

- 1Medical Faculty, Department of Basic Neurosciences, University of Geneva, Geneva, Switzerland
- 2Clinic of Neurology, Department of Clinical Neurosciences, Geneva University Hospital, Geneva, Switzerland
As in other parts of the central nervous system (CNS) of the mouse, glutamatergic synapses onto dopamine (DA) neurons in the ventral tegmental area (VTA) mature postnatally. At birth many AMPA receptors (AMPARs) lack GluA2R subunit and most NMDARs contain the GluN2B subunit. Within 2 weeks these receptors are replaced with GluA2- and GluN2A- containing AMPARs and NMDARs, respectively. Recent data suggest that a single injection of cocaine (or another drug of addiction) triggers glutamate receptor redistribution with the reappearance of the subunits typically present in immature synapses, as if addictive drugs reopen the developmental critical period. Here we review the experimental evidence for this hypothesis and discuss the implications for circuit function.
Introduction
Fast excitatory synaptic transmission in the mammalian central nervous system (CNS) is mediated predominantly by ionotropic glutamate receptors of the AMPA- and NMDA-type. These receptors are composed of different subunits that determine the biophysical and trafficking properties of the channel. Depending on the particular subunit composition of the ionotropic receptors both synaptic transmission and synaptic plasticity can vary considerably. The AMPA receptors (AMPARs) are the workhorses of fast excitatory transmission in the CNS. They are tetrameric receptor assemblies of GluA1–4 subunits, which are differently expressed in the brain and are encoded by separate genes (Hollmann and Heinemann, 1994; Kato et al., 2008; Traynelis et al., 2010). The subunits that are predominantly expressed in the CNS are the GluA1 and GluA2 subunits and the majority of receptors at the synapse are heteromers incorporating both. The GluA2 subunit is critical in determining the properties of AMPARs; the presence of an arginine (R) residue in its pore membrane domain, introduced by post-translational editing, renders the receptor impermeable to Ca2+ (CI-AMPARs) and therefore results in a linear current-voltage (I–V) curve that reverses at 0 mV. Conversely, when the GluA2 subunit is absent, the receptors are Ca2+-permeable (CP-AMPARs) and the I–V curve becomes inwardly rectifying because the currents at positive potentials are inhibited by endogenous polyamines (Donevan and Rogawski, 1995; Liu and Zukin, 2007) and because the unitary current is larger. Many reversible post-translational modifications of both GluA1 and GluA2 subunits regulate AMPAR expression and function; among them subunit phosphorylation as well as palmitoylation and ubiquitination have been shown to modulate both biophysical properties and receptor trafficking (Lu and Roche, 2011). How this post-translational modification affects the trafficking of AMPARs in a subunit-specific manner, and how this determines synaptic plasticity, remains elusive.
Tight control of CP-AMPAR expression ensures that the majority of cells in the brain contain CI-AMPARs (Shin et al., 2005). Although the CP-AMPARs may mediate excitotoxicity associated with different neurological disorders (Liu and Zukin, 2007) it has been shown that these receptors also play an important role in synaptic plasticity (Plant et al., 2006; Adesnik and Nicoll, 2007). Moreover there are several synapses in which CP-AMPARs are developmentally expressed, in contrast to the adult where CP-AMPARs are mainly found on GABAegic interneurons (Goldberg et al., 2003). Because of their characteristics, the CP-AMPARs confer novel properties to the synapses where they are expressed and their specific role in synaptic function as well as synaptic plasticity is beginning to be elucidated.
NMDARs are heteromeric receptors containing GluN1 as an obligatory subunit. GluN1 is expressed both in the neonatal as well as the adult brain. Two other NMDAR subunits exist: GluN2 and GluN3 (Moriyoshi et al., 1991; Traynelis et al., 2010). Given that they each can be expressed in distinct subunit isoforms (e.g., GluN2A/2B) their combination gives rise to multiple functionally distinct NMDARs (Paoletti, 2011). It has been shown that NMDAR subunit composition is developmentally regulated; at many synapses GluN2B is the predominant isoform during the first 2 postnatal weeks (Monyer et al., 1994). The GluN2A isoform gradually increases after birth while the GluN2B and GluN2D expression progressively decrease. This developmental switch results in a change in the kinetics and subunit-specific modulation of NMDAR currents. The GluN3 family can be expressed as two isoforms (GluN3A and GluN3B) that arise from two different genes that have different expression profiles. GluN3A is mainly expressed during early postnatal life and decreases in adulthood while GluN3B expression is low at early stages and increases in adult animals (Henson et al., 2010). There is accumulating evidence suggesting that NMDARs are not a static component of the postsynaptic density but that their function and expression could be regulated by activity-dependent mechanisms (Bellone and Nicoll, 2007). Further investigations are now necessary for understanding the subunit-specific control of receptor activity both during development and in neurological disorders.
Postnatal Maturation of Glutamate Transmission
During brain development, the prenatal and postnatal periods are characterized by rapid changes in neuronal organization, thus providing a critical period during which environmental experiences can lead to long-term changes of both the brain and behavior. At the cellular level changes in synaptic morphology and receptor expression are associated with changes in different functional properties that influence the synaptic transmission. While the regulation of NMDAR composition during development has been studied in detail in many brain regions, AMPAR subunit regulation has been less thoroughly investigated. Compelling evidence has shown that GluA2 content and consequently the expression of CP-AMPARs is developmentally regulated in principal neurons in different systems (Pellegri et al., 1998; Rohrbough and Spitzer, 1999; Lawrence and Trussell, 2000; Pickard et al., 2000). The principal neurons in the cortex have been extensively studied and it has been shown that the functional properties of AMPARs on layer 5 pyramidal neurons change with respect to the GluA2 subunit during early postnatal development (Kumar et al., 2002). These results are based on the observation of a rectifying I–V curve, Ca2+ permeability and blockade of the AMPAR-EPSC by either intracellular or extracellular polyamines. It has been shown that the developmental switch of GluA2 happens in layer 2/3 pyramidal cells as well as in layer 4 stellate cells. Although the change in AMPAR subunit composition seems to be a general property of principal neocortical neurons, the timing of the switch is different among the layers (Brill and Huguenard, 2008). The switch occurs between P14–P16 in layer 5 pyramidal neurons, but between P12–P14 in layer 2/3 pyramidal cells and between P7–P8 in layer 4 stellate cells. Interestingly the timing of the switch corresponds to their positions within the canonical neocortical circuits rather that the ontogenetic age of the layer (Crair and Malenka, 1995; Isaac et al., 1997). Moreover the timing of the GluA2 switch in layer 2/3 pyramidal neurons coincides with the critical period for the developmental plasticity in somatosensory cortex (Stern et al., 2001). What still needs to be determined is the role of CP-AMPARs at developmental synapses. It has been suggested that in the neocortex the early low expression of GluA2 subunits could underlie the increased seizure susceptibility of the immature brain (Sensi et al., 1999).
A similar developmental expression of CP-AMPARs has also been observed at mossy fiber-CA3 pyramidal synapses where they dominate the transmission up to P17. Here it has been shown that the expression of CP-AMPARs is regulated by the PDZ (postsynaptic density-95/Disc large/zona occludens-1) domain-containing protein interacting with C kinase 1 (PICK1) since PICK1 knock-out mice have a linear I–V curves and are insensitive to Phylantotoxin 433, a specific GluA2-lacking AMPAR blocker (Ho et al., 2007). Interestingly CP-AMPARs are removed by depolarization-induced long-term depression (DiLTD), a form of plasticity specifically observed only at young synapses that relies on postsynaptic calcium influx through L-type voltage gated calcium channels and release from intracellular stores (Ho et al., 2007). What was still unknown is the physiological trigger for the developmental loss of CP-AMPARs.
Recently we have observed that at excitatory synapses onto dopaminergic (DA) neurons in the ventral tegmental area (VTA) the postnatal maturation of AMPARs and NMDARs develops in parallel. Indeed during the first week the transmission is dominated by CP-AMPARs and GluN2B-containing NMDARs. Subsequently metabotropic glutamate receptor 1 (mGluR1) drives the insertion of CI-AMPARs and GluN2A-containing NMDARs through a signaling cascade that involves phospholipase C (PLC) (Bellone et al., 2011), indicating that glutamatergic synaptic transmission in the VTA relies on the activation of these metabotropic receptors. Recently it has been proposed that in the hippocampus the activity-dependent GluN2B to GluN2A subunit switch during development is mediated by group-I mGluRs (Matta et al., 2011). Taken together these data suggest a common role for mGluRs in the postnatal development of excitatory synapses. Future studies will have to address whether at the other synapses the mechanism implicated in the AMPAR switch during synaptic maturation is an active mechanism similar to the one in the VTA or whether it is a passive one in which GluA2 expression slowly increases.
The switch in the expression of the GluA2 subunit has important functional implications since the permeability of Ca2+ through the receptors changes during development. Interestingly, imaging data in the VTA have suggested that synaptic calcium entry was almost exclusively mediated by CP-AMPARs at neonatal synapses, despite the fact that substantial NMDAR-EPSCs were observed in simultaneous whole-cell recordings (Bellone et al., 2011). One explanation for these data is that the imaging and electrophysiological methods monitored distinct pools of synapses with contrasting properties. Otherwise these results may reflect the presence of NMDARs that are calcium impermeable. In fact, it has been shown that calcium-impermeable GluN3A-containing receptors are preferentially expressed during a narrow temporal window in postnatal development at certain synapses (Roberts et al., 2009).
Many developing synapses have been described as silent, where the transmission is mediated by NMDARs, and become functional with the insertion of AMPARs during the first postnatal week (Isaac, 2003). The presence of CP-AMPARs could represent another form of synaptic maturation and could have strong implications for synaptic transmission and plasticity. Calcium influx in response to synaptic activity triggers many intracellular signaling cascades including activity-dependent forms of synaptic plasticity. In order for calcium to enter the neurons through NMDARs the membrane needs to be depolarized as Mg2+ blocks NMDARs at rest. Conversely, CP-AMPARs can flux calcium most efficiently when they are hyperpolarized. In other words the activity requirements for synaptic calcium entry change radically once the cell switches from CP-AMPARs to NMDARs.
Expression of CP-AMPARs in the Adult Brain
Beside principal cells during development, CP-AMPARs have been found in naive brain mostly in interneurons and it has been shown that they participate in novel forms of long-lasting synaptic plasticity. Parallel fiber to stellate cell synapses express GluA2-lacking AMPARs and it has been shown that repetitive activation of these synapses produces a long-lasting switch to GluA2-containing AMPARs. As this subunit is Ca2+ impermeable and shows slow channel kinetics, the switch in AMPARs could affect the stellate cell waveform shape and modify the efficacy of inhibitory synaptic transmission between stellate and Purkinje cells (Liu and Cull-Candy, 2000; Liu and Zukin, 2007). The presence of CP-AMPARs has also been shown in interneurons from the amygdala. At these synapses a novel form of long-term potentiation (LTP), independent of NMDARs, has been described. The induction of this form of plasticity was dependent on postsynaptic calcium and also, while not directly proven, has been suggested to depend on Ca2+ entry through CP-AMPARs (Mahanty and Sah, 1998). Recently Kullmann and colleagues described a novel form of plasticity at interneurons in the stratum oriens of hippocampus (Lamsa et al., 2007). This form of LTP doesn't follow the hebbian rules since it can be induced by a hyperpolarization of the postsynaptic terminal in concomitance with presynaptic glutamate release. Therefore, this type of plasticity has been called anti-hebbian LTP. CP-AMPARs play a major role in this type of plasticity as was demonstrated by showing that blocking these receptors during the induction protocol abolished the LTP. The functional role of this type of plasticity is not yet clear. Although many forms of learning are NMDAR-dependent, certain types of memories and learning processes occur independently of these receptors, yet are clearly experience-dependent. Supporting this hypothesis it has been suggested that GluA2 deletion in CA1 selectively impairs NMDAR-dependent learning and that the NMDAR-independent learning requires the activation of CP-AMPARs (Wiltgen et al., 2010). This finding strongly suggests that CP-AMPARs play a functional role in NMDAR-independent learning.
Experience-Dependent Trafficking of CP-AMPARs and NMDARs
Experience-dependent trafficking of both AMPARs and NMDARs in the CNS play an important role in many learning processes. It has been suggested that in adult brain the NMDAR trafficking occurs at much slower rates compared to AMPARs. This finding was supported by the relative stability of NMDARs at the synapses in contrast to the rapid AMPAR turnover (Bredt and Nicoll, 2003). Moreover with standard LTP protocols little or no change in the NMDAR-mediated synaptic transmission has been observed. In the last few years this idea has been challenged by many lines of evidence suggesting a change in NMDAR function as a result of different behavioral experiences. It has been shown that while visual deprivation changes the ratio between GluN2A/GluN2B leading to an increase in the GluN2B expression (He et al., 2006), sleep deprivation shifts the ratio and increases the GluN2A subunit (Kopp et al., 2006). Although further studies are needed to investigate the mechanisms controlling the NMDAR subunit composition and their role in synaptic plasticity, this dynamic regulation of NMDARs in adulthood demonstrates that the adult brain is continuously modified by experiences.
There is a wealth of evidence indicating AMPAR trafficking at glutamatergic synapses but only recently has it been shown that CP-AMPARs can be trafficked to the synapse after experience-dependent plasticity in different brain regions. One very elegant study has demonstrated that a single whisker experience protocol in vivo can induce a pathway-specific strengthening of neocortical excitatory synapses with a delivery of Glu2A-lacking receptors at spared but not deprived inputs. It has been hypothesized that the presence of CP-AMPARs at layer 4 to layer 2/3 synapses may alter the rules for plasticity facilitating an NMDAR-independent form of plasticity (Clem and Barth, 2006). In the hippocampus the insertion of CP-AMPARs following an LTP protocol is debated; it has been observed that pairing-induced LTP resulted in a transient incorporation of synaptic CP-AMPARs (Plant et al., 2006; Guire et al., 2008), whereas two other reports did not see this subunit recomposition using either pairing-induced LTP or HFS-LTP (Adesnik and Nicoll, 2007; Gray et al., 2007). It has been suggested that the ability of HFS to induce AMPAR subunit reassembly may be dependent on the age of the rats (Lu et al., 2007). What still is not clear is the importance of the transient expression of CP-AMPARs for the LTP and it has been suggested that these types of receptors, due to their ability to flux Ca+, may regulate the signaling cascade events important for the maintenance of the plasticity. Taken together these findings suggest that experience-dependent insertion of CP-AMPARs could alter synaptic plasticity and engage different forms of learning that are independent of NMDARs (Tayler et al., 2011).
Drug-Dependent Trafficking of Glutamate Receptors
In the mesocorticolimbic DA system, which originates from the DA neurons of VTA that project onto the nucleus accumbens (NAc) and prefrontal cortex (PFC), many common cellular adaptations have been described following drug exposure (Nestler, 2005). In particular addictive drugs, following acute and chronic exposure, are able to leave a trace on transmission at many synapse of the mesocorticolimbic system, a phenomenon termed “drug-evoked synaptic plasticity” (Lüscher and Malenka, 2011). Although many different forms of drug-evoked synaptic plasticity have been described in the PFC and NAc, for the purpose of this review we will focus on the effects of addictive drugs at the excitatory afferents onto DA neurons of the VTA.
A day after the exposure to a single dose of addictive substance it has been shown that at least two parameters of synaptic transmission are altered at excitatory afferents onto DA neurons in the VTA. First the AMPA/NMDA ratio is increased (Ungless et al., 2001). Second the current-voltage relationship of AMPAR-EPSCs is no longer linear, but is inwardly rectifying (Bellone and Lüscher, 2006). After drug treatment the rectification index (ratio of the chord conductance at negative over positive potential) as well as electron microscopy experiments have suggested the presence of a fraction of AMPARs that lack the Glu2A subunit (Bellone and Lüscher, 2006; Mameli et al., 2007). It is still not clear if auxiliary subunits such as the TARPs could contribute to the rectification (Soto et al., 2007) or which are the mechanisms that underlie the insertion of GluA2-lacking AMPARs instead of GluA2-containing. But, regardless of the exact molecular mechanism, the appearance of rectifying AMPAR-EPSCs after drug exposure suggested that in some synapses AMPARs are redistributed.
How can the AMPA/NMDA ratio increase (particularly when both components are measured at +40 mV) if the inserted AMPARs do not conduct current at positive potentials? Initial studies indeed suggested that the NMDAR component remained unchanged (Ungless et al., 2001). This was based on the observation that the bath application of NMDA elicited currents of similar magnitude in rat brain slices regardless of whether they were exposed to cocaine or saline. This approach however has the disadvantage of activating extrasynaptic as well as synaptic receptors. Using a two-photon laser to photolyse caged glutamate we compared EPSC amplitudes in slices from saline- with cocaine-treated animals (Mameli et al., 2011). This approach not only confirmed the existence of rectifying unitary AMPAR-EPSCs but also showed that unitary NMDAR-EPSCs were significantly smaller after the drug treatment. While the molecular mechanisms underlying drug-evoked plasticity of NMDAR-mediated transmission have yet to be investigated, preliminary data from our laboratory suggest that the overall reduction of the amplitude is indeed due to a subunit switch such that after the drug treatment the receptors are enhanced in GluN2B content (unpublished data). In summary, at excitatory synapses onto DA neurons, after a single injection of different addictive drugs, GluA2-lacking (and maybe GluN2B-containing NMDARs) are driven into the synapses and contribute to the transmission up to 5 days after the exposure (Bellone and Lüscher, 2006; Mameli et al., 2009; Brown et al., 2010) (Figure 1).
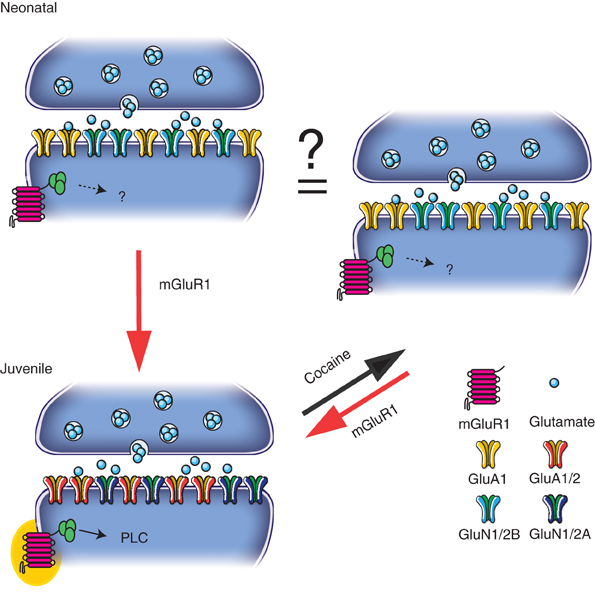
Figure 1. During the first postnatal week (“Neonatal”), transmission is dominated by CP-AMPARs and GluN2B-containing NMDARs. Subsequently (“Juvenile”) mGluR1 receptors play a role in the synaptic insertion of CI-AMPARs and GluN2A-containing NMDAR. At juvenile synapses, a single cocaine injection exchanges CP-AMPARs for CI ones and causes decrease in NMDAR function changing the relative contribution of GluN2A- and GluN2B-containing NMDARs. This switch in the glutamatergic transmission may contribute to the re-opening of a critical period seen during the development. Cocaine-induced plasticity can then be reverted by the activation of mGluR1.
We have previously described a form of synaptic plasticity induced by mGluR1 that is responsible for reverting drug-evoked synaptic plasticity in the VTA (Bellone and Lüscher, 2006). After a single cocaine injection, both pharmacological and electrophysiological activation of mGluR1 drive the insertion of GluA2-containing and the removal of GluA2-lacking AMPARs (Mameli et al., 2007). Interestingly a similar form of plasticity has been described in the NAc where activation of mGluR1 reversed the accumulation of GluA2-lacking AMPARs after cue-induced cocaine craving (McCutcheon et al., 2011). Moreover mGluR1 mediated removal of GluA2-lacking AMPARs is also involved in modulation of fear memory in the amygdala (Clem and Barth, 2006).
An emerging hypothesis in the field is that in different brain regions, mGluR1 receptors control the basal level of GluA2-containing AMPARs and that any reduction of their function leads to an up-regulation of GluA2-lacking AMPARs. It remains to be determined how cocaine can alter mGluR1. We don't know if cocaine induces a change in the number of mGluR1 or whether the drug interferes with the intracellular signaling pathway.
Recently we have demonstrated that in utero cocaine exposure leads to a delay in the normal maturation of both AMPARs and NMDARs (Bellone et al., 2011). Since the postnatal synaptic maturation of excitatory transmission onto DA neurons of the VTA relies on the activation of mGluR1, a consequence of in utero cocaine exposure is that a substantial fraction of AMPARs in adolescent and young adult mice remains calcium permeable while the GluN2B subunit still contributes to transmission during the first 2 months of life (Figure 2). It will now be necessary to determine the consequences of such a delay in postnatal maturation of excitatory synapses onto DA VTA neurons for functional plasticity.
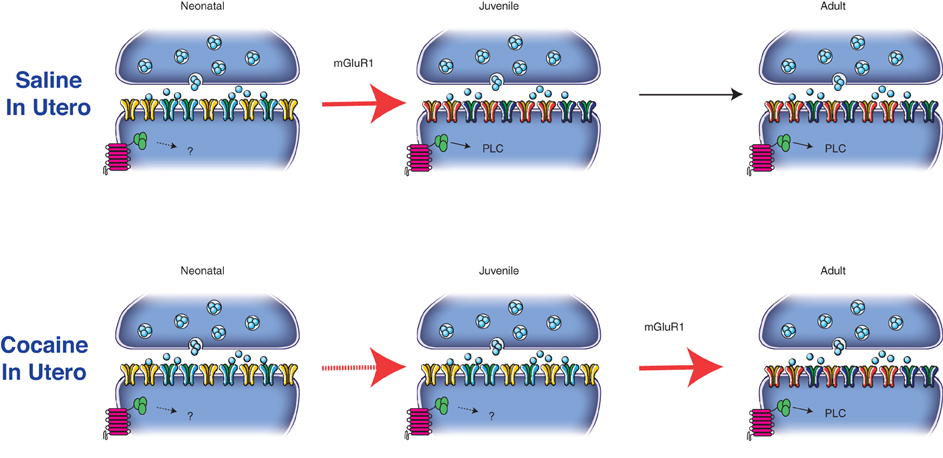
Figure 2. mGluR1 activation drives the postnatal maturation of glutamatergic synapses onto DA neurons in the VTA. In particular the activation of the metabotropic receptors drives the insertion of GluN2A-containing NMDARs and GluA2-containing AMPARs from neonatal (P2–6) to juvenile synapses (P14–26). The composition of glutamatergic synapses then remains unaltered from juvenile to adult age (P > 60). In utero cocaine exposure interfere with the mGluR1-signaling pathway delaying the postnatal maturation of excitatory transmission at these synapses.
Over the last few years, the idea has emerged that changes in the excitatory transmission onto DA VTA neurons are permissive for further changes in the mesolimbic system. While a single injection of cocaine is sufficient to causes a rapid, but transient, potentiation of excitatory inputs in the VTA, several injections are required to induce plasticity in the NAc (Mameli et al., 2009). Moreover in this area, CP-AMPARs appear after prolonged withdrawal from cocaine and have been implicated in a behavioral model of cue-triggered relapse, termed the incubation of craving (Conrad et al., 2008). Although no apparent changes in the NMDAR expression have been seen after prolonged withdrawal in the NAc, it has been suggested that new GluN2B-containing silent synapses are replace by GluN2A-containing synapses after that period (Ferrario et al., 2010). Further studies are needed to better characterize the NMDAR-dependent trafficking in the ventral striatum both during development and after addictive drugs.
In conclusion, both in the VTA and in the NAc, mGluR1 activation is sufficient to reverse the cocaine-induced plasticity with similar mechanisms (Bellone and Lüscher, 2006; Mameli et al., 2007; McCutcheon et al., 2011). Therefore, we can speculate that recruitment of mGluR1 could function as a protective mechanism to counteract drug exposure and those individuals with a deficient mGluR1-dependent LTD mechanism may be particularly at risk of addiction.
Conclusions
Many questions remain unsolved. Which are the mechanisms that enable the synaptic incorporation and retention of CP-AMPARs after a single cocaine injection in the VTA and after prolonged withdrawal in the NAc? What is the significance of the insertion of CP-AMPARs and the concomitant decrease in NMDAR function at these synapses? Previous findings in hippocampal interneurons expressing CP-AMPARs show that pairing of a hyperpolarizing current injection with synaptic release can enhance synaptic transmission (Lamsa et al., 2007). In the VTA, it has recently been shown that the cocaine exposure enables the induction of a similar form of anti-hebbian plasticity that also relies on calcium entrance from CP-AMPARs and is independent on NMDARs. In contrast, in drug naïve or saline-treated mice, where the transmission is mediated by CI-AMPARs, the LTP induction follows hebbian rules, i.e., LTP is induced by concomitant glutamate release and postsynaptic depolarization. It has therefore been suggested that the exchange of GluA2-containing AMPARs for Glu2A-lacking ones and a decrease in NMDAR function is sufficient to invert the rules of plasticity induction (Mameli et al., 2011). Therefore, the drug-evoked plasticity in the VTA is merely permissive for a specific form of activity-dependent synaptic plasticity. Future studies will have to address whether anti-hebbian plasticity can occur in vivo, for example during a hyperpolarization of DA neurons when upstream GABA neurons are strongly activated. It is important to underline that the drug-related adaptations here described may represent an aberrant form of plasticity that drives long-term adaptations in the downstream reward circuit.
Conversely, during early development this form of plasticity may be the default and enhance special forms of learning. It is conceivable that during development DA neurons do not need to discriminate among the inputs they receive and it is only with the time that they learn to differentiate by strengthening certain connections. The CP-AMPARs and the plasticity they are associated with could be important for this type of learning. The capacity to discriminate among different stimuli is important in order to select the most valuable reward during the decision-making process. It has been well described in adult brain that DA VTA neurons respond to neutral, aversive and explicit non-reward predicting stimuli in a similar manner (Schultz, 2011). We could speculate that during the development, since the brain has not been yet exposed to different stimuli, the DA neurons are prone to generalization and discriminate poorly. Subsequently with advanced development various stimuli will acquire a valence and therefore the DA VTA neurons will be activated only by salient stimuli. Drug of abuse enhanced generalized dopamine activation, resulting in decreased reward discrimination. In this context after drug exposure, neutral or aversive stimuli would lead to stronger phasic striatal dopamine changes and would increase dopamine-dependent postsynaptic responsiveness and plasticity. When we give cocaine at an adult age, the drugs re-open a critical period previously seen during development and induce a switch in the AMPAR subunits composition with an insertion of CP-AMPARs. We can speculate that this switch could contribute to the re-programming of the DA system resulting in reduced reward discrimination.
While we are only beginning to understand the repercussions of drug-evoked receptor redistribution, the striking resemblance with the processes occurring during early postnatal development brings a new, exciting perspective that may help us to understand both normal development and the development of addiction.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Matthew Brown for critical reading of the manuscript.
References
Adesnik, H., and Nicoll, R. A. (2007). Conservation of glutamate receptor 2-containing AMPA receptors during long-term potentiation. J. Neurosci. 27, 4598–4602.
Bellone, C., and Lüscher, C. (2006). Cocaine triggered AMPA receptor redistribution is reversed in vivo by mGluR-dependent long-term depression. Nat. Neurosci. 9, 636–641.
Bellone, C., Mameli, M., and Lüscher, C. (2011). In utero exposure to cocaine delays postnatal synaptic maturation of glutamatergic transmission in the VTA. Nat. Neurosci. 14, 1439–1446.
Bellone, C., and Nicoll, R. A. (2007). Rapid bidirectional switching of synaptic NMDA receptors. Neuron 55, 779–785.
Bredt, D. S., and Nicoll, R. A. (2003). AMPA receptor trafficking at excitatory synapses. Neuron 40, 361–379.
Brill, J., and Huguenard, J. R. (2008). Sequential changes in AMPA receptor targeting in the developing neocortical excitatory circuit. J. Neurosci. 28, 13918–13928.
Brown, M. T. C., Bellone, C., Mameli, M., Labouèbe, G., Bocklisch, C., Balland, B., Dahan, L., Luján, R., Deisseroth, K., and Luscher, C. (2010). Drug-driven AMPA receptor redistribution mimicked by selective dopamine neuron stimulation. PLoS ONE 5:e15870. doi: 10.1371/journal.pone.0015870
Clem, R. L., and Barth, A. (2006). Pathway-specific trafficking of native AMPARs by in vivo experience. Neuron 49, 663–670.
Conrad, K. L., Tseng, K. Y., Uejima, J. L., Reimers, J. M., Heng, L.-J., Shaham, Y., Marinelli, M., and Wolf, M. E. (2008). Formation of accumbens GluR2-lacking AMPA receptors mediates incubation of cocaine craving. Nature 454, 118–121.
Crair, M. C., and Malenka, R. C. (1995). A critical period for long-term potentiation at thalamocortical synapses. Nature 375, 325–328.
Donevan, S. D., and Rogawski, M. A. (1995). Intracellular polyamines mediate inward rectification of Ca(2+)-permeable alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors. Proc. Natl. Acad. Sci. U.S.A. 92, 9298–9302.
Ferrario, C. R., Li, X., Wang, X., Reimers, J. M., Uejima, J. L., and Wolf, M. E. (2010). The role of glutamate receptor redistribution in locomotor sensitization to cocaine. Neuropsychopharmacology 35, 818–833.
Goldberg, J. H., Tamas, G., Aronov, D., and Yuste, R. (2003). Calcium microdomains in aspiny dendrites. Neuron 40, 807–821.
Gray, E. E., Fink, A. E., Sariñana, J., Vissel, B., and O'Dell, T. J. (2007). Long-term potentiation in the hippocampal CA1 region does not require insertion and activation of GluR2-lacking AMPA receptors. J. Neurophysiol. 98, 2488–2492.
Guire, E. S., Oh, M. C., Soderling, T. R., and Derkach, V. A. (2008). Recruitment of calcium-permeable AMPA receptors during synaptic potentiation is regulated by CaM-kinase I. J. Neurosci. 28, 6000–6009.
He, H.-Y., Hodos, W., and Quinlan, E. M. (2006). Visual deprivation reactivates rapid ocular dominance plasticity in adult visual cortex. J. Neurosci. 26, 2951–2955.
Henson, M. A., Roberts, A. C., Pérez-Otaño, I., and Philpot, B. D. (2010). Influence of the NR3A subunit on NMDA receptor functions. Prog. Neurobiol. 91, 23–37.
Ho, M. T. W., Pelkey, K. A., Topolnik, L., Petralia, R. S., Takamiya, K., Xia, J., Huganir, R. L., Lacaille, J. C., and McBain, C. J. (2007). Developmental expression of Ca2+-permeable AMPA receptors underlies depolarization-induced long-term depression at mossy fiber CA3 pyramid synapses. J. Neurosci. 27, 11651–11662.
Hollmann, M., and Heinemann, S. (1994). Cloned glutamate receptors. Annu. Rev. Neurosci. 17, 31–108.
Isaac, J. T., Crair, M. C., Nicoll, R. A., and Malenka, R. C. (1997). Silent synapses during development of thalamocortical inputs. Neuron 18, 269–280.
Isaac, J. T. R. (2003). Postsynaptic silent synapses: evidence and mechanisms. Neuropharmacology 45, 450–460.
Kato, A. S., Siuda, E. R., Nisenbaum, E. S., and Bredt, D. S. (2008). AMPA receptor subunit-specific regulation by a distinct family of type II TARPs. Neuron 59, 986–996.
Kopp, C., Longordo, F., Nicholson, J. R., and Lüthi, A. (2006). Insufficient sleep reversibly alters bidirectional synaptic plasticity and NMDA receptor function. J. Neurosci. 26, 12456–12465.
Kumar, S. S., Bacci, A., Kharazia, V., and Huguenard, J. R. (2002). A developmental switch of AMPA receptor subunits in neocortical pyramidal neurons. J. Neurosci. 22, 3005–3015.
Lamsa, K. P., Heeroma, J. H., Somogyi, P., Rusakov, D. A., and Kullmann, D. M. (2007). Anti-Hebbian long-term potentiation in the hippocampal feedback inhibitory circuit. Science 315, 1262–1266.
Lawrence, J. J., and Trussell, L. O. (2000). Long-term specification of AMPA receptor properties after synapse formation. J. Neurosci. 20, 4864–4870.
Liu, S. J., and Zukin, R. S. (2007). Ca2+-permeable AMPA receptors in synaptic plasticity and neuronal death. Trends Neurosci. 30, 126–134.
Liu, S. Q., and Cull-Candy, S. G. (2000). Synaptic activity at calcium-permeable AMPA receptors induces a switch in receptor subtype. Nature 405, 454–458.
Lu, W., and Roche, K. W. (2011). Posttranslational regulation of AMPA receptor trafficking and function. Curr. Opin. Neurobiol. 22, 1–10.
Lu, Y., Allen, M., Halt, A. R., Weisenhaus, M., Dallapiazza, R. F., Hall, D. D., Usachev, Y. M., McKnight, G. S., and Hell, J. W. (2007). Age-dependent requirement of AKAP150-anchored PKA and GluR2-lacking AMPA receptors in LTP. EMBO J. 26, 4879–4890.
Lüscher, C., and Malenka, R. C. (2011). Drug-evoked synaptic plasticity in addiction: from molecular changes to circuit remodeling. Neuron 69, 650–663.
Mahanty, N. K., and Sah, P. (1998). Calcium-permeable AMPA receptors mediate long-term potentiation in interneurons in the amygdala. Nature 394, 683–687.
Mameli, M., Balland, B., Lujan, R., and Lüscher, C. (2007). Rapid synthesis and synaptic insertion of GluR2 for mGluR-LTD in the ventral tegmental area. Science 317, 530–533.
Mameli, M., Bellone, C., Brown, M. T. C., and Lüscher, C. (2011). Cocaine inverts rules for synaptic plasticity of glutamate transmission in the ventral tegmental area. Nat. Neurosci. 14, 414–416.
Mameli, M., Halbout, B., Creton, C., Engblom, D., Parkitna, J. R., Spanagel, R., and Lüscher, C. (2009). Cocaine-evoked synaptic plasticity: persistence in the VTA triggers adaptations in the NAc. Nat. Neurosci. 12, 1036–1041.
Matta, J. A., Ashby, M. C., Sanz-Clemente, A., Roche, K. W., and Isaac, J. T. R. (2011). mGluR5 and NMDA receptors drive the experience- and activity-dependent NMDA receptor NR2B to NR2A subunit switch. Neuron 70, 339–351.
McCutcheon, J. E., Wang, X., Tseng, K. Y., Wolf, M. E., and Marinelli, M. (2011). Calcium-permeable AMPA receptors are present in nucleus accumbens synapses after prolonged withdrawal from cocaine self-administration but not experimenter-administered cocaine. J. Neurosci. 31, 5737–5743.
Monyer, H., Burnashev, N., Laurie, D. J., Sakmann, B., and Seeburg, P. H. (1994). Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron 12, 529–540.
Moriyoshi, K., Masu, M., Ishii, T., Shigemoto, R., Mizuno, N., and Nakanishi, S. (1991). Molecular cloning and characterization of the rat NMDA receptor. Nature 354, 31–37.
Nestler, E. J. (2005). Is there a common molecular pathway for addiction? Nat. Neurosci. 8, 1445–1449.
Paoletti, P. (2011). Molecular basis of NMDA receptor functional diversity. Eur. J. Neurosci. 33, 1351–1365.
Pellegri, G., Magistretti, P. J., and Martin, J. L. (1998). VIP and PACAP potentiate the action of glutamate on BDNF expression in mouse cortical neurones. Eur. J. Neurosci. 10, 272–280.
Pickard, L., Noël, J., Henley, J. M., Collingridge, G. L., and Molnar, E. (2000). Developmental changes in synaptic AMPA and NMDA receptor distribution and AMPA receptor subunit composition in living hippocampal neurons. J. Neurosci. 20, 7922–7931.
Plant, K., Pelkey, K. A., Bortolotto, Z. A., Morita, D., Terashima, A., McBain, C. J., Collingridge, G. L., and Isaac, J. T. R. (2006). Transient incorporation of native GluR2-lacking AMPA receptors during hippocampal long-term potentiation. Nat. Neurosci. 9, 602–604.
Roberts, A. C., DIez-GarcIa, J., Rodriguiz, R. M., López, I. P., Luján, R., Martínez-Turrillas, R., Pic, O. E., Henson, M. A., Bernardo, D. R., Jarrett, T. M., Clendeninn, D. J., López-Mascaraque, L., Feng, G., Lo, D. C., Wesseling, J. F., Wetsel, W. C., Philpot, B. D., and Pérez-Otaño, I. (2009). Downregulation of NR3A-containing NMDARs is required for synapse maturation and memory consolidation. Neuron 63, 342–356.
Rohrbough, J., and Spitzer, N. C. (1999). Ca(2+)-permeable AMPA receptors and spontaneous presynaptic transmitter release at developing excitatory spinal synapses. J. Neurosci. 19, 8528–8541.
Schultz, W. (2011). Potential vulnerabilities of neuronal reward, risk, and decision mechanisms to addictive drugs. Neuron 69, 603–617.
Sensi, S. L., Yin, H. Z., Carriedo, S. G., Rao, S. S., and Weiss, J. H. (1999). Preferential Zn2+ influx through Ca2+-permeable AMPA/kainate channels triggers prolonged mitochondrial superoxide production. Proc. Natl. Acad. Sci. U.S.A. 96, 2414–2419.
Shin, J., Shen, F., and Huguenard, J. R. (2005). Polyamines modulate AMPA receptor-dependent synaptic responses in immature layer v pyramidal neurons. J. Neurophysiol. 93, 2634–2643.
Soto, D., Coombs, I. D., Kelly, L., Farrant, M., and Cull-Candy, S. G. (2007). Stargazin attenuates intracellular polyamine block of calcium-permeable AMPA receptors. Nat. Neurosci. 10, 1260–1267.
Stern, E. A., Maravall, M., and Svoboda, K. (2001). Rapid development and plasticity of layer 2/3 maps in rat barrel cortex in vivo. Neuron 31, 305–315.
Tayler, K. K., Lowry, E., Tanaka, K., Levy, B., Reijmers, L., Mayford, M., and Wiltgen, B. J. (2011). Characterization of NMDAR-independent learning in the hippocampus. Front. Behav. Neurosci. 5:28. doi: 10.3389/fnbeh.2011.00028
Traynelis, S. F., Wollmuth, L. P., McBain, C. J., Menniti, F. S., Vance, K. M., Ogden, K. K., Hansen, K. B., Yuan, H., Myers, S. J., and Dingledine, R. (2010). Glutamate receptor ion channels: structure, regulation, and function. Pharmacol. Rev. 62, 405–496.
Ungless, M. A., Whistler, J. L., Malenka, R. C., and Bonci, A. (2001). Single cocaine exposure in vivo induces long-term potentiation in dopamine neurons. Nature 411, 583–587.
Wiltgen, B. J., Royle, G. A., Gray, E. E., Abdipranoto, A., Thangthaeng, N., Jacobs, N., Saab, F., Tonegawa, S., Heinemann, S. F., O'Dell, T. J., Fanselow, M. S., and Vissel, B. (2010). A role for calcium-permeable AMPA receptors in synaptic plasticity and learning. PLoS ONE 5:e12818. doi: 10.1371/journal.pone.0012818
Keywords: CP-AMPARs, VTA, addictive drugs, postnatal development
Citation: Bellone C and Lüscher C (2012) Drug-evoked plasticity: do addictive drugs reopen a critical period of postnatal synaptic development?. Front. Mol. Neurosci. 5:75. doi: 10.3389/fnmol.2012.00075
Received: 13 January 2012; Accepted: 29 May 2012;
Published online: 15 June 2012.
Edited by:
R. Suzanne Zukin, Albert Einstein College of Medicine, USAReviewed by:
Carlo Sala, CNR Institute of Neuroscience, ItalyEnrico Tongiorgi, University of Trieste, Italy
Copyright: © 2012 Bellone and Lüscher. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Camilla Bellone, Medical Faculty, Department of Basic Neuroscience, University of Geneva, 1, Michel-Servet, 1211 Geneve, Switzerland. e-mail:Y2FtaWxsYS5iZWxsb25lQHVuaWdlLmNo