK-3 activity maintains cognitive function, whereas patients with dementia show excessive activation of GSK-3.
The Role of GSK-3β in Synaptic Plasticity
GSK-3 exists as two isoforms, α and β, which share high sequence identity and are encoded by genes on chromosomes 19 and 3 respectively, in humans (Woodgett, 1990). GSK-3β and GSK-3α localize to different compartments. GSK-3β, but not GSK-3α localizes to the mitochondria and synaptosomes (Hoshi et al., 1995). Therefore it is likely that GSK-3β may be directly involved in synaptic plasticity, while GSK-3α may act indirectly, via the regulation of gene expression (more details in this Research Topics series, as reviewed by Beaulieu et al., 2011; Polter and Li, 2011). These isoforms share common substrates including tau, but they also have distinct functions. While knockout of GSK α in mice induces increased insulin sensitivity, knockout of GSK-3β in mice is embryonically lethal (Hoeflich et al., 2000; MacAulay et al., 2007).
GSK-3β is pivotal in the cascade leading to NFT formation, which in turn drives dementia in AD. GSK-3β could be seen as a time-delayed ignition switch in the brain, which in old age triggers the process of dementia. As mentioned previously, patients with a long history of lithium therapy, and consequently suppressed levels of GSK-3, show a higher risk for developing dementia compared with lithium naïve patients (Dunn et al., 2005). These observations imply that controlled levels of GSK-3 activity are required for maintaining normal brain function, and as we already know, excessive activation of GSK-3β, drives NFT formation, leading to disease. Unraveling the dual role of GSK-3β requires an understanding of the physiological function of this protein in healthy adult brains and how this changes with aging. As we know, GSK-3β is required for NMDA-dependent LTD induction (Peineau et al., 2007). It is this requirement for GSK-3β in synaptic plasticity that fuels the analysis of GSK-3β in memory formation.
GSK-3β Activation is Required for Memory Reconsolidation
Learning stimuli first lead to short-term memory formation, which lasts a few hours and is then converted to long-term memory, through a process of memory consolidation. Active memory is formed by recalling and updating long-term memory. This updated memory becomes long-term memory through a process of memory reconsolidation. Reconsolidation is required for updating reactivated memory, and maintaining long-term memory (Figure 1). Although memory consolidation and reconsolidation are thought to have distinct molecular pathways, both are protein synthesis-dependent (Nader et al., 2000; Riccio et al., 2002; Eisenberg et al., 2003; Biedenkapp and Rudy, 2004; Dudai and Eisenberg, 2004; Lee et al., 2006; Morris et al., 2006). We used GSK-3β heterozygous knockout mice (+/-) to understand how GSK-3β fits into these pathways. While the homozygous GSK-3β mutation is embryonically lethal, heterozygous mice express GSK-3β at approximately 50%, and a relative activity was about 70% of wild type mice (Kimura et al., 2008). For GSK-3α, the paralog of GSK-3β, the total amount and relative activity of GSK-3α did not differ between GSK-3β+/- and wild type mice. As previously reported (Hoeflich et al., 2000), GSK-3β+/- mice are healthy and fertile, with normal circadian rhythms, life span, and locomotor activity, compared to wild type mice.
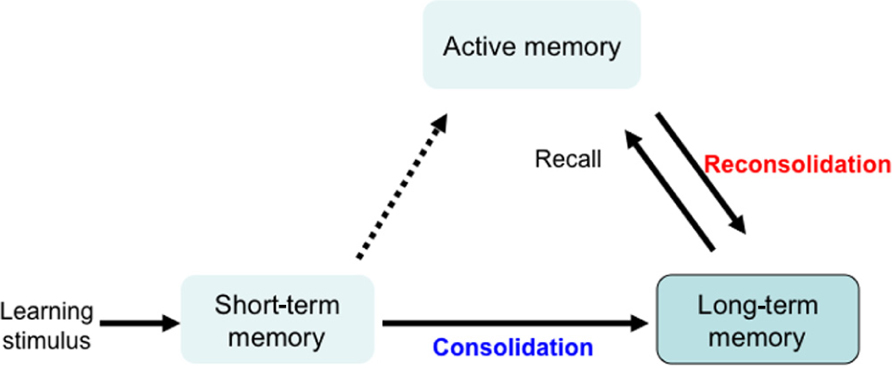
FIGURE 1. Mechanism for memory formation and maintenance. Learning stimulation leads to short-term memory, which lasts a few hours and is converted to long-term memory through a process of consolidation. Active memory is formed by recalling and updating long-term memory. This updated memory becomes long-term memory through a process of memory reconsolidation. Reconsolidation is a protein synthesis-dependent process that is required for updating the reactivated memory and for maintaining long-term memory.
In the contextual fear conditioning paradigm, GSK-3β+/-mice showed similar freezing times in response to unconditioned stimuli as wild type mice, and there was no difference in the freeze times between GSK-3β+/- and WT mice in the consolidation test (Figure 2A; Kimura et al., 2008). This suggests no impairment in the ability of GSK-3β+/- mice to form and consolidate memories, and that these memories can be maintained for at least 7 days, the time period examined in this study (Kimura et al., 2008). In reconsolidation however, GSK-3β+/- mice, showed significantly less freeze time compared with wild type mice, at day 7 (Figure 2B; Kimura et al., 2008). These results indicate that GSK-3β heterozygotes are capable of learning and stabilizing long-term memory for 7 days, if memory is not reactivated. However, GSK-3β+/- mice failed to achieve reconsolidation when memory was reactivated once before testing (Kimura et al., 2008). The retrograde amnesia exhibited by these mice in the reconsolidation test of contextual fear conditioning, points to possible impaired memory reconsolidation, in keeping with the theory that GSK-3β activation is required for memory reconsolidation or maintenance.
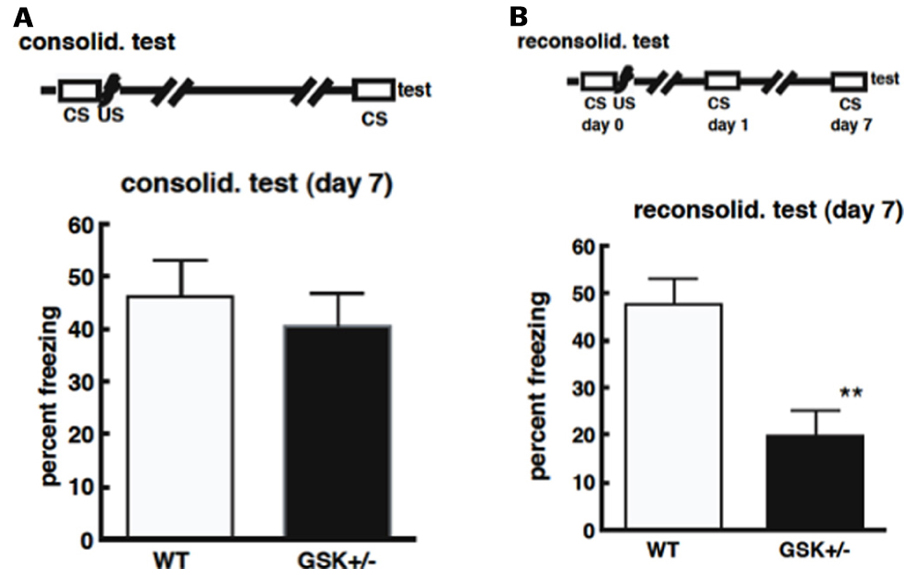
FIGURE 2. (A) Deficiencies in GSK-3β do not affect the process of memory consolidation. Mice were placed in a novel environment for 5 min (CS) and then subjected to three sequential foot-shocks (US). On day 7 after conditioning, animals were placed in the original environment, without shocks, for 5 min. The freezing time was recorded for these animals. (B) Deficiencies in GSK-3β impair memory reconsolidation. Mice were placed in the environmental for 5 min (CS), and then subjected to three sequential foot-shocks (US). One day after conditioning, animals were exposed to the same environment for 5 min. Six days after their first exposure to conditioning, animals were re-exposed again and the freezing time was recorded.
Prospective Role of GSK-3β in Brain Aging
GSK-3β is involved in NMDA-dependent LTD induction and memory reconsolidation (Peineau et al., 2007; also reviewed in this Research Topic series by Bradley et al., 2012). However, the relationship between LTD induction and memory reconsolidation is unclear, although there are reports that LTD is important for memory formation or new object recognition. Focusing on synaptic plasticity, particularly LTP, genetic ablation of the NMDA receptor impaired place learning in a LTD dependent manner (Tsien et al.,1996). Further analysis using CaMKIV knockout mice indicates that late LTP is involved in the consolidation process of memory formation (Kang et al., 2001). Thus, both LTP and LTD contribute to memory formation, in which the memory consolidation processes may preferentially depend on LTP, and memory reconsolidation processes require LTD. In memory reconsolidation, LTD maintains a prior potentiated circuit by competitive synaptic maintenance (Diamond et al., 2005) and protects stable memory traces. This may explain why activation of GSK-3β is required in reconsolidation but not in consolidation processes in normal brain function. GSK-3β activation in the entorhinal cortex and hippocampus is required for spatial recognition, in aged but not young brains (unpublished result). While this process is required for maintaining normal brain function in old age, the frequent activation of GSK-3β induces NFTs in the entorhinal cortex (Braak and Braak, 1996) and hippocampus. It would therefore appear that GSK-3β activation is an early event in normal brain aging as well as AD.
We put forward that, generally, as we age, we learn and accumulate many memories. When we are confronted with a new idea or task, we draw on our experiences, that is, we recall related memories to help us understand new information. The frequent need to recall and reconsolidate memories relies on increased activation of GSK-3β and consequently, tau phosphorylation. Over time, NFTs accumulate in the entorhinal cortex, which is a very early pathological change in sporadic AD.
Conclusion
Tau hyperphosphorylation and NFT formation are early features of dementia associated with AD. This major change in the phosphorylation state of tau leads to deposition of pathological tau in NFTs, and these tangles are formed in a specific spatial and temporal pattern within the brain. It is the formation rather than the presence of these NFTs that induces neuronal dysfunction and death, leading to tauopathies.
GSK-3β is a major kinase for tau phosphorylation associated with both physiological brain function and AD pathophysiology. GSK-3β is also required for synaptic plasticity. Reduced GSK-3 expression in GSK-3β+/- mice results in impaired memory reconsolidation emphasizing the importance of GSK-3 in promoting memory maintenance via reconsolidation. A greater understanding of how synaptic plasticity changes with aging, through the analysis of AD-related molecules such as GSK-3β and tau, would provide a solid platform of knowledge, from which new therapeutic targets and innovative agents could be developed for AD.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Beaulieu, J. M., Del’Guidice, T., Sotnikova, T. D., Lemasson, M., and Gainetdinov, R. R. (2011). Beyond cAMP: the regulation of Akt and GSK3 by dopamine receptors. Front. Mol. Neurosci. 4:38. doi: 10.3389/ fnmol.2011.00038
Biedenkapp, J. C., and Rudy, J. W. (2004). Context memories and reactivation: constraints on the reconsolidation hypothesis. Behav. Neurosci. 118, 956–964.
Braak, H., and Braak, E. (1990). Neurofibrillary changes confined to the entorhinal region and an abundance of cortical amyloid in cases of presenile and senile dementia. Acta Neuropathol. 80, 479–486.
Braak, H., and Braak, E. (1996). Evolution of the neuropathology of Alzheimer’s disease. Acta Neurol. Scand. Suppl. 165, 3–12.
Bradley, C. A., Peineau, S., Taghibiglou, C., Nicolas, C. S., Whitcomb, D. J., Bortolotto, Z. A., Kaang, B. K., Cho, K., Wang, Y. T., and Collingridge, G. L. (2012). A pivotal role of GSK-3 in synaptic plasticity. Front. Mol. Neurosci. 5:13. doi: 10.3389/fnmol. 2012.00013
de Barreda, E. G, Perez, M., Gomez Ramos, P., De Cristobal, J., Martin-Maestro, P., Moran, A., Dawson, H. N., Vitek, M. P., Lucas, J. J., Hernandez, F., and Avila, J. (2010). Tau-knockout mice show reduced GSK3-induced hippocampal degeneration and learning deficits. Neurobiol. Dis. 37, 622–629.
Diamond, D. M., Park, C. R., Campbell, A. M., and Woodson, J. C. (2005). Competitive interactions between endogenous LTD and LTP in the hippocampus underlie the storage of emotional memories and stress-induced amnesia. Hippocampus 15, 1006–1025.
Dudai, Y., and Eisenberg, M. (2004). Rites of passage of the engram: reconsolidation and the lingering consolidation hypothesis. Neuron 44, 93–100.
Dunn, N., Holmes, C., and Mullee, M. (2005). Does lithium therapy protect against the onset of dementia? Alzheimer Dis. Assoc. Disord. 19, 20–22.
Eisenberg, M., Kobilo, T., Berman, D. E., and Dudai, Y. (2003). Stability of retrieved memory: inverse correlation with trace dominance. Science 301, 1102–1104.
Eldar-Finkelman, H., and Martinez, A. (2011). GSK-3 inhibitors: preclinical and clinical focus on CNS. Front. Mol. Neurosci. 4:32. doi: 10.3389/ fnmol.2011.00032
Engel, T., Goni-Oliver, P., Lucas, J. J., Avila, J., and Hernandez, F. (2006). Chronic lithium administration to FTDP-17 tau and GSK-3beta over-expressing mice prevents tau hyperphosphorylation and neurofibrillary tangle formation, but pre-formed neurofibrillary tangles do not revert. J. Neurochem. 99, 1445–1455.
Forlenza, O. V., Diniz, B. S., Radanovic, M., Santos, F. S., Talib, L. L., and Gattaz, W. F. (2011). Disease-modifying properties of long-term lithium treatment for amnestic mild cognitive impairment: randomised controlled trial. Br. J. Psychiatry 198, 351–356.
Freland, L., and Beaulieu, J. M. (2012). Inhibition of GSK3 by lithium, from single molecules to signaling networks. Front. Mol. Neurosci. 5:14. doi: 10.3389/fnmol.2012.00014
Goate, A., and Hardy, J. (2011). Twenty years of Alzheimer’s disease-causing mutations. J. Neurochem. Suppl. 1, 3–8.
Gomez-Isla, T., Hollister, R., West, H., Mui, S., Growdon, J. H., Petersen, R. C., Parisi, J. E., and Hyman, B. T. (1997). Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer’s disease. Ann. Neurol. 41, 17–24.
Gotz, J., and Ittner, L. M. (2008). Animal models of Alzheimer’s disease and frontotemporal dementia. Nat. Rev. Neurosci. 9, 532–544.
Gould, T. D., Picchini, A. M., Einat, H., and Manji, H. K. (2006). Targeting glycogen synthase kinase-3 in the CNS: implications for the development of new treatments for mood disorders. Curr. Drug Targets 7, 1399–1409.
Hampel, H., Ewers, M., Burger, K., Annas, P., Mortberg, A., Bogstedt, A., Frolich, L., Schroder, J., Schonknecht, P., Riepe, M. W., Kraft, I., Gasser, T., Leyhe, T., Moller, H. J., Kurz, A., and Basun, H. (2009). Lithium trial in Alzheimer’s disease: a randomized, single-blind, placebo-controlled, multicenter 10-week study. J. Clin. Psychiatry 70, 922–931.
Hardy, J., and Selkoe, D. J. (2002). The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297, 353–356.
Hernandez, F., Borrell, J., Guaza, C., Avila, J., and Lucas, J. J. (2002). Spatial learning deficit in transgenic mice that conditionally over-express GSK-3beta in the brain but do not form tau filaments. J. Neurochem. 83, 1529–1533.
Hoeflich, K. P., Luo, J., Rubie, E. A., Tsao, M. S., Jin, O., and Woodgett, J. R. (2000). Requirement for glycogen synthase kinase-3beta in cell survival and NF-kappaB activation. Nature 406, 86–90.
Holmes, C., Boche, D., Wilkinson, D., Yadegarfar, G., Hopkins, V., Bayer, A., Jones, R. W., Bullock, R., Love, S., Neal, J. W., Zotova, E., and Nicoll, J. A. (2008). Long-term effects of Abeta42 immunisation in Alzheimer’s disease: follow-up of a randomised, placebo-controlled phase I trial. Lancet 372, 216–223.
Hooper, C., Markevich, V., Plattner, F., Killick, R., Schofield, E., Engel, T., Hernandez, F., Anderton, B., Rosenblum, K., Bliss, T., Cooke, S. F., Avila, J., Lucas, J. J., Giese, K. P., Stephenson, J., and Lovestone, S. (2007). Glycogen synthase kinase-3 inhibition is integral to long-term potentiation. Eur. J. Neurosci. 25, 81–86.
Hoshi, M., Sato, M., Kondo, S., Takashima, A., Noguchi, K., Takahashi, M., Ishiguro, K., and Imahori, K. (1995). Different localization of tau protein kinase I/glycogen synthase kinase-3 beta from glycogen synthase kinase-3 alpha in cerebellum mitochondria. J. Biochem. 118, 683–685.
Ishiguro, K., Ihara, Y., Uchida, T., and Imahori, K. (1988). A novel tubulin-dependent protein kinase forming a paired helical filament epitope on tau. J. Biochem. 104, 319–321.
Ishiguro, K., Shiratsuchi, A., Sato, S., Omori, A., Arioka, M., Kobayashi, S., Uchida, T., and Imahori, K. (1993). Glycogen synthase kinase 3 beta is identical to tau protein kinase I generating several epitopes of paired helical filaments. FEBS Lett. 325, 167–172.
Kang, H., Sun, L. D., Atkins, C. M., Soderling, T. R., Wilson, M. A., and Tonegawa, S. (2001). An important role of neural activity-dependent CaMKIV signaling in the consolidation of long-term memory. Cell 106, 771–783.
Kimura, T., Fukuda, T., Sahara, N., Yamashita, S., Murayama, M., Mizoroki, T., Yoshiike, Y., Lee, B., Sotiropoulos, I., Maeda, S., and Takashima, A. (2010). Aggregation of detergent-insoluble tau is involved in neuronal loss but not in synaptic loss. J. Biol. Chem. 285, 38692–38699.
Kimura, T., Yamashita, S., Fukuda, T., Park, J. M., Murayama, M., Mizoroki, T., Yoshiike, Y., Sahara, N., and Takashima, A. (2007). Hyperphosphorylated tau in parahippocampal cortex impairs place learning in aged mice expressing wild-type human tau. EMBO J. 26, 5143–5152.
Kimura, T., Yamashita, S., Nakao, S., Park, J. M., Murayama, M., Mizoroki, T., Yoshiike, Y., Sahara, N., and Takashima, A. (2008). GSK-3β is required for memory reconsolidation in adult brain. PLoS ONE 3, e3540. doi: 10.1371/journal.pone.0003540
Klein, P. S., and Melton, D. A. (1996). A molecular mechanism for the effect of lithium on development. Proc. Natl. Acad. Sci. U.S.A. 93, 8455–8459.
Lee, J. L. C., Milton, A. L., and Everitt, B. J. (2006). Reconsolidation and extinction of conditioned fear: inhibition and potentiation. J. Neurosci. 26, 10051–10056.
Leroy, K., Ando, K., Heraud, C., Yilmaz, Z., Authelet, M., Boeynaems, J. M., Buee, L., De Decker, R., and Brion, J. P. (2010). Lithium treatment arrests the development of neurofibrillary tangles in mutant tau transgenic mice with advanced neurofibrillary pathology. J. Alzheimers Dis. 19, 705–719.
Lucas, J. J., Hernandez, F., Gomez-Ramos, P., Moran, M. A., Hen, R., and Avila, J. (2001). Decreased nuclear beta-catenin, tau hyperphosphorylation and neurodegeneration in GSK-3beta conditional transgenic mice. EMBO J. 20, 27–39.
MacAulay, K., Doble, B. W., Patel, S., Hansotia, T., Sinclair, E. M., Drucker, D. J., Nagy, A., and Woodgett, J. R. (2007). Glycogen synthase kinase 3alpha-specific regulation of murine hepatic glycogen metabolism. Cell Metab. 6, 329–337.
Maeda, S., Sahara, N., Saito, Y., Murayama, M., Yoshiike, Y., Kim, H., Miyasaka, T., Murayama, S., Ikai, A., and Takashima, A. (2007). Granular tau oligomers as intermediates of tau filaments. Biochemistry 46, 3856–3861.
Masliah, E., Ellisman, M., Carragher, B., Mallory, M., Young, S., Hansen, L., Deteresa, R., and Terry, R. D. (1992). Three-dimensional analysis of the relationship between synaptic pathology and neuropil threads in Alzheimer disease. J. Neuropathol. Exp. Neurol. 51, 404–414.
Morishima-Kawashima, M., Hasegawa, M., Takio, K., Suzuki, M., Yoshida, H., Titani, K., and Ihara, Y. (1995). Proline-directed and non-proline-directed phosphorylation of PHF-tau. J. Biol. Chem. 270, 823–829.
Morris, R. G. M., Inglis, J., Ainge, J. A., Olverman, H. J., Tulloch, J., Dudai, Y., and Kelly, P. A. T. (2006). Memory reconsolidation: sensitivity of spatial memory to inhibition of protein synthesis in dorsal hippocampus during encoding and retrieval. Neuron 50, 479–489.
Nader, K., Schafe, G. E., and Ledoux, J. E. (2000). The labile nature of consolidation theory. Nat. Rev. Neurosci. 1, 216–219.
Peineau, S., Taghibiglou, C., Bradley, C., Wong, T. P., Liu, L., Lu, J., Lo, E., Wu, D., Saule, E., Bouschet, T., Matthews, P., Isaac, J. T., Bortolotto, Z. A., Wang, Y. T., and Collingridge, G. L. (2007). LTP inhibits LTD in the hippocampus via regulation of GSK3beta. Neuron 53, 703–717.
Poissonnet, A., Henry-Feugeas, M. C., Drunat, O., Wolmark, Y., Delpierre, S., and Koskas, P. (2012). Evaluation of visual recognition memory for the early diagnosis of Alzheimer’s disease in patients over 75 years. Rev. Neurol. doi: 10.1016/j.neurol.2011.11.004.
Polter, A. M., and Li, X. (2011). Glycogen synthase kinase-3 is an intermediate modulator of serotonin neurotransmission. Front. Mol. Neurosci. 4:31. doi: 10.3389/fnmol. 2011.00031
Ramsden, M., Kotilinek, L., Forster, C., Paulson, J., Mcgowan, E., Santacruz, K., Guimaraes, A., Yue, M., Lewis, J., Carlson, G., Hutton, M., and Ashe, K. H. (2005). Age-dependent neurofibrillary tangle formation, neuron loss, and memory impairment in a mouse model of human tauopathy (P301L). J. Neurosci. 25, 10637–10647.
Riccio, D. C., Moody, E. W., and Millin, P. M. (2002). Reconsolidation reconsidered. Integr. Physiol. Behav. Sci. 37, 245–253.
Roberson, E. D., Scearce-Levie, K., Palop, J. J., Yan, F., Cheng, I. H., Wu, T., Gerstein, H., Yu, G. Q., and Mucke, L. (2007). Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science 316, 750–754.
Santacruz, K., Lewis, J., Spires, T., Paulson, J., Kotilinek, L., Ingelsson, M., Guimaraes, A., Deture, M., Ramsden, M., Mcgowan, E., Forster, C., Yue, M., Orne, J., Janus, C., Mariash, A., Kuskowski, M., Hyman, B., Hutton, M., and Ashe, K. H. (2005). Tau suppression in a neurodegenerative mouse model improves memory function. Science 309, 476–481.
Sato, S., Tatebayashi, Y., Akagi, T., Chui, D. H., Murayama, M., Miyasaka, T., Planel, E., Tanemura, K., Sun, X., Hashikawa, T., Yoshioka, K., Ishiguro, K., and Takashima, A. (2002). Aberrant tau phosphorylation by glycogen synthase kinase-3beta and JNK3 induces oligomeric tau fibrils in COS-7 cells. J. Biol. Chem. 277, 42060–42065.
Selkoe, D. J. (1986). Altered structural proteins in plaques and tangles: what do they tell us about the biology of Alzheimer’s disease? Neurobiol. Aging 7, 425–432.
Sereno, L., Coma, M., Rodriguez, M., Sanchez-Ferrer, P., Sanchez, M. B., Gich, I., Agullo, J. M., Perez, M., Avila, J., Guardia-Laguarta, C., Clarimon, J., Lleo, A., and Gomez-Isla, T. (2009). A novel GSK-3beta inhibitor reduces Alzheimer’s pathology and rescues neuronal loss in vivo. Neurobiol. Dis. 35, 359–367.
Spires, T. L., Orne, J. D., Santacruz, K., Pitstick, R., Carlson, G. A., Ashe, K. H., and Hyman, B. T. (2006). Region-specific dissociation of neuronal loss and neurofibrillary pathology in a mouse model of tauopathy. Am. J. Pathol. 168, 1598–1607.
Takashima, A. (2008). Hyperphosphorylated tau is a cause of neuronal dysfunction in tauopathy. J. Alzheimers Dis. 14, 371–375.
Takashima, A., Noguchi, K., Michel, G., Mercken, M., Hoshi, M., Ishiguro, K., and Imahori, K. (1996). Exposure of rat hippocampal neurons to amyloid beta peptide (25–35) induces the inactivation of phosphatidyl inositol-3 kinase and the activation of tau protein kinase I/glycogen synthase kinase-3 beta. Neurosci. Lett. 203, 33–36.
Takashima, A., Noguchi, K., Sato, K., Hoshino, T., and Imahori, K. (1993). Tau protein kinase I is essential for amyloid beta-protein-induced neurotoxicity. Proc. Natl. Acad. Sci. U.S.A. 90, 7789–7793.
Tandon, A., Rogaeva, E., Mullan, M., and St George-Hyslop, P. H. (2000). Molecular genetics of Alzheimer’s disease: the role of beta-amyloid and the presenilins. Curr. Opin. Neurol. 13, 377–384.
Tsien, J. Z., Huerta, P. T., and Tonegawa, S. (1996). The essential role of hippocampal CA1 NMDA receptor-dependent synaptic plasticity in spatial memory. Cell 87, 1327–1338.
Walsh, D. M., Klyubin, I., Fadeeva, J. V., Cullen, W. K., Anwyl, R., Wolfe, M. S., Rowan, M. J., and Selkoe, D. J. (2002). Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416, 535–539.
Westerman, M. A., Cooper-Blacketer, D., Mariash, A., Kotilinek, L., Kawarabayashi, T., Younkin, L. H., Carlson, G. A., Younkin, S. G., and Ashe, K. H. (2002). The relationship between Abeta and memory in the Tg2576 mouse model of Alzheimer’s disease. J. Neurosci. 22, 1858–1867.
Woodgett, J. R. (1990). Molecular cloning and expression of glycogen synthase kinase-3/factor A. EMBO J. 8, 2431–2438.
Keywords: Alzheimer’s disease, aging, memory formation, memory impairment, tau
Citation: Takashima A (2012) GSK-3β and memory formation. Front. Mol. Neurosci. 5:47. doi: 10.3389/fnmol.2012. 00047
Received: 17 September 2011; Accepted: 22 March 2012;
Published online: 23 April 2012.
Edited by:
Jim Robert Woodgett, Mount Sinai Hospital, CanadaReviewed by:
Oksana Kaidanovich-Beilin, Samuel Lunenfeld Research Institute, CanadaKenichi Okamoto, Samuel Lunenfeld Research Institute of Mount Sinai Hospital, Canada
Copyright: © 2012 Takashima. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Akihiko Takashima, Department of Aging Neurobiology, Center for Development of Advanced Medicine for Dementia, National Center for Geriatrics and Gerontology, 35 Gengo Morioka, Obu-shi, Aichi 474-8511, Japan. e-mail:a2VubmV0aEBuY2dnLmdvLmpw