 Lilian Enriquez-Barreto1,2†
Lilian Enriquez-Barreto1,2† Cecilia Palazzetti1 Leann H. Brennaman2 Patricia F. Maness2*‡
Cecilia Palazzetti1 Leann H. Brennaman2 Patricia F. Maness2*‡ Alfonso Fairén1*‡
Alfonso Fairén1*‡- 1Instituto de Neurociencias, Consejo Superior de Investigaciones Científicas—Universidad Miguel Hernández, San Juan de Alicante, Spain
- 2Department of Biochemistry and Biophysics, University of North Carolina School of Medicine, Chapel Hill, NC, USA
To study the potential role of neural cell adhesion molecule (NCAM) in the development of thalamocortical (TC) axon topography, wild type, and NCAM null mutant mice were analyzed for NCAM expression, projection, and targeting of TC afferents within the somatosensory area of the neocortex. Here we report that NCAM and its α-2,8-linked polysialic acid (PSA) are expressed in developing TC axons during projection to the neocortex. Pathfinding of TC axons in wild type and null mutant mice was mapped using anterograde DiI labeling. At embryonic day E16.5, null mutant mice displayed misguided TC axons in the dorsal telencephalon, but not in the ventral telencephalon, an intermediate target that initially sorts TC axons toward correct neocortical areas. During the early postnatal period, rostrolateral TC axons within the internal capsule along the ventral telencephalon adopted distorted trajectories in the ventral telencephalon and failed to reach the neocortex in NCAM null mutant animals. NCAM null mutants showed abnormal segregation of layer IV barrels in a restricted portion of the somatosensory cortex. As shown by Nissl and cytochrome oxidase staining, barrels of the anterolateral barrel subfield (ALBSF) and the most distal barrels of the posteromedial barrel subfield (PMBSF) did not segregate properly in null mutant mice. These results indicate a novel role for NCAM in axonal pathfinding and topographic sorting of TC axons, which may be important for the function of specific territories of sensory representation in the somatosensory cortex.
Introduction
Establishment of topographic connectivity between thalamic nuclei and neocortical areas is fundamental to sensory function, yet the molecular signals that govern thalamocortical (TC) axon guidance and synaptic targeting are incompletely understood. In the developing TC pathway, axons from motor, somatosensory, and visual thalamic nuclei project topographically to primary motor and sensory cortical areas along strict rostro-caudal and medio-lateral axes. Once arriving in the neocortex, TC axons are guided to area- and laminar-specific synaptic targets by poorly defined cortical cues, and then synapses are formed and further refined by activity (reviewed in López-Bendito and Molnár, 2003; Molnár et al., 2012). One emerging concept is that cell adhesion molecules guide proper TC axon guidance. The L1 family cell adhesion molecules, L1, CHL1, and NrCAM, are key determinants of guidance for specific subpopulations of TC axons at the ventral telencephalon and in the cortex (Ohyama et al., 2004; Wiencken-Barger et al., 2004; Wright et al., 2007; Demyanenko et al., 2011a,b). L1 family cell adhesion molecules are structurally related to the neural cell adhesion molecule (NCAM). However, a role for NCAM has not been established in TC axon guidance.
NCAM is a pivotal regulator of axon growth, cell adhesion, and migration. NCAM polymorphisms and altered expression have been associated with schizophrenia, bipolar disorder, and Alzheimer's disease. In addition, the entire extracellular region of NCAM is excessively released by proteolysis as a soluble fragment in affected brain regions in schizophrenia. NCAM null mutant mice lacking the three principal NCAM isoforms (NCAM180, 140, and 120) exhibit impaired learning, working memory, and hippocampal long-term potentiation (LTP). Mice expressing the soluble NCAM extracellular domain demonstrate impaired sensory gating (Pillai-Nair et al., 2005), working memory, and prefrontal cortical LTP (Brennaman et al., 2011). NCAM is also highly glycosylated, and is the principal carrier of α-2,8 polysialic acid (PSA). PSA is highly expressed during embryonic and early postnatal development and persists at low levels in the adult for plasticity (Bonfanti, 2006; Gascon et al., 2007; Rutishauser, 2008). PSA decreases NCAM–NCAM homophilic and heterophilic adhesion (Johnson et al., 2005). Mice lacking the polysialyltransferases that attach PSA to NCAM, ST8SiaII or ST8SiaIV, have decreased social interaction and motivation, impaired fear conditioning, increased exploration and aggression and impaired hippocampal LTP (Angata et al., 2004; Calandreau et al., 2010). Similar to NCAM, changes in PSA expression or polymorphisms in ST8SiaII have been observed in schizophrenia (Barbeau et al., 1995; Arai et al., 2006; Tao et al., 2007; Isomura et al., 2011), bipolar disorder (Lee et al., 2011), and autism (Anney et al., 2010). Interestingly, mice lacking PSA but not NCAM exhibit TC pathfinding defects, such that TC axons defasciculate and fail to enter the ventral telencephalon or cortex (Schiff et al., 2011). Additional deletion of NCAM partially restored TC axon guidance, suggesting that NCAM may have distinct roles from PSA in this trajectory.
Here, we report that NCAM null mutant mice exhibited aberrant TC axon guidance and an altered somatosensory map. However, projection through the ventral telencephalon was normal prenatally and demonstrates selective roles for NCAM vs. PSA in TC axon guidance.
Materials and Methods
Mice
All procedures were conducted in accordance with the National Institutes of Health and European Union guidelines and with the approval of the institutional animal care and use committees of University of North Carolina and the Instituto de Neurociencias. Homozygous-null NCAM mutant mice (C57Bl/6 background) were provided by Dr. H. Cremer (Developmental Biology Institute of Marseille Luminy, Marseille, France). NCAM deficient (KO) mice and wild type (WT) littermates were generated by crossing heterozygotes. Midday of the vaginal plug was considered as E0.5, and postnatal day 0 (P0) as day of birth.
Immunohistochemistry
Timed-pregnant dams were anaesthetized by intraperitoneal injection of ketamine (110 mg/kg) and xylazine (10 mg/kg) and the embryos were extracted by caesarean section. Embryos (E13.5, E14.5, E16.5, and E18.5) were fixed in 4% paraformaldehyde in PBS. Postnatal (P0, P2, P4, and P6) and adult (P30) mice were anaesthetized similarly and were fixed by intracardiac perfusion. Vibratome sections, 100 μm-thick, were blocked in 4% bovine serum albumin (BSA), 3% NHS, 0.2% Triton X-100 in PBS at room temperature for 1 h and incubated with primary antibodies diluted in blocking solution at room temperature overnight. Primary antibodies were: rat monoclonal antibody P61 to an intracellular epitope of the 140/180 isoforms of NCAM (gift of Drs. C. Goridis and H. Cremer, 1:50), mouse monoclonal antibodies 12E3 to α2-8 linked neuraminic acid (NeuAc-α2-8)n with n = 5 (oligo/polysialic acid; gift from Dr. T. Seki; 1:1000); rabbit polyclonal antibodies to L1 (gift from Dr. F. G. Rathjen, 1:1000) and serotonin (DiaSorin, Stillwater, IL, USA; 1:2000). The specificity of these antibodies in mouse forebrain from E13.5 through adulthood has been demonstrated (Gennarini et al., 1984; Rathjen and Schachner, 1984; Seki and Arai, 1991; Nadanaka et al., 2001). Secondary antibodies were as follows: ImmunoPure biotinylated goat anti mouse IgM (Pierce, Rockford, IL, USA; 1:100); Cy3 conjugated streptavidin (Jackson ImmunoResearch, West Grove, PA, USA; 1:500); AlexaFluor 488 anti-mouse IgM, AlexaFluor 555 goat anti rat IgG, AlexaFluor 546 anti-rabbit IgG (Molecular Probes–Invitrogen; 1:500). Sections were mounted on microscope slides and covered with AF2 (Citifluor Ltd., London, UK) or Vectashield (Vector Labs) mounting media.
DiI Injections and Analysis
Fixed brains of E14.5, E16.5, E18.5, P0, P2, and P4 wild type and NCAM-deficient mice were vibratome sectioned from the caudal end of the cerebrum. Once the dorsal thalamus was reached, the brains were removed from the vibratome and a crystal of DiI (Molecular Probes–Invitrogen) was inserted at mid rostrocaudal levels of the dorsal thalamus, comprising the primordium of the ventrobasal complex, to anterogradely label TC axons that project to the somatosensory cortex.
Mice remained in 0.05 % azide in PBS for 3–4 weeks. Brains were then sectioned at 100 μm in a vertical plane oriented 45° with respect to the coronal plane, in order to recover the maximum number of TC axons in each single section (Agmon and Connors, 1991; see schematics in Figure 4). For combined DiI—immunofluorescence staining, selected sections from DiI injected brains were processed without detergents. Sections were mounted onto glass slides with Citifluor.
The geometry of individual TC axons in the early postnatal cortex was studied in P2 and P4 wild type and null-mutant brains implanted with small DiI crystals in the dorsal thalamus. DiI labeled axons in obliquely oriented sections were imaged in a confocal microscope traced using Neurolucida 7.0 and analyzed using NeuroExplorer software (MicroBrightField, Inc.). For the analysis we considered the following parameters: number of nodes, branch order, and planar angle of bifurcations. We used the unpaired Student's t-test with statistical significance set at p < 0.05.
Nissl Staining and Cytochrome Oxidase Histochemistry
For Nissl staining, vibratome sections from adult mouse brains were mounted on gelatin-coated slides and dried overnight. Sections were defatted in an ascending series of ethanol and xylene, rehydrated again and stained in 1% aqueous cresyl violet.
For cytochrome oxidase histochemistry, adult wild type and null-mutant mice were perfusion fixed with 4% paraformaldehyde, 0.2 glutaraldehyde and 4% glucose in 0.1 phosphate buffer, stored in fixative overnight and sectioned at 100 μm in the coronal plane or in a plane parallel to the pial surface in flattened tissue blocks. Sections were processed to reveal cytochrome oxidase activity as described (Kageyama and Wong-Riley, 1982).
Sections were analyzed in a Leica DM5000 epifluorescence microscope and in confocal microscopes Leica TCS-SL and Olympus FV500.
Results
Expression of PSA-NCAM and NCAM in Thalamocortical Axons
We assessed the presence of PSA and NCAM in thalamocortical (TC) axons during prenatal development using axonal tract-tracing and immunohistochemistry. As a first step to verify that TCAs expressed NCAM and its polysialilated form PSA-NCAM, small crystals of DiI were inserted in the dorsal thalamus of E14.5 wild type mice. DiI is a lipophilic tracer that diffuses within the lipid bilayer of the plasma membrane and accurately labels axonal projections in brain tissues fixed with aldehydes (Godement et al., 1987). DiI-labeled TC axons were immunofluorescently labeled with antibodies to the cytoplasmic domain of transmembrane NCAM isoforms (140 and 180 kDa) and PSA. DiI labeled TC axons displayed both NCAM and PSA (Figures 1A,B). DiI was excited with a HeNe 543 nm laser, while an Ar/Kr 488 nm laser excited AlexaFluor 488 used to detect NCAM and PSA immunoreactivities. Comparison of Figures 1A',A”,B',B” shows that no bleeding occurred between the two detection channels in the confocal microscope, thus confirming the localization of NCAM or PSA detection signals in DiI-labeled TC axons.
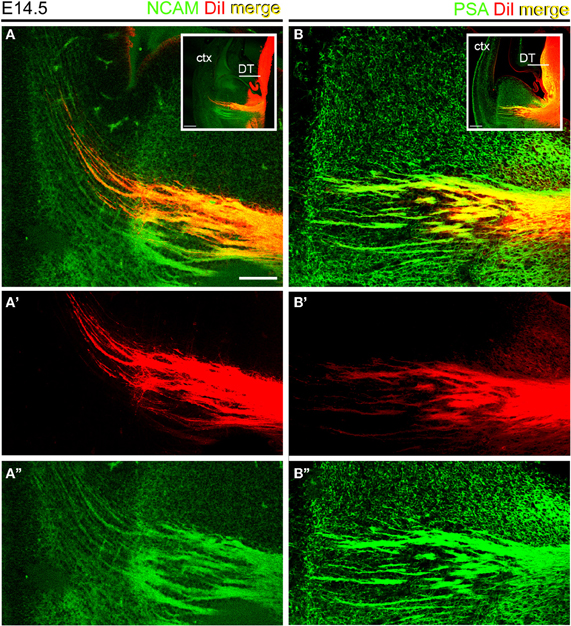
Figure 1. Thalamocortical axons expressed NCAM and PSA at E14.5. Sections containing DiI-labeled TC axons were labeled with antibodies to the intracellular domains of large NCAM isoforms (A) and PSA (B). Insets show the regions analyzed. A', A” and B and B” show the detection channels separately. Note that the red signal of DiI labeled axons in the ventral telencephalon coincided in all cases with the green signal corresponding to NCAM (A–A”) or PSA (B–B”) immunoreactivities. Maximum projections of confocal microscope stacks. DT, dorsal thalamus; ctx, cortex. Bars in A, B, 200 μm; insets, 100 μm.
Next, wild type and NCAM null mutant E13.5 and E15.5 mice (Cremer et al., 1994) were immunolabeled with antibodies to NCAM or PSA along with L1, which labels TC axons (Chung et al., 1991; Fukuda et al., 1997; Demyanenko et al., 2011b). L1-immunoreactive axons expressed transmembrane NCAM isoforms, as detected with a monoclonal antibody (mAb P61) that recognizes the cytoplasmic domain of both NCAM140 and NCAM180 (Figures 2A–A” and 3A–A”; Gennarini et al., 1984), but not NCAM120. NCAM immunoreactivity was absent in NCAM null mutant mice (Figures 2B–B” and 3B–B”), demonstrating the specificity of this antibody. Using monoclonal antibody 12E3 that reacts with small chains of PSA (Seki and Arai, 1991; Nadanaka et al., 2001), we found that L1-expressing TC axons displayed immunoreactivity for PSA of low sialic acid content in wild type mice (Figures 2C–C” and 3C–C”). In NCAM null mutant mice, 12E3 immunoreactivity was abolished (Figures 2D–D” and 3D–D”), suggesting that NCAM was the only carrier of PSA in TC axons at this stage (Seki and Arai, 1993; Cremer et al., 1997; Dityatev et al., 2004). NCAM and PSA exhibited similar patterns of immunoreactivity. PSA showed more extensive labeling than NCAM, which may be due to antibody sensitivity or PSA linkage to NCAM120, secreted NCAM, or other splice variants not recognized by our NCAM antibody. However, very little NCAM120 is present at this stage (Edelman and Chuong, 1982; Brennaman and Maness, 2008).
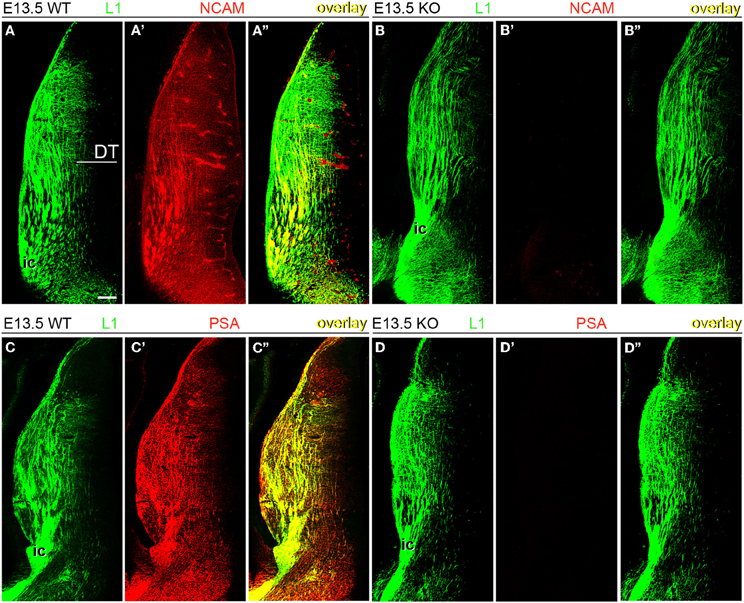
Figure 2. Expression of NCAM and oligopolysialylated NCAM in thalamocortical axons in the dorsal thalamus at E13.5. (A–A”) TC axons co-labeled by L1 and NCAM antibodies in wild type mice. (B–B”) In NCAM null mutant mice, NCAM immunostaining was absent, confirming the specificity of monoclonal antibody P61 in our material. (C–C”,D–D”) PSA immunoreactivity was present in L1-immunoreactive axons in the dorsal thalamus of wild type mice (C) but absent in NCAM null mice (D), suggesting that PSA is linked to NCAM in these axons. L1-immunoreactive TC axons formed descending fascicles that converged into the caudal limb of the internal capsule (ic) leading through the ventral telencephalon to the developing cortex. Single confocal optical sections. DT, dorsal thalamus; ic, internal capsule. Bar (in A): 100 μm.
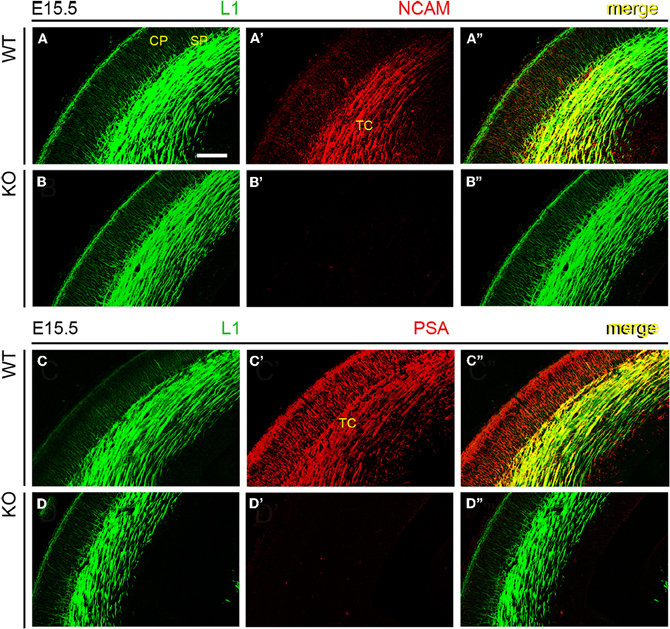
Figure 3. NCAM and PSA expression in thalamocortical axons. An antibody to adhesion molecule L1 labeled TC axons approaching the subplate in E15.5 mouse embryos, irrespective of phenotype. (A–A”) Monoclonal antibody P61, directed against an intracellular epitope of NCAM, showed the co-expression of L1 and NCAM in TC axons. (B–B”) Absence of NCAM immunoreactivity in NCAM null mutant mice. (C–C”) Monoclonal antibody 12E3 recognized oligopolysialylated chains of PSA in L1-immunoreactive TC axons. (D–D”) Absence of PSA-NCAM immunoreactivity in NCAM null mutant mice. The absence of NCAM (B”) and PSA (D”) immunoreactivities in null mutant mice suggested that, under our experimental conditions, NCAM is the sole carrier of polysialic acid in the developing cortex. Single confocal optical sections. CP, cortical plate; SP, subplate; TC, thalamocortical axons. Bar: 200 μm.
The Genetic Deletion of All Three Principal NCAM Isoforms Causes Topographically Localized Defects in Thalamocortical Axonal Guidance
In the mouse, the TC axonal projection courses through the ventral telencephalon within a narrow vertical parallelepiped oriented at 45° from the midsagittal plane (Agmon and Connors, 1991). Thus, to further study possible defects in axonal fasciculation and pathfinding in the TC axonal pathway of NCAM null mutant mice, we examined the anatomical distributions of DiI labeled TC axons in tissue sections oriented to include the maximum rostrolateral-to-caudomedial extent of this axonal projection. The sections analyzed were controlled for similar injection size and location to minimize artifacts. In these sections (Figures 4A,B, see inset), the laterally located axons were rostral and medial axons were caudal in comparative terms.
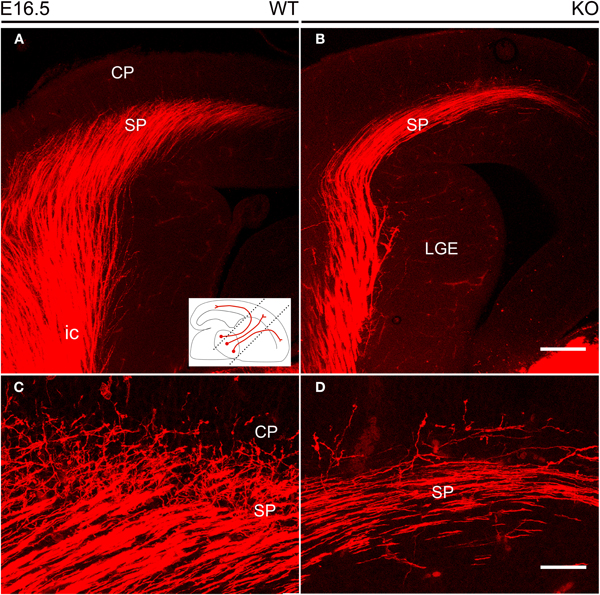
Figure 4. Thalamocortical axons labeled by DiI inserted into the dorsal thalamus at E16.5. (A,B) DiI-labeled TC axons followed the internal capsule in the ventral forebrain to reach the subplate. The inset shows the orientation of the vertical slices at 45° from the midsagittal plane used to image DiI labeling. In these sections, TC axons in the lateral part of the internal capsule are more anterior than those located medially. (C,D) TC axons started their entry into the cortical plate, but these axonal arborizations were visibly more profuse in wild type (C) than in NCAM null mice (D). Images are maximum projections of confocal optical sections, covering total thicknesses of 29 μm (A,B) or 11 μm (C,D). CP, cortical plate; ic, internal capsule; LGE, lateral ganglionic eminence; SP, subplate. Bars: 200 μm (A,B); 50 μm (C,D).
The analysis of wild type and null mutant mouse embryos during the prenatal period revealed no major changes in the anatomical trajectory of TC fibers along their subpallial course through the ventral telencephalon. At E16.5 (Figure 4), the TC axonal projection along the subpallium was sparser in null mutant mice as compared to wild type mice (Figures 4A,B), and fewer TC axons were detected in the cortical subplate impinging into the lower levels of the cortical plate (Figures 4C,D).
Defects in TC Axonal Guidance through the Ventral Telencephalon in Early Postnatal Null Mutant Mice
NCAM removal caused a more visible phenotype of altered guidance of TC axons during early postnatal development. As TC axons course through the ventral telencephalon, their characteristic fan-like distribution as seen in the obliquely oriented vertical sections (Figure 5A) was altered in early postnatal NCAM null mice with respect to wild type animals. In null mutant P0 mice, TC axons navigated in a disorderly manner and, unlike in wild type mice, gave rise to local axon collaterals in the ventral telencephalon (Figures 5A,B). In addition, NCAM null TC axons in the rostrolateral tier of the internal capsule became disoriented within the ventral telencephalon, bent abruptly at right angles (Figure 5B, arrows), and failed to reach the cortex unlike wild type mice (Figure 5A). The packing density of TC axons in the cortex in null mutant mice (P0) was also visibly decreased, consistent with the observed disorientation (Figures 5C,D).
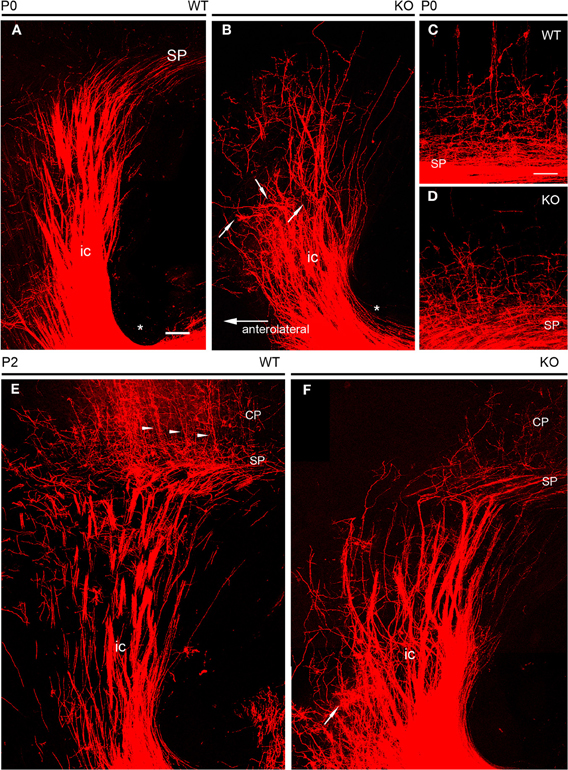
Figure 5. DiI tracing of thalamocortical fibers at early postnatal ages. (A,B) At P0, thick fascicles of DiI-labeled TC axons traversed the ventral telencephalon in their way to the developing cortex. The course of TC axons along the internal capsule was straight in wild type embryos (A), where few axon collaterals dispersed laterally. In null mutant mice (B), TC axon fascicles in the lateral parts of the pathway bent abruptly, failing to reach the cortex; arrows point at selected examples of dispersed axon fascicles. Axons at more medial locations, although less dispersed, navigated in a disorderly manner, giving rise to abundant axon collaterals in the ventral telencephalon. Asterisks in A and B mark the inferior level of the internal capsule in the ventral telencephalon. (C,D) TC axons entering the lower tier of the cortical plate in wild type and knockout mice at P0. Null mutant mice showed less densely packed axonal arborizations than wild type mice. (E,F) At P2, compared to wild type mice (C), TC axons in the ventral telencephalon of null mutant mice (D) dispersed laterally; one extreme example is marked with an arrow. Null mutant mice showed sparser TC axons in the cortex than wild type mice. Arrowheads in E point to retrogradely DiI labeled neurons in the cortical plate. The subplate was identified as the level TC axons bend medially to adopt trajectories parallel to the palial surface. Maximum confocal projections. Bar: 100 μm.
The localized anatomical changes in the subpallial course of TC axons persisted by P2 (Figures 5E,F), at the time TC axons start to achieve their final distribution within the cerebral cortex, and cortical layers start their cytoarchitectonic differentiation (Agmon et al., 1993; López-Bendito and Molnár, 2003). Thus, in P2 null mutant mice, the most medially located axons seemed unchanged. On the contrary, TC axons located in the rostrolateral sector of the internal capsule dispersed laterally at difference to wild type axons (Figures 5C,D).
DiI crystals were inserted into the middle third of the dorsal thalamus where the ventrobasal complex of the thalamus that projects to the somatosensory cortex will develop, and avoided the lateral geniculate primordium located more caudally. Thus, the somatosensory cortex was the probable cortical area of destination of the DiI labeled TC axons. This prompted the question as to whether localized changes in the TC axons as they course through the ventral telencephalon during the early postnatal period might result in alterations in the distribution of TC axons within the cerebral cortex and localized alterations in the cytoarchitectonic organization of this target cortical area.
Intracortical Arborizations of Thalamocortical Axons in NCAM Null Mutant Mice
NCAM null mutant mice consistently showed reduced packing density of DiI labeled TC axons in the developing cortex (Figures 6A,B) in spite of the variability of DiI labeling. However, it was unclear whether this was accompanied by changes in the TC axon arborizations within the postnatal cerebral cortex. Prompted by the finding by Yamamoto et al. (2000) that PSA removal alters the orientation and layer targeting of TC axons in slice culture experiments, we analyzed whether NCAM deletion likewise causes alterations in the branching patterns of preterminal TC axons labeled anterogradely with DiI. We studied two different ages corresponding to the first week after birth when segregation into barrels occurs, at P2 (WT, n = 6 hemispheres; KO, n = 6 hemispheres) and at P4 (WT, n = 7 hemispheres; KO, n = 10 hemispheres).
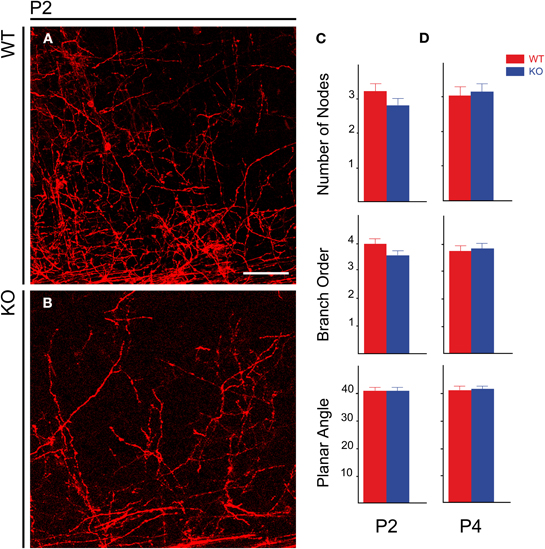
Figure 6. Distribution of TC axons in the lower layers of the developing cerebral cortex in postnatal animals. (A,B) Distribution of TC preterminal axons in the cortical plate of P2 wild type (A) and mutant (B) mice. (C,D) Bar graphs showing the absence of modifications in the geometry of TC axons within the cortex in null mutant mice at P2 (C) and P4 (D). Bar: 50 μm.
For the analysis of the geometry of individual TC axons in the cortex, we considered the following parameters: number of nodes, branch order, and planar angle of preterminal TC axons. The number of nodes and the branch order give idea of the complexity of the axonal trees. The Student's t-test detected no differences between wild type and null mutant mice. At P2 (Figure 6C), the values for number of nodes were 3.21 ± 0.22 per axon for wild type mice vs. 2.79 ± 0.21 for null mutant mice (mean ± s.e.m.), p =0.09. At P4 (Figure 6D), values were 3.04 ± 0.25 for WT mice vs. 3.15 ± 0.24 for null mutant mice, p = 0.38 (Figure 6D). Branch order was also unchanged: at P2—WT, 3.96 ± 0.19 vs. null mutant, 3.54 ± 0.18 (Figure 6C), p = 0.06; at P4—WT, 3.74 ± 0.21 vs. null mutant, 3.83 ± 0.18, p = 0.38 (Figure 6D) The planar angle is that formed in the section plane by the two segments uniting an n order node with the next order (n +1) node in an arborization, and measures a characteristic of the branching pattern of the axons disregarding the axonal trajectory. Planar angles were also unchanged at P2 and P4: P2—WT, 40.97° ± 1.17 vs. null mutant, 40.69° ± 1.45, p = 0.44 (Figure 6C); P4—WT, 41.22° ± 1.55 vs. null mutant, 41.48° ± 1.31, p = 0.45 (Figure 6D). Together, these results indicated that, contrary to PSA removal, NCAM removal does not alter the branching patterns of TC axons in the cortex.
The Genetic Deletion of NCAM Alters the Somatosensory Map in the Cerebral Cortex
Layer IV of the rodent somatosensory cortex contains multicellular units called barrels so arranged as to form a visible map of the sensory periphery (Woolsey and Van der Loos, 1970; Van der Loos and Woolsey, 1973). This distinctive feature makes the somatosensory system a remarkable model to study molecular mechanisms implicated in the topographic sorting of TC axons to neocortical areas. We focused on the anatomical representation of the sensory map in the somatosensory cortex, whose refinement along development requires the correct targeting of TC axons.
We first analyzed in early postnatal mice the distribution of TC axons within layer IV of the somatosensory cortex (Figure 7). During the first postnatal week, thalamic fibers navigate through layers V and VI and invade layer IV, which is incompletely differentiated. TC axons assume their characteristic periphery-related pattern and impose a barrel arrangement onto layer IV (Agmon et al., 1993; López-Bendito and Molnár, 2003). Developing TC axons ascend to layer IV where they form spatially periodic clusters of terminations of gradually increasing complexity that correspond to the barrels, rather than branching profusely within lower cortical layers with subsequent retraction (Agmon et al., 1993, 1995). To see how TC axons segregate in NCAM null mice, we analyzed the pattern of distribution of thalamic terminals by means of serotonin (5-HT) immunohistochemistry. Thalamic relay neurons transiently express the plasma 5-HT transporter (5-HTT) and the vesicular monoamine transporter type 2 (VMAT2), allowing 5-HT to concentrate in the TC axons and axon terminals during early postnatal life (Lebrand et al., 1996). 5-HT immunostaining was continuous in the forming layer IV of P2 mice, but at P6 it suggested a discrete pattern in wild type mice, but not NCAM null mice (Figure 7).
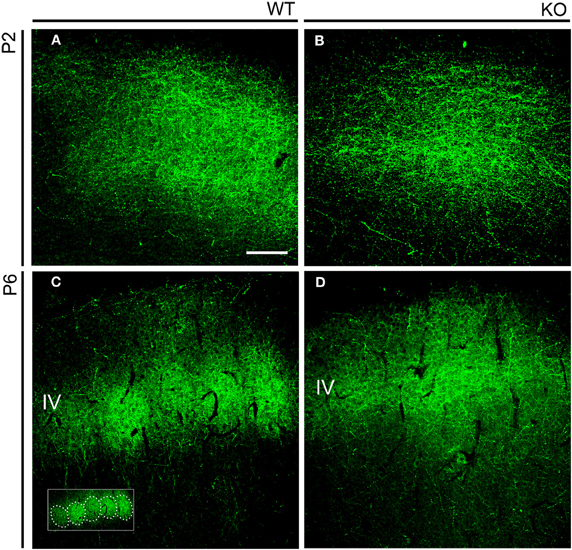
Figure 7. Serotonin immunohistochemistry in the early postnatal somatosensory cortex. (A,B). Serotonin immunostaining highlighted the distribution of TC axons in the developing cortex. At P2, at a time barrel units are not yet formed, immunostaining revealed a uniform distribution of axons in both wild type and mutant mice. (C,D). At P6, serotonin immunoreactivity clustered in layer IV in a manner reminiscent of barrels (highlighted in the inset) in wild type animals (C). In P6 NCAM null mutant mice, TC axons also localized to layer IV but showed a continuous distribution (D). Confocal optical sections. IV, cortical layer IV. Bar: 100 μm.
Nissl staining of coronal sections of adult brains revealed mild disruptions of the somatosensory map in NCAM null mutant mice (Figures 8A–D). As compared to the well-defined barrels of wild type animals (n = 5) due to the presence of distinct barrel septa, null mutant mice (n = 5) showed indistinct barrels with no discernible septa between them (Figures 8A,C) and the Nissl stain revealed a microlaminar deposition of cells within layer IV (Figures 8C,D). These changes occurred through all antero-posterior levels of the somatosensory area of the cortex. Layer IV of the somatosensory cortex in null mutant mice remained prominent, suggesting reorganization of TC axons (Figures 8A–B). Cytochrome oxidase histochemistry (Wong-Riley and Welt, 1980) in coronal sections highlighted individual barrels in null mutant mice (n = 3), though the staining was fainter in null mice as compared to their wild type (n = 3) littermates, as shown for rostral barrels (Figures 8E,F). This was taken as an indication that the overall organization of the somatosensory cortex was preserved in null mutant mice, although the refined cytoarchitectonic organization of the barrels was somewhat altered, indicating that layer IV neurons could be abnormally deployed in the barrels and septa.
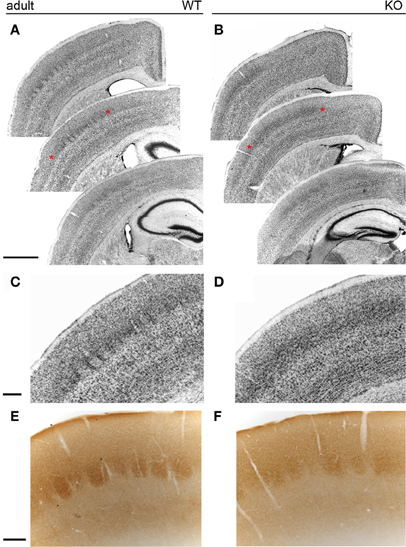
Figure 8. The barrelfield in the adult somatosensory cortex. In wild type animals (A), Nissl staining revealed individual cytoarchitectonic units (barrels) in layer IV of the somatosensory cortex. In contrast, Nissl staining of NCAM mutant mice (B) showed less distinct barrels. (C,D) Enlarged views of the areas marked with asterisks in (A,B). While septa were evident in wild type animals (C), null mutant mice (D) lacked well-delineated septa and layer IV cells formed horizontally placed microlaminae in knockout mice. (E,F) Cytochrome oxidase histochemistry of coronal sections revealed no obvious changes between wild type and null mutant mice. Bars: A, B, 1 mm (in A); C, D, 200 μm (in C); E, F, 200 μm (in E).
Then, we analyzed the somatosensory map in tangential sections of the cortex for the occurrence of topographic changes in adult NCAM null mutants. The posteromedial barrel subfield (PMBSF) contains a group of large, ovoid barrels whose topographic distribution is homeomorphic to that of the largest vibrissa follicles of the animal's snout. The anterolateral barrel subfield (ALBSF) contains similar yet smaller barrels, which correspond to the smallest and less prominent whiskers of the animal's face. As shown in tangential reconstructions of Nissl staining (Figures 9A,B), the barrel septa were less prominent in null mutant mice (n = 4) than in wild type mice (n = 4) and were indiscernible at rostral positions within the PMBSF of null mutant mice (Figure 9B). Cytochrome oxidase staining delineated distinct barrels in both genotypes (wild type, n = 4; null mutant, n = 6). However, in null mutant mice, only the most caudomedial part of the PMBSF that contains the straddler barrels (α–δ) and the first large barrels of rows A–E, showed such a clear delineation and their overall topography seemed unchanged (Figures 9C,D). The other barrels in the PMBSF and in the entire ALBSF were profoundly altered in null mutant mice, and were virtually indistinct. Both Nissl staining and cytochrome oxidase histochemistry unveiled a prominent phenotype trait, the disruption of barrels in the ALBSF and of the most rostral barrels in the PMBSF. These alterations in the barrel field of the somatosensory cortex suggested that the regional alteration of NCAM null TC axons might contribute to disrupted topographic mapping, and that NCAM may be required for the topographical anatomical distribution of TC axons in the somatosensory cortex.
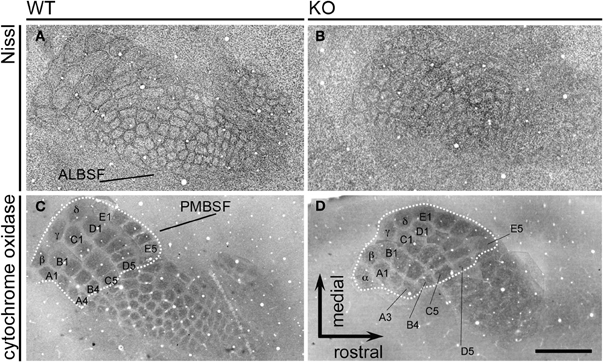
Figure 9. The adult barrelfield in tangential sections. Nissl staining (A,B) of tangential sections show the PosteroMedial Barrel SubField (PMBSF) and the AnteroLateral Barrel SubField (ALBSF) subdivisions of the primary somatosensory area. The PMBSF barrels displayed blurred septa in null mutant mice, and ALBSF barrels were less distinct in mutant than in wild type mice. Cytochrome oxidase histochemistry (C,D) was used to assess possible changes in null mutant mice. The dotted lines delineate the part of the PMBSF containing the straddler barrels (α–δ) and the first posteromedial barrels of each row A to E (A1–A3, B1–B4, C1–C5, D1–D5, E1–E5); these barrels seemed unmodified in mutant animals. On the contrary, the rostrolateral barrels in the PMBSF and the barrels in the ALBSF were blurred and indistinct in mutant mice, suggesting that the genetic deletion of NCAM causes selective topographical changes in the somatosensory map. Images are reconstructions of serial sections. Bar: 1 mm.
Discussion
We have shown here that NCAM participates in the guidance of TC axons to the somatosensory cortex, and provide the first evidence for a role for NCAM in TC topography. PSA and NCAM were expressed on developing TC axons from E13.5 to E15.5. In embryonic stages, NCAM null TC axons traversed the ventral telencephalon normally, but a contingent of axons appeared to ectopically branch in the ventral telencephalon and misproject into the dorsal telencephalon in early postnatal stages. In anterograde DiI labeling at E16.5, NCAM null mutant mice appeared to have fewer axons projecting from the middle third of the dorsal thalamus which forms the ventrobasal complex and sends axons to the somatosensory cortex. The rostral third of the thalamus will give rise to ventroanterior and ventrolateral nuclei which project to the motor cortex, while the caudal third of the thalamus will give rise to the dorsal lateral geniculate nucleus that projects to the visual cortex. Accordingly, rostral TC axons in the internal capsule failed to appropriately target the dorsal telencephalon. Together, these results suggest that TC axon guidance to the motor and somatosensory cortices may be altered in NCAM null mice. In accordance with these findings, NCAM null mutant mice displayed aberrant barrel architecture at P6 and in adult somatosensory cortex.
Our findings are distinct from those observed with PSA-deficient mice. TC axons from mice lacking both polysialyltransferases (ST8SiaII and ST8SiaIV) failed to enter the ventral telencephalon at E15.5, and instead terminated in the reticular thalamic nucleus (Weinhold et al., 2005; Schiff et al., 2011). The consequence of PSA removal is increased and precocious NCAM-dependent adhesion (Hildebrandt et al., 2007). The prominent role of PSA elimination as the origin of this phenotype contrasted with the absence of major phenotype differences in the TC axonal projection in triple knockout animals for ST8SiaII, ST8SiaIV, and NCAM (Schiff et al., 2011), suggesting that additional NCAM removal can restore normal TC axon trajectory in the ventral telencephalon. NCAM null TC axons enter the ventral telencephalon and sort normally at embryonic stages. However, our study also shows that NCAM null TC axons showed pathfinding defects in the ventral telencephalon during the early postnatal life, at the time TC axons establish their connections in the cerebral cortex, and also regional defects in the establishment of the somatosensory map in the cerebral cortex that are described here for the first time. Thus, our new results on a postnatal role for NCAM in TC axon projections complement the role of PSA, by identifying specific roles for NCAM in this trajectory.
The role of other members of the immunoglobulin superfamily of neural cell adhesion molecules in TC axonal pathfinding has been studied intensively in the last few years. L1 family cell adhesion molecules also show shifts of contingents of rostral thalamic axons to caudal regions in the developing and final topographic map. Close homolog of L1, CHL1, directs TC axons to the somatosensory cortex (Wright et al., 2007), and cooperates with L1 to guide axons to the motor cortex (Demyanenko et al., 2011b). Additionally, NrCAM also participates in topographic mapping of TC axons to the motor and somatosensory cortices (Demyanenko et al., 2011a). Together these results suggest that Ig superfamily cell adhesion molecules are critical for proper TC axon guidance to motor and somatosensory cortices. However, this also raises the question as to whether the adhesion molecules cooperate on the same axon or guide subpopulations of axons from the thalamus. Of particular interest in this regard is that L1, CHL1, and NrCAM respond to different guidance cues to promote proper targeting of TC axons. Repellant guidance cues such as ephrins (Dufour et al., 2003) and semaphorins (Wright et al., 2007; Demyanenko et al., 2011a) are expressed in gradients within the ventral telencephalon and allow for preliminary sorting of thalamic axons based on their expression of Eph and Neuropilin receptors. CHL1 interacts with Neuropilin 1 to respond to Sema3A, which is required for proper topographic mapping to the somatosensory cortex (Wright et al., 2007). In mapping motor axons, CHL1, and L1 differentially interact with EphA7 and EphA3, 4 or 7, respectively, to mediate repellant axon guidance in response to ephrinA5 (Demyanenko et al., 2011b). NrCAM associates with Neuropilin 2 and Sema3F to promote proper mapping of motor and somatosensory axons (Demyanenko et al., 2011a). While we did not identify which repellent guidance cue cooperates with NCAM, a recent study has shown that NCAM can associate with EphA3 to promote proper inhibitory synaptic targeting in response to ephrinA5 (Brennaman et al., 2012). The differential responses of these adhesion molecules to repellent guidance cues, suggest that while some axons may express L1, CHL1, and NCAM to respond to ephrinA5, others may only express NrCAM or CHL1. This combinatorial effect would allow all axons from individual thalamic nuclei to project to their correct targets.
A limitation of this study is that retrograde TC labeling analysis was not performed in P5 mice once the final map is established. While our results suggest that motor and somatosensory axons misproject at early time points, only the somatosensory barrel field was examined in adult animals. TC axons shape the barrel field into the pattern of the whiskers (Erzurumlu and Jhaveri, 1990; Agmon et al., 1993; Schlaggar and O'Leary, 1994; Agmon et al., 1995; Wu et al., 2011), followed by the organization of layer IV neurons into discrete barrel units 1–2 days later (Rice and Van der Loos, 1977; Jhaveri et al., 1991). Therefore, our findings that the formation of barrels in layer IV at P6 were altered and the disorganization of the ALBSF and of the rostrolateral tier of the PMBSF in NCAM null mutant mice is likely a result of TC axon misguidance. However, we cannot determine whether these observations are a result of misprojection of axons to the visual cortex or if the axons degenerated.
The results presented here provide insight into a novel role for NCAM in TC topographic mapping. This may have profound implications in neuropsychiatric disorders where mutations in NCAM or changes in expression are observed, suggesting that TC mapping may be disturbed resulting in impaired sensory function.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Supported by grants from National Institutes of Health Grants NS049109 (Patricia F. Maness) and MH064056 (Silvio Conte Center for Neuroscience of Mental Disorders) (Patricia F. Maness) and the Spanish Ministerio de Ciencia e Innovación/Fondo Europeo para el Desarrollo Regional (FEDER), BFU2004-04660, BFU2007-60263, and BFU2010-17305 (Alfonso Fairén). Thanks to H. Cremer, C. Goridis, F. J. Rathjen, and T. Seki for antibodies, Galina P. Demyanenko and Cristina Gil-Sanz for advice and Belén Andrés for excellent technical help.
References
Agmon, A., and Connors, B. W. (1991). Thalamocortical responses of mouse somatosensory (barrel) cortex in vitro. Neuroscience 41, 365–379.
Agmon, A., Yang, L. T., Jones, E. G., and O'Dowd, D. K. (1995). Topological precision in the thalamic projection to neonatal mouse barrel cortex. J. Neurosci. 15, 549–561.
Agmon, A., Yang, L. T., O'Dowd, D. K., and Jones, E. G. (1993). Organized growth of thalamocortical axons from the deep tier of terminations into layer IV of developing mouse barrel cortex. J. Neurosci. 13, 5365–5382.
Angata, K., Long, J. M., Bukalo, O., Lee, W., Dityatev, A., Wynshaw-Boris, A., Schachner, M., Fukuda, M., and Marth, J. D. (2004). Sialyltransferase ST8Sia-II assembles a subset of polysialic acid that directs hippocampal axonal targeting and promotes fear behavior. J. Biol. Chem. 279, 32603–32613.
Anney, R., Klei, L., Pinto, D., Regan, R., Conroy, J., Magalhaes, T. R., Correia, C., Abrahams, B. S., Sykes, N., Pagnamenta, A. T., Almeida, J., Bacchelli, E., Bailey, A. J., Baird, G., Battaglia, A., Berney, T., Bolshakova, N., Bölte, S., Bolton, P. F., Bourgeron, T., Brennan, S., Brian, J., Carson, A. R., Casallo, G., Casey, J., Chu, S. H., Cochrane, L., Corsello, C., Crawford, E. L., Crossett, A., Dawson, G., de Jonge, M., Delorme, R., Drmic, I., Duketis, E., Duque, F., Estes, A., Farrar, P., Fernandez, B. A., Folstein, S. E., Fombonne, E., Freitag, C. M., Gilbert, J., Gillberg, C., Glessner, J. T., Goldberg, J., Green, J., Guter, S. J., Hakonarson, H., Heron, E. A., Hill, M., Holt, R., Howe, J. L., Hughes, G., Hus, V., Igliozzi, R., Kim, C., Klauck, S. M., Kolevzon, A., Korvatska, O., Kustanovich, V., Lajonchere, C. M., Lamb, J. A., Laskawiec, M., Leboyer, M., Le Couteur, A., Leventhal, B. L., Lionel, A. C., Liu, X. Q., Lord, C., Lotspeich, L., Lund, S. C., Maestrini, E., Mahoney, W., Mantoulan, C., Marshall, C. R., McConachie, H., McDougle, C. J., McGrath, J., McMahon, W. M., Melhem, N. M., Merikangas, A., Migita, O., Minshew, N. J., Mirza, G. K., Munson, J., Nelson, S. F., Noakes, C., Noor, A., Nygren, G., Oliveira, G., Papanikolaou, K., Parr, J. R., Parrini, B., Paton, T., Pickles, A., Piven, J., Posey, D. J., Poustka, A., Poustka, F., Prasad, A., Ragoussis, J., Renshaw, K., Rickaby, J., Roberts, W., Roeder, K., Roge, B., Rutter, M. L., Bierut, L. J., Rice, J. P., Salt, J., Sansom, K., Sato, D., Segurado, R., Senman, L., Shah, N., Sheffield, V. C., Soorya, L., Sousa, I., Stoppioni, V., Strawbridge, C., Tancredi, R., Tansey, K., Thiruvahindrapduram, B., Thompson, A. P., Thomson, S., Tryfon, A., Tsiantis, J., Van Engeland, H., Vincent, J. B., Volkmar, F., Wallace, S., Wang, K., Wang, Z., Wassink, T. H., Wing, K., Wittemeyer, K., Wood, S., Yaspan, B. L., Zurawiecki, D., Zwaigenbaum, L., Betancur, C., Buxbaum, J. D., Cantor, R. M., Cook, E. H., Coon, H., Cuccaro, M. L., Gallagher, L., Geschwind, D. H., Gill, M., Haines, J. L., Miller, J., Monaco, A. P., Nurnberger, J. I. Jr., Paterson, A. D., Pericak-Vance, M. A., Schellenberg, G. D., Scherer, S. W., Sutcliffe, J. S., Szatmari, P., Vicente, A. M., Vieland, V. J., Wijsman, E. M., Devlin, B., Ennis, S., and Hallmayer, J. (2010). A genome-wide scan for common alleles affecting risk for autism. Hum. Mol. Genet. 19, 4072–4082.
Arai, M., Yamada, K., Toyota, T., Obata, N., Haga, S., Yoshida, Y., Nakamura, K., Minabe, Y., Ujike, H., Sora, I., Ikeda, K., Mori, N., Yoshikawa, T., and Itokawa, M. (2006). Association between polymorphisms in the promoter region of the sialyltransferase 8B (SIAT8B) gene and schizophrenia. Biol. Psychiatry 59, 652–659.
Barbeau, D., Liang, J. J., Robitalille, Y., Quirion, R., and Srivastava, L. K. (1995). Decreased expression of the embryonic form of the neural cell adhesion molecule in schizophrenic brains. Proc. Natl. Acad. Sci. U.S.A. 92, 2785–2789.
Bonfanti, L. (2006). PSA-NCAM in mammalian structural plasticity and neurogenesis. Prog. Neurobiol. 80, 129–164.
Brennaman, L. H., Kochlamazashvili, G., Stoenica, L., Nonneman, R. J., Moy, S. S., Schachner, M., Dityatev, A., and Maness, P. F. (2011). Transgenic mice overexpressing the extracellular domain of NCAM are impaired in working memory and cortical plasticity. Neurobiol. Dis. 43, 372–378.
Brennaman, L. H., and Maness, P. F. (2008). Developmental regulation of GABAergic interneuron branching and synaptic development in the prefrontal cortex by soluble neural cell adhesion molecule. Mol. Cell. Neurosci. 37, 781–793.
Brennaman, L. H., Zhang, X., Guan, H., Triplett, J. W., Brown, A., Demyanenko, G. P., Manis, P. B., Landmesser, L., and Maness, P. F. (2012). Polysialylated NCAM and ephrinA/EphA regulate synaptic development of GABAergic interneurons in prefrontal cortex. Cereb. Cortex. doi: 10.1093/cercor/bhr392. [Epub ahead of print].
Calandreau, L., Márquez, C., Bisaz, R., Fantin, M., and Sandi, C. (2010). Differential impact of polysialyltransferase ST8SiaII and ST8SiaIV knockout on social interaction and aggression. Genes Brain Behav. 9, 958–967.
Chung, W. W., Lagenaur, C. F., Yan, Y. M., and Lund, J. S. (1991). Developmental expression of neural cell adhesion molecules in the mouse neocortex and olfactory bulb. J. Comp. Neurol. 314, 290–305.
Cremer, H., Chazal, G., Goridis, C., and Represa, A. (1997). NCAM is essential for axonal growth and fasciculation in the hippocampus. Mol. Cell. Neurosci. 8, 323–335.
Cremer, H., Lange, R., Christoph, A., Plomann, M., Vopper, G., Roes, J., Brown, R., Baldwin, S., Kraemer, P., Scheff, S., Barthels, D., Rajewsky, K., and Wille, W. (1994). Inactivation of the N-CAM gene in mice results in size reduction of the olfactory bulb and deficits in spatial learning. Nature 367, 455–459.
Demyanenko, G. P., Riday, T. T., Tran, T. S., Dalal, J., Darnell, E. P., Brennaman, L. H., Sakurai, T., Grumet, M., Philpot, B. D., and Maness, P. F. (2011a). NrCAM deletion causes topographic mistargeting of thalamocortical axons to the visual cortex and disrupts visual acuity. J. Neurosci. 31, 1545–1558.
Demyanenko, G. P., Siesser, P. F., Wright, A. G., Brennaman, L. H., Bartsch, U., Schachner, M., and Maness, P. F. (2011b). L1 and CHL1 cooperate in thalamocortical axon targeting. Cereb. Cortex 21, 401–412.
Dityatev, A., Dityateva, G., Sytnyk, V., Delling, M., Toni, N., Nikonenko, I., Muller, D., and Schachner, M. (2004). Polysialylated neural cell adhesion molecule promotes remodeling and formation of hippocampal synapses. J. Neurosci. 24, 9372–9382.
Dufour, A., Seibt, J., Passante, L., Depaepe, V., Ciossek, T., Frisen, J., Kullander, K., Flanagan, J. G., Polleux, F., and Vanderhaeghen, P. (2003). Area specificity and topography of thalamocortical projections are controlled by ephrin/Eph genes. Neuron 39, 453–465.
Edelman, G. M., and Chuong, C. M. (1982). Embryonic to adult conversion of neural cell adhesion molecules in normal and staggerer mice. Proc. Natl. Acad. Sci. U.S.A. 79, 7036–7040.
Erzurumlu, R. S., and Jhaveri, S. (1990). Thalamic axons confer a blueprint of the sensory periphery onto the developing rat somatosensory cortex. Dev. Brain Res. 56, 229–234.
Fukuda, T., Kawano, H., Ohyama, K., Li, H. P., Takeda, Y., Oohira, A., and Kawamura, K. (1997). Immunohistochemical localization of neurocan and L1 in the formation of thalamocortical pathway of developing rats. J. Comp. Neurol. 382, 141–152.
Gascon, E., Vutskits, L., and Kiss, J. Z. (2007). Polysialic acid-neural cell adhesion molecule in brain plasticity: from synapses to integration of new neurons. Brain Res. Rev. 56, 101–118.
Gennarini, G., Rougon, G., Deagostini-Bazin, H., Hirn, M., and Goridis, C. (1984). Studies on the transmembrane disposition of the neural cell adhesion molecule N-CAM. A monoclonal antibody recognizing a cytoplasmic domain and evidence for the presence of phosphoserine residues. Eur. J. Biochem. 142, 57–64.
Godement, P., Vanselow, J., Thanos, S., and Bonhoeffer, F. (1987). A study in developing visual systems with a new method of staining neurones and their processes in fixed tissue. Development 101, 697–713.
Hildebrandt, H., Mühlenhoff, M., Weinhold, B., and Gerardy-Schahn, R. (2007). Dissecting polysialic acid and NCAM functions in brain development. J. Neurochem. 103(Suppl. 1), 56–64.
Isomura, R., Kitajima, K., and Sato, C. (2011). Structural and functional impairments of polysialic acid by a mutated polysialyltransferase found in schizophrenia. J. Biol. Chem. 286, 21535–21545.
Jhaveri, S., Erzurumlu, R. S., and Crossin, K. (1991). Barrel construction in rodent neocortex: role of thalamic afferents versus extracellular matrix molecules. Proc. Natl. Acad. Sci. U.S.A. 88, 4489–4493.
Johnson, C. P., Fujimoto, I., Rutishauser, U., and Leckband, D. E. (2005). Direct evidence that neural cell adhesion molecule (NCAM) polysialylation increases intermembrane repulsion and abrogates adhesion. J. Biol. Chem. 280, 137–145. Erratum in J. Biol. Chem. 280, 23424.
Kageyama, G. H., and Wong-Riley, M. T. (1982). Histochemical localization of cytochrome oxidase in the hippocampus: correlation with specific neuronal types and afferent pathways. Neuroscience 7, 2337–2361.
Lebrand, C., Cases, O., Adelbrecht, C., Doye, A., Alvarez, C., El Mestikawy, S., Seif, I., and Gaspar, P. (1996). Transient uptake and storage of serotonin in developing thalamic neurons. Neuron 17, 823–835.
Lee, M. T., Chen, C. H., Lee, C. S., Chen, C. C., Chong, M. Y., Ouyang, W. C., Chiu, N. Y., Chuo, L. J., Chen, C. Y., Tan, H. K., Lane, H. Y., Chang, T. J., Lin, C. H., Jou, S. H., Hou, Y. M., Feng, J., Lai, T. J., Tung, C. L., Chen, T. J., Chang, C. J., Lung, F. W., Chen, C. K., Shiah, I. S., Liu, C. Y., Teng, P. R., Chen, K. H., Shen, L. J., Cheng, C. S., Chang, T. P., Li, C. F., Chou, C. H., Chen, C. Y., Wang, K. H., Fann, C. S., Wu, J. Y., Chen, Y. T., and Cheng, A. T. (2011). Genome-wide association study of bipolar I disorder in the Han Chinese population. Mol. Psychiatry 16, 548–556.
López-Bendito, G., and Molnár, Z. (2003). Thalamocortical development: how are we going to get there? Nat. Rev. Neurosci. 4, 276–289.
Molnár, Z., Garel, S., López-Bendito, G., Maness, P., and Price, D. J. (2012). Mechanisms controlling the guidance of thalamocortical axons through the embryonic forebrain. Eur. J. Neurosci. 35, 1573–1585.
Nadanaka, S., Sato, C., Kitajima, K., Katagiri, K., Irie, S., and Yamagata, T. (2001). Occurrence of oligosialic acids on integrin alpha 5 subunit and their involvement in cell adhesion to fibronectin. J. Biol. Chem. 276, 33657–33664.
Ohyama, K., Tan-Takeuchi, K., Kutsche, M., Schachner, M., Uyemura, K., and Kawamura, K. (2004). Neural cell adhesion molecule L1 is required for fasciculation and routing of thalamocortical fibres and corticothalamic fibres. Neurosci. Res. 48, 471–475.
Pillai-Nair, N., Panicker, A. K., Rodriguiz, R. M., Gilmore, K. L., Demyanenko, G. P., Huang, J. Z., Wetsel, W. C., and Maness, P. F. (2005). Neural cell adhesion molecule-secreting transgenic mice display abnormalities in GABAergic interneurons and alterations in behavior. J. Neurosci. 25, 4659–4671.
Rathjen, F. G., and Schachner, M. (1984). Immunocytological and biochemical characterization of a new neuronal cell surface component (L1 antigen) which is involved in cell adhesion. EMBO J. 3, 1–10.
Rice, F. L., and Van der Loos, H. (1977). Development of the barrels and barrel field in the somatosensory cortex of the mouse. J. Comp. Neurol. 171, 545–460.
Rutishauser, U. (2008). Polysialic acid in the plasticity of the developing and adult vertebrate nervous system. Nat. Rev. Neurosci. 9, 26–35.
Schiff, M., Röckle, I., Burkhardt, H., Weinhold, B., and Hildebrandt, H. (2011). Thalamocortical pathfinding defects precede degeneration of the reticular thalamic nucleus in polysialic acid-deficient mice. J. Neurosci. 31, 1302–1312.
Schlaggar, B. L., and O'Leary, D. D. (1994). Early development of the somatotopic map and barrel patterning in rat somatosensory cortex. J. Comp. Neurol. 346, 80–96.
Seki, T., and Arai, Y. (1991). Expression of highly polysialylated NCAM in the neocortex and piriform cortex of the developing and the adult rat. Anat. Embryol. (Berl.) 184, 395–401.
Seki, T., and Arai, Y. (1993). Distribution and possible roles of the highly polysialylated neural cell adhesion molecule (NCAM-H) in the developing and adult central nervous system. Neurosci. Res. 17, 265–290.
Tao, R., Li, C., Zheng, Y., Qin, W., Zhang, J., Li, X., Xu, Y., Shi, Y. Y., Feng, G., and He, L. (2007). Positive association between SIAT8B and schizophrenia in the Chinese Han population. Schizophr. Res. 90, 108–114.
Van der Loos, H., and Woolsey, T. A. (1973). Somatosensory cortex: structural alterations following early injury to sense organs. Science 179, 395–398.
Weinhold, B., Seidenfaden, R., Röckle, I., Mühlenhoff, M., Schertzinger, F., Conzelmann, S., Marth, J. D., Gerardy-Schahn, R., and Hildebrandt, H. (2005). Genetic ablation of polysialic acid causes severe neurodevelopmental defects rescued by deletion of the neural cell adhesion molecule. J. Biol. Chem. 280, 42971–42977.
Wiencken-Barger, A. E., Mavity-Hudson, J., Bartsch, U., Schachner, M., and Casagrande, V. A. (2004). The role of L1 in axon pathfinding and fasciculation. Cereb. Cortex 14, 121–131.
Wong-Riley, M. T., and Welt, C. (1980). Histochemical changes in cytochrome oxidase of cortical barrels after vibrissal removal in neonatal and adult mice. Proc. Natl. Acad. Sci. U.S.A. 70, 2333–2337.
Woolsey, T. A., and Van der Loos, H. (1970). The structural organization of layer IV in the somatosensory region (SI) of mouse cerebral cortex. The description of a cortical field composed of discrete cytoarchitectonic units. Brain Res. 17, 205–242.
Wu, C. S., Ballester Rosado, C. J., and Lu, H. C. (2011). What can we get from ‘barrels’: the rodent barrel cortex as a model for studying the establishment of neural circuits. Eur. J. Neurosci. 34, 1663–1676.
Wright, A. G., Demyanenko, G. P., Powell, A., Schachner, M., Enriquez-Barreto, L., Tran, T. S., Polleux, F., and Maness, P. F. (2007). Close homolog of L1 (CHL1) and neuropilin1 mediate guidance of thalamocortical axons at the ventral telencephalon. J. Neurosci. 27, 13667–13679.
Keywords: thalamocortical axons, axonal pathfinding, axonal tract tracing, barrel cortex, adhesion and signaling molecules, NCAM
Citation: Enriquez-Barreto L, Palazzetti C, Brennaman LH, Maness PF and Fairén A (2012) Neural cell adhesion molecule, NCAM, regulates thalamocortical axon pathfinding and the organization of the cortical somatosensory representation in mouse. Front. Mol. Neurosci. 5:76. doi: 10.3389/fnmol.2012.00076
Received: 01 March 2012; Accepted: 05 June 2012;
Published online: 20 June 2012.
Edited by:
Joshua A. Weiner, The University of Iowa, USAReviewed by:
Hansjürgen Volkmer, Universität Tübingen, GermanyJoshua A. Weiner, The University of Iowa, USA
Copyright: © 2012 Enriquez-Barreto, Palazzetti, Brennaman, Maness and Fairén. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Patricia F. Maness, Department of Biochemistry and Biophysics, University of North Carolina School of Medicine, Genetic Medicine Building, 120 Mason Farm Road, Suite 3010, Campus Box #7260, Chapel Hill, NC 27599-7260, USA. e-mail:cGF0cmljaWFfbWFuZXNzQG1lZC51bmMuZWR1
Alfonso Fairén, Instituto de Neurociencias, Consejo Superior de Investigaciones Científicas and Universidad Miguel Hernández, Campus de San Juan, Avenida Ramón y Cajal, s/n, 03550 San Juan de Alicante, Spain. e-mail:ZmFpcmVuQHVtaC5lcw==
†Present address: Centro de Investigación Biomédica de La Rioja, 26006 Logroño, Spain.
‡ Senior co-authors.