 Laura E. Jensen1
Laura E. Jensen1 Geert Bultynck2Tomas Luyten2Hozeefa Amijee3
Geert Bultynck2Tomas Luyten2Hozeefa Amijee3 Martin D. Bootman1†
Martin D. Bootman1† H. Llewelyn Roderick1*
H. Llewelyn Roderick1*- 1Babraham Institute, Babraham Research Campus, Babraham, Cambridge, UK
- 2Laboratory of Molecular and Cellular Signaling, Department Molecular Cell Biology, K.U. Leuven, Leuven, Belgium
- 3Senexis, Babraham Research Campus, Babraham, Cambridge, UK
Dysregulation of Ca2+ homeostasis is considered to contribute to the toxic action of the Alzheimer's disease (AD)-associated amyloid-β-peptide (Aβ). Ca2+ fluxes across the plasma membrane and release from intracellular stores have both been reported to underlie the Ca2+ fluxes induced by Aβ42. Here, we investigated the contribution of Ca2+ release from the endoplasmic reticulum (ER) to the effects of Aβ42 upon Ca2+ homeostasis and the mechanism by which Aβ42 elicited these effects. Consistent with previous reports, application of soluble oligomeric forms of Aβ42 induced an elevation in intracellular Ca2+. The Aβ42-stimulated Ca2+ signals persisted in the absence of extracellular Ca2+ indicating a significant contribution of Ca2+ release from the ER Ca2+ store to the generation of these signals. Moreover, inositol 1,4,5-trisphosphate (InsP3) signaling contributed to Aβ42-stimulated Ca2+ release. The Ca2+ mobilizing effect of Aβ42 was also observed when applied to permeabilized cells deficient in InsP3 receptors, revealing an additional direct effect of Aβ42 upon the ER, and a mechanism for induction of toxicity by intracellular Aβ42.
Introduction
Alzheimer's disease (AD) is a progressive and irreversible brain disorder, which results in severe memory loss, behavioral as well as personality changes and a decline in cognitive abilities. While the most common type of AD remains idiopathic in origin, with age the most significant risk factor for disease onset (sporadic AD, sAD), ~5% of cases show a Mendelian pattern of inheritance (familial AD, fAD). The amyloid β-peptide (Aβ) is hypothesized to be central to the pathogenesis of both sporadic and familial AD (Hardy and Selkoe, 2002). Aβ is a small, hydrophobic polypeptide, consisting of 39–42 amino acid residues, which occurs principally as a 40 or 42 amino acid peptide, Aβ40 and Aβ42, respectively (Zhang et al., 2011). An imbalance between the production and clearance of Aβ, as occurs in fAD and sAD, respectively, leads to the accumulation of Aβ and, in turn, to its aggregation. This aggregation process represents a critical step in the pathogenesis of AD because the neurotoxic properties of Aβ are associated only with aggregated forms of the peptide (Kuperstein et al., 2010). Protein aggregation is highly dynamic and involves a wide range of intermediate structures such as oligomers, comprising dimers, trimers, dodecamers, and higher-molecular weight complexes, before aggregating into protofibrils and finally into mature amyloid fibrils (Dobson, 2003).
A mounting body of evidence now suggests that soluble oligomeric forms of Aβ constitute the primary neurotoxic species rather than monomers or fibrils (Lambert et al., 1998; Chromy et al., 2003; Gong et al., 2003; Demuro et al., 2005; Klyubin et al., 2005). Soluble oligomers have proved toxic when applied to cultured cells and primary neuronal cultures in vitro (Lambert et al., 1998; Bucciantini et al., 2002; Dahlgren et al., 2002; Kayed et al., 2003; Whalen et al., 2005). In addition, they are capable of inducing cognitive deficits when administered in vivo (Cleary et al., 2005; Rowan et al., 2007) and adversely affect hippocampal LTP in vivo (Walsh et al., 2002; Cleary et al., 2005; Klyubin et al., 2009, 2011).
Dysregulation of intracellular Ca2+ homeostasis is associated with cell exposure to Aβ and likely underlies its neurotoxic effects (Bezprozvanny and Mattson, 2008; Green and Laferla, 2008; Berridge, 2010; Demuro et al., 2010). A number of mechanisms by which Aβ elicits its effects on intracellular Ca2+ homeostasis have been put forward. These include direct effects on the plasma membrane, where it has been proposed to destabilize its structure (Mueller et al., 1995; Mason et al., 1996), to induce a generalized increase in membrane permeability (Bucciantini et al., 2002; Kayed et al., 2003) or to insert into the membrane forming cation-conducting pores (Arispe et al., 1993; Mueller et al., 1995; Mason et al., 1996; Bucciantini et al., 2002; Kayed et al., 2003; Kawahara, 2004; Simakova and Arispe, 2006; Arispe et al., 2007; Demuro et al., 2011). Aβ has also been reported to activate plasma membrane receptors, including N-methyl-d-aspartate (NMDA) receptors coupled to Ca2+ influx (Guo et al., 1996; Dobson, 2003; Blanchard et al., 2004; De Felice et al., 2007), to alter neuronal excitability which, in turn, influences the extent of Ca2+ influx (Good et al., 1996) and to induce dysregulation of endoplasmic reticulum (ER) Ca2+ homeostasis (Ferreiro et al., 2004, 2006; Resende et al., 2008). In addition to acting from the extracellular space, where it accumulates in the diseased brain, Aβ also has an intracellular site of action (Wirths et al., 2004). Indeed, as a result of uptake from the extracellular space or via its intracellular synthesis and processing, Aβ has been reported to accumulate within the cell (Pierrot et al., 2004; Bayer and Wirths, 2011; Kaminski Schierle et al., 2011). This intracellular Aβ is also neurotoxic and has been shown to target the ER and the mitochondria, inducing a stress response and causing permeability transition, respectively (Yao et al., 2009; Umeda et al., 2011).
In this study, we investigated (1) the contribution of Ca2+ mobilization from the ER to the increase in intracellular Ca2+ induced by oligomeric Aβ42, (2) the mechanism (s) by which Aβ42 elicited this effect, (3) the capacity for Aβ42 to mobilize Ca2+ directly from the ER. To allow isolation of effects on the ER from other plasma membrane targets of Aβ42, model cells systems were used that allowed fundamental aspects of ER Ca2+ regulation to be studied. We determined that Ca2+ release from the intracellular ER substantially contributed to the increase in intracellular Ca2+ concentration induced by oligomeric Aβ42. The Aβ42-induced Ca2+ elevation comprised InsP3 dependent and independent components. Using DT40 cells deficient in the three InsP3R isoforms that were permeabilized to allow direct access of Aβ42 to the ER, we also demonstrated that it had the capacity to release Ca2+ from the ER independent of InsP3Rs. Together, these data place the ER and Ca2+ released from it as central to the actions of both extracellular Aβ and Aβ that has reached an intracellular location.
Materials and Methods
Materials
Peptides were purchased from The American Peptide Company and rPeptide. Cell culture reagents and chemicals were from Invitrogen or Sigma, unless otherwise stated.
Cell Culture
Human neuroblastoma SH-SY5Y cells were cultured in F-12 Nutrient Mixture (Ham) containing FBS (10%), penicillin (100 units/ml), streptomycin (100 μg/ml), non-essential amino acids (0.1 mM), and L-glutamine (2 mM). Prior to all experiments, SH-SY5Y cells were cultured overnight in Opti-MEM Reduced Serum Medium, containing FBS (1.5%), penicillin (100 units/ml), streptomycin (1.0 μg/ml), non-essential amino acids (0.1 mM), and L-glutamine (2 mM). For live-cell Ca2+ imaging experiments, cells were plated onto poly-L-lysine-coated coverslips at a density of 3.2 × 104 cell/cm2. For the MTT reduction assay, cells were plated at a density of 9 × 103 cells/cm2. To overexpress GFP-tagged type 1 InsP3 5′-Phosphatase (GFP-5′P) or GFP (Peppiatt et al., 2004; Higazi et al., 2009), cells were infected with adenovirus for 8 h prior to overnight culture. Culture of DT40 cells and DT40 cells deficient in the three InsP3R isoforms (DT40 TKO) was performed as previously described (Tovey et al., 2006).
Preparation of Aβ42 Oligomers
Wild type and scrambled Aβ42 were obtained at a purity of >95%. Peptide mass was verified by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry and peptides from the same batch were used throughout. Samples of synthetic Aβ42 oligomers were prepared as previously described (Demuro et al., 2005) and remained stable for at least 3 weeks. Samples of Aβ 1–42 scrambled peptide (KVKGLIDGAHIGDLVYEFMDSNSAIFREGVGAGHVHVAQVEF) were prepared in the same way as Aβ42 oligomers. All Aβ samples were stored at 4°C and were used within 10–15 days of preparation. Toxicity of Aβ42 preparations was confirmed by MTT assay before use in Ca2+ imaging experiments (Figure S1A). The oligomeric nature of the Aβ42 preparation was established by surface plasmon resonance (SPR) spectroscopy using an antibody specific to oligomeric Aβ42 (Figure S1B). All Aβ42 concentrations stated are based on the molar mass of the peptide.
Live Cell Ca2+ Imaging
Methods for single cell analysis of intracellular Ca2+ concentration were as previously described (Peppiatt et al., 2003). Cells were loaded at 37°C with 2 μM of the acetoxymethyl (AM) ester form of fura-2 for 30 min followed by an equivalent period in dye free media to allow de-esterification of the indicator. Imaging experiments were performed using either Ca2+-containing (121 mM NaCl, 5.4 mM KCl, 0.8 mM MgCl2, 1.8 mM CaCl2, 6 mM NaHCO3, 25 mM HEPES, 5.5 mM glucose, pH 7.3) or Ca2+ free (as for Ca2+ containing with 1.8 mM CaCl2 replaced with 1 mM EGTA) buffer as indicated. Fura-2 imaging was carried out using an imaging system configured around a Nikon TE300 inverted epi-fluorescence microscope equipped with a 20× 0.75 NA multi-immersion objective. Samples were illuminated by alternate excitation at 340 and 380 nm using a Sutter filter changer (340HT15 and 380HT15; Sutter Industries) and emitted light was filtered at >460 nm (1 ratio pair per 2 s). Images were captured using a Hamamatsu ORCA ER CCD camera. The imaging system was controlled with Ultraview software (PerkinElmer Life Sciences Ltd., UK). Acquired images were processed with Ultraview software and analyzed in MATLAB. Background subtracted fura-2 ratios were calibrated according to standard procedures (Grynkiewicz et al., 1985), using the maximum and minimum ratio values obtained through exposing cells sequentially to Ca2+ free and Ca2+ containing imaging buffer to which 2 μM ionomycin had been added. Parameters analyzed from the Ca2+ responses included the peak amplitude, the time to peak and the integral of the response (the area under the curve) and the percentage of responding cells.
InsP3-induced Ca2+ release (IICR) from permeabilized wild type and InsP3R null DT40 cells (three InsP3R isoforms deleted by homologous recombination; DT40 TKO) (Sugawara et al., 1997) was performed as previously described (Tovey et al., 2006). Briefly, the ER of cells was loaded with the low-affinity Ca2+ indicator mag-fluo-4 and Aβ-induced Ca2+ release was measured from the saponin-permeabilized cells using a fluorescence plate reader (FlexStation 3, Molecular Devices).
MTT Reduction Assay
The Cell Titer 96 Non-Radioactive Cell Proliferation Assay (Promega) was used to validate the cytotoxic effect of Aβ42 on SH-SY5Y cells and was performed according to manufacturer's instructions. Briefly, cells were incubated with Aβ42 (n = 4) for 24 h prior to the addition of the MTT dye solution and a further 4 h incubation at 37°C, 5% CO2. Thereafter, the solubilization/stop solution was added and incubated overnight at room temperature. Absorbances were read at 570 nm with a reference wavelength of 650 nm using a fluorescence plate reader (Synergy HT, BIO-TEK). The data is expressed as the percentage of MTT reduction relative to both live- and dead-cell controls and thus represents the percentage of viable cells. Aβ42 samples were considered to be toxic if 25–40% of cells remained metabolically healthy at an Aβ42 concentration of 1 μ M and if more than 50% remained metabolically healthy at a concentration of 100 nM.
Statistical Analysis
Data is presented as the mean value of the combined datasets ± SEM. Statistical significance was determined by Student's t-test (two-tailed). Data was accepted as significant when p < 0.05 and is denoted by *p < 0.05, **p < 0.01, or ***p < 0.001.
Results
Intracellular ca2+ is elevated in cells exposed to oligomeric Aβ42
Experiments were first performed to validate the Ca2+ mobilizing properties of oligomeric Aβ42 over the concentration range of its toxicity. Application of Aβ42 spanning its cytotoxic range (1, 5 and 10 μM) caused an elevation in intracellular Ca2+ (Figure 1A). The increase in cytosolic Ca2+ concentration immediately followed the addition of Aβ42, developed to a peak within minutes of application and subsequently returned to baseline, despite the continued presence of the peptide. No Ca2+ responses were detected when Aβ42 below 1 μM was applied (data not shown). Between 1 μM and 10 μM Aβ, the number of responding cells, the peak amplitude and the integral of the Ca2+ responses increased in a concentration-dependent manner. The number of responding cells reached 100% at 5 μM Aβ42 (Figures 1Bi,iii,v). To test cell viability as well as to determine whether metabotropic Ca2+ signaling was affected by Aβ, carbachol (CCH) was applied subsequent to Aβ. CCH elicited Ca2+ responses in 100% of cells pre-exposed to 1 or 5 μM oligomeric Aβ42 or to a vehicle control (10%) (Figures 1Bii,iv,vi). At 10 μM Aβ, however, the number of cells responding to CCH was significantly reduced (Figure 1Bii). The peak amplitude and integral of the Ca2+ responses to CCH subsequently applied were inversely related to the magnitude of the Ca2+ responses elicited by oligomeric Aβ42 (Figures 1Biv,vi). This observation suggested that exposure to Aβ42 oligomers was depleting the intracellular CCH-sensitive ER Ca2+ store. These Ca2+ mobilizing effects of oligomeric Aβ42 were significantly greater than observed in cells exposed to Aβ42 that had been prepared in a manner to yield a monomeric form of the peptide (Figure S2, S1B). From these results, due to its potency in mobilizing Ca2+ whilst preserving agonist responses, a concentration of 5 μM oligomeric Aβ42 was selected for use in subsequent experiments.
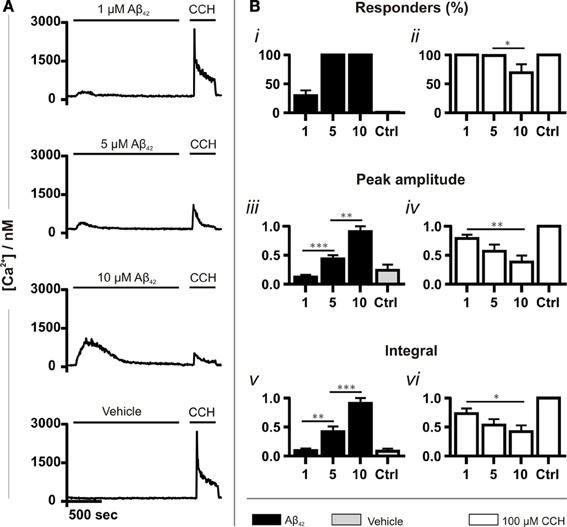
Figure 1. Aβ42 oligomers induce Ca2+ transients in a concentration-dependent manner. (A) Example fura-2 Ca2+ traces of SH-SY5Y cells exposed to a concentration range of Aβ42 oligomers followed by 100 μM CCH. A trace taken from cells in which Aβ42 oligomers were substituted with double-distilled water (dd H2O; vehicle) is also shown (for each group, n > 744 cells). (B) Quantitative analysis of the Ca2+ responses illustrated in A. The magnitude of Ca2+ responses elicited by Aβ42 oligomers, dd H2O and CCH is presented as (Bi,ii) percentage of responding cells, (Biii,iv) peak amplitude and (Bv,vi) integral of the response. Aβ42 oligomer-induced Ca2+ transients were normalized to the responses induced with the highest concentration (10 μM) of the respective Aβ42 preparation. CCH-induced Ca2+ responses were normalized to control experiments conducted on the same experimental day. Bar graphs are mean ± SEM from at least three independent experiments. *p < 0.05; **p < 0.01; ***p < 0.001.
Aβ42 Oligomer-Induced Ca2+ transients are peptide sequence specific
As a control for the application of peptide, experiments were also performed using a scrambled Aβ sequence, which had been prepared in the same manner as the wild type Aβ42. Although significantly less toxic than the wild type sequence (Figure S1A), scrambled Aβ peptide also evoked Ca2+ responses in all cells (Figure 2Ai). However, consistent with its lower toxicity, both the amplitude and the integral of the Ca2+ transients elicited by scrambled Aβ were significantly lower than those induced by oligomeric Aβ42 and, in addition, they required a significantly longer time to reach peak (Figures 2Bi,Ci,Di). Furthermore, concordant with the less potent effect of scrambled Aβ in mobilizing intracellular Ca2+, the amplitude and integral of CCH-induced Ca2+ transients elicited following prior exposure to scrambled Aβ were significantly greater than those stimulated following prior exposure to oligomeric Aβ42 (Figures 2Bii,Cii,Dii).
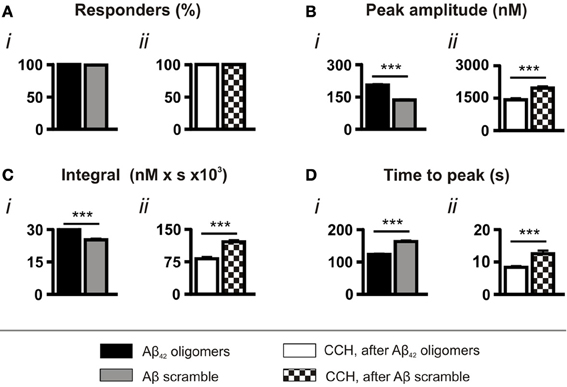
Figure 2. Aβ42 oligomer-induced Ca2+ transients are sequence specific. Bar charts illustrating the magnitude of Ca2+ responses elicited by SH-SY5Y cells following the application of 5 μM Aβ42 oligomers or Aβ scramble and 100 μM CCH (n > 370 cells). Data is presented as (A) percentage of responding cells, (B) peak amplitude, (C) integral of the response, (D) time to peak. Bar graphs are mean ± SEM from at least three independent experiments. ***p < 0.001.
Taken together, the comparison of the effects of Aβ scramble and oligomeric Aβ42 demonstrates that the amino acid sequence of Aβ42 has potent Ca2+ mobilizing properties, which are distinct from the action of Aβ scramble.
Aβ42 Oligomers Mobilize Ca2+ from Intracellular Stores
The reduced magnitude of CCH-induced Ca2+ signals observed in cells previously exposed to oligomeric Aβ42 suggested that this form of Aβ42 was exerting an effect on intracellular Ca2+ stores. Therefore, we tested the relative contributions of Ca2+ influx from the extracellular space and its release from intracellular stores to Aβ42-induced Ca2+ transients.
To determine the contribution of extracellular Ca2+ and Ca2+ influx to Aβ42 oligomer-induced Ca2+ transients, we performed experiments using Ca2+-free imaging buffer. Under these conditions, Aβ42 oligomers retained their ability to induce Ca2+ responses, with 100% of cells responding (Figure 3Ai). While no significant difference was observed in the peak amplitude (Figure 3Aiii) of Aβ42 oligomer-induced Ca2+ transients, the integral of the response was significantly decreased in the absence of extracellular Ca2+ (Figure 3Av).
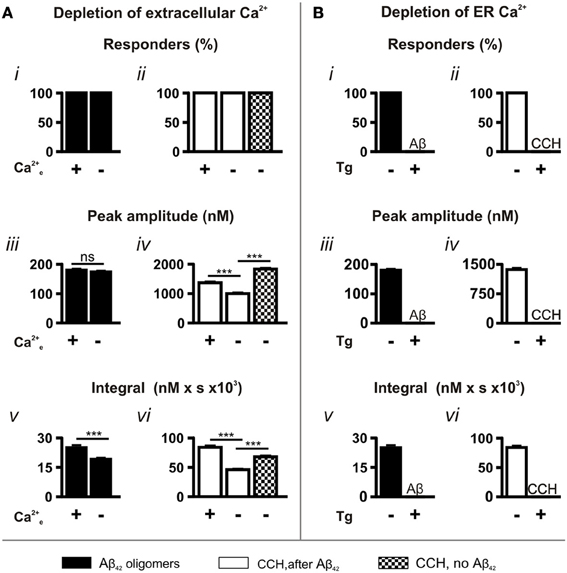
Figure 3. Aβ42 oligomer-induced Ca2+ transients arise through release from the ER Ca2+ store. Bar charts illustrating the magnitude of Ca2+ responses elicited following the manipulation of (A) extracellular (n > 218 cells) and (B) ER Ca2+ concentrations (n > 512 cells). The magnitude of Ca2+ responses elicited by 5 μM Aβ42 oligomers and 100 μM CCH is presented as (i,ii) percentage of responding cells, (iii,iv) peak amplitude and (v,vi) integral of the response. Bar graphs are mean ± SEM from at least three independent experiments. ***P < 0.001.
In contrast to the Aβ42 oligomer-induced Ca2+ response, the peak amplitude and the integral of the Ca2+ responses to CCH applied following Aβ42 oligomer exposure were significantly decreased by removal of extracellular Ca2+ from the imaging buffer (CCH, after Aβ42; Figures 3Aiv,vi). This effect on the CCH-induced Ca2+ responses is likely due to lack of store-operated Ca2+ entry, which would replenish the Ca2+ released from stores by Aβ42. Indeed, the peak amplitude and the integral of CCH-induced Ca2+ responses elicited in Ca2+ free buffer were significantly greater in naïve cells (CCH, no Aβ42) than when Aβ42 oligomers were previously applied (Figures 3Aiv,vi). Since Aβ42 oligomer-induced Ca2+ transients were not significantly affected by removal of extracellular Ca2+, these results suggest that oligomeric Aβ42 and CCH mobilize Ca2+ from a common intracellular Ca2+ pool.
The requirement of Ca2+ release from the ER Ca2+ store for the Ca2+ transients elicited by Aβ-induced was next investigated. To this end, ER Ca2+ stores were depleted by exposure of cells the SERCA pump inhibitor thapsigargin (Tg; 2 μM, 15 min) prior to the application of Aβ42. In the absence of replete ER Ca2+ stores, Aβ42-induced Ca2+ transients were completely abrogated (Figures 3Bi,iii,v). Similarly, CCH-induced Ca2+ responses were eliminated in Tg-treated cells (Figures 3Bii,Biv,Bvi), confirming the effect of Tg. Taken together, these experiments establish that Aβ42 oligomers mobilize Ca2+ from the ER.
Aβ42-Induced Ca2+ Release Occurs in Part through InsP3Rs
Having determined that Aβ42 oligomers mobilize Ca2+ from the intracellular ER Ca2+ store, we aimed to identify the mechanism by which Ca2+ release occurs. We therefore tested whether Aβ42 was causing Ca2+ release from the ER through activation of InsP3R or ryanodine receptor (RyR) Ca2+ release channels localized to this organelle.
Although SH-SY5Y cells have been reported to express functional RyRs, application of caffeine (10 mM), an agonist of the three RyR isoforms (10 mM) did not elicit a Ca2+ response in the SH-SY5Y cells used in this study (Figure S2A). Furthermore, the neuronally-expressed type 2 RyR could not be detected by immunoblot analysis (Figure S2B). Based on these observations, a role for RyR2 in Aβ42 oligomer-mediated Ca2+ release was ruled out.
SH-SY5Y cells express InsP3Rs and elicit robust Ca2+ responses to InsP3-generating agonists including CCH (Tovey et al., 2001) (Figures 1–3). Therefore, we focused our investigation on the contribution of InsP3Rs to Aβ42-induced Ca2+ transients. To abrogate InsP3-mediated Ca2+ responses, InsP3 signaling was inhibited pharmacologically with 10 mM caffeine (Parker and Ivorra, 1991; Bezprozvanny et al., 1994) or was prevented by adenoviral-mediated overexpression of GFP-5′P, which metabolizes the second messenger InsP3 to inactive InsP2 (Higazi et al., 2009). To exclude the contribution of Ca2+ influx to the Aβ42 oligomer-induced Ca2+ transients, these experiments were performed in the absence of extracellular Ca2+.
Caffeine application did not affect the number of cells exhibiting Ca2+ responses following Aβ42 oligomer application, with 100% of cells responding (Figure 4B). However, caffeine significantly decreased the peak amplitude and the integral of the Aβ42 oligomer-induced Ca2+ transients (Figure 4B). In contrast, Aβ scramble-induced Ca2+ transients were unaffected by caffeine application (Figure 4C). Ca2+ responses to 0.5 μM CCH were abolished by caffeine, demonstrating its inhibitory effect upon IICR (Figures 4A–C).
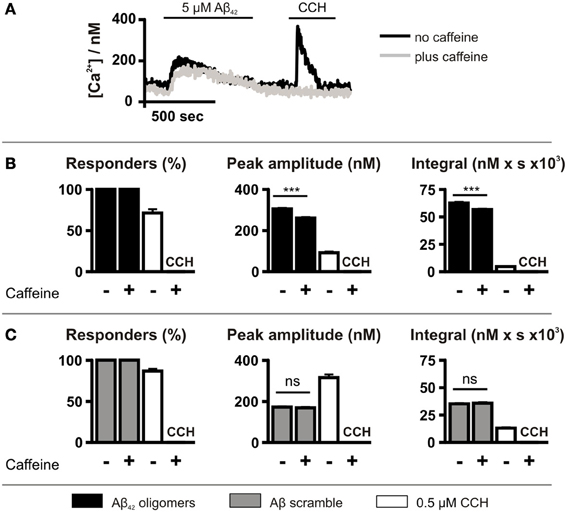
Figure 4. Aβ42 oligomer-induced Ca2+ release is sensitive to caffeine. (A) Imaging protocol employed to investigate the involvement of InsP3Rs in Aβ42 oligomer-mediated Ca2+ release from the ER. InsP3Rs were inhibited by co-administration of caffeine. (B,C) Bar charts illustrating the magnitude of Ca2+ responses elicited by SH-SY5Y cells following the application of 5 μM Aβ42 oligomers (n > 780 cells) (B) or Aβ scramble (n > 144 cells) (C) and 0.5 μM CCH (n = 512 cells) in the presence or absence of 10 mM caffeine. Data is presented as percentage of responding cells, peak amplitude and integral of the response. Bar graphs are mean ± SEM from at least three independent experiments. ***P < 0.001.
Although caffeine inhibits InsP3Rs (Bezprozvanny et al., 1994), it also acts on targets other than the InsP3R such as cyclic nucleotide phosphodiesterases and phospholipase C (PLC) (Toescu et al., 1992; Taylor and Broad, 1998). Therefore, to investigate further the role of InsP3 signaling in the generation of Aβ42 oligomer-induced Ca2+ transients, InsP3 signaling was inhibited by GFP-5′P overexpression. Using this strategy, InsP3-mediated Ca2+ signals induced by CCH were prevented, validating this approach for suppression of InsP3 signaling (Figure 5A). As observed for caffeine, however, GFP-5′P overexpression did not prevent Aβ42 oligomer-induced Ca2+ transients, with 100% of cells responding (Figure 5B). However, the peak amplitude and the integral of Aβ42 oligomer-induced Ca2+ transients were significantly decreased by overexpression of GFP-5′P (Figure 5B) when compared to the magnitude of Ca2+ transients in control cells, expressing GFP alone. Significantly, Aβ scramble-induced Ca2+ transients were not affected by GFP-5′P overexpression with no significant impact of its expression upon the peak amplitude or the integral of Aβ scramble-induced Ca2+ transients (Figure 5C). Taken together, these results demonstrate that Ca2+ transients elicited by Aβ42 oligomers arise as a result of release from the ER intracellular Ca2+ store and that activation of InsP3Rs contributes to this effect.
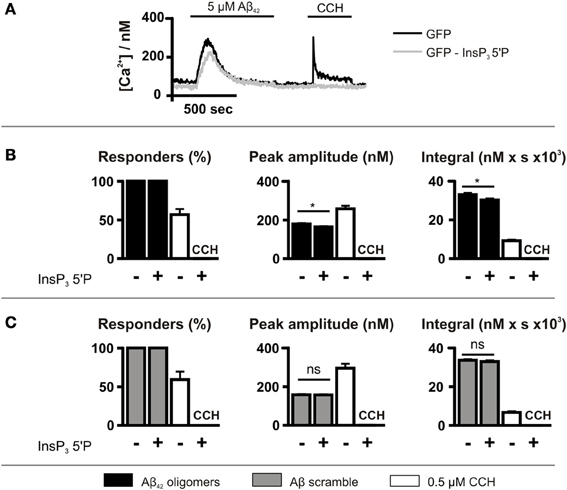
Figure 5. Aβ42 oligomer-induced Ca2+ release occurs is reduced by InsP3 5′P expression. (A) Imaging protocol employed to investigate the involvement of InsP3Rs in Aβ42 oligomer-mediated Ca2+ release from the ER. InsP3 was metabolized by overexpression of InsP3 5′P. (B,C) Bar charts illustrating the magnitude of Ca2+ responses elicited by SH-SY5Y cells infected with InsP3 5′P or GFP alone following the application of 5 μM Aβ42 oligomers (n > 207 cells) (B) or Aβ scramble (n > 115 cells) (C) and 0.5 μM CCH (n > 55 cells). Data is presented as percentage of responding cells, peak amplitude and integral of the response. Bar graphs are mean ± SEM from at least three independent experiments. *P < 0.05.
Aβ42 Oligomer-Induced Ca2+ Leak from the ER
The data presented above indicates that externally applied Aβ42 rapidly induces an increase on cytosolic Ca2+ that involves InsP3-dependent and -independent Ca2+ release from the ER. Since Aβ42 has also been shown to elicit some of its cytotoxic effects as a result of intracellular accumulation (Wirths et al., 2004), we investigated whether it mobilized Ca2+ from the ER when directly applied. We also tested whether InsP3Rs were required for its intracellular action.
To this end, an established permeabilized cell high-throughput functional assay of ER Ca2+ release was used (Tovey et al., 2006). This model uses as substrate for specific analysis of ER Ca2+ release, a plasma membrane-permeabilized preparation of the DT40 chicken B-lymphocyte cell line. A derivative of this cell line in which the 3 InsP3R isoforms have been deleted by homologous recombination (DT40 TKO), allows the requirement for InsP3Rs for Ca2+ release to be tested (Sugawara et al., 1997). Cell permeabilization and substantial dilution in intracellular buffer rules out the contribution of endogenously generated InsP3 to signaling in this assay. Using this assay, a significantly greater InsP3 independent ER Ca2+ leak was observed in both wild-type (p = 0.002) and DT40 TKO cells (p = 0.0195) exposed to Aβ42 oligomers compared to the passive Ca2+ leak detected in each cell type (Figures 6A,B). The maximal Ca2+ leak rate induced by Aβ42 oligomers was not significantly different between wild-type and DT40 TKO cells (p = 0.2606, Figure 6C), suggesting that InsP3Rs were not required for Aβ42 oligomers to trigger Ca2+ release.
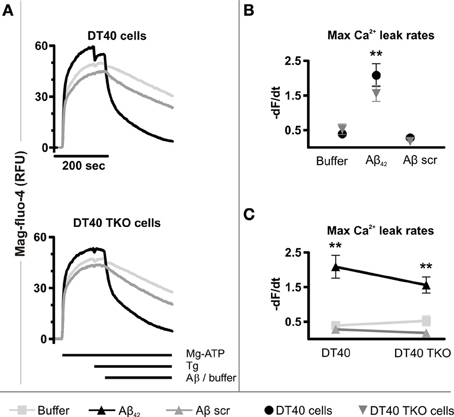
Figure 6. Aβ42 oligomers stimulate Ca2+ release from the ER of permeabilized cells. (A) Representative traces of mag-fluo-4 fluorescence (relative fluorescence units, RFU) in permeabilized DT40 cells and DT40 TKO cells, illustrating changes in ER luminal Ca2+ levels as a function of time in response to Aβ42 oligomers, Aβ scramble and buffer alone. Data represents the mean of three measurements. (B,C) Initial quantitative analysis of the maximum Ca2+ leak rates from the ER of permeabilized DT40 cells and DT40 TKO cells triggered by Aβ42 oligomers, Aβ scramble and buffer, respectively. The maximal Ca2+ leak rate was calculated by taking the maximal value of the first derivative of the fluorescence values to the time. Data were calculated as −dF/dt and represents the mean Ca2+ leak rate obtained from three measurements in RFU over time (RFU/s). **p < 0.01.
Aβ scramble did not increase the rate of the Ca2+ leak in DT40 cells (p = 0.2528) or in DT40 TKO cells (p = 0.0993) compared to the passive Ca2+ leak observed in each cell type (Figure 6B), and thus there was no significant difference in the maximal Ca2+ leak rate following Aβ scramble application between these two cell types (p = 0.2522, Figure 6C). Importantly, a significant difference between the Ca2+ leak rates triggered by exposure to Aβ42 oligomers and Aβ scramble in wild-type DT40 cells (p = 0.0056) and DT40 TKO cells (p = 0.0045) was observed, indicating that Aβ-induced Ca2+ leak from the ER is dependent and specific to the amino acid sequence of Aβ42. Taken together, these results suggest that Aβ42 oligomers trigger a Ca2+ leak from the ER, which does not depend upon a direct interaction with InsP3Rs.
Discussion
Here we show that the oligomeric form of the AD-associated peptide Aβ42 has potent Ca2+ mobilizing properties and we identify mechanisms responsible for its action. Using both intact and permeabilized cell assays to investigate the effects of extracellular and internalized Aβ42, respectively, we establish that Ca2+ release from the ER makes the greatest contribution to the Ca2+ mobilizing effects of Aβ42. The InsP3 signaling pathway also contributes to the Ca2+ mobilizing properties of oligomeric Aβ42 in intact cells. InsP3Rs were not required for Aβ42-stimulated Ca2+ flux in permeabilized cells ruling out a direct regulation of InsP3Rs by Aβ42.
Central to the Ca2+ hypothesis of amyloid toxicity is the property of Aβ to induce Ca2+ elevations in its target cells. This sets in motion a cascade of events, which culminates in neuronal death. Ever since this hypothesis was put forward more than 20 years ago (Khachaturian, 1989, 1994), numerous reports have described Aβ-induced changes in intracellular Ca2+ in a number of cell types including primary neurons and astrocytes as well as neuroblastoma cell lines (Abramov et al., 2004b; Demuro et al., 2005). While there is general consensus that Aβ affects Ca2+ homeostasis, the mechanisms underlying this action of Aβ are many. Contributing to this diversity are the different experimental models used, the peptide sequence applied, the conformational state of the peptide and the method used for peptide preparation. Indeed, a number of shorter Aβ sequences have been employed in in vitro studies and depletion of ER Ca2+ store content reported (Ferreiro et al., 2004, 2008). Since Aβ42 is considered to be more relevant to the pathology of AD, we focused on its effects on intracellular Ca2+ homeostasis. Not only is an accumulation of Aβ42 observed in AD, this longer and more hydrophobic peptide is also more prone to self-assemble than Aβ 40, the other principle length at which Aβ occurs. As a result, Aβ42 exerts a greater degree of neurotoxicity (Jarrett and Lansbury, 1993). Consistent with the growing body of evidence that soluble oligomeric forms of Aβ constitute the primary neurotoxic species (Walsh et al., 2002; Gong et al., 2003; Cleary et al., 2005; Klyubin et al., 2005), this species of Aβ42 potently induced Ca2+ fluxes and cytotoxicity in this study (Figures 1, 2 and Figure S2). Highlighting the requirement for appropriate peptide controls when studying Aβ42, Ca2+ release and cytotoxicity was also induced by a scrambled peptide sequence of Aβ42, although the magnitude of these responses was significantly lower than that induced by the wild type sequence. From these results, we concluded that the peptide sequence of Aβ42 was the major contributor to the toxicity and Ca2+ mobilizing properties. The temporal properties of the Ca2+ transients we observed were reminiscent of those reported elsewhere, being relatively slow in reaching peak and returning to baseline levels after a few minutes (Demuro et al., 2005; Simakova and Arispe, 2006). The return of these Ca2+ signals to baseline does, however, suggest that the Ca2+ elevations induced by Aβ42 were not immediately toxic. The Ca2+ mobilizing properties of the scrambled peptide, however, may reflect the previously described intrinsic properties of an oligomeric/amyloid peptide (Bucciantini et al., 2002; Yoshiike et al., 2008). For example, oligomeric forms of polyQ and insulin have been shown to induce Ca2+ transients (Demuro et al., 2005). The solvent HFIP used for preparation of the peptide has also previously been shown to exhibit cytotoxicity and to affect ion conductance of membranes (Capone et al., 2009).
Both Ca2+ influx from the extracellular space and release from ER-localized intracellular stores have been reported to be induced by Aβ and involved in its toxic action (Blanchard et al., 2004; Ferreiro et al., 2004, 2006; Kayed et al., 2004; Demuro et al., 2005, 2011; Kelly and Ferreira, 2006; Simakova and Arispe, 2006; Arispe et al., 2007; De Felice et al., 2007; Resende et al., 2008; Demuro and Parker, 2013). Although Ca2+ entry from the extracellular space was a component of the Ca2+ elevation induced by Aβ42 in this study, the greatest contribution was due to release from the ER. Moreover the lack of an effect of removal of extracellular Ca2+ upon the initial peak of the Ca2+ response or the number of responding cells suggested that Ca2+ entry across the plasma membrane was secondary to Ca2+ release from the ER. Since Aβ42 was acting to deplete the ER stores, the Ca2+ influx could arise via a store-operated Ca2+ entry pathway. These observations are not, however, incompatible with an additional mechanism for Ca2+ entry via plasma membrane pores formed by Aβ42, which have been shown to require a longer period to develop (Demuro et al., 2011). Whether the Ca2+ fluxes associated with the formation of membrane pores, which were generally local to the pore and were of a relatively small magnitude, contribute to the global Ca2+ transient is not clear (Demuro et al., 2011).
Analysis of the mechanisms underlying Ca2+ release from the ER revealed that while InsP3Rs contributed to Aβ42-induced Ca2+ release from the ER in intact cells, the greater part of the Ca2+ elevation induced by Aβ42 was due to an alternative mechanism. However, IICR did not contribute to the Ca2+ responses induced by scrambled peptide. From these results, we concluded that Aβ42-induced Ca2+ release from the ER comprises an Aβ42 sequence-specific component, which is InsP3-dependent, and a second component, which is peptide sequence- and InsP3-independent. Comparison of these Aβ42 and Aβ42 scrambled datasets reveals that although the InsP3-dependent component of the total Aβ42 signal is relatively minor, when considered as a fraction of the Aβ42-specific Ca2+ signal (i.e., Aβ42—Aβ42 scrambled Ca2+ transient), its importance is increased.
Our demonstration of the participation of InsP3 signaling in Aβ42-induced Ca2+ responses provides robust evidence in support of this pathway in Aβ42-mediated Ca2+ signals thus far. In particular, the use of InsP3 5′phosphatase overexpression to suppress InsP3 signaling is a highly selective strategy, overcoming issues regarding incomplete knockdown of InsP3Rs and contribution of the isoforms not targeted when using siRNA approaches. The inhibition of Ca2+ signals by caffeine is also consistent with a role for the InsP3 signaling pathway in the Ca2+ mobilizing effects of Aβ (Parker and Ivorra, 1991; Bezprozvanny et al., 1994). Not only does caffeine inhibit InsP3Rs directly (Bezprozvanny et al., 1994), by also inhibiting PLC, caffeine is a potent inhibitor of InsP3 generation (Taylor and Broad, 1998). These findings are consistent with the reduction in the Aβ42-induced Ca2+ transient observed following application of the PLC inhibitor U73122 (Resende et al., 2008) although U73122 has numerous non-specific effects. The mechanism by which InsP3 signaling is engaged by Aβ42 in this study remains to be established. Since the effects of inhibition of InsP3 signaling persist in the absence of extracellular Ca2+, activation of PLC and InsP3 generation by Aβ42-stimulated Ca2+ influx can be excluded. Thus, a more likely scenario would involve Aβ42 engagement of a PLC-coupled G protein coupled-receptor (GPCR). Indeed, a number of different GPCRs, including metabotropic glutamate receptors, are activated by Aβ42, contributing to modulation of LTP, Aβ42 synthesis and processing and cytotoxicity (Wang et al., 2004; Thathiah and De Strooper, 2011).
The internalization of Aβ from the extracellular space (Bucciantini et al., 2004; Pierrot et al., 2004; Wirths et al., 2004; Kaminski Schierle et al., 2011) raises a further possibility that Aβ acts to either directly activate/sensitize InsP3Rs or to alter InsP3 generation/metabolism. Since significant intracellular Aβ42 accumulation would require up to 1 h (Bucciantini et al., 2004; Kaminski Schierle et al., 2011), it is unlikely that this endocytosed Aβ42 contributes to the acute modulation of Ca2+ fluxes observed in this study and elsewhere in intact cells. Endocytosis of Aβ42 may, however, contribute to the more chronic effects on Ca2+ homeostasis as well as cytotoxicity previously reported (Ferreiro et al., 2004, 2006; Resende et al., 2008). The possibility that Aβ42 could directly affect ER Ca2+ homeostasis from an intracellular location was therefore also considered. Using a permeabilized cell assay to allow control of cytosolic conditions and access of Aβ42 to the ER, an Aβ42-stimulated Ca2+ efflux from the ER was observed. Unlike that observed for intact cells, the difference between Aβ42 and Aβ42 scrambled was dramatic, revealing a highly specific effect of Aβ42 upon ER Ca2+ mobilization. These effects were observed in the absence of exogenous InsP3 suggesting that the effects were InsP3R-independent. The extensive dilution of cytosol following permeabilization of the DT40 cells would also likely preclude a contribution of Aβ42-stimulated InsP3 generation. More significantly, InsP3Rs were not required for the Ca2+ mobilizing properties of Aβ42, since deficiency in all three InsP3R isoforms did not affect the Ca2+ mobilizing properties of Aβ42. The absence of a requirement for InsP3Rs for Aβ42-stimulated Ca2+ flux in the permeabilized cell system does not rule out the possibility that IICR contributes to Ca2+ fluxes and toxicity mediated by intracellular Aβ 42. Indeed, by activating Ca2+-sensitive PLC and generation of InsP3, Ca2+ mobilized by Aβ42 could promote IICR. Consistent with this notion, microinjected Aβ42 was recently shown to promote Ca2+ signals in Xenopus oocytes in a manner that involved InsP3 generation (Demuro and Parker, 2013).
The depletion of the ER Ca2+ store by Aβ42 has important implications for the mechanisms of its toxicity. Depletion of ER Ca2+ stores results in the accumulation of unfolded proteins and activation of the ER stress response, which via caspase 12 activation and Bap31 cleavage can subsequently induce mitochondrial apoptotic cascades (Verkhratsky, 2005; Xu et al., 2005; Mekahli et al., 2011). The engagement of InsP3Rs during Aβ42-stimulated depletion of ER Ca2+ may be of greater consequence. Specifically, InsP3R-induced Ca2+ release from the ER and its subsequent sequestration by neighboring mitochondria could lead to mitochondrial Ca2+ overload, permeability transition and death (Csordas et al., 2006). These pathways also lead to increased reactive oxygen species generation, which is commonly observed in AD (Ferreiro et al., 2004, 2008; Arduino et al., 2009; Clark et al., 2010).
The use of SH-SY5Y neuroblastoma cell line and permeabilized DT40 B-lymphocytes in this study, rather than primary neurons allowed careful dissection of the role of ER Ca2+ signaling to Aβ-induced Ca2+ signals independent from Ca2+ fluxes that may arise in neurons as a result of electrical or synaptic activity. Moreover, using this cell line, contributions from other Aβ targets described in neurons such as NMDA receptors are excluded. Analogous to a number of other studies in electrically non-excitable primary and cultured cells including Xenopus oocytes (Demuro and Parker, 2013) astrocytes and PC12 cells (Abramov et al., 2003, 2004a; Simakova and Arispe, 2006), our data indicates that certain of the Ca2+ mobilizing properties of Aβ42 are neuron-independent and do not require the expression of any other of its reported targets. Fundamental aspects of the Ca2+ mobilizing properties of Aβ42 were further revealed and exemplified by the Aβ42-stimulated Ca2+ flux from the InsP3R-deficient ER of permeabilized DT40 B-lymphocytes. These latter data demonstrate for the first time that Aβ42 has the capacity to directly induce Ca2+ flux from the ER. Given the importance of the ER and InsP3Rs in neuronal functions, future studies will be required to test whether InsP3Rs contribute to Aβ-mediated neuronal pathology.
Author Contributions
Laura E. Jensen: substantial contributions to conception and design, acquisition, analysis and interpretation of data as well as writing of manuscript. H. Llewelyn Roderick: substantial contributions to conception and design, interpretation of data as well as writing of manuscript. Geert Bultynck and Tomas Luyten: designed, acquired, analysed and interpreted data of Figure 6. Hozeefa Amijee: designed, acquired and interpreted data of Figure S2. Martin D. Bootman: proof-read manuscript.
Conflict of Interest Statement
Some of this work was supported by a grant from Senexis, Babraham Research Campus and Hozefa Amijee was employed by Senexis at the time this work was carried out.
Acknowledgments
This study was supported by The Babraham Institute, Senexis, The Royal Society (University Research Fellowship to H. Llewelyn Roderick) and The Gates Cambridge Trust [Gates Cambridge Scholarship to Laura E. Jensen (née Allan)]. Work in the Geert Bultynck's laboratory was supported by the Research Council of the KU Leuven via the Concerted Actions program (GOA/09/012) and via an OT START (STRT1/10/044), and by the Interuniversity Attraction Poles Program (Belgian Science Policy; P6/28 and P7/13).
Supplementary Material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/journal/10.3389/fnmol.2013.00036/abstract
Figure S1. Validating the toxicity and conformation of Aβ42 oligomers. (A) Assessment of toxicity of homogeneous Aβ preparations. Bar chart illustrating the cytotoxic effects of Aβ42 preparations upon SH-SY5Y cells determined using the MTT assay. Data is expressed as a percentage of MTT reduced by test samples to the dead cell controls following 24-h treatment with Aβ42 oligomers and scrambled Aβ at the respective concentrations. (Bi) Schematic diagram illustrating the epitopes of Aβ42 recognized by the conformation dependent, anti-oligomer antibody, A11 (Kayed et al., 2003), and the sequence dependent, anti-amyloid antibody, 12F4 (Parvathy et al., 2001). (Bii,iii) Sensorgrams obtained from surface plasmon resonance spectroscopy, as described (Maezawa et al., 2008). A Biacore T-100, equipped with four flow cells on a sensor chip, was used for these real-time binding studies. Biotinylated Aβ42was prepared by mixing a 1:10 molar ratio of biotinylated and unbiotinylated Aβ42. In preparation for the binding studies, Aβ42 was injected onto a streptavidin chip at a concentration of 10 μM to immobilize Aβ42by streptavidin-biotin coupling. The streptavidin chip of flow cell (Fc) 2 was partially (50%) and of Fc-4 fully saturated (100%) with Aβ42 oligomers. As a control, the surface of Fc-3 was partially saturated (50%) with Aβ42monomers. Antibodies (Bii) A11 and (Biii) 12F4 were injected over the immobilized Aβ42 of each flow cell at a concentration of 50 μg/ml and 10 μg/ml, respectively. The injection of the anti-oligomer antibody, A11, was followed by a regeneration step prior to injection of 12F4. The binding of injected antibodies, present in the flow phase, to the immobilized Aβ42was measured by response units (RU) elicited. All values were corrected for the RU obtained from the reference cell, flow cell 1, which was saturated with biotinylated Aβ42 only.
Figure S2. Comparison of Ca2+ responses elicited by Aβ42 oligomers and monomers in SH-SY5Y cells. (A) Imaging protocol employed to assess the effects of homogeneous preparations of Aβ42 on the Ca2+ signaling capacity of fluo-4-loaded SH-SY5Y cells. Cellular Ca2+ responses were recorded by wide-field epifluorescence. The magnitude of Ca2+ responses elicited by 5 μM Aβ42 monomers and oligomers and the subsequent application of 100 μM CCH is presented as (B) percentage of responding cells, (C) peak amplitude and (D) integral of the response. Soluble Aβ monomers and Aβ oligomers were prepared as previously described (Demuro et al., 2005). This method of Aβ preparation reportedly results in homogeneous populations of Aβ monomers and oligomers (also Figure S1B). All Aβ42 concentrations stated were based on the molar mass of the peptide.
Figure S3. Human neuroblastoma SH-SY5Y cells lack RyR expression. (A) Representative Ca2+ trace illustrating that SH-SY5Y cells do not elicit Ca2+ responses following the application of 10 mM caffeine, indicating that cells lack RyRs (n = 239 cells). However, SH-SY5Y cells do exhibit InsP3-mediated Ca2+ responses. (B) Immunoblot analysis corroborating the lack of RyR2 expression in SH-SY5Y cells. RyR2 expression is observed in control samples of adult hippocampal tissue and primary hippocampal cultures maintained for 4, 8, 11, and 15 days in vitro (DIVs).
References
Abramov, A. Y., Canevari, L., and Duchen, M. R. (2003). Changes in intracellular calcium and glutathione in astrocytes as the primary mechanism of amyloid neurotoxicity. J. Neurosci. 23, 5088–5095.
Abramov, A. Y., Canevari, L., and Duchen, M. R. (2004a). Beta-amyloid peptides induce mitochondrial dysfunction and oxidative stress in astrocytes and death of neurons through activation of NADPH oxidase. J. Neurosci. 24, 565–575. doi: 10.1523/JNEUROSCI.4042-03.2004
Abramov, A. Y., Canevari, L., and Duchen, M. R. (2004b). Calcium signals induced by amyloid beta peptide and their consequences in neurons and astrocytes in culture. Biochim. Biophys. Acta 1742, 81–87. doi: 10.1016/j.bbamcr.2004.09.006
Arduino, D. M., Esteves, A. R., Cardoso, S. M., and Oliveira, C. R. (2009). Endoplasmic reticulum and mitochondria interplay mediates apoptotic cell death: relevance to Parkinson's disease. Neurochem. Int. 55, 341–348. doi: 10.1016/j.neuint.2009.04.004
Arispe, N., Diaz, J. C., and Simakova, O. (2007). Abeta ion channels. Prospects for treating Alzheimer's disease with Abeta channel blockers. Biochim. Biophys. Acta 1768, 1952–1965. doi: 10.1016/j.bbamem.2007.03.014
Arispe, N., Pollard, H. B., and Rojas, E. (1993). Giant multilevel cation channels formed by Alzheimer disease amyloid beta-protein Abeta1-40 in bilayer membranes. Proc. Natl. Acad. Sci. U.S.A. 90, 10573–10577. doi: 10.1073/pnas.90.22.10573
Bayer, T. A., and Wirths, O. (2011). Intraneuronal Abeta as a trigger for neuron loss: can this be translated into human pathology? Biochem. Soc. Trans. 39, 857–861. doi: 10.1042/BST0390857
Berridge, M. J. (2010). Calcium hypothesis of Alzheimer's disease. Pflugers Arch. 459, 441–449. doi: 10.1007/s00424-009-0736-1
Bezprozvanny, I., Bezprozvannaya, S., Ehrlich, B. E. (1994). Caffeine-induced inhibition of inositol(1,4,5)-trisphosphate-gated calcium channels from cerebellum. Mol. Biol. Cell 5, 97. doi: 10.1091/mbc.5.1.97
Bezprozvanny, I., and Mattson, M. P. (2008). Neuronal calcium mishandling and the pathogenesis of Alzheimer's disease. Trends Neurosci. 31, 454–463. doi: 10.1016/j.tins.2008.06.005
Blanchard, B. J., Chen, A., Rozeboom, L. M., Stafford, K. A., Weigele, P., and Ingram, V. M. (2004). Efficient reversal of Alzheimer's disease fibril formation and elimination of neurotoxicity by a small molecule. Proc. Natl. Acad. Sci. U.S.A. 101, 14326–14332. doi: 10.1073/pnas.0405941101
Bucciantini, M., Calloni, G., Chiti, F., Formigli, L., Nosi, D., Dobson, C. M., et al. (2004). Prefibrillar amyloid protein aggregates share common features of cytotoxicity. J. Biol. Chem. 279, 31374–31382. doi: 10.1074/jbc.M400348200
Bucciantini, M., Giannoni, E., Chiti, F., Baroni, F., Formigli, L., Zurdo, J., et al. (2002). Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 416, 507–511. doi: 10.1038/416507a
Capone, R., Quiroz, F. G., Prangkio, P., Saluja, I., Sauer, A. M., Bautista, M. R., et al. (2009). Amyloid-beta-induced ion flux in artificial lipid bilayers and neuronal cells: resolving a controversy. Neurotox. Res. 16, 1–13. doi: 10.1007/s12640-009-9033-1
Chromy, B. A., Nowak, R. J., Lambert, M. P., Viola, K. L., Chang, L., Velasco, P. T., et al. (2003). Self-assembly of Abeta1-42 into globular neurotoxins. Biochemistry 42, 12749–12760. doi: 10.1021/bi030029q
Clark, T. A., Lee, H. P., Rolston, R. K., Zhu, X., Marlatt, M. W., Castellani, R. J., et al. (2010). Oxidative stress and its implications for future treatments and management of Alzheimer disease. Int. J. Biomed. Sci. 6, 225–227.
Cleary, J. P., Walsh, D. M., Hofmeister, J. J., Shankar, G. M., Kuskowski, M. A., Selkoe, D. J., et al. (2005). Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat. Neurosci. 8, 79–84. doi: 10.1038/nn1372
Csordas, G., Renken, C., Varnai, P., Walter, L., Weaver, D., Buttle, K. F., et al. (2006). Structural and functional features and significance of the physical linkage between ER and mitochondria. J. Cell Biol. 174, 915–921. doi: 10.1083/jcb.200604016
Dahlgren, K. N., Manelli, A. M., Stine, W. B. Jr., Baker, L. K., Krafft, G. A., and Ladu, M. J. (2002). Oligomeric and fibrillar species of amyloid-beta peptides differentially affect neuronal viability. J. Biol. Chem. 277, 32046–32053. doi: 10.1074/jbc.M201750200
De Felice, F. G., Velasco, P. T., Lambert, M. P., Viola, K. L., Fernandez, S. J., Ferreira, S. T., et al. (2007). Abeta oligomers induce neuronal oxidative stress through an NMDA receptor-dependent mechanism that is blocked by the Alzheimer's drug memantine. J. Biol. Chem. 282, 11590–11601. doi: 10.1074/jbc.M607483200
Demuro, A., Mina, E., Kayed, R., Milton, S. C., Parker, I., and Glabe, C. G. (2005). Calcium dysregulation and membrane disruption as a ubiquitous neurotoxic mechanism of soluble amyloid oligomers. J. Biol. Chem. 280, 17294–17300. doi: 10.1074/jbc.M500997200
Demuro, A., and Parker, I. (2013). Cytotoxicity of intracellular aβ 42 amyloid oligomers involves Ca2+ release from the endoplasmic reticulum by stimulated production of inositol trisphosphate. J. Neurosci. 33, 3824–3833. doi: 10.1523/JNEUROSCI.4367-12.2013
Demuro, A., Parker, I., and Stutzmann, G. E. (2010). Calcium signaling and amyloid toxicity in Alzheimer disease. J. Biol. Chem. 285, 12463–12468. doi: 10.1074/jbc.R109.080895
Demuro, A., Smith, M., and Parker, I. (2011). Single-channel Ca(2+) imaging implicates Aβ 1-42 amyloid pores in Alzheimerandapos's disease pathology. J. Cell Biol. 195, 515–524. doi: 10.1083/jcb.201104133
Ferreiro, E., Oliveira, C. R., and Pereira, C. (2004). Involvement of endoplasmic reticulum Ca2+ release through ryanodine and inositol 1,4,5-triphosphate receptors in the neurotoxic effects induced by the amyloid-beta peptide. J. Neurosci. Res. 76, 872–880. doi: 10.1002/jnr.20135
Ferreiro, E., Oliveira, C. R., and Pereira, C. M. (2008). The release of calcium from the endoplasmic reticulum induced by amyloid-beta and prion peptides activates the mitochondrial apoptotic pathway. Neurobiol. Dis. 30, 331–342. doi: 10.1016/j.nbd.2008.02.003
Ferreiro, E., Resende, R., Costa, R., Oliveira, C. R., and Pereira, C. M. (2006). An endoplasmic-reticulum-specific apoptotic pathway is involved in prion and amyloid-beta peptides neurotoxicity. Neurobiol. Dis. 23, 669–678. doi: 10.1016/j.nbd.2006.05.011
Gong, Y., Chang, L., Viola, K. L., Lacor, P. N., Lambert, M. P., Finch, C. E., et al. (2003). Alzheimer's disease-affected brain: presence of oligomeric Abeta ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proc. Natl. Acad. Sci. U.S.A. 100, 10417–10422. doi: 10.1073/pnas.1834302100
Good, T. A., Smith, D. O., and Murphy, R. M. (1996). Beta-amyloid peptide blocks the fast-inactivating K+ current in rat hippocampal neurons. Biophys. J. 70, 296–304. doi: 10.1016/S0006-3495(96)79570-X
Green, K. N., and Laferla, F. M. (2008). Linking calcium to Abeta and Alzheimer's disease. Neuron 59, 190–194. doi: 10.1016/j.neuron.2008.07.013
Grynkiewicz, G., Poenie, M., and Tsien, R. Y. (1985). A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 260, 3440–3450.
Guo, Q., Furukawa, K., Sopher, B. L., Pham, D. G., Xie, J., Robinson, N., et al. (1996). Alzheimer's PS-1 mutation perturbs calcium homeostasis and sensitizes PC12 cells to death induced by amyloid beta-peptide. Neuroreport 8, 379–383. doi: 10.1097/00001756-199612200-00074
Hardy, J., and Selkoe, D. J. (2002). The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297, 353–356. doi: 10.1126/science.1072994
Higazi, D. R., Fearnley, C. J., Drawnel, F. M., Talasila, A., Corps, E. M., Ritter, O., et al. (2009). Endothelin-1-stimulated InsP3-induced Ca2+ release is a nexus for hypertrophic signaling in cardiac myocytes. Mol. Cell 33, 472–482. doi: 10.1016/j.molcel.2009.02.005
Jarrett, J. T., and Lansbury, P. T. Jr. (1993). Seeding “one-dimensional crystallization” of amyloid: a pathogenic mechanism in Alzheimer's disease and scrapie? Cell 73, 1055–1058. doi: 10.1016/0092-8674(93)90635-4
Kaminski Schierle, G. S., Van De Linde, S., Erdelyi, M., Esbjorner, E. K., Klein, T., Rees, E., et al. (2011). In situ measurements of the formation and morphology of intracellular beta-amyloid fibrils by super-resolution fluorescence imaging. J. Am. Chem. Soc. 133, 12902–12905. doi: 10.1021/ja201651w
Kawahara, M. (2004). Disruption of calcium homeostasis in the pathogenesis of Alzheimer's disease and other conformational diseases. Curr. Alzheimer Res. 1, 87–95. doi: 10.2174/1567205043332234
Kayed, R., Head, E., Thompson, J. L., McIntire, T. M., Milton, S. C., Cotman, C. W., et al. (2003). Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300, 486–489. doi: 10.1126/science.1079469
Kayed, R., Sokolov, Y., Edmonds, B., McIntire, T. M., Milton, S. C., Hall, J. E., et al. (2004). Permeabilization of lipid bilayers is a common conformation-dependent activity of soluble amyloid oligomers in protein misfolding diseases. J. Biol. Chem. 279, 46363–46366. doi: 10.1074/jbc.C400260200
Kelly, B. L., and Ferreira, A. (2006). beta-Amyloid-induced dynamin 1 degradation is mediated by N-methyl-D-aspartate receptors in hippocampal neurons. J. Biol. Chem. 281, 28079–28089. doi: 10.1074/jbc.M605081200
Khachaturian, Z. S. (1989). Calcium, membranes, aging, and Alzheimer's disease. Introduction and overview. Ann. N.Y. Acad. Sci. 568, 1–4. doi: 10.1111/j.1749-6632.1989.tb12485.x
Khachaturian, Z. S. (1994). Calcium hypothesis of Alzheimer's disease and brain aging. Ann. N.Y. Acad. Sci. 747, 1–11. doi: 10.1111/j.1749-6632.1994.tb44398.x
Klyubin, I., Walsh, D. M., Lemere, C. A., Cullen, W. K., Shankar, G. M., Betts, V., et al. (2005). Amyloid beta protein immunotherapy neutralizes Abeta oligomers that disrupt synaptic plasticity in vivo. Nat. Med. 11, 556–561. doi: 10.1038/nm1234
Klyubin, I., Wang, Q., Reed, M. N., Irving, E. A., Upton, N., Hofmeister, J., et al. (2009). Protection against Abeta-mediated rapid disruption of synaptic plasticity and memory by memantine. Neurobiol. Aging 32, 614–623. doi: 10.1016/j.neurobiolaging.2009.04.005
Klyubin, I., Wang, Q., Reed, M. N., Irving, E. A., Upton, N., Hofmeister, J., et al. (2011). Protection against Abeta-mediated rapid disruption of synaptic plasticity and memory by memantine. Neurobiol. Aging 32, 614–623. doi: 10.1016/j.neurobiolaging.2009.04.005
Kuperstein, I., Broersen, K., Benilova, I., Rozenski, J., Jonckheere, W., Debulpaep, M., et al. (2010). Neurotoxicity of Alzheimer's disease Abeta peptides is induced by small changes in the Abeta42 to Abeta40 ratio. EMBO J. 29, 3408–3420. doi: 10.1038/emboj.2010.211
Lambert, M. P., Barlow, A. K., Chromy, B. A., Edwards, C., Freed, R., Liosatos, M., et al. (1998). Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. U.S.A. 95, 6448–6453. doi: 10.1073/pnas.95.11.6448
Maezawa, I., Hong, H.-S., Liu, R., Wu, C.-Y. I., Cheng, R. H., Kung, M.-P., et al. (2008). Congo red and thioflavin-T analogs detect Abeta oligomers. J. Neurochem. 104, 457–468.
Mason, R. P., Trumbore, M. W., and Pettegrew, J. W. (1996). Molecular membrane interactions of a phospholipid metabolite. Implications for Alzheimer's disease pathophysiology. Ann. N.Y. Acad. Sci. 777, 368–373. doi: 10.1111/j.1749-6632.1996.tb34447.x
Mekahli, D., Bultynck, G., Parys, J. B., De Smedt, H., and Missiaen, L. (2011). Endoplasmic-reticulum calcium depletion and disease. Cold Spring Harb. Perspect. Biol. 3, 461–490. doi: 10.1101/cshperspect.a004317
Mueller, W. E., Koch, S., Eckert, A., Hartmann, H., and Scheuer, K. (1995). beta-Amyloid peptide decreases membrane fluidity. Brain Res. 674, 133–136. doi: 10.1016/0006-8993(94)01463-R
Parker, I., and Ivorra, I. (1991). Caffeine inhibits inositol trisphosphate-mediated liberation of intracellular calcium in Xenopus oocytes. J Physiol 433, 229–240.
Parvathy, S., Davies, P., Haroutunian, V., Purohit, D. P., Davis, K. L., Mohs, R. C., et al. (2001). Correlation between Abetax-40-, Abetax-42-, and Abetax-43-containing amyloid plaques and cognitive decline. Arch. Neurol. 58, 2025–2032. doi: 10.1001/archneur.58.12.2025
Peppiatt, C. M., Collins, T. J., Mackenzie, L., Conway, S. J., Holmes, A. B., Bootman, M. D., et al. (2003). 2-Aminoethoxydiphenyl borate (2-APB) antagonises inositol 1,4,5-trisphosphate-induced calcium release, inhibits calcium pumps and has a use-dependent and slowly reversible action on store-operated calcium entry channels. Cell Calcium 34, 97–108. doi: 10.1016/S0143-4160(03)00026-5
Peppiatt, C. M., Holmes, A. M., Seo, J. T., Bootman, M. D., Collins, T. J., McDonald, F., et al. (2004). Calmidazolium and arachidonate activate a calcium entry pathway that is distinct from store-operated calcium influx in HeLa cells. Biochem. J. 381, 929–939. doi: 10.1042/BJ20040097
Pierrot, N., Ghisdal, P., Caumont, A. S., and Octave, J. N. (2004). Intraneuronal amyloid-beta1-42 production triggered by sustained increase of cytosolic calcium concentration induces neuronal death. J. Neurochem. 88, 1140–1150. doi: 10.1046/j.1471-4159.2003.02227.x
Resende, R., Ferreiro, E., Pereira, C., and Resende De Oliveira, C. (2008). Neurotoxic effect of oligomeric and fibrillar species of amyloid-beta peptide 1-42: involvement of endoplasmic reticulum calcium release in oligomer-induced cell death. Neuroscience 155, 725–737. doi: 10.1016/j.neuroscience.2008.06.036
Rowan, M. J., Klyubin, I., Wang, Q., Hu, N. W., and Anwyl, R. (2007). Synaptic memory mechanisms: Alzheimer's disease amyloid beta-peptide-induced dysfunction. Biochem. Soc. Trans. 35, 1219–1223. doi: 10.1042/BST0351219
Simakova, O., and Arispe, N. J. (2006). Early and late cytotoxic effects of external application of the Alzheimer's Abeta result from the initial formation and function of Abeta ion channels. Biochemistry 45, 5907–5915. doi: 10.1021/bi060148g
Sugawara, H., Kurosaki, M., Takata, M., and Kurosaki, T. (1997). Genetic evidence for involvement of type 1, type 2 and type 3 inositol 1,4,5-trisphosphate receptors in signal transduction through the B-cell antigen receptor. EMBO J. 16, 3078–3088. doi: 10.1093/emboj/16.11.3078
Taylor, C. W., and Broad, L. M. (1998). Pharmacological analysis of intracellular Ca2+ signalling: problems and pitfalls. Trends Pharmacol. Sci. 19, 370–375. doi: 10.1016/S0165-6147(98)01243-7
Thathiah, A., and De Strooper, B. (2011). The role of G protein-coupled receptors in the pathology of Alzheimer's disease. Nat. Rev. Neurosci. 12, 73–87. doi: 10.1038/nrn2977
Toescu, E. C., O'neill, S. C., Petersen, O. H., and Eisner, D. A. (1992). Caffeine inhibits the agonist-evoked cytosolic Ca2+ signal in mouse pancreatic acinar cells by blocking inositol trisphosphate production. J. Biol. Chem. 267, 23467–23470.
Tovey, S. C., De Smet, P., Lipp, P., Thomas, D., Young, K. W., Missiaen, L., et al. (2001). Calcium puffs are generic InsP(3)-activated elementary calcium signals and are downregulated by prolonged hormonal stimulation to inhibit cellular calcium responses. J. Cell. Sci. 114, 3979–3989.
Tovey, S. C., Sun, Y., and Taylor, C. W. (2006). Rapid functional assays of intracellular Ca2+ channels. Nat. Protoc. 1, 259–263. doi: 10.1038/nprot.2006.40
Umeda, T., Tomiyama, T., Sakama, N., Tanaka, S., Lambert, M. P., Klein, W. L., et al. (2011). Intraneuronal amyloid beta oligomers cause cell death via endoplasmic reticulum stress, endosomal/lysosomal leakage, and mitochondrial dysfunction in vivo. J. Neurosci. Res. 89, 1031–1042. doi: 10.1002/jnr.22640
Verkhratsky, A. (2005). Physiology and pathophysiology of the calcium store in the endoplasmic reticulum of neurons. Physiol. Rev. 85, 201–279. doi: 10.1152/physrev.00004.2004
Walsh, D. M., Klyubin, I., Fadeeva, J. V., Cullen, W. K., Anwyl, R., Wolfe, M. S., et al. (2002). Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416, 535–539. doi: 10.1038/416535a
Wang, Q., Walsh, D. M., Rowan, M. J., Selkoe, D. J., and Anwyl, R. (2004). Block of long-term potentiation by naturally secreted and synthetic amyloid beta-peptide in hippocampal slices is mediated via activation of the kinases c-Jun N-terminal kinase, cyclin-dependent kinase 5, and p38 mitogen-activated protein kinase as well as metabotropic glutamate receptor type 5. J. Neurosci. 24, 3370–3378. doi: 10.1523/JNEUROSCI.1633-03.2004
Whalen, B. M., Selkoe, D. J., and Hartley, D. M. (2005). Small non-fibrillar assemblies of amyloid beta-protein bearing the Arctic mutation induce rapid neuritic degeneration. Neurobiol. Dis. 20, 254–266. doi: 10.1016/j.nbd.2005.03.007
Wirths, O., Multhaup, G., and Bayer, T. A. (2004). A modified beta-amyloid hypothesis: intraneuronal accumulation of the beta-amyloid peptide–the first step of a fatal cascade. J. Neurochem. 91, 513–520. doi: 10.1111/j.1471-4159.2004.02737.x
Xu, C., Bailly-Maitre, B., and Reed, J. C. (2005). Endoplasmic reticulum stress: cell life and death decisions. J. Clin. Invest. 115, 2656–2664. doi: 10.1172/JCI26373
Yao, J., Irwin, R. W., Zhao, L., Nilsen, J., Hamilton, R. T., and Brinton, R. D. (2009). Mitochondrial bioenergetic deficit precedes Alzheimer's pathology in female mouse model of Alzheimer's disease. Proc. Natl. Acad. Sci. U.S.A. 106, 14670–14675. doi: 10.1073/pnas.0903563106
Yoshiike, Y., Minai, R., Matsuo, Y., Chen, Y. R., Kimura, T., and Takashima, A. (2008). Amyloid oligomer conformation in a group of natively folded proteins. PLoS ONE 3:e3235. doi: 10.1371/journal.pone.0003235
Keywords: Alzheimer's disease, Aβ oligomers, calcium/Ca2+, InsP3/IP3, InsP3 receptors/InsP3Rs, endoplasmic reticulum/ER
Citation: Jensen LE, Bultynck G, Luyten T, Amijee H, Bootman MD and Roderick HL (2013) Alzheimer's disease-associated peptide Aβ42 mobilizes ER Ca2+ via InsP3R-dependent and -independent mechanisms. Front. Mol. Neurosci. 6:36. doi: 10.3389/fnmol.2013.00036
Received: 17 August 2013; Accepted: 14 October 2013;
Published online: 05 November 2013.
Edited by:
Gaiti Hasan, National Centre for Biological Sciences, TIFR, IndiaReviewed by:
David S. Greenberg, The Hebrew University of Jerusalem, IsraelDavid Yule, University of Rochester, USA
Copyright © 2013 Jensen, Bultynck, Luyten, Amijee, Bootman and Roderick. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: H. Llewelyn Roderick, Babraham Institute, Babraham, Cambridge, CB22 3AT, UK e-mail:bGxld2VseW4ucm9kZXJpY2tAYmFicmFoYW0uYWMudWs=
†Present address: Martin D. Bootman, Department of Life, Health and Chemical Sciences, The Open University, Milton Keynes, UK