 Ilse I. G. M. van de Vondervoort1,2*†
Ilse I. G. M. van de Vondervoort1,2*† Peter M. Gordebeke2,3†
Peter M. Gordebeke2,3† Nima Khoshab3
Nima Khoshab3 Paul H. E. Tiesinga2,3
Paul H. E. Tiesinga2,3 Jan K. Buitelaar1
Jan K. Buitelaar1 Tamas Kozicz2,4
Tamas Kozicz2,4 Armaz Aschrafi2,3 †
Armaz Aschrafi2,3 † Jeffrey C. Glennon1,2†
Jeffrey C. Glennon1,2†- 1Department of Cognitive Neuroscience, RadboudUMC, Nijmegen, Netherlands
- 2Centre for Neuroscience, Donders Institute for Brain, Cognition, and Behavior, Nijmegen, Netherlands
- 3Department of Neuroinformatics, Radboud University, Nijmegen, Netherlands
- 4Department of Anatomy, Radboud University, Nijmegen, Netherlands
Recent studies have emphasized an important role for long non-coding RNAs (lncRNA) in epigenetic regulation, development, and disease. Despite growing interest in lncRNAs, the mechanisms by which lncRNAs control cellular processes are still elusive. Improved understanding of these mechanisms is critical, because the majority of the mammalian genome is transcribed, in most cases resulting in non-coding RNA products. Recent studies have suggested the involvement of lncRNA in neurobehavioral and neurodevelopmental disorders, highlighting the functional importance of this subclass of brain-enriched RNAs. Impaired expression of lnRNAs has been implicated in several forms of intellectual disability disorders. However, the role of this family of RNAs in cognitive function is largely unknown. Here we provide an overview of recently identified mechanisms of neuronal development involving lncRNAs, and the consequences of lncRNA deregulation for neurodevelopmental disorders.
Introduction
Therapeutic strategies for the amelioration of neurobehavioral dysfunction in neurodevelopmental disorders such as intellectual disabilities (ID), or autism spectrum disorders (ASD) are often insufficient for a large patient population. These disorders have complex behavioral and cognitive phenotypes that are thought to develop through disturbances in neural circuitry and synaptic function. Moreover, genetic epidemiology and population genetic studies suggested that a spectrum of allelic risk underlies complex traits like ID (Geschwind, 2008). However, the existence of risk alleles rarely confers diagnostic specificity (Hitzemann et al., 2013). One possible explanation for this may involve dysregulation of the rate of gene transcription/translation by small or long non-coding (nc)RNAs, leading to abnormal expression of ID-risk genes of phenotypic relevance (Olde Loohuis et al., 2012). Several studies have now indicated altered levels of brain-specific small and long ncRNA in ID and other neurodevelopmental disorders (Willemsen et al., 2011). lncRNAs constitute a large fraction of the total ncRNA pool, each exceeding 200 nucleotides in length. It was initially assumed that lncRNAs merely act as primary or precursor transcripts for the production of short ncRNAs (sncRNAs) such as microRNAs (miRNAs) or small nucleolar RNAs (snoRNAs; Aschrafi et al., 2008). Conversely to snoRNAs genes, however, the evolutionary conservation of lncRNAs often extends beyond the overlapping sncRNA segments (Wang et al., 2004). They have been shown to either act solely, or together with proteins, exerting a wide range of cellular roles, e.g., their regulation of transcription and RNA processing (Wang et al., 2008). The purpose of this review is to emphasize the role of lncRNAs in regulating neuronal molecular pathways, and to highlight their putative role in dysregulation of these mechanisms in neurodevelopmental disorders.
Mechanisms of Action of lncRNAs
LncRNA Transcription Modulates the Expression of other Genes
Transcription of lncRNAs from alternative transcription start sites in the vicinity of other genes may interfere with the transcription efficiency of that gene (Martens et al., 2005; Martianov et al., 2007). These transcriptional interference mechanisms have been shown to regulate key developmental pathways, such as those involving Hox-genes expression (Wang et al., 2011). A complete overview of potential regulatory mechanisms of lncRNAs is provided in (Guttman and Rinn, 2012) or (Ponting et al., 2009). A schematic overview of lncRNAs cellular function is depicted in Figure 1.
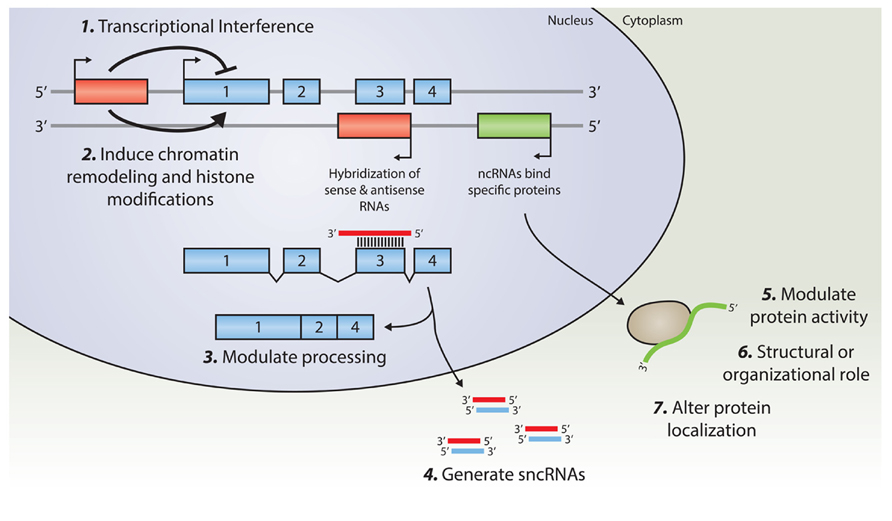
FIGURE 1. An overview of known regulatory mechanisms for lncRNA. Transcription from an upstream promoter can negatively or positively affect the expression of a downstream gene via (1) transcriptional interference mediated by inhibition of RNA Polymerase 2 recruitment, or by (2) inducing chromatin remodeling and histone modification. Alternatively, an antisense transcript is able to hybridize to the overlapping sense transcript and modulate further processing (3), or provide a substrate for Dicer, or other nucleases, in order to generate various small non-coding RNAs (4). By binding to specific protein partners, a long noncoding transcript may modulate the activity of that particular protein (5), serve as a structural component that allows the formation of a larger RNA–protein complex (6), or alter the cellular localization of the protein (7).
LncRNAs may Regulate RNA-Processing and Protein Activity
Initial research suggested that the functions of lncRNAs relate to their interactions with the RNA-binding proteins (RBPs), a protein family highly abundant in the brain (Smart et al., 2007). Due to the long sequence and structural characteristics of lncRNAs, along with various RBPs and RNA-binding domains, numerous combinations of lncRNA/RNA-binding proteins can be formed. This allows the recruitment of various protein-complexes and a multitude of “downstream” functions. Previous studies suggested that lncRNAs, in concert with RBPs and different protein-complexes, have the capacity to induce chromatin remodeling and histone modification, as well as modulating alternative splicing and protein-activity. For example, Alu RNA and B2 RNA may directly affect RNA polymerase II activity. Both are transcribed from Short Interspersed Nuclear Elements (SINEs; Yakovchuk et al., 2009). Alu RNA and B2 RNA block binding of RNA polymerase II to the promoter and change the conformation of the transcription initiation complex significantly.
Chromatin Remodeling and Histone Modification can be Induced by lncRNAs
LncRNAs are capable of mediating the activity of proteins involved in chromatin remodeling and histone modification, including those at the Polycomb Repressive Complex 2 (PRC2) complex (Khalil et al., 2009; Tsai et al., 2010) and the CBP/p300 complex (Wang et al., 2008). A genome-wide study revealed that approximately one third of conserved intergenic lncRNAs associates with either the PRC2 complex or the CoREST/REST or SCMX proteins, all known chromatin-modifying proteins (Khalil et al., 2009). A prominent epigenetic mechanism exerted by lncRNAs is the X-chromosome inactivation. The extent of this control is unique among the chromosomes and is disrupted in X-linked IDs. X-chromosome inactivation is mediated via the lncRNA Xist that binds to one of the X-chromosomes (Zhao et al., 2008). RepA was found to be both part of the Xist lncRNA, as well as expressed by itself (Zhao et al., 2008). The RepA lncRNA is able to bind the histone methyltransferase Enhancer of Zester Homolog 2 (Ezh2), which is a subunit of the PRC2. The recruitment of the PRC2 complex by Xist, via the RepA sequence, allows trimethylation on lysine-27 of H3 histones (H3K27), effectively repressing gene expression, and inactivating the X-chromosome (Zhao et al., 2008). Very Recently, Xist was found to function in a two-step mechanism, though targeting of gene-rich islands before gene-poor domains (Simon et al., 2013).
Functional Roles of LncRNAs in Nervous System Development and Function
Multiple lines of evidence suggest that dysregulated processes as seen in neurodevelopmental disorders are based on mechanisms that are under tight regulation by lncRNAs (see below). A number of ncRNAs were found to be specifically expressed within the hippocampus (Mercer et al., 2008b), a region involved in processing and consolidation of memories. Several lncRNAs originate from genomic regions associated with protein-coding genes involved in memory formation and maintenance, such as an lncRNA transcribed antisense to Camkk1, which is involved in male-specific memory formation (Mercer et al., 2008a).
During brain development, differentiation of neural stem cells and progenitors is crucial. Recently, various lncRNAs have been linked to these events, implying a key role for lncRNAs not only during development, but also in several neuropathologies (reviewed by e.g., Qureshi et al., 2010). For example, a subset of lncRNAs are specifically associated with genes from the Dlx-family, known to be involved in brain development in mammals and Drosophila. Two of the differentially expressed lncRNAs, Evx1as and Hox5b/6as were shown to be associated with trimethylated H3K4 histones and histone methyltransferases (Dinger et al., 2008). In addition, embryonic ventral forebrain-2 (Evf2) is transcribed from the Dlx5/Dlx6 locus, antisense to the Dlx6 gene (Feng et al., 2006). Dlx6 is a homeobox-containing transcription factor important in forebrain neurogenesis (Stenman et al., 2003). Furthermore, 659 evolutionary conserved murine lncRNAs have been identified of which the brain-specific lncRNAs are preferentially (2 to 3-fold increase) located adjacent to brain-expressed protein-coding genes, involved in transcriptional regulation, or in nervous system development (Ponjavic et al., 2009).
Recent studies identified 945 lncRNAs, of which 174 were differentially expressed in the mouse embryoid bodies; and that are annotated to developmentally important events relating to stem cell pluripotency (Dinger et al., 2008). One of these RNAs, Sox2OT (Sox2 Overlapping Transcript) is a highly conserved lncRNA that overlaps the Sox2 gene (Fantes et al., 2003). Sox2 is a transcription-factor critical in maintaining self-renewal properties of neural stem cells (Mizuseki et al., 1998). Similar to Sox2, Sox2OT is present in neural stem cells and is downregulated during differentiation (Amaral et al., 2009).
During fate-specification from neuronal oligodendrocyte bipotent progenitors into GABAergic interneurons, 56 lncRNAs were found to be upregulated, including Gtl2, Rian, Evf2 and Copg2as, but also the novel AK044422 (Mercer et al., 2010). Interestingly, AK044422 overlaps with miR-124a, a highly conserved and highly expressed brain-specific miRNA previously implicated in regulating neuronal specification and differentiation (Makeyev et al., 2007; Visvanathan et al., 2007). Synaptogenesis is a pivotal process during neuronal development, which is altered in various neurodevelopmental disorders (reviewed by e.g., (Zoghbi, 2003; Ecker et al., 2013)). Metastasis-associated lung adenocarcinoma transcript 1 (Malat1) is an lncRNA that is enriched in nuclear speckles (Hutchinson et al., 2007; Clemson et al., 2009). There, it co-localizes with splicing factors to controls the expression of genes involved in synapse function and synaptogenesis (Bernard et al., 2010).
LncRNAs are involved in Neurodevelopmental Disorders
Several lncRNAs are either differentially expressed in or associated with neurodevelopmental disorders, such as Prader–Willi syndrome (PWS), Angelman syndrome (AS), ID, and ASD (Table 1). The role of lncRNAs is possibly best understood in genomic imprinting disorders such as PWS (Wevrick and Francke, 1997; Jong et al., 1999) and AS (Runte et al., 2004), both of which feature learning difficulties but otherwise have different symptoms (further discussed below).
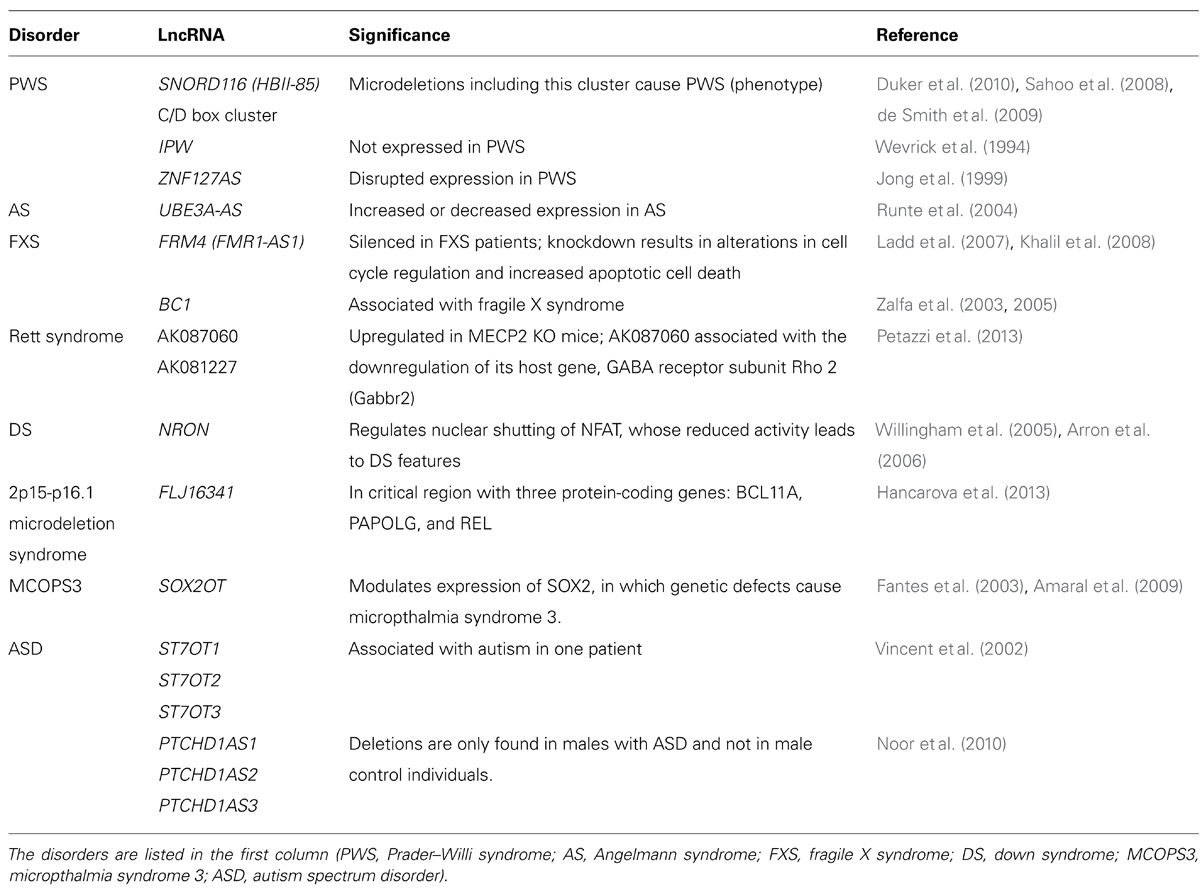
TABLE 1. An overview of the lncRNAs identified in neurodevelopmental disorders.
Imprinting Disorders
Genomic imprinting is mediated by various processes such as DNA methylation and histone modification, but also by ncRNAs (Bartolomei, 2009). PWS (MIM 176270) is characterized by infantile hypotonia, early childhood obesity, short stature, hypogenitalism/hypogonadism, ID, and other behavioral problems including temper tantrums. The genetic cause of the disorder lies in a disruption of the paternal chromosome 15q11.2q13, since the maternal chromosome is inactive through imprinting (Horsthemke and Wagstaff, 2008). To date, two genes have functionally been associated with the pathology of the disorder: NECDIN and small nuclear ribonucleoprotein polypeptide N (SNRPN). Necdin deficient mice show a subset of the multiple clinical manifestations of PWS (Muscatelli et al., 2000). SNRPN encodes the SmN splicing factor, the SNRPN upstream reading frame (SNURF) and partially overlaps the UBE3A gene. Importantly, the downstream introns of SNRPN contain C/D box-containing SNORD116 (HBII-85) snoRNA clusters whose expression is under control of the SNRPN promoter (Runte et al., 2001). Several case reports indicated that paternally inherited microdeletions of this cluster cause PWS (Sahoo et al., 2008; de Smith et al., 2009; Duker et al., 2010). Moreover, two mouse models with targeted deletions in the MBII-85 snoRNA cluster exhibited a similar phenotype as other PWS models, which included decreased activity, hypotonia at birth, and postnatal growth retardation (Skryabin et al., 2007; Ding et al., 2008).
IPW (Imprinted gene in the PWS region) is located in the proximal chromosome 15q, merely 150 kb distal to SNRPN and is not expressed in patients with 15q11-q13 deletions (Wevrick et al., 1994). Additionally, ZNF127 is located in the same region and has been reported to have a disrupted expression in PWS. This gene has a potentially non-coding antisense gene, ZNF127AS, which might be regulating the imprinting of ZNF127 gene (Jong et al., 1999).
Angelman syndrome (MIM 105830) is caused by a disruption of the maternal allele of chromosome 15q11-q13, covering the same genomic location as PWS. However, the symptoms are different and include intellectual disability, movement or balance disorder, typical abnormal behaviors, and severe limitations in speech and language. The genetic underpinning of the disorder is thought to be a disruption in the UBE3A gene (Matsuura et al., 1997). The UBE3A-AS gene is transcribed antisense to the UBE3A gene and repression of UBE3A is dependent on UBE3A-AS (Chamberlain and Brannan, 2001; Johnstone et al., 2006). However, another study suggests that silencing of the paternal UBE3A can also occur when UBE3A-AS is not present, indicating that the regulation is more complex (Le Meur et al., 2005).
Intellectual Disability
Despite the highly variable genetic etiology in ID, only a limited number of molecular and cellular pathways appear to be affected by the magnitude of different gene mutations. ID genes have been shown to cluster in pathways underlying neurogenesis, neural migration, neuronal outgrowth, and synaptic function (van Bokhoven, 2011). Numerous studies have suggested that synaptogenesis and normal synaptic function is dependent on the activity of a large number of proteins, and that disturbance of individual components within the network, or alterations of their activities causes synaptic dysfunction, phenotypically culminating in ID (Aschrafi et al., 2005). Regulation of gene transcripts by small and large ncRNAs may underlie epigenetic control of synaptic activity in ID and other neurodevelopmental disorders. Previous studies have indicated that disruption of lncRNA expression and signaling impairs synaptic plasticity, and results in severe cognitive impairment in mice, and human, which are detailed below.
Fragile X Syndrome
Fragile X syndrome (FXS, MIM 300624) is inherited via an X-linked dominant pattern and characterized by moderate to severe mental retardation, macro-orchidism, and distinct facial features. The disorder is caused by an unstable expansion of a CGG repeat in the FMR1 gene leading to silencing of the gene by methylation of repeat and promoter (Sutcliffe et al., 1992), resulting in decreased FMRP protein levels in the brain (Devys et al., 1993). Accumulating evidence suggests that the etiology of the disorder is influenced by lncRNAs. The promoter of FMR1 is bidirectional and can also give rise to the lncRNA FMR4 or FMR1-AS1, a gene transcribed in the antisense orientation and overlaps the CGG repeat region. FMR4 is similar to FRM1 in being silenced in FXS patients and upregulated in permutation carriers (Ladd et al., 2007; Khalil et al., 2008). Following siRNA knockdown of FMR4, alterations in cell cycle and apoptosis were reported. Conversely, overexpression of FMR4 resulted in increased cell proliferation. Additionally, knockdown of FMR4 did not influence FMR1 expression and vice versa, suggesting an independent mechanism from FMR1 (Khalil et al., 2008). Together, these findings points toward a contribution of FMR4 in the pathology of FXS.
Recently, Pastori et al. (2013) discovered two new transcripts in the FMR1 gene locus: FMR5 and FMR6. FMR5 was similarly expressed in brain regions from unaffected and permutation individuals and full mutation patients, whereas FMR6 was silenced in full mutation and permutation carriers. According to the authors, this might suggest an abnormal transcription or chromatin remodeling prior to transition to the full mutation. In addition to the finding that both FMR5 and FMR6 are expressed in blood leukocytes, these lncRNAs are potentially useful as biomarkers in FXS.
FMRP, the protein that is encoded by FMR1, acts as a translational repressor of specific mRNAs at the synapse and associates with the dendritic RNA BC1 (Zalfa et al., 2003). BC1 enables the interaction of FMRP with the target mRNAs; and FMRP can directly bind to BC1 and its human analog BC200 via its N-terminus. Of note, the 5′ stem loop of BC1 is involved in FMRP recognition and this region is complementary to FMRP target mRNAs (Zalfa et al., 2005). Taken together, the studies suggested that BC1 is a ncRNA that is essential for the repression of mRNAs via FMRP and loss of this repression in FXS patients could result in synaptic dysfunction. It should be noted that, In Iacoangeli et al. (2008), five independent groups reported that results published by Zalfa et al. (2003) are not reproducible. Thus, there is no confirmation, independent of the Bagni group, of a specific physical link between FMRP and BC1 RNA.
Rett syndrome
Rett syndrome (MIM 312750) is characterized by arrested development between 6 and 18 months of age in females, regression of acquired skills, loss of speech, stereotypical movements, seizures, and ID. Mutations in the MECP2, which binds methylated CpGs and can both activate and repress transcription, were first described to be the cause of the disorder (Amir et al., 1999). While assessing the transcriptome of male Mecp2 hemizygous knockout mouse brains (Petazzi et al., 2013), it was revealed that the lncRNAs AK081227 and AK087060 were both significantly upregulated as compared to wild-type littermates. Importantly, overexpression of AK08127 was associated with the downregulation of its host coding protein gene, the gamma-aminobutyric acid receptor subunit Rho 2. This suggest that transcriptional dysregulation of lncRNAs may have the capacity to contribute to the etiology of Rett syndrome.
Down syndrome
Down syndrome (DS) or Trisomy 21 (MIM 190685) is characterized by ID, distinct facial characteristics and congenital heart defects. The lncRNA NRON may be involved in DS, since NRON modulates cytoplasmic-to-nuclear transport of NFAT (Willingham et al., 2005). Decreased nuclear NFAT activity leads to DS-like characteristics in animal models, suggesting a possible role for NRON in DS (Arron et al., 2006). Recently, an inducible XIST was introduced on chromosome 21 using genome editing (Jiang et al., 2013). This approach created a model to investigate genomic expression changes and cellular pathologies of trisomy 21. Notably, deficits in proliferation and neural rosette formation are rapidly reversed upon silencing one chromosome 21, representing a major step toward potential development of “chromosome therapy” (see Figure 2 for a proposed approach).
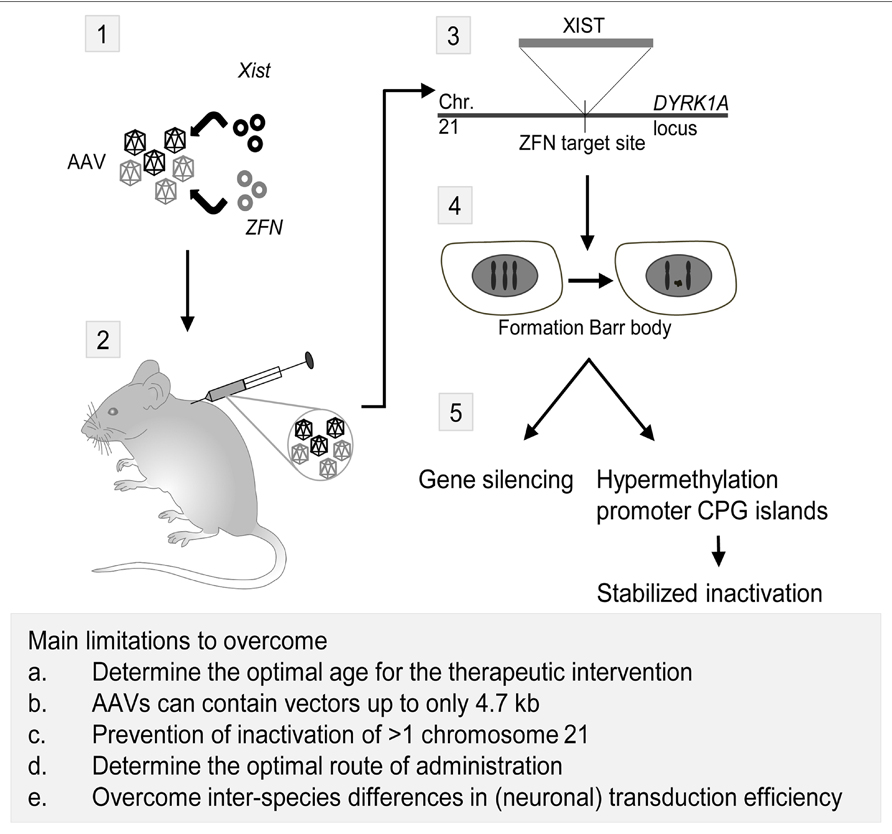
FIGURE 2. Proposed strategy for a therapeutic application of Xist and zinc finger nucleases (ZFN) to treat trisomy 21. Adeno-associated viruses (AAVs) are currently the most promising CNS gene delivery vector (for review, see Gray, 2013). As shown in this scheme, the first step in the approach would be incorporation of plasmids containing Xist and ZFN targeted to the DYRK1A locus on chromosome 21 in AAVs (1). Next, injection of the viruses in rodents can be performed intracranial, intravenous or in the cerebrospinal fluid (2). Intracranial injections have been successfully performed in mammals as large as cats, but an estimated number of 20–30 required injections per hemisphere in human infants rendered this technique unfavorable over alternatives (Vite et al., 2005). AAV9 vectors have the capacity to cross the blood-brain barrier and transduce neurons and astrocytes (Foust et al., 2009), making intravascular injection of viral vectors for CNS targeted gene therapy a possibility. The third possible route of administration is injecting the viral vectors in the cerebrospinal fluid (CSF), thus transducing the central nervous system effectively even in non-human primates (Samaranch et al., 2012). After injection of the AAVs and transduction of the viruses in the cells, Xist will be incorporated on the ZFN target site in the DYRK1A locus on chromosome 21 (3). Here, it will induce the formation of a chromosome 21 Barr body (4), which will lead to gene silencing and hypermethylation of promoter CPG islands, effectively stabilizing the inactivation of the chromosome (5; Jiang et al., 2013). Eventually, this approach may lead to a CNS-wide inactivation of the third chromosome 21, thereby reducing the symptoms of the trisomy 21 disorder. However, several major limitations have to be overcome in order to translate this proposed approach to humans. First, the optimal age for the therapeutic intervention should be established. The majority of the in vivo gene therapy studies have been performed in juvenile or adult animals, but starting the treatment at an earlier age should be considered in order to achieve the best therapeutic effect. Moreover, the optimal route of delivery for CNS gene therapy is currently not established yet, with possibilities being intravascular injection, injection in the CSF, and to a lesser extend, intracranial injections. Third, a practical issue of using AAVs in the therapeutic approach is the limitation of AAVs to contain vectors up to only 4.7 kb in length. This is insufficient for the Xist containing vector used in the proof-of-principle study by Jiang et al. (2013). Last, comparing intravascular injection of vectors with a CNS target revealed that both neuronal and overall transduction efficiency in primates is considerably lower than in rodents, the latter most likely due to circulating pre-existing neutralizing AAV antibodies (Gray et al., 2011).
Other syndromic neurodevelopmental disorders
In the last decade, several new rare microdeletion syndromes were identified. One of these is the 2p15-p16.1 microdeletion syndrome (Rajcan-Separovic et al., 2007), characterized by ID, autistic features, microcephaly, short stature, and various dysmorphic facial features. The genomic cause of this disorder remains to be elucidated, but the susceptibility candidate genes include BCL11A, PAPOLG and REL and one lncRNA gene FLJ16341, although the function of this lncRNA is still elusive.
Autism Spectrum Disorder
Autism spectrum disorders is an umbrella term for various developmental disorders, including autism, pervasive developmental disorder not otherwise specified (PDD-NOS) and the Asperger syndrome. Common symptoms of the various ASD disorders include problems of reciprocal social interactions, verbal and non-verbal communication, and rigid and stereotyped behaviors. ASD is a clinically and etiologically heterogeneous disorder with a complex genetic architecture. Not only multiple common genetic variants appear to be involved, each with small effect size, but also rare variants with strong effect size (Devlin and Scherer, 2012). The latter are mostly de novo mutations, as evidenced by whole-exome and genome sequencing studies in ASD patients (Talkowski et al., 2012; Vulto-van Silfhout et al., 2013), or copy number variations (CNVs; Poelmans et al., 2013). Microarray analysis shows that 5–10% of subjects with ASD have an identifiable genetic etiology in recurrent or de novo chromosomal rearrangements (Marshall et al., 2008). In the last decade, several studies reported aberrant expression of lncRNAs, suggesting that these might be important in the etiology of the disorder. Recently, Ziats and Rennert (2013) showed that over 200 lncRNAs were differentially expressed in a microarray of postmortem prefrontal cortex and cerebellum tissue of ASD patients. A decade earlier, Vincent et al. (2002) identified a novel autism locus, which includes the gene RAY1/ST7. This locus contains at least four non-coding genes (ST7OT1-4), both on the sense and antisense strands that potentially regulate RAY1/ST7. Several rare variants were detected in autism patients on either the RAY1/ST7 or the ST7OT1-3 genes that were not observed in a control population.
Mutations in the X-chromosome PTCHD1 gene have been reported to involve X-linked ID and ASD (Noor et al., 2010; Filges et al., 2011). Although the exact function of the gene is still unknown, several lines of evidence suggest that it might have a causative role in a subset of ID and/or ASD patients (Filges et al., 2011). On the antisense strand of the PTCHD1 gene, several overlapping lncRNAs (PTCHD1AS1, PTCHD1AS2 and PTCHD1AS3) were detected, which may serve as regulators for PTCHD1, since the 5′ exons are adjacent on opposite strands.
Conclusion
Regulation of epigenetics processes during brain development and in activity-dependent brain functions are key to the symptomology underlying many neurodevelopmental disorders. In recent time, a wide range of cutting-edge “omics” and bioinformatics based technologies vastly accelerated our understanding of the key molecular players and mechanisms involved in regulating these epigenetic processes. In contrast to the earlier held view that lncRNAs were merely transcriptional noise, it is now apparent that lncRNAs exert important regulatory functions in the brain, both during adult and developmental stages and represent a key epigenetic mediator of these processes. The interplay between lncRNAs and chromatin remodeling factors may be key to understanding the role of epigenetics in neurodevelopmental disorders (Kramer and van Bokhoven, 2009). LncRNAs are now believed to modulate molecular events during neurogenesis, cell-fate decisions, differentiation and maturation, but are also involved in higher brain functions such as memory formation. The large number of brain-expressed lncRNAs suggests that many more such higher-order functions might also be modulated by lncRNA-mediated mechanisms, which remain to be more fully illustrated in future research efforts. Animal models of lncRNA function, e.g. knockout mice for Malat1 (Zhang et al., 2012) and Neat1 (Nakagawa et al., 2011), have been developed recently and might provide a better insight in lncRNA-mediated mechanisms. However, already at this stage it is clear that lncRNAs may offer a unique approach to modulate pathogenetic events in the causation of neurodevelopmental disorders.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The research of the authors is supported by fundings from the European Community’s Seventh Framework Programme (FP7/2007-2013) under grant agreement no. 278948, and the Marie Curie International Reintegration Grant.
References
Amaral, P. P., Neyt, C., Wilkins, S. J., Askarian-Amiri, M. E., Sunkin, S. M., Perkins, A. C., et al. (2009). Complex architecture and regulated expression of the Sox2ot locus during vertebrate development. RNA 15, 2013–2027. doi: 10.1261/rna.1705309
Amir, R. E., Van Den Veyver, I. B., Wan, M., Tran, C. Q., Francke, U., and Zoghbi, H. Y. (1999). Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 23, 185–188. doi: 10.1038/13810
Arron, J. R., Winslow, M. M., Polleri, A., Chang, C. P., Wu, H., Gao, X., et al. (2006). NFAT dysregulation by increased dosage of DSBR1 and DYRK1A on chromosome 21. Nature 441, 595–600. doi: 10.1038/nature04678
Aschrafi, A., Cunningham, B. A., Edelman, G. M., and Vanderklish, P. W. (2005). The fragile X mental retardation protein and group I metabotropic glutamate receptors regulate levels of mRNA granules in brain 8. Proc. Natl. Acad. Sci. U.S.A. 102, 2180–2185. doi: 10.1073/pnas.0409803102
Aschrafi, A., Schwechter, A. D., Mameza, M. G., Natera-Naranjo, O., Gioio, A. E., and Kaplan, B. B. (2008). MicroRNA-338 regulates local cytochrome c oxidase IV mRNA levels and oxidative phosphorylation in the axons of sympathetic neurons. J. Neurosci. 28, 12581–12590. doi: 10.1523/JNEUROSCI.3338-08.2008
Bartolomei, M. S. (2009). Genomic imprinting: employing and avoiding epigenetic processes. Genes Dev. 23, 2124–2133. doi: 10.1101/gad.1841409
Bernard, D., Prasanth, K. V., Tripathi, V., Colasse, S., Nakamura, T., Xuan, Z., et al. (2010). A long nuclear-retained non-coding RNA regulates synaptogenesis by modulating gene expression. EMBO J. 29, 3082–3093. doi: 10.1038/emboj.2010.199
Chamberlain, S. J., and Brannan, C. I. (2001). The Prader-Willi syndrome imprinting center activates the paternally expressed murine Ube3a antisense transcript but represses paternal Ube3a. Genomics 73, 316–322. doi: 10.1006/geno.2001.6543
Clemson, C. M., Hutchinson, J. N., Sara, S. A., Ensminger, A. W., Fox, A. H., Chess, A., et al. (2009). An architectural role for a nuclear noncoding RNA: NEAT1 RNA is essential for the structure of paraspeckles. Mol. Cell 33, 717–726. doi: 10.1016/j.molcel.2009.01.026
de Smith, A. J., Purmann, C., Walters, R. G., Ellis, R. J., Holder, S. E., Van Haelst, M. M., et al. (2009). A deletion of the HBII-85 class of small nucleolar RNAs (snoRNAs) is associated with hyperphagia, obesity and hypogonadism. Hum. Mol. Genet. 18, 3257–3265. doi: 10.1093/hmg/ddp263
Devlin, B., and Scherer, S. W. (2012). Genetic architecture in autism spectrum disorder. Curr. Opin. Genet. Dev. 22, 229–237. doi: 10.1016/j.gde.2012.03.002
Devys, D., Lutz, Y., Rouyer, N., Bellocq, J. P., and Mandel, J. L. (1993). The FMR-1 protein is cytoplasmic, most abundant in neurons and appears normal in carriers of a fragile X premutation. Nat. Genet. 4, 335–340. doi: 10.1038/ng0893-335
Ding, F., Li, H. H., Zhang, S., Solomon, N. M., Camper, S. A., Cohen, P., et al. (2008). SnoRNA Snord116 (PwBR1/MBII-85) deletion causes growth deficiency and hyperphagia in mice. PLoS ONE 3:e1709. doi: 10.1371/journal.pone.0001709
Dinger, M. E., Amaral, P. P., Mercer, T. R., Pang, K. C., Bruce, S. J., Gardiner, B. B., et al. (2008). Long noncoding RNAs in mouse embryonic stem cell pluripotency and differentiation. Genome Res. 18, 1433–1445. doi: 10.1101/gr.078378.108
Duker, A. L., Ballif, B. C., Bawle, E. V., Person, R. E., Mahadevan, S., Alliman, S., et al. (2010). Paternally inherited microdeletion at 15q11.2 confirms a significant role for the SNORD116 C/D box snoRNA cluster in Prader-Willi syndrome. Eur. J. Hum. Genet. 18, 1196–1201. doi: 10.1038/ejhg.2010.102
Ecker, C., Spooren, W., and Murphy, D. G. (2013). Translational approaches to the biology of Autism: false dawn or a new era? Mol. Psychiatry 18, 435–442. doi: 10.1038/mp.2012.102
Fantes, J., Ragge, N. K., Lynch, S. A., Mcgill, N. I., Collin, J. R., Howard-Peebles, P. N., et al. (2003). Mutations in SOX2 cause anophthalmia. Nat. Genet. 33, 461–463. doi: 10.1038/ng1120
Feng, J., Bi, C., Clark, B. S., Mady, R., Shah, P., and Kohtz, J. D. (2006). The Evf-2 noncoding RNA is transcribed from the Dlx-5/6 ultraconserved region and functions as a Dlx-2 transcriptional coactivator. Genes Dev. 20, 1470–1484. doi: 10.1101/gad.1416106
Filges, I., Rothlisberger, B., Blattner, A., Boesch, N., Demougin, P., Wenzel, F., et al. (2011). Deletion in Xp22.11: PTCHD1 is a candidate gene for X-linked intellectual disability with or without autism. Clin. Genet. 79, 79–85. doi: 10.1111/j.1399-0004.2010.01590.x
Foust, K. D., Nurre, E., Montgomery, C. L., Hernandez, A., Chan, C. M., and Kaspar, B. K. (2009). Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat. Biotechnol. 27, 59–65. doi: 10.1038/nbt.1515
Geschwind, D. H. (2008). Autism: many genes, common pathways? Cell 135, 391–395. doi: 10.1016/j.cell.2008.10.016
Gray, S. J. (2013). Gene therapy and neurodevelopmental disorders. Neuropharmacology 68, 136–142. doi: 10.1016/j.neuropharm.2012.06.024
Gray, S. J., Matagne, V., Bachaboina, L., Yadav, S., Ojeda, S. R., and Samulski, R. J. (2011). Preclinical differences of intravascular AAV9 delivery to neurons and glia: a comparative study of adult mice and nonhuman primates. Mol. Ther. 19, 1058–1069. doi: 10.1038/mt.2011.72
Guttman, M., and Rinn, J. L. (2012). Modular regulatory principles of large non-coding RNAs. Nature 482, 339–346. doi: 10.1038/nature10887
Hancarova, M., Simandlova, M., Drabova, J., Mannik, K., Kurg, A., and Sedlacek, Z. (2013). A patient with de novo 0.45 Mb deletion of 2p16.1: the role of BCL11A, PAPOLG, REL, and FLJ16341 in the 2p15-p16.1 microdeletion syndrome. Am. J. Med. Genet. A 161A, 865–870. doi: 10.1002/ajmg.a.35783
Hitzemann, R., Bottomly, D., Darakjian, P., Walter, N., Iancu, O., Searles, R., et al. (2013). Genes, behavior and next-generation RNA sequencing. Genes Brain Behav. 12, 1–12. doi: 10.1111/gbb.12007
Horsthemke, B., and Wagstaff, J. (2008). Mechanisms of imprinting of the Prader-Willi/Angelman region. Am. J. Med. Genet. A 146A, 2041–2052. doi: 10.1002/ajmg.a.32364
Hutchinson, J. N., Ensminger, A. W., Clemson, C. M., Lynch, C. R., Lawrence, J. B., and Chess, A. (2007). A screen for nuclear transcripts identifies two linked noncoding RNAs associated with SC35 splicing domains. BMC Genomics 8:39. doi: 10.1186/1471-2164-8-39
Iacoangeli, A., Rozhdestvensky, T. S., Dolzhanskaya, N., Tournier, B., Schutt, J., Brosius, J., et al. (2008). On BC1 RNA and the fragile X mental retardation protein. Proc. Natl. Acad. Sci. U.S.A. 105, 734–739. doi: 10.1073/pnas.0710991105
Jiang, J., Jing, Y., Cost, G. J., Chiang, J. C., Kolpa, H. J., Cotton, A. M., et al. (2013). Translating dosage compensation to trisomy 21. Nature 500, 296–300. doi: 10.1038/nature12394
Johnstone, K. A., Dubose, A. J., Futtner, C. R., Elmore, M. D., Brannan, C. I., and Resnick, J. L. (2006). A human imprinting centre demonstrates conserved acquisition but diverged maintenance of imprinting in a mouse model for Angelman syndrome imprinting defects. Hum. Mol. Genet. 15, 393–404. doi: 10.1093/hmg/ddi456
Jong, M. T., Gray, T. A., Ji, Y., Glenn, C. C., Saitoh, S., Driscoll, D. J., et al. (1999). A novel imprinted gene, encoding a RING zinc-finger protein, and overlapping antisense transcript in the Prader-Willi syndrome critical region. Hum. Mol. Genet. 8, 783–793. doi: 10.1093/hmg/8.5.783
Khalil, A. M., Faghihi, M. A., Modarresi, F., Brothers, S. P., and Wahlestedt, C. (2008). A novel RNA transcript with antiapoptotic function is silenced in fragile X syndrome. PLoS ONE 3:e1486. doi: 10.1371/journal.pone.0001486
Khalil, A. M., Guttman, M., Huarte, M., Garber, M., Raj, A., Rivea Morales, D., et al. (2009). Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc. Natl. Acad. Sci. U.S.A. 106, 11667–11672. doi: 10.1073/pnas.0904715106
Kramer, J. M., and van Bokhoven, H. (2009). Genetic and epigenetic defects in mental retardation. Int. J. Biochem. Cell Biol. 41, 96–107. doi: 10.1016/j.biocel.2008.08.009
Ladd, P. D., Smith, L. E., Rabaia, N. A., Moore, J. M., Georges, S. A., Hansen, R. S., et al. (2007). An antisense transcript spanning the CGG repeat region of FMR1 is upregulated in premutation carriers but silenced in full mutation individuals. Hum. Mol. Genet. 16, 3174–3187. doi: 10.1093/hmg/ddm293
Le Meur, E., Watrin, F., Landers, M., Sturny, R., Lalande, M., and Muscatelli, F. (2005). Dynamic developmental regulation of the large non-coding RNA associated with the mouse 7C imprinted chromosomal region. Dev. Biol. 286, 587–600. doi: 10.1016/j.ydbio.2005.07.030
Makeyev, E. V., Zhang, J., Carrasco, M. A., and Maniatis, T. (2007). The MicroRNA miR-124 promotes neuronal differentiation by triggering brain-specific alternative pre-mRNA splicing. Mol. Cell 27, 435–448. doi: 10.1016/j.molcel.2007.07.015
Marshall, C. R., Noor, A., Vincent, J. B., Lionel, A. C., Feuk, L., Skaug, J., et al. (2008). Structural variation of chromosomes in autism spectrum disorder. Am. J. Hum. Genet. 82, 477–488. doi: 10.1016/j.ajhg.2007.12.009
Martens, J. A., Wu, P.-Y. J., and Winston, F. (2005). Regulation of an intergenic transcript controls adjacent gene transcription in Saccharomyces cerevisiae. Genes Dev. 19, 2695–2704. doi: 10.1101/gad.1367605
Martianov, I., Ramadass, A., Serra Barros, A., Chow, N., and Akoulitchev, A. (2007). Repression of the human dihydrofolate reductase gene by a non-coding interfering transcript. Nature 445, 666–670. doi: 10.1038/nature05519
Matsuura, T., Sutcliffe, J. S., Fang, P., Galjaard, R. J., Jiang, Y. H., Benton, C. S., et al. (1997). De novo truncating mutations in E6-AP ubiquitin-protein ligase gene (UBE3A) in Angelman syndrome. Nat. Genet. 15, 74–77. doi: 10.1038/ng0197-74
Mercer, T. R., Dinger, M. E., Mariani, J., Kosik, K. S., Mehler, M. F., and Mattick, J. S. (2008a). Noncoding RNAs in long-term memory formation. Neuroscientist 14, 434–445. doi: 10.1177/1073858408319187
Mercer, T. R., Dinger, M. E., Sunkin, S. M., Mehler, M. F., and Mattick, J. S. (2008b). Specific expression of long noncoding RNAs in the mouse brain. Proc. Natl. Acad. Sci. U.S.A. 105, 716–721. doi: 10.1073/pnas.0706729105
Mercer, T. R., Qureshi, I. A., Gokhan, S., Dinger, M. E., Li, G., Mattick, J. S., et al. (2010). Long noncoding RNAs in neuronal-glial fate specification and oligodendrocyte lineage maturation. BMC Neurosci. 11:14. doi: 10.1186/1471-2202-11-14
Mizuseki, K., Kishi, M., Matsui, M., Nakanishi, S., and Sasai, Y. (1998). Xenopus Zic-related-1 and Sox-2, two factors induced by chordin, have distinct activities in the initiation of neural induction. Development 125, 579–587.
Muscatelli, F., Abrous, D. N., Massacrier, A., Boccaccio, I., Le Moal, M., Cau, P., et al. (2000). Disruption of the mouse Necdin gene results in hypothalamic and behavioral alterations reminiscent of the human Prader-Willi syndrome. Hum. Mol. Genet. 9, 3101–3110. doi: 10.1093/hmg/9.20.3101
Nakagawa, S., Naganuma, T., Shioi, G., and Hirose, T. (2011). Paraspeckles are subpopulation-specific nuclear bodies that are not essential in mice. J. Cell Biol. 193, 31–39. doi: 10.1083/jcb.201011110
Noor, A., Whibley, A., Marshall, C. R., Gianakopoulos, P. J., Piton, A., Carson, A. R., et al. (2010). Disruption at the PTCHD1 Locus on Xp22.11 in Autism spectrum disorder and intellectual disability. Sci. Transl. Med. 2, 49ra68. doi: 10.1126/scitranslmed.3001267
Olde Loohuis, N. F., Kos, A., Martens, G. J., Van Bokhoven, H., Nadif Kasri, N., and Aschrafi, A. (2012). MicroRNA networks direct neuronal development and plasticity. Cell. Mol. Life Sci. 69, 89–102. doi: 10.1007/s00018-011-0788-1
Pastori, C., Peschansky, V. J., Barbouth, D., Mehta, A., Silva, J. P., and Wahlestedt, C. (2013). Comprehensive analysis of the transcriptional landscape of the human FMR1 gene reveals two new long noncoding RNAs differentially expressed in Fragile X syndrome and Fragile X-associated tremor/ataxia syndrome. Human Genet. doi: 10.1007/s00439-013-1356-6 [Epub ahead of print].
Petazzi, P., Sandoval, J., Szczesna, K., Jorge, O. C., Roa, L., Sayols, S., et al. (2013). Dysregulation of the long non-coding RNA transcriptome in a Rett syndrome mouse model. RNA Biol. 10, 1197–1203. doi: 10.4161/rna.24286
Poelmans, G., Franke, B., Pauls, D. L., Glennon, J. C., and Buitelaar, J. K. (2013). AKAPs integrate genetic findings for autism spectrum disorders. Transl. Psychiatry 3, e270. doi: 10.1038/tp.2013.48
Ponjavic, J., Oliver, P. L., Lunter, G., and Ponting, C. P. (2009). Genomic and transcriptional co-localization of protein-coding and long non-coding RNA pairs in the developing brain. PLoS Genet. 5:e1000617: doi: 10.1371/journal.pgen.1000617
Ponting, C. P., Oliver, P. L., and Reik, W. (2009). Evolution and functions of long noncoding RNAs. Cell 136, 629–641. doi: 10.1016/j.cell.2009.02.006
Qureshi, I. A., Mattick, J. S., and Mehler, M. F. (2010). Long non-coding RNAs in nervous system function and disease. Brain Res. 1338, 20–35. doi: 10.1016/j.brainres.2010.03.110
Rajcan-Separovic, E., Harvard, C., Liu, X., Mcgillivray, B., Hall, J. G., Qiao, Y., et al. (2007). Clinical and molecular cytogenetic characterisation of a newly recognised microdeletion syndrome involving 2p15-16.1. J. Med. Genet. 44, 269–276. doi: 10.1136/jmg.2006.045013
Runte, M., Huttenhofer, A., Gross, S., Kiefmann, M., Horsthemke, B., and Buiting, K. (2001). The IC-SNURF-SNRPN transcript serves as a host for multiple small nucleolar RNA species and as an antisense RNA for UBE3A. Hum. Mol. Genet. 10, 2687–2700. doi: 10.1093/hmg/10.23.2687
Runte, M., Kroisel, P. M., Gillessen-Kaesbach, G., Varon, R., Horn, D., Cohen, M. Y., et al. (2004). SNURF-SNRPN and UBE3A transcript levels in patients with Angelman syndrome. Hum. Genet. 114, 553–561. doi: 10.1007/s00439-004-1104-z
Sahoo, T., Del Gaudio, D., German, J. R., Shinawi, M., Peters, S. U., Person, R. E., et al. (2008). Prader-Willi phenotype caused by paternal deficiency for the HBII-85 C/D box small nucleolar RNA cluster. Nat. Genet. 40, 719–721. doi: 10.1038/ng.158
Samaranch, L., Salegio, E. A., San Sebastian, W., Kells, A. P., Foust, K. D., Bringas, J. R., et al. (2012). Adeno-associated virus serotype 9 transduction in the central nervous system of nonhuman primates. Hum. Gene Ther. 23, 382–389. doi: 10.1089/hum.2011.200
Simon, M. D., Pinter, S. F., Fang, R., Sarma, K., Rutenberg-Schoenberg, M., Bowman, S. K., et al. (2013). High-resolution Xist binding maps reveal two-step spreading during X-chromosome inactivation. Nature. doi: 10.1038/nature12719 [Epub ahead of print].
Skryabin, B. V., Gubar, L. V., Seeger, B., Pfeiffer, J., Handel, S., Robeck, T., et al. (2007). Deletion of the MBII-85 snoRNA gene cluster in mice results in postnatal growth retardation. PLoS Genet. 3:e235. doi: 10.1371/journal.pgen.0030235
Smart, F., Aschrafi, A., Atkins, A., Owens, G. C., Pilotte, J., Cunningham, B. A., et al. (2007). Two isoforms of the cold-inducible mRNA-binding protein RBM3 localize to dendrites and promote translation 6. J. Neurochem. 101, 1367–1379. doi: 10.1111/j.1471-4159.2007.04521.x
Stenman, J., Toresson, H., and Campbell, K. (2003). Identification of two distinct progenitor populations in the lateral ganglionic eminence: implications for striatal and olfactory bulb neurogenesis. J. Neuroscience 23, 167–174.
Sutcliffe, J. S., Nelson, D. L., Zhang, F., Pieretti, M., Caskey, C. T., Saxe, D., et al. (1992). DNA methylation represses FMR-1 transcription in fragile X syndrome. Hum. Mol. Genet. 1, 397–400. doi: 10.1093/hmg/1.6.397
Talkowski, M. E., Rosenfeld, J. A., Blumenthal, I., Pillalamarri, V., Chiang, C., Heilbut, A., et al. (2012). Sequencing chromosomal abnormalities reveals neurodevelopmental loci that confer risk across diagnostic boundaries. Cell 149, 525–537. doi: 10.1016/j.cell.2012.03.028
Tsai, M.-C., Manor, O., Wan, Y., Mosammaparast, N., Wang, J. K., Lan, F., et al. (2010). Long noncoding RNA as modular scaffold of histone modification complexes. Science 329, 689–693. doi: 10.1126/science.1192002
van Bokhoven, H. (2011). Genetic and epigenetic networks in intellectual disabilities. Annu. Rev. Genet. 45, 81–104. doi: 10.1146/annurev-genet-110410-132512
Vincent, J. B., Petek, E., Thevarkunnel, S., Kolozsvari, D., Cheung, J., Patel, M., et al. (2002). The RAY1/ST7 tumor-suppressor locus on chromosome 7q31 represents a complex multi-transcript system. Genomics 80, 283–294. doi: 10.1006/geno.2002.6835
Visvanathan, J., Lee, S., Lee, B., Lee, J. W., and Lee, S. K. (2007). The microRNA miR-124 antagonizes the anti-neural REST/SCP1 pathway during embryonic CNS development. Genes Dev. 21, 744–749. doi: 10.1101/gad.1519107
Vite, C. H., Mcgowan, J. C., Niogi, S. N., Passini, M. A., Drobatz, K. J., Haskins, M. E., et al. (2005). Effective gene therapy for an inherited CNS disease in a large animal model. Ann. Neurol. 57, 355–364. doi: 10.1002/ana.20392
Vulto-van Silfhout, A. T., De Vries, B. B., Van Bon, B. W., Hoischen, A., Ruiterkamp-Versteeg, M., Gilissen, C., et al. (2013). Mutations in MED12 Cause X-Linked Ohdo Syndrome. Am. J. Human Genet. 92, 401–406. doi: 10.1016/j.ajhg.2013.01.007
Wang, J., Zhang, J., Zheng, H., Li, J., Liu, D., Li, H., et al. (2004). Mouse transcriptome: neutral evolution of ‘non-coding’complementary DNAs. Nature 431, 1. doi: 10.1038/nature03016
Wang, X., Arai, S., Song, X., Reichart, D., Du, K., Pascual, G., et al. (2008). Induced ncRNAs allosterically modify RNA-binding proteins in cis to inhibit transcription. Nature 454, 126–130. doi: 10.1038/nature06992
Wang, X., Song, X., Glass, C. K., and Rosenfeld, M. G. (2011). The long arm of long noncoding RNAs: roles as sensors regulating gene transcriptional programs. Cold Spring Harb. Perspect. Biol. 3, a003756. doi: 10.1101/cshperspect.a003756
Wevrick, R., and Francke, U. (1997). An imprinted mouse transcript homologous to the human imprinted in Prader-Willi syndrome (IPW) gene. Hum. Mol. Genet. 6, 325–332. doi: 10.1093/hmg/6.2.325
Wevrick, R., Kerns, J. A., and Francke, U. (1994). Identification of a novel paternally expressed gene in the Prader-Willi syndrome region. Hum. Mol. Genet. 3, 1877–1882. doi: 10.1093/hmg/3.10.1877
Willemsen, M. H., Valles, A., Kirkels, L. A., Mastebroek, M., Olde Loohuis, N., Kos, A., et al. (2011). Chromosome 1p21.3 microdeletions comprising DPYD and MIR137 are associated with intellectual disability. J. Med. Genet. 48, 810–818. doi: 10.1136/jmedgenet-2011-100294
Willingham, A. T., Orth, A. P., Batalov, S., Peters, E. C., Wen, B. G., Aza-Blanc, P., et al. (2005). A strategy for probing the function of noncoding RNAs finds a repressor of NFAT. Science 309, 1570–1573. doi: 10.1126/science.1115901
Yakovchuk, P., Goodrich, J. A., and Kugel, J. F. (2009). B2 RNA and Alu RNA repress transcription by disrupting contacts between RNA polymerase II and promoter DNA within assembled complexes. Proc. Natl. Acad. Sci. U.S.A. 106, 5569–5574. doi: 10.1073/pnas.0810738106
Zalfa, F., Adinolfi, S., Napoli, I., Kuhn-Holsken, E., Urlaub, H., Achsel, T., et al. (2005). Fragile X mental retardation protein (FMRP) binds specifically to the brain cytoplasmic RNAs BC1/BC200 via a novel RNA-binding motif. J. Biol. Chem. 280, 33403–33410. doi: 10.1074/jbc.M504286200
Zalfa, F., Giorgi, M., Primerano, B., Moro, A., Di Penta, A., Reis, S., et al. (2003). The fragile X syndrome protein FMRP associates with BC1 RNA and regulates the translation of specific mRNAs at synapses. Cell 112, 317–327. doi: 10.1016/S0092-8674(03)00079-5
Zhang, B., Arun, G., Mao, Y. S., Lazar, Z., Hung, G., Bhattacharjee, G., et al. (2012). The lncRNA Malat1 is dispensable for mouse development but its transcription plays a cis-regulatory role in the adult. Cell Rep. 2, 111–123. doi: 10.1016/j.celrep.2012.06.003
Zhao, J., Sun, B. K., Erwin, J. A., Song, J.-J., and Lee, J. T. (2008). Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science 322, 750–756. doi: 10.1126/science.1163045
Ziats, M. N., and Rennert, O. M. (2013). Aberrant expression of long noncoding RNAs in autistic brain. J. Mol. Neurosci. 49, 589–593. doi: 10.1007/s12031-012-9880-8
Keywords: long non-coding RNA, nervous system development, fragile X syndrome, genomic imprinting, autism spectrum disorders, intellectual disability, schizophrenia
Citation: van de Vondervoort IIGM, Gordebeke PM, Khoshab N, Tiesinga PHE, Buitelaar JK, Kozicz T, Aschrafi A and Glennon JC (2013) Long non-coding RNAs in neurodevelopmental disorders. Front. Mol. Neurosci. 6:53. doi: 10.3389/fnmol.2013.00053
Received: 29 July 2013; Accepted: 09 December 2013;
Published online: 30 December 2013.
Edited by:
Hermona Soreq, The Hebrew University of Jerusalem, IsraelReviewed by:
Hermona Soreq, The Hebrew University of Jerusalem, IsraelDusan Bartsch, Heidelberg University, Germany
Copyright © 2013 van de Vondervoort, Gordebeke, Khoshab, Tiesinga, Buitelaar, Kozicz, Aschrafi and Glennon. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ilse I. G. M. van de Vondervoort, Department of Cognitive Neuroscience, RadboudUMC, Geert Grooteplein-Noord 21, Nijmegen 6525 EZ, Netherlands e-mail:aWxzZS52YW5kZXZvbmRlcnZvb3J0QHJhZGJvdWR1bWMubmw=
† Ilse I. G. M. van de Vondervoort, Peter M. Gordebeke, Armaz Aschrafi and Jeffrey C. Glennon have contributed equally to this work.