 Magdalena Czeredys
Magdalena Czeredys Joanna Gruszczynska-Biegala
Joanna Gruszczynska-Biegala Teresa Schacht
Teresa Schacht Axel Methner
Axel Methner Jacek Kuznicki
Jacek Kuznicki- 1Laboratory of Neurodegeneration, International Institute of Molecular and Cell Biology, Warsaw, Poland
- 2Focus Program Translational Neuroscience, Rhine Main Neuroscience Network, Department of Neurology, Johannes Gutenberg University Medical Center Mainz, Mainz, Germany
- 3Laboratory of Calcium Binding Proteins, Nencki Institute of Experimental Biology, Warsaw, Poland
Huntington's disease (HD) is a hereditary neurodegenerative disease caused by the expansion of a polyglutamine stretch in the huntingtin (HTT) protein and characterized by dysregulated calcium homeostasis. We investigated whether these disturbances are correlated with changes in the mRNA level of the genes that encode proteins involved in calcium homeostasis and signaling (i.e., the calciosome). Using custom-made TaqMan low-density arrays containing probes for 96 genes, we quantified mRNA in the striatum in YAC128 mice, a model of HD, and wildtype mice. HTT mutation caused the increased expression of some components of the calcium signalosome, including calretinin, presenilin 2, and calmyrin 1, and the increased expression of genes indirectly involved in calcium homeostasis, such as huntingtin-associated protein 1 and calcyclin-binding protein. To verify these findings in a different model, we used PC12 cells with an inducible expression of mutated full-length HTT. Using single-cell imaging with Fura-2AM, we found that store-operated Ca2+ entry but not endoplasmic reticulum (ER) store content was changed as a result of the expression of mutant HTT. Statistically significant downregulation of the Orai calcium channel subunit 2, calmodulin, and septin 4 was detected in cells that expressed mutated HTT. Our data indicate that the dysregulation of calcium homeostasis correlates with changes in the gene expression of members of the calciosome. These changes, however, differed in the two models of HD used in this study. Our results indicate that each HD model exhibits distinct features that may only partially resemble the human disease.
Introduction
Cellular calcium transients control a vast array of cellular functions, from short-term responses (e.g., contraction and secretion) to the long-term regulation of cell growth and proliferation. Cytosolic Ca2+ increases in response to activation of cell-surface receptors, either directly after the activation of ionotropic glutamate channels or indirectly after generation of the Ca2+-mobilizing second messenger inositol 1,4,5-trisphosphate (IP3). IP3 interacts with its receptors (IP3R1-3) on the membrane of the endoplasmic reticulum (ER), the major store of Ca2+ in the cell. The opening of these IP3R Ca2+ channels releases Ca2+ stored in the ER (Foskett et al., 2007). Increased cytosolic Ca2+ can also cause Ca2+-induced release of Ca2+ from ryanodine receptors (RyR1-3) (Hamilton, 2005). The depletion of ER Ca2+ stores results in activation of store-operated channels (SOCs) at the plasma membrane, mediating store-operated calcium entry (SOCE) from the extracellular space, followed by the removal of cytosolic Ca2+ and replenishment of luminal Ca2+ through sarcoplasmic/ER Ca2+-adenosine triphosphatases (ATPases; SERCA1-3). Considering their electrophysiological and molecular properties, two main types of SOCs can be described. The first are the highly Ca2+-selective calcium release activated calcium currents (ICRAC) mediated by the Orai family of proteins, which carry highly Ca2+-selective ICRAC currents (Vig et al., 2006). The second type of SOCs are non-selective Ca2+-permeable TRPC (C-type transient receptor potential) channels (Huang et al., 2006). The Ca2+ sensor's stromal interaction molecules 1 and 2 (STIM1 and STIM2) detect changes in Ca2+ concentration in the ER via an EF-hand Ca2+-binding domain, in response to store depletion, they rearrange into punctate structures that are close to the plasma membrane. This activates members of the Orai family (Orai1, Orai2, and Orai3) of Ca2+-influx channels (Liou et al., 2005; Roos et al., 2005), resulting in Ca2+ entry by ICRAC into the cell. STIM1 and Orai1 have also been implicated with cell death and mitochondrial bioenergetics (Henke et al., 2012, 2013). STIM1 and STIM2 are expressed in the brain (Klejman et al., 2009; Skibinska-Kijek et al., 2009; Ng et al., 2011; Steinbeck et al., 2011). The main function of STIM1 in neurons appears to be the activation of SOCE (Gruszczynska-Biegala et al., 2011). STIM1 directly suppresses the depolarization-induced opening of the voltage-gated Ca2+ channel CaV1.2 by binding to the C-terminus of this channel, leading to the inhibition of gating and long-term internalization of the channel from the membrane (Park et al., 2010). The primary function of STIM2 in neurons was suggested to be the regulation of the resting level of Ca2+ in the ER and Ca2+ leakage (Gruszczynska-Biegala et al., 2011; Gruszczynska-Biegala and Kuznicki, 2013). To maintain the spatial and temporal properties of Ca2+ signals, neurons express numerous Ca2+-binding proteins that act as Ca2+ buffers (Nikoletopoulou and Tavernarakis, 2012). In the cytoplasm, parvalbumin (PV), calbindin D-28k (CALB1), and calretinin (CALB2) (Billing-Marczak and Kuznicki, 1999) act as Ca2+ buffers, whereas buffer proteins localized within the lumen of the ER, including calsequestrin (CASQ) and calreticulin (CALR) (Groenendyk et al., 2004; Michalak et al., 2009), allow the store to accumulate the large amounts of Ca2+ necessary for rapid cell signaling responses. These proteins involved in calcium signaling and homeostasis represent a special toolkit referred to as the Ca2+ signalosome (Berridge et al., 2003; Berridge, 2012).
Proper action of the Ca2+ signalosome is crucial for the functioning of neuronal cells and during aging the Ca2+ signaling machinery undergoes significant changes (Toescu and Verkhratsky, 2007; Puzianowska-Kuznicka and Kuznicki, 2009). Moreover, conclusive evidence indicates that neuronal Ca2+ signaling is abnormal in many neurodegenerative disorders (Wojda et al., 2008; Bezprozvanny, 2010). Notably, SOCE is dysregulated in epilepsy (Steinbeck et al., 2011) and AD (Jaworska et al., 2013; Ryazantseva et al., 2013). One of the neurodegenerative disorders with disrupted Ca2+ homeostasis is Huntington's disease (HD), which is characterized clinically by chorea, dementia, and psychiatric symptoms. This genetic disorder is caused by the expansion of a CAG trinucleotide repeat in exon 1 of the huntingtin gene (HTT), which is translated into polyglutamine residues (polyQ) in the huntingtin protein (HTT). Huntingtin is toxic to cells when the number of CAG repeats exceeds 36, a point at which HTT forms aggregates in the nuclei of neurons where it might inhibit the function of various proteins, including key transcriptional factors. This leads to transcriptional dysfunction and may cause neuronal degeneration (Sugars and Rubinsztein, 2003; Li and Li, 2004). HD is characterized at the molecular level by various changes, including disturbances in calcium homeostasis and signaling components, and these changes are considered to be involved in various processes that lead to the neurodegeneration observed in HD (Giacomello et al., 2011, 2013).
It is probable that destabilization of neuronal Ca2+ signaling is one of the toxic functions of the mutant HTT. To test this hypothesis, we used two models of HD: a cellular model and a transgenic model overexpressing mutant HTT. Inducible PC12 cells overexpress an expanded HTT with 73 glutamines (Apostol et al., 2003) and are useful for studying early changes in HD, since they accumulate aggregates of mutant HTT upon treatment with the steroid ponasterone A. Although others have used this inducible PC12 model successfully to study HD pathogenesis, no disturbances in the expression or function of the calcium signalosome have yet been described. However, it was shown that the first 17 amino-acid residues of mutated HTT modulate its sub-cellular localization, aggregation and effects on Ca2+ homeostasis in glutamate-challenged PC12 cells (Rockabrand et al., 2007). Our HD animal model, YAC128 transgenic mice express the full-length human HTT protein with 128 CAG repeats and display an age-dependent loss of striatal neurons, similar to those seen in human HD (Slow et al., 2003). YAC128 mice initially exhibit hyperactivity, followed by the onset of motor deficits and finally hypokinesis. Motor deficits in YAC128 mice are highly correlated with striatal neuronal loss. There is compelling evidence that supports Ca2+ dysfunction playing a central role in the pathogenesis of HD. In medium spiny neurons (MSNs) from YAC128 transgenic mice but not control mice, the activity of SOCE was enhanced (Wu et al., 2011). Moreover, in a yeast two-hybrid screen and YAC128 mice, an interaction between mutant HTT and the 5C-terminus of IP3R1 was shown to be mediated by huntingtin-associated protein-1A (Tang et al., 2003). Repeated application of glutamate to MSNs in these mice elevated cytosolic Ca2+ levels, resulting in apoptosis mediated by the activation of Gq-coupled metabotropic glutamate receptors (Tang et al., 2005). Similar results were shown in the HdH (Q111/Q111) mouse model, in which agonist-induced Ca2+ release in neurons from these mice was increased relative to (Q20/Q20) control mice. Basal protein kinase B (PKB) activation was also higher in HdH (Q111/Q111) neurons, and this process was dependent on metabotropic glutamate receptor 5 (mGluR5) (Ribeiro et al., 2010).
Although the effect of mutant HTT on SOCE was previously studied in transgenic YAC128 mice (Wu et al., 2011), there is no data concerning calcium signalosome alterations, which could be considered to be involved in HD pathogenesis. Therefore, the aim of our study was to investigate whether mutated HTT is responsible for the changes in the expression of calciosome genes, and if so, which of such changes might explain the alterations observed in HD. Using custom-made TaqMan low-density arrays and individual real-time quantitative polymerase chain reaction (qRT-PCR), we studied changes in gene expression in two HD models. Our arrays contained most of the genes involved in calcium signaling and homeostasis, some genes responsible for amyloid beta precursor protein (APP) processing and insulin signaling, as well as a few genes that encode protein kinases or other proteins connected with HD. We found increased expression of few components of the calcium signalosome and genes indirectly involved in calcium homeostasis in YAC128 brains. In PC12 cells with induced expression of mutated full-length HTT, we detected downregulation of the genes that encode some proteins involved in the calcium signalosome. Our data indicate that the dysregulation of calcium homeostasis correlates with changes in the gene expression of members of the calciosome, but the two models used in this study exhibit distinct changes in gene expression.
Materials and Methods
Animals
Female transgenic YAC128 mice that overexpressed human HTT (128 Gln) and non-transgenic littermates that were applied as controls aged between 3 and 6 months (Slow et al., 2003) were used. The animals were kept under normal laboratory conditions (12 h/12 h dark/light cycle; 50–60% relative humidity; 22°C) with food and water available ad libitum. Animal care was in accordance with the European Communities Council Directive (86/609/EEC). The experimental procedures were approved by the Local Commission for the Ethics of Animal Experimentation No. 1 in Warsaw (approval no. 305/212).
Examined Brain Regions
The striatum (head of the caudate nucleus), the primary (M1) and secondary (M2) motor cortex, and the cerebellum were identified macroscopically according to anatomical landmarks (Paxinos and Franklin, 2004) in YAC128 and control mice brains. These brain structures were isolated and kept in −80°C until the future experiments.
Cell Cultures
PC12 cells with an inducible expression of full-length human HTT containing 73 polyglutamine repeats translated from a random codon with a TagRFP tag at the C-terminus were obtained from the Coriell Institute for Medical Research. The PC12 cells were grown to 90% confluence at 37°C in a 5% CO2, 96% relative humidity incubator in cell-culture plates (100 cm2; BD Biosciences) treated with 10 μg/μl poly-L-lysine (Sigma) in a cell culture medium that contained 15% horse serum (HS; H1270, Sigma), 2.5% fetal bovine serum (FBS; F7524, Sigma), and phenol red-free Dulbecco's Modified Eagle Medium (DMEM; 41965-039, Gibco) supplemented with 1% penicillin-streptomycin (P0781, Sigma), 0.1 mg/ml zeocin (R25001, Invitrogen), and 0.1 mg/ml geneticine (G8168, Sigma). The cells were passaged with a 1:5 dilution at least three times prior to the experiments. Each passage consisted of 5 min incubation with 2 ml of trypsin replacement (Sigma), the addition of 8 ml of cell media to inactivate the enzyme, and brief centrifugation (2 min, 300 × g), followed by media replacement. Cell suspensions were then added to the cell-culture plates and pipetted into single cells. The cells grew to a monolayer (> 50% confluence in 1–2 days), whereupon differentiation into neuron-like cells was achieved by treatment with neural growth factor (NGF 2.5S, 100 ng/ml; 13257-019, Invitrogen) for 48 h in low serum medium that contained DMEM, 1% HS, and 0.25% FBS supplemented by antibiotics as above. To induce the expression of human HTT, 2 μM ponasterone A (P3490, Sigma) was added to the differentiated cells for 48 h. As ponasterone A was dissolved in 100% ethanol, control cells were treated with the same amount of 100% ethanol in parallel with the ponasterone A-treated cells.
Single-Cell Ca2+ Measurements
Single-cell Ca2+ imaging was performed using the ratiometric Ca2+ indicator dye Fura-2 acetoxymethyl ester (Fura-2 AM). The cells (17,000 cells/well) were differentiated into neuron-like cells in 96-well imaging plates (BD Bioscience) for 48 h and induced to express human HTT 48 h before the experiment. The cells were loaded with 5 μM Fura-2 AM (F1201, Molecular Probes) for 30 min at 37°C in Hank's Balanced Salt Solution supplemented with 2 mM CaCl2 and then rinsed and left undisturbed for 30 min to allow for de-esterification. Calcium-free medium instead of 2 mM CaCl2 contained 0.5 mM ethylene glycol tetraacetic acid (EGTA). The experiments were performed using a BD Pathway 855 High Content Imaging System (BD Biosciences). Images were acquired at 340 and 380 nm excitation wavelengths, and the ratio was calculated every 5 s for every cell.
RNA Isolation from Brain Tissue and PC12 Cells
Brain samples were homogenized separately using a Potter-Elvehjem homogenizer. RNA was extracted using the RNeasy Lipid Tissue Mini kit with additional DNase treatment (Qiagen). To isolate RNA from PC12 cells, homogenization was performed using Qiashredders (Qiagen), and RNA was extracted using the RNeasy Plus Mini kit (Qiagen). cDNA was synthesized by reverse transcription (SuperScript III RnaseH, Invitrogen).
Gene Expression Analysis
All gene expression analysis were performed using the 7900HT system (Life Technologies). Gene profiling in the brain was performed using custom-designed TaqMan low-density arrays (Life Technologies), hereinafter referred to as RT-qPCR arrays. For each array (384 assays), 1.6 μg of cDNA was loaded. The obtained data were analyzed using the relative quantification method and 2−ΔCT formula (ΔCT = CTtarget − CTGapdh, where CT denotes the cycle threshold). The list of genes and their annotations included in the RT-qPCR arrays is shown in Table 1. For RT-qPCR arrays TaqMan chemistry (4369016, Life Technologies) was used.
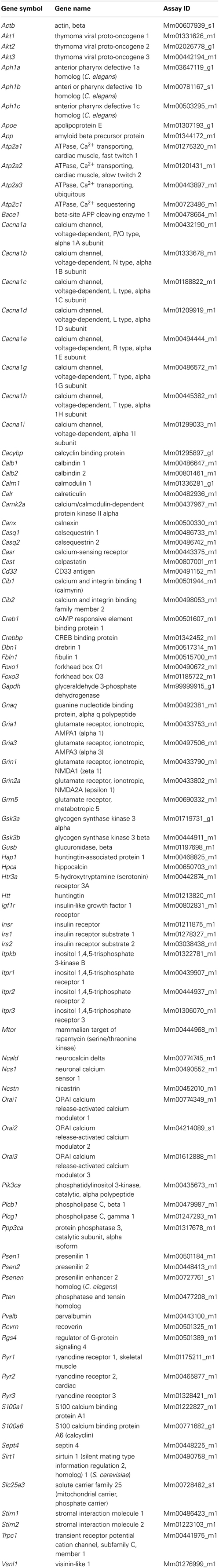
Table 1. List of genes and their annotations included in the RT-qPCR array.
Single analyses of gene expression levels using cDNA preparations from PC12 cell cultures were performed in individual real-time PCR reactions, hereinafter referred to as qRT-PCR. The obtained data were quantified using the relative standard curve method. For each gene, standard cDNAs were diluted and amplified along with sample cDNAs in the same PCR run. Standard curves were generated using the 7900HT system software (Life Technologies). The quantity of mRNA in each sample (R0) was determined from the relative standard curve (using sample CT values) and expressed in arbitrary units corresponding to the dilution factors of the standard RNA preparation. The amplification efficiency (E) of each PCR reaction was determined using the equation of the standard curve:
Rat-specific primers were designed using Primer Blast (National Center for Biotechnology Information), and their sequences are shown in Table 2. For individual qRT-PCR in PC12 cells SYBR Green chemistry (4385612, Life Technologies) was used. The expression of the Tmem66, Cracr2a (Efcab4b), Ryr3, and Atp2a1 genes was verified using FAM dye-labeled TaqMan probes (Life Technologies) and TaqMan chemistry (4369016, Life Technologies). The expression of Cracr2a (Efcab4b), Ryr3, and Atp2a1 was undetectable in PC12 cells using both hand-made and custom-made primers.

Table 2. List of primers used in individual qRT-PCR for PC12 cells.
Immunoblotting
Cells were extracted in ice-cold RIPA buffer: 50 mM Tris, pH 7.5, 150 mM NaCl, 1% NP-40, 0.5% NaDOC, 0.1% sodium dodecyl sulfate (SDS), and 1 mM ethylenediaminetetraacetic acid (EDTA) that contained mini complete protease inhibitor cocktail (Roche) and phosphatase inhibitors (Sigma). The lysed cells were centrifuged at 16,000 × g for 30 min at 4°C. After measuring the protein concentrations with the BC Assay Protein Quantification Kit (Interchim), 100 μg protein was loaded on 3–8% Tris-acetate gel (Invitrogen). The proteins were transferred to a nitrocellulose membrane using the iBlot system (Invitrogen), fixed with Ponceau S solution (Sigma) for 20 s, and then blocked for 1 h with 3% dry non-fat milk in phosphate-buffered saline with 0.01% Tween-20 (PBS-T) at room temperature under gentle shaking. The membranes were incubated overnight at 4°C against actin (MAB1501, 1:4000, Millipore) and huntingtin (1756-1, 1:1000, Epitomics) diluted in PBS-T that contained 3% dry non-fat milk. After washing three times with PBS-T, the membranes were incubated with the secondary antibody IRDye 800 goat anti-rabbit immunoglobulin G (IgG) and IRDye 680 goat anti-mouse IgG, (both from Licor, 926-32211 and 926-32220, respectively) for 1 h at room temperature. The secondary antibodies were diluted to 1:30000 in 3% dry non-fat milk in PBS-T. Afterward, the membranes were washed three times with PBS-T and scanned for infrared fluorescence at 680 or 800 nm using an Odyssey scanner system (Licor). Brain tissue was homogenized on ice using a glass-homogenizer (70 strokes) and cleared by centrifugation at 12,000 × g for 20 min. Protein extracts (20 μg) were separated on 10% SDS-polyacrylamide gel electrophoresis (PAGE), transferred to a Protran nitrocellulose membrane (Whatman), and blocked for 2 h at room temperature in TBS-T: 50 mM Tris-HCl, pH 7.5, 150 mM NaCl, and 0.1% Tween 20 plus 5% dry non-fat milk. Nitrocellulose sheets were then incubated at 4°C overnight in blocking solution with primary monoclonal antibodies against HAP1 (611302, DB Transduction Laboratories) diluted to 1:300; CacyBP/SIP (ab51288, Abcam), 1:1000; Cib2 (H00010518-A01, Abnova), 1:100; pan-cadherin (ab6528, Abcam), 1:2500; as well as with primary polyclonal antibodies against CIB1 (11823-1-AP, Proteintech Europe), 1:100; CALB2 (7699/3H, Swant), 1:500; and glyceraldehyde 3-phosphate dehydrogenase (sc-25778, GAPDH) (Santa Cruz), 1:1000. The appropriate horseradish peroxidase-conjugated secondary antibody anti-mouse IgG (A9044) and anti-rabbit IgG (A0545), both from Sigma, was added at a dilution of 1:10000 for 1 h at room temperature. The peroxidase was detected with enhanced chemiluminescence (Amersham Biosciences). The intensity of the bands was measured using an ImageQuant LAS 4000 (GE Healthcare) and Quantity One software (Bio-Rad). Densitometry was then performed using the intensity of GAPDH or pan-cadherin bands as an internal standard depending on the molecular weight of the analyzed protein.
Statistical Analysis
To calculate p-values for differences in gene expression levels between the same brain regions (striatum or motor cortex or cerebellum) in wildtype and transgenic mice, we applied two-tailed paired Student's t-test using ΔCT values for the results obtained with the RT-qPCR arrays (Figure 1; n = 3) or relative values for the results obtained with individual qRT-PCRs (Table 4; n = 3). The degree of significance vs. control is indicated by asterisks: *p < 0.05, **p < 0.005, ***p < 0.0005 (ns, not significant, p > 0.05). To make the criteria for statistical significance more stringent, the Bonferroni correction (p < 0.00052) was applied. This was calculated as the p-value (0.05) divided by 96, which is the number of studied genes (Noble, 2009).
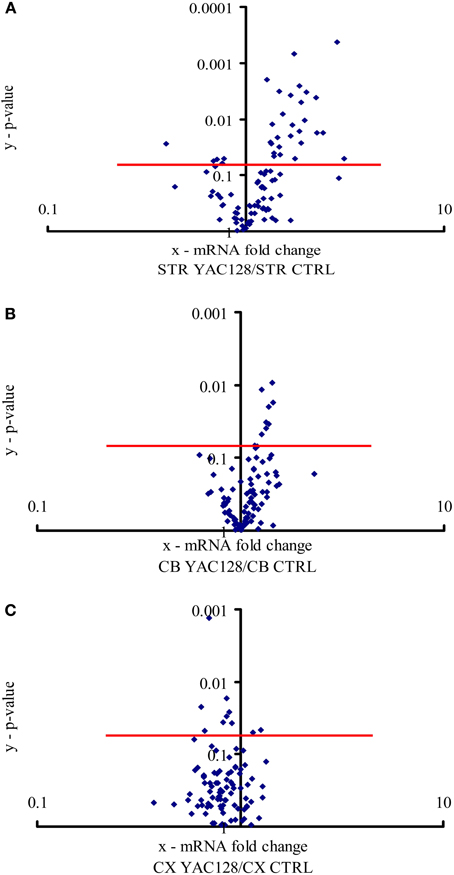
Figure 1. Expression of calcium signaling and homeostasis genes in the striatum, cerebellum and motor cortex. The volcano plot arranges genes along the dimensions of (x) mean expression fold difference between the analyzed brain structures: (A) striatum (STR), (B) cerebellum (CB), and (C) motor cortex (CX) in YAC128 mice and control mice (CTRL), and (y) p-value (Student's t-test). A logarithmic scale is used. Points located above the red lines represent genes whose expression was significantly changed (p < 0.05). The results represent data based on three independent mRNA preparations from the studied brain structures of 3-month-old mice.
Results
Differences in Gene Expression between Transgenic HD Mice (YAC128) and Wildtype Mice in the Striatum, Cerebellum and Motor Cortex
To estimate the relative mRNA levels of the ensemble of genes that encode the calciosome, custom-made RT-qPCR arrays were prepared containing genes expressed in the mouse brain according to the Allen Brain Atlas (www.brain-map.org; accessed July 2, 2013; Table 1). We dissected the striatum from 3-month-old YAC128 mice and non-transgenic controls and analyzed the expression of four standards, 57 calciosome genes, as well as genes implicated in HD and Alzheimer's disease (AD) using two technical replicates. For comparison, we also performed these analyses using preparations of the cerebellum and motor cortex.
All 96 genes present in our custom-made PCR arrays were detected in the forebrain. In the striatum, ~31 of the analyzed genes exhibited a statistically significant difference (p < 0.05) in expression between YAC128 mice and control mice (Figure 1A). Twenty-seven genes were expressed at a higher level in HD transgenic mice compared with non-transgenic mice, and four genes were expressed at a lower level. Statistically significant differences between gene expression in YAC128 mice and control mice were observed in the cerebellum and motor cortex. In the cerebellum, eight genes exhibited enhanced expression in HD transgenic mice, and in the cortex, two had enhanced expression and eight reduced expression relative to control animals (Figures 1B,C).
Expression of Genes Potentially Implicated in HD in YAC128 Mice
To identify genes with potential significance in this HD animal model, we focused on genes whose expression was changed at least 1.5-fold in the striatum; namely, Hap1 (huntingtin-associated protein 1), Calb2 (calretinin), Aph1 (anterior pharynx defective 1 homolog) isoforms a, b and c, Psenen (presenilin enhancer 2 homolog), Psen2 (presenilin 2), Cib1 (calmyrin 1), CacyBP/SIP (calcyclin-binding protein), Calr (calreticulin), and Cib2 (calmyrin 2).
The expression of Hap1, Calb2, and Aph1b was approximately three-times higher in the striatum of HD mice compared to control mice. When we employed the Bonferroni correction, of the 31 genes whose mRNA levels were significantly changed in the striatum in YAC128 mice, only the Aph1b gene met the criterion of p < 0.00052.
The most prominent changes in gene expression between 3-month-old YAC128 mice and non-transgenic mice were observed in the striatum (Figure 1A). This is consistent with the fact that the first changes that occur in the brains of patients suffering from HD also appear in the striatum (Vonsattel et al., 1985). Since the clinical and pathological changes become more apparent with age, we analyzed the same brain structure in 6-month-old YAC128 mice and wildtype animals. Equal amounts of cDNA obtained from the striatum in three transgenic HD mice and three control mice were prepared, and real-time PCR was performed using our RT-qPCR array. The most significant changes that were observed in the striatum in 3-month-old YAC128 mice were also observed in 6-month-old HD animals. The genes whose expression exhibited at least a 1.3-fold increase in 6-month-old mice were compared to the list of genes that were overexpressed in the striatum in 3-month-old mice. Eleven genes were identified that had significantly higher expression in the striatum in mice YAC128 of both ages (Table 3) comparing to control mice.
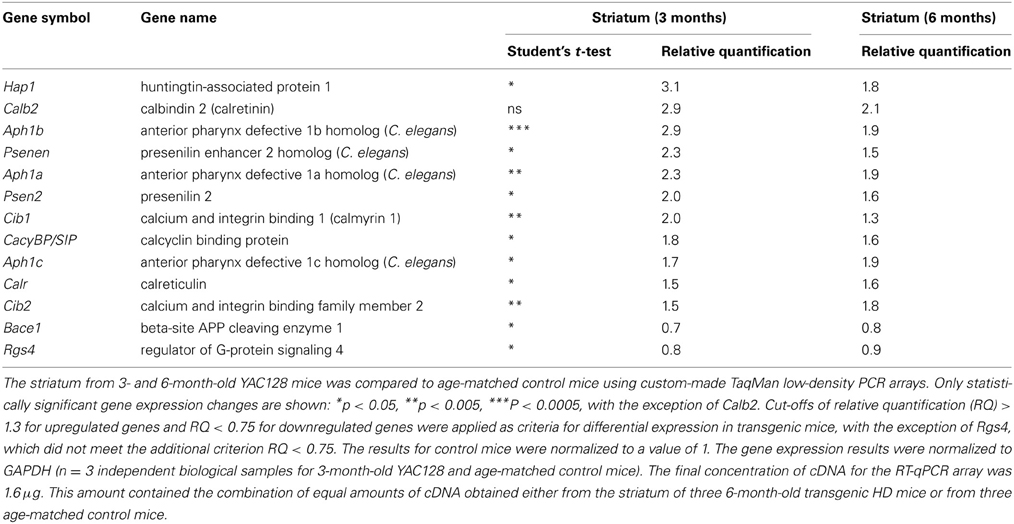
Table 3. Gene expression analysis in the striatum in YAC128 mice.
In addition to the genes that had higher expression in the striatum in YAC128 mice, two genes were downregulated in both age cohorts: Bace1, a gene that encodes the β-secretase component (β-site APP cleaving enzyme 1), and Rgs4, a member of the family of guanosine triphosphatase (GTPase)-activating proteins that regulate Gq and Gi proteins (Huang et al., 1997). However, in contrast to the up to three-fold increases associated with the other genes, the level of these two mRNAs decreased by only 10–30% (0.7–0.9-fold).
In the group of genes presented in Table 3, only Aph1b had p < 0.00052, as calculated using the Bonferroni correction. However, we also identified 12 other genes that did not meet the Bonferroni correction criterion. Nevertheless, for these genes, we observed high relative quantification and the same extent of mRNA changes in the striatum in both HD mice groups compared with control mice. We therefore chose to check some of these results at the protein level.
Verification of Gene Expression Data at the Protein Level Determined by Western Blot
Protein extracts from the striatum of 3-, 4-, and 6-month-old YAC128 and age-matched control mice were prepared, separated by SDS-PAGE, and incubated with primary antibodies against HAP1, CacyBP/SIP, CALB2, CIB2, CIB1 and GAPDH or pan-cadherin as loading controls. In six independent western blot experiments, we observed increase in CacyBP/SIP and CIB2 proteins in the striatum of YAC128 mice compared to control mice (Figures 2B,D). In five out of six blots there was an increase in HAP1 protein (Figure 2A) and a decrease in CIB1 (Figure 2E). In four out of six blots we observed increase in CALB2 (Figure 2C). This confirms that screening for changes at the mRNA level is feasible but must be followed by verification at the protein level.

Figure 2. Protein expression analysis in the striatum in YAC128 mice. Immunoblots of HAP1 (A), CacyBP/SIP (B), CALB2 (C), CIB2 (D), and CIB1 (E) in the striatum of YAC128 mice and age-matched control mice (CTRL) are shown. First two blots show the data from 3- month-old mice, third and fourth blots—from 4-month-old mice, and fifth and sixth blots from 6-month-old animals. 20 μg of protein was loaded on the gel. HAP1 densitometry was performed using the intensity of GAPDH (A). Pan-cadherin bands were used as an internal standard for CacyBP/SIP (B), CALB2 (C), CIB2 (D), and CIB1 (E). The fold change of the studied proteins is shown above the immunoblots.
Reduction of SOCE by Expression of Mutated Huntingtin in PC12 Cells
To functionally evaluate our results at the cellular level, we adopted a cellular model of HD; namely, inducible PC12 cells (Apostol et al., 2003), in which an expanded HTT with 73 glutamines is expressed upon treatment with the insect steroid ponasterone A (Figure 3A). We first investigated the effects of mutated HTT on intracellular Ca2+ homeostasis and the expression of the genes identified in the first part of the study. Cultured PC12 cells were separated into two portions; one was treated with ponasterone A for 48 h to induce the expression of mutated human HTT and the other one with vehicle. For the calcium imaging experiments, both cultures were incubated with Fura-2AM. SOCE was examined by incubating the cells in a calcium-free medium (0.5 mM EGTA) followed by store depletion with 2 μM of the SERCA pump inhibitor thapsigargin (TG). The reintroduction of extracellular Ca2+ evoked a robust Ca2+ influx that corresponded to SOCE in both types of PC12 cells. However, cells with human HTT exhibited a significant reduction in Ca2+ reentry compared with control cells (Figure 3C).
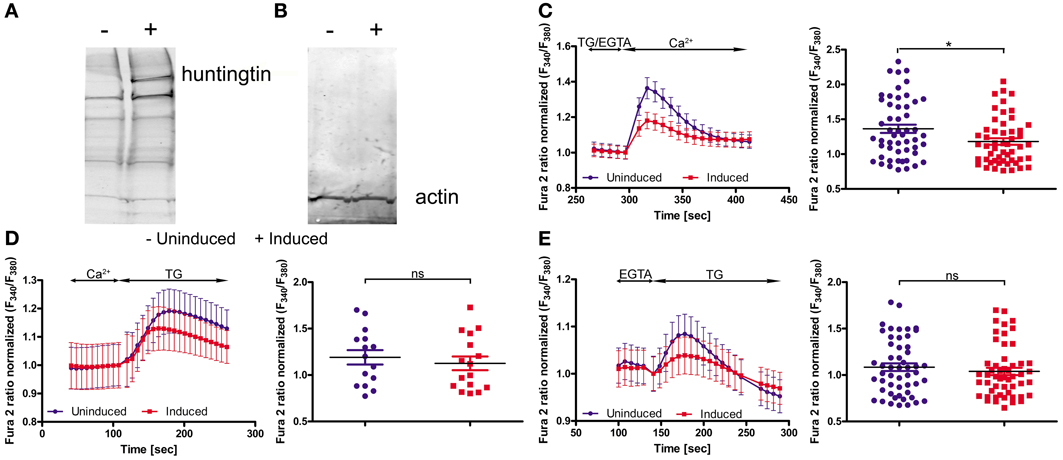
Figure 3. Huntingtin expression in PC12 cells reduces SOCE. Immunoblots of huntingtin (A) and actin (B) in induced (+) and uninduced (−) PC12 cells are shown. Ratiometric Fura-2 analysis of uninduced and induced PC12 cells was performed on a BD Pathway high-content imaging system. (C) SOCE measurements began in a buffer supplemented with 0.5 mM EGTA, which was then replaced by a buffer with 0.5 mM EGTA and 2 μM thapsigargin (TG). After 2.5 min, the readdition of 2 mM Ca2+ to the extracellular media resulted in Ca2+ influx. The traces show only Ca2+ readdition after store depletion. F340/F380 values beginning just before the readdition of Ca2+ were normalized to the same values (1). The ER calcium stores were depleted by the addition of 2 μM TG in the presence (D) or absence (E) of extracellular Ca2+. F340/F380 values beginning just before the addition of TG were normalized to the same values (1) (C–E, left panels). The data from different experiments were averaged (C–E, right panels). Summary data present the maximum (peak) of the F340/F380 ratio after the addition of Ca2+ or TG are expressed as the mean ± SD of the data shown in the left panels. *p < 0.05; ns, not significant (p > 0.05). Averaged traces from (C) 52 individually measured wells that contained a total of ~2,000 cells per trace, (D) 52 individually measured wells that contained a total of ~2,000 cells per trace, and (E) 16 individually measured wells that contained a total of ~500 cells per trace.
In order to check if mutant HTT affect the ER Ca2+ store content in PC12 cells, we incubated cells either in the absence of extracellular Ca2+ or in the presence of 2 mM Ca2+. Next, 2 μM TG was added to inhibit Ca2+-ATPase and the amount of calcium ions released from ER were detected. The TG-releasable pool was smaller under both conditions in the cells with induced expression of HTT, but the difference did not reach statistical significance (Figures 3D,E). PC12 cells expressing mutant HTT exhibited significant differences in SOCE, which is in line with our hypothesis that HD is caused or accompanied by changes in the calciosome.
Next, we tried to verify the expression of those calciosome components that were regulated in YAC128 mice using induced PC12 cells. Surprisingly, we did not detect changes in Hap1, Calb2, Aph1b, or CacyBP/SIP gene expression in this model. We then chose genes that encode proteins known to be directly involved in SOCE or genes that encoded proteins that regulate SOCE activity to find a possible explanation for the reduced SOCE in inducible huntingtin-expressing PC12 cells. As shown in Table 4, we detected a statistically significant downregulation of Orai calcium release-activated calcium modulator 2 (Orai2), calmodulin 3 (Calm3), and septin 4 (Sept4). No changes in other genes that encode proteins that directly participate in SOCE, such as STIMs, RYRs, IP3Rs, SERCAs, and other Orais, were detected. No changes were found in other genes that encode proteins that indirectly regulate SOCE, such as TRPCs, CALMs, Golli, Post, and TMEM66. These data suggest that different mechanisms contribute to changes in calcium homeostasis in inducible huntingtin-expressing PC12 cells and transgenic YAC128 mice.
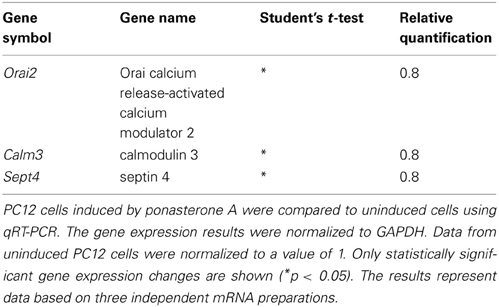
Table 4. Gene expression analysis of induced and uninduced PC12 cells.
Discussion
Several studies have investigated gene expression in HD models and HD patients using microarray analysis and discovered genes whose expression is altered (Luthi-Carter et al., 2000, 2002a,b; Desplats et al., 2006; Hodges et al., 2006). We expanded these studies to the YAC128 transgenic mice because they exhibit a slow, long-term progression of symptoms characteristic of HD (Slow et al., 2003), which may help identify the primary effects of mutant HTT. We focused on genes that encode members of calcium signalosomes (calciosome) because changes in calcium signaling and homeostasis components have been suggested to be early steps in the development of HD (Giacomello et al., 2011, 2013).
We found that ~32% of the analyzed genes in the striatum exhibited statistically significant changes in expression in YAC128 mice compared with control mice (Figure 1A). In the cerebellum and motor cortex, the levels of expression of fewer genes differed between YAC128 mice and wild type mice (Figures 1B,C), which is consistent with changes occurring first in the striatum of patients suffering from HD (Vonsattel et al., 1985). In the striatum, one gene, Aph1b, was positively regulated, even after a Bonferroni correction (p < 0.00052) to correct for multiple testing. Nevertheless, ~13% of genes in the striatum met the arbitrary criteria after cut-offs of relative quantification results (RQ > 1.3 for upregulated genes and RQ < 0.75 for downregulated genes) were applied (Table 3). They were characterized by the same extent of mRNA changes in the striatum in both groups, 3- and 6-month-old HD mice, as compared with control mice. Western blots were performed to check, if the change of specific mRNA level affect the level of a particular protein (Figure 2).
At the top of our list, presenting the highest score for relative quantification in the striatum in transgenic YAC128 mice compared with control mice, is the mRNA coding for huntingtin-associated protein 1 (Table 3). Our results revealed a three-fold increase in the expression of Hap1 in the striatum in YAC128 mice compared with control mice. This is consistent with data obtained using another transgenic mouse model, R6/2. The enrichment of several mRNAs, including Hap1 (although not statistically significant), was observed in neuronal nitric-oxide-synthase-positive interneurons compared with medium spiny projection neurons (Zucker et al., 2005).
Hap1 did not meet the criterion of the Bonferroni correction (p < 0.00052), however, an increase in HAP1 protein was observed in brain extracts from five out of six studied YAC128 mice using western blotting (Figure 2A). In these five samples about 1.8–2.8-fold increase at the protein level of HAP1 was observed, except one sample, which exhibited 0.46-fold decrease of HAP1. Also, this sample showed unusually high level of CIB2 protein as compared to other five samples (Figure 2D). HAP1 is known to play a role in signal transduction, the regulation of vesicular transport, gene transcription, and the regulation of membrane receptor recycling (Wu and Zhou, 2009). It was identified as an HTT-interacting protein in yeast two-hybrid screens (Li et al., 1995). HAP1 protein binds more tightly to HTT with an expanded glutamine repeat than to wildtype HTT, and the binding is enhanced by lengthening the glutamine repeat (Li et al., 1998). A IP3R1-HAP1A-HTT ternary complex was identified in the brain that facilitates IP3R1-mediated intracellular Ca2+ release in MSNs and is activated only by mutant HTT (Tang et al., 2003). Interactions between HTT and the IP3R1 C-terminus depend on both, the presence of HAP1 and polyglutamine expansion. Mutant HTT can bind to the IP3R1 C-terminus either directly or indirectly through HAP1. The functional effects of mutant HTT on IP3R1-mediated Ca2+ release are attenuated in medium spiny striatal neurons in Hap1 knockout mice compared with MSNs in wildtype mice (Tang et al., 2004).
Members of calcium signalosomes, which were significantly upregulated in the striatum in YAC128 mice, were Calb2, CacyBP/SIP, Cib1, Cib2, and Carl. An increase at the protein level in samples from six animals was observed for CacyBP/SIP protein and CIB2 (Figures 2B,D). CALB2 protein increased in four samples (1.5, 2.5, 2.25, and 1.3-fold), and two were characterized by a slightly lower expression of CALB2 in the striatum (0.79 and 0.7-fold). Taking into account the possible fluctuations in neuronal calcium, this result can be explained by the fact that some antibodies to calcium-binding proteins, including CALB2, preferentially recognize particular calcium-induced protein conformations (Winsky and Kuznicki, 1996). CALB2 is a member of the EF-hand family of calcium-binding proteins, which interacts with wildtype HTT but preferentially with mutant HTT (Dong et al., 2012). In neuronal cell models of HD, the overexpression of CALB2 reduced mutant HTT-induced cytotoxicity, whereas knockdown of Calb2 enhanced mutant HTT-induced neuronal cell death (Dong et al., 2012). It was suggested that CALB2 may protect only medium-sized neurons against neurodegeneration in HD (Cicchetti and Parent, 1996). However, neurodegenerative processes that play a role in HD led to a decrease in the density of CALB2 in large striatal interneurons, without causing their death (Massouh et al., 2008). An increase in the expression of Calb2 in the striatum in YAC128 mice may indicate the presence of a defense mechanism against mutant HTT-induced cytotoxicity. However, high-throughput analysis did not identify an increase in Calb2 in HD patients (Hodges et al., 2006). In contrast, a decrease in the expression of Calb1, which encodes another 6 EF-hands calcium binding protein, calbindin, was found in post-mortem brain samples from the caudate of HD patients (Hodges et al., 2006). This is consistent with the substantial loss of neurons in the neostriatum that contain calbindin, suggesting that a failure of calcium buffering may contribute to cell death in HD (Seto-Ohshima et al., 1988).
The expression of CacyBP/SIP was also significantly upregulated in the striatum in YAC128 mice relative to control mice. This result was confirmed by western blotting, which showed an increase in a 30-kDa calcyclin (S100A6)-binding protein in all six studied samples. The CacyBP/SIP protein may play a role in the organization of microtubules and in calcium-dependent ubiquitination of target proteins (Filipek and Kuznicki, 1998; Matsuzawa and Reed, 2001; Filipek et al., 2002, 2008; Jurewicz et al., 2013).
The statistically significant upregulation of Cib1 mRNA in YAC128 mice is consistent with a seven-fold increase in this gene's mRNA in microarray studies of the striatum in HD patients (Hodges et al., 2006). However, at the protein level this cannot be confirmed (Figure 2E), since we observed downregulation of CIB1 in samples from the striatum of five animals (0.15, 0.7, 0.85, 0.78, and 0.5-fold) and an increase in one (1.6-fold). We detected a statistically significant increase in Cib2 mRNA and this result was confirmed by six independent western blotting experiments, showing an increase in CIB2 protein (Figure 2D). CIBs are EF-hand Ca2+ binding proteins identified in the brain (Bernstein et al., 2005; Blazejczyk et al., 2009). CIB1 has been implicated in neurodegenerative processes in AD, in which it was shown to interact with presenilin 2, but not presenilin 1 (Blazejczyk et al., 2006), but the role of CIBs in HD has not yet been confirmed.
Another gene that was significantly upregulated on the mRNA level in the striatum of YAC128 mice compared with control mice was Calr, however, the microarray studies did not detect upregulation of Calr in brain structures in HD patients (Hodges et al., 2006). Calreticulin is a multifunctional protein that acts as a transcription factor in the nucleus or binds Ca2+ in the ER (Michalak et al., 2009).
We detected statistically significant changes in the gene expression of Psen2 mRNA in YAC128 mice and other components of γ-secretase complex, such as Psenen and Aph1 isoforms. Considering that only the Aph1b gene met the criterion for Bonferroni correction, we speculate that this gene may by involved in the pathogenesis of HD. APH1 encoded by the Aph1 gene is required for proteolytic activity and binds to the γ-secretase complex (Lee et al., 2004). Presenilins and Psenen are involved in the cleavage of the Notch receptor and APP processing (Francis et al., 2002; Goutte et al., 2002; Luo et al., 2003). Moreover, PSENs participate also in the modulation of Ca2+ signaling in the ER (Leissring et al., 2000; Supnet and Bezprozvanny, 2011). Thus, one can associate the altered gene expression of γ-secretase components with abnormal proteolytic activity in HD (Tarlac and Storey, 2003). It was suggested that HTT-interacting protein HIP1 (huntingtin-interacting protein 1) may provide a functional link between non-canonical Notch signaling-mediated neurogenesis through a deltex-dependent pathway (Moores et al., 2008).
In our experiments, most of the genes exhibited upregulation, in contrast to the results obtained from Affymetrix oligonucleotide array studies in HD R6/2 mice (Desplats et al., 2006), in which a decrease in the expression level of a majority of the genes was observed. R6/2 mice express exon 1 of human HD gene carrying 116 CAG repeats and exhibit a more progressive neurological phenotype (Mangiarini et al., 1996). However, comparing the results of high-throughput studies with our present study, which focused on a small group of genes, we identified some similarities. We and Desplats et al. (2006) detected a decrease in the expression of the Rgs4 gene that encodes a protein involved in the regulation of G-protein signaling. Similarly, statistically significant downregulation of Rgs4 was found in the striatum and motor cortex in HD patients using Affymetrix GeneChip microarrays (Hodges et al., 2006). The RGS4 protein inhibits Gq, which, in turn, attenuates intracellular calcium signaling via phospholipase C (Berman et al., 1996). This is in line with results suggesting a cytosolic and mitochondrial Ca2+ overload in HD (Bezprozvanny and Hayden, 2004).
Cellular models of HD are another tool to detect the early, direct effects of mutant HTT without the influence of other secondary processes in advanced stages of the disease. We used PC12 cells with inducible expression of mutant HTT. Others have characterized this model in regard to HTT aggregates: 1 day after induction, only a few cells that expressed mutant HTT contained aggregates, whereas 5 days after induction, nearly all of the cells contained visible aggregates (Cong et al., 2005). We analyzed the cells 2 days after induction of HTT expression, which exhibited overexpression of expanded HTT protein (Figure 3A) and unchanged level of actin protein (Figure 3B) as found by immunoblotting. We observed a statistically significant reduction of calcium influx during SOCE, but not calcium content in the ER, in inducible huntingtin-expressing PC12 cells compared with control uninduced cells. This indicates that despite the likely association of mutant HTT with the ER (Rockabrand et al., 2007), the Ca2+ content of PC12 cells is not changed. Reduced activity of the SOCE in induced PC12 cells might be explained by our finding that Orai2, Sept4 and Calm3 are downregulated. The Orai calcium channel is activated during SOCE (Frischauf et al., 2008). Septin 4 has been shown to facilitate interactions between STIM1 and Orai1 proteins (Sharma et al., 2013). Microarray studies of the caudate from HD patients detected a seven-fold decrease in the expression of Calm3 (Hodges et al., 2006). In induced PC12 cells, a decrease in the expression of another isoform of calmodulin, Calm1, has been described (van Roon-Mom et al., 2008). Calmodulins are multifunctional intermediate messenger proteins that transduce calcium signals by binding calcium ions and then modifying their interactions with various target proteins (Chin and Means, 2000). A previous study found that HTT interacts with calmodulin and that the expansion of polyglutamine altered this interaction (Bao et al., 1996).
In contrast to the observed decrease in SOCE in PC12 cells (this work) in cultured MSNs from YAC128 mice, an increase in calcium influx during SOCE was reported by Wu and coworkers. They demonstrated a key role of TRPC1 channels in supporting the SOC pathway in HD neurons (Wu et al., 2011). However, we did not find any differences in Trpc1 gene expression in induced PC12 cells. The differences in SOCE dysregulation observed between the YAC128 mouse model by Wu et al. and our observation in PC12 cells may result from the length of mutated HTT, which can selectively affect gene expression. Alternatively, the difference could be due to the length of time mutated HTT was expressed. MSNs from YAC128 mice used by Wu et al. were cultured from 10–14 days, while PC12 cells were analyzed 2 days after HTT induction.
Although inducible PC12 cells and transgenic YAC128 mice show different changes in calcium influx during SOCE, each of these models has characteristic features of HD, such as the deposition of HTT aggregates mainly in the nuclei of inducible PC12 cells (van Roon-Mom et al., 2008) and nuclei of MSNs in the striatum in YAC128 mice (Van Raamsdonk et al., 2007). In both models, some similarities with the results obtained from samples from HD patients as well as other HD transgenic and cellular models were observed. In our experiments, the expression of Rgs4, Hap1, Cib1, and Calm3 changed in the same direction as the changes reported by other studies.
This work presents for the first time convincing data demonstrating an increased expression of huntingtin-associated protein 1 in the striatum of HD mouse model at the mRNA and protein level. Our data showing upregulation of HAP1 are in line with those of Wu et al. (2011) when the observations of Tang et al. are taken into account (Tang et al., 2003, 2004). They found that mutant Htt via the interaction with HAP1 can activate IP3R1 in vivo. Thus, activation of SOCE reported by Wu et al. (2011) could be explained by the increased release of Ca2+ from ER due to the facilitated opening of the IP3R1. Since the dysregulation of neuronal Ca2+ represents an early event in the pathogenesis of models of HD (Zeron et al., 2002; Tang et al., 2003), the HAP1 protein might be considered as a potential therapeutic target in HD.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was supported by The National Centre for Research and Development in Poland in the framework of the project titled “Pathways of Store-Operated Calcium Entry (SOCE) as a novel therapeutic target in neurodegenerative diseases” (ERA.Net RUS project no NCBR/ERA NET RUS/03/2012). We thank Darragh O'Neill for excellent proofreading of the manuscript.
References
Apostol, B. L., Kazantsev, A., Raffioni, S., Illes, K., Pallos, J., Bodai, L., et al. (2003). A cell-based assay for aggregation inhibitors as therapeutics of polyglutamine-repeat disease and validation in Drosophila. Proc. Natl. Acad. Sci. U.S.A. 100, 5950–5955. doi: 10.1073/pnas.2628045100
Bao, J., Sharp, A. H., Wagster, M. V., Becher, M., Schilling, G., Ross, C. A., et al. (1996). Expansion of polyglutamine repeat in huntingtin leads to abnormal protein interactions involving calmodulin. Proc. Natl. Acad. Sci. U.S.A. 93, 5037–5042. doi: 10.1073/pnas.93.10.5037
Berman, D. M., Kozasa, T., and Gilman, A. G. (1996). The GTPase-activating protein RGS4 stabilizes the transition state for nucleotide hydrolysis. J. Biol. Chem. 271, 27209–27212. doi: 10.1074/jbc.271.44.27209
Bernstein, H. G., Blazejczyk, M., Rudka, T., Gundelfinger, E. D., Dobrowolny, H., Bogerts, B., et al. (2005). The Alzheimer disease-related calcium-binding protein Calmyrin is present in human forebrain with an altered distribution in Alzheimer's as compared to normal ageing brains. Neuropathol. Appl. Neurobiol. 31, 314–324. doi: 10.1111/j.1365-2990.2005.00646.x
Berridge, M. J. (2012). Calcium signalling remodelling and disease. Biochem. Soc. Trans. 40, 297–309. doi: 10.1042/BST20110766
Berridge, M. J., Bootman, M. D., and Roderick, H. L. (2003). Calcium signalling: dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 4, 517–529. doi: 10.1038/nrm1155
Bezprozvanny, I., and Hayden, M. R. (2004). Deranged neuronal calcium signaling and Huntington disease. Biochem. Biophys. Res. Commun. 322, 1310–1317. doi: 10.1016/j.bbrc.2004.08.035
Billing-Marczak, K., and Kuznicki, J. (1999). Calretinin—sensor or buffer—function still unclear. Pol. J. Pharmacol. 51, 173–178.
Blazejczyk, M., Sobczak, A., Debowska, K., Wisniewska, M. B., Kirilenko, A., Pikula, S., et al. (2009). Biochemical characterization and expression analysis of a novel EF-hand Ca2+ binding protein calmyrin2 (Cib2) in brain indicates its function in NMDA receptor mediated Ca2+ signaling. Arch. Biochem. Biophys. 487, 66–78. doi: 10.1016/j.abb.2009.05.002
Blazejczyk, M., Wojda, U., Sobczak, A., Spilker, C., Bernstein, H. G., Gundelfinger, E. D., et al. (2006). Ca2+-independent binding and cellular expression profiles question a significant role of calmyrin in transduction of Ca2+-signals to Alzheimer's disease-related presenilin 2 in forebrain. Biochim. Biophys. Acta 1762, 66–72. doi: 10.1016/j.bbadis.2005.09.006
Chin, D., and Means, A. R. (2000). Calmodulin: a prototypical calcium sensor. Trends Cell Biol. 10, 322–328. doi: 10.1016/S0962-8924(00)01800-6
Cicchetti, F., and Parent, A. (1996). Striatal interneurons in Huntington's disease: selective increase in the density of calretinin-immunoreactive medium-sized neurons. Mov. Disord. 11, 619–626. doi: 10.1002/mds.870110605
Cong, S. Y., Pepers, B. A., Evert, B. O., Rubinsztein, D. C., Roos, R. A., van Ommen, G. J., et al. (2005). Mutant huntingtin represses CBP, but not p300, by binding and protein degradation. Mol. Cell. Neurosci. 30, 560–571. doi: 10.1016/j.mcn.2005.05.003
Desplats, P. A., Kass, K. E., Gilmartin, T., Stanwood, G. D., Woodward, E. L., Head, S. R., et al. (2006). Selective deficits in the expression of striatal-enriched mRNAs in Huntington's disease. J. Neurochem. 96, 743–757. doi: 10.1111/j.1471-4159.2005.03588.x
Dong, G., Gross, K., Qiao, F., Ferguson, J., Callegari, E. A., Rezvani, K., et al. (2012). Calretinin interacts with huntingtin and reduces mutant huntingtin-caused cytotoxicity. J. Neurochem. 123, 437–446. doi: 10.1111/j.1471-4159.2012.07919.x
Filipek, A., Jastrzebska, B., Nowotny, M., and Kuznicki, J. (2002). CacyBP/SIP, a calcyclin and Siah-1-interacting protein, binds EF-hand proteins of the S100 family. J. Biol. Chem. 277, 28848–28852. doi: 10.1074/jbc.M203602200
Filipek, A., and Kuznicki, J. (1998). Molecular cloning and expression of a mouse brain cDNA encoding a novel protein target of calcyclin. J. Neurochem. 70, 1793–1798. doi: 10.1046/j.1471-4159.1998.70051793.x
Filipek, A., Schneider, G., Mietelska, A., Figiel, I., and Niewiadomska, G. (2008). Age-dependent changes in neuronal distribution of CacyBP/SIP: comparison to tubulin and the tau protein. J. Neural Transm. 115, 1257–1264. doi: 10.1007/s00702-008-0062-3
Foskett, J. K., White, C., Cheung, K. H., and Mak, D. O. (2007). Inositol trisphosphate receptor Ca2+ release channels. Physiol. Rev. 87, 593–658. doi: 10.1152/physrev.00035.2006
Francis, R., McGrath, G., Zhang, J., Ruddy, D. A., Sym, M., Apfeld, J., et al. (2002). aph-1 and pen-2 are required for Notch pathway signaling, gamma-secretase cleavage of betaAPP, and presenilin protein accumulation. Dev. Cell 3, 85–97. doi: 10.1016/S1534-5807(02)00189-2
Frischauf, I., Schindl, R., Derler, I., Bergsmann, J., Fahrner, M., and Romanin, C. (2008). The STIM/Orai coupling machinery. Channels (Austin) 2, 261–268. doi: 10.4161/chan.2.4.6705
Giacomello, M., Hudec, R., and Lopreiato, R. (2011). Huntington's disease, calcium, and mitochondria. Biofactors 37, 206–218. doi: 10.1002/biof.162
Giacomello, M., Oliveros, J. C., Naranjo, J. R., and Carafoli, E. (2013). Neuronal Ca(2+) dyshomeostasis in Huntington disease. Prion 7, 76–84. doi: 10.4161/pri.23581
Goutte, C., Tsunozaki, M., Hale, V. A., and Priess, J. R. (2002). APH-1 is a multipass membrane protein essential for the Notch signaling pathway in Caenorhabditis elegans embryos. Proc. Natl. Acad. Sci. U.S.A. 99, 775–779. doi: 10.1073/pnas.022523499
Groenendyk, J., Lynch, J., and Michalak, M. (2004). Calreticulin, Ca2+, and calcineurin - signaling from the endoplasmic reticulum. Mol. Cells 17, 383–389.
Gruszczynska-Biegala, J., and Kuznicki, J. (2013). Native STIM2 and ORAI1 proteins form a calcium-sensitive and thapsigargin-insensitive complex in cortical neurons. J. Neurochem. 126, 727–738. doi: 10.1111/jnc.12320
Gruszczynska-Biegala, J., Pomorski, P., Wisniewska, M. B., and Kuznicki, J. (2011). Differential roles for STIM1 and STIM2 in store-operated calcium entry in rat neurons. PLoS ONE 6:e19285. doi: 10.1371/journal.pone.0019285
Hamilton, S. L. (2005). Ryanodine receptors. Cell Calcium 38, 253–260. doi: 10.1016/j.ceca.2005.06.037
Henke, N., Albrecht, P., Bouchachia, I., Ryazantseva, M., Knoll, K., Lewerenz, J., et al. (2013). The plasma membrane channel ORAI1 mediates detrimental calcium influx caused by endogenous oxidative stress. Cell Death Dis. 4:e470. doi: 10.1038/cddis.2012.216
Henke, N., Albrecht, P., Pfeiffer, A., Toutzaris, D., Zanger, K., and Methner, A. (2012). Stromal interaction molecule 1 (STIM1) is involved in the regulation of mitochondrial shape and bioenergetics and plays a role in oxidative stress. J. Biol. Chem. 287, 42042–42052. doi: 10.1074/jbc.M112.417212
Hodges, A., Strand, A. D., Aragaki, A. K., Kuhn, A., Sengstag, T., Hughes, G., et al. (2006). Regional and cellular gene expression changes in human Huntington's disease brain. Hum. Mol. Genet. 15, 965–977. doi: 10.1093/hmg/ddl013
Huang, C., Hepler, J. R., Gilman, A. G., and Mumby, S. M. (1997). Attenuation of Gi- and Gq-mediated signaling by expression of RGS4 or GAIP in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 94, 6159–6163. doi: 10.1073/pnas.94.12.6159
Huang, G. N., Zeng, W., Kim, J. Y., Yuan, J. P., Han, L., Muallem, S., et al. (2006). STIM1 carboxyl-terminus activates native SOC, I(crac) and TRPC1 channels. Nat. Cell Biol. 8, 1003–1010. doi: 10.1038/ncb1454
Jaworska, A., Dzbek, J., Styczynska, M., and Kuznicki, J. (2013). Analysis of calcium homeostasis in fresh lymphocytes from patients with sporadic Alzheimer's disease or mild cognitive impairment. Biochim. Biophys. Acta 1833, 1692–1699. doi: 10.1016/j.bbamcr.2013.01.012
Jurewicz, E., Ostrowska, Z., Jozwiak, J., Redowicz, M. J., Lesniak, W., Moraczewska, J., et al. (2013). CacyBP/SIP as a novel modulator of the thin filament. Biochim. Biophys. Acta 1833, 761–766. doi: 10.1016/j.bbamcr.2012.12.010
Klejman, M. E., Gruszczynska-Biegala, J., Skibinska-Kijek, A., Wisniewska, M. B., Misztal, K., Blazejczyk, M., et al. (2009). Expression of STIM1 in brain and puncta-like co-localization of STIM1 and ORAI1 upon depletion of Ca(2+) store in neurons. Neurochem. Int. 54, 49–55. doi: 10.1016/j.neuint.2008.10.005
Lee, S. F., Shah, S., Yu, C., Wigley, W. C., Li, H., Lim, M., et al. (2004). A conserved GXXXG motif in APH-1 is critical for assembly and activity of the gamma-secretase complex. J. Biol. Chem. 279, 4144–4152. doi: 10.1074/jbc.M309745200
Leissring, M. A., Akbari, Y., Fanger, C. M., Cahalan, M. D., Mattson, M. P., and LaFerla, F. M. (2000). Capacitative calcium entry deficits and elevated luminal calcium content in mutant presenilin-1 knockin mice. J. Cell Biol. 149, 793–798. doi: 10.1083/jcb.149.4.793
Li, S. H., Hosseini, S. H., Gutekunst, C. A., Hersch, S. M., Ferrante, R. J., and Li, X. J. (1998). A human HAP1 homologue. Cloning, expression, and interaction with huntingtin. J. Biol. Chem. 273, 19220–19227. doi: 10.1074/jbc.273.30.19220
Li, S. H., and Li, X. J. (2004). Huntingtin-protein interactions and the pathogenesis of Huntington's disease. Trends Genet. 20, 146–154. doi: 10.1016/j.tig.2004.01.008
Li, X. J., Li, S. H., Sharp, A. H., Nucifora, F. C. Jr., Schilling, G., et al. (1995). A huntingtin-associated protein enriched in brain with implications for pathology. Nature 378, 398–402. doi: 10.1038/378398a0
Liou, J., Kim, M. L., Heo, W. D., Jones, J. T., Myers, J. W., Ferrell, J. E., et al. (2005). STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr. Biol. 15, 1235–1241. doi: 10.1016/j.cub.2005.05.055
Luo, W. J., Wang, H., Li, H., Kim, B. S., Shah, S., Lee, H. J., et al. (2003). PEN-2 and APH-1 coordinately regulate proteolytic processing of presenilin 1. J. Biol. Chem. 278, 7850–7854. doi: 10.1074/jbc.C200648200
Luthi-Carter, R., Hanson, S. A., Strand, A. D., Bergstrom, D. A., Chun, W., Peters, N. L., et al. (2002a). Dysregulation of gene expression in the R6/2 model of polyglutamine disease: parallel changes in muscle and brain. Hum. Mol. Genet. 11, 1911–1926. doi: 10.1093/hmg/11.17.1911
Luthi-Carter, R., Strand, A. D., Hanson, S. A., Kooperberg, C., Schilling, G., La Spada, A. R., et al. (2002b). Polyglutamine and transcription: gene expression changes shared by DRPLA and Huntington's disease mouse models reveal context-independent effects. Hum. Mol. Genet. 11, 1927–1937. doi: 10.1093/hmg/11.17.1927
Luthi-Carter, R., Strand, A., Peters, N. L., Solano, S. M., Hollingsworth, Z. R., Menon, A. S., et al. (2000). Decreased expression of striatal signaling genes in a mouse model of Huntington's disease. Hum. Mol. Genet. 9, 1259–1271. doi: 10.1093/hmg/9.9.1259
Mangiarini, L., Sathasivam, K., Seller, M., Cozens, B., Harper, A., Hetherington, C., et al. (1996). Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 87, 493–506. doi: 10.1016/S0092-8674(00)81369-0
Massouh, M., Wallman, M. J., Pourcher, E., and Parent, A. (2008). The fate of the large striatal interneurons expressing calretinin in Huntington's disease. Neurosci. Res. 62, 216–224. doi: 10.1016/j.neures.2008.08.007
Matsuzawa, S. I., and Reed, J. C. (2001). Siah-1, SIP, and Ebi collaborate in a novel pathway for beta-catenin degradation linked to p53 responses. Mol. Cell 7, 915–926. doi: 10.1016/S1097-2765(01)00242-8
Michalak, M., Groenendyk, J., Szabo, E., Gold, L. I., and Opas, M. (2009). Calreticulin, a multi-process calcium-buffering chaperone of the endoplasmic reticulum. Biochem. J. 417, 651–666. doi: 10.1042/BJ20081847
Moores, J. N., Roy, S., Nicholson, D. W., and Staveley, B. E. (2008). Huntingtin interacting protein 1 can regulate neurogenesis in Drosophila. Eur. J. Neurosci. 28, 599–609. doi: 10.1111/j.1460-9568.2008.06359.x
Ng, A. N., Krogh, M., and Toresson, H. (2011). Dendritic EGFP-STIM1 activation after type I metabotropic glutamate and muscarinic acetylcholine receptor stimulation in hippocampal neuron. J. Neurosci. Res. 89, 1235–1244. doi: 10.1002/jnr.22648
Nikoletopoulou, V., and Tavernarakis, N. (2012). Calcium homeostasis in aging neurons. Front. Genet. 3:200. doi: 10.3389/fgene.2012.00200
Noble, W. S. (2009). How does multiple testing correction work? Nat. Biotechnol. 27, 1135–1137. doi: 10.1038/nbt1209-1135
Park, C. Y., Shcheglovitov, A., and Dolmetsch, R. (2010). The CRAC channel activator STIM1 binds and inhibits L-type voltage-gated calcium channels. Science 330, 101–105. doi: 10.1126/science.1191027
Paxinos, G., and Franklin, K. B. J. (2004). The Mouse Brain in Stereotaxic Coordinates. San Diego, CA: Academic Press.
Puzianowska-Kuznicka, M., and Kuznicki, J. (2009). The ER and ageing II: calcium homeostasis. Ageing Res. Rev. 8, 160–172. doi: 10.1016/j.arr.2009.05.002
Ribeiro, F. M., Paquet, M., Ferreira, L. T., Cregan, T., Swan, P., Cregan, S. P., et al. (2010). Metabotropic glutamate receptor-mediated cell signaling pathways are altered in a mouse model of Huntington's disease. J. Neurosci. 30, 316–324. doi: 10.1523/JNEUROSCI.4974-09.2010
Rockabrand, E., Slepko, N., Pantalone, A., Nukala, V. N., Kazantsev, A., Marsh, J. L., et al. (2007). The first 17 amino acids of Huntingtin modulate its sub-cellular localization, aggregation and effects on calcium homeostasis. Hum. Mol. Genet. 16, 61–77. doi: 10.1093/hmg/ddl440
Roos, J., DiGregorio, P. J., Yeromin, A. V., Ohlsen, K., Lioudyno, M., Zhang, S., et al. (2005). STIM1, an essential and conserved component of store-operated Ca2+ channel function. J. Cell Biol. 169, 435–445. doi: 10.1083/jcb.200502019
Ryazantseva, M., Skobeleva, K., and Kaznacheyeva, E. (2013). Familial Alzheimer's disease-linked presenilin-1 mutation M146V affects store-operated calcium entry: does gain look like loss? Biochimie 95, 1506–1509. doi: 10.1016/j.biochi.2013.04.009
Seto-Ohshima, A., Emson, P. C., Lawson, E., Mountjoy, C. Q., and Carrasco, L. H. (1988). Loss of matrix calcium-binding protein-containing neurons in Huntington's disease. Lancet 1, 1252–1255. doi: 10.1016/S0140-6736(88)92073-9
Sharma, S., Quintana, A., Findlay, G. M., Mettlen, M., Baust, B., Jain, M., et al. (2013). An siRNA screen for NFAT activation identifies septins as coordinators of store-operated Ca2+ entry. Nature 499, 238–242. doi: 10.1038/nature12229
Skibinska-Kijek, A., Wisniewska, M. B., Gruszczynska-Biegala, J., Methner, A., and Kuznicki, J. (2009). Immunolocalization of STIM1 in the mouse brain. Acta Neurobiol. Exp. (Wars) 69, 413–428.
Slow, E. J., van Raamsdonk, J., Rogers, D., Coleman, S. H., Graham, R. K., Deng, Y., et al. (2003). Selective striatal neuronal loss in a YAC128 mouse model of Huntington disease. Hum. Mol. Genet. 12, 1555–1567. doi: 10.1093/hmg/ddg169
Steinbeck, J. A., Henke, N., Opatz, J., Gruszczynska-Biegala, J., Schneider, L., Theiss, S., et al. (2011). Store-operated calcium entry modulates neuronal network activity in a model of chronic epilepsy. Exp. Neurol. 232, 185–194. doi: 10.1016/j.expneurol.2011.08.022
Sugars, K. L., and Rubinsztein, D. C. (2003). Transcriptional abnormalities in Huntington disease. Trends Genet. 19, 233–238. doi: 10.1016/S0168-9525(03)00074-X
Supnet, C., and Bezprozvanny, I. (2011). Presenilins function in ER calcium leak and Alzheimer's disease pathogenesis. Cell Calcium 50, 303–309. doi: 10.1016/j.ceca.2011.05.013
Tang, T. S., Slow, E., Lupu, V., Stavrovskaya, I. G., Sugimori, M., Llinas, R., et al. (2005). Disturbed Ca2+ signaling and apoptosis of medium spiny neurons in Huntington's disease. Proc. Natl. Acad. Sci. U.S.A. 102, 2602–2607. doi: 10.1073/pnas.0409402102
Tang, T. S., Tu, H., Chan, E. Y., Maximov, A., Wang, Z., Wellington, C. L., et al. (2003). Huntingtin and huntingtin-associated protein 1 influence neuronal calcium signaling mediated by inositol-(1,4,5) triphosphate receptor type 1. Neuron 39, 227–239. doi: 10.1016/S0896-6273(03)00366-0
Tang, T. S., Tu, H., Orban, P. C., Chan, E. Y., Hayden, M. R., and Bezprozvanny, I. (2004). HAP1 facilitates effects of mutant huntingtin on inositol 1,4,5-trisphosphate-induced Ca release in primary culture of striatal medium spiny neurons. Eur. J. Neurosci. 20, 1779–1787. doi: 10.1111/j.1460-9568.2004.03633.x
Tarlac, V., and Storey, E. (2003). Role of proteolysis in polyglutamine disorders. J. Neurosci. Res. 74, 406–416. doi: 10.1002/jnr.10746
Toescu, E. C., and Verkhratsky, A. (2007). The importance of being subtle: small changes in calcium homeostasis control cognitive decline in normal aging. Aging Cell 6, 267–273. doi: 10.1111/j.1474-9726.2007.00296.x
Van Raamsdonk, J. M., Warby, S. C., and Hayden, M. R. (2007). Selective degeneration in YAC mouse models of Huntington disease. Brain Res. Bull. 72, 124–131. doi: 10.1016/j.brainresbull.2006.10.018
van Roon-Mom, W. M., Pepers, B. A., t Hoen, P. A., Verwijmeren, C. A., den Dunnen, J. T., Dorsman, J. C., et al. (2008). Mutant huntingtin activates Nrf2-responsive genes and impairs dopamine synthesis in a PC12 model of Huntington's disease. BMC Mol. Biol. 9:84. doi: 10.1186/1471-2199-9-84
Vig, M., Peinelt, C., Beck, A., Koomoa, D. L., Rabah, D., Koblan-Huberson, M., et al. (2006). CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science 312, 1220–1223. doi: 10.1126/science.1127883
Vonsattel, J. P., Myers, R. H., Stevens, T. J., Ferrante, R. J., Bird, E. D., and Richardson, E. P. Jr. (1985). Neuropathological classification of Huntington's disease. J. Neuropathol. Exp. Neurol. 44, 559–577. doi: 10.1097/00005072-198511000-00003
Winsky, L., and Kuznicki, J. (1996). Antibody recognition of calcium-binding proteins depends on their calcium-binding status. J. Neurochem. 66, 764–771. doi: 10.1046/j.1471-4159.1996.66020764.x
Wojda, U., Salinska, E., and Kuznicki, J. (2008). Calcium ions in neuronal degeneration. IUBMB Life 60, 575–590. doi: 10.1002/iub.91
Wu, J., Shih, H. P., Vigont, V., Hrdlicka, L., Diggins, L., Singh, C., et al. (2011). Neuronal store-operated calcium entry pathway as a novel therapeutic target for Huntington's disease treatment. Chem. Biol. 18, 777–793. doi: 10.1016/j.chembiol.2011.04.012
Wu, L. L., and Zhou, X. F. (2009). Huntingtin associated protein 1 and its functions. Cell Adh. Migr. 3, 71–76. doi: 10.4161/cam.3.1.7511
Zeron, M. M., Hansson, O., Chen, N., Wellington, C. L., Leavitt, B. R., Brundin, P., et al. (2002). Increased sensitivity to N-methyl-D-aspartate receptor-mediated excitotoxicity in a mouse model of Huntington's disease. Neuron 33, 849–860. doi: 10.1016/S0896-6273(02)00615-3
Zucker, B., Luthi-Carter, R., Kama, J. A., Dunah, A. W., Stern, E. A., Fox, J. H., et al. (2005). Transcriptional dysregulation in striatal projection- and interneurons in a mouse model of Huntington's disease: neuronal selectivity and potential neuroprotective role of HAP1. Hum. Mol. Genet. 14, 179–189. doi: 10.1093/hmg/ddi014
Keywords: calcium signalosome, store-operated calcium entry, transgenic mice, TaqMan low-density arrays, Huntington's disease, huntingtin, huntingtin-associated protein 1, calcyclin-binding protein
Citation: Czeredys M, Gruszczynska-Biegala J, Schacht T, Methner A and Kuznicki J (2013) Expression of genes encoding the calcium signalosome in cellular and transgenic models of Huntington's disease. Front. Mol. Neurosci. 6:42. doi: 10.3389/fnmol.2013.00042
Received: 18 July 2013; Accepted: 05 November 2013;
Published online: 25 November 2013.
Edited by:
Gaiti Hasan, National Centre for Biological Sciences, IndiaReviewed by:
Nancy A. Muma, University of Kansas, USAIlya Bezprozvanny, UT Southwestern Medical Center at Dallas, USA
Copyright © 2013 Czeredys, Gruszczynska-Biegala, Schacht, Methner and Kuznicki. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jacek Kuznicki, Laboratory of Neurodegeneration, International Institute of Molecular and Cell Biology, Trojdena 4, 02-109 Warsaw, Poland e-mail:amt1em5pY2tpQGlpbWNiLmdvdi5wbA==