

- 1Ernest Gallo Clinic and Research Center at the University of California San Francisco, Emeryville, CA, USA
- 2Clinical Pharmacology and Experimental Therapeutics, School of Medicine, University of California San Francisco, San Francisco, CA, USA
- 3Translational Research Institute and the Institute for Health and Biomedical Innovation, Queensland University of Technology, Brisbane, QLD, Australia
Corticotropin releasing factor (CRF) has been shown to induce various behavioral changes related to adaptation to stress. Dysregulation of the CRF system at any point can lead to a variety of psychiatric disorders, including substance use disorders (SUDs). CRF has been associated with stress-induced drug reinforcement. Extensive literature has identified CRF to play an important role in the molecular mechanisms that lead to an increase in susceptibility that precipitates relapse to SUDs. The CRF system has a heterogeneous role in SUDs. It enhances the acute effects of drugs of abuse and is also responsible for the potentiation of drug-induced neuroplasticity evoked during the withdrawal period. We present in this review the brain regions and circuitries where CRF is expressed and may participate in stress-induced drug abuse. Finally, we attempt to evaluate the role of modulating the CRF system as a possible therapeutic strategy for treating the dysregulation of emotional behaviors that result from the acute positive reinforcement of substances of abuse as well as the negative reinforcement produced by withdrawal.
Introduction
Drug addiction is a chronic condition characterized by periods of abstinence and relapse. The effects of drugs of abuse on brain function have been extensively evaluated with the intention of developing therapies that can prevent relapse and facilitate the treatment of substance use disorders (SUDs). An extensive literature has shown that addictive drugs affect systems that govern reward pathways (mesolimbic dopaminergic pathway), learning and memory processes (hippocampus), emotion (amygdala), and cognitive functions (prefrontal cortex). The reinforcing effects of drug of abuse have been attributed to actions in the limbic system that in turn influence motivational, emotional and affective behaviors (Rezayof et al., 2002; David et al., 2008; Martin et al., 2008; Nielsen et al., 2011; Xue et al., 2012) and for reviews see (Koob, 1992; Pierce and Kumaresan, 2006; Feltenstein and See, 2008). Specifically, the alteration of reward processing (Wise, 1998, 2005) has been identified as a critical factor that leads to an increase in the chance of relapse (Koob and Le Moal, 1997; Everitt et al., 1999; Koob et al., 2004; Everitt and Robbins, 2005). The development of SUDs is a progression that commences with the first exposure to the drug and ends with physiological and psychological dependence.
Although substances of abuse have different mechanisms of action, repeated exposure has been shown to lead to similar neural adaptations. Addiction to any class of drugs has been described as a learning process. Individuals learn associations between the rewarding effects of the drugs and the environmental cues that predict drug availability. Neuroadaptations in areas associated with learning and memory (hippocampus and amygdala) are affected after a single episode of any drug use by influencing synaptic transmission. Following chronic drug use, the compulsive seeking and uncontrollable use leads to long-lasting alterations in synaptic plasticity, such as changes in synaptic strength.
Human studies (Gawin and Kleber, 1986; Wallace, 1989) and experiments with preclinical models (Thatcher-Britton and Koob, 1986; Piazza et al., 1990; Goeders and Guerin, 1994; Kreibich et al., 2009) have identified stress as a critical factor in the drug addiction process, including triggering relapse. Corticotropin releasing factor (CRF) has been implicated in neuroendocrine and behavioral responses to stress (Britton et al., 1982; Koob and Bloom, 1985). It has been shown to be activated during stress-induced drug reinstatement, where it acts to facilitate relapse and increase anxiety during acute and chronic withdrawal (Shaham et al., 1995; Ambrosio et al., 1997; Koob, 1999) and see (Sarnyai et al., 2001; George et al., 2011) for extensive review.
CRF-induced neuroplastic changes have been studied both in mesolimbic brain circuits that include the ventral tegmental area (VTA) and nucleus accumbens (NAcc) (Ungless et al., 2003; Wang et al., 2007a; Hahn et al., 2009) and also in brain regions associated with emotion, such as the amygdala (Fudge and Emiliano, 2003; Pollandt et al., 2006; Fu et al., 2007; Kash et al., 2008; Francesconi et al., 2009). Despite extensive research supporting the role of CRF in drug addiction, the specific participation of CRF on drug-induced synaptic plasticity that leads to relapse remains undetermined.
This review will attempt to examine recent research on the role of CRF and its interaction with drug-mediated synaptic plasticity. The VTA and the amygdalar nuclei where CRF is highly expressed will be described. We will discuss whether CRF facilitates or inhibits synaptic strength from the basal condition. Finally, we will attempt to integrate the neurobiological changes that result from the interaction of substances of abuse with stress to evaluate alternative drug targets for experimental therapeutics to prevent relapse and facilitate the treatment of SUDs.
Substance Use Disorders (SUDs) and Stress
SUDs are a chronic and relapsing condition characterized by an intense desire for drug intake during the withdrawal period. This craving process leads to a progression from the initial impulsive consumption to a subsequent compulsive and habit forming consumption that result in loss of control in limiting intake and subsequent inability to change the habit developed over time. One of the main challenges in preclinical addiction research has been to elucidate the pathways that lead to the loss of control of drug use and the predisposition to relapse (Koob and Le Moal, 1997). As described by the Opponent Process Model, the repetitive use of addictive substances alters the reward circuits by decreasing the intense pleasure state and by increasing the following unpleasant state. After discontinuation of repeated exposure to addictive drugs, compensatory reactions develop that oppose the primary effects of the drug—the withdrawal symptoms. The reduction of the withdrawal symptoms would therefore represent negative reinforcement. The reduction of the unpleasant state of the withdrawal symptoms becomes the major drive in continued drug use. In a simplified view of the dopamine theory (Wise, 1978, 2008; Berridge and Robinson, 1998; Everitt and Robbins, 2005; Diana, 2011), the acute euphoric process obtained by binge-intoxication represents the activation of the dopaminergic system, while the negative component resulting from the withdrawal period is marked by the reduction of dopamine function (Tomkins and Sellers, 2001). The introduction of functional toxicity (Weiss and Koob, 2001), which is associated with the unpleasant withdrawal state powered by the recruitment of the stress neurotransmitter, CRF, further expanded the dopamine theory as it applies to addiction.
Corticotropin Releasing Factor (CRF) System
CRF, also known as corticotropin releasing hormone (CRH), has been shown to induce various behavioral changes related to adaptation to stress. Dysregulation of the CRF system at any point can lead to a variety of psychiatric disorders such as depression, obsessive compulsive disorder, post-traumatic stress disorder and SUDs (Cole et al., 1990; Sarnyai et al., 1992, 2001; Cador et al., 1993; Koob and Kreek, 2007; Koob and Le Moal, 2008a). Footshock-induced stress has been shown to be effective in inducing reinstatement of alcohol (Le et al., 1998, 2000; Gass and Olive, 2007; Richards et al., 2008), nicotine (Buczek et al., 1999), cocaine (Erb et al., 1996), opiate and psycostimulants (Lu et al., 2003) and heroin (Shaham et al., 1997) seeking. Specifically CRF has been associated with drug reinstatement (Shaham et al., 1997; Le et al., 2002; Liu and Weiss, 2002; Funk et al., 2006). CRF has also been shown to produce anxiety-like behaviors during withdrawal from chronic ethanol (Baldwin et al., 1991; Overstreet et al., 2004) and may be responsible for persistent vulnerability and eventual relapse.
The CRF system consists of four ligands: CRF, urocortin (UCN) (Vaughan et al., 1995) 1, 2, and 3, two G-protein-coupled receptors (GPCR), CRF-receptor 1 (CRF-R1) and CRF-receptor 2 (CRF-R2), as well as a secreted CRF binding protein (CRF-BP); see Table 1 and (Bale and Vale, 2004) for CRF system review.
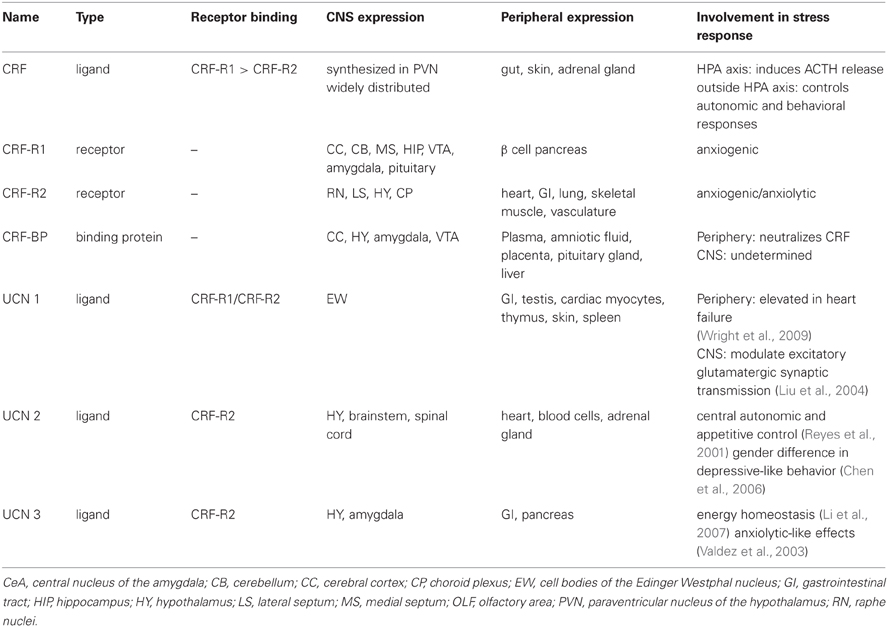
Table 1. Corticotropin Releasing Factor (CRF) system.
It was originally identified as a hypothalamic factor responsible for stimulating adrenocorticotropic hormone (ACTH) secretion from the anterior pituitary (Guillemin and Rosenberg, 1955; Saffran et al., 1955) where it stimulates glucocorticoid synthesis and secretion form the adrenal cortex (Turnbull and Rivier, 1997). Its name was established thirty years before its biochemical identification in the 1980's (Vale et al., 1981) while its gene identifier in the National Center for Biotechnology Information (NCBI) is CRH. It is a 4.7-kilo-Dalton (kDa) peptide and consists of 41-amino acid residues. Neurosecretory neurons of the paraventricular nucleus (PVN) of the hypothalamus synthesize CRF (Meloni et al., 2005). CRF is then released into the afferent portal blood vessels to the anterior pituitary gland where it induces ACTH release in the systemic circulation. The hypothalamic-pituitary-adrenal (HPA) axis is regulated by negative feedback from glucocorticoids that activate glucocorticoid receptors specifically in the PVN and hippocampus. CRF is also expressed outside the HPA axis to control autonomic and behavioral responses to stressors (Palkovits et al., 1983; Swanson et al., 1983) including stress-induced reinstatement of drug seeking.
CRF mediates physiological stress responses by activating CRF-R1 and CRF-R2, which are distributed throughout the periphery and the brain (De Souza, 1995; Bale and Vale, 2004). It is believed that the binding of CRF to CRF-Rs is a two-step mechanism. The N-terminus of the receptor initially binds to the C-terminus of CRF, which initiates a rearrangement of the receptor (Grace et al., 2007). The CRF N-terminus contacts the other sites on the receptor to initiate cellular signaling (Vale et al., 1981; Rivier et al., 1984) and consequently activate the G-protein (Nielsen et al., 2000; Grace et al., 2004; Rijkers et al., 2004; Yamada et al., 2004; Hoare, 2005). The CRF system comprises other peptides with structural homology to CRF. UCN 1 shows 45% sequence identity with CRF and binds with high affinity to both CRF receptor subtypes (Perrin et al., 1995), whereas CRF binds with highest affinity to CRF-R1 (Vaughan et al., 1995; Burnett, 2005). UCN 2, also known as stresscopin related peptide, and UCN 3, also known as stresscopin bind specifically to CRF-R2 (Hsu and Hsueh, 2001; Lewis et al., 2001; Reyes et al., 2001).
CRF-R1 has 415 amino acid residues and it is expressed in the periphery and in the CNS (Chang et al., 1993; Chen et al., 1993; Vita et al., 1993; Potter et al., 1994; Tsai-Morris et al., 1996; Sanchez et al., 1999; Van Pett et al., 2000). Chronic stress mediated by activation of CRF-R1 by CRF has been associated with the development of anxiety disorders (Arborelius et al., 1999); CRF-R1 antagonists have been shown to reduce anxiety-like behaviors (Funk et al., 2007). Transgenic mice with deletion of CRF-R1 (CRF-R1 knock out (KO) mice) have reduced reaction to both stress and anxiety, for comprehensive review see (Bale and Vale, 2004). This anxiolytic effect, however may be attributed to the reduction in circulating glucocorticoids in preclinical models (Tronche et al., 1999). A conditional KO mouse line was generated to differentiate the behavioral from the neuroendocrine CRF-mediated CRF-R1 signaling pathways. The selective inactivation of the limbic structures, but not of the HPA system has shown that CRF-R1 modulates anxiety-like behaviors and it is independent of the HPA (Muller et al., 2003). Furthermore, CRF-R1 is thought to increase susceptibility to alcohol relapse behaviors (Hansson et al., 2006; Heilig and Koob, 2007). A recent study evaluated the role of CRF both within and outside the HPA has shown that CRF via CRF-R1 signaling may have opposite effects on stress-related alcohol consumption (Molander et al., 2012).
CRF-R2 has three variants: α,β, and γ. The α is comprised of 411 amino acid residues and the β is comprised of 413–418 amino acid residues. Both are found in the brain and periphery; however, CRF-R2β is predominantly found in the heart and vasculature (Lovenberg et al., 1995a,b; Kimura et al., 2002; Burnett, 2005). The γ variant is a smaller peptide containing only 397 amino acid residues, and is found only in the human brain (Kostich et al., 1998). The precise role of CRF-R2 in the regulation of the stress response is a subject of intense investigation. Genetic mouse model studies with deletion of CRF-R2 (CRF-R2 KO mice) have demonstrated that CRF activation of CRF-R2 can lead to either an increased or decreased response to stressors (Bale et al., 2000, 2002; Coste et al., 2000; Kishimoto et al., 2000).
The lack of specific antisera that support immunohistochemical experiments and the low resolution of ligand binding approaches have limited the studies to elucidate the CRF-Rs distribution and limit the analysis at the mRNA level. To overcome this impediment, a transgenic mouse that reports expression of CRF-R1 with green fluorescent protein (GFP) has been successfully generated providing a novel tool to investigate the role of CRF-R1 signaling in stress adaptation (Justice et al., 2008).
CRF-BP is a water-soluble, 37 kD protein and consists of 322 amino acid residues (Bale and Vale, 2004). It is a secreted glycoprotein, efficiently stored into secretory granules and released into the extracellular space through exocytosis (Blanco et al., 2011). It contains aspargine N-linked-type oligosaccharides that are critical for CRF-BP binding to CRF (Suda et al., 1989). Previous attempts to identify small molecule inhibitors of CRF-BP have produced limited success due in part to the high affinity (picomolar) of CRF binding to CRF-BP (Behan et al., 1995a) and also because CRF-BP full length (FL) is susceptible to autocatalytic proteolysis (Woods et al., 1999). The spontaneous proteolytic cleavage yields a larger N-terminal fragment of 27 kD, CRF-BP (27 kD), which retains the binding site for CRF and a smaller, 9.6 kD C-terminal fragment, CRF-BP (10 kD) (Woods et al., 1999) with no apparent physiological or pathological role. The unique cleavage site in CRF-BP (FL) has been identified between amino acid residues serine 234 and alanine 235. The generation of two fragments has made it extremely difficult to successfully purify sufficient quantities of CRF-BP (FL) to study the physiological properties of the native protein. CRF-BP is distributed in plasma, amniotic and synovial fluid, the placenta, the pituitary gland, the liver, and in several distinct brain regions, including the cerebral cortex, the hippocampus (Behan et al., 1995a), the amygdala (Herringa et al., 2004) and the VTA (Wang and Morales, 2008). In the periphery, circulating CRF-BP neutralizes the physiological actions of CRF (Kemp et al., 1998). Because of the high affinity with CRF, it is believed that CRF-BP plays a buffer role by reducing the amount of free CRF. In the brain, however, CRF-BP is mostly membrane-bound and expressed in different amounts in neurons and neuroglial cells (Behan et al., 1995b). Within neuronal cells, recent findings demonstrated that discrete subpopulations of VTA dopaminergic and γ-aminobutyric acid (GABAergic) neurons express CRF-BP (Wang and Morales, 2008). The physiological role of CRF-BP in the central nervous system (CNS) is still unclear. Additionally, theories suggest the possibility that CRF-BP may assist the clearance of CRF from the body and may also protect CRF from degradation (Seasholtz et al., 2002). Genetic mouse model studies with deletion of CRF-BP (CRF-BP KO mice) have shown there is an increase in anxiety-like behavior (Karolyi et al., 1999). Electrophysiology studies have shown that CRF signals through CRF-R2 to potentiate N-Methyl-D-aspartate (NMDA)-mediated excitatory postsynaptic currents (EPSCs) in the VTA (Ungless et al., 2003). Furthermore, using CRF (6–33), a peptide that competes with CRF at the CRF-BP binding site, but does not bind to CRF-R2, it was shown that it blocked CRF-induced potentiation of NMDAR-mediated EPSCs (Ungless et al., 2003). Taken together, these results suggest that CRF-BP possesses a diverse role in modulating the CRF-system. As described by in vitro and in vivo studies, purifying human CRF-BP (FL) in sufficient quantities for investigation has not been successful to date (Woods et al., 1997). There have not been any research tools available to characterize the role of CRF-BP in the CNS by expressing CRF-BP on the cell surface. Therefore, it has not been possible to determine whether CRF-BP participates specifically in the CRF-R2 signaling. A summary of the involvement of the CRF binding in addictive behavior is described in Table 2.
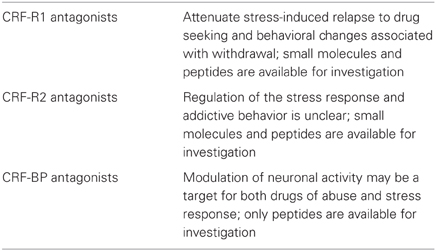
Table 2. Involvement of the CRF binding in addictive behaviors.
Stress-Induced Drug Addiction: CRF-Mediated Neurotransmission and Plasticity
Reinforcement: Ventral Tegmental Area (VTA) and Nucleus Accumbens (NAcc)
Addictive drugs have been shown to increase the concentration of dopamine in the NAcc. Furthermore, the increase of dopamine has been associated with the amplification of the hedonic impact of positive reinforcers (Fibiger, 1978; Berridge et al., 1989) and the development of addictive behaviors (Yokel and Wise, 1975; Bonci and Malenka, 1999; Wise, 2008). The NAcc receives input from the VTA and it is thought that this pathway may be responsible not only for the acute pleasure effect of drug intake, but also for the negative reinforcement and the effects of cues on drug-seeking behaviors (Koob and Nestler, 1997).
CRF cellular involvement in the VTA
The VTA receives CRF projections mostly from the limbic forebrain and PVN of the hypothalamus (Rodaros et al., 2007) that form glutamatergic synapses and symmetric GABAergic synapses (Tagliaferro and Morales, 2008). The PVN is the site for CRF synthesis (Meloni et al., 2005) and the majority of asymmetric synapses (glutamatergic) are expressed in CRF- and dopaminergic-containing neurons. VTA dopaminergic neurons express CRF-R1 (Van Pett et al., 2000) and a more recent study has shown that the majority of VTA neurons expressing CRF-BP are dopaminergic (Wang and Morales, 2008). The CRF system modulates dopaminergic neurons by activating CRF-R1 and CRF-R2; however, CRF is not only involved in the neuroexcitability of the dopaminergic system. It may also be responsible for modulating excitatory and inhibitory synaptic inputs since the VTA receive inputs from both CRF-glutamatergic- and CRF-GABAergic-containing neurons (Tagliaferro and Morales, 2008) and for review see Borgland et al. (2010).
CRF increases the firing rate of VTA dopaminergic neurons (Korotkova et al., 2006; Wanat et al., 2008) via CRF-R1, and involves the phospholipase C (PLC)–protein kinase C (PKC) signaling pathway with enhancement of Ih (hyperpolarization-activated inward current) (Wanat et al., 2008). CRF can also induce a transient slowly developing potentiation of NMDA-mediated synaptic transmission via CRF-R2 and activation of the PLC-PKC signaling pathway. CRF-R2-mediated potentiation has been shown to require the presence of CRF-BP (Ungless et al., 2003). The mechanism of action of CRF-R2 and CRF-BP is still under investigation as the research tools needed to study CRF-BP and antisera that specifically target CRF-R2 have not been available.
CRF appears to have both excitatory and inhibitory actions on the dopaminergic neurons in the VTA. Studies using cocaine and methamphetamine have shown that the excitatory effect of CRF on dopaminergic neurons involves fast events, for example action potential firing rate and NMDAR-mediated synaptic transmission, while the inhibitory effects of CRF involve slow forms of synaptic transmission that would result in long-term plasticity (Beckstead et al., 2009). Those observations demonstrated that CRF may have different actions on receptors that mediate the synaptic action on dopamine. This cellular mechanism may refine the role of stress by CRF actions on dopamine-mediated behaviors (Beckstead et al., 2009).
As it has been shown that potentiation of CRF-R2, but not CRF-R1, signaling requires the presence of CRF-BP (Ungless et al., 2003), it has been proposed that CRF-BP and CRF-R2 mediate longer-lasting forms of synaptic plasticity (Bonci and Malenka, 1999). Both behavioral sensitization and long-term potentiation (LTP) share many characteristics such as the involvement of NMDAR activation for the induction of LTP in VTA dopaminergic neurons (Bonci and Malenka, 1999; Ungless et al., 2001). As a consequence, it has been suggested that synaptic plasticity at excitatory synapses on VTA dopaminergic neurons may play a principal role in triggering behavioral change. Since NMDAR activation is required for the induction of LTP in VTA dopaminergic neurons, CRF-Rs activation may modulate longer-lasting forms of plasticity (Bonci and Malenka, 1999; Ungless et al., 2001; Bonci and Borgland, 2009).
CRF-mediated neurotransmission and plasticity
Synaptic adaptations observed in remodeling of neuronal circuits in addictive drug studies have been shown to have implications in behavior and memory traits that characterize SUDs. The neuroplasticity underlying drug-induced sensitization has produced a growing body of evidence that suggests it may represent the molecular effect that is critical in modulating addictive behaviors and would contribute to stress-induced compulsive behaviors in addiction.
Axon terminals of CRF neurons synapse onto VTA neuronal dendrites (Tagliaferro and Morales, 2008) and it appears that stress affects the CRF release in this region (Wang et al., 2006). Electrophysiological studies have shown that CRF-BP is required for a slowly developing, transient potentiation of NMDAR-mediated synaptic transmission elicited by CRF via CRF-R2 specifically (Ungless et al., 2003). These results have been corroborated by behavioral studies that determined the effectiveness of stress in triggering glutamate and dopamine release in cocaine seeking of drug-experienced rats (Wang et al., 2007b). Using chronic cocaine preclinical models, the study has shown the positive reinforcement associated with CRF, specifically CRF/CRF-R2/CRF-BP interaction with the dopaminergic system. Those findings support additional research efforts to develop novel approaches that probe CRF-BP on the cell surface.
In conclusion, CRF increases VTA glutamatergic synaptic function, which may facilitate VTA burst firing or induction of synaptic plasticity that may result from repeated exposure to drugs of abuse. This process may produce long-term neuroadaptations that alter stress responses and enhance drug seeking. Electrophysiological studies combined with behavioral studies have proposed that previous experience with drugs of abuse may facilitate the ability of stress to drive drug seeking and, therefore, relapse. These results suggest that CRF may be important for drug-evoked synaptic plasticity in VTA dopaminergic neurons and may represent the molecular substrate that explains the anxiety and stress response during withdrawal from substances of abuse.
Cellular Involvement of CRF in the Amygdala
The amygdala is believed to be a pivotal brain region for emotional response and it is critical for providing affective salience to sensory information (Adolphs et al., 1994; LeDoux, 2003; Phelps and LeDoux, 2005). Negative affective responses have been studied in specific nuclei of the amygdala by studying the conditioned fear response (Davis, 1992a,b). The amygdala is widely connected to other limbic regions where it participates in integrating sensory and cognitive information (LeDoux, 1992, 1993). Experimental evidence strongly suggests drugs of abuse act on this system and can modify synaptic events especially during withdrawal. While the VTA has been associated with the reinforcing effects of ethanol (Gatto et al., 1994), the activation of the GABAergic system has been associated with alcohol's anxiolytic effect (Frye and Breese, 1982). In addition to the rewarding circuits of the shell of the NAcc, and brain regions activated by pharmacological stressor, such as yohimbine and footshock were found to be specific in the basolateral and central amygdalar nuclei, and the bed nucleus of the stria terminalis (BNST) (Funk et al., 2006). Preclinical studies demonstrated that exposure and withdrawal from ethanol induces functional and biochemical changes in the amygdala of rats, demonstrating that this circuit is involved in long-term increases in anxiety-like behavior following chronic ethanol exposure (Christian et al., 2012).
The amygdala mediates conditioned and unconditioned responses to aversive stimuli (Davis and Whalen, 2001) and it has been investigated using Pavlovian fear conditioning by pairing a conditioned stimulus with an aversive unconditioned stimulus. The re-exposure of the unconditioned stimulus elicits a conditioned fear response derived by the conditioned-unconditioned association (Pitts et al., 2009). The association signal takes place in the basolateral amygdala (BLA) and is then transmitted to the central nucleus of the amygdala (CeA) (McDonald, 1998; Maren, 1999; Davis and Shi, 2000; Pitkanen et al., 2000; Pare et al., 2004). This transmission process involves both positive and negative associations.
All components of the CRF system, CRF, CRF-Rs and CRF-BP are expressed in the amygdala (Potter et al., 1994). Furthermore, the amygdala is a major extrahypothalamic source of CRF-containing neurons (Palkovits et al., 1983; Van Pett et al., 2000). Both BLA and CeA nuclei play a role in the stress response (Richter et al., 1995; Merali et al., 1998; Koob and Heinrichs, 1999). Extensive studies have shown that the CRF system participates in memory consolidation that involves the BLA-CeA circuit (Roozendaal et al., 2002; Hubbard et al., 2007). It has been observed that CRF release in the amygdala is increased during acute withdrawal (Richter and Weiss, 1999); therefore, it has been hypothesized that CRF may modulate drug-evoked synaptic plasticity (Ungless et al., 2001, 2003) and for a recent review, see (Luscher and Malenka, 2011). The neuronal basis for negative reinforcement is less well-understood; however, more recent behavioral studies have shown that CRF is capable of potentiating excitatory synaptic currents via CRF-R1 in the CeA two weeks following withdrawal from cocaine (Pollandt et al., 2006).
A recent study has shown that CRF-R1 specifically possess a bidirectional role in anxiety (Refojo et al., 2011). While deletion of CRF-R1 in the mid brain dopaminergic neurons increases anxiety-like behaviors and reduces dopamine release in the prefrontal cortex, deletion of CRF-R1 in the forebrain glutamanergic neuronal network reduces anxiety and disrupts transmission in the amygdala and hippocampus (Refojo et al., 2011).
The role of CRF was also evaluated extensively in voluntary ethanol consumption using gene expression and genetic variation in preclinical models see (Bjork et al., 2010) for extensive review. In ethanol-exposed animals, ethanol intake was reduced by administration of CRF-R1 antagonist, and tested using pharmacological interventions that reduce anxiety-like behaviors (Logrip et al., 2011; Zorrilla and Koob, 2012). The reduction of ethanol intake was also observed in transgenic mice with deletion of CRF-R1 (CRF-R1 KO) (Chu et al., 2007). CRF-R1 antagonists reduce drug withdrawal-associated anxiety and attenuate the negative reinforcing effects of ethanol associated with prolonged ethanol exposure (Ghitza et al., 2006; Marinelli et al., 2007; Li et al., 2007; Koob and Le Moal, 2008b; Richards et al., 2008). CRF-R1 inhibitors have shown to attenuate stress-induced relapse to cocaine and heroin in trained animals (Shaham et al., 1998) and to reduce stress-induced reinstatement and stress-induced reactivation of conditioned place preference in many addictive drugs (Koob and Zorrilla, 2010).
The extended amygdala
Among the extrahypothalamic structures that contain CRF expressing neurons there is the “extended amygdala.” The extended amygdala is comprised by the BNST, the central medial amygdala (CeA), the sublenticular sustantia innominata and a transition zone forming the posterior part of the NAcc (Heimer and Alheid, 1991). It represents the brain circuit involved in processing the aversive stimuli produced by ethanol withdrawal (Koob and Le Moal, 2001), in which the GABA system has been altered and the CRF system in the adjacent CeA has been shown to be activated (Roberts et al., 1996). Those observations indicate that GABAergic activity within interneurons of the extended amygdala may play a prominent role in the chronic negative emotion-like state of motivational significance for drug seeking in alcohol dependence (Koob and Le Moal, 2001; Koob, 2003, 2009a,b). In addition, an in situ hybridization study has shown that recruitment of CRF-R1 signaling, in the components of the extended amygdala, may be responsible of driving the excessive voluntary alcohol intake and may be linked to increase stress activity (Hansson et al., 2007).
The BNST (as well as distinct regions of the CeA) has been associated with stress and anxiety (Walker and Davis, 2008) and is involved specifically with CRF signaling (Davis et al., 1997). The CeA and BNST have direct projections to many brain regions that have been studied to elucidate the symptoms of fear or anxiety (Davis, 1992b). The BNST has been identified as a possible regulator of VTA dopaminergic neuron firing (Georges and Aston-Jones, 2002) and consequently involved in the regulation of acute actions of alcohol, nicotine, and cocaine (Watkins et al., 1999; Carboni et al., 2000; Eiler et al., 2003).
The BNST possesses an extensive network of dopaminergic fibers (Fudge and Emiliano, 2003) and is connected to the reward pathway by extensive projections to the VTA, thus influencing the excitatory input through both NMDA and non-NMDA receptors (Georges and Aston-Jones, 2001, 2002). This dopaminergic excitatory transmission in the VTA requires the presence of CRF (Kash et al., 2008). Acute cocaine administration has been shown to induce dopamine signaling through a specific CRF-R1-dependent enhancement of NMDA excitatory transmission (Kash et al., 2008). This mechanism was described as a short-term form of plasticity in the BNST, which may be responsible for the acute effects of addictive drugs (Kash et al., 2008). These findings suggested that glutamatergic neurotransmission in BNST may be functionally involved with acute reinforcing actions of drug of abuse (Walker and Davis, 2008).
Basolateral amygdala (BLA)
The basolateral nucleus of the amygdala (BLA) is critically implicated in emotional learning (LeDoux, 2000), and in reward (Balleine and Killcross, 2006; Tye et al., 2008). Neurons from the BLA project directly to the CeA as well as to the BNST. The BLA is mostly composed by glutamatergic pyramidal neurons and provides the main excitatory input to the CeA and other limbic and cortical structures (Sah et al., 2003); however, the excitatory transmission is believed to be modulated by the relatively small number of GABAergic interneurons found there (Washburn and Moises, 1992). GABAergic interneurons have been identified as regulators of stress and anxiety (Silberman et al., 2009).
CRF is present abundantly in the BLA, in addition to CRF-R1 and CRF-BP, (Sakanaka et al., 1986; Potter et al., 1992; Van Pett et al., 2000); however, the effects of CRF in the BLA have been studied far less than the other nuclei of the amygdala. The BLA has been shown to be a critical nucleus for the consolidation of fear and memory and, therefore, is a possible target for dampening emotional memories. It has been shown that intra BLA infusions of CRF increase anxiety-like behaviors (anorexia and grooming) that are blocked by the administration of a CRF-R1 antagonist (Jochman et al., 2005). Another BLA microinfusion study showed that CRF-R1 activates fear memory consolidation and that this effect is blocked by administration of another CRF-R1 antagonist. The fear memory consolidation process seems specifically regulated by the CRF-R1 activation since CRF-R2 antagonist in the BLA disrupted neither the contextual fear conditioning nor performance of contextual freezing in the drug-free conditioned fear test (Hubbard et al., 2007). BLA CRF-R1 activation has been described as induced synaptic plasticity, and demonstrating that BLA CRF-R1 activation can be pharmacologically blocked by small molecules, the possibility to compromise the consolidation of fear memory suggests a potential therapeutic opportunity to ease the development of intense emotional memories.
Central nucleus amygdala (CeA)
The CeA has been identified as locus for both acute positive reinforcement of ethanol self-administration and for the negative reinforcement associated with ethanol withdrawal (Baldwin et al., 1991; Heinrichs et al., 1992, 1995; Koob and Le Moal, 1997, 2001; Zorrilla et al., 2001). The CeA has also been identified as a critical locus for reversing many behavioral effects associated with ethanol intoxication (Hyytia and Koob, 1995).
In the CeA, most neurons are GABAergic (Sun and Cassell, 1993), and CRF is highly co-expressed with GABAergic neurons (Veinante et al., 1997; Day et al., 1999). The CeA abundantly expresses CRF, CRF-R1 and CRF-BP (Sakanaka et al., 1986; Potter et al., 1992; Van Pett et al., 2000). Moreover, in the CeA the action of CRF and ethanol has been shown to increase GABA release (Nie et al., 2004) and the amount of CRF release is increased in preclinical models of ethanol dependence (Merlo Pich et al., 1995). Protein kinase C epsilon (PKCε) has been shown to modulate CRF-R1 signaling in the CeA (Choi et al., 2002) and transgenic mice with deletion of PKCε (PKCε KO mice) have shown reduced anxiety-like behaviors (Hodge et al., 2002). Electrophysiological studies have shown that ethanol-induced GABA release in the amygdala is regulated by CRF-R1 (Nie et al., 2004) and that ethanol-stimulated vesicular GABA release depends on PKCε models (Bajo et al., 2008). PKCε signaling pathway in the CeA is activated by CRF-R1 activation and modulates GABAergic neurotransmission that may contribute to the anxiogenic effects of ethanol (Smith et al., 1998; Timpl et al., 1998). This functional link between ethanol, CRF and PKCε that modulates GABAergic neurotransmission in the CeA may contribute to the dysregulation of emotional behaviors that regulate acute positive reinforcement of ethanol consumption and the negative reinforcement produced by ethanol withdrawal.
It has been shown that there is a critical difference between CRF effects in low/moderate ethanol-exposed animals (binge-like ethanol consumption) and ethanol-dependent animals (chronic-like ethanol exposure). While binge-like ethanol (Lowery-Gionta et al., 2012) may cause transient perturbations of the CRF system which may be able to return to its homeostatic state, the chronic-like ethanol exposure (Roberto et al., 2003, 2004) may be responsible for the CRF neuroadaptation that would influence the allostatic state. An allostatic state is defined as a state of chronic deviation of the regulatory network from their normal process and the establishment of a different set point of “apparent stability” (Koob and Le Moal, 2001). This chronic deviation of reward set point is critically altered during drug withdrawal and may contribute to subsequent neuroadaptation that produces vulnerability to addiction and relapses (Koob and Le Moal, 2001). Acute stress does not increase the mRNA expression of any components of the CRF system in the CeA (Herringa et al., 2004), however, in the CeA of animals exposed to ethanol, there was a significant increase in CRF mRNA expression (Lack et al., 2005) as well as in ethanol-dependent animals during withdrawal (Sommer et al., 2008).
The recruitment of CRF in the CeA during early drinking episodes, before dependence, may initiate neuroplastic changes in the system that may become more intense with additional ethanol exposures (Lowery-Gionta et al., 2012). It has been proposed that this CRF-dependent change contributes to the transition from binge-drinking to ethanol dependence (Lowery-Gionta et al., 2012). The authors also found that ethanol enhances GABAergic transmission in the amygdala at both pre- and post-synaptic sites in ethanol naïve animals, while binge ethanol consumption blunts the CRF-mediated GABAergic transmission (Lowery-Gionta et al., 2012). This study revealed that drinking reduced the effect CRF has on GABAergic transmission. In contrast, others have found that animals dependent on ethanol showed enhanced GABAergic transmission in the CeA (Roberto et al., 2004).
CRF and norepinephrine have been shown to increase GABAergic activity measured by GABAA inhibitory postsynaptic potential (IPSCs) in whole-cell recording from the CeA. This effect was blocked by CRF-R1 antagonists and blocked in CRF-R1 knockout mice (Nie et al., 2004; Kash and Winder, 2006). The augmented GABA release produced by ethanol in the CeA in dependent animals was observed both in electrophysiological and in vivo microdialysis experiments (Roberto et al., 2003). Later studies in ethanol-dependent rats corroborated that CRF-alcohol interaction on GABAergic transmission in the CeA is more pronounced during alcohol dependence (Roberto et al., 2004).
Conclusions
This review has summarized the multiple mechanisms that underlie persistent changes in synaptic efficacy following administration of addictive drugs. It is evident that the CRF system significantly facilitates the induction and maintenance of plasticity in the VTA and amygdala, with resulting enhancement of glutamate-mediated excitation and reduction of GABA-mediated inhibition, thus contributing to the molecular basis of drug addiction.
Neuroplasticity in brain reward circuitry following a history of ethanol dependence has been shown (Hansson et al., 2008). Experimental data illustrated in this review support the hypothesis that stress induces plasticity within the VTA and amygdala nuclei and may participate in the development of a chronic anxiety state that could lead to the development of SUDs. These changes in the limbic neuronal network may represent the trigger that may lead to loss of control of drug use. Addictive drugs have been shown to induce behavioral sensitization and there is a large body of literature that evaluates the role of stress and addictive behaviors. Studies of long-term neuroadaptation in alcohol addiction have shown that brain stress and fear systems become activated (Heilig et al., 2010); however, there is still much to be elucidated pertaining to drugs' actions on the CRF system, both in regard to synaptic plasticity and behavioral responses. Several blood–brain barrier-penetrating CRF-R1 antagonists have been developed, however while some compounds have shown efficacy in animal models to treat alcoholism (Gehlert et al., 2007, 2012), CRF-R1 antagonists have still not succeeded in clinical trials (Koob and Zorrilla, 2012).
Preventing all exposure to substances of abuse is almost impossible, as many psychoactive substances (alcohol, nicotine, caffeine, and prescription medications) are generally accepted in our society. There are many medications that are FDA approved or used off-label for alcohol dependence that focus on the treatment of symptom reduction (disulfuram, naltrexone), assistance with withdrawal (benzodiazepines, valporic acid, varenicline), and relapse prevention (acamprosate, ondansetron, baclofen, topiramate, varenicline, methadone) and others FDA approved medications for other indications are at the preclinical stage (mifepristone) (Simms et al., 2011), however, the recidivism in drug abuse is still a major problem for SUDs. Although different classes of substances of abuse have different mechanisms of action, repeated drug use leads to stimulation of the HPA axis and the abrupt cessation of chronic drug use increases activation of CRF. Medications that modulate stress responses may offer a novel pharmacotherapeutic approach for SUDs. Regulating stress outcomes by acting on the CRF system may offer the possibility to develop that novel therapeutic directed to diminish the effect of CRF in synaptic transmissions. By easing the stress-induced drug seeking, it may be possible to reduce relapse and facilitate the formation of memories with less deleterious behavioral consequences.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank J. Simms, S. Srinivasan and L. Daitch for their contribution to the editing of the manuscript. This work was supported by funding from the State of California Medical Research on Alcohol & Substance Abuse through UCSF to Selena E. Bartlett, the National Institutes of Health: 1R21DA029966-01 and NIH Fast Track award to screen the MLSMR collection to Selena E. Bartlett, UCSF School of Pharmacy (Dean's Office and Clinical Pharmacy) and the School of Medicine (Clinical Pharmacology and Experimental Therapeutics) to Carolina L. Haass-Koffler.
References
Adolphs, R., Tranel, D., Damasio, H., and Damasio, A. (1994). Impaired recognition of emotion in facial expressions following bilateral damage to the human amygdala. Nature 372, 669–672.
Ambrosio, E., Sharpe, L. G., and Pilotte, N. S. (1997). Regional binding to corticotropin releasing factor receptors in brain of rats exposed to chronic cocaine and cocaine withdrawal. Synapse 25, 272–276.
Arborelius, L., Owens, M. J., Plotsky, P. M., and Nemeroff, C. B. (1999). The role of corticotropin-releasing factor in depression and anxiety disorders. J. Endocrinol. 160, 1–12.
Bajo, M., Cruz, M. T., Siggins, G. R., Messing, R., and Roberto, M. (2008). Protein kinase C epsilon mediation of CRF- and ethanol-induced GABA release in central amygdala. Proc. Natl. Acad. Sci. U.S.A. 105, 8410–8415.
Baldwin, H. A., Rassnick, S., Rivier, J., Koob, G. F., and Britton, K. T. (1991). CRF antagonist reverses the “anxiogenic” response to ethanol withdrawal in the rat. Psychopharmacology (Berl.) 103, 227–232.
Bale, T. L., Contarino, A., Smith, G. W., Chan, R., Gold, L. H., Sawchenko, P. E., Koob, G. F., Vale, W. W., and Lee, K. F. (2000). Mice deficient for corticotropin-releasing hormone receptor-2 display anxiety-like behaviour and are hypersensitive to stress. Nat. Genet. 24, 410–414.
Bale, T. L., Lee, K. F., and Vale, W. W. (2002). The role of corticotropin-releasing factor receptors in stress and anxiety. Integr. Comp. Biol. 42, 552–555.
Bale, T. L., and Vale, W. W. (2004). CRF and CRF receptors: role in stress responsivity and other behaviors. Annu. Rev. Pharmacol. Toxicol. 44, 525–557.
Balleine, B. W., and Killcross, S. (2006). Parallel incentive processing: an integrated view of amygdala function. Trends Neurosci. 29, 272–279.
Beckstead, M. J., Gantz, S. C., Ford, C. P., Stenzel-Poore, M. P., Phillips, P. E., Mark, G. P., and Williams, J. T. (2009). CRF enhancement of GIRK channel-mediated transmission in dopamine neurons. Neuropsychopharmacology 34, 1926–1935.
Behan, D. P., De Souza, E. B., Lowry, P. J., Potter, E., Sawchenko, P., and Vale, W. W. (1995a). Corticotropin releasing factor (CRF) binding protein: a novel regulator of CRF and related peptides. Front. Neuroendocrinol. 16:362–382. doi: 10.1006/frne.1995.1013
Behan, D. P., Maciejewski, D., Chalmers, D., and De Souza, E. B. (1995b). Corticotropin releasing factor binding protein (CRF-BP) is expressed in neuronal and astrocytic cells. Brain Res. 698, 259–264.
Berridge, K. C., and Robinson, T. E. (1998). What is the role of dopamine in reward: hedonic impact, reward learning, or incentive salience? Brain Res. Brain Res. Rev. 28, 309–369.
Berridge, K. C., Venier, I. L., and Robinson, T. E. (1989). Taste reactivity analysis of 6-hydroxydopamine-induced aphagia: implications for arousal and anhedonia hypotheses of dopamine function. Behav. Neurosci. 103, 36–45.
Bjork, K., Hansson, A. C., and Sommer, W. H. (2010). Genetic variation and brain gene expression in rodent models of alcoholism implications for medication development. Int. Rev. Neurobiol. 91, 129–171.
Blanco, E. H., Zuniga, J. P., Andres, M. E., Alvarez, A. R., and Gysling, K. (2011). Corticotropin-releasing factor binding protein enters the regulated secretory pathway in neuroendocrine cells and cortical neurons. Neuropeptides 45, 273–279.
Bonci, A., and Borgland, S. (2009). Role of orexin/hypocretin and CRF in the formation of drug-dependent synaptic plasticity in the mesolimbic system. Neuropharmacology 56(Suppl. 1), 107–111.
Bonci, A., and Malenka, R. C. (1999). Properties and plasticity of excitatory synapses on dopaminergic and GABAergic cells in the ventral tegmental area. J. Neurosci. 19, 3723–3730.
Borgland, S. L., Ungless, M. A., and Bonci, A. (2010). A convergent actions of orexin/hypocretin and CRF on dopamine neurons: emerging players in addiction. Brain Res. 1314, 139–144.
Britton, D. R., Koob, G. F., Rivier, J., and Vale, W. (1982). Intraventricular corticotropin-releasing factor enhances behavioral effects of novelty. Life Sci. 31, 363–367.
Buczek, Y., Le, A. D., Wang, A., Stewart, J., and Shaham, Y. (1999). Stress reinstates nicotine seeking but not sucrose solution seeking in rats. Psychopharmacology (Berl.) 144, 183–188.
Burnett, J. C. Jr. (2005). Urocortin: advancing the neurohumoral hypothesis of heart failure. Circulation 112, 3544–3546.
Cador, M., Cole, B. J., Koob, G. F., Stinus, L., and Le Moal, M. (1993). Central administration of corticotropin releasing factor induces long-term sensitization to D-amphetamine. Brain Res. 606, 181–186.
Carboni, E., Silvagni, A., Rolando, M. T., and Di Chiara, G. (2000). Stimulation of in vivo dopamine transmission in the bed nucleus of stria terminalis by reinforcing drugs. J. Neurosci. 20, RC102.
Chang, C. P., Pearse, R. V. 2nd., O'Connell, S., and Rosenfeld, M. G. (1993). Identification of a seven transmembrane helix receptor for corticotropin-releasing factor and sauvagine in mammalian brain. Neuron 11, 1187–1195.
Chen, A., Zorrilla, E., Smith, S., Rousso, D., Levy, C., Vaughan, J., Donaldson, C., Roberts, A., Lee, K. F., and Vale, W. (2006). Urocortin 2-deficient mice exhibit gender-specific alterations in circadian hypothalamus-pituitary-adrenal axis and depressive-like behavior. J. Neurosci. 26, 5500–5510.
Chen, R., Lewis, K. A., Perrin, M. H., and Vale, W. W. (1993). Expression cloning of a human corticotropin-releasing-factor receptor. Proc. Natl. Acad. Sci. U.S.A. 90, 8967–8971.
Choi, D. S., Wang, D., Dadgar, J., Chang, W. S., and Messing, R. O. (2002). Conditional rescue of protein kinase C epsilon regulates ethanol preference and hypnotic sensitivity in adult mice. J. Neurosci. 22, 9905–9911.
Christian, D. T., Alexander, N. J., Diaz, M. R., Robinson, S., and McCool, B. A. (2012). Chronic intermittent ethanol and withdrawal differentially modulate basolateral amygdala AMPA-type glutamate receptor function and trafficking. Neuropharmacology 62, 2429–2438.
Chu, K., Koob, G. F., Cole, M., Zorrilla, E. P., and Roberts, A. J. (2007). Dependence-induced increases in ethanol self-administration in mice are blocked by the CRF1 receptor antagonist antalarmin and by CRF1 receptor knockout. Pharmacol. Biochem. Behav. 86, 813–821.
Cole, B. J., Cador, M., Stinus, L., Rivier, J., Vale, W., Koob, G. F., and Le Moal, M. (1990). Central administration of a CRF antagonist blocks the development of stress-induced behavioral sensitization. Brain Res. 512, 343–346.
Coste, S. C., Kesterson, R. A., Heldwein, K. A., Stevens, S. L., Heard, A. D., Hollis, J. H., Murray, S. E., Hill, J. K., Pantely, G. A., Hohimer, A. R., Hatton, D. C., Phillips, T. J., Finn, D. A., Low, M. J., Rittenberg, M. B., Stenzel, P., and Stenzel-Poore, M. P. (2000). Abnormal adaptations to stress and impaired cardiovascular function in mice lacking corticotropin-releasing hormone receptor-2. Nat. Genet. 24, 403–409.
David, V., Matifas, A., Gavello-Baudy, S., Decorte, L., Kieffer, B. L., and Cazala, P. (2008). Brain regional Fos expression elicited by the activation of mu- but not delta-opioid receptors of the ventral tegmental area: evidence for an implication of the ventral thalamus in opiate reward. Neuropsychopharmacology 33, 1746–1759.
Davis, L. L., Trivedi, M., Choate, A., Kramer, G. L., and Petty, F. (1997). Growth hormone response to the GABAB agonist baclofen in major depressive disorder. Psychoneuroendocrinology 22, 129–140.
Davis, M. (1992a). The role of the amygdala in fear-potentiated startle: implications for animal models of anxiety. Trends Pharmacol. Sci. 13, 35–41.
Day, H. E., Curran, E. J., Watson, S. J. Jr., and Akil, H. (1999). Distinct neurochemical populations in the rat central nucleus of the amygdala and bed nucleus of the stria terminalis: evidence for their selective activation by interleukin-1beta. J. Comp. Neurol. 413, 113–128.
De Souza, E. B. (1995). Corticotropin-releasing factor receptors: physiology, pharmacology, biochemistry and role in central nervous system and immune disorders. Psychoneuroendocrinology 20, 789–819.
Diana, M. (2011). The dopamine hypothesis of drug addiction and its potential therapeutic value. Front. Psychiatry 2:64. doi: 10.3389/fpsyt.2011.00064
Eiler, W. J. 2nd., Seyoum, R., Foster, K. L., Mailey, C., and June, H. L. (2003). D1 dopamine receptor regulates alcohol-motivated behaviors in the bed nucleus of the stria terminalis in alcohol-preferring (P) rats. Synapse 48, 45–56.
Erb, S., Shaham, Y., and Stewart, J. (1996). Stress reinstates cocaine-seeking behavior after prolonged extinction and a drug-free period. Psychopharmacology (Berl.) 128, 408–412.
Everitt, B. J., Parkinson, J. A., Olmstead, M. C., Arroyo, M., Robledo, P., and Robbins, T. W. (1999). Associative processes in addiction and reward. The role of amygdala-ventral striatal subsystems. Ann. N.Y. Acad. Sci. 877, 412–438.
Everitt, B. J., and Robbins, T. W. (2005). Neural systems of reinforcement for drug addiction: from actions to habits to compulsion. Nat. Neurosci. 8, 1481–1489.
Feltenstein, M. W., and See, R. E. (2008). The neurocircuitry of addiction: an overview. Br. J. Pharmacol. 154, 261–274.
Fibiger, H. C. (1978). Drugs and reinforcement mechanisms: a critical review of the catecholamine theory. Annu. Rev. Pharmacol. Toxicol. 18, 37–56.
Francesconi, W., Berton, F., Repunte-Canonigo, V., Hagihara, K., Thurbon, D., Lekic, D., Specio, S. E., Greenwell, T. N., Chen, S. A., Rice, K. C., Richardson, H. N., O'Dell, L. E., Zorrilla, E. P., Morales, M., Koob, G. F., and Sanna, P. P. (2009). Protracted withdrawal from alcohol and drugs of abuse impairs long-term potentiation of intrinsic excitability in the juxtacapsular bed nucleus of the stria terminalis. J. Neurosci. 29, 5389–5401.
Frye, G. D., and Breese, G. R. (1982). GABAergic modulation of ethanol-induced motor impairment. J. Pharmacol. Exp. Ther. 223, 750–756.
Fu, Y., Pollandt, S., Liu, J., Krishnan, B., Genzer, K., Orozco-Cabal, L., Gallagher, J. P., and Shinnick-Gallagher, P. (2007). Long-term potentiation (LTP) in the central amygdala (CeA) is enhanced after prolonged withdrawal from chronic cocaine and requires CRF1 receptors. J. Neurophysiol. 97, 937–941.
Fudge, J. L., and Emiliano, A. B. (2003). The extended amygdala and the dopamine system: another piece of the dopamine puzzle. J. Neuropsychiatry Clin. Neurosci. 15, 306–316.
Funk, C. K., Zorrilla, E. P., Lee, M. J., Rice, K. C., and Koob, G. F. (2007). Corticotropin-releasing factor 1 antagonists selectively reduce ethanol self-administration in ethanol-dependent rats. Biol. Psychiatry 61, 78–86.
Funk, D., Li, Z., and Le, A. D. (2006). Effects of environmental and pharmacological stressors on c-fos and corticotropin-releasing factor mRNA in rat brain: relationship to the reinstatement of alcohol seeking. Neuroscience 138, 235–243.
Gass, J. T., and Olive, M. F. (2007). Reinstatement of ethanol-seeking behavior following intravenous self-administration in Wistar rats. Alcohol. Clin. Exp. Res. 31, 1441–1445.
Gatto, G. J., McBride, W. J., Murphy, J. M., Lumeng, L., and Li, T. K. (1994). Ethanol self-infusion into the ventral tegmental area by alcohol-preferring rats. Alcohol 11, 557–564.
Gawin, F. H., and Kleber, H. D. (1986). Abstinence symptomatology and psychiatric diagnosis in cocaine abusers. Clinical observations. Arch. Gen. Psychiatry 43, 107–113.
Gehlert, D. R., Cippitelli, A., Thorsell, A., Le, A. D., Hipskind, P. A., Hamdouchi, C., Lu, J., Hembre, E. J., Cramer, J., Song, M., McKinzie, D., Morin, M., Ciccocioppo, R., and Heilig, M. (2007). 3-(4-Chloro-2-morpholin-4-yl-thiazol-5-yl)-8-(1-ethylpropyl)-2, 6-dime thyl-imidazo [1, 2-b]pyridazine: a novel brain-penetrant, orally available corticotropin-releasing factor receptor 1 antagonist with efficacy in animal models of alcoholism. J. Neurosci. 27, 2718–2726.
Gehlert, D. R., Cramer, J., and Morin, S. M. (2012). Effects of corticotropin-releasing factor 1 receptor antagonism on the hypothalamic-pituitary-adrenal axis of rodents. J. Pharmacol. Exp. Ther. 341, 672–680.
George, O., Le Moal, M., and Koob, G. F. (2011). Allostasis and addiction: role of the dopamine and corticotropin-releasing factor systems. Physiol. Behav. 106, 58–64.
Georges, F., and Aston-Jones, G. (2001). Potent regulation of midbrain dopamine neurons by the bed nucleus of the stria terminalis. J. Neurosci. 21, RC160.
Georges, F., and Aston-Jones, G. (2002). Activation of ventral tegmental area cells by the bed nucleus of the stria terminalis: a novel excitatory amino acid input to midbrain dopamine neurons. J. Neurosci. 22, 5173–5187.
Ghitza, U. E., Gray, S. M., Epstein, D. H., Rice, K. C., and Shaham, Y. (2006). The anxiogenic drug yohimbine reinstates palatable food seeking in a rat relapse model: a role of CRF1 receptors. Neuropsychopharmacology 31, 2188–2196.
Goeders, N. E., and Guerin, G. F. (1994). Non-contingent electric footshock facilitates the acquisition of intravenous cocaine self-administration in rats. Psychopharmacology (Berl.) 114, 63–70.
Grace, C. R., Perrin, M. H., DiGruccio, M. R., Miller, C. L., Rivier, J. E., Vale, W. W., and Riek, R. (2004). NMR structure and peptide hormone binding site of the first extracellular domain of a type B1 G protein-coupled receptor. Proc. Natl. Acad. Sci. U.S.A. 101, 12836–12841.
Grace, C. R., Perrin, M. H., Gulyas, J., Digruccio, M. R., Cantle, J. P., Rivier, J. E., Vale, W. W., and Riek, R. (2007). Structure of the N-terminal domain of a type B1 G protein-coupled receptor in complex with a peptide ligand. Proc. Natl. Acad. Sci. U.S.A. 104, 4858–4863.
Guillemin, R., and Rosenberg, B. (1955). Humoral hypothalamic control of anterior pituitary: a study with combined tissue cultures. Endocrinology 57, 599–607.
Hahn, J., Hopf, F. W., and Bonci, A. (2009). Chronic cocaine enhances corticotropin-releasing factor-dependent potentiation of excitatory transmission in ventral tegmental area dopamine neurons. J. Neurosci. 29, 6535–6544.
Hansson, A. C., Cippitelli, A., Sommer, W. H., Ciccocioppo, R., and Heilig, M. (2007). Region-specific down-regulation of Crhr1 gene expression in alcohol-preferring msP rats following ad lib access to alcohol. Addict. Biol. 12, 30–34.
Hansson, A. C., Cippitelli, A., Sommer, W. H., Fedeli, A., Bjork, K., Soverchia, L., Terasmaa, A., Massi, M., Heilig, M., and Ciccocioppo, R. (2006). Variation at the rat Crhr1 locus and sensitivity to relapse into alcohol seeking induced by environmental stress. Proc. Natl. Acad. Sci. U.S.A. 103, 15236–15241.
Hansson, A. C., Rimondini, R., Neznanova, O., Sommer, W. H., and Heilig, M. (2008). Neuroplasticity in brain reward circuitry following a history of ethanol dependence. Eur. J. Neurosci. 27, 1912–1922.
Heilig, M., and Koob, G. F. (2007). A key role for corticotropin-releasing factor in alcohol dependence. Trends Neurosci. 30, 399–406.
Heilig, M., Thorsell, A., Sommer, W. H., Hansson, A. C., Ramchandani, V. A., George, D. T., Hommer, D., and Barr, C. S. (2010). Translating the neuroscience of alcoholism into clinical treatments: from blocking the buzz to curing the blues. Neurosci. Biobehav. Rev. 35, 334–344.
Heimer, L., and Alheid, G. F. (1991). Piecing together the puzzle of basal forebrain anatomy. Adv. Exp. Med. Biol. 295, 1–42.
Heinrichs, S. C., Menzaghi, F., Schulteis, G., Koob, G. F., and Stinus, L. (1995). Suppression of corticotropin-releasing factor in the amygdala attenuates aversive consequences of morphine withdrawal. Behav. Pharmacol. 6, 74–80.
Heinrichs, S. C., Pich, E. M., Miczek, K. A., Britton, K. T., and Koob, G. F. (1992). Corticotropin-releasing factor antagonist reduces emotionality in socially defeated rats via direct neurotropic action. Brain Res. 581, 190–197.
Herringa, R. J., Nanda, S. A., Hsu, D. T., Roseboom, P. H., and Kalin, N. H. (2004). The effects of acute stress on the regulation of central and basolateral amygdala CRF-binding protein gene expression. Brain Res. Mol. Brain Res. 131, 17–25.
Hoare, S. R. (2005). Mechanisms of peptide and nonpeptide ligand binding to Class B G-protein-coupled receptors. Drug Discov. Today 10, 417–427.
Hodge, C. W., Raber, J., McMahon, T., Walter, H., Sanchez-Perez, A. M., Olive, M. F., Mehmert, K., Morrow, A. L., and Messing, R. O. (2002). Decreased anxiety-like behavior, reduced stress hormones, and neurosteroid supersensitivity in mice lacking protein kinase Cepsilon. J. Clin. Invest. 110, 1003–1010.
Hsu, S. Y., and Hsueh, A. J. (2001). Human stresscopin and stresscopin-related peptide are selective ligands for the type 2 corticotropin-releasing hormone receptor. Nat. Med. 7, 605–611.
Hubbard, D. T., Nakashima, B. R., Lee, I., and Takahashi, L. K. (2007). Activation of basolateral amygdala corticotropin-releasing factor 1 receptors modulates the consolidation of contextual fear. Neuroscience 150, 818–828.
Hyytia, P., and Koob, G. F. (1995). GABAA receptor antagonism in the extended amygdala decreases ethanol self-administration in rats. Eur. J. Pharmacol. 283, 151–159.
Jochman, K. A., Newman, S. M., Kalin, N. H., and Bakshi, V. P. (2005). Corticotropin-releasing factor-1 receptors in the basolateral amygdala mediate stress-induced anorexia. Behav. Neurosci. 119, 1448–1458.
Justice, N. J., Yuan, Z. F., Sawchenko, P. E., and Vale, W. (2008). Type 1 corticotropin-releasing factor receptor expression reported in BAC transgenic mice: implications for reconciling ligand-receptor mismatch in the central corticotropin-releasing factor system. J. Comp. Neurol. 511, 479–496.
Karolyi, I. J., Burrows, H. L., Ramesh, T. M., Nakajima, M., Lesh, J. S., Seong, E., Camper, S. A., and Seasholtz, A. F. (1999). Altered anxiety and weight gain in corticotropin-releasing hormone-binding protein-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 96, 11595–11600.
Kash, T. L., Nobis, W. P., Matthews, R. T., and Winder, D. G. (2008). Dopamine enhances fast excitatory synaptic transmission in the extended amygdala by a CRF-R1-dependent process. J. Neurosci. 28, 13856–13865.
Kash, T. L., and Winder, D. G. (2006). Neuropeptide Y and corticotropin-releasing factor bi-directionally modulate inhibitory synaptic transmission in the bed nucleus of the stria terminalis. Neuropharmacology 51, 1013–1022.
Kemp, C. F., Woods, R. J., and Lowry, P. J. (1998). The corticotrophin-releasing factor-binding protein: an act of several parts. Peptides 19, 1119–1128.
Kimura, Y., Takahashi, K., Totsune, K., Muramatsu, Y., Kaneko, C., Darnel, A. D., Suzuki, T., Ebina, M., Nukiwa, T., and Sasano, H. (2002). Expression of urocortin and corticotropin-releasing factor receptor subtypes in the human heart. J. Clin. Endocrinol. Metab. 87, 340–346.
Kishimoto, T., Radulovic, J., Radulovic, M., Lin, C. R., Schrick, C., Hooshmand, F., Hermanson, O., Rosenfeld, M. G., and Spiess, J. (2000). Deletion of crhr2 reveals an anxiolytic role for corticotropin-releasing hormone receptor-2. Nat. Genet. 24, 415–419.
Koob, G., and Kreek, M. J. (2007). Stress, dysregulation of drug reward pathways, and the transition to drug dependence. Am. J. Psychiatry 164, 1149–1159.
Koob, G. F. (1992). Drugs of abuse: anatomy, pharmacology and function of reward pathways. Trends Pharmacol. Sci. 13, 177–184.
Koob, G. F. (1999). Stress, corticotropin-releasing factor, and drug addiction. Ann. N.Y. Acad. Sci. 897, 27–45.
Koob, G. F. (2003). Neuroadaptive mechanisms of addiction: studies on the extended amygdala. Eur. Neuropsychopharmacol. 13, 442–452.
Koob, G. F. (2009a). Dynamics of neuronal circuits in addiction: reward, antireward, and emotional memory. Pharmacopsychiatry 42(Suppl. 1), S32–S41.
Koob, G. F. (2009b). Neurobiological substrates for the dark side of compulsivity in addiction. Neuropharmacology 56(Suppl. 1), 18–31.
Koob, G. F., Ahmed, S. H., Boutrel, B., Chen, S. A., Kenny, P. J., Markou, A., O'Dell, L. E., Parsons, L. H., and Sanna, P. P. (2004). Neurobiological mechanisms in the transition from drug use to drug dependence. Neurosci. Biobehav. Rev. 27, 739–749.
Koob, G. F., and Bloom, F. E. (1985). Corticotropin-releasing factor and behavior. Fed. Proc. 44(1 Pt 2), 259–263.
Koob, G. F., and Heinrichs, S. C. (1999). A role for corticotropin releasing factor and urocortin in behavioral responses to stressors. Brain Res. 848, 141–152.
Koob, G. F., and Le Moal, M. (1997). Drug abuse: hedonic homeostatic dysregulation. Science 278, 52–58.
Koob, G. F., and Le Moal, M. (2001). Drug addiction, dysregulation of reward, and allostasis. Neuropsychopharmacology 24, 97–129.
Koob, G. F., and Le Moal, M. (2008a). Addiction and the brain antireward system. Annu. Rev. Psychol. 59, 29–53.
Koob, G. F., and Le Moal, M. (2008b). Review. Neurobiological mechanisms for opponent motivational processes in addiction. Philos. Trans. R. Soc. Lond. B Biol. Sci. 363, 3113–3123.
Koob, G. F., and Nestler, E. J. (1997). The neurobiology of drug addiction. J. Neuropsychiatry Clin. Neurosci. 9, 482–497.
Koob, G. F., and Zorrilla, E. P. (2010). Neurobiological mechanisms of addiction: focus on corticotropin-releasing factor. Curr. Opin. Investig. Drugs 11, 63–71.
Koob, G. F., and Zorrilla, E. P. (2012). Update on corticotropin-releasing factor pharmacotherapy for psychiatric disorders: a revisionist view. Neuropsychopharmacology 37, 308–309.
Korotkova, T. M., Brown, R. E., Sergeeva, O. A., Ponomarenko, A. A., and Haas, H. L. (2006). Effects of arousal- and feeding-related neuropeptides on dopaminergic and GABAergic neurons in the ventral tegmental area of the rat. Eur. J. Neurosci. 23, 2677–2685.
Kostich, W. A., Chen, A., Sperle, K., and Largent, B. L. (1998). Molecular identification and analysis of a novel human corticotropin-releasing factor (CRF) receptor: the CRF2gamma receptor. Mol. Endocrinol. 12, 1077–1085.
Kreibich, A. S., Briand, L., Cleck, J. N., Ecke, L., Rice, K. C., and Blendy, J. A. (2009). Stress-induced potentiation of cocaine reward: a role for CRF R1 and CREB. Neuropsychopharmacology 34, 2609–2617.
Lack, A. K., Floyd, D. W., and McCool, B. A. (2005). Chronic ethanol ingestion modulates proanxiety factors expressed in rat central amygdala. Alcohol 36, 83–90.
Le, A. D., Harding, S., Juzytsch, W., Fletcher, P. J., and Shaham, Y. (2002). The role of corticotropin-releasing factor in the median raphe nucleus in relapse to alcohol. J. Neurosci. 22, 7844–7849.
Le, A. D., Harding, S., Juzytsch, W., Watchus, J., Shalev, U., and Shaham, Y. (2000). The role of corticotrophin-releasing factor in stress-induced relapse to alcohol-seeking behavior in rats. Psychopharmacology (Berl.) 150, 317–324.
Le, A. D., Quan, B., Juzytch, W., Fletcher, P. J., Joharchi, N., and Shaham, Y. (1998). Reinstatement of alcohol-seeking by priming injections of alcohol and exposure to stress in rats. Psychopharmacology (Berl.) 135, 169–174.
LeDoux, J. E. (1992). Brain mechanisms of emotion and emotional learning. Curr. Opin. Neurobiol. 2, 191–197.
Lewis, K., Li, C., Perrin, M. H., Blount, A., Kunitake, K., Donaldson, C., Vaughan, J., Reyes, T. M., Gulyas, J., Fischer, W., Bilezikjian, L., Rivier, J., Sawchenko, P. E., and Vale, W. W. (2001). Identification of urocortin III, an additional member of the corticotropin-releasing factor (CRF) family with high affinity for the CRF2 receptor. Proc. Natl. Acad. Sci. U.S.A. 98, 7570–7575.
Li, C., Chen, P., Vaughan, J., Lee, K. F., and Vale, W. (2007). Urocortin 3 regulates glucose-stimulated insulin secretion and energy homeostasis. Proc. Natl. Acad. Sci. U.S.A. 104, 4206–4211.
Liu, J., Yu, B., Neugebauer, V., Grigoriadis, D. E., Rivier, J., Vale, W. W., Shinnick-Gallagher, P., and Gallagher, J. P. (2004). Corticotropin-releasing factor and Urocortin I modulate excitatory glutamatergic synaptic transmission. J. Neurosci. 24, 4020–4029.
Liu, X., and Weiss, F. (2002). Additive effect of stress and drug cues on reinstatement of ethanol seeking: exacerbation by history of dependence and role of concurrent activation of corticotropin-releasing factor and opioid mechanisms. J. Neurosci. 22, 7856–7861.
Logrip, M. L., Koob, G. F., and Zorrilla, E. P. (2011). Role of corticotropin-releasing factor in drug addiction: potential for pharmacological intervention. CNS Drugs 25, 271–287.
Lovenberg, T. W., Chalmers, D. T., Liu, C., and De Souza, E. B. (1995a). CRF2 alpha and CRF2 beta receptor mRNAs are differentially distributed between the rat central nervous system and peripheral tissues. Endocrinology 136, 4139–4142.
Lovenberg, T. W., Liaw, C. W., Grigoriadis, D. E., Clevenger, W., Chalmers, D. T., De Souza, E. B., and Oltersdorf, T. (1995b). Cloning and characterization of a functionally distinct corticotropin-releasing factor receptor subtype from rat brain. Proc. Natl. Acad. Sci. U.S.A. 92, 836–840.
Lowery-Gionta, E. G., Navarro, M., Li, C., Pleil, K. E., Rinker, J. A., Cox, B. R., Sprow, G. M., Kash, T. L., and Thiele, T. E. (2012). Corticotropin releasing factor signaling in the central amygdala is recruited during binge-like ethanol consumption in C57BL/6J mice. J. Neurosci. 32, 3405–3413.
Lu, L., Shepard, J. D., Hall, F. S., and Shaham, Y. (2003). Effect of environmental stressors on opiate and psychostimulant reinforcement, reinstatement and discrimination in rats: a review. Neurosci. Biobehav. Rev. 27, 457–491.
Luscher, C., and Malenka, R. C. (2011). Drug-evoked synaptic plasticity in addiction: from molecular changes to circuit remodeling. Neuron 69, 650–663.
Maren, S. (1999). Long-term potentiation in the amygdala: a mechanism for emotional learning and memory. Trends Neurosci. 22, 561–567.
Marinelli, P. W., Funk, D., Juzytsch, W., Harding, S., Rice, K. C., Shaham, Y., and Le, A. D. (2007). The CRF1 receptor antagonist antalarmin attenuates yohimbine-induced increases in operant alcohol self-administration and reinstatement of alcohol seeking in rats. Psychopharmacology (Berl.) 195, 345–355.
Martin, T. J., Coller, M., Co, C., and Smith, J. E. (2008). Micro-opioid receptor alkylation in the ventral pallidum and ventral tegmental area, but not in the nucleus accumbens, attenuates the effects of heroin on cocaine self-administration in rats. Neuropsychopharmacology 33, 1171–1178.
Meloni, E. G., Jackson, A. V., Cohen, B. M., and Carlezon, W. A. Jr. (2005). Corticotropin-releasing factor from the rat brain measured by protein immunoblot. Peptides 26, 2252–2256.
Merali, Z., McIntosh, J., Kent, P., Michaud, D., and Anisman, H. (1998). Aversive and appetitive events evoke the release of corticotropin-releasing hormone and bombesin-like peptides at the central nucleus of the amygdala. J. Neurosci. 18, 4758–4766.
Merlo Pich, E., Lorang, M., Yeganeh, M., Rodriguez de Fonseca, F., Raber, J., Koob, G. F., and Weiss, F. (1995). Increase of extracellular corticotropin-releasing factor-like immunoreactivity levels in the amygdala of awake rats during restraint stress and ethanol withdrawal as measured by microdialysis. J. Neurosci. 15, 5439–5447.
Molander, A., Vengeliene, V., Heilig, M., Wurst, W., Deussing, J. M., and Spanagel, R. (2012). Brain-specific inactivation of the Crhr1 gene inhibits post-dependent and stress-induced alcohol intake, but does not affect relapse-like drinking. Neuropsychopharmacology 37, 1047–1056.
Muller, M. B., Zimmermann, S., Sillaber, I., Hagemeyer, T. P., Deussing, J. M., Timpl, P., Kormann, M. S., Droste, S. K., Kuhn, R., Reul, J. M., Holsboer, F., and Wurst, W. (2003). Limbic corticotropin-releasing hormone receptor 1 mediates anxiety-related behavior and hormonal adaptation to stress. Nat. Neurosci. 6, 1100–1107.
Nie, Z., Schweitzer, P., Roberts, A. J., Madamba, S. G., Moore, S. D., and Siggins, G. R. (2004). Ethanol augments GABAergic transmission in the central amygdala via CRF1 receptors. Science 303, 1512–1514.
Nielsen, C. K., Simms, J. A., Bito-Onon, J. J., Li, R., Ananthan, S., and Bartlett, S. E. (2011). The delta opioid receptor antagonist, SoRI-9409, decreases yohimbine stress-induced reinstatement of ethanol-seeking. Addict. Biol. 17, 224–234.
Nielsen, S. M., Nielsen, L. Z., Hjorth, S. A., Perrin, M. H., and Vale, W. W. (2000). Constitutive activation of tethered-peptide/corticotropin-releasing factor receptor chimeras. Proc. Natl. Acad. Sci. U.S.A. 97, 10277–10281.
Overstreet, D. H., Knapp, D. J., and Breese, G. R. (2004). Modulation of multiple ethanol withdrawal-induced anxiety-like behavior by CRF and CRF1 receptors. Pharmacol. Biochem. Behav. 77, 405–413.
Palkovits, M., Brownstein, M. J., and Vale, W. (1983). Corticotropin releasing factor (CRF) immunoreactivity in hypothalamic and extrahypothalamic nuclei of sheep brain. Neuroendocrinology 37, 302–305.
Pare, D., Quirk, G. J., and Ledoux, J. E. (2004). New vistas on amygdala networks in conditioned fear. J. Neurophysiol. 92, 1–9.
Perrin, M., Donaldson, C., Chen, R., Blount, A., Berggren, T., Bilezikjian, L., Sawchenko, P., and Vale, W. (1995). Identification of a second corticotropin-releasing factor receptor gene and characterization of a cDNA expressed in heart. Proc. Natl. Acad. Sci. U.S.A. 92, 2969–2973.
Phelps, E. A., and LeDoux, J. E. (2005). Contributions of the amygdala to emotion processing: from animal models to human behavior. Neuron 48, 175–187.
Piazza, P. V., Deminiere, J. M., Le Moal, M., and Simon, H. (1990). Stress- and pharmacologically-induced behavioral sensitization increases vulnerability to acquisition of amphetamine self-administration. Brain Res. 514, 22–26.
Pierce, R. C., and Kumaresan, V. (2006). The mesolimbic dopamine system: the final common pathway for the reinforcing effect of drugs of abuse? Neurosci. Biobehav. Rev. 30, 215–238.
Pitkanen, A., Jolkkonen, E., and Kemppainen, S. (2000). Anatomic heterogeneity of the rat amygdaloid complex. Folia. Morphol. (Warsz.) 59, 1–23.
Pitts, M. W., Todorovic, C., Blank, T., and Takahashi, L. K. (2009). The central nucleus of the amygdala and corticotropin-releasing factor: insights into contextual fear memory. J. Neurosci. 29, 7379–7388.
Pollandt, S., Liu, J., Orozco-Cabal, L., Grigoriadis, D. E., Vale, W. W., Gallagher, J. P., and Shinnick-Gallagher, P. (2006). Cocaine withdrawal enhances long-term potentiation induced by corticotropin-releasing factor at central amygdala glutamatergic synapses via CRF, NMDA receptors and PKA. Eur. J. Neurosci. 24, 1733–1743.
Potter, E., Behan, D. P., Linton, E. A., Lowry, P. J., Sawchenko, P. E., and Vale, W. W. (1992). The central distribution of a corticotropin-releasing factor (CRF)-binding protein predicts multiple sites and modes of interaction with CRF. Proc. Natl. Acad. Sci. U.S.A. 89, 4192–4196.
Potter, E., Sutton, S., Donaldson, C., Chen, R., Perrin, M., Lewis, K., Sawchenko, P. E., and Vale, W. (1994). Distribution of corticotropin-releasing factor receptor mRNA expression in the rat brain and pituitary. Proc. Natl. Acad. Sci. U.S.A. 91, 8777–8781.
Refojo, D., Schweizer, M., Kuehne, C., Ehrenberg, S., Thoeringer, C., Vogl, A. M., Dedic, N., Schumacher, M., von Wolff, G., Avrabos, C., Touma, C., Engblom, D., Schutz, G., Nave, K. A., Eder, M., Wotjak, C. T., Sillaber, I., Holsboer, F., Wurst, W., and Deussing, J. M. (2011). Glutamatergic and dopaminergic neurons mediate anxiogenic and anxiolytic effects of CRHR1. Science 333, 1903–1907.
Reyes, T. M., Lewis, K., Perrin, M. H., Kunitake, K. S., Vaughan, J., Arias, C. A., Hogenesch, J. B., Gulyas, J., Rivier, J., Vale, W. W., and Sawchenko, P. E. (2001). Urocortin II: a member of the corticotropin-releasing factor (CRF) neuropeptide family that is selectively bound by type 2 CRF receptors. Proc. Natl. Acad. Sci. U.S.A. 98, 2843–2848.
Rezayof, A., Zarrindast, M. R., Sahraei, H., and Haeri-Rohani, A. H. (2002). Involvement of dopamine D2 receptors of the central amygdala on the acquisition and expression of morphine-induced place preference in rat. Pharmacol. Biochem. Behav. 74, 187–197.
Richards, J. K., Simms, J. A., Steensland, P., Taha, S. A., Borgland, S. L., Bonci, A., and Bartlett, S. E. (2008). Inhibition of orexin-1/hypocretin-1 receptors inhibits yohimbine-induced reinstatement of ethanol and sucrose seeking in Long-Evans rats. Psychopharmacology (Berl.) 199, 109–117.
Richter, R. M., Pich, E. M., Koob, G. F., and Weiss, F. (1995). Sensitization of cocaine-stimulated increase in extracellular levels of corticotropin-releasing factor from the rat amygdala after repeated administration as determined by intracranial microdialysis. Neurosci. Lett. 187, 169–172.
Richter, R. M., and Weiss, F. (1999). In vivo CRF release in rat amygdala is increased during cocaine withdrawal in self-administering rats. Synapse 32, 254–261.
Rijkers, D. T., Kruijtzer, J. A., van Oostenbrugge, M., Ronken, E., den Hartog, J. A., and Liskamp, R. M. (2004). Structure-activity studies on the corticotropin releasing factor antagonist astressin, leading to a minimal sequence necessary for antagonistic activity. Chembiochem 5, 340–348.
Rivier, J., Rivier, C., and Vale, W. (1984). Synthetic competitive antagonists of corticotropin-releasing factor: effect on ACTH secretion in the rat. Science 224, 889–891.
Roberto, M., Madamba, S. G., Moore, S. D., Tallent, M. K., and Siggins, G. R. (2003). Ethanol increases GABAergic transmission at both pre- and postsynaptic sites in rat central amygdala neurons. Proc. Natl. Acad. Sci. U.S.A. 100, 2053–2058.
Roberto, M., Madamba, S. G., Stouffer, D. G., Parsons, L. H., and Siggins, G. R. (2004). Increased GABA release in the central amygdala of ethanol-dependent rats. J. Neurosci. 24, 10159–10166.
Roberts, A. J., Cole, M., and Koob, G. F. (1996). Intra-amygdala muscimol decreases operant ethanol self-administration in dependent rats. Alcohol. Clin. Exp. Res. 20, 1289–1298.
Rodaros, D., Caruana, D. A., Amir, S., and Stewart, J. (2007). Corticotropin-releasing factor projections from limbic forebrain and paraventricular nucleus of the hypothalamus to the region of the ventral tegmental area. Neuroscience 150, 8–13.
Roozendaal, B., Brunson, K. L., Holloway, B. L., McGaugh, J. L., and Baram, T. Z. (2002). Involvement of stress-released corticotropin-releasing hormone in the basolateral amygdala in regulating memory consolidation. Proc. Natl. Acad. Sci. U.S.A. 99, 13908–13913.
Saffran, M., Schally, A. V., and Benfey, B. G. (1955). Stimulation of the release of corticotropin from the adenohypophysis by a neurohypophysial factor. Endocrinology 57, 439–444.
Sah, P., Faber, E. S., Lopez De Armentia, M., and Power, J. (2003). The amygdaloid complex: anatomy and physiology. Physiol. Rev. 83, 803–834.
Sakanaka, M., Shibasaki, T., and Lederis, K. (1986). Distribution and efferent projections of corticotropin-releasing factor-like immunoreactivity in the rat amygdaloid complex. Brain Res. 382, 213–238.
Sanchez, M. M., Young, L. J., Plotsky, P. M., and Insel, T. R. (1999). Autoradiographic and in situ hybridization localization of corticotropin-releasing factor 1 and 2 receptors in nonhuman primate brain. J. Comp. Neurol. 408, 365–377.
Sarnyai, Z., Hohn, J., Szabo, G., and Penke, B. (1992). Critical role of endogenous corticotropin-releasing factor (CRF) in the mediation of the behavioral action of cocaine in rats. Life Sci. 51, 2019–2024.
Sarnyai, Z., Shaham, Y., and Heinrichs, S. C. (2001). The role of corticotropin-releasing factor in drug addiction. Pharmacol. Rev. 53, 209–243.
Seasholtz, A. F., Valverde, R. A., and Denver, R. J. (2002). Corticotropin-releasing hormone-binding protein: biochemistry and function from fishes to mammals. J. Endocrinol. 175, 89–97.
Shaham, Y., Erb, S., Leung, S., Buczek, Y., and Stewart, J. (1998). CP-154 526, a selective, non-peptide antagonist of the corticotropin-releasing factor1 receptor attenuates stress-induced relapse to drug seeking in cocaine- and heroin-trained rats. Psychopharmacology (Berl.) 137, 184–190.
Shaham, Y., Funk, D., Erb, S., Brown, T. J., Walker, C. D., and Stewart, J. (1997). Corticotropin-releasing factor, but not corticosterone, is involved in stress-induced relapse to heroin-seeking in rats. J. Neurosci. 17, 2605–2614.
Shaham, Y., Kelsey, J. E., and Stewart, J. (1995). Temporal factors in the effect of restraint stress on morphine-induced behavioral sensitization in the rat. Psychopharmacology (Berl.) 117, 102–109.
Silberman, Y., Bajo, M., Chappell, A. M., Christian, D. T., Cruz, M., Diaz, M. R., Kash, T., Lack, A. K., Messing, R. O., Siggins, G. R., Winder, D., Roberto, M., McCool, B. A., and Weiner, J. L. (2009). Neurobiological mechanisms contributing to alcohol-stress-anxiety interactions. Alcohol 43, 509–519.
Simms, J. A., Haass-Koffler, C. L., Bito-Onon, J., Li, R., and Bartlett, S. E. (2011). Mifepristone in the central nucleus of the amygdala reduces yohimbine stress-induced reinstatement of ethanol-seeking. Neuropsychopharmacology 37, 906–918.
Smith, G. W., Aubry, J. M., Dellu, F., Contarino, A., Bilezikjian, L. M., Gold, L. H., Chen, R., Marchuk, Y., Hauser, C., Bentley, C. A., Sawchenko, P. E., Koob, G. F., Vale, W., and Lee, K. F. (1998). Corticotropin releasing factor receptor 1-deficient mice display decreased anxiety, impaired stress response, and aberrant neuroendocrine development. Neuron 20, 1093–1102.
Sommer, W. H., Rimondini, R., Hansson, A. C., Hipskind, P. A., Gehlert, D. R., Barr, C. S., and Heilig, M. A. (2008). Upregulation of voluntary alcohol intake, behavioral sensitivity to stress, and amygdala crhr1 expression following a history of dependence. Biol. Psychiatry 63, 139–145.
Suda, T., Sumitomo, T., Tozawa, F., Ushiyama, T., and Demura, H. (1989). Corticotropin-releasing factor-binding protein is a glycoprotein. Biochem. Biophys. Res. Commun. 165, 703–707.
Sun, N., and Cassell, M. D. (1993). Intrinsic GABAergic neurons in the rat central extended amygdala. J. Comp. Neurol. 330, 381–404.
Swanson, L. W., Sawchenko, P. E., Rivier, J., and Vale, W. W. (1983). Organization of ovine corticotropin-releasing factor immunoreactive cells and fibers in the rat brain: an immunohistochemical study. Neuroendocrinology 36, 165–186.
Tagliaferro, P., and Morales, M. (2008). Synapses between corticotropin-releasing factor-containing axon terminals and dopaminergic neurons in the ventral tegmental area are predominantly glutamatergic. J. Comp. Neurol. 506, 616–626.
Thatcher-Britton, K., and Koob, G. F. (1986). Alcohol reverses the proconflict effect of corticotropin-releasing factor. Regul. Pept. 16, 315–320.
Timpl, P., Spanagel, R., Sillaber, I., Kresse, A., Reul, J. M., Stalla, G. K., Blanquet, V., Steckler, T., Holsboer, F., and Wurst, W. (1998). Impaired stress response and reduced anxiety in mice lacking a functional corticotropin-releasing hormone receptor 1. Nat. Genet. 19, 162–166.
Tomkins, D. M., and Sellers, E. M. (2001). Addiction and the brain: the role of neurotransmitters in the cause and treatment of drug dependence. CMAJ 164, 817–821.
Tronche, F., Kellendonk, C., Kretz, O., Gass, P., Anlag, K., Orban, P. C., Bock, R., Klein, R., and Schutz, G. (1999). Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat. Genet. 23, 99–103.
Tsai-Morris, C. H., Buczko, E., Geng, Y., Gamboa-Pinto, A., and Dufau, M. L. (1996). The genomic structure of the rat corticotropin releasing factor receptor. A member of the class II G protein-coupled receptors. J. Biol. Chem. 271, 14519–14525.
Turnbull, A. V., and Rivier, C. (1997). Corticotropin-releasing factor (CRF) and endocrine responses to stress: CRF receptors, binding protein, and related peptides. Proc. Soc. Exp. Biol. Med. 215, 1–10.
Tye, K. M., Stuber, G. D., de Ridder, B., Bonci, A., and Janak, P. H. (2008). Rapid strengthening of thalamo-amygdala synapses mediates cue-reward learning. Nature 453, 1253–1257.
Ungless, M. A., Singh, V., Crowder, T. L., Yaka, R., Ron, D., and Bonci, A. (2003). Corticotropin-releasing factor requires CRF binding protein to potentiate NMDA receptors via CRF receptor 2 in dopamine neurons. Neuron 39, 401–407.
Ungless, M. A., Whistler, J. L., Malenka, R. C., and Bonci, A. (2001). Single cocaine exposure in vivo induces long-term potentiation in dopamine neurons. Nature 411, 583–587.
Valdez, G. R., Zorrilla, E. P., Rivier, J., Vale, W. W., and Koob, G. F. (2003). Locomotor suppressive and anxiolytic-like effects of urocortin 3, a highly selective type 2 corticotropin-releasing factor agonist. Brain Res. 980, 206–212.
Vale, W., Spiess, J., Rivier, C., and Rivier, J. (1981). Characterization of a 41-residue ovine hypothalamic peptide that stimulates secretion of corticotropin and beta-endorphin. Science 213, 1394–1397.
Van Pett, K., Viau, V., Bittencourt, J. C., Chan, R. K., Li, H. Y., Arias, C., Prins, G. S., Perrin, M., Vale, W., and Sawchenko, P. E. (2000). Distribution of mRNAs encoding CRF receptors in brain and pituitary of rat and mouse. J. Comp. Neurol. 428, 191–212.
Vaughan, J., Donaldson, C., Bittencourt, J., Perrin, M. H., Lewis, K., Sutton, S., Chan, R., Turnbull, A. V., Lovejoy, D., Rivier, C., Rivier, J., Sawchenko, P. E., and Vale, W. (1995). Urocortin, a mammalian neuropeptide related to fish urotensin I and to corticotropin-releasing factor. Nature 378, 287–292.
Veinante, P., Stoeckel, M. E., and Freund-Mercier, M. J. (1997). GABA- and peptide-immunoreactivities co-localize in the rat central extended amygdala. Neuroreport 8, 2985–2989.
Vita, N., Laurent, P., Lefort, S., Chalon, P., Lelias, J. M., Kaghad, M., Le Fur, G., Caput, D., and Ferrara, P. (1993). Primary structure and functional expression of mouse pituitary and human brain corticotrophin releasing factor receptors. FEBS Lett. 335, 1–5.
Walker, D. L., and Davis, M. (2008). Role of the extended amygdala in short-duration versus sustained fear: a tribute to Dr. Lennart Heimer. Brain Struct. Funct. 213, 29–42.
Wallace, B. C. (1989). Psychological and environmental determinants of relapse in crack cocaine smokers. J. Subst. Abuse Treat. 6, 95–106.
Wanat, M. J., Hopf, F. W., Stuber, G. D., Phillips, P. E., and Bonci, A. (2008). Corticotropin-releasing factor increases mouse ventral tegmental area dopamine neuron firing through a protein kinase C-dependent enhancement of Ih. J. Physiol. 586, 2157–2170.
Wang, B., You, Z. B., Rice, K. C., and Wise, R. A. (2007a). Stress-induced relapse to cocaine seeking: roles for the CRF(2) receptor and CRF-binding protein in the ventral tegmental area of the rat. Psychopharmacology (Berl.) 193, 283–294.
Wang, H., Aodon g, Shu, Y., Momotani, Y., Wang, X., Mori, Y., and Momotani, E. (2007b). Corticotropin-releasing hormone and urocortin expression in peripheral blood cells from experimentally infected cattle with Mycobacterium avium subsp. paratuberculosis. Microbes Infect. 9, 1061–1069.
Wang, H. L., and Morales, M. (2008). Corticotropin-releasing factor binding protein within the ventral tegmental area is expressed in a subset of dopaminergic neurons. J. Comp. Neurol. 509, 302–318.
Wang, J., Fang, Q., Liu, Z., and Lu, L. (2006). Region-specific effects of brain corticotropin-releasing factor receptor type 1 blockade on footshock-stress- or drug-priming-induced reinstatement of morphine conditioned place preference in rats. Psychopharmacology (Berl.) 185, 19–28.
Washburn, M. S., and Moises, H. C. (1992). Electrophysiological and morphological properties of rat basolateral amygdaloid neurons in vitro. J. Neurosci. 12, 4066–4079.
Watkins, S. S., Epping-Jordan, M. P., Koob, G. F., and Markou, A. (1999). Blockade of nicotine self-administration with nicotinic antagonists in rats. Pharmacol. Biochem. Behav. 62, 743–751.
Weiss, F., and Koob, G. F. (2001). Drug addiction: functional neurotoxicity of the brain reward systems. Neurotox. Res. 3, 145–156.
Wise, R. A. (2008). Dopamine and reward: the anhedonia hypothesis 30 years on. Neurotox. Res. 14, 169–183.
Woods, R. J., Kemp, C. F., David, J., and Lowry, P. J. (1997). Heterogeneity of the human corticotropin-releasing factor-binding protein. J. Clin. Endocrinol. Metab. 82, 1566–1571.
Woods, R. J., Kemp, C. F., David, J., Sumner, I. G., and Lowry, P. J. (1999). Cleavage of recombinant human corticotropin-releasing factor (CRF)-binding protein produces a 27-kilodalton fragment capable of binding CRF. J. Clin. Endocrinol. Metab. 84, 2788–2794.
Wright, S. P., Doughty, R. N., Frampton, C. M., Gamble, G. D., Yandle, T. G., and Richards, A. M. (2009). Plasma urocortin 1 in human heart failure. Circ. Heart Fail. 2, 465–471.
Xue, Y., Steketee, J. D., and Sun, W. (2012). Inactivation of the central nucleus of the amygdala reduces the effect of punishment on cocaine self-administration in rats. Eur. J. Neurosci. 35, 775–783.
Yamada, Y., Mizutani, K., Mizusawa, Y., Hantani, Y., Tanaka, M., Tanaka, Y., Tomimoto, M., Sugawara, M., Imai, N., Yamada, H., Okajima, N., and Haruta, J. (2004). New class of corticotropin-releasing factor (CRF) antagonists: small peptides having high binding affinity for CRF receptor. J. Med. Chem. 47, 1075–1078.
Yokel, R. A., and Wise, R. A. (1975). Increased lever pressing for amphetamine after pimozide in rats: implications for a dopamine theory of reward. Science 187, 547–549.
Zorrilla, E. P., and Koob, G. F. (2012). Progress in corticotropin-releasing factor-1 antagonist development. Drug Discov. Today 15, 371–383.
Keywords: neuroplasticity, addiction, corticotropin releasing factor system, ethanol, anxiety, stress-induced
Citation: Haass-Koffler CL and Bartlett SE (2012) Stress and addiction: contribution of the corticotropin releasing factor (CRF) system in neuroplasticity. Front. Mol. Neurosci. 5:91. doi: 10.3389/fnmol.2012.00091
Received: 01 May 2012; Accepted: 15 August 2012;
Published online: 06 September 2012.
Edited by:
Ildikó Rácz, University of Bonn, GermanyReviewed by:
Hansen Wang, University of Toronto, CanadaAnita Hansson, Central Institute of Mental Health, Germany
Copyright © 2012 Haass-Koffler and Bartlett. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Selena E. Bartlett, Translational Research Institute and the Institute for Health and Biomedical Innovation, Queensland University of Technology, Brisbane, QLD 4059, Australia. e-mail:c2VsZW5hLmJhcnRsZXR0QHF1dC5lZHUuYXU=