


- 1Centre for Cellular and Molecular Biology, CSIR, Hyderabad, India
- 2RG Neuroplasticity, Leibniz Institute for Neurobiology, Magdeburg, Germany
Dendritic spines are believed to be micro-compartments of Ca2+ regulation. In a recent study, it was suggested that the ubiquitous and evolutionarily conserved Ca2+ sensor, calmodulin (CaM), is the first to intercept Ca2+ entering the spine and might be responsible for the fast decay of Ca2+ transients in spines. Neuronal calcium sensor (NCS) and neuronal calcium-binding protein (nCaBP) families consist of Ca2+ sensors with largely unknown synaptic functions despite an increasing number of interaction partners. Particularly how these sensors operate in spines in the presence of CaM has not been discussed in detail before. The limited Ca2+ resources and the existence of common targets create a highly competitive environment where Ca2+ sensors compete with each other for Ca2+ and target binding. In this review, we take a simple numerical approach to put forth possible scenarios and their impact on signaling via Ca2+ sensors of the NCS and nCaBP families. We also discuss the ways in which spine geometry and properties of ion channels, their kinetics and distribution, alter the spatio-temporal aspects of Ca2+ transients in dendritic spines, whose interplay with Ca2+ sensors in turn influences the race for Ca2+.
Introduction
In the human brain, spinous synapses on pyramidal neurons are the most abundant synapse type in the cerebrum and Ca2+ signaling in spines has been extensively studied. A largely overlooked area of neuronal Ca2+ signaling, though, is the functional role of EF-hand Ca2+-binding proteins of the calmodulin (CaM) superfamily in dendritic spines. Traditionally, these proteins have been assigned to the neuronal calcium sensor (NCS) and neuronal calcium-binding protein (nCaBP) families, all of which are evolutionarily related to the ancestral CaM (Figure 1). Being particularly abundant in brain and retina, members of the NCS and nCaBP family have been implicated in a plethora of different cellular events (see Burgoyne, 2007; and Mikhaylova et al., 2011), although their exact synaptic function is largely unknown.
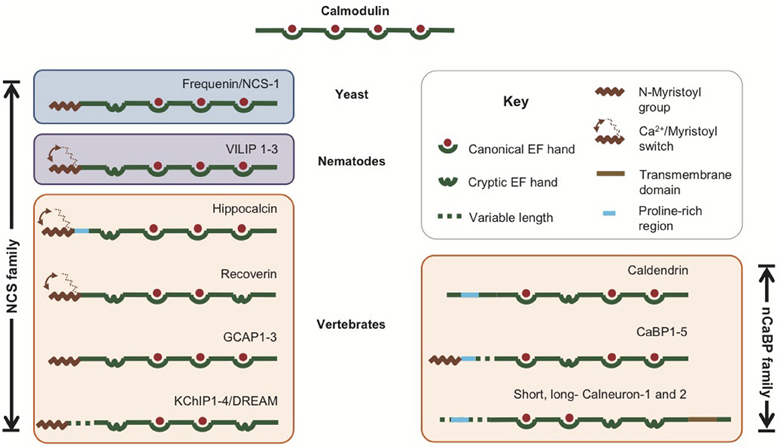
Figure 1. The NCS and nCaBP families of Ca2+ sensors. A cartoon representation of the different proteins of these families and the various motifs present in them are shown. The proteins have been placed in boxes based on the group of organisms where they first evolved. The ancestral CaM is also shown for comparison.
Evolution of NCS and nCaBP Families of Proteins
The NCS family of Ca2+ sensors (Figure 1) has been named after a group of proteins initially thought to be specifically expressed in neurons (De Castro et al., 1995). This group originated from the ancestral frequenin/NCS-1 and has diversified during evolution. Several reviews cover the topics of evolution and function of the NCS family of proteins (Burgoyne, 2007; Mikhaylova et al., 2011). Briefly, on the basis of sequence analysis, these proteins have been grouped into five classes, labeled in the order of their appearance during evolution (Burgoyne and Weiss, 2001; Burgoyne, 2007). Class A consists of NCS-1 or frequenin which appeared first in yeast. Visinin-like proteins or VILIPs evolved first in Caenorhabditis elegans and constitute the class B. With the evolution of the vertebrate eye, two new classes—C and D arose which comprise recoverin and guanylate cyclase activating proteins (GCAPs). Class E includes the voltage-gated K+ channel (Kv) interacting proteins or KChIPs and appeared first in insects. The mammalian genome encodes a single NCS-1, five VILIPS (hippocalcin, neurocalcin-δ, VILIPs1-3), a single recoverin, three GCAPs (GCAP1–3) and four KChIPs (KChIP 1–4) which exist in multiple isoforms. The various proteins of the NCS family show roughly <20% sequence identity with CaM. They possess four EF-hands out of which only two or three are capable of binding Ca2+. All of the members except KChIP2 and KChIP3 show an N-terminal myristoylation consensus sequence (Figure 1). This post-translational modification is important for their membrane localization. The Ca2+ binding is generally cooperative in most of the members and they show a much higher affinity for Ca2+ compared to CaM, while many of them also bind Mg2+ (Mikhaylova et al., 2011).
The nCaBP family of proteins (Seidenbecher et al., 1998; Haeseleer et al., 2000; Wu et al., 2001; Laube et al., 2002; Mikhaylova et al., 2006, 2009, 2011; McCue et al., 2010a,b) comprising caldendrin/CaBPs 1–5 and calneurons-1 and -2 arose much later during evolution and are found only in vertebrates (Figure 1). With respect to their EF-hands, they show a greater similarity to CaM than the NCS-1 family. It is, therefore, believed that the nCaBP family has evolved directly from the ancestral CaM (Seidenbecher et al., 1998; Haeseleer et al., 2000; Wu et al., 2001; Mikhaylova et al., 2006, 2011; McCue et al., 2010a). Like the NCS family, the nCaBPs too possess cryptic EF-hands and a few members are also N-myristoylated (CaBPs 1 and 2). A common distinctive feature is the presence of four extra amino acids in the linker region between the two EF-hand pairs in Caldendrin/CaBPs (Haeseleer et al., 2000). Besides this common feature, the family members show diversity in the N-terminal region, which, in case of caldendrin, CaBP1 and CaBP2, is due to alternate splicing (Haeseleer et al., 2000; Laube et al., 2002; Mikhaylova et al., 2011). Calneurons (also called CaBP7 and 8) are a subfamily that has evolved independently from caldendrin/CaBPs with a different EF-hand organization, much higher Ca2+-binding affinities and a carboxy-terminal transmembrane domain (Wu et al., 2001; Mikhaylova et al., 2006, 2009; McCue et al., 2009, 2011; Hradsky et al., 2011). They have been assigned to the nCaBP family largely based on sequence similarity (Mikhaylova et al., 2006, 2011; McCue et al., 2010a).
The Race for Ca2+: NCS and nCaBPs in Dendritic Spines
Dendritic spines are considered as microcompartments of Ca2+ signaling (Yuste and Denk, 1995; Yuste et al., 2000; Sabatini et al., 2001) with faster Ca2+ decay kinetics than their parent dendrites (Cornelisse et al., 2007). “Fast” Ca2+ buffers such as calbindin D28K are thought to be important for this increased rate of decay of Ca2+-transients in spines immediately after the closing of Ca2+ channels (Keller et al., 2008). It has also been assumed that these fast buffers are the first to intercept Ca2+ entering the spine. In a landmark study by Faas et al. (2011), it was found utilizing 1-(2-Nitro-4,5-dimethoxyphenyl)-N,N,N′,N′-tetrakis[(oxycarbonyl)methyl]-1,2 ethanediamine (DM-nitrophen)-Ca2+ uncaging experiments, that CaM binds Ca2+ at a faster rate than previously thought and the Ca2+-association to the N-terminal lobe turned out to be even faster than those of calbindin. Notably, calbindin D28K is absent in CA3 pyramidal cells and is expressed only at very low levels in a sub-population of CA1 pyramidal cells (Sloviter, 1989; Czarnecki et al., 2005; Jinno and Kosaka, 2010). Other important Ca2+ buffers, like parvalbumin and calretinin are also not expressed in CA1 and CA3 pyramidal neurons of the hippocampus (Sloviter, 1989; Baimbridge et al., 1992; Résibois and Rogers, 1992; Czarnecki et al., 2005). Thus, CaM, with its fast Ca2+-binding ability, high abundance, and ubiquitous expression, is most likely the principal buffer in these neurons (also discussed in Kubota et al., 2008). Its Ca2+-dependent targets are numerous and regulate diverse cellular events, making it a very important Ca2+ sensor as well. An important question thus arises—how do other NCS and nCaBP proteins function in the presence of CaM? With respect to the abundance and fast association rate of CaM and the steep and short Ca2+ transients in spines, would other sensors have a chance at all to compete for Ca2+ binding? In many cases, CaM and NCS/nCaBP proteins associate with the same target with different functional outcomes (Figure 2). However, it is still essentially unclear how they can compete with CaM and with each other for target interactions particularly in dendritic spines. In this opinion type review, we focus on these questions and provide some numerical reasoning which might be useful for future experiments. Keeping non-specialist readers in mind, we approach these questions in a step-wise manner and although some of the initial assumptions present an over-simplified view of this very complex and dynamic system, we hope that this approach will help to better appreciate the complexity of neuronal Ca2+ signaling and the race for Ca2+.
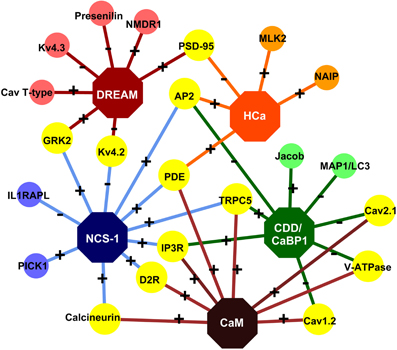
Figure 2. The NCS/nCaBP interactome. Some of the known targets of the NCS and nCaBP family members in the spine are shown. Specific interaction partners are color-coded according to the color of the sensor. Common interaction partners are shown in yellow. “+” and “−“ indicate Ca2+ -dependence and -independence of an interaction, respectively. AP2: Clathrin adaptor protein 2 [Haynes et al. (2006)]; Calcineurin [Schaad et al. (1996); Xia and Storm (2005)]; Cav1.2: Voltage-gated (L-type) Ca2+ channel [Zhou et al. (2004), (2005); Tippens and Lee (2007); Dick et al. (2008)]; Cav2.1: Voltage-gated (P/Q-type) Ca2+ channel [Lee et al. (2000), (2006); Few et al. (2005)]; CavT-type: Voltage-gated (T-type) Ca2+ channel [Anderson et al. (2010)]; CDD: Caldendrin; D2R: Dopamine D2 receptor [Bofill-Cardona et al. (2000); Kabbani et al. (2002); Woll et al. (2011)]; GRK2: G protein-coupled receptor kinase 2 [Kabbani et al. (2002); Ruiz-Gomez et al. (2007)]; HCa: Hippocalcin; IL1RAPL: Interleukin like-1 receptor accessory protein-like protein [Bahi et al. (2003)]; IP3R: Inositol 1,4,5-Trisphosphate Receptor [Hirota et al. (1999); Yang et al. (2002); Schlecker et al. (2006)]; Jacob [Dieterich et al. (2008)]; Kv4.2: Voltage-gated potassium channel 4.2 [An et al. (2000); Nakamura et al. (2001)]; Kv4.3: Voltage-gated potassium channel 4.3 [An et al. (2000)]; MAP1/LC3: microtubule-associated protein 1A/1B light chain 3 [Seidenbecher et al. (2004)]; MLK2: Mixed-Lineage Kinase 2 [Nagata et al. (1998)]; NAIP: Neuronal apoptosis inhibitory protein [Mercer et al. (2000); Lindholm et al. (2002)]; NMDR1: NMDA Receptor NR1 subunit [Zhang et al. (2010)]; PDE: cyclic nucleotide phosphodiesterase [Schaad et al. (1996); Haynes et al. (2006)]; PICK1: Protein Interacting with C-Kinase 1 [Jo et al. (2008)]; Presenilin [Buxbaum et al. (1998)]; PSD-95: postsynaptic density-95 protein [Jo et al. (2010); Wu et al. (2010)]; TRPC5: Transient receptor potential channel 5 [Kinoshita-Kawada et al. (2005); Ordaz et al. (2005); Hui et al. (2006)]; V-ATPase: Vacuolar Type H+-Adenosine 5-Triphosphatase [Haynes et al. (2006)]. The figure was created with the help of Cytoscape 2.8 [Cline et al. (2007)].
Abundance and Affinities of Ca2+ Sensors
Table 1A provides an estimate of the abundance and Ca2+-binding affinity of five important EF-hand Ca2+ sensors found in dendritic spines of hippocampal pyramidal neurons. The precise protein concentrations of these sensors in neuronal subcompartments such as the synapse are unknown. A detailed discussion on the concentration of CaM in spines is available in Faas et al., 2011. For most brain regions including cerebral cortex, hippocampus, caudate nucleus, striatum, and amygdala, an average CaM concentration of about 100 μM has been reported (Vargas and Guidotti, 1980; Kakiuchi et al., 1982; Klee and Vanaman, 1982; Kitajima et al., 1983; Sano and Kitajima, 1983; Teolato et al., 1983; Biber et al., 1984). The values reported for the hippocampus range from 74 to 156 μM (Kakiuchi et al., 1982; Klee and Vanaman, 1982; Biber et al., 1984). With the exception of hippocalcin, which is very abundant in hippocampus (Table 1), other sensors will most likely be expressed at much lower levels with an average cellular concentration that is estimated to range between 1 and 10 μM (Furuta et al., 1999; Burgoyne, 2007; Mikhaylova et al., 2011).
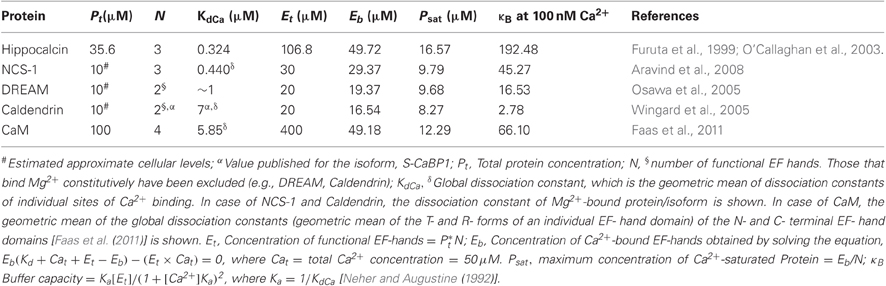
Table 1A. Concentration, affinity, and other parameters of selected neuronal Ca2+ sensors and CaM.
Following excessive synaptic activity, the induction of back-propagating dendritic action potentials (bAPs) may result in Ca2+ levels up to 50 μM within a dendritic spine (Faas et al., 2011). As a starting point, we therefore, considered a hypothetical situation in which Ca2+ sensors equilibrate with the 50 μM Ca2+ that enters the spine. This assumption is most likely not valid for all sensors as Ca2+ transients occurring during a single action potential are very brief and favor fast buffers (Markram et al., 1998). The actual time required for equilibration depends on the association and dissociation rates of Ca2+ binding (Markram et al., 1998) and this has to be correlated with the rate of Ca2+ influx. We discuss this aspect in detail in a later section. Thus, an equilibrium will possibly even not be reached during slower transients and high frequency dendritic spiking. In addition, since the structural unit that binds Ca2+ is a single EF-hand motif, we have initially treated the spine as a bag full of many EF-hands with different affinities for Ca2+, corresponding to the global affinity of the parent protein. This is also not a realistic assumption as EF-hand motifs pair up to form EF-hand domains and these domains, even within a single protein, show distinct affinities, binding and dissociation rates as well as cooperativity in Ca2+-binding (Grabarek, 2006; Gifford et al., 2007). Based on these simplistic assumptions, we calculated the parameters, Eb, which is the concentration of Ca2+-bound EF hands of a particular protein, and Psat, the maximum concentration of that protein that can possibly get saturated with Ca2+. These calculations were done for each protein separately and independent of the others. We have also calculated the buffer capacities (κB; Neher and Augustine, 1992) of these proteins at resting [Ca2+] of 100 nM in order to give a general idea about the steady state distribution of their Ca2+-bound forms during smaller Ca2+ transients. The Ca2+ buffer capacity of a spine, which is a function of the cumulative buffer capacities of its Ca2+-binding proteins, determines the peak amplitude and the decay rate of its Ca2+ transients. The kinetic profile of a Ca2+ transient in the spine with a higher buffer capacity has a smaller peak amplitude and a lower decay rate than the spine with lower buffer capacity. Based on the calculations in Table 1A, we ranked the sensors in different categories, shown in Table 1B. Since this review focuses mainly on the rising phase of Ca2+ transients, we have skipped the category of buffer capacity in Table 1B.

Table 1B. Ranking of various Ca2+ sensors under different categories (Absolute values calculated in Table 1A are shown in brackets).
Dendritic spine heads of hippocampal pyramidal neurons have an average diameter of 0.5 μm and a volume of 0.062 fL (Harris and Stevens, 1989), and contain Ca2+ buffers at 210 μM concentration (Cornelisse et al., 2007). If [Ca2+] increases up to 50 μM in the spine, this means that ∼2000 Ca2+ ions enter the spine at which point, >6000 Ca2+ binding protein molecules, roughly accounting for >20,000 EF hands, out of which ∼4000 belong to CaM alone, must compete with each other to bind Ca2+. Under the category of total abundance (Pt) in Table 1B, CaM appears to be a clear winner in the race for Ca2+ by a very large margin, followed by hippocalcin. However, owing to its much greater affinity to Ca2+, the concentration of Ca2+-saturated hippocalcin exceeds that of Ca2+-saturated CaM (given as parameter Psat). Provided Ca2+ saturation is essential for the activation of a sensor, in the competition for binding a low abundant Ca2+-dependent target having the same affinity for all the sensors, hippocalcin would out-compete CaM. Other sensors, which include NCS-1, DREAM, and caldendrin with a low Psat value, would have a much lesser chance to interact with this target under these conditions.
As stated earlier, the validity of the equilibrium assumption made above depends on the Ca2+ binding kinetics of the Ca2+ sensors (or their EF-hands) and the Ca2+ influx rate. Differences in the binding kinetics of Ca2+ sensors could result in a non-equilibrium concentration distribution of Ca2+-bound proteins leading to Psat values different from those in Table 1A. Obtaining this non-equilibrium concentration distribution necessitates a thorough understanding of the kinetics of all these sensors. Unfortunately, the kinetic data for most of these sensors is not available in the literature.
Enrichment and Sequestration of Ca2+ Sensors
While CaM is soluble and probably uniformly distributed in the cytosol (but see below), most NCS and nCaBP proteins are enriched in specific sub-cellular compartments, such as plasma membrane, golgi, endoplasmic reticulum (ER), and post-synaptic density (PSD; an electron-dense region with post-synaptic membrane thickening and enriched with cytoskeletal elements, scaffolding proteins and neurotransmitter receptors). The mechanism for membrane attachment is largely based on an N-terminal myristoyl group that provides a lipid anchor that interacts with certain phospholipids unique to the membranes of the organelle (O'Callaghan et al., 2005; Mikhaylova et al., 2011). While in case of hippocalcin, this myristoyl group is buried in the apo protein and gets exposed in a Ca2+-dependent manner—the so called Ca2+-myristoyl switch-, the myristoyl group in NCS-1 is probably constitutively exposed and membrane-bound. Caldendrin gets enriched at the PSD by a yet unknown mechanism (Seidenbecher et al., 1998; Laube et al., 2002).
The protein concentration (Pt) stated in Table 1A estimates the global concentration of the proteins in neurons. Could accumulation at a specific organelle be effective enough to increase the concentration of these proteins to levels greater than CaM? In general, a diffusible Ca2+ sensor with at least two canonical EF-hands and at a global concentration of 10 μM (which is thought to be roughly the expression level of major EF-hand Ca2+ sensors other than CaM in brain), would have to get enriched at synapses by at least a factor of 20 in order to match the abundance of CaM. Is this degree of accumulation achievable? The PSD has a thickness of ∼40 nm (Takashima et al., 2011) and an area of 0.08 μm2 (Arellano et al., 2007). The average volume of the PSD and the postsynaptic membrane is therefore ∼0.003 μm3, which is ∼20 times lesser than the spine head volume. Caldendrin, which gets enriched at the PSD in an activity-dependent manner (Smalla et al., 2003) probably meets this criterion in the PSD but not in the entire spine. Hippocalcin, which is already more abundant in the hippocampus than other EF-hand sensors, can get concentrated by dozens of times upon translocation to the membrane and as a consequence might even surpass the abundance of CaM at synaptic membranes (Dovgan et al., 2010).
In parallel to the above enrichment, sequestration of CaM might be another way to increase the relative abundance of NCS and nCaBPs as compared to CaM. RC3/neurogranin is a protein belonging to the IQ-motif family of CaM binding proteins with an estimated abundance of ∼60 μM (Huang et al., 2004) in dendritic spines of CA1 pyramidal neurons. It preferentially binds to apo-CaM with a high affinity and, therefore, sequesters it at low [Ca2+] (Gerendasy et al., 1994) and upon binding it reduces the affinity of CaM for Ca2+ (Gaertner et al., 2004). The affinity of RC3 and CaM binding reduces significantly at higher [Ca2+] and gets completely abolished upon phosphorylation of RC3 by protein kinase C (Gerendasy et al., 1994). Due to these properties, the interaction between RC3 and CaM at low resting [Ca2+] has important implications on the availability of free CaM for Ca2+ dependent as well as independent targets (Gerendasy et al., 1994). Ca2+/calmodulin-dependent protein kinase II (CaMKII) is one of the most abundant proteins in the PSD that might similarly sequester CaM. The concentration of its subunits ranges from 100 to 200 μM (Lisman and Zhabotinsky, 2001). It has been shown that autophosphorylation of CaMKII causes a 100-fold reduction in dissociation rate of CaM due to which CaM stays bound to phosphorylated CaMKII long after [Ca2+] returns to basal levels (Meyer et al., 1992). Since NCS and nCaBPS have not been reported to associate with CaMKII, it will be interesting to experimentally test if chelation of CaM by CaMKII could create open slots for target interactions of other Ca2+ sensors.
Ca2+-Independent Pre-Association of NCS and nCaBPs with Target Molecules
Another way to circumvent the problem of limited Ca2+ resources for target interactions is a Ca2+-independent pre-association with a binding partner. Caldendrin is the best example for this mode of operation. Regarding its EF-hand containing C-terminal domains it is the closest relative of CaM and shares this region with its shorter splice isoforms. Interestingly, caldendrin modulates the activity of Cav1.2 (L-type) Ca2+ channels via different molecular determinants than the shorter splice isoform, caldendrin-S1 (also called S-CaBP1), which is, however, barely expressed in brain (Laube et al., 2002; Zhou et al., 2005; Tippens and Lee, 2007), indicating that the structures of the isoforms may be very different. An important feature of caldendrin and S-CaBP1 is that they bind many of their targets, e.g., Cav1.2, Cav2.1 Ca2+-channels, LC3, V-ATPase, and Inositol 1,4,5-trisphosphate receptors (InsP(3)Rs) in a Ca2+-independent manner (Figure 2; Kasri et al., 2004; Seidenbecher et al., 2004; Zhou et al., 2004, 2005; Few et al., 2005; Haynes et al., 2006; Lee et al., 2006; Tippens and Lee, 2007) whereas Ca2+-binding increases the affinity of the association and triggers the actual signaling event. Although CaM is also a subunit of complexes with many enzymes and ion channels, such a pre-association would be very advantageous to convey a signal faster than other Ca2+ sensors, given that there is no other pre-association with another sensor at the target site and thereby could provide a molecular mechanism by which signals can be transduced to a specific target interaction irrespective of Ca2+-concentrations and CaM levels.
The idea of a signalosome-like protein preassembly that provides a clear advantage in terms of accessibility of a target site within Ca2+-nanodomains is not experimentally supported yet. A Ca2+-dependent increase in the binding affinity for a target within such a pre-associated signalosome could overrule all advantages of CaM in the race of Ca2+-binding in spines. This is clearly conceivable since Ca2+-affinities of EF-hand domains can increase in the target-bound form (Dukhanina et al., 1997; Peersen et al., 1997). In this respect, it is also worth mentioning that levels of macromolecular crowding impact the conformation of EF-hand domains and potentially their Ca2+-affinity (Wang et al., 2011). The impact of macromolecular complexes in spines could be substantial, given the high protein content and the compact structure of the PSD.
Variability and Inhomogeneity of Dendritic Spines and their Influence on the Race
Dendritic spines display spatio-temporal gradients in cytosolic Ca2+ concentration and Ca2+ amplitudes. This variability reflects the diversity of various factors including the kind, number, and distribution of Ca2+-channels and pumps, the mechanisms that regulate their activity, and the diffusability of Ca2+ and Ca2+-bound buffers. These factors can influence the race for Ca2+ and will be described in more detail below.
The Interplay Between Ca2+ Ion Channels and Ca2+ Sensors
The principal sources of Ca2+ in spines are voltage-gated Ca2+ channels (VGCCs), InsP(3)Rs, ryanodine receptors (RyRs), and Ca2+ permeable glutamate receptors, such as α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPAR) and N-methyl-D-aspartate receptors (NMDARs). VGCCs, NMDARs, and AMPARs are located on the plasma membrane, whereas, InsP(3)Rs and RyRs line the membranes of smooth ER invading the spine.
The opening of VGCCs leads to a fast rise in Ca2+ concentration with a rise time constant of 3.24 ms (Cornelisse et al., 2007) which decays slowly with a time constant of ∼15 ms (Sabatini et al., 2002). On the other hand, NMDARs open slowly (rise time of Ca2+ >100 ms) and allow for longer-lasting and much larger Ca2+ influxes. Kubota and Waxham (2010) elegantly described the critical impact of Ca2+ injection rates of VGCCs and NMDARs on Ca2+ dynamics. VGCCs have a high injection rate (∼1.4 ions/μs) but stay open for a very short time. Therefore, the opening of a VGCC leads to an accumulation of Ca2+ very close to the mouth of the channel before it diffuses away (Figure 3A). On the other hand, NMDARs have a much lower injection rate (∼0.07 ions/μs) and remain open for a longer time. Ca2+ that enters the spine following the opening of these receptors can diffuse to a considerable distance (∼140 nm from the channel) before the next ion enters (Kubota and Waxham, 2010). What properties should a Ca2+ sensor have, in order to respond to these diverse signals? The answer lies in the binding kinetics of the individual EF-hands.
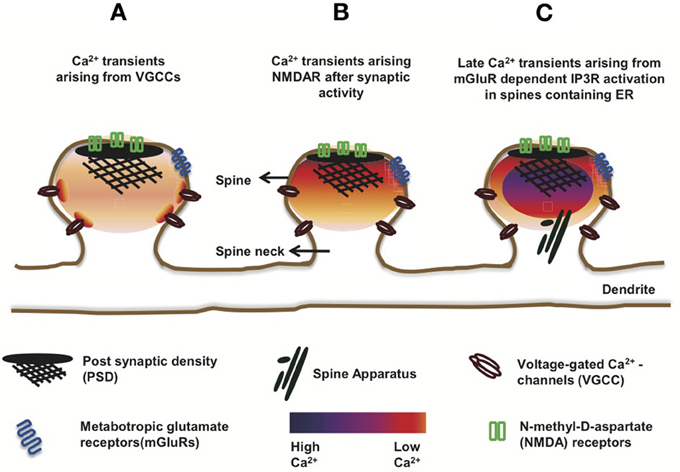
Figure 3. Ca2+ transients in dendritic spines. The schematic shows mushroom spines displaying variability in the amplitudes and spatial patterns of Ca2+ transients, depending on the type of ion channels involved. (A) Opening of VGCCs leads to a fast rise and decay of Ca2+-transients in the spine. Except near the mouth of the channel where [Ca2+] reaches a very high level, the distribution of the ion is largely uniform, reflecting the uniform distribution of VGCCs over the spine membrane. (B) Activated NMDARs allow slower, larger, and longer-lasting Ca2+-transients than the VGCCs. Unlike the latter, NMDARs are clustered at the PSD. Therefore, the Ca2+-transients arising from NMDARs show a stronger spatial gradient than the transients arising from open VGCCs. (C) In a subset of ER-containing spines, delayed Ca2+-transients with several folds higher amplitude than the NMDAR-mediated ones, have been observed and attributed to mGluR-dependent Ca2+-induced Ca2+ release (CICR) from IP3Rs, located on ER membranes.
In the previous paragraphs, we considered only the global Ca2+ affinity of a Ca2+-binding protein which indicates only an average of affinities of its individual EF-hands. The dissociation constant (inverse of affinity) is an equilibrium constant, given by the ratio between the OFF and ON rate constants of a reversible reaction.
Due to subtle variations in structure, individual EF-hands have different ON and OFF rates of Ca2+-binding and, therefore, are fine-tuned to different Ca2+-binding affinities (Grabarek, 2006; Gifford et al., 2007). To be able to respond to a Ca2+ signal, the binding kinetics of EF-hands must match the rate of Ca2+ entry. CaM has four EF hands organized into two domains. These EF-hands display cooperativity and allosterism in Ca2+-binding. An apo-EF hand is in the tensed or T-state, which binds the first ion to change to the relaxed or R-state. Although it was previously estimated that the N-terminal domain has a faster ON-rate of Ca2+ binding than the C-terminal domain (Kubota et al., 2007), an important finding by Faas et al. (2011) was that the ON-rate of Ca2+ binding of the N-terminal lobe of CaM [kon(T)=7.7 × 108 M−1s−1] was faster than important Ca2+ buffers like calbindin [kon(T)=7.5 × 107 M−1s−1] and calretinin [1.8 × 106 M−1s−1; Faas et al., 2007], making it the prime “fast” buffer. In spite of the fast binding rate, this domain has a much lower affinity (Kd = 12.7 μM) than the C-terminal domain (Kd = 2.7 μM) owing to its faster OFF-rate. When a VGCC opens, the instantaneous rise in Ca2+ concentration and the short duration for which the channel stays open would enable only those sensors with a fast binding capacity without much regard to their affinity since the Ca2+ levels attained are reasonably high. These features match perfectly with the fast binding N-terminal EF-hands of CaM. Along these lines, the N-terminal EF-hand domain could also be important for the role of CaM in long-term potentiation (LTP), which is associated with a steep rise in [Ca2+] to high amplitudes (Byrne et al., 2009). However, the function of CaM or its individual domains as Ca2+ sensors also depends on the kinetics of downstream target interaction. Only those targets are physiologically relevant whose association rate with CaM or one of its EF-hand domains is greater than the dissociation rate of Ca2+ from that domain. This is especially important in the case of the N-terminal domain of CaM, since it has a faster dissociation rate [koff(R) = 2.2 × 104 s−1] than the C-terminal domain [koff(R) = 6.5 s−1] (Faas et al., 2011). Unfortunately, although a number of Ca2+-dependent targets of CaM have been reported in the literature, the interaction kinetics of only a few of them is known.
The C-terminal lobe is unlikely to participate during the fast Ca2+ transients described above, due to its slow association rate. The possibility of a selective participation of the C-terminal domain during longer-lasting Ca2+ transients of long-term depression has been pointed out earlier (Byrne et al., 2009). The fast-binding of the N-terminal EF hands of CaM would be of no particular advantage in case of NMDAR-mediated Ca2+ influx. Their fast OFF-rates make CaM a low affinity sensor, rendering it less sensitive toward changes in Ca2+ concentration during NMDAR-mediated Ca2+ transients. The slow kinetics of NMDARs would not only engage the slower Ca2+ binding EF-hands/proteins, but might preferentially activate those Ca2+-binding proteins which are more sensitive to gradual changes in [Ca2+], such as the high affinity proteins of the NCS family, e.g., NCS-1 (KdCa = 440 nM; Aravind et al., 2008) and hippocalcin (KdCa = 324 nM; O'Callaghan et al., 2003), assuming that their Ca2+ association rate is not lower than the diffusion rate of Ca2+. Though their binding rates have not been determined, an advantage remains with these sensitive proteins even if they are slower in binding Ca2+ than CaM. It is hence not surprising that these two proteins play a crucial role in mGluR-dependent and NMDAR-dependent synaptic plasticity (Palmer et al., 2005; Jo et al., 2008, 2010). Importantly, it has also been shown that the Ca2+ influx through NMDARs, and not through VGCCs, is mainly responsible for the translocation of hippocalcin (Dovgan et al., 2010).
The VGCCs and NMDARs also differ in their distribution over the spine membrane. Based on a model by Keller et al. (2008), the VGCCs are uniformly distributed over the membrane. Therefore, their opening results in a uniform distribution of Ca2+ over the entire spine volume (Figure 3A). Although [Ca2+] can reach more than 10 μM at the mouth of an open VGCC (Simon and Llinás, 1985), these microdomains of high Ca2+ exist only within a few nanometer around the channel and dissipate within microseconds of channel closing (Sabatini et al., 2002). Intriguingly, the N- and C-terminal domains of CaM complexed to a VGCC (Cav1–2) might show distinct selectivity toward these local (nano domain of the complex) and global changes in [Ca2+] arising from the “host” VGCC (Tadross et al., 2008). The C-terminal domain might transduce local (nano domain) Ca2+ signals and the N-terminal domain global signals and is reportedly also capable of switching its selectivity between local (Cav1) and global(Cav2) changes in [Ca2+] (Dick et al., 2008; Tadross et al., 2008).
In contrast to VGCCs, NMDARs are clustered at the PSD. This leads to a longer lasting Ca2+ gradient extended across the spine during an excitatory post-synaptic potential (EPSP) (Keller et al., 2008; Figure 3B). This scenario might favor caldendrin, which is also enriched in the PSD. Although caldendrin and CaM bind Ca2+ with similar affinity, it is conceivable that the greater physical proximity of caldendrin might render it a strong competitor for CaM.
Interaction partners can influence the Ca2+ binding kinetics and affinity of Ca2+-binding proteins. RC3/Neurogranin interacts with apo-CaM (discussed in an earlier section) and increases the koff of the C-terminal lobe of CaM (Gaertner et al., 2004), thereby reducing its affinity. On the other hand, another CaM target- CaMKII decreases the koff of both N- and C-terminal lobes of CaM (Gaertner et al., 2004). The regulation of Ca2+-binding kinetics of CaM by its targets could explain how it is able to decode a variety of Ca2+ signals. Alteration of Ca2+- binding kinetics and affinity of CaM can not only influence Ca2+/CaM-dependent signaling pathways, but also regulate the amounts and duration for which free [Ca2+] is available for other Ca2+ sensors; an idea that is supported by mathematical models (Kubota et al., 2007, 2008).
Depolarization of spines is the key event that regulates the Ca2+ entry via VGCCs and NMDARs, which, as discussed above, is very influential in the race for Ca2+. During synaptic activity, AMPARs function as the major source of spine depolarization, necessary to activate VGCCs as well as to remove the Mg2+ blockage of NMDARs (Bloodgood et al., 2009; Holbro et al., 2010). The arrival of bAPs to a spine is another source of spine depolarization. Co-incident pre- and post-synaptic activity which involves glutamate release at the synapse preceding the arrival of bAPs in a millisecond time window, is responsible for the non-linear amplification of Ca2+ transients that underlie LTP (Helias et al., 2008; Holbro et al., 2010; Hao and Oertner, 2011). NMDARs, with their dependence on both glutamate binding and voltage-dependent unblocking have been considered to play the role of a co-incidence detector for the induction of LTP. However, it has also been suggested that NMDARs alone are not sufficient for co-incidence detection, but that it is the spine that acts as a co-incident detector of pre- and post-synaptic activity and the degree of depolarization of the whole spine is the key element for the induction of LTP (Hao and Oertner, 2011). The depolarizing currents from AMPARs sensitize the NMDARs and VGCCs, so that their I-V curves reach the steepest zone where even a slight further depolarization drastically increases Ca2+ influx into the spine (Holbro et al., 2010). A negative feedback loop provided by small conductance Ca2+-activated K+ (SK) channels controls the depolarization of spines. The Cav2.3 class of VGCCs activate SK channels, which shunt the synaptic current and dampen Ca2+ fluxes through NMDARs by promoting Mg2+ blockage of these receptors (Ngo-Anh et al., 2005; Bloodgood and Sabatini, 2007; Bloodgood et al., 2009). Spine head depolarization and the non-linear amplification of Ca2+ transients upon co-incident pre- and post-synaptic activity have been observed to be sharpest in spines that were well isolated from the dendrite (Holbro et al., 2010). This suggests the importance of spine neck dimensions in synaptic plasticity. The spine geometry also correlates well with the number of ion channels present on its membrane. Both the AMPAR and NMDAR-mediated currents increase with increase in the size of spine head. Intriguingly, the postsynaptic Ca2+ increase is lower in larger spines (Nimchinsky et al., 2004; Hayashi and Majewska, 2005; Noguchi et al., 2005). The importance of spine geometry in biochemical and electrical compartmentalization of spine heads is discussed in detail later.
The Role of Smooth Endoplasmic Reticulum (SER)
The smooth endoplasmic reticulum (SER), which is also called spine apparatus in spines, is a major internal store of Ca2+ and houses Ca2+ pumps such as sarco/ER Ca2+-ATPase (SERCA) and Ca2+- sensitive Ca2+ channels, namely, InsP(3)Rs and RyRs. While the SER is, undoubtedly, a major player in the regulation of cytosolic Ca2+ in a cell in general, its precise role in shaping the Ca2+ dynamics in dendritic spines in particular, is a matter of debate.
Emptage et al. (1999) found that although SER does not play any role in bAP-stimulated Ca2+ transients, they have a significant contribution in NMDAR-dependent Ca2+ dynamics. Upon depletion of the internal Ca2+ store by SERCA blockers and by application of RyR antagonists, a significant reduction in NMDAR-dependent Ca2+ transients was observed. This led the authors to conclude that during a single synaptic event, the Ca2+ current through activated NMDARs is too small in itself to be detected. What is actually detected is a much larger Ca2+ influx through RyRs that were triggered by the small Ca2+ currents coming from the NMDARs (Emptage et al., 1999). This phenomenon in which the SER releases the stored Ca2+ through its Ca2+-sensitive channels, is known as Ca2+-induced Ca2+ release or CICR (for review, see Rose and Konnerth, 2001).
Contradictory to this observation, other groups later found no significant role of CICR in either bAP- or NMDAR-dependent Ca2+ transients (Sabatini et al., 2002; Holbro et al., 2009). Both spines, that contained or lacked ER, showed no difference in NMDAR-dependent Ca2+ signals (Holbro et al., 2009); however, the authors observed delayed Ca2+ transients in a subset of ER-positive spines with more than five-fold larger amplitude arising in the order of 100 ms later than the NMDAR-dependent transient (Figure 3C). These large Ca2+ waves were abolished in the presence of group I metabotropic glutamate receptor (mGluR) or InsP(3)R blockers as well as upon depletion of Ca2+ stores, but persisted even in the presence of NMDAR blockers. Group I mGluRs are located in a perisynaptic zone surrounding the AMPARs and NMDARs and their role in mGluR-dependent long-term depression (mGluR-LTD) has been well studied (Lüscher and Huber, 2010). Holbro et al. (2009) observed that the induction of mGluR-dependent LTD was limited to ER-positive spines. Therefore, they suggested that the ER plays a role in mGluR-dependent LTD which involves CICR by activated IP3Rs. Notably, only about 20% of the CA1 dendritic spines contain SER (Spacek and Harris, 1997; Toresson and Grant, 2005; Holbro et al., 2009). One can, therefore, speculate that in ER containing spines, upon the arrival of large Ca2+ waves, the concentration of Ca2+ might rise to a level where all Ca2+ sensors can be completely saturated, probably obsoleting the race for Ca2+, albeit further intensifying the race for targets.
Spine Geometry and the Race for Ca2+
A spine is considered to be a micro-compartment of depolarization and Ca2+-dynamics distinct from the dendrite owing to the diffusional resistance of the spine neck. The idea that the spine neck acts as an electrical resistor is supported by the finding that (1) VGCCs present on the spines get activated by synaptic but not dendritic depolarization (Bloodgood et al., 2009); (2) voltage pulses arriving at a spine head from the soma get attenuated linearly with increasing length of the spine neck (Araya et al., 2006); (3) potential changes arising in a spine due to synaptic activity are largely restricted to its volume and their invasion into the parent dendrite are limited by the spine neck (Araya et al., 2006; Bloodgood et al., 2009). Therefore, an electrical resistance to movement of ions provided by the spine neck controls the activation of depolarization-dependent Ca2+ channels and thereby controls the amplitude of Ca2+ transients within the spine boundaries.
Besides electrical resistance, the spine neck also provides a diffusional barrier for ions and other molecules (Hayashi and Majewska, 2005; Biess et al., 2007; Grunditz et al., 2008; Schmidt and Eilers, 2009; Sabatini et al., 2002). The residing actin meshwork within the neck could, therefore, act as a molecular sieve for the entry and exit of NCS and nCaBPs from spines in a manner similar to CaMKII (Byrne et al., 2011). The spine neck diameter has been found to be correlated with spine head volume and it has been observed that Ca2+ dynamics varies with spine volume, in part due to the faster diffusion allowed by wider spine necks, and in part because of the relationship between the number of AMPA and NMDA receptors and the spine head size (discussed above; Hayashi and Majewska, 2005). Neuronal activity has been shown to regulate the diffusional properties of the spine neck (Bloodgood and Sabatini, 2005), the plasticity of which, can further control the spatio-temporal aspects of Ca2+ dynamics in spines (Segal, 2001; Grunditz et al., 2008). Future studies with super-resolution microscopy and live-imaging will resolve these issues and potentially also answer the question how dynamic the spine geometry actually is.
Conclusions and Future Directions
In the next coming years, it will be imperative to learn more about the biophysical features including their precise ion binding properties, their actual concentration in spines and the sequence of filling of EF-hand motifs of NCS and nCaBPs, to judge the physiological relevance of their synaptic protein interactions. How the presence of one Ca2+ sensor would influence the Ca2+-binding to another sensor or target is another interesting question to be addressed. Super resolution microscopy to identify nanodomains, fast spectroscopic methods to study in vitro protein kinetics and advanced modeling might also help to address at least some of the unresolved issues. Finally, a systematic analysis of the synaptic interactome of NCS and nCaBPs will help to appreciate their synaptic role. Compelling evidence for this synaptic role is, with the exception of hippocalcin, caldendrin, and NCS-1, still lacking and the conditions under which these proteins will eventually “meet” Ca2+ in the synapse still remain to be established.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grants from the Deutsche Forschungsgemeinschaft (DFG/Kr1879/3-1; SFB 779 TPB8; SFB 854 TP7), DIP grant and the MC-ITN NPlast (EU-FP7). Vijeta Raghuram thanks Indian Council of Medical Research, India for senior research fellowship (SRF). Yogendra Sharma thanks for a neuroscience grant from the DBT, India.
References
An, W. F., Bowlby, M. R., Betty, M., Cao, J., Ling, H. P., Mendoza, G., Hinson, J. W., Mattsson, K. I., Strassle, B. W., Trimmer, J. S., and Rhodes, K. J. (2000). Modulation of A-type potassium channels by a family of calcium sensors. Nature 403, 553–556.
Anderson, D., Mehaffey, W. H., Iftinca, M., Rehak, R., Engbers, J. D., Hameed, S., Zamponi, G. W., and Turner, R. W. (2010). Regulation of neuronal activity by Cav3-Kv4 channel signaling complexes. Nat. Neurosci. 13, 333–337.
Aravind, P., Chandra, K., Reddy, P. P., Jeromin, A., Chary, K. V., and Sharma, Y. (2008). Regulatory and structural EF-hand motifs of neuronal calcium sensor-1, Mg2+ modulates Ca2+ binding, Ca2+ -induced conformational changes, and equilibrium unfolding transitions. J. Mol. Biol. 376, 1100–1115.
Araya, R., Jiang, J., Eisenthal, K. B., and Yuste, R. (2006). The spine neck filters membrane potentials. Proc. Natl. Acad. Sci. U.S.A. 103, 17961–17966.
Arellano, J. I., Benavides-Piccione, R., Defelipe, J., and Yuste, R. (2007). Ultrastructure of dendritic spines: correlation between synaptic and spine morphologies. Front. Neurosci. 1:10. doi: 10.3389/neuro.01.1.1.010.2007
Bahi, N., Friocourt, G., Carrié, A., Graham, M. E., Weiss, J. L., Chafey, P., Fauchereau, F., Burgoyne, R. D., and Chelly, J. (2003). IL1 receptor accessory protein like, a protein involved in X-linked mental retardation, interacts with Neuronal Calcium Sensor-1 and regulates exocytosis. Hum. Mol. Genet. 12, 1415–1425.
Baimbridge, K. G., Celio, M. R., and Rogers, J. H. (1992). Calcium-binding proteins in the nervous system. Trends Neurosci. 15, 303–308.
Berridge, M. J., Lipp, P., and Bootman, M. D. (2000). The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell. Biol. 1, 11–21.
Biber, A., Schmid, G., and Hempel, K. (1984). Calmodulin content in specific brain areas. Exp. Brain Res. 56, 323–326.
Biess, A., Korkotian, E., and Holcman, D. (2007). Diffusion in a dendritic spine: the role of geometry. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 76, 021922.
Bloodgood, B. L., Giessel, A. J., and Sabatini, B. L. (2009). Biphasic synaptic Ca influx arising from compartmentalized electrical signals in dendritic spines. PLoS Biol. 7:e1000190. doi: 10.1371/journal.pbio.1000190
Bloodgood, B. L., and Sabatini, B. L. (2005). Neuronal activity regulates diffusion across the neck of dendritic spines. Science 310, 866–869.
Bloodgood, B. L., and Sabatini, B. L. (2007). Nonlinear regulation of unitary synaptic signals by CaV(2.3) voltage-sensitive calcium channels located in dendritic spines. Neuron 53, 249–260.
Bofill-Cardona, E., Kudlacek, O., Yang, Q., Ahorn, H., Freissmuth, M., and Nanoff, C. (2000). Binding of calmodulin to the D2-dopamine receptor reduces receptor signaling by arresting the G protein activation switch. J. Biol. Chem. 275, 32672–32680.
Burgoyne, R. D. (2007). Neuronal calcium sensor proteins: generating diversity in neuronal Ca2+ signalling. Nat. Rev. Neurosci. 8, 182–193.
Burgoyne, R. D., and Weiss, J. L. (2001). The neuronal calcium sensor family of Ca2+-binding proteins. Biochem. J. 353, 1–12.
Buxbaum, J. D., Choi, E. K., Luo, Y., Lilliehook, C., Crowley, A. C., Merriam, D. E., and Wasco, W. (1998). Calsenilin: a calcium-binding protein that interacts with the presenilins and regulates the levels of a presenilin fragment. Nat. Med. 4, 1177–1181.
Byrne, M. J., Putkey, J. A., Waxham, M. N., and Kubota, Y. (2009). Dissecting cooperative calmodulin binding to CaM kinase II: a detailed stochastic model. J. Comput. Neurosci. 27, 621–638.
Byrne, M. J., Waxham, M. N., and Kubota, Y. (2011). The impacts of geometry and binding on CaMKII diffusion and retention in dendritic spines. J. Comput. Neurosci. 31, 1–12.
Cline, M. S., Smoot, M., Cerami, E., Kuchinsky, A., Landys, N., Workman, C., Christmas, R., Avila-Campilo, I., Creech, M., Gross, B., Hanspers, K., Isserlin, R., Kelley, R., Killcoyne, S., Lotia, S., Maere, S., Morris, J., Ono, K., Pavlovic, V., Pico, A. R., Vailaya, A., Wang, P. L., Adler, A., Conklin, B. R., Hood, L., Kuiper, M., Sander, C., Schmulevich, I., Schwikowski, B., Warner, G. J., Ideker, T., and Bader, G. D. (2007). Integration of biological networks and gene expression data using Cytoscape. Nat. Protoc. 2, 2366–2382.
Cornelisse, L. N., van Elburg, R. A., Meredith, R. M., Yuste, R., and Mansvelder, H. D. (2007). High speed two-photon imaging of calcium dynamics in dendritic spines: consequences for spine calcium kinetics and buffer capacity. PLoS One 2:e1073. doi: 10.1371/journal.pone.0001073
Czarnecki, K., Haas, C. A., Bas Orth, C., Deller, T., and Frotscher, M. (2005). Postnatal development of synaptopodin expression in the rodent hippocampus. J. Comp. Neurol. 490, 133–144.
De Castro, E., Nef, S., Fiumelli, H., Lenz, S. E., Kawamura, S., and Nef, P. (1995). Regulation of rhodopsin phosphorylation by a family of neuronal calcium sensors. Biochem. Biophys. Res. Commun. 216, 133–140.
Dick, I. E., Tadross, M. R., Liang, H., Tay, L. H., Yang, W., and Yue, D. T. (2008). A modular switch for spatial Ca2+ selectivity in the calmodulin regulation of CaV channels. Nature 451, 830–834.
Dieterich, D. C., Karpova, A., Mikhaylova, M., Zdobnova, I., König, I., Landwehr, M., Kreutz, M., Smalla, K. H., Richter, K., Landgraf, P., Reissner, C., Boeckers, T. M., Zuschratter, W., Spilker, C., Seidenbecher, C. I., Garner, C. C., Gundelfinger, E. D., and Kreutz, M. R. (2008). Caldendrin-Jacob: a protein liaison that couples NMDA receptor signalling to the nucleus. PLoS Biol. 6:e34. doi: 10.1371/journal.pbio.0060034
Dovgan, A. V., Cherkas, V. P., Stepanyuk, A. R., Fitzgerald, D. J., Haynes, L. P., Tepikin, A. V., Burgoyne, R. D., and Belan, P. V. (2010). Decoding glutamate receptor activation by the Ca2+ sensor protein hippocalcin in rat hippocampal neurons. Eur. J. Neurosci. 32, 347–358.
Dukhanina, E. A., Dukhanin, A. S., Lomonosov, M. Y., Lukanidin, E. M., and Georgiev, G. P. (1997). Spectral studies on the calcium-binding properties of Mtsl protein and its interaction with target protein. FEBS Lett. 410, 403–406.
Emptage, N., Bliss, T. V., and Fine, A. (1999). Single synaptic events evoke NMDA receptor-mediated release of calcium from internal stores in hippocampal dendritic spines. Neuron 22, 115–124.
Faas, G. C., Raghavachari, S., Lisman, J. E., and Mody, I. (2011). Calmodulin as a direct detector of Ca2+ signals. Nat. Neurosci. 14, 301–304.
Faas, G. C., Schwaller, B., Vergara, J. L., and Mody, I. (2007). Resolving the fast kinetics of cooperative binding: Ca2+ buffering by calretinin. PLoS Biol. 5:e311. doi: 10.1371/journal.pbio.0050311
Few, A. P., Lautermilch, N. J., Westenbroek, R. E., Scheuer, T., and Catterall, W. A. (2005). Differential regulation of CaV2.1 channels by calcium-binding protein 1 and visinin-like protein-2 requires N-terminal myristoylation. J. Neurosci. 25, 7071–7080.
Furuta, Y., Kobayashi, M., Masaki, T., and Takamatsu, K. (1999). Age-related changes in expression of hippocalcin and NVP2 in rat brain. Neurochem. Res. 24, 651–658.
Gaertner, T. R., Putkey, J. A., and Waxham, M. N. (2004). RC3/Neurogranin and Ca2+/calmodulin dependent protein kinase II produce opposing effects on the affinity of calmodulin for calcium. J. Biol. Chem. 279, 39374–39382.
Gerendasy, D. D., Herron, S. R., Watson, J. B., and Sutcliffe, J. G. (1994). Mutational and biophysical studies suggest C3/neurogranin regulates calmodulin availability. J. Biol. Chem. 269, 22420–22426.
Gierke, P., Zhao, C., Brackmann, M., Linke, B., Heinemann, U., and Braunewell, K. H. (2004). Expression analysis of members of the neuronal calcium sensor protein family: combining bioinformatics and Western Blot analysis. Biochem. Biophys. Res. Commun. 323, 38–43.
Gifford, J. L., Walsh, M. P., and Vogel, H. J. (2007). Structures and metal-ion-binding properties of the Ca2+-binding helix-loop-helix EF-hand motifs. Biochem. J. 405, 199–221.
Grabarek, Z. (2006). Structural basis for diversity of the EF-hand calcium-binding proteins. J. Mol. Biol. 359, 509–525.
Grunditz, A., Holbro, N., Tian, L., Zuo, Y., and Oertner, T. G. (2008). Spine neck plasticity controls postsynaptic calcium signals through electrical compartmentalization. J. Neurosci. 28, 13457–13466.
Haeseleer, F., Sokal, I., Verlinde, C. L., Erdjument-Bromage, H., Tempst, P., Pronin, A. N., Benovic, J. L., Fariss, R. N., and Palczewski, K. (2000). Five members of a novel Ca2+-binding protein (CABP) subfamily with similarity to calmodulin. J. Biol. Chem. 275, 1247–1260.
Hao, J., and Oertner, T. G. (2011). Depolarization gates spine calcium transients and spike-timing-dependent potentiation. Curr. Opin. Neurobiol. doi: 10.1016/j.conb.2011.10.004. [Epub ahead of print].
Harris, K. M., and Stevens, J. K. (1989). Dendritic spines of CA1 pyramidal cells in the rat hippocampus: serial electron microscopy with reference to their biophysical characteristics. J. Neurosci. 9, 2982–2997.
Hayashi, Y., and Majewska, A. K. (2005). Dendritic spine geometry: functional implication and regulation. Neuron 46, 529–532.
Haynes, L. P., Fitzgerald, D. J., Wareing, B., O'Callaghan, D. W., Morgan, A., and Burgoyne, R. D. (2006). Analysis of the interacting partners of the neuronal calcium-binding proteins L-CaBP1, hippocalcin, NCS-1 and neurocalcin delta. Proteomics 6, 1822–1832.
Helias, M., Rotter, S., Gewaltig, M. O., and Diesmann, M. (2008). Structural plasticity controlled by calcium based correlation detection. Front. Comput. Neurosci. 2:7. doi: 10.3389/neuro.10.007.2008
Henzl, M. T., and Tanner, J. J. (2007). Solution structure of Ca2+-free rat beta-parvalbumin (oncomodulin). Protein Sci. 16, 1914–1926.
Hirota, J., Michikawa, T., Natsume, M., Furuichi, T., and Mikoshiba, K. (1999). Calmodulin inhibits inositol 1,4,5-trisphosphate-induced calcium release through the purified and reconstituted inositol 1,4,5-trisphosphate receptor type 1. FEBS Lett. 456, 322–326.
Holbro, N., Grunditz, A., and Oertner, T. G. (2009). Differential distribution of endoplasmic reticulum controls metabotropic signaling and plasticity at hippocampal synapses. Proc. Natl. Acad. Sci. U.S.A. 106, 15055–15060.
Holbro, N., Grunditz, A., Wiegert, J. S., and Oertner, T. G. (2010). AMPA receptors gate spine Ca(2+) transients and spike-timing-dependent potentiation. Proc. Natl. Acad. Sci. U.S.A. 107, 15975–15980.
Hradsky, J., Raghuram, V., Reddy, P. P., Navarro, G., Hupe, M., Casado, V., McCormick, P. J., Sharma, Y., Kreutz, M. R., and Mikhaylova, M. (2011). Post-translational membrane insertion of tail-anchored transmembrane EF-hand Ca2+ sensor calneurons requires the TRC40/Asna1 protein chaperone. J. Biol. Chem. 286, 36762–36776.
Huang, K. P., Huang, F. L., Jäger, T., Li, J., Reymann, K. G., and Balschun, D. (2004). Neurogranin/RC3 enhances long-term potentiation and learning by promoting calcium-mediated signaling. J. Neurosci. 24, 10660–10669.
Hui, H., McHugh, D., Hannan, M., Zeng, F., Xu, S. Z., Khan, S. H., Levenson, R., Beech, D. J., and Weiss, J. L. (2006). Calcium-sensing mechanism in TRPC5 channels contributing to retardation of neurite outgrowth. J. Physiol. 572, 165–172.
Jinno, S., and Kosaka, T. (2010). Stereological estimation of numerical densities of glutamatergic principal neurons in the mouse hippocampus. Hippocampus 20, 829–840.
Jo, J., Heon, S., Kim, M. J., Son, G. H., Park, Y., Henley, J. M., Weiss, J. L., Sheng, M., Collingridge, G. L., and Cho, K. (2008). Metabotropic glutamate receptor-mediated LTD involves two interacting Ca2+ sensors, NCS-1 and PICK1. Neuron 60, 1095–1111.
Jo, J., Son, G. H., Winters, B. L., Kim, M. J., Whitcomb, D. J., Dickinson, B. A., Lee, Y. B., Futai, K., Amici, M., Sheng, M., Collingridge, G. L., and Cho, K. (2010). Muscarinic receptors induce LTD of NMDAR EPSCs via a mechanism involving hippocalcin, AP2 and PSD-95. Nat. Neurosci. 13, 1216–1224.
Kabbani, N., Negyessy, L., Lin, R., Goldman-Rakic, P., and Levenson, R. (2002). Interaction with neuronal calcium sensor NCS-1 mediates desensitization of the D2 dopamine receptor. J. Neurosci. 22, 8476–8486.
Kakiuchi, S., Yasuda, S., Yamazaki, R., Teshima, Y., Kanda, K., Kakiuchi, R., and Sobue, K. (1982). Quantitative determinations of calmodulin in the supernatant and particulate fractions of mammalian tissues. J. Biochem. 92, 1041–1048.
Kapp-Barnea, Y., Melnikov, S., Shefler, I., Jeromin, A., and Sagi-Eisenberg, R. (2003). Neuronal calcium sensor-1 and phosphatidylinositol 4-kinase β regulate IgE receptor-triggered exocytosis in cultured mast cells. J. Immunol. 171, 5320–5327.
Kasri, N. N., Holmes, A. M., Bultynck, G., Parys, J. B., Bootman, M. D., Rietdorf, K., Missiaen, L., McDonald, F., De Smedt, H., Conway, S. J., Holmes, A. B., Berridge, M. J., and Roderick, H. L. (2004). Regulation of InsP3 receptor activity by neuronal Ca2+-binding proteins. EMBO J. 23, 312–321.
Keller, D. X., Franks, K. M., Bartol, T. M. Jr., and Sejnowski, T. J. (2008). Calmodulin activation by calcium transients in the postsynaptic density of dendritic spines. PLoS One 3:e2045. doi: 10.1371/journal.pone.0002045
Kinoshita-Kawada, M., Tang, J., Xiao, R., Kaneko, S., Foskett, J. K., and Zhu, M. X. (2005). Inhibition of TRPC5 channels by Ca2+-binding protein 1 in Xenopus oocytes. Pflugers Arch. 450, 345–354.
Kitajima, S., Seto-Ohshima, A., Sano, M., and Kato, K. (1983). Production of antibodies to calmodulin in rabbits and enzyme immunoassays for calmodulin and anti-calmodulin. J. Biochem. 94, 559–564.
Kubota, Y., Putkey, J. A., Shouval, H. Z., and Waxham, M. N. (2008). IQ-motif proteins influence intracellular free Ca2+ in hippocampal neurons through their interactions with calmodulin. J. Neurophysiol. 99, 264–276.
Kubota, Y., Putkey, J. A., and Waxham, M. N. (2007). Neurogranin controls the spatiotemporal pattern of postsynaptic Ca2+/CaM signaling. Biophys. J. 93, 3848–3859.
Kubota, Y., and Waxham, M. N. (2010). Lobe specific Ca2+-calmodulin nano-domain in neuronal spines: a single molecule level analysis. PLoS Comput. Biol. 6:e1000987. doi: 10.1371/journal.pcbi.1000987
Kuo, H. C., Cheng, C. F., Clark, R. B., Lin, J. J., Lin, J. L., Hoshijima, M., Nguyêñ-Trân, V. T., Gu, Y., Ikeda, Y., Chu, P. H., Ross, J., Giles, W. R., and Chien, K. R. (2001). A defect in the Kv channel-interacting protein 2 (KChIP2) gene leads to a complete loss of I(to) and confers susceptibility to ventricular tachycardia. Cell 107, 801–813.
Laube, G., Seidenbecher, C. I., Richter, K., Dieterich, D. C., Hoffmann, B., Landwehr, M., Smalla, K. H., Winter, C., Böckers, T. M., Wolf, G., Gundelfinger, E. D., and Kreutz, M. R. (2002). The neuron-specific Ca2+- binding protein caldendrin: gene structure, splice isoforms and expression in the rat central nervous system. Mol. Cell. Neurosci. 19, 459–475.
Lee, A., Scheuer, T., and Catterall, W. A. (2000). Ca2+/calmodulin dependent facilitation and inactivation of P/Q-type Ca2+ channels. J. Neurosci. 20, 6830–6838.
Lee, A., Westenbroek, R. E., Haeseleer, F., Palczewski, K., Scheuer, T., and Catterall, W. A. (2006). Differential modulation of Cav2.1 channels by calmodulin and Ca2+-binding protein 1. Nat. Neurosci. 5, 210–217.
Lindholm, D., Mercer, E. A., Yu, L. Y., Chen, Y., Kukkonen, J., Korhonen, L., and Arumäe, U. (2002). Neuronal apoptosis inhibitory protein: structural requirements for hippocalcin binding and effects on survival of NGF-dependent sympathetic neurons. Biochim. Biophys. Acta 1600, 138–147.
Lisman, J. E., and Zhabotinsky, A. M. (2001). A model of synaptic memory: a CaMKII/PP1 switch that potentiates transmission by organizing an AMPA receptor anchoring assembly. Neuron 31, 191–201.
Lüscher, C., and Huber, K. M. (2010). Group 1 mGluR-dependent synaptic long-term depression: mechanisms and implications for circuitry and disease. Neuron 65, 445–459.
Markram, H., Roth, A., and Helmchen, F. (1998). Competitive calcium binding: implications for dendritic calcium signaling. J. Comput. Neurosci. 5, 331–348.
McCue, H. V., Burgoyne, R. D., and Haynes, L. P. (2009). Membrane targeting of the EF-hand containing calcium-sensing proteins CaBP7 and CaBP8. Biochem. Biophys. Res. Commun. 380, 825–831.
McCue, H. V., Burgoyne, R. D., and Haynes, L. P. (2011). Determination of the membrane topology of the small EF-hand Ca2+-sensing proteins CaBP7 and CaBP8. PLoS One 6:e17853. doi: 10.1371/journal.pone.0017853
McCue, H. V., Haynes, L. P., and Burgoyne, R. D. (2010a). Bioinformatic analysis of CaBP/calneuron proteins reveals a family of highly conserved vertebrate Ca2+-binding proteins. BMC Res. Notes 3, 118.
McCue, H. V., Haynes, L. P., and Burgoyne, R. D. (2010b). The diversity of calcium sensor proteins in the regulation of neuronal function. Cold Spring Harb. Perspect. Biol. 2, a004085.
McFerran, B. W., Graham, M. E., and Burgoyne, R. D. (1998). Neuronal Ca2+ sensor 1, the mammalian homologue of frequenin, is expressed in chromaffin and PC12 cells and regulates neurosecretion from dense-core granules. J. Biol. Chem. 273, 22768–22772.
Mercer, E. A., Korhonen, L., Skoglösa, Y., Olsson, P. A., Kukkonen, J. P., and Lindholm, D. (2000). NAIP interacts with hippocalcin and protects neurons against calcium-induced cell death through caspase-3-dependent and -independent pathways. EMBO J. 19, 3597–3607.
Meyer, T., Hanson, P. I., Stryer, L., and Schulman, H. (1992). Calmodulin trapping by calcium-calmodulin-dependent protein kinase. Science 256, 1199–1202.
Mikhaylova, M., Hradsky, J., and Kreutz, M. R. (2011). Between promiscuity and specificity: novel roles of EF-hand calcium sensors in neuronal Ca2+ signalling. J. Neurochem. 118, 695–713.
Mikhaylova, M., Reddy, P. P., Munsch, T., Landgraf, P., Suman, S. K., Smalla, K. H., Gundelfinger, E. D., Sharma, Y., and Kreutz, M. R. (2009). Calneurons provide a calcium threshold for trans-Golgi network to plasma membrane trafficking. Proc. Natl. Acad. Sci. U.S.A. 106, 9093–9098.
Mikhaylova, M., Sharma, Y., Reissner, C., Nagel, F., Aravind, P., Rajini, B., Smalla, K. H., Gundelfinger, E. D., and Kreutz, M. R. (2006). Neuronal Ca2+ signaling via caldendrin and calneurons. Biochim. Biophys. Acta 1763, 1229–1237.
Nagata, K., Puls, A., Futter, C. L., Aspenstrom, P., Schaefer, E., Nakata, T., Hirokawa, N., and Hall, A. (1998). The MAP kinase kinase kinase MLK2 co-localizes with activated JNK along microtubules and associates with kinesin superfamily motor KIF3. EMBO J. 17, 149–158.
Nakamura, T. Y., Pountney, D. J., Ozaita, A., Nandi, S., Ueda, S., Rudy, B., and Coetzee, W. A. (2001). A role for frequenin, a Ca2+-binding protein, as a regulator of Kv4 K-currents. Proc. Natl. Acad. Sci. U.S.A. 98, 12808–12813.
Neher, E., and Augustine, G. J. (1992). Calcium gradients and buffers in bovine chromaffin cells. J. Physiol. 450, 273–301.
Ngo-Anh, T. J., Bloodgood, B. L., Lin, M., Sabatini, B. L., Maylie, J., and Adelman, J. P. (2005). SK channels and NMDA receptors form a Ca2+-mediated feedback loop in dendritic spines. Nat. Neurosci. 8, 642–649.
Nimchinsky, E. A., Yasuda, R., Oertner, T. G., and Svoboda, K. (2004). The number of glutamate receptors opened by synaptic stimulation in single hippocampal spines. J. Neurosci. 24, 2054–2064.
Noguchi, J., Matsuzaki, M., Ellis-Davies, G. C., and Kasai, H. (2005). Spine-neck geometry determines NMDA receptor-dependent Ca2+ signaling in dendrites. Neuron 46, 609–622.
O'Callaghan, D. W., Haynes, L. P., and Burgoyne, R. D. (2005). High-affinity interaction of the N-terminal myristoylation motif of the neuronal calcium sensor protein hippocalcin with phosphatidylinositol 4,5-bisphosphate. Biochem. J. 391, 231–238.
O'Callaghan, D. W., Tepikin, A. V., and Burgoyne, R. D. (2003). Dynamics and calcium sensitivity of the Ca2+/myristoyl switch protein hippocalcin in living cells. J. Cell. Biol. 163, 715–721.
Ordaz, B., Tang, J., Xiao, R., Salgado, A., Sampieri, A., Zhu, M. X., and Vaca, L. (2005). Calmodulin and calcium interplay in the modulation of TRPC5 channel activity. J. Biol. Chem. 280, 30788–30796.
Osawa, M., Dace, A., Tong, K. I., Valiveti, A., Ikura, M., and Ames, J. B. (2005). Mg2+ and Ca2+ differentially regulate DNA binding and dimerization of DREAM. J. Biol. Chem. 280, 18008–18014.
Palmer, C. L., Lim, W., Hastie, P. G., Toward, M., Korolchuk, V. I., Burbidge, S. A., Banting, G., Collingridge, G. L., Isaac, J. T., and Henley, J. M. (2005). Hippocalcin functions as a calcium sensor in hippocampal LTD. Neuron 47, 487–494.
Peersen, O. B., Madsen, T. S., and Falke, J. J. (1997). Intermolecular tuning of calmodulin by target peptides and proteins: differential effects on Ca2+ binding and implications for kinase activation. Protein Sci. 6, 794–807.
Redecker, P., Kreutz, M. R., Bockmann, J., Gundelfinger, E. D., and Boeckers, T. M. (2003). Brain synaptic junctional proteins at the acrosome of rat testicular germ cells. J. Histochem. Cytochem. 51, 809–819.
Résibois, A., and Rogers, J. H. (1992). Calretinin in rat brain: an immunohistochemical study. Neuroscience 46, 101–134.
Rose, C. R., and Konnerth, A. (2001). Stores not just for storage: intracellular calcium release and synaptic plasticity. Neuron 31, 519–522.
Ruiz-Gomez, A., Mellström, B., Tornero, D., Morato, E., Savignac, M., Holguín, H., Aurrekoetxea, K., González, P., González-García, C., Ceña, V., Mayor, F. Jr., and Naranjo, J. R. (2007). G protein-coupled receptor kinase 2-mediated phosphorylation of downstream regulatory element antagonist modulator regulates membrane trafficking of Kv4.2 potassium channel. J. Biol. Chem. 282, 1205–1215.
Sabatini, B. L., Maravall, M., and Svoboda, K. (2001). Ca2+ signaling in dendritic spines. Curr. Opin. Neurobiol. 11, 349–356.
Sabatini, B. L., Oertner, T. G., and Svoboda, K. (2002). The life cycle of Ca2+ ions in dendritic spines. Neuron 33, 439–452.
Sano, M., and Kitajima, S. (1983). Ontogeny of calmodulin and calmodulin-dependent adenylate cyclase in rat brain. Brain Res. 283, 215–220.
Schaad, N. C., De Castro, E., Nef, S., Hegi, S., Hinrichsen, R., Martone, M. E., Ellisman, M. H., Sikkink, R., Rusnak, F., Sygush, J., and Nef, P. (1996). Direct modulation of calmodulin targets by the neuronal calcium sensor NCS-1. Proc. Natl. Acad. Sci. U.S.A. 93, 9253–9258.
Schlecker, C., Boehmerle, W., Jeromin, A., DeGray, B., Varshney, A., Sharma, Y., Szigeti-Buck, K., and Ehrlich, B. E. (2006). Neuronal calcium sensor-1 enhancement of InsP3 receptor activity is inhibited by therapeutic levels of lithium. J. Clin. Invest. 116, 1668–1674.
Schmidt, H., and Eilers, J. (2009). Spine neck geometry determines spino-dendritic cross-talk in the presence of mobile endogenous calcium binding proteins. J. Comput. Neurosci. 27, 229–243.
Schmidt, H., Schwaller, B., and Eilers, J. (2005). Calbindin D28k targets myo-inositol monophosphatase in spines and dendrites of cerebellar Purkinje neurons. Proc. Natl. Acad. Sci. U.S.A. 102, 5850–5855.
Schwaller, B. (2011). The use of transgenic mouse models to reveal the functions of Ca(2+) buffer proteins in excitable cells. Biochim. Biophys. Acta doi: 10.1016/j.bbagen.2011.11.008. [Epub ahead of print].
Segal, M. (2001). Rapid plasticity of dendritic spine: hints to possible functions? Prog. Neurobiol. 63, 61–70.
Seidenbecher, C. I., Landwehr, M., Smalla, K. H., Kreutz, M., Dieterich, D. C., Zuschratter, W., Reissner, C., Hammarback, J. A., Böckers, T. M., Gundelfinger, E. D., and Kreutz, M. R. (2004). Caldendrin but not calmodulin binds to light chain 3 of MAP1A/B: an association with the microtubule cytoskeleton highlighting exclusive binding partners for neuronal Ca2+-sensor proteins. J. Mol. Biol. 336, 957–970.
Seidenbecher, C. I., Langnaese, K., Sanmartí-Vila, L., Boeckers, T. M., Smalla, K. H., Sabel, B. A., Garner, C. C., Gundelfinger, E. D., and Kreutz, M. R. (1998). Caldendrin, a novel neuronal calcium-binding protein confined to the somato-dendritic compartment. J. Biol. Chem. 273, 21324–21331.
Simon, S. M., and Llinás, R. R. (1985). Compartmentalization of the submembrane calcium activity during calcium influx and its significance in transmitter release. Biophys. J. 48, 485–498.
Sloviter, R. S. (1989). Calcium-binding protein (calbindin-D28k) and parvalbumin immunocytochemistry: localization in the rat hippocampus with specific reference to the selective vulnerability of hippocampal neurons to seizure activity. J. Comp. Neurol. 280, 183–196.
Smalla, K. H., Seidenbecher, C. I., Tischmeyer, W., Schicknick, H., Wyneken, U., Böckers, T. M., Gundelfinger, E. D., and Kreutz, M. R. (2003). Kainate-induced epileptic seizures induce a recruitment of caldendrin to the postsynaptic density in rat brain. Mol. Brain Res. 116, 159–162.
Spacek, J., and Harris, K. M. (1997). Three-dimensional organization of smooth endoplasmic reticulum in hippocampal CA1 dendrites and dendritic spines of the immature and mature rat. J. Neurosci. 17, 190–203.
Stuart, R. O., Bush, K. T., and Nigam, S. K. (2003). Changes in gene expression patterns in the ureteric bud and metanephric mesenchyme in models of kidney development. Kidney Int. 64, 1997–2008.
Tadross, M. R., Dick, I. E., and Yue, D. T. (2008). Mechanism of local and global Ca2+ sensing by calmodulin in complex with a Ca2+ channel. Cell 133, 1228–1240.
Takashima, N., Odaka, Y. S., Sakoori, K., Akagi, T., Hashikawa, T., Morimura, N., Yamada, K., and Aruga, J. (2011). Impaired cognitive function and altered hippocampal synapse morphology in mice lacking Lrrtm1, a gene associated with schizophrenia. PLoS One 6:e22716. doi: 10.1371/journal.pone.0022716
Teolato, S., Calderini, G., Bonetti, A. C., and Toffano, G. (1983). Calmodulin content in different brain areas of aging rats. Neurosci. Lett. 38, 57–60.
Tippens, A. L., and Lee, A. (2007). Caldendrin, a neuron-specific modulator of Cav/1.2 (L-type) Ca2+ channels. J. Biol. Chem. 282, 8464–8473.
Toresson, H., and Grant, S. G. (2005). Dynamic distribution of endoplasmic reticulum in hippocampal neuron dendritic spines. Eur. J. Neurosci. 22, 1793–1798.
Vargas, F., and Guidotti, A. (1980). Calmodulin in brain of schizophrenics. Neurochem. Res. 5, 673–681.
Wang, Q., Liang, K. C., Czader, A., Waxham, M. N., and Cheung, M. S. (2011). The effect of macromolecular crowding, ionic strength and calcium binding on calmodulin dynamics. PLoS Comput. Biol. 7:e1002114. doi: 10.1371/journal.pcbi.1002114
Wingard, J. N., Chan, J., Bosanac, I., Haeseleer, F., Palczewski, K., Ikura, M., and Ames, J. B. (2005). Structural analysis of Mg2+ and Ca2+ binding to CaBP1, a neuron-specific regulator of calcium channels. J. Biol. Chem. 280, 37461–37470.
Woll, M. P., De Cotiis, D. A., Bewley, M. C., Tacelosky, D. M., Levenson, R., and Flanagan, J. M. (2011). Interaction between the D2 dopamine receptor and neuronal calcium sensor-1 analyzed by fluorescence anisotropy. Biochemistry 50, 8780–8791.
Wu, L. J., Mellström, B., Wang, H., Ren, M., Domingo, S., Kim, S. S., Li, X. Y., Chen, T., Naranjo, J. R., and Zhuo, M. (2010). DREAM (Downstream Regulatory Element Antagonist Modulator) contributes to synaptic depression and contextual fear memory. Mol. Brain 3, 3.
Wu, Y. Q., Lin, X., Liu, C. M., Jamrich, M., and Shaffer, L. G. (2001). Identification of a human brain-specific gene, calneuron 1, a new member of the calmodulin superfamily. Mol. Genet. Metab. 72, 343–350.
Xia, Z., and Storm, D. R. (2005). The role of calmodulin as a signal integrator for synaptic plasticity. Nat. Rev. Neurosci. 6, 267–276.
Yang, J., McBride, S., Mak, D. O., Vardi, N., Palczewski, K., Haeseleer, F., and Foskett, J. K. (2002). Identification of a family of calcium sensors as protein ligands of inositol trisphosphate receptor Ca2+ release channels. Proc. Natl. Acad. Sci. U.S.A. 99, 7711–7716.
Yuste, R., Majewska, A., and Holthoff, K. (2000). From form to function: calcium compartmentalization in dendritic spines. Nat. Neurosci. 3, 653–659.
Yuste, R., and Denk, W. (1995). Dendritic spines as basic functional units of neuronal integration. Nature 375, 682–684.
Zhang, Y., Su, P., Liang, P., Liu, T., Liu, X., Liu, X. Y., Zhang, B., Han, T., Zhu, Y. B., Yin, D. M., Li, J., Zhou, Z., Wang, K. W., and Wang, Y. (2010). The DREAM protein negatively regulates the NMDA receptor through interaction with the NR1 subunit. J. Neurosci. 30, 7575–7586.
Zhou, H., Kim, S. A., Kirk, E. A., Tippens, A. L., Sun, H., Haeseleer, F., and Lee, A. (2004). Ca2+-binding protein-1 facilitates and forms a postsynaptic complex with Cav1.2 (L-type) Ca2+ channels. J. Neurosci. 24, 4698–4708.
Keywords: Ca2+, neuronal calcium signaling, neuronal calcium sensor, calcium-binding protein, dendritic spine, binding affinity, calcium dynamics, protein-protein interaction
Citation: Raghuram V, Sharma Y and Kreutz MR (2012) Ca2+ sensor proteins in dendritic spines: a race for Ca2+. Front. Mol. Neurosci. 5:61. doi: 10.3389/fnmol.2012.00061
Received: 12 February 2012; Accepted: 18 April 2012;
Published online: 08 May 2012.
Edited by:
Beat Schwaller, University of Fribourg, SwitzerlandReviewed by:
Miou Zhou, University of California, Los Angeles, USAGuido C. Faas, University of California, Los Angeles, USA; Hartmut Schmidt, University of Leipzig, Germany
Copyright: © 2012 Raghuram, Sharma and Kreutz. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Michael R. Kreutz, RG Neuroplasticity, Leibniz Institute for Neurobiology, Brenneckestr. 6, 39118 Magdeburg, Germany. e-mail:a3JldXR6QGxpbi1tYWdkZWJ1cmcuZGU=