
 William S. Kean1
William S. Kean1

- 1Department of Neurology, Uniformed Services University of the Health Sciences, Bethesda, MD, USA
- 2Department of Anatomy, Physiology and Genetics, Uniformed Services University of the Health Sciences, Bethesda, MD, USA
Brain cells expend large amounts of energy sequestering calcium (Ca2+), while loss of Ca2+ compartmentalization leads to cell damage or death. Upon cell entry, glucose is converted to glucose-6-phosphate (G6P), a parent substrate to several metabolic major pathways, including glycolysis. In several tissues, G6P alters the ability of the endoplasmic reticulum (ER) to sequester Ca2+. This led to the hypothesis that G6P regulates Ca2+ accumulation by acting as an endogenous ligand for sarco-endoplasmic reticulum calcium ATPase (SERCA). Whole brain ER microsomes were pooled from adult male Sprague-Dawley rats. Using radio-isotopic assays, 45Ca2+ accumulation was quantified following incubation with increasing amounts of G6P, in the presence or absence of thapsigargin, a potent SERCA inhibitor. To qualitatively assess SERCA activity, the simultaneous release of inorganic phosphate (Pi) coupled with Ca2+ accumulation was quantified. Addition of G6P significantly and decreased Ca2+ accumulation in a dose-dependent fashion (1–10 mM). The reduction in Ca2+ accumulation was not significantly different that seen with addition of thapsigargin. Addition of glucose-1-phosphate or fructose-6-phosphate, or other glucose metabolic pathway intermediates, had no effect on Ca2+ accumulation. Further, the release of Pi was markedly decreased, indicating G6P-mediated SERCA inhibition as the responsible mechanism for reduced Ca2+ uptake. Simultaneous addition of thapsigargin and G6P did decrease inorganic phosphate in comparison to either treatment alone, which suggests that the two treatments have different mechanisms of action. Therefore, G6P may be a novel, endogenous regulator of SERCA activity. Additionally, pathological conditions observed during disease states that disrupt glucose homeostasis, may be attributable to Ca2+ dystasis caused by altered G6P regulation of SERCA activity.
Introduction
Cell function and survival relies on precisely regulated and distributed calcium (Ca2+) stores. To prevent unregulated Ca2+-dependent activity, cells sequester Ca2+ behind biomembranes in organellar compartments maintaining total cellular Ca2+ concentration in the low millimolar range. Most Ca2+ is sequestered within organelles, including the endoplasmic reticulum (ER), Golgi, mitochondria, and nucleus (Carafoli, 1987; Dong et al., 2006; Vandecaetsbeek et al., 2011), while resting cytosolic Ca2+ concentration ([Ca2+]cyt) is maintained in the low nanomolar range (Usachev et al., 1995).
Compartmentalizing Ca2+ requires constant cellular energy expenditure, and glucose energy metabolism requires Ca2+-dependent enzymes. These two processes are thus interdependent. Structurally, soluble glycolytic enzymes in erythrocytes co-localize with membrane-bound ATPase transporters, an organization that provides readily available ATP for transport activity (Schrier, 1966; Jeremy et al., 1992; Nakamura et al., 1993; Xu et al., 1995; Xu and Becker, 1998). In myocardium, glycolytic enzymes are anchored next to sarco-endoplasmic reticulum calcium ATPase (SERCA), allowing direct energy support for the transport of Ca2+ from the myoplasm into the ER lumen (Xu et al., 1995), while inhibition of glycolysis interrupts Ca2+ transport by decreasing ATP availability (Paul et al., 1989). In pancreatic beta cells, exposure to elevated extracellular glucose resulted in a transient increase followed by an decrease in [Ca2+]cyt, possibly due to influx of extracellular Ca2+ via capacitative entry followed by organellar uptake (Gylfe, 1988). However, the mechanism by which alterations in glucose metabolism result in altered Ca2+ homeostasis has not been fully determined, although current data suggest a more rapid, directly involved process than simply changes in energy supplies with downstream Ca2+ ramifications. Over the past two decades, research efforts have begun to focus on glucose-6-phosphate (G6P), a key metabolite of glucose, as the link between glucose metabolism and Ca2+ homeostasis.
Once glucose enters cells, it is rapidly phosphorylated to produce G6P, which “traps” the charged metabolite within the cell. Depending on tissue type and needs of the cell, this “hub” metabolite can then enter one of three major metabolic pathways: glycolysis, the pentose phosphate shunt or glycogenesis. One of the first investigations into the effect of G6P on Ca2+ accumulation used rat liver ER microsomes (Benedetti et al., 1985). In comparison to oxalate, addition of G6P, which can act as an anion, significantly reduced Ca2+ accumulation. Further experiments comparing G6P-mediated changes in Ca2+ accumulation in comparison to those observed by addition of oxalate alone were not performed, which makes interpretation of the results difficult. In contrast, Chen et al. (1998) reported increased SR/ER microsomal Ca2+ accumulation in whole rat brain, liver, and heart microsomal preparations after an extremely short time period of G6P incubation (Chen et al., 1998). No mechanism was asserted, and, unfortunately, the parameters of these fluorometric and light scattering assays produced somewhat ambiguous results and made speculation regarding possible mechanisms of G6P-mediated alterations in Ca2+ accumulation difficult. Therefore, while a thorough review of the current literature supports the hypothesis that G6P regulates Ca2+ accumulation in SR/ER, the precise nature of the effect remains unknown.
The volume of scientific literature devoted singularly to glucose energy metabolism and Ca2+ homeostasis under physiological and pathological conditions is impressive. Surprisingly, however, there have been few investigations into mechanisms by which these two interdependent physiological pathways are coupled or linked pathophysiologically. Attempts to mechanistically link glucose metabolism with Ca2+ homeostasis have led to promising advances that could open major avenues into the understanding of numerous diseases. Characterizing the exact mechanism by which G6P directly regulates Ca2+ homeostasis is fundamental in this regard.
Materials and Methods
Materials
All chemicals and reagents were laboratory grade, and purchased from Sigma-Aldrich chemical company (St. Louis, MO) unless otherwise stated. The vacuum manifold and 96-well filter plates were from Millipore (Bedford, MA).
Animals
Male Sprague-Dawley (SD) rats (Taconic) aged 12–16 weeks were used for all experiments. Animal procedures were approved by the Uniformed Services University of the Health Sciences Institution for Animal Care and Use Committee in accordance with international guidelines on the ethical use of animals. Rats received ad libitum access to standard rodent chow (Harlan Teklad, Frederick, MD) and maintained on a 12 h light/dark cycle until euthanized.
Preparation of Microsomes
Whole rat brain microsomes were prepared by differential density centrifugation as previously described (Verma et al., 1992). Briefly, rats were sedated by CO2 narcosis to absence of toe-pinch and decapitated. Brains were rapidly removed and immediately placed on a glass plate on ice while minced in ice-cold homogenization buffer (250 mM sucrose, 20 mM HEPES, pH 7.35) supplemented with 100 μM EDTA and a protease inhibitor cocktail (Sigma, St. Louis, MO) that contained 4-(2-aminoethyl) benzenesulfonyl fluoride (AEBSF), pepstatin A, E-64, bestatin, leupeptin, and aprotinin. Eight minced brains per microsomal preparation were pooled and homogenized in 10 volumes of ice-cold buffer using a motor-driven glass/Teflon homogenizer. The following was performed at 4°C. The chilled homogenate was centrifuged at 1000 × g for 10 min and the supernatant decanted and centrifuged at 10,000 × g for 15 min. The supernatant was retained for subsequent ultracentrifugation at 100,000 × g for 1 h. The supernatant was discarded and the pellet rinsed twice with ice-cold homogenization buffer to remove all trace of EDTA and protease inhibitors before resuspending in 2 mL of ice-cold homogenization buffer. Protein quantification was determined using the BCA method (Pierce Biotech, Rockford, IL) and the supernatant diluted to a final concentration of 2.5 mg/mL of protein and stored at -70°C in 1.2 mL aliquots until use.
45Ca2+ Uptake in Microsomes
The Ca2+ uptake assay is a protocol developed in our lab performed in a 96-well filter microplate format as described previously with some modifications (Verma et al., 1992; Watson et al., 2003). To help ensure reproducibility and validity, Ca2+ uptake was studied using buffers prepared in bulk by World Precision Instruments, Inc. (WPI, Sarasota, FL) that contained 20 mM HEPES (pH 7.35), 10 mM KCl, 5 mM NaN3, 3% (w/v) polyethylene glycol (mw 10,000), 25 mM K2 oxalate, and 50 μM CaCl2. Buffer Ca2+ was EGTA-chelated by WPI from 50 μM total to desired free concentrations. Unless otherwise stated, free Ca2+ concentration used was 300 nM. All reactions were initiated by the addition of ATP. Vacuum filtration halted reactions, and microplate filters were washed twice with ice-cold wash buffer (100 mM KCl, 10 mM K2 oxalate, 3% (w/v) PEG, 5 mM MgCl2, 10mM HEPES-KOH (pH 7.3), and 2 mM EGTA). Unless otherwise indicated, all reactions proceeded at 37°C for 60 min. Treatment with the Ca2+ ionophore A23187 was used to determine non-specific activity. Scintillation fluid was added to filter plate wells and amount of 45Ca2+ per well-measured using a 1450 Microbeta Trilux 2 (Perkin Elmer, San Jose, CA). Once collected, the data for each treatment was normalized to the control outcome.
45Ca2+ Uptake in Fresh Frozen Sections
Radiotracer 45Ca2+ uptake was also studied autoradiographically in fresh frozen brain sections. Thawed sections from −70°C were incubated in permeabilization buffer (10 mM HEPES, pH 7.3, 100 mM KCl, 5 mM NaN3, 3% (w/v) PEG, 1 mM DTT, and 50 μM digitonin) for 10 min at room temperature (21°C). The sections were then transferred to slide mailers containing 12 mL of radiotracer Ca2+ uptake buffer used in the microsomal Ca2+ assays without or with indicated treatments and incubated at 37°C for 60 min. Reactions were halted by placing slides in ice-cold wash buffer (100 mM KCl, 10 mM K2 oxalate, 3% (w/v) PEG, 5 mM MgCl2, 10 mM HEPES-KOH (pH 7.3), and 2 mM EGTA) for two separate 5 min rinses. Slides were then air-vacuum dried before placing in a cassette with beta-particle sensitive film until developed.
Ca2+ Pre-Load and Leak Assay
Microsomal uptake and release of Ca2+ were measured using radiotracer 45Ca2+ in uptake assays performed in 96-well filter plates as described above with modifications. Microsomes were pre-loaded with 45Ca2+ for 45 min, as described above. After 45 min of uptake, treatments were added including control (no treatment), and the assay allowed to continue another 15 min. Microsomal Ca2+ accumulation was subsequently measured by vacuum filtration at 60 min followed by scintillation counting. Once again, non-specific background was considered 45Ca2+ measured in the presence of 10 μM A23187. Calcium leakage was defined as 45Ca2+ accumulated in the presence of a SERCA inhibitor, such as thapsigargin (thapsigargin), subtracted from control uptake at 60 min. Since the addition of a SERCA inhibitor after 45 min of uptake would prevent virtually all further microsomal uptake by definition, any decrease in microsomal Ca2+ accumulation compared to control measured at 60 min would be considered leakage via passive or non-stimulated cycling resident ER receptor channels [1,3,4-Inositol-triphosphate receptors (IP3Rs), or ryanodine receptors (RYRs)].
Quantification of SERCA Hydrolytic Activity
In microsomes, SERCA activity is the primary mechanism for the hydrolysis of ATP to release Pi. Quantification of Pi release allows for an indirect measurement of SERCA activity in conditions which optimize Ca2+ uptake into the ER lumen. A previously described colorimetric assay (Ekman and Jager, 1993) adapted from the ammonium molybdate/malachite green method quantified Pi production by SERCA activity. Colorimetric reagent was prepared by mixing one volume of 10% (w/v) (NH4)6Mo7O24–4 H2O in 4M HCl with three volumes of 0.2% (w/v) malachite green in 4M HCl, followed by stirring (30 min), then gravity filtration (Whatmann filter paper, pore size six microns, Millipore, Billerica, MA) to remove large sediment containing (NH4)6Mo7O24 (ammonium molybdate, added to reagent in excess) and contaminants. A 1–12 nmol standard curve of potassium phosphate was generated to correlate color development with Pi concentration.
SERCA activity was determined by measuring Pi release in eight separate conditions: control, G6P, G1P, and F6P (all at 10 mM), in the absence and presence of thapsigargin (1 μM). For each experiment, all treatment conditions were simultaneously analyzed in 1.5 mL centrifuge tubes containing 440 uL pre-warmed (37°C) Ca2+ uptake buffer (see above) supplemented with 1 mM ouabain and 40 μL microsomal preparation (2.5 mg protein/mL) for 10 min at 37°C. The addition of 3 mM Mg2+ and 2 mM ATP prepared in Ca2+ uptake buffer (19 μL) initiated SERCA activity. Aliquots (25 μL) were collected and mixed with 775 μL ultrapure water and 200 μL colorimetric reagents. After 10 min incubation at room temperature, samples (200 μL) were then read (OD660) in a FLUOstar Omega microplate reader (BMG Labtech, Cary, NC). Since an ATP-regenerating system was not used, ATP was the limiting factor in SERCA-mediated Ca2+ uptake, and SERCA hydrolytic activity was determined after 6 min to prevent ATP reduction secondary to ATPase activity from affecting reproducibility. Each sample was measured in triplicate and mean activity calculated (nmol phosphate released/min/mg soluble protein). SERCA activity for each treatment was then calculated as the difference between measured activity in the absence and presence of thapsigargin.
Statistical Analyses
Collected data were analyzed via ANOVA using SigmaStat (Systat Software, Chicago, IL). When normality tests failed, a Kruskal Wallis ANOVA on Ranks was performed. Significance was based on two-tailed tests, with p < 0.05. For post hoc comparisons, Holm–Sidak Tests were used. Data analysis was performed at the conclusion of the project, after all results were collected. For all experiments, an “n” value of 5–7 was used, with each single “n” consisting of eight pooled brains.
Results
Glucose-6-Phosphate Significantly Reduced Endoplasmic Reticulum Calcium Accumulation
In whole rat brain ER microsomes, G6P (10 mM) significantly reduced Ca2+ accumulation when compared to control (0 mM), while two other cytosolic hexose phosphates did not affect Ca2+ accumulation (Figure 1). Increasing concentrations of G6P caused a dose-dependent decrease in Ca2+ accumulation in ER microsomes with the maximum experimental concentration of G6P (10 mM) reducing microsomal Ca2+ accumulation in comparison to control. At low concentrations (1.25 and 2.5 mM), reduction did not reach statistical significance. In contrast, two G6P 6-carbon metabolites from different pathways, glucose-1-phosphate (glycogenesis) and fructose-6-phosphate (glycolysis), did not affect Ca2+ accumulation (p > 0.05) at any concentration.
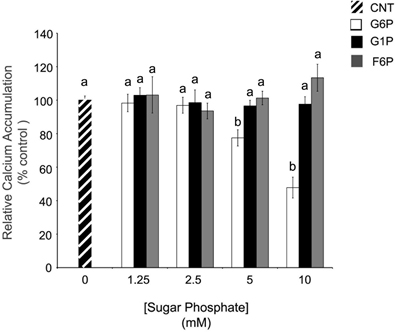
Figure 1. In comparison to other hexose phosphates, glucose-6-phosphate reduces the accumulation of 45Ca2+ in brain ER microsomes. Addition of glucose-6-phosphate (10 mM), but not glucose-1-phosphate (10 mM) or fructose-6-phosphate (10 mM), significantly reduced (p < 0.05) calcium accumulation. Data are means ± SEM for n = 5. Each microsomal preparation contained a minimum of eight whole brains pooled together. abMeans with differing superscripts are significantly different, p < 0.05, while means that share superscripts are not significantly different.
Glucose Metabolites did not Alter Endoplasmic Reticulum Calcium Accumulation
To verify the specificity of the response to G6P, the ER Ca2+ accumulation assay was repeated using nine other metabolites that are ultimately derived from glucose, or that are intimately involved in glucose metabolism (Table 1). Most interestingly, glucose alone had no effect (p > 0.05) on ER Ca2+ accumulation, nor did other glycolytic intermediates (fructose-1,6-bisphosphate and pyruvate). Intermediates of the TCA cycle (aspartate, glutamate, malate, and oxaloacetate) were likewise without effect (p > 0.05) on ER Ca2+ accumulation. Since G6P can also be used in the pentose phosphate shunt, phosphoglucolactone, and gluconolactone were also tested, with no effect (p > 0.05) on Ca2+ accumulation.
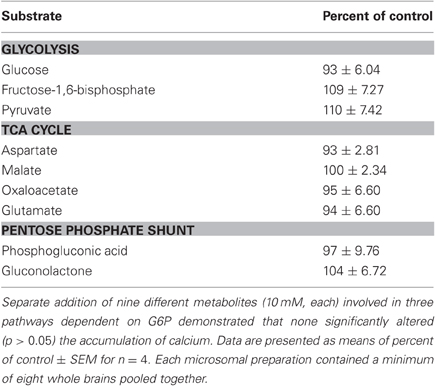
Table 1. After 60 min of incubation, intermediates in glycolysis, the TCA cycle, and the pentose phosphate shunt did not affect the accumulation of 45Ca2+.
Reduction in Calcium Accumulation Caused by Glucose-6-Phosphate is not Regionally Specific
Since ER microsomes were prepared from whole brain homogenates, brain regional variability could not be evaluated. Fresh frozen sections were analyzed autoradiographically to determine the anatomical regional distribution of G6P-sensitive pools of ER Ca2+ accumulation (Figure 2). In comparison to control, G6P and thapsigargin caused generalized reduction in Ca2+ accumulation in remarkably similar patterns with no apparent gray matter regional heterogeneity noted. Additionally, white matter areas also demonstrated reduced accumulation indicating treatment response is a fundamental mechanism ubiquitously expressed throughout all brain regions, including gray and white matter. Rat forebrain treated with low (1 mM) and high (10 mM) concentrations of G6P demonstrated a dose-dependent reduction in Ca2+ accumulation (Figure 2A), while thapsigargin also reduced Ca2+ accumulation (Figure 2B). Overall response appeared minimal at 1 mM, however, 10 mM G6P markedly and diffusely reduced Ca2+ accumulation compared to control. Focused examination of the hippocampus formation demonstrated noticeable decreased Ca2+ accumulation compared to control even at 1 mM (Figure 2C). At the larger, 10 mM concentration, G6P reduces Ca2+ accumulation to such an extent the hippocampal formation becomes unrecognizable.
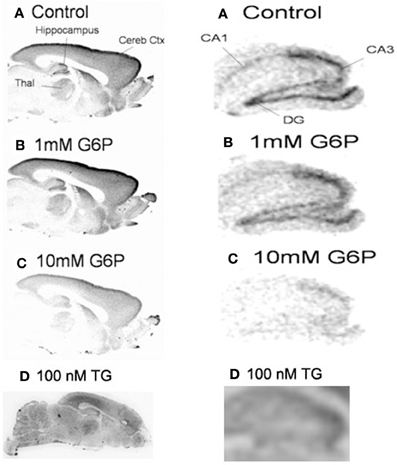
Figure 2. In fresh frozen rat brain sections, glucose-6-phosphate caused a generalized reduction in the accumulation of 45Ca2+. Treatment of fresh frozen brain sections with glucose-6-phosphate (10 mM) or thapsigargin (1 μM), revealed very similar pattern of generalized reduction in 45Ca2+ accumulation in gray and white matter. No regional specificity was observed, as the effect of both treatments appeared to be a diffuse response. Images are representative of n = 4.
Glucose-6-Phosphate Appears to Reduce Calcium Accumulation via Inhibition of SERCA
Since the pattern of accumulation in sections following G6P treatment appeared to mimic that seen following thapsigargin-induced inhibition of SERCA, further experiments examining G6P and thapsigargin inhibition were performed. Comparison of Ca2+ accumulation in microsomes was performed, following incubation with G6P, thapsigargin, or G6P plus thapsigargin (Figure 3). Both G6P and thapsigargin significantly reduced (p < 0.05) microsomal Ca2+ accumulation, while simultaneous addition of G6P and thapsigargin did not further decrease Ca2+ accumulation (p > 0.05).
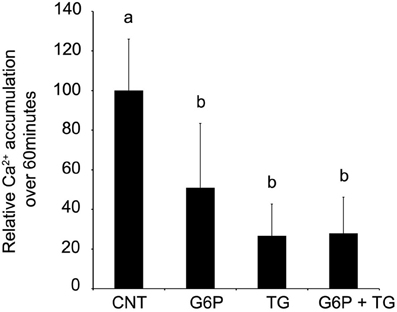
Figure 3. After 60 min of incubation, glucose-6-phosphate reduces the accumulation of 45Ca2+ in brain ER microsomes. Addition of glucose-6-phosphate (10 mM), thapsigargin (1 μM), or the two in combination significantly reduced (p < 0.05) calcium accumulation. Individually, glucose-6-phosphate and thapsigargin reduced calcium accumulation by approximately 50%. When the two treatments were combined, there was no additive effect. Data are means ± SEM for n = 5. Each microsomal preparation contained a minimum of eight whole brains pooled together. abMeans with differing superscripts are significantly different, p < 0.05, while means that share superscripts are not significantly different.
SERCA is an Mg2+, ATP-dependent enzyme, using energy yielded from ATP hydrolysis to transport, via conformational change, cytosolic Ca2+ against its electrochemical gradient into the ER lumen while releasing ADP and inorganic phosphate (Pi) into the cytosol (buffer, experimentally). Taking advantage of this enzymatic process allows for indirect quantification of SERCA activity by colorimetrically quantifying Pi release in ER microsomal preparations following experimental treatment. Release of Pi was quantified after incubation in four separate conditions: control, 1 μM thapsigargin, 10 mM G6P, or 1 μM thapsigargin plus 10 mM G6P. Maximal Pi release, which is indicative of maximal SERCA activity and hence the largest Ca2+ accumulation, was observed in the control condition (Figure 4). A net decrease in Pi release, indicative of diminished SERCA activity and thus reduced Ca2+ accumulation was observed following separate addition of G6P and thapsigargin. While the combination of G6P and thapsigargin also decreased net Pi release in comparison to control (p < 0.05), this effect was not additive or synergistic and was, in fact, no different (p > 0.05) from thapsigargin treatment alone.
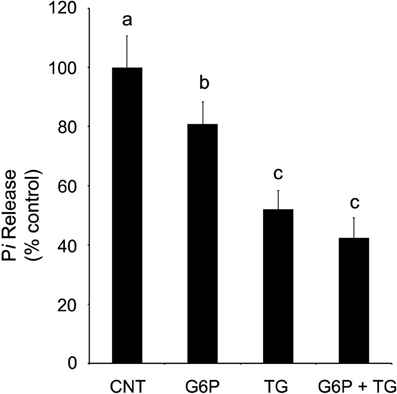
Figure 4. Glucose-6-phosphate reduces SERCA activity, as quantified by the release of inorganic phosphate from rat brain ER microsomes, in comparison to control. Glucose-6-phosphate significantly reduced (p < 0.05) the release of inorganic phosphate from brain ER microsome preparations after incubation for 6 min. However, the decrease in SERCA activity was not as great as that observed with the addition of thapsigargin (1 μM), a known potent, irreversible SERCA inhibitor. Treatment by both glucose-6-phosphate and thapsigargin did not further reduce accumulation below that of thapsigargin alone. Data are means ± SEM for n = 7. abcMeans with differing superscripts are significantly different, p < 0.05, while means that share superscripts are not significantly different.
While previously it was demonstrated that G6P is the only glucose metabolite having a significant reducing effect on Ca2+ accumulation, the specificity of the inhibitory effect on SERCA activity was confirmed by comparison to treatment with G1P (10 mM) and F6P (10 mM). As before, SERCA activity was reduced (p < 0.05) following the addition of G6P, but neither G1P nor F1P had an effect (p > 0.05) on Pi release (Figure 5).
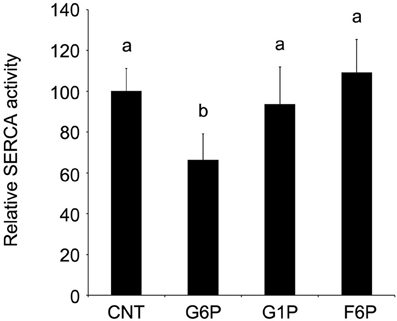
Figure 5. Glucose-6-phosphate reduces microsomal SERCA hydrolytic activity, as quantified by decreased release of inorganic phosphate in comparison to other hexose phosphates tested. Glucose-6-phosphate (10 mM) significantly reduced (p < 0.05) the release of inorganic phosphate from brain ER microsomes after 6 min incubation. Neither glucose-1-phosphate (10 mM) nor fructose-6-phosphate (10 mM) altered phosphate release (p > 0.05) in comparison to control. Data are means ± SEM for n = 7. abMeans with differing superscripts are significantly different, p < 0.05, while means that share superscripts are not significantly different.
Since the original Ca2+ accumulation was measured after 60 min of incubation, while Pi release was quantified at 6 min due to the necessary exclusion of ATPase regenerating systems (described in Methods), Ca2+ accumulation assays were repeated at 6 min to allow for a direct comparison (Figure 6). As with the 60 min assay, thapsigargin alone significantly reduced (p < 0.05) microsomal Ca2+ accumulation, which G6P showed a trend toward reduced (p < 0.10) Ca2+ accumulation. Interestingly, when quantified after only 6 min of incubation, simultaneous addition of G6P and thapsigargin additively reduced Ca2+ accumulation (p < 0.05) compared to individual treatment.
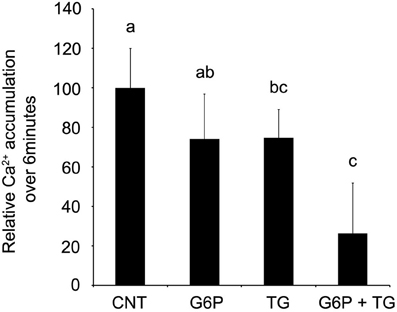
Figure 6. After 6 min of incubation, glucose-6-phosphate demonstrated a trend toward reduced accumulation of 45Ca2+ in brain ER microsomes. Addition of glucose-6-phosphate (10 mM) demonstrated a trend toward reduced accumulation of calcium (p < 0.10), while thapsigargin (1 μM) reduced ER Ca2+ accumulation (p < 0.05). When combined, the treatments additively reduced ER Ca2+ accumulation (p < 0.05) in comparison to control. Data are means ± SEM for n = 5. Each microsomal preparation contained a minimum of eight whole brains pooled together. abcMeans with differing superscripts are significantly different, p < 0.05, while means that share superscripts are not significantly different.
Glucose-6-Phosphate does not Cause Release of Calcium from ER Pools
To investigate the possibility that G6P could be stimulating release from ER microsomal pools, Ca2+ accumulation protocols were used to pre-load ER microsomes for 45 min with radio-tracer 45Ca2+ before application of one of three treatments (G6P, thapsigargin, A23187) for an additional 15 min (Figure 7). Net accumulation at 60 min was then compared between treatments. Neither G6P nor thapsigargin, a known potent irreversible SERCA inhibitor, caused significant Ca2+ release (p > 0.05), although both treatments markedly decreased the rate of Ca2+ accumulation between 45 and 60 min (Figure 7). In contrast, the Ca2+ ionophore A23187 caused a rapid, significant release of Ca2+ with a profile far different from that of G6P and thapsigargin, with net ER microsomal Ca2+ concentration at 60 min significantly reduced (p < 0.05) compared to that at 45 min.
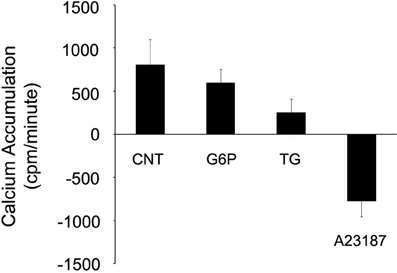
Figure 7. Neither glucose-6-phosphate nor thapsigargin cause release of 45Ca2+ from rat brain ER microsomes. After pre-loading ER microsomes with radiotracer 45Ca2+ uptake for 45 min, glucose-6-phosphate (10 mM), thapsigargin (1 μM), or the calcium ionophore A23187 (10 μM) treatment was added for 15 min. Only A23187 caused release of 45Ca2+ from the rat brain microsomes when measured by beta counter. Both glucose-6-phosphate and thapsigargin reduced the rate of accumulation, but did not cause release. Data are means ± SEM for n = 7.
Discussion
The key finding in this study is G6P dose-dependently reduces Ca2+ accumulation in rat brain ER. Further, the mechanism of reduction is via inhibition of SERCA activity and not stimulation of classical or novel release channels. More specifically, G6P is an endogenous inhibitor of the thapsigargin-sensitive ER Ca2+ pool. These data suggest G6P, the first product in glucose metabolism, regulates Ca2+ homeostasis and directly links these two physiological processes. Further, this effect is unique to G6P, as neither glucose, which is an uncharged or neutral molecule, nor other phosphohexoses which are anionic compounds alter the ability of microsomes to sequester Ca2+.
This study is not the first to examine the effects of G6P on Ca2+ accumulation in isolated microsomes; however, it is the first to propose a mechanism. Previous studies provided ambiguous data regarding the effect of G6P on Ca2+ accumulation in microsomes isolated from rat kidney, liver, heart, brain, and permeabilized pancreatic islet cells (Benedetti et al., 1985; Wolf et al., 1986; Fulceri et al., 1990; Chen et al., 1998). A major difference between our experiments and those mentioned previously is the predominant presence of a permeable anion in the buffer, in addition to the anion G6P. The assay buffer used in these experiments included dipotassium oxalate (25 mM) to maximize the accumulation of 45Ca2+ in isolated microsomes. Sequestration of luminal free Ca2+ by oxalate eliminates negative feedback inhibition on SERCA activity (Wells and Abercrombie, 1998) and provides greater sensitivity for quantifying the effects of G6P (Benedetti et al., 1985). If G6P acted solely as an anion to complex with intra-luminal Ca2+ in the radiotracer uptake microsomal assay, then accumulation in ER lumens during incubation would be expected to increase compared to control (absence of G6P). However, 10 mM G6P causes dramatic reductions in microsomal Ca2+ accumulation (∼60% reduction compared to control in whole rat brain microsomes) even in the presence of a potent intraluminal Ca2+ complexing anion (oxalate). The advantage of using ER microsomal preparations, in combination with fresh frozen slices for comparison, is that the glycolytic enzymes and mitochondrial function have been removed or extinguished. Therefore, G6P cannot simply be used for the provision of energy to the SERCA, since the cellular machinery needed for ATP production is absent. Benedetti et al. (1985) reported that G6P significantly reduced Ca2+ accumulation in ER microsomes when compared to assay buffers containing oxalate (Benedetti et al., 1985). However, G6P increased intraluminal Ca2+ in the absence of buffering anions. Inclusion of buffering anions in Ca2+ accumulation assays, which mimics in vivo conditions, provide the necessary sensitivity to elucidate mechanism(s) by which G6P regulates Ca2+ uptake into the ER. Future work utilizing an in vivo model, with a full complement of anions, cations, enzymes, and transporters, is required to confirm the exact mechanism of G6P-mediated changes in ER Ca2+ homeostasis under physiological conditions.
In nearly every experiment conducted G6P and thapsigargin resulted in identical outcomes, notably in experiments designed to test the effects of G6P on SERCA. SERCA is a Ca2+ ATPase that transports Ca2+, while producing ADP and Pi from ATP. Thapsigargin reduces Ca2+ accumulation by irreversibly binding to the SERCA and reducing ATPase activity, thus limiting the production of ADP + Pi (Thastrup et al., 1990; Lytton et al., 1991; Caspersen and Treiman, 1995; Poulsen et al., 1995). Therefore, to investigate if G6P similarly inhibits SERCA activity, Pi release from microsomes was quantified following treatment with G6P and thapsigargin, individually and combined. As with other measures, G6P and thapsigargin had an individual effect (reduced Pi hydrolysis by inhibiting SERCA activity), but did not have an additive/synergistic effect. However, there was a minor difference in the amount of Pi released, as well as in the Ca2+ accumulation when measured at 6 and 60 min, the only experiments with this minor discrepancy between the two treatments. However, it should be noted that thapsigargin binds irreversibly to SERCA. In contrast, G6P, an endogenous and ubiquitous metabolite, would not be expected to bind SERCA in an irreversible fashion due to the negative bioenergetic consequences and tumorigenic potential. Quite likely, this difference in irreversible and reversible binding, between thapsigargin and G6P, respectively, explains these minor discrepancies. Comparisons of Ca2+ accumulation in fresh frozen brain sections following incubation with thapsigargin or G6P revealed an identical diffuse, regionally non-specific reduction in Ca2+ accumulation. In contrast, previously published data examining the effects of IP3 and RYR antagonists demonstrate a regionally specific, and complementary, pattern of reduced Ca2+ accumulation (Verma et al., 1990a,b, 1992).
These results, while convincing evidence that G6P and thapsigargin reduce ER Ca2+ accumulation via a similar mechanism(s) (i.e., inhibition of SERCA), were not conclusive. Therefore, a Ca2+ release assay was performed using thapsigargin and G6P. Microsomes were loaded for 45 min with Ca2+, then G6P and thapsigargin separately or together. A23187, an ionophore that causes rapid near emptying of Ca2+ from microsomes to equilibrium with the buffer was used to measure non-specific background. While the ionophore (A23187) caused rapid near total emptying of the microsomes, both G6P and thapsigargin appeared to decrease the rate of uptake and not causing a direct release of accumulated Ca2+. ER Ca2+ accumulation is a continuing balance between SERCA-mediated uptake and release via Ca2+ channels and leak pathways. By reducing SERCA activity without altering release mechanisms, G6P (and thapsigargin) lower the setpoint of ER Ca2+ content by reducing accumulation.
A demonstrable link between glucose metabolism and regulation of Ca2+ sequestration mechanism will influence not just understanding of basic cellular physiology, but may have major implications for the study of a variety of clinically relevant neuropathologies. For example, diabetes and hyperglycemia are well-documented to cause significantly worse outcome for patients who suffer traumatic brain injury or stroke. Further, many of the clinical manifestations of these neuropathologies are strongly associated with altered Ca2+ homeostasis. However, the underlying mechanism(s) of this relationship has not been determined. By offering a direct, mechanistic link between G6P and disruptions in Ca2+ accumulation commonly observed in brain injury, such as after stroke or traumatic brain injury, these results suggest new approaches to large scale, old problems with important clinical ramifications.
Conflict of Interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Disclaimer
The opinions expressed herein belong solely to the authors. They do not nor should they be interpreted as representative of or endorsed by the Uniformed Services University of the Health Sciences, US Army, US Navy, Department of Defense or any other agency of the federal government. Products mentioned by name within this study do not imply endorsement of these products nor condemnation of similar products not used.
References
Benedetti, A., Fulceri, R., and Comporti, M. (1985). Calcium sequestration activity in rat liver microsomes. Evidence for a cooperation of calcium transport with glucose-6-phosphatase. Biochim. Biophys. Acta. 816, 267.
Caspersen, C., and Treiman, M. (1995). Thapsigargin discriminates strongly between Ca(2+)-ATPase phosphorylated intermediates with different subcellular distributions in bovine adrenal chromaffin cells. FEBS Lett. 377, 31.
Chen, P. Y., Csutora, P., Veyna-Burke, N. A., and Marchase, R. B. (1998). Glucose-6-phosphate and Ca2+ sequestration are mutually enhanced in microsomes from liver, brain, and heart. Diabetes 47, 874.
Dong, Z., Saikumar, P., Weinberg, J. M., and Venkatachalam, M. A. (2006). Calcium in cell injury and death. Annu. Rev. Pathol. 1, 405.
Ekman, P., and Jager, O. (1993). Quantification of subnanomolar amounts of phosphate bound to seryl and threonyl residues in phosphoproteins using alkaline hydrolysis and malachite green. Anal. Biochem. 214, 138.
Fulceri, R., Romani, A., Pompella, A., and Benedetti, A. (1990). Glucose 6-phosphate stimulation of MgATP-dependent Ca2+ uptake by rat kidney microsomes. Biochim. Biophys. Acta 1022, 129.
Gylfe, E. (1988). Glucose-induced early changes in cytoplasmic calcium of pancreatic beta-cells studied with time-sharing dual-wavelength fluorometry. J. Biol. Chem. 263, 5044.
Jeremy, R. W., Koretsune, Y., Marban, E., and Becker, L. C. (1992). Relation between glycolysis and calcium homeostasis in postischemic myocardium. Circ. Res. 70, 1180.
Lytton, J., Westlin, M., and Hanley, M. R. (1991). Thapsigargin inhibits the sarcoplasmic or endoplasmic reticulum Ca-ATPase family of calcium pumps. J. Biol. Chem. 266, 17067.
Nakamura, K., Kusuoka, H., Ambrosio, G., and Becker, L. C. (1993). Glycolysis is necessary to preserve myocardial Ca2+ homeostasis during beta-adrenergic stimulation. Am. J. Physiol. 264 (3 Pt 2), H670.
Paul, R. J., Hardin, C. D., Raeymaekers, L., Wuytack, F., and Casteels, R. (1989). Preferential support of Ca2+ uptake in smooth muscle plasma membrane vesicles by an endogenous glycolytic cascade. FASEB J. 3, 2298.
Poulsen, J. C., Caspersen, C., Mathiasen, D., East, J. M., Tunwell, R. E., Lai, F. A., Maeda, N., Mikoshiba, K., and Treiman, M. (1995). Thapsigargin-sensitive Ca(2+)-ATPases account for Ca2+ uptake to inositol 1,4,5-trisphosphate-sensitive and caffeine-sensitive Ca2+ stores in adrenal chromaffin cells. Biochem. J. 307(Pt 3), 749.
Schrier, S. L. (1966). Organization of enzymes in human erythrocyte membranes. Am. J. Physiol. 210, 139.
Thastrup, O., Cullen, P. J., Drobak, B. K., Hanley, M. R., and Dawson, A. P. (1990). Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca(2+)-ATPase. Proc. Natl. Acad. Sci. U.S.A. 87, 2466.
Usachev, Y. M., Marchenko, S. M., and Sage, S. O. (1995). Cytosolic calcium concentration in resting and stimulated endothelium of excised intact rat aorta. J. Physiol. 489 (Pt 2), 309.
Vandecaetsbeek, I., Vangheluwe, P., Raeymaekers, L., Wuytack, F., and Vanoevelen, J. (2011). The Ca2+ pumps of the endoplasmic reticulum and Golgi apparatus. Cold Spring Harb. Perspect. Biol. 3.
Verma, A., Hirsch, D. J., Hanley, M. R., Thastrup, O., Christensen, S. B., and Snyder, S. H. (1990a). Inositol trisphosphate and thapsigargin discriminate endoplasmic reticulum stores of calcium in rat brain. Biochem. Biophys. Res. Commun. 172, 811.
Verma, A., Hirsch, D. J., and Snyder, S. H. (1992). Calcium pools mobilized by calcium or inositol 1,4,5-trisphosphate are differentially localized in rat heart and brain. Mol. Biol. Cell. 3, 621.
Verma, A., Ross, C. A., Verma, D., Supattapone, S., and Snyder, S. H. (1990b). Rat brain endoplasmic reticulum calcium pools are anatomically and functionally segregated. Cell Regul. 1, 781.
Watson, W. D., Facchina, S. L., Grimaldi, M., and Verma, A. (2003). Sarco-endoplasmic reticulum Ca2+ ATPase (SERCA) inhibitors identify a novel calcium pool in the central nervous system. J. Neurochem. 87, 30.
Wells, K. M., and Abercrombie, R. F. (1998). Luminal Ca2+ protects against thapsigargin inhibition in neuronal endoplasmic reticulum. J. Biol. Chem. 273, 5020.
Wolf, B. A., Colca, J. R., Comens, P. G., Turk, J., and McDaniel, M. L. (1986). Glucose 6-phosphate regulates Ca2+ steady state in endoplasmic reticulum of islets. A possible link in glucose-induced insulin secretion. J. Biol. Chem. 261, 16284.
Xu, K. Y., and Becker, L. C. (1998). Ultrastructural localization of glycolytic enzymes on sarcoplasmic reticulum vesticles. J. Histochem. Cytochem. 46, 419.
Keywords: glucose-6-phosphate, G6P, calcium, SERCA, microsome, endoplasmic reticulum, thapsigargin, brain
Citation: Cole JT, Kean WS, Pollard HB, Verma A and Watson WD (2012) Glucose-6-phosphate reduces calcium accumulation in rat brain endoplasmic reticulum. Front. Mol. Neurosci. 5:51. doi: 10.3389/fnmol.2012.00051
Received: 09 January 2012; Accepted: 01 April 2012;
Published online: 20 April 2012.
Edited by:
Hans-Georg Breitinger, The German University in Cairo, EgyptReviewed by:
Herman Moreno, Columbia University Medical School, USAJens Schwamborn, University Muenster, Germany
Copyright: © 2012 Cole, Kean, Pollard, Verma and Watson. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Jeffrey T. Cole, Department of Neurology, Uniformed Services University of the Health Sciences, B-3012, 4301 Jones Bridge Road, Bethesda, MD 20814-4712, USA. e-mail:SmVmZnJleS5jb2xlQHVzdWhzLmVkdQ==