


- 1 Henry Wellcome Laboratories for Integrative Neuroscience and Endocrinology, School of Clinical Sciences, Faculty of Medicine and Dentistry, University of Bristol, Bristol, UK
- 2 MRC Centre for Synaptic Plasticity, University of Bristol, Bristol, UK
Neuronal calcium sensors (NCS) readily bind calcium and undergo conformational changes enabling them to interact and regulate specific target molecules. These interactions lead to dynamic alterations in protein trafficking that significantly impact upon synaptic function. Emerging evidence suggests that NCS and alterations in Ca2+ mobilization modulate glutamate receptor trafficking, subsequently determining the expression of different forms of synaptic plasticity. In this review, we aim to discuss the functional relevance of NCS in protein trafficking and their emerging role in synaptic plasticity. Their significance within the concept of “translational neuroscience” will also be highlighted, by assessing their potential as key molecules in neurodegeneration.
Introduction
Ca2+ signaling plays an important role in diverse biological processes, ranging from gene expression to cellular development (Sheng et al., 1991; Means, 1994; Park et al., 2007). Cellular Ca2+ sources are abundant and include the mitochondria, endoplasmic reticulum, lysosome, and extracellular environment. Changes in Ca2+ mobilization from this array of “Ca2+ stores” serve as the primary factor in the regulation of Ca2+ sensors and the subsequent activity of various substrates (Rosen et al., 1994; Moldoveanu et al., 2002; Burgoyne, 2007). Accordingly, uncovering the mechanisms underlying the activation and function of various Ca2+ sensors is fundamental to developing our understanding of dynamic neuronal responses to Ca2+; controlling synaptic transmission, modulating neuronal excitability, and, the particular focus of this review, regulating synaptic plasticity (Berridge, 2000).
Although a few exceptional cases of Ca2+-independent forms of synaptic plasticity have been reported (Fitzjohn et al., 2001; Dickinson et al., 2009), it is widely accepted that the majority of synaptic long-term plasticity operates through Ca2+-dependent mechanisms. Tetanic high frequency stimulation of presynaptic regions in the hippocampus induces a rise in postsynaptic Ca2+, leading to long-term potentiation (LTP) (Malenka et al., 1986; Lisman, 1989). Conversely, low frequency stimulation induces a low-to-moderate rise in free intracellular Ca2+, producing long-term depression (LTD) (Mulkey et al., 1994). These different and specific effects suggest that Ca2+ is involved in the induction of LTP as well as LTD, and that the magnitudes of activity-dependent rises in free Ca2+ and Ca2+ mobilization from different sources determines the induction of LTP and LTD (Lisman, 1989; Artola and Singer, 1993; Cho et al., 2001).
Thought to be central to this functional dichotomy are Ca2+-regulated enzymes. For example, LTP-inducing Ca2+ rises are detected by calmodulin (CaM; Mulkey et al., 1993) and activate Ca2+/calmodulin-dependent kinases (CaMKs; Malenka et al., 1986), while LTD-inducing Ca2+ signals activate a calcineurin/inhibitor-1 phosphatase cascade (Mulkey et al., 1994). These Ca2+-sensitive molecules play a key role in neuronal function through the regulation of glutamate receptor trafficking and synaptic plasticity in various regions of the brain (Palmer et al., 2005; Burgoyne, 2007; Jo et al., 2008,2010). This is achieved either through direct interaction with cargo molecules, or through regulation of protein membrane trafficking (Palmer et al., 2005; Jo et al., 2008,2010).
Recently, neuronal calcium sensors (NCS) have been shown to interact with endocytic molecules involved in glutamate receptor trafficking Figures 1A,B; Palmer et al., 2005; Jo et al., 2008,2010). More specifically, these Ca2+ sensors interact with several downstream effectors involved in AMPAR trafficking, including ABP/GRIP (Chung et al., 2000), adaptor protein 2 (AP2; Lee et al., 2002; Palmer et al., 2005), the Arp2/3 complex (Rocca et al., 2008), and PSD-95 (Kim et al., 2007). Here we will discuss how NCS proteins serve to orchestrate LTD signaling, and what makes them unique to one another in their roles in synaptic plasticity.
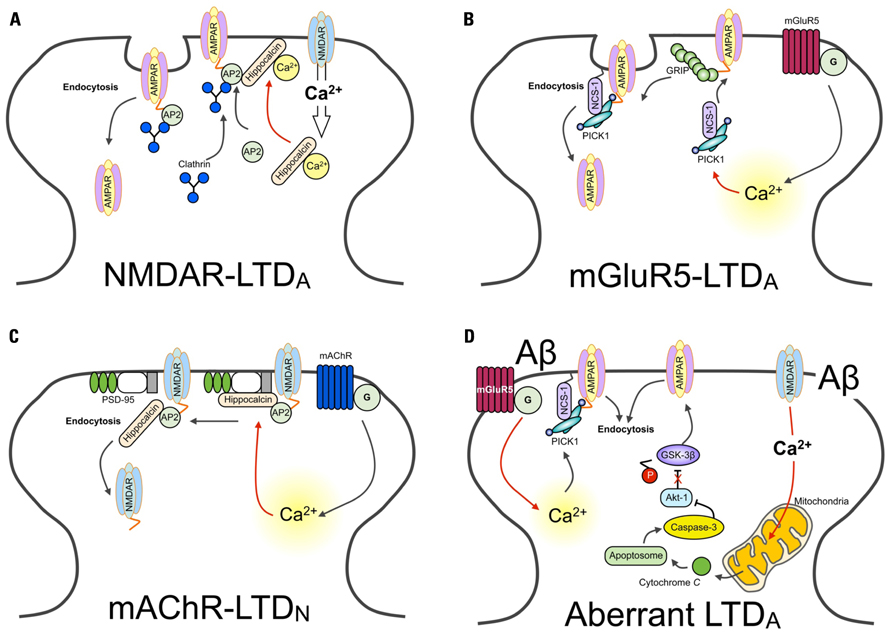
FIGURE 1. Ca2+ sensors and LTD. Schematic diagram showing different Ca2+ sensors regulate distinct forms of LTD. (A) The activation of NMDARs results in Ca2+ entry in the neuron. Ca2+ entry through NMDARs is sensed by hippocalcin, activating the myristoyl switch and stimulating the binding to β-adaptin of the AP2 complex, resulting in its translocation to the plasma membrane. AP2 is then able to bind with the GluA2 subunit of AMPARs, recruiting clathrin. Finally, hippocalcin is displaced by clathrin, and AMPARs are internalized. (B) AMPARs are stabilized at the synapse through interactions with GRIP. Activation of the G-protein coupled metabotropic glutamate receptor (mGluR), specifically the mGluR5-isoform containing receptor, induces the release of Ca2+ from intracellular stores. Increased levels of Ca2+ trigger the association of PICK1 with NCS-1. PICK1 interacts with PKC. NCS-1 localizes PICK1 in close proximity with AMPARs, facilitating the PKC-mediated phosphorylation of GluA2. This releases AMPARs from the GRIP interaction, mobilizing them for synaptic removal via endocytosis. (C) Hippocalcin in bound with the SH3 domain of PSD-95. This interacts with the NMDAR subunit GluN2B. These interactions prevent the binding of AP2, required for dynamin and clathrin-dependent endocytosis. Activation of the G-protein coupled mAChR induces the release of Ca2+ from intracellular stores. Increased levels of Ca2+are sensed by hippocalcin. As a consequence of this, PSD-95 disassociates from NMDARs. AP2 is now free to bind with NMDARs and initiate their endocytosis. (D) The hypothetical model of Ca2+-mediated Aβ toxicity. The aberrant activation of synaptic receptors leads to enhanced Ca2+ influx from both extracellular sites and intracellular Ca2+ stores. Ca2+ entry via NMDARs can impair mitochondrial function, leading to the release of cytochrome c and the formation of the apoptosome. This activates caspase-3, which cleaves and inhibits Akt-1. Given that Akt-1 ordinarily functions to phosphorylate GSK-3β and thereby downregulate the activity of GSK-3β, without this constitutive inhibition GSK-3β is now able to induce AMPAR endocytosis (Lopez et al., 2008; Li et al., 2009; Jo et al., 2011). Similarly, sustained release of Ca2+ from intracellular stores is likely to be sensed by, for example, PICK1. One likely consequence of this is the induction of LTD-signaling mechanisms and the endocytosis of AMPARs.
Neuronal Calcium Sensors
Neuronal calcium sensors proteins are a subgroup of proteins belonging to the EF-hand super family (Pongs et al., 1993; for detail of their structural and functional properties, we refer the reader to a number of excellent comprehensive reviews that cover these issues in great depth; Burgoyne, 2007; Ames et al., 2012; Burgoyne and Haynes, 2012). NCS proteins are widely expressed in neurons throughout the nervous system, and are able to regulate axonal outgrowth and synaptic transmission (Pongs et al., 1993; Olafsson et al., 1997). Upon Ca2+ binding, they exhibit the distinct property of being able to associate with the plasma membrane, via the post-translational addition of a myristoyl group (Ames et al., 1997). Such functional characteristics (among others to be discussed) render these proteins particularly adept at regulating synaptic receptor movement in response to neuronal activation, a fundamental prerequisite for the regulation of synaptic plasticity.
NCS and LTD
Activation of NMDAR and metabotropic glutamate receptor (mGluR) induces both NMDAR-dependent and mGluR-dependent LTD (NMDAR-LTD, mGluR-LTD respectively; see review Anwyl, 2006). Importantly, induction mechanisms of NMDAR- and mGluR-LTD are mediated by different Ca2+-dependent signaling pathways, involving different Ca2+ sensors (Jo et al., 2008; Figure 1B). These two distinct forms of LTD are conferred by different Ca2+ sensitivities and/or conformational changes of particular intracellular Ca2+ binding proteins. Accordingly, whilst NMDAR-LTD requires CaM and hippocalcin, mGluR-LTD involves NCS-1, protein kinase C (PKC), and IP3 (Jo et al., 2008). This suggests that distinct properties of Ca2+ sensors not only control the induction of LTD, but also maintain and regulate specificity of various signaling cascades. Given the physiological importance of different forms of Ca2+ sensors in LTD, the selective behavior of these proteins is undoubtedly significant in receptor trafficking, particularly receptor endocytosis.
NCS-1, PICK1, and AMPA Receptor Endocytosis
Neuronal calcium sensor-1, first described as a regulator of synaptic transmission at the neuromuscular junction in Drosophila and Xenopus (Pongs et al., 1993; Olafsson et al., 1997), is highly expressed throughout the brain (Paterlini et al., 2000). NCS-1 interacts with protein kinase interacting with C kinase 1 (PICK1) and regulates synaptic plasticity in the perirhinal cortex (Jo et al., 2008). NCS-1 binds directly to PICK1 via its Bin/Amphiphysin/Rvs (BAR) domain, in a Ca2+-dependent manner. The PICK1-BAR domain dimerizes, forming a concave arrangement. This unique conformation is thought to act as a “curvature sensor” (Peter et al., 2004), serving as a means of interaction between PICK1 and curved lipid membranes, like those of endocytic vesicles (He et al., 2011). The surface of the PICK1-BAR domain consists of positively charged regions, which mediate non-covalent interactions with negatively charged lipids. Accordingly, changes in membrane charges could dynamically regulate the membrane-localization of PICK1 (Jin et al., 2006), a possible crucial factor in synaptic plasticity.
PICK1 plays a key role in mediating the interaction between GluA2/3 of AMPARs and synaptic stabilizing structures, and accordingly can function to promote receptor endocytosis (Chung et al., 2000; Xia et al., 2000; Hanley and Henley, 2005). Again, the BAR domain plays a central part here; PICK1 binds with phosphoinositide lipids through the BAR domain, and this lipid/BAR interaction is essential for the synaptic targeting of PICK1 (Jin et al., 2006). Specifically, the BAR domain interacts with lipids of endocytic vesicles, mediating the internalization of PICK1 and associated synaptic receptors. Accordingly, it was shown that expression of a mutant BAR domain-containing PICK1 (K266, 268E) prevented the endocytosis of GluA2-containing AMPARs and enhanced AMPAR-mediated synaptic transmission (Jin et al., 2006). Interestingly, PICK1 itself is also a Ca2+ sensor (Hanley and Henley, 2005), and can regulate AMPAR endocytosis through actin depolymerization (Rocca et al., 2008). Thus, it is thought that the association of PICK1 with NCS-1 might serve to target PICK1 to the vicinity of AMPARs to initiate their removal from the synapse, providing a distinctive role for NCS-1 in LTD (Jo et al., 2008).
Hippocalcin and LTD
Emerging findings have outlined an important role for hippocalcin, a member of the visinin-like (VSNL) family proteins (VSNLs), in regulating dynamic neuronal synaptic change. It has previously been shown that NMDAR-mediated Ca2+ entry into neurons results in the hippocalcin-dependent internalization of AMPARs (Palmer et al., 2005). Here, it was shown that hippocalcin interacts with the AP2 adaptor complex subunit β2-adaptin Figure 1A). This, in turn, binds with the GluA2/3 AMPAR subunit – an interaction that is Ca2+-dependent – and promotes its clathrin-mediated endocytosis. In this study, the infusion of a dominant negative truncated form of hippocalcin (Hip2-72), an N-terminal region of the protein that does not include Ca2+ binding domains and is required for β2-adaptin interaction, inhibits the induction of LTD. Critically, this hippocalcin-mediated mechanism appears to be specific for LTD, as there was no effect found on the induction of LTP, though the same NMDAR-mediated Ca2+ influx is involved in LTP and LTD.
A more recent study has found evidence to suggest that under basal conditions, hippocalcin binds with the SH3 region of PSD-95, and that muscarinic acetylcholine receptor (mAChR)-induced intracellular Ca2+ release induces the translocation of hippocalcin to the plasma membrane Figure 1C). This leads to the dissociation of PSD-95 from NMDARs, allowing for the binding of AP2 to NMDARs to result in their endocytosis (Jo et al., 2010). Therefore, given the associate relationship between hippocalcin and the endocytosis of AMPARs, it is likely that hippocalcin could discriminate and respond to two distinct forms of intracellular Ca2+ mobilization (i.e., NMDAR- and mAChR-mediated). It is clear, therefore, that NCS-1 and hippocalcin are central regulators of receptor trafficking, pivotal in the expression of physiological LTD. Here, these NCS proteins activate key LTD molecules to induce both AMPAR and NMDAR internalization. Further work is required, however, to fully characterize how the same Ca2+ sensor can detect two distinct Ca2+ mobilizations and induce distinct receptor trafficking.
Ca2+ Dysregulation and Neurodegeneration: Are Calcium Sensors the Key?
Dysregulation of Ca2+ is well documented in the “Ca2+ theory of neurodegenerative disease,” involving excitatory toxicity and mitochondria-mediated apoptosis (Khachaturian, 1987; Schneider et al., 2001). For example, changes in [Ca2+]i can induce a concomitant change in mitochondrial Ca2+ ([Ca2+]m), leading to an increase of reactive oxygen species (ROS) production and the release of cytochrome c (Jiang et al., 2001; Brustovetsky et al., 2003). Released cytochrome c binds apoptotic protease activating factor 1 (Apaf-1) and triggers the caspase cascade and cell death (Hengartner, 2000). Given the significance of disrupted Ca2+ homeostasis to enhanced oxidative stress and neuronal loss evident in neurodegenerative diseases, here we discuss how Ca2+ and Ca2+ sensor-mediated receptor trafficking may affect synaptic function during Alzheimer’s disease (AD).
Large bodies of evidence support that amyloid-beta peptide (Aβ) induces the dysregulation of Ca2+ homeostasis and leads to activation of pro-apoptotic signal cascades (Ekinci et al., 2000; Smith et al., 2005; Lopez et al., 2008). Surprisingly however, a role for Ca2+ sensors in this pathogenesis has not yet been unambiguously demonstrated. Aβ -induced [Ca2+]i rises have been shown to regulate calsenilin, a KChIP subfamily of NCS, and its binding with the pro-apoptotic C-terminus of presenilin-2 (PS2; Buxbaum et al., 1998; Jo et al., 2005). The calsenilin–PS2 association leads to an increase in apoptosis and APP production (Jo et al., 2005; Jang et al., 2011). Additionally, the Ca2+ sensor visinin-like protein (VILIP) has been shown to associate with amyloid plaques and its expression enhances phosphorylation of tau, an additional hallmark of AD brains (Schnurra et al., 2001). In contrast to this finding, however, expression of VILIP-1 was reduced in AD brains compared with age-matched brain samples (Braunewell et al., 2001). Together, such studies currently paint a somewhat undefined picture as the exact role of NCS in AD pathology. Nevertheless, these studies do indicate that the aberrant regulation of Ca2+ sensors could underlie the development of AD, and this concept certainly warrants future investigation.
Caspase has been implicated as a key LTD molecule in the hippocampus and is involved in Aβ-mediated synaptic dysfunction (Li et al., 2010; Jo et al., 2011). Recently, it has been revealed that synaptic impairment caused by Aβ is mediated by a caspase–Akt-1–GSK3β signal cascade (termed the CAG cascade; Li et al., 2010; Jo et al., 2011). Interestingly, Aβ induces aberrant synaptic plasticity, leads to the inhibition of LTP but facilitation of LTD, and causes AMPAR endocytosis (Kim et al., 2001; Walsh et al., 2002; Hsieh et al., 2006; Shankar et al., 2007,2008; Li et al., 2009). Thus, it is perhaps not surprising that the Aβ -mediated activation of the CAG cascade leads to the facilitation of LTD Figure 1D). As we have described in this review, NCS-1, hippocalcin, and PICK1 are key molecules in the signaling underlying the induction of LTD and AMPAR and NMDAR endocytosis (Hanley and Henley, 2005; Citri et al., 2010; Jo et al., 2010). It would therefore be of great interest to investigate whether NCS could be aberrantly regulated during AD pathology.
As suggested in Figure 1C, activation of mAChR regulates NMDAR trafficking through a hippocalcin and PSD-95-mediated mechanism. Given the importance of enhancing cholinergic transmission and downregulating NMDAR transmission – strategies used as clinically approved AD treatments (e.g., memantine) – the role played by this NCS in receptor trafficking could provide a potential therapeutic target for Aβ-mediated synaptic dysfunction.
Concluding Remarks
Ca2+ signals can be “detected (sensor)” and “translated (switch)” to effectors. “Sensing” and “switching” should be tightly controlled to maintain effective homeostatic regulation in neurons. Growing evidence supports the notion that NSC and PICK1 have a key role in the endocytosis of glutamate receptors, a major molecular mechanism of LTD at excitatory synapses. Interestingly, Aβ-mediated neurotoxicity has been linked with excessive intracellular Ca2+ and aberrant synaptic plasticity. Thus our assumption is that overactive AMPAR endocytosis (or excessive LTD) caused by hyperactive NCS is likely to be found in AD or Aβ-induced neurotoxicity models. Therefore, it is of great interest to examine how NCS are involved in neurotoxicity and synaptic dysfunction. What is evident is the fact that intracellular Ca2+ mobilization, which includes mitochondrial Ca2+ flux and Ca2+ sensing, is a fundamental process in both physiological and pathological states. Through this review, we have aimed to bring new insight into NCS and synaptic plasticity, and provide a potential translation to synaptic disease models.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by BBSRC, Wellcome Trust, MRC, and Alzheimer’s Research UK (to Kwangwook Cho).
References
Ames, J. B., Ishima, R., Tanaka, T., Gordon, J. I., Stryer, L., and Ikura, M. (1997). Molecular mechanics of calcium-myristoyl switches. Nature 389, 198–202.
Ames, J. B., Sunghyuk, L., and Ikura, M. (2012). Molecular structure and target recognition of neuronal calcium sensor proteins. Front. Mol. Neurosci. 5:10. doi: 10.3389/fnmol.2012.00010
Anwyl, R. (2006). Induction and expression mechanisms of postynaptic NMDA receptor-independent homosynaptic long-term depression. Prog. Neurobiol. 78, 17–37.
Artola, A., and Singer, W. (1993). Long-term depression of excitatory synaptic transmission and its relationship to long-term potentiation. Trends Neurosci. 16, 480–487.
Braunewell, K., Riederer, P., Spilker, C., Gundelfinger, E. D., Bogerts, B., and Bernstein, H. G. (2001). Abnormal localization of two neuronal calcium sensor proteins, visinin-like proteins (vilips)-1 and -3, in neocortical brain areas of Alzheimer disease patients. Dement. Geriatr. Cogn. Disord. 12, 110–116.
Brustovetsky, N., Dubinsky, J. M., Antonsson, B., and Jemmerson, R. (2003). Two pathways for tBID-induced cytochrome c release from rat brain mitochondria: BAK- versus BAX-dependence. J. Neurochem. 84, 196–207.
Burgoyne, R. D. (2007). Neuronal calcium sensor proteins: generating diversity in neuronal signalling. Nat. Rev. Neurosci. 8, 182–193.
Burgoyne, R. D., and Haynes, L. P. (2012). Understanding the physiological roles of the neuronal calcium sensor proteins. Mol. Brain 5, 2.
Buxbaum, J. D., Choi, E. K., Luo, Y., Lilliehook, C., Crowley, A. C., Merriam, D. E., and Wasco, W. (1998). Calsenilin: a calcium-binding protein that interacts with the presenilins and regulates the levels of a presenilin fragment. Nat. Med. 4, 1177–1181.
Cho, K., Aggleton, J. P., Brown, M. W., and Bashir, Z. I. (2001). An experimental test of the role of postsynaptic calcium levels in determining synaptic strength using perirhinal cortex of rat. J. Physiol. 532, 459–466.
Chung, H. J., Xia, J., Scannevin, R. H., Zhang, X., and Huganir, R. L. (2000). Phosphorylation of the AMPA receptor subunit GluR2 differentially regulates its interaction with PDZ domain-containing proteins. J. Neurosci. 20, 7258–7267.
Citri, A., Bhattacharyya, S., Ma, C., Morishita, W., Fang, S., Rizo, J., and Malenka, R. C. (2010). Calcium binding to PICK1 is essential for the intracellular retention of AMPA receptors underlying long-term depression. J. Neurosci. 30, 16437–16452.
Dickinson, B. A., Jo, J., Seok, H., Son, G. H., Whitcomb, D. J., Davies, C. H., Sheng, M., Collingridge, G. L., and Cho, K. (2009). A novel mechanism of hippocampal LTD involving muscarinic receptor-triggered interactions between AMPARs, GRIP and liprin-alpha. Mol. Brain 2, 18.
Ekinci, F. J., Linsley, M. D., and Shea, T. B. (2000). Beta-amyloid-induced calcium influx induces apoptosis in culture by oxidative stress rather than tau phosphorylation. Brain Res. Mol. Brain Res. 76, 389–395.
Fitzjohn, S. M., Palmer, M. J., May, J. E., Neeson, A., Morris, S. A., and Collingridge, G. L. (2001). A characterisation of long-term depression induced by metabotropic glutamate receptor activation in the rat hippocampus in vitro. J. Physiol. 537, 421–430.
Hanley, J. G., and Henley, J. M. (2005). PICK1 is a calcium-sensor for NMDA-induced AMPA receptor trafficking. Embo. J. 24, 3266–3278.
He, Y., Liwo, A., Weinstein, H., and Scheraga, H. A. (2011). PDZ binding to the BAR domain of PICK1 is elucidated by coarse-grained molecular dynamics. J. Mol. Biol. 405, 298–314.
Hsieh, H., Boehm, J., Sato, C., Iwatsubo, T., Tomita, T., Sisodia, S., and Malinow, R. (2006). AMPAR removal underlies Abeta-induced synaptic depression and dendritic spine loss. Neuron 52, 831–843.
Jang, C., Choi, J. K., Na, Y. J., Jang, B., Wasco, W., Buxbaum, J. D., Kim, Y. S., and Choi, E. K. (2011). Calsenilin regulates presenilin 1/gamma-secretase-mediated N-cadherin epsilon-cleavage and beta-catenin signaling. FASEB J. 25, 4174–4183.
Jiang, D., Sullivan, P. G., Sensi, S. L., Steward, O., and Weiss, J. H. (2001). Zn2 induces permeability transition pore opening and release of pro-apoptotic peptides from neuronal mitochondria. J. Biol. Chem. 276, 47524–47529.
Jin, W., Ge, W. P., Xu, J., Cao, M., Peng, L., Yung, W., Liao, D., Duan, S., Zhang, M., and Xia, J. (2006). Lipid binding regulates synaptic targeting of PICK1, AMPA receptor trafficking, and synaptic plasticity. J. Neurosci. 26, 2380–2390.
Jo, D. G., Jang, J., Kim, B. J., Lundkvist, J., and Jung, Y. K. (2005). Overexpression of calsenilin enhances gamma-secretase activity. Neurosci. Lett. 378, 59–64.
Jo, J., Heon, S., Kim, M. J., Son, G. H., Park, Y., Henley, J. M., Weiss, J. L., Sheng, M., Collingridge, G. L., and Cho, K. (2008). Metabotropic glutamate receptor-mediated LTD involves two interacting Ca2+ sensors, NCS-1 and PICK1. Neuron 60, 1095–1111.
Jo, J., Son, G. H., Winters, B. L., Kim, M. J., Whitcomb, D. J., Dickinson, B. A., Lee, Y. B., Futai, K., Amici, M., Sheng, M., Collingridge, G. L., and Cho, K. (2010). Muscarinic receptors induce LTD of NMDAR EPSCs via a mechanism involving hippocalcin, AP2 and PSD-95. Nat. Neurosci. 13, 1216–1224.
Jo, J., Whitcomb, D. J., Olsen, K. M., Kerrigan, T. L., Lo, S. C., Bru-Mercier, G., Dickinson, B., Scullion, S., Sheng, M., Collingridge, G., and Cho, K. (2011). Abeta(1–42) inhibition of LTP is mediated by a signaling pathway involving caspase-3, Akt1 and GSK-3beta. Nat. Neurosci. 14, 545–547.
Khachaturian, Z. S. (1987). Hypothesis on the regulation of cytosol calcium concentration and the aging brain. Neurobiol. Aging 8, 345–346.
Kim, J. H., Anwyl, R., Suh, Y. H., Djamgoz, M. B., and Rowan, M. J. (2001). Use-dependent effects of amyloidogenic fragments of (beta)-amyloid precursor protein on synaptic plasticity in rat hippocampus in vivo. J. Neurosci. 21, 1327–1333.
Kim, M. J., Futai, K., Jo, J., Hayashi, Y., Cho, K., and Sheng, M. (2007). Synaptic accumulation of PSD-95 and synaptic function regulated by phosphorylation of serine-295 of PSD-95. Neuron 56, 488–502.
Lee, S. H., Liu, L., Wang, Y. T., and Sheng, M. (2002). Clathrin adaptor AP2 and NSF interact with overlapping sites of GluR2 and play distinct roles in AMPA receptor trafficking and hippocampal LTD. Neuron 36, 661–674.
Li, S., Hong, S., Shepardson, N. E., Walsh, D. M., Shankar, G. M., and Selkoe, D. (2009). Soluble oligomers of amyloid Beta protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron 62, 788–801.
Li, Z., Jo, J., Jia, J. M., Lo, S. C., Whitcomb, D. J., Jiao, S., Cho, K., and Sheng, M. (2010). Caspase-3 activation via mitochondria is required for long-term depression and AMPA receptor internalization. Cell 141, 859–871.
Lisman, J. (1989). A mechanism for the Hebb and the anti-Hebb processes underlying learning and memory. Proc. Natl. Acad. Sci. U.S.A. 86, 9574–9578.
Lopez, J. R., Lyckman, A., Oddo, S., Laferla, F. M., Querfurth, H. W., and Shtifman, A. (2008). Increased intraneuronal resting Ca2+ in adult Alzheimer’s disease mice. J. Neurochem. 105, 262–271.
Malenka, R. C., Madison, D. V., and Nicoll, R. A. (1986). Potentiation of synaptic transmission in the hippocampus by phorbol esters. Nature 321, 175–177.
Moldoveanu, T., Hosfield, C. M., Lim, D., Elce, J. S., Jia, Z., and Davies, P. L. (2002). A Ca2+ switch aligns the active site of calpain. Cell 108, 649–660.
Mulkey, R. M., Endo, S., Shenolikar, S., and Malenka, R. C. (1994). Involvement of a calcineurin/inhibitor-1 phosphatase cascade in hippocampal long-term depression. Nature 369, 486–488.
Mulkey, R. M., Herron, C. E., and Malenka, R. C. (1993). An essential role for protein phosphatases in hippocampal long-term depression. Science 261, 1051–1055.
Olafsson, P., Soares, H. D., Herzog, K. H., Wang, T., Morgan, J. I., and Lu, B. (1997). The Ca2+ binding protein, frequenin is a nervous system-specific protein in mouse preferentially localized in neurites. Brain Res. Mol. Brain Res. 44, 73–82.
Palmer, C. L., Lim, W., Hastie, P. G., Toward, M., Korolchuk, V. I., Burbidge, S. A., Banting, G., Collingridge, G. L., Isaac, J. T., and Henley, J. M. (2005). Hippocalcin functions as a calcium sensor in hippocampal LTD. Neuron 47, 487–494.
Park, H., Varadi, A., Seok, H., Jo, J., Gilpin, H., Liew, C. G., Jung, S., Andrews, P. W., Molnar, E., and Cho, K. (2007). mGluR5 is involved in dendrite differentiation and excitatory synaptic transmission in NTERA2 human embryonic carcinoma cell-derived neurons. Neuropharmacology 52, 1403–1414.
Paterlini, M., Revilla, V., Grant, A. L., and Wisden, W. (2000). Expression of the neuronal calcium sensor protein family in the rat brain. Neuroscience 99, 205–216.
Peter, B. J., Kent, H. M., Mills, I. G., Vallis, Y., Butler, P. J., Evans, P. R., and McMahon, H. T. (2004). BAR domains as sensors of membrane curvature: the amphiphysin BAR structure. Science 303, 495–499.
Pongs, O., Lindemeier, J., Zhu, X. R., Theil, T., Engelkamp, D., Krah-Jentgens, I., Lambrecht, H. G., Koch, K. W., Schwemer, J., Rivosecchi, R., Mallart, A., Galceran, J., Canal, I., Barbas, J. A., and FerrÚs, A. (1993). Frequenin – a novel calcium-binding protein that modulates synaptic efficacy in the Drosophila nervous system. Neuron 11, 15–28.
Rocca, D. L., Martin, S., Jenkins, E. L., and Hanley, J. G. (2008). Inhibition of Arp2/3-mediated actin polymerization by PICK1 regulates neuronal morphology and AMPA receptor endocytosis. Nat. Cell Biol. 10, 259–271.
Rosen, L. B., Ginty, D. D., Weber, M. J., and Greenberg, M. E. (1994). Membrane depolarization and calcium influx stimulate MEK and MAP kinase via activation of Ras. Neuron 12, 1207–1221.
Schneider, I., Reverse, D., Dewachter, I., Ris, L., Caluwaerts, N., Kuiperi, C., Gilis, M., Geerts, H., Kretzschmar, H., Godaux, E., Moechars, D., Van Leuven, F., and Herms, J. (2001). Mutant presenilins disturb neuronal calcium homeostasis in the brain of transgenic mice, decreasing the threshold for excitotoxicity and facilitating long-term potentiation. J. Biol. Chem. 276, 11539–11544.
Schnurra, I., Bernstein, H. G., Riederer, P., and Braunewell, K. H. (2001). The neuronal calcium sensor protein VILIP-1 is associated with amyloid plaques and extracellular tangles in Alzheimer’s disease and promotes cell death and tau phosphorylation in vitro: a link between calcium sensors and Alzheimer’s disease? Neurobiol. Dis. 8, 900–909.
Shankar, G. M., Bloodgood, B. L., Townsend, M., Walsh, D. M., Selkoe, D. J., and Sabatini, B. L. (2007). Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J. Neurosci. 27, 2866–2875.
Shankar, G. M., Li, S., Mehta, T. H., Garcia-Munoz, A., Shepardson, N. E., Smith, I., Brett, F. M., Farrell, M. A., Rowan, M. J., Lemere, C. A., Regan, C. M., Walsh, D. M., Sabatini, B. L., and Selkoe, D. J. (2008). Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 14, 837–842.
Sheng, M., Thompson, M. A., and Greenberg, M. E. (1991). CREB: a Ca2-regulated transcription factor phosphorylated by calmodulin-dependent kinases. Science 252, 1427–1430.
Smith, I. F., Green, K. N., and LaFerla, F. M. (2005). Calcium dysregulation in Alzheimer’s disease: recent advances gained from genetically modified animals. Cell Calcium 38, 427–437.
Walsh, D. M., Klyubin, I., Fadeeva, J. V., Cullen, W. K., Anwyl, R., Wolfe, M. S., Rowan, M. J., and Selkoe, D. J. (2002). Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416, 535–539.
Keywords: neuronal calcium sensor, long-term synaptic plasticity, Alzheimer’s disease
Citation: Kerrigan TL, Whitcomb DJ, Regan PL and Cho K. (2012) The role of neuronal calcium sensors in balancing synaptic plasticity and synaptic dysfunction. Front. Mol. Neurosci. 5:57. doi:10.3389/fnmol.2012.00057
Received: 01 March 2012; Accepted: 07 April 2012;
Published online: 04 May 2012.
Edited by:
Michael R. Kreutz, Leibniz-Institute for Neurobiology, GermanyReviewed by:
Michael R. Kreutz, Leibniz-Institute for Neurobiology, GermanyJose R. Naranjo, Centro Nalcional de Biotecnologia/Consejo Superior de Investigaciones Cientificas, Spain
Copyright: © 2012 Kerrigan, Whitcomb, Regan and Cho. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Daniel J. Whitcomb and Kwangwook Cho, Henry Wellcome Laboratories for Integrative Neuroscience and Endocrinology, School of Clinical Sciences, Faculty of Medicine and Dentistry, University of Bristol, Whitson Street, Bristol BS1 3NY, Bristol, UK. e-mail:ZC5qLndoaXRjb21iQGJyaXN0b2wuYWMudWs=; a2VpLmNob0BicmlzdG9sLmFjLnVr