 Rachel T. Uren
Rachel T. Uren Ann M. Turnley
Ann M. Turnley- Neural Regeneration Laboratory, Centre for Neuroscience Research and Department of Anatomy and Neuroscience, The University of Melbourne, Melbourne, VIC, Australia
The classic neurotrophins Nerve Growth Factor (NGF), Brain Derived Neurotrophic Factor (BDNF) and Neurotrophins NT-3 and NT-4 are well known to regulate various aspects of neuronal differentiation, survival and growth. They do this by binding to their cognate receptors, members of the Tropomyosin-related kinase (Trk) receptor tyrosine kinase family, namely TrkA, TrkB, and TrkC. These receptors are then internalized and localized to different cellular compartments, where signal transduction occurs. Conversely, members of the suppressor of cytokine signaling (SOCS) family are best known as negative regulators of signaling via the JAK/STAT pathway. Some members of the family, and in particular SOCS2, have roles in the nervous system that at least partially overlap with that of neurotrophins, namely neuronal differentiation and neurite outgrowth. Recent evidence suggests that SOCS2 is a novel regulator of NGF signaling, altering TrkA cellular localization and downstream signaling to affect neurite growth but not neuronal survival. This review first discusses regulation of Trk receptor signaling, followed by the role of SOCS2 in the nervous system and finishes with a discussion of possible mechanisms by which SOCS2 may regulate TrkA function.
Introduction
A great deal of interest has focused on the therapeutic potential of neurotrophins to prevent neurodegeneration and promote neural regeneration in a range of neurological diseases and neurotrauma (Allen et al., 2011; Colafrancesco and Villoslada, 2011; Ossipov, 2011; Duman and Voleti, 2012). Although administration of neurotrophins, such as NGF, has promoted improvement in many studies, their use as a therapeutic option is limited by undesirable side-effects, such as neuropathic pain. Therefore, other approaches to target neurotrophin receptors is required for therapeutic purposes, to modulate specific aspects of neurotrophin signaling that are beneficial and avoid those that are detrimental. To achieve this, a deeper understanding of the mechanisms that regulate neurotrophin receptor function and signaling is required. Here we describe the potential role of SOCS2 as a novel regulator of NGF receptor (TrkA) signaling that differentially affects NGF-mediated neurite outgrowth but not neuron survival, altering TrkA protein levels, cellular localization and extent and duration of specific NGF-induced signal transduction pathways.
The Classical Neurotrophins and Their Receptors
The presence of a soluble factor capable of supporting the survival of motor and sensory neurons was first demonstrated in 1949 and prompted the characterization of NGF (Hamburger and Levi-Montalcini, 1949; Levi-Montalcini, 1987), one of the earliest identified growth factors and the first member of the “classical neurotrophin” family. In addition to its role in the peripheral nervous system, NGF has been shown to have actions in the central nervous system and the immune system (Dreyfus, 1989; Dreyfus et al., 1989; Snider, 1994; Ernsberger, 2009; Scuri et al., 2010; Allard et al., 2012). Three additional mammalian neurotrophic factors have since been identified; brain-derived neurotrophic factor (BDNF), neurotrophin-3 (NT-3) and neurotrophin-4/5 (NT-4) (reviewed by (Reichardt, 2006; Skaper, 2012).
Neurotrophins signal through two-types of cell surface receptor; the p75 neurotrophin receptor (p75NTR) (Radeke et al., 1987; Bibel et al., 1999) and the Tropomyosin-related kinase (Trk) receptor tyrosine kinases (Kaplan et al., 1991a,b; Klein et al., 1991a). They bind with equal affinity to p75NTR (Radeke et al., 1987; Rodriguez-Tebar et al., 1990; Hallbook et al., 1991; Rodriguez-Tebar et al., 1992), however, the neurotrophins are more selective in their interactions with the Trk receptors; TrkA binds NGF, TrkB binds BDNF and NT-4/5 and TrkC binds NT-3 (Kaplan et al., 1991a,b; Klein et al., 1991a; Dechant et al., 1993; Mahadeo et al., 1994). The mature neurotrophins form homodimers that can simultaneously bind two receptor molecules (Barbacid et al., 1991; Heymach and Shooter, 1995). The Trk receptors can form homodimers or can associate with p75NTR (Bibel et al., 1999), thus a single neurotrophin dimer can simultaneously bind a Trk:Trk or Trk:p75NTR complex. When the Trks form a co-receptor complex with p75NTR, lower concentrations of the neurotrophic ligand are required to initiate signaling via the Trk receptor than if the Trk receptors were solely expressed (Mahadeo et al., 1994). The co-receptor Sortilin can also bind the Trk receptors and has been implicated in the transport of TrkA along axons (Vaegter et al., 2011). Beyond its involvement in Trk and sortilin signaling, p75NTR has been implicated in the inhibition of axon growth and dendritic complexity by regulating the GTPase RhoA (Yamashita et al., 1999, 2002; Zagrebelsky et al., 2005).
Signaling Events Downstream of the Trk Neurotrophin Receptors
A diverse range of cell responses can be initiated upon neurotrophin binding. The type of signal is influenced by the combination of neurotrophin receptors presented on the cell membrane and the unique collection of intracellular effectors that can be recruited by the different receptors upon activation (Lee et al., 2001; Huang and Reichardt, 2003). Signaling downstream of the Trk receptors is a complex affair (Reichardt, 2006). The Trk receptors possess a single transmembrane domain and an extracellular ligand-binding domain comprising leucine-rich motifs (LRR1-3), two cysteine clusters (C1 and C2) and two immunoglobulin-like domains (Ig1 and Ig2) (Johnson et al., 1986; Radeke et al., 1987; Schneider and Schweiger, 1991; Windisch et al., 1995a,b) (Figure 1). The second immunoglobulin-like domain largely dictates the neurotrophin-binding specificity of the Trk receptors and there is some evidence that the immunoglobulin-like domains serve to inhibit spontaneous dimerization of the Trk receptors in the absence of ligand (Arevalo et al., 2000). Ligand binding to the Trk receptors triggers receptor dimerization and activation of the intrinsic cytoplasmic kinase domain and receptor autophosphorylation. There are 10 known conserved tyrosine residues on the cytoplasmic tail of the mammalian Trk receptors, some of which are phosphorylated upon receptor activation (Stephens et al., 1994; Inagaki et al., 1995). Three of these sites are located in the auto-regulatory loop of the tyrosine kinase domain and thus modulate kinase activity when phosphorylated. The remaining phosphorylated tyrosine residues form binding sites for scaffold proteins and enzymes that contain phosphotyrosine binding (PTB) domains or Src-homology-2 (SH2) domains and the specificity of these interactions is tailored by the flanking amino acids. The best characterized of these docking sites are Y490 and Y785 as per the nomenclature of human TrkA, also known as Y499 and Y794 in rat TrkA. The other conserved sites have been shown to display some degree of functional redundancy with respect to downstream signaling.
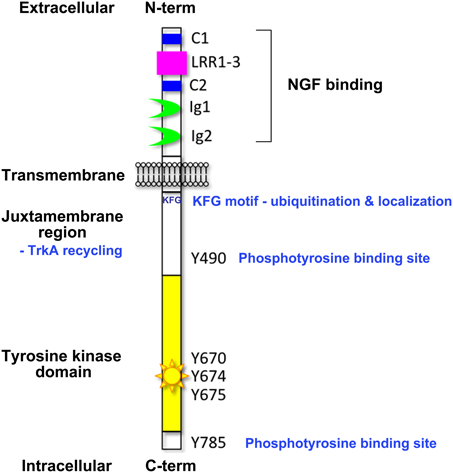
Figure 1. Domain organization of the Trk neurotrophin receptors. This figure highlights the key structural elements of the Trk neurotrophin receptors; the extracellular ligand-binding domain comprising leucine-rich motifs (LRR1-3), two cysteine clusters (C1 and C2) and two immunoglobulin-like domains (Ig1 and Ig2), the transmembrane domain, and the intracellular tyrosine kinase domain. Critical tyrosine residues of the human TrkA intracellular domain are highlighted. Tyrosine residues at positions 670, 674, and 675 are located in the activation loop of the human TrkA tyrosine kinase domain. Tyrosine 490 forms a PTB binding motif (NPXpY) known to bind SHC and FRS2 (Obermeier et al., 1993b; Ong et al., 2000) and tyrosine 785 binds to PLCγ (Obermeier et al., 1993a). The juxtamembrane KFG motif is an ubiquitination site involved in receptor localization.
Activation of signaling pathways
The activated Trk receptors initiate diverse signaling pathways downstream of Ras, phosphatidylinositol 3-kinase (PI3-kinase) and phospholipase C- γ1 (PLC-γ1) (Figure 2) (Obermeier et al., 1993a,b; Ong et al., 2000). These signaling components are common to many other growth factor and cytokine stimuli, thus there is obviously scope for crosstalk between these diverse signaling pathways. Both the Ras-MAPK and PI3-K/AKT pathways converge on the common upstream scaffold protein, Src homologous and collagen-like (Shc), which is known to recognize phosphorylated Y490 of the Trk receptors. This tyrosine residue is also well characterized as a FRS2 binding site which can also promote the prolonged activation of MAP kinases via recruitment of GRB2, Crk and Src. PLC γ1 binds the activated TrkA receptor at Y785 and prompts increased Ca2+ levels which can influence synaptic plasticity and activation of protein kinase C (PKC) signaling cascades to effect changes in transcription. The neurotrophins are also known to activate members of the Rho GTPase family such as Cdc42 and Rac that are regulators of the cytoskeletal rearrangements required for cell motility and the formation of growth cones (Yuan et al., 2003).
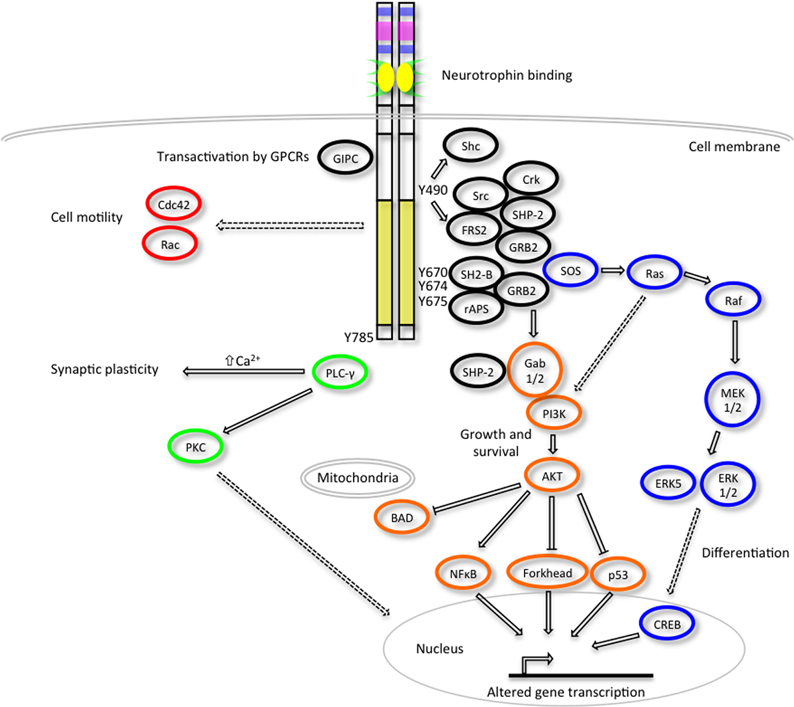
Figure 2. Trk receptor signaling pathways. Upon neurotrophin binding, the activated Trk receptors engage various intracellular signaling pathways such as those mediated by extracellular signal-regulated kinases (ERKs), phosphatidylinositol 3-kinase (PI3K) and phospholipase Cγ (PLC-γ) to promote cellular differentiation, growth, survival, synaptic plasticity and changes to cell motility (adapted from Huang and Reichardt, 2003). Critical tyrosine (Y) residues of the human TrkA intracellular domain are shown. Direct, indirect and inhibitory pathways are denoted by solid arrows, dashed arrows and blunt arrows respectively.
A less established pathway downstream of the Trk receptors is the activation of the signal transducer and activator of transcription-3 (STAT3) transcription factor. Early studies demonstrated that dominant negative STAT3 can promote PC12 neurite outgrowth in the absence of NGF (Ihara et al., 1997) and STAT3 was identified as a downstream effector of Trk neurotrophins in both PC12 cells and primary hippocampal neurons (Ng et al., 2006). BDNF can induce proliferation of neural stem cells via a mechanism involving MAP kinase, AKT and STAT3 activation (Islam et al., 2009) and constitutively active Trk receptors can induce STAT3 activation (Miranda et al., 2010).
A variety of phosphatases are recruited to the Trk receptors. The protein tyrosine phosphatase SH2 domain-containing phosphatase -2 (SHP-2) has been shown to have a transient association with the Trk receptors upon neurotrophin binding (Okada et al., 1996; Goldsmith and Koizumi, 1997) and provide an important counterbalance to TrkB activation (Rusanescu et al., 2005). SHP-2 may also be indirectly recruited to the Trk receptors via adapter molecules such as FRS2 (Yamamoto et al., 2005; Easton et al., 2006) and NOMA-GAP (Rosario et al., 2007). The related SHP-1 phosphatase has also been shown to bind TrkA at Y490 and dephosphorylate residues Y674 and Y675 in the TrkA tyrosine kinase activation loop (Marsh et al., 2003). The receptor-like protein tyrosine phosphatase sigma (PTPsigma) can bind stably to TrkA and TrkC via its transmembrane domain and dephosphorylate all the Trk receptors upon neurotrophin stimulation (Faux et al., 2007). Another receptor-like phosphatase, the protein tyrosine phosphatase receptor zeta (PTPRz), can inhibit phosphorylation of the activation loop tyrosine residues of the TrkA receptor upon NGF stimulation (Shintani and Noda, 2008). Interestingly, some phosphatases have a positive regulatory role in TrkA signal transduction. The protein phosphatase 2A has been shown to enhance the tyrosine kinase activity of the TrkA receptor leading to sustained activation and downstream AKT and ERK1/2 activity, possibly mediated by dephosphorylation of inhibitory serine and threonine phosphorylation sites (Van Kanegan and Strack, 2009).
The activated Trk receptors can modulate the signaling of various ion channels and, even in the absence of neurotrophins, the Trk receptors can be transactivated upon association with membrane associated G-protein coupled receptors (GPCRs) (Chao, 2003). The juxtamembrane domain of TrkA has been shown to interact with a PDZ domain-containing scaffold protein GIPC that is also a component of a GPCR complex (Lou et al., 2001). More recent studies have shown that GIPC is required for the efficient trafficking and endocytosis of TrkA (Varsano et al., 2006). This finding points to the critical role of receptor trafficking in the modulation of Trk-mediated signaling.
Neurotrophin Receptor Dynamics and the Unique Cell Biology of Neurotrophin Signaling
The retrograde transport of neurotrophic stimuli has been most extensively studied in the developing peripheral nervous system and is critical for the formation and refinement of neuronal circuits during development and for the regulation of synaptic plasticity and neuronal survival in the mature nervous system (Huang and Reichardt, 2001; Zweifel et al., 2005). The mechanics of this receptor trafficking have been scrutinized since retrograde axonal transport of NGF was first demonstrated in 1974 (Hendry et al., 1974; Paravicini et al., 1975; Johnson and Yip, 1985). Target tissues secrete limiting amounts of neurotrophins and neighboring nerve terminals will transport this trophic signal to the distant neuronal cell bodies and thus support the survival of the innervating neuron. The complex morphology of neurons poses specific challenges for the transport of molecules from one part of the cell to another, as axon lengths can be in excess of 1 meter in some instances. Passive diffusion cannot account for the speed and distance travelled by these signals, and several different mechanisms have been proposed for the propagation of NGF signals, however, a mounting body of evidence now supports a model of the active retrograde transport of the neurotrophin signal in neurons via “signaling endosomes” (reviewed by Howe and Mobley, 2005; Ibanez, 2007; Wu et al., 2009; Ascano et al., 2012).
Neurotrophin receptor endocytosis
Clathrin-mediated endocytosis upon ligand binding is an important mechanism for both the attenuation and propagation of signals received by many receptor tyrosine kinases (McPherson et al., 2001). A point of difference between the Trk receptors and other receptor tyrosine kinases may be their ability to form long-lived signaling endosomes due to a selective and specific process of clathrin-independent macroendocytosis (Valdez et al., 2005, 2007). These specialized endosomes appear to retain association with markers of early endosomes, and thus fail to mature to late endosomes or progress to lysosomal degradation (Valdez et al., 2005).
In neurons, neurotrophin binding to p75NTR or Trk receptors at the axon terminal induces receptor-mediated endocytosis of the activated receptor complex, and a subset of these internalized receptors are retrogradely transported to the cell body in vesicles that resemble early endosomes (Ehlers et al., 1995; Grimes et al., 1996; Bhattacharyya et al., 1997; Tsui-Pierchala and Ginty, 1999; Watson et al., 1999; Jullien et al., 2002, 2003; Delcroix et al., 2003). These endosomes containing activated Trk receptors can associate with the dynein motor and move along the axon toward the cell body (Bhattacharyya et al., 2002). Data from compartmentalized cultures revealed that NGF applied to the distal axons of sympathetic neurons can support their survival. However, the addition of the specific Trk kinase inhibitor K252a to either the distal axon or the cell body compartment induced cell death suggesting that activated TrkA was required for propagation of the survival signal into the cell and that the downstream effectors of TrkA signaling could not propagate the signal in the absence of activated TrkA and NGF must be retained in the signaling endosomes for the TrkA survival signal to persist (Ye et al., 2003).
Intracellular Localization of Internalized Trk Receptors
Upon receptor internalization, the exposed cytoplasmic domains of receptor tyrosine kinases can interact with different intermediates found in different subcellular locations, and thereby fine-tune the signaling output of the activated receptors (reviewed by Romanelli and Wood, 2008; Acconcia et al., 2009; Disanza et al., 2009; Sadowski et al., 2009). An example of differential signaling based on altered subcellular localization of activated NGF-TrkA complexes comes from the PC12 cell line that is a useful model of neuronal-like differentiation. PC12 cells express both TrkA and p75NTR and upon serum withdrawal and treatment with NGF, undergo mitotic arrest and extend neurites (Greene and Tischler, 1976; Greene, 1978). It has been demonstrated that in PC12 cells internalization of the activated NGF: TrkA complex is required for the induction of neurite outgrowth but not NGF-mediated survival, suggesting that the intracellular localization of the activated receptors can influence the type of signaling output (Zhang et al., 2000). Activated Trk receptors located at the cell surface promote cell survival signaling via PI3-K/AKT activation whereas Trk receptors signaling from internalized endosomes promote cell differentiation by preferential activation of MEK/ERK pathways (Zhang et al., 2000). A conflicting report using sciatic nerves showed that the p85 subunit of the PI3-K signaling cascade is associated with internal membrane compartments such as early endosomes (Delcroix et al., 2003). It appears that activated Trk receptors are localized to different membrane fractions when comparing PC12 cells and primary neurons (Yano and Chao, 2005), so this may underlie the apparent discrepancy regarding the types of Trk signaling observed in different subcellular locations.
Specific sequences in the cytoplasmic tail of the receptor tyrosine kinases can regulate the targeting of receptors to particular compartments within the cell. In the case of TrkA, a juxtamembrane portion of the receptor between amino acids 473 and 493, that is not present in TrkB or TrkC, is required for NGF-induced recycling of the rat TrkA receptor (Chen et al., 2005). Treatment of primary sympathetic neurons with NGF induced recycling of the TrkA receptor, however application of BDNF induced the degradation of the TrkB receptor. Modification of the TrkB receptor to include this juxtamembrane region from TrkA resulted in the ligand-induced recycling of the modified TrkB receptor.
Ubiquitination of Trk receptors
Ubiquitination has also emerged as a very important molecular identifier for the intracellular trafficking of receptor tyrosine kinases including TrkA (Acconcia et al., 2009). Ubiquitin is a polypeptide that can be coupled to proteins via lysine residue modification catalyzed by ubiquitin-activating enzymes, ubiquitin-conjugating enzymes, and ubiquitin ligase enzymes (Komander and Rape, 2012). The different molecular conformations adopted by the different types of ubiquitin labeling can encode various outcomes such as protein degradation, protein activation, altered protein-protein interactions and altered intracellular trafficking (Komander and Rape, 2012). Mono-ubiquitination is defined as the coupling of a single ubiquitin moiety to lysine residues on the target protein. Poly-ubiquitination is the coupling of chains of ubiquitin and the topology of these ubiquitin chains has been thought to dictate proteolytic versus non-proteolytic downstream consequences. The internalization of NGF-TrkA has been shown to be regulated by the poly-ubiquitination of TrkA by the ubiquitin ligase TRAF6 in PC12 cells (Geetha et al., 2005) whereas stimulation of PC12 cells and primary cortical neurons with NGF and BDNF has been shown to promote ubiquitination of both TrkA and TrkB, primarily taking the form of multiple sites of mono-ubiquitination rather than poly-ubiquitination (Arevalo et al., 2006). Proteasome inhibition appears to induce neurite outgrowth through TrkA receptor ubiquitination (Song et al., 2009) and recent studies have also highlighted that the proper trafficking of the activated TrkA receptor is essential for NGF-mediated signaling in cultured dorsal root ganglion (DRG) neurons and is regulated by multiple sites of mono-ubiquitination by the ubiquitin ligase Nedd4-2 (Georgieva et al., 2011; Yu et al., 2011). The ubiquitin ligase Cbl has also been implicated in the ligand-dependent ubiquitination of TrkA and subsequent targeting of the receptor to lysosomes for degradation (Takahashi et al., 2011). Clearly ubiquitination and endosomal sorting both have important roles to play in neurotrophin signaling, however, there is still scope for more detailed exploration of the precise molecular mechanisms involved.
Members of the suppressor of cytokine signaling (SOCS) family have recently emerged as ubiquitin ligases regulating expression of a variety of receptors and their signaling pathways. One member of this family in particular, SOCS2, has a variety of functions in the nervous system, many of which overlap with the biological effects of TrkA signaling, suggesting a possible regulatory role for SOCS2 in TrkA function.
The Suppressors of Cytokine Signaling (SOCS) in the Nervous System
The SOCS proteins are a family of intracellular proteins implicated in the negative regulation of a variety of cytokine, growth factor and hormone signals, particularly those mediated by the Janus kinase/signal transducer and activator of transcription (JAK/STAT) signaling pathway (O'Sullivan et al., 2007; Croker et al., 2008; Piessevaux et al., 2008a).
The SOCS proteins have pleiotropic effects in the healthy and diseased nervous system (Wang and Campbell, 2002; Campbell, 2005). They have been implicated in the regulation of diverse cellular processes including neurodevelopment (Turnley et al., 2001; Feng et al., 2007), adult neurogenesis (Ransome and Turnley, 2007), neuroinflammation (Turnley et al., 2002a; Baker et al., 2009; Campbell et al., 2010; Gilli et al., 2010, 2011) and neurotrauma (Stark and Cross, 2006; Qin et al., 2008; Choi et al., 2009; Girolami et al., 2010; Hellstrom et al., 2011). Consistent with the emerging link between excessive inflammation and neurodegenerative disorders (Frank-Cannon et al., 2009), there is now evidence that the SOCS proteins might also have a role to play in the progression of neurodegenerative disorders (Ghosh and Pahan, 2012). In many of the studies cited, the SOCS proteins have been implicated as regulators of inflammatory responses in the central nervous system, however it is important to note that the SOCS proteins also demonstrate neural specific functions in neuronal differentiation. SOCS2 is one member of the SOCS family that is recognized as an important regulator of neuronal function.
Biochemistry of the SOCS Proteins—General
The role of the SOCS proteins has been most extensively studied in the context of cytokine signaling via receptors that lack intrinsic tyrosine kinase activity that recruit effectors such as the cytoplasmic Janus kinases (JAKs) (O'Sullivan et al., 2007). Cytokine binding to the receptor at the cell surface promotes association of the receptor subunits and signals a cascade of downstream phosphorylation events. This signaling pathway begins with the cross-phosphorylation and activation of the receptor-associated JAKs. The JAKs in turn phosphorylate sites on the cytoplasmic tails of the activated receptors thereby creating docking sites for the STAT proteins. Recruitment of these transcription factors is followed by the phosphorylation and dimerization of the STATs. Activated STAT dimers translocate to the nucleus where they can initiate transcription of a variety of genes responsible for survival and proliferation. STAT activation also promotes transcription of negative regulators such as the SOCS family that can suppress further signaling and thus restore sensitivity of the cell to future cytokine stimuli.
The SOCS proteins were first identified as cytokine-inducible inhibitors of signaling with the characterization of CIS (Yoshimura et al., 1995) and SOCS1 (Endo et al., 1997; Naka et al., 1997; Starr et al., 1997). There are several modes by which the SOCS proteins can inhibit cytokine signaling; competition for STAT binding sites, binding to activated receptors or JAKs and promoting their degradation by the proteasome or direct inhibition of the catalytic site of the JAKs (Figure 3). SOCS proteins can inhibit the downstream effectors of the cytokine stimulus that promoted their initial induction (i.e., “negative feedback”) or inhibit signaling intermediates downstream of independent cytokine stimuli (i.e., “cross-talk”).
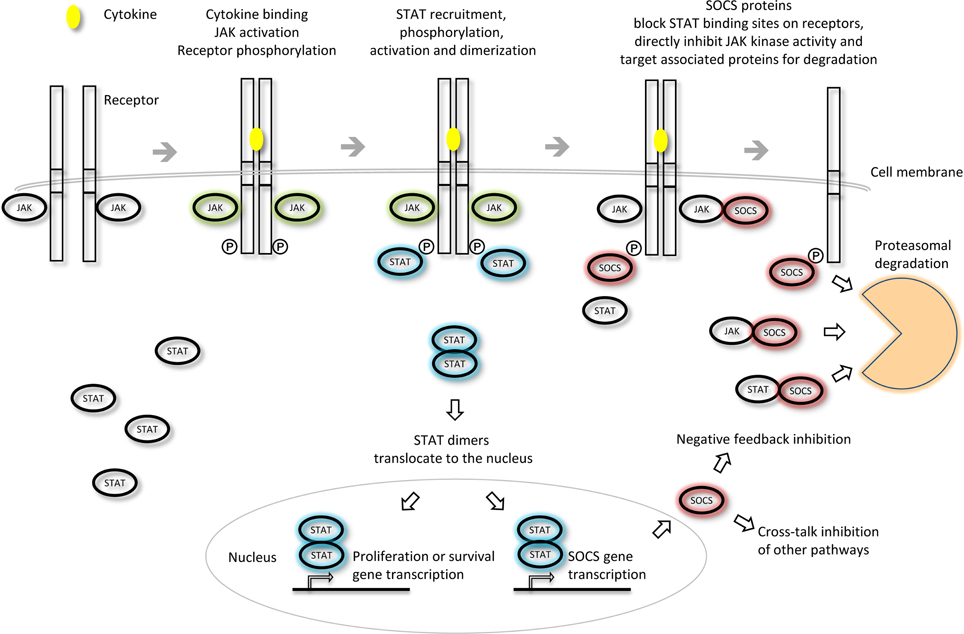
Figure 3. Inhibition of JAK-STAT cytokine signaling by the SOCS proteins. The different SOCS proteins can inhibit JAK-STAT signaling via different mechanisms including competition for STAT binding sites on activated receptors, direct inhibition of JAK kinase activity or targeting of associated signaling molecules for proteasomal degradation. Adapted from Palmer and Restifo (2009).
Each of the 8 SOCS proteins (SOCS1-7 and CIS) comprise a variable N-terminal region, a central Src Homology 2 (SH2) domain and a highly conserved C-terminal SOCS box (Figure 4). SH2 domains recognize and bind to specific tyrosine phosphorylated motifs within proteins whilst the SOCS box motif interacts with E3 ubiquitin ligases thereby targeting any SOCS-associated proteins for degradation by the proteasome (Kamura et al., 1998; Zhang et al., 1999; Krebs et al., 2002). Pairs of SOCS proteins show varying degrees of similarity in their primary amino acid sequence, suggestive of a degree of functional redundancy amongst members of the family (Hilton et al., 1998). The crystal structures of SOCS2 (Bullock et al., 2006), SOCS3 (Babon et al., 2008) and SOCS4 (Bullock et al., 2007) in complex with elongin BC have been solved. In addition, in vitro studies utilizing recombinant purified proteins have demonstrated that the SOCS box motifs of all the SOCS proteins recruit elongin BC before binding Cullin5, an E3 ubiquitin ligase scaffold protein (Babon et al., 2009). A region immediately upstream of the classical SH2 domain, known as the extended SH2 subdomain (ESS), appears crucial to the formation of a stable interface between the SOCS box and the SH2 domain in a subset of the SOCS family comprising CIS, SOCS1, SOCS2, and SOCS3 (Bullock et al., 2006).
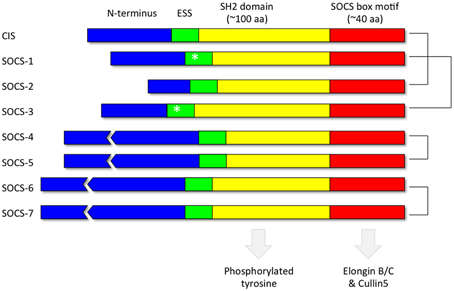
Figure 4. Domain organization of the SOCS proteins. Each of the 8 SOCS proteins (SOCS1-7 and CIS) comprise a variable N-terminal region, a central Src Homology 2 (SH2) domain to recognize motifs containing phosphorylated tyrosine residues and a highly conserved C-terminal SOCS box to facilitate association with components of the ubiquitin ligase machinery such as elongin BC and cullin5. The kinase inhibitory region (KIR) (Sasaki et al., 1999; Yasukawa et al., 1999; Kershaw et al., 2013) within the extended SH2 subdomain (ESS) of SOCS1 and SOCS3 are denoted by an asterisk. The most closely related pairs of SOCS proteins according to amino acid sequence similarity are denoted on the right.
The SOCS proteins couple specific phosphotyrosine-motif recognition with ubiquitin-ligase activity and are thus potent inhibitors of a variety of cytokine signaling pathways. As previously mentioned, the SOCS proteins can participate in “negative feedback” inhibition or “cross-talk” suppression of independent cytokine stimuli. This cross-talk has been shown to extend to the cross regulation amongst the SOCS proteins. SOCS2, SOCS6, and SOCS7 appear to form a subgroup of the SOCS family that have been shown to interact with the SOCS box of all members of the SOCS family and may be required to regulate the stability of other SOCS proteins (Piessevaux et al., 2006). This cross regulation might enable late-induction SOCS proteins (such as SOCS2) to down regulate early-induction SOCS proteins (such as CIS, SOCS1, and SOCS3) so the cell can restore sensitivity to future cytokine stimulation (Tannahill et al., 2005; Piessevaux et al., 2006).
The regulation of receptor trafficking by the SOCS proteins
The SOCS proteins typically act by regulating the ubiquitin tagging of signaling intermediates to promote their proteasome-dependent degradation and thus suppress further signaling. As previously mentioned, a more complex role for post-translational modifications such as ubiquitination has emerged whereby the type and extent of ubiquitin tagging can also dictate the sorting of membrane-associated proteins to early endosomes which can continue to signal after internalization. It is intriguing to speculate the important role that ubiquitin-ligases such as the SOCS proteins may play in both determining the strength or duration of signaling events, and fine-tuning the type of responses of cells such as neurons to extracellular cytokine stimuli. One example that implicates the SOCS proteins in this receptor trafficking role has been observed in the SOCS3 dependent transport of the granulocyte colony stimulating factor receptor (G-CSFR) (Irandoust et al., 2007). G-CSFR transport from the early endosomes to the lysosomes was impaired when a membrane-proximal lysine residue of the G-CSFR was mutated to prevent ubiquitin conjugation at this site and altered ubiquitin conjugation of this receptor resulted in altered intracellular signaling outcomes.
The Biochemistry of SOCS2
SOCS2 was isolated in a yeast-two-hybrid screen in which the cytoplasmic domain of the insulin-like growth factor-1 (IGF-1) receptor was used as bait to screen a cDNA library of proteins expressed in the human fetal brain (Dey et al., 1998). IGF-1 is a second messenger of GH and the essential role of SOCS2 in the negative regulation of the GH/IGF-1 pathway was subsequently validated in vivo by the phenotype of the SOCS2KO mice (Metcalf et al., 2000). The activated GHR engages the JAK/STAT pathway and accordingly the observed induction of SOCS2 by GH (Adams et al., 1998; Tollet-Egnell et al., 1999) is supported by the presence of putative STAT5 transcription factor binding sites, amongst others, in the vicinity of the Socs2 gene (Laz et al., 2009).
The transfection of 293T cells with a series of SOCS2 deletion constructs in addition to the GH receptor highlighted the structural motifs required for the SOCS2 downregulation of signaling via the GH receptor (Greenhalgh et al., 2005). Removal of the SOCS2 N-terminus relieved inhibition of GH signaling and point mutation of critically conserved residues within the phosphotyrosine-binding pocket of the SH2 domain produced partial or complete loss of inhibitory function. Furthermore, deletion of the SOCS box relieved inhibition of GH signaling and actually resulted in enhanced signaling even at low concentrations. The inhibitory role of SOCS2 has been shown to rely on the C-terminal SOCS box in the case of proteasomal degradation of the phosphorylated proline-rich tyrosine kinase 2 (Pyk2) (Lee et al., 2010). The most recent development in our understanding of SOCS2 biochemistry came with the demonstration that SOCS2 can directly regulate the levels of the growth hormone receptor in a 293T overexpression system (Vesterlund et al., 2011). The SOCS2 ubiquitin ligase complex was shown to directly ubiquitinate the GH receptor and co-expression of SOCS2 decreased the half-life of the GHR. This effect of SOCS2 overexpression was removed by inhibition of the proteasome and mutation of GHR at Y487 and an intact SOCS box was required for the stabilization of the SOCS2-ubiquitin ligase complex.
Overexpression of SOCS2, both in vivo and in vitro, has unveiled a complex role for SOCS2 in the modulation of GH signaling and it is clear that SOCS2 can both inhibit and enhance GH signaling (Favre et al., 1999; Greenhalgh et al., 2002a). There are several explanations for the enhancement of GH signaling observed when SOCS2 is overexpressed. It is possible that the overexpressed FLAG-SOCS2 used in the in vitro studies is non-functional and inhibiting the role of endogenous SOCS2, thus the overexpressed SOCS2 is acting as a dominant-negative mutant (Greenhalgh et al., 2002a). Alternatively, at high concentrations SOCS2 might be competing for SOCS3 binding sites on the GH receptor (residues Y332 and Y487) and since SOCS3 is considered a more potent inhibitor of GH signaling, displacing SOCS3 from these binding sites might indirectly enhance GH signaling (Greenhalgh et al., 2002a). Other groups (Tannahill et al., 2005; Piessevaux et al., 2008a) propose that since SOCS2 can bind all members of the SOCS family, SOCS2 may accelerate the turnover of other SOCS proteins via a “cross-regulatory mechanism” that would allow SOCS2 to enhance cytokine responses by accelerating proteasome-dependent turnover of SOCS3. In addition, binding to elongin BC has been shown to be essential for the proper folding and substrate recognition by the SOCS protein CIS (Piessevaux et al., 2008b). If the pool of available elongin BC were depleted by binding to overexpressed SOCS2, this may indirectly enhance cytokine responses that would normally be suppressed by other SOCS proteins.
SOCS2 and Mouse Biology—General
SOCS2 was initially characterized as a negative regulator of growth hormone (GH) signaling (Metcalf et al., 2000). The SOCS2 mediated downregulation of GH signaling is best demonstrated by the gigantism phenotype of mice lacking SOCS2 (Metcalf et al., 2000). The SOCS2KO mice were approximately 40% heavier by 6 weeks of age and this corresponded to an increase in bone length and the size of most organs, although no hematological abnormalities were observed. The difference in body size was accelerated at about 3–4 weeks of age, and this was noted as the time point at which growth hormone receptor is upregulated in many tissues (Shoba et al., 1999). Other aspects of the SOCS2KO phenotype that suggested a role for SOCS2 in GH signaling included an increase in the levels of the GH second messenger insulin-like growth factor-1 (IGF-1), decreased levels of major urinary protein and thickening of the skin due to collagen accumulation.
Mice lacking both STAT5b and SOCS2 and were found to grow normally (Greenhalgh et al., 2002b) which suggested that the SOCS2 modulation of JAK/STAT signaling downstream of the GH receptor depends on the activity of STAT5b. Accordingly, SOCS2 has been shown to interact with the GH receptor in vivo and with STAT5b binding sites on the cytoplasmic tail of the GH receptor (Y487 and Y495) in vitro (Greenhalgh et al., 2005). The importance of SOCS2 as a negative regulator of GH signaling in vivo was further exemplified by the absence of the SOCS2KO phenotype when endogenous GH was also removed (Greenhalgh et al., 2005).
Given that the SOCS2KO mice were hyper-responsive to GH signaling, it was presumed that increased SOCS2 expression would yield mice hypo-responsive to GH signaling with a phenotype similar to the dwarfism observed in GH receptor knockout mice (Zhou et al., 1997). On the contrary, transgenic mice that overexpress SOCS2 (SOCS2TG) displayed a modest overgrowth phenotype indicating that when SOCS2 is either absent or expressed at high levels there is enhanced signaling via the GH receptor (Greenhalgh et al., 2002a). Whilst the SOCS2TG mice were approximately 15% heavier at 3 weeks of age, they did not display any other abnormalities with respect to specific tissues or hematopoietic profile.
SOCS2 and Neurobiology
Neuronal expression profile of SOCS2; probable roles in neuronal proliferation and differentiation
SOCS2 is notable for the high levels of RNA expression in neurons of the developing and adult mouse brain, and interestingly the onset of SOCS2 expression appears to coincide with that of neuronal differentiation (Polizzotto et al., 2000). The neural specific expression of SOCS2 during development prompted investigation into the role of SOCS2 in neuronal differentiation. SOCS2 is expressed in neural stem cells and neurons most highly at E14 which is a time point that coincides with a peak in neuronal generation (Polizzotto et al., 2000). More detailed in vitro analysis of the neuronal expression profile of SOCS2 revealed that it is expressed in cultured embryonic day 10 (E10) neuroepithelial cells and E17 cortical neurons but not cortical astrocytes or adult multipotent stem cells (Turnley et al., 2002b). In the neuroepithelial cultures, Socs2 gene expression was increased above basal levels by the addition of LIF, interferon gamma, OsM and CNTF, and LIF promoted Socs2 gene expression in E17 cortical neuron cultures (Turnley et al., 2002b). Notably, growth hormone (GH) did not influence Socs2 expression in either cell type (Turnley et al., 2002b) unlike previous studies in which GH induced expression of several SOCS proteins including CIS, SOCS1, SOCS2 and SOCS3 in non-neuronal cell lines and tissues (Adams et al., 1998; Tollet-Egnell et al., 1999). A link between SOCS2 and the transcriptional regulation of neurogenesis has been found in the GH regulation of the proneurogenic basic helix loop helix transcription factor Neurogenin-1 (Turnley et al., 2002b).
Neuroanatomical consequences of SOCS2 overexpression
Mice with altered growth hormone responsiveness, including SOCS2KO (hyper-responsive), SOCS2TG (hypo-responsive) and GH receptor null (GHRKO) (non-responsive) mice, exhibit a variety of alterations in neural architecture (Ransome et al., 2004; Ransome and Turnley, 2005). The changes in neural architecture observed in SOCS2TG and SOCS2KO mice implicate this gene in neuronal differentiation and neurite outgrowth. Interestingly, despite the fact that loss of SOCS2 has an effect on mouse body and organ size (Metcalf et al., 2000), it has no effect on brain size, although there was an effect on neuronal density and composition (Ransome et al., 2004; Ransome and Turnley, 2005). The SOCS2KO mice have decreased cortical neuron density whereas the SOCS2TG and GHRKO mice exhibit an increased cortical neuron density. The SOCS2TG mice have increased synaptic density and dendritic branching in the cortex, in contrast to the sparse dendritic branching of pyramidal neurons in the cortex of the SOCS2KO and GHRKO mice. Further, SOCS2TG mice and GHRKO mice had markedly increased numbers of Calretinin and Calbindin positive cortical interneurons, although expression of other markers, such as Parvalbumin and somatostatin were not significantly altered. Furthermore, the overall number of gamma-aminobutyric acid positive interneurons did not appear to change between genotypes. The altered expression of cortical interneuron markers observed in SOCS2TG and SOCS2KO mice suggests that SOCS2 may regulate expression of specific transcription factors required for interneuron subtype specification or that expression of the interneuron markers might be modulated by the amount of synaptic activity within a cell and thus the altered levels of expression in the SOCS2TG and SOCS2KO mice might simply arise from changes in neuronal circuitry and synaptic activity (Ransome and Turnley, 2005). Juxtaposition of the neuroanatomical phenotypes of mice with altered responsiveness to GH signaling (SOCS2KO hyper-responsive > SOCS2TG hypo-responsive > GHRKO non-responsive) reveals that whilst cortical neuron density correlates with increasing responsiveness to GH, the measures of dendritic branching or some interneuron populations do not. This invites speculation that SOCS2 can also modulate other signaling events independent of GH signaling in neural cells.
SOCS2 and neurite outgrowth
The proper formation of connections between cells of the nervous system is critically dependent upon the extension of processes or “neurites” at the surface of neurons. As mentioned previously, a useful model of neuronal-like differentiation is the PC12 cell line derived from a rat pheochromocytoma that undergoes mitotic arrest and extends neurites when serum is withdrawn and NGF is applied (Greene and Tischler, 1976; Greene, 1978). Recent evidence suggests that SOCS2 may play a role in neurite initiation as well as regulating neurite length. Overexpression of SOCS2 in PC12 cells induces neurite outgrowth under non-differentiating conditions (Goldshmit et al., 2004a) and primary cortical neurons derived from transgenic mice that overexpress SOCS2 demonstrate an increase in neurite length and neurite number (Goldshmit et al., 2004b). Despite the fact that SOCS2 is required to overcome the inhibitory effects of GH on neuronal differentiation, SOCS2 has also been shown to enhance neurite outgrowth in neurons derived from neural progenitor cells and this activity was not inhibited by the addition of GH (Scott et al., 2006). These data suggest that the mechanisms by which SOCS2 regulates neuronal differentiation and neurite outgrowth may be independent.
Since these initial reports of SOCS2 involvement in neurite outgrowth, there have been other studies implicating various members of the SOCS family in neurite outgrowth including SOCS3 and SOCS6 (Miao et al., 2006; Gupta et al., 2011). SOCS7 may also emerge as an important regulator of neurite outgrowth as it has been identified as an Nck interacting protein (Kremer et al., 2007) and overexpression of Nck in the PC12 cell line has been shown to enhance proliferation and block NGF and bFGF induced differentiation by a MAPK independent mechanism (Rockow et al., 1996).
SOCS2 as a Novel Regulator of TrkA Localization and Signaling
While SOCS2 has broad ranging effects on neuronal differentiation, maturation and survival, the molecular basis for SOCS2 activity in neurons is still largely unresolved. An altered responsiveness to GH does not appear to address all the changes observed in the neural architecture of SOCS2KO and SOCS2TG mice or the enhanced neurite outgrowth in neurons derived from neural progenitor cells. Previous studies showed that overexpression of SOCS2 in PC12 cells resulted in enhanced neurite outgrowth, and this response was enhanced by the addition of NGF (Goldshmit et al., 2004a). Thus, the possibility that SOCS2 might be regulating the neuronal response to NGF prompted further investigation.
Neonatal dorsal root ganglia (DRG) contain a large population of TrkA+ neurons that require NGF for survival and promotion of neurite outgrowth. Analysis of DRG neuron cultures derived from SOCS2TG and SOCS2KO mice showed that increased or no expression SOCS2 respectively, regulated NGF-mediated neurite outgrowth, with SOCS2 overexpression increasing and lack of SOCS2 decreasing neurite length and complexity (Uren et al., 2014). Surprisingly however, SOCS2 overexpression did not influence NGF-induced survival of DRG neurons, suggesting that SOCS2 differentially regulates TrkA-related signal transduction pathways. In order to try to dissect the mechanisms by which this differential regulation occurred, a range of binding studies, expression and signal transduction analyses were performed using SOCS2 and TrkA transfected 293T cells (contain no endogenous TrkA), SOCS2 transfected PC12 cells (contain endogenous TrkA and SOCS2) and SOCS2TG and wildtype DRG neurons (contain endogenous TrkA and SOCS2).
Co-immunoprecipitation of SOCS2 and TrkA mutant proteins in 293T cells showed that the juxtamembrane region of TrkA was required for binding of SOCS2, with possible involvement of the kinase domain as well. Overexpression of SOCS2 increased total TrkA levels in transfected 293T cells and PC12 cells and increased cell surface expression of TrkA in PC12 cells and SOCS2TG DRG neurons (Uren et al., 2014), suggesting that SOCS2 regulates TrkA protein turnover and cellular localization. Further, SOCS2 overexpression increased the extent and duration of NGF-induced activation of signal transduction pathways involved in neurite outgrowth such as ERK1/2 and pAKT (Uren et al., 2014). These effects of SOCS2 on NGF mediated neurite outgrowth but not survival, with altered TrkA surface expression and enhanced signal transduction pathways are summarized in Table 1.
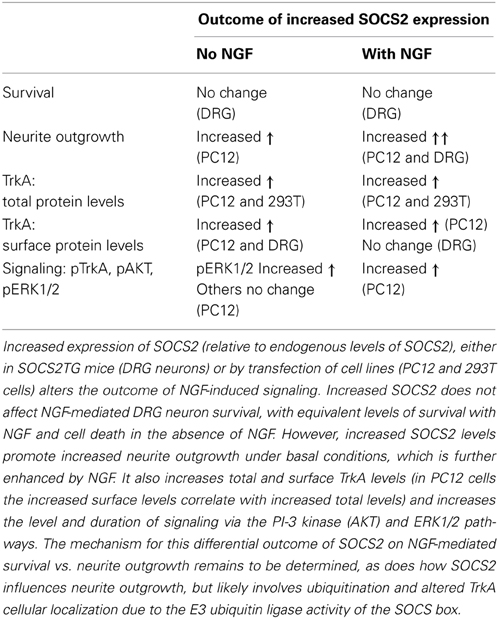
Table 1. SOCS2:TrkA interaction and outcome summary.
While the mechanism by which SOCS2 regulates TrkA signaling and functional outcome remains to be determined, it most likely revolves around altered TrkA localization induced by SOCS2-mediated ubiquitination. This could regulate the amount of TrkA receptor at the cell surface and influence the rate at which it is internalized, recycled, or transported to different intracellular compartments, thereby modulating sensitivity of a cell to NGF and altering downstream signal transduction pathways. Ubiquitination of the lysine in the TrkA juxtamembrane KFG motif regulates TrkA expression levels and localization, with deletion of this domain leading to decreased ubiquitination, increased TrkA levels and increased phosphorylation of the AKT and MAP kinase pathways (Kiris et al., 2014), mirroring the effects of SOCS2 overexpression (Uren et al., 2014). While SOCS2 was able to bind to TrkA with the KFG domain deleted (TrkAΔ450–452) further functional effects of this deletion on TrkA expression, localization or functional effects such as neurite outgrowth were not examined (Uren et al., 2014). However, the similarity between findings when SOCS2 is overexpressed or the TrkA KFG domain is deleted suggests that regulation of ubiquitination by SOCS2, either directly or indirectly, at the KFG motif may be at least part of the mechanism regulating TrkA localization, expression levels and downstream signal transduction. Interestingly, the converse effects of decreased surface TrkA levels have also been described. Depletion of NHE5 [a Na(+)/H(+) exchanger that acidifies recycling endosomes to promote cell surface expression] resulted in decreased levels of TrkA at the cell surface in PC12 cells, with concomitant decreased phosphorylation of AKT and ERK1/2 and decreased neurite outgrowth in response to NGF treatment (Diering et al., 2013).
Conclusions
Neurotrophins and their receptors are expressed in specific areas throughout the nervous system. Their expression levels, signal transduction pathways and localization are tightly regulated by a range of different mechanisms, including dephosphorylation and degradation. SOCS proteins are best known for their role as negative feedback inhibitors of cytokine signaling but have been shown to have a range of other roles, depending on cell type and receptor under study. Their structure, which includes an SH2 interaction domain and the SOCS box ubiquitin ligase interaction domain, suggests that they may interact with many more proteins. This certainly appears to be the case for the TrkA receptor and SOCS2. However, given that SOCS2 has also been shown to bind to the BDNF and NT-3 neurotrophin receptors (Uren et al., 2014), it is possible that SOCS2 plays even broader roles in regulation of neurotrophin signaling. Further, such crosstalk is not necessarily confined to the nervous system and may play a role in hematopoiesis, given the expression and function of Trk receptors in early hematopoietic cells (Chevalier et al., 1994; Ip, 1998; Li et al., 2009; Rezaee et al., 2010). While the mechanisms remain to be determined, SOCS2 and perhaps other SOCS molecules may provide new therapeutic targets for regulation of pathological signaling via neurotrophin receptors and suggests that further research into the field is warranted.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by an NHMRC Fellowship (#628344) to Ann M. Turnley, an Australian Postgraduate Award to Rachel T. Uren and a Melbourne Faculty Research Grant. The authors declare no conflict of interest.
References
Acconcia, F., Sigismund, S., and Polo, S. (2009). Ubiquitin in trafficking: the network at work. Exp. Cell Res. 315, 1610–1618. doi: 10.1016/j.yexcr.2008.10.014
Adams, T. E., Hansen, J. A., Starr, R., Nicola, N. A., Hilton, D. J., and Billestrup, N. (1998). Growth hormone preferentially induces the rapid, transient expression of SOCS-3, a novel inhibitor of cytokine receptor signaling. J. Biol. Chem. 273, 1285–1287. doi: 10.1074/jbc.273.3.1285
Allard, S., Leon, W. C., Pakavathkumar, P., Bruno, M. A., Ribeiro-da-Silva, A., and Cuello, A. C. (2012). Impact of the NGF maturation and degradation pathway on the cortical cholinergic system phenotype. J. Neurosci. 32, 2002–2012. doi: 10.1523/JNEUROSCI.1144-11.2012
Allen, S. J., Watson, J. J., and Dawbarn, D. (2011). The neurotrophins and their role in Alzheimer's disease. Curr. Neuropharmacol. 9, 559–573. doi: 10.2174/157015911798376190
Arevalo, J. C., Conde, B., Hempstead, B. L., Chao, M. V., Martin-Zanca, D., and Perez, P. (2000). TrkA immunoglobulin-like ligand binding domains inhibit spontaneous activation of the receptor. Mol. Cell Biol. 20, 5908–5916. doi: 10.1128/MCB.20.16.5908-5916.2000
Arevalo, J. C., Waite, J., Rajagopal, R., Beyna, M., Chen, Z. Y., Lee, F. S., et al. (2006). Cell survival through Trk neurotrophin receptors is differentially regulated by ubiquitination. Neuron 50, 549–559. doi: 10.1016/j.neuron.2006.03.044
Ascano, M., Bodmer, D., and Kuruvilla, R. (2012). Endocytic trafficking of neurotrophins in neural development. Trends Cell Biol. 22, 266–273. doi: 10.1016/j.tcb.2012.02.005
Babon, J. J., Sabo, J. K., Soetopo, A., Yao, S., Bailey, M. F., Zhang, J. G., et al. (2008). The SOCS box domain of SOCS3: structure and interaction with the elonginBC-cullin5 ubiquitin ligase. J. Mol. Biol. 381, 928–940. doi: 10.1016/j.jmb.2008.06.038
Babon, J. J., Sabo, J. K., Zhang, J. G., Nicola, N. A., and Norton, R. S. (2009). The SOCS box encodes a hierarchy of affinities for Cullin5: implications for ubiquitin ligase formation and cytokine signalling suppression. J. Mol. Biol. 387, 162–174. doi: 10.1016/j.jmb.2009.01.024
Baker, B. J., Akhtar, L. N., and Benveniste, E. N. (2009). SOCS1 and SOCS3 in the control of CNS immunity. Trends Immunol. 30, 392–400. doi: 10.1016/j.it.2009.07.001
Barbacid, M., Lamballe, F., Pulido, D., and Klein, R. (1991). The trk family of tyrosine protein kinase receptors. Biochim. Biophys. Acta 1072, 115–127. doi: 10.1016/0304-419X(91)90010-I
Bhattacharyya, A., Watson, F. L., Bradlee, T. A., Pomeroy, S. L., Stiles, C. D., and Segal, R. A. (1997). Trk receptors function as rapid retrograde signal carriers in the adult nervous system. J. Neurosci. 17, 7007–7016.
Bhattacharyya, A., Watson, F. L., Pomeroy, S. L., Zhang, Y. Z., Stiles, C. D., and Segal, R. A. (2002). High-resolution imaging demonstrates dynein-based vesicular transport of activated Trk receptors. J. Neurobiol. 51, 302–312. doi: 10.1002/neu.10062
Bibel, M., Hoppe, E., and Barde, Y. A. (1999). Biochemical and functional interactions between the neurotrophin receptors trk and p75NTR. EMBO J. 18, 616–622. doi: 10.1093/emboj/18.3.616
Bullock, A. N., Debreczeni, J. E., Edwards, A. M., Sundstrom, M., and Knapp, S. (2006). Crystal structure of the SOCS2-elongin C-elongin B complex defines a prototypical SOCS box ubiquitin ligase. Proc. Natl. Acad. Sci. U.S.A. 103, 7637–7642. doi: 10.1073/pnas.0601638103
Bullock, A. N., Rodriguez, M. C., Debreczeni, J. E., Songyang, Z., and Knapp, S. (2007). Structure of the SOCS4-ElonginB/C complex reveals a distinct SOCS box interface and the molecular basis for SOCS-dependent EGFR degradation. Structure 15, 1493–1504. doi: 10.1016/j.str.2007.09.016
Campbell, I. L. (2005). Cytokine-mediated inflammation, tumorigenesis, and disease-associated JAK/STAT/SOCS signaling circuits in the CNS. Brain Res. Brain Res. Rev. 48, 166–177. doi: 10.1016/j.brainresrev.2004.12.006
Campbell, I. L., Hofer, M. J., and Pagenstecher, A. (2010). Transgenic models for cytokine-induced neurological disease. Biochim. Biophys. Acta 1802, 903–917. doi: 10.1016/j.bbadis.2009.10.004
Chao, M. V. (2003). Neurotrophins and their receptors: a convergence point for many signalling pathways. Nat. Rev. Neurosci. 4, 299–309. doi: 10.1038/nrn1078
Chen, Z. Y., Ieraci, A., Tanowitz, M., and Lee, F. S. (2005). A novel endocytic recycling signal distinguishes biological responses of Trk neurotrophin receptors. Mol. Biol. Cell 16, 5761–5772. doi: 10.1091/mbc.E05-07-0651
Chevalier, S., Praloran, V., Smith, C., MacGrogan, D., Ip, N. Y., Yancopoulos, G. D., et al. (1994). Expression and functionality of the trkA proto-oncogene product/NGF receptor in undifferentiated hematopoietic cells. Blood 83, 1479–1485.
Choi, J. S., Shin, Y. J., Lee, J. Y., Choi, J. Y., Cha, J. H., Chun, M. H., et al. (2009). Enhanced expression of SOCS-2 in the rat hippocampus after transient forebrain ischemia. J. Neurotrauma 26, 2097–2106. doi: 10.1089/neu.2008.0793
Colafrancesco, V., and Villoslada, P. (2011). Targeting NGF pathway for developing neuroprotective therapies for multiple sclerosis and other neurological diseases. Arch. Ital. Biol. 149, 183–192. doi: 10.4449/aib.v149i2.1376
Croker, B. A., Kiu, H., and Nicholson, S. E. (2008). SOCS regulation of the JAK/STAT signalling pathway. Semin. Cell Dev. Biol. 19, 414–422. doi: 10.1016/j.semcdb.2008.07.010
Dechant, G., Biffo, S., Okazawa, H., Kolbeck, R., Pottgiesser, J., and Barde, Y. A. (1993). Expression and binding characteristics of the BDNF receptor chick trkB. Development 119, 545–558.
Delcroix, J. D., Valletta, J. S., Wu, C., Hunt, S. J., Kowal, A. S., and Mobley, W. C. (2003). NGF signaling in sensory neurons: evidence that early endosomes carry NGF retrograde signals. Neuron 39, 69–84. doi: 10.1016/S0896-6273(03)00397-0
Dey, B. R., Spence, S. L., Nissley, P., and Furlanetto, R. W. (1998). Interaction of human suppressor of cytokine signaling (SOCS)-2 with the insulin-like growth factor-I receptor. J. Biol. Chem. 273, 24095–24101. doi: 10.1074/jbc.273.37.24095
Diering, G. H., Numata, Y., Fan, S., Church, J., and Numata, M. (2013). Endosomal acidification by Na+/H+ exchanger NHE5 regulates TrkA cell-surface targeting and NGF-induced PI3K signaling. Mol. Biol. Cell 24, 3435–3448. doi: 10.1091/mbc.E12-06-0445
Disanza, A., Frittoli, E., Palamidessi, A., and Scita, G. (2009). Endocytosis and spatial restriction of cell signaling. Mol. Oncol. 3, 280–296. doi: 10.1016/j.molonc.2009.05.008
Dreyfus, C. F. (1989). Effects of nerve growth factor on cholinergic brain neurons. Trends Pharmacol. Sci. 10, 145–149. doi: 10.1016/0165-6147(89)90166-1
Dreyfus, C. F., Bernd, P., Martinez, H. J., Rubin, S. J., and Black, I. B. (1989). GABAergic and cholinergic neurons exhibit high-affinity nerve growth factor binding in rat basal forebrain. Exp. Neurol. 104, 181–185. doi: 10.1016/S0014-4886(89)80012-3
Duman, R. S., and Voleti, B. (2012). Signaling pathways underlying the pathophysiology and treatment of depression: novel mechanisms for rapid-acting agents. Trends Neurosci. 35, 47–56. doi: 10.1016/j.tins.2011.11.004
Easton, J. B., Royer, A. R., and Middlemas, D. S. (2006). The protein tyrosine phosphatase, Shp2, is required for the complete activation of the RAS/MAPK pathway by brain-derived neurotrophic factor. J. Neurochem. 97, 834–845. doi: 10.1111/j.1471-4159.2006.03789.x
Ehlers, M. D., Kaplan, D. R., Price, D. L., and Koliatsos, V. E. (1995). NGF-stimulated retrograde transport of trkA in the mammalian nervous system. J. Cell Biol. 130, 149–156. doi: 10.1083/jcb.130.1.149
Endo, T. A., Masuhara, M., Yokouchi, M., Suzuki, R., Sakamoto, H., Mitsui, K., et al. (1997). A new protein containing an SH2 domain that inhibits JAK kinases. Nature 387, 921–924. doi: 10.1038/43213
Ernsberger, U. (2009). Role of neurotrophin signalling in the differentiation of neurons from dorsal root ganglia and sympathetic ganglia. Cell Tissue Res. 336, 349–384. doi: 10.1007/s00441-009-0784-z
Faux, C., Hawadle, M., Nixon, J., Wallace, A., Lee, S., Murray, S., et al. (2007). PTPsigma binds and dephosphorylates neurotrophin receptors and can suppress NGF-dependent neurite outgrowth from sensory neurons. Biochim. Biophys. Acta 1773, 1689–1700. doi: 10.1016/j.bbamcr.2007.06.008
Favre, H., Benhamou, A., Finidori, J., Kelly, P. A., and Edery, M. (1999). Dual effects of suppressor of cytokine signaling (SOCS-2) on growth hormone signal transduction. FEBS Lett. 453, 63–66. doi: 10.1016/S0014-5793(99)00681-X
Feng, L., Allen, N. S., Simo, S., and Cooper, J. A. (2007). Cullin 5 regulates Dab1 protein levels and neuron positioning during cortical development. Genes Dev. 21, 2717–2730. doi: 10.1101/gad.1604207
Frank-Cannon, T. C., Alto, L. T., McAlpine, F. E., and Tansey, M. G. (2009). Does neuroinflammation fan the flame in neurodegenerative diseases? Mol. Neurodegener. 4:47. doi: 10.1186/1750-1326-4-47
Geetha, T., Jiang, J., and Wooten, M. W. (2005). Lysine 63 polyubiquitination of the nerve growth factor receptor TrkA directs internalization and signaling. Mol. Cell 20, 301–312. doi: 10.1016/j.molcel.2005.09.014
Georgieva, M. V., de Pablo, Y., Sanchis, D., Comella, J. X., and Llovera, M. (2011). Ubiquitination of TrkA by Nedd4-2 regulates receptor lysosomal targeting and mediates receptor signaling. J. Neurochem. 117, 479–493. doi: 10.1111/j.1471-4159.2011.07218.x
Ghosh, A., and Pahan, K. (2012). Gemfibrozil, a lipid-lowering drug, induces suppressor of cytokine signaling 3 in glial cells: implications for neurodegenerative disorders. J. Biol. Chem. 287, 27189–27203. doi: 10.1074/jbc.M112.346932
Gilli, F., Lindberg, R. L., Valentino, P., Marnetto, F., Malucchi, S., Sala, A., et al. (2010). Learning from nature: pregnancy changes the expression of inflammation-related genes in patients with multiple sclerosis. PLoS ONE 5:e8962. doi: 10.1371/journal.pone.0008962
Gilli, F., Navone, N. D., Perga, S., Marnetto, F., Caldano, M., Capobianco, M., et al. (2011). Loss of braking signals during inflammation: a factor affecting the development and disease course of multiple sclerosis. Arch Neurol. 68, 879–888. doi: 10.1001/archneurol.2011.32
Girolami, E. I., Bouhy, D., Haber, M., Johnson, H., and David, S. (2010). Differential expression and potential role of SOCS1 and SOCS3 in Wallerian degeneration in injured peripheral nerve. Exp. Neurol. 223, 173–182. doi: 10.1016/j.expneurol.2009.06.018
Goldshmit, Y., Greenhalgh, C. J., and Turnley, A. M. (2004b). Suppressor of cytokine signalling-2 and epidermal growth factor regulate neurite outgrowth of cortical neurons. Eur. J. Neurosci. 20, 2260–2266. doi: 10.1111/j.1460-9568.2004.03698.x
Goldshmit, Y., Walters, C. E., Scott, H. J., Greenhalgh, C. J., and Turnley, A. M. (2004a). SOCS2 induces neurite outgrowth by regulation of epidermal growth factor receptor activation. J. Biol. Chem. 279, 16349–16355. doi: 10.1074/jbc.M312873200
Goldsmith, B. A., and Koizumi, S. (1997). Transient association of the phosphotyrosine phosphatase SHP-2 with TrkA is induced by nerve growth factor. J. Neurochem. 69, 1014–1019. doi: 10.1046/j.1471-4159.1997.69031014.x
Greene, L. A. (1978). Nerve growth factor prevents the death and stimulates the neuronal differentiation of clonal PC12 pheochromocytoma cells in serum-free medium. J. Cell Biol. 78, 747–755. doi: 10.1083/jcb.78.3.747
Greene, L. A., and Tischler, A. S. (1976). Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc. Natl. Acad. Sci. U.S.A. 73, 2424–2428. doi: 10.1073/pnas.73.7.2424
Greenhalgh, C. J., Bertolino, P., Asa, S. L., Metcalf, D., Corbin, J. E., Adams, T. E., et al. (2002b). Growth enhancement in suppressor of cytokine signaling 2 (SOCS-2)-deficient mice is dependent on signal transducer and activator of transcription 5b (STAT5b). Mol. Endocrinol. 16, 1394–1406. doi: 10.1210/mend.16.6.0845
Greenhalgh, C. J., Metcalf, D., Thaus, A. L., Corbin, J. E., Uren, R., Morgan, P. O., et al. (2002a). Biological evidence that SOCS-2 can act either as an enhancer or suppressor of growth hormone signaling. J. Biol. Chem. 277, 40181–40184. doi: 10.1074/jbc.C200450200
Greenhalgh, C. J., Rico-Bautista, E., Lorentzon, M., Thaus, A. L., Morgan, P. O., Willson, T. A., et al. (2005). SOCS2 negatively regulates growth hormone action in vitro and in vivo. J. Clin. Invest. 115, 397–406. doi: 10.1172/JCI200522710
Grimes, M. L., Zhou, J., Beattie, E. C., Yuen, E. C., Hall, D. E., Valletta, J. S., et al. (1996). Endocytosis of activated TrkA: evidence that nerve growth factor induces formation of signaling endosomes. J. Neurosci. 16, 7950–7964.
Gupta, S., Mishra, K., Surolia, A., and Banerjee, K. (2011). Suppressor of cytokine signalling-6 promotes neurite outgrowth via JAK2/STAT5-mediated signalling pathway, involving negative feedback inhibition. PLoS ONE 6:e26674. doi: 10.1371/journal.pone.0026674
Hallbook, F., Ibanez, C. F., and Persson, H. (1991). Evolutionary studies of the nerve growth factor family reveal a novel member abundantly expressed in Xenopus ovary. Neuron 6, 845–858. doi: 10.1016/0896-6273(91)90180-8
Hamburger, V., and Levi-Montalcini, R. (1949). Proliferation, differentiation and degeneration in the spinal ganglia of the chick embryo under normal and experimental conditions. J. Exp. Zool. 111, 457–501. doi: 10.1002/jez.1401110308
Hellstrom, M., Muhling, J., Ehlert, E. M., Verhaagen, J., Pollett, M. A., Hu, Y., et al. (2011). Negative impact of rAAV2 mediated expression of SOCS3 on the regeneration of adult retinal ganglion cell axons. Mol. Cell Neurosci. 46, 507–515. doi: 10.1016/j.mcn.2010.12.003
Hendry, I. A., Stockel, K., Thoenen, H., and Iversen, L. L. (1974). The retrograde axonal transport of nerve growth factor. Brain Res. 68, 103–121. doi: 10.1016/0006-8993(74)90536-8
Heymach, J. V. Jr., and Shooter, E. M. (1995). The biosynthesis of neurotrophin heterodimers by transfected mammalian cells. J. Biol. Chem. 270, 12297–12304. doi: 10.1074/jbc.270.20.12297
Hilton, D. J., Richardson, R. T., Alexander, W. S., Viney, E. M., Willson, T. A., Sprigg, N. S., et al. (1998). Twenty proteins containing a C-terminal SOCS box form five structural classes. Proc. Natl. Acad. Sci. U.S.A. 95, 114–119. doi: 10.1073/pnas.95.1.114
Howe, C. L., and Mobley, W. C. (2005). Long-distance retrograde neurotrophic signaling. Curr. Opin. Neurobiol. 15, 40–48. doi: 10.1016/j.conb.2005.01.010
Huang, E. J., and Reichardt, L. F. (2001). Neurotrophins: roles in neuronal development and function. Annu. Rev. Neurosci. 24, 677–736. doi: 10.1146/annurev.neuro.24.1.677
Huang, E. J., and Reichardt, L. F. (2003). Trk receptors: roles in neuronal signal transduction. Annu. Rev. Biochem. 72, 609–642. doi: 10.1146/annurev.biochem.72.121801.161629
Ibanez, C. F. (2007). Message in a bottle: long-range retrograde signaling in the nervous system. Trends Cell Biol. 17, 519–528. doi: 10.1016/j.tcb.2007.09.003
Ihara, S., Nakajima, K., Fukada, T., Hibi, M., Nagata, S., Hirano, T., et al. (1997). Dual control of neurite outgrowth by STAT3 and MAP kinase in PC12 cells stimulated with interleukin-6. EMBO J. 16, 5345–5352. doi: 10.1093/emboj/16.17.5345
Inagaki, N., Thoenen, H., and Lindholm, D. (1995). TrkA tyrosine residues involved in NGF-induced neurite outgrowth of PC12 cells. Eur. J. Neurosci. 7, 1125–1133. doi: 10.1111/j.1460-9568.1995.tb01102.x
Ip, N. Y. (1998). The neurotrophins and neuropoietic cytokines: two families of growth factors acting on neural and hematopoietic cells. Ann. N.Y. Acad. Sci. 840, 97–106. doi: 10.1111/j.1749-6632.1998.tb09553.x
Irandoust, M. I., Aarts, L. H., Roovers, O., Gits, J., Erkeland, S. J., and Touw, I. P. (2007). Suppressor of cytokine signaling 3 controls lysosomal routing of G-CSF receptor. EMBO J. 26, 1782–1793. doi: 10.1038/sj.emboj.7601640
Islam, O., Loo, T. X., and Heese, K. (2009). Brain-derived neurotrophic factor (BDNF) has proliferative effects on neural stem cells through the truncated TRK-B receptor, MAP kinase, AKT, and STAT-3 signaling pathways. Curr. Neurovasc. Res. 6, 42–53. doi: 10.2174/156720209787466028
Johnson, D., Lanahan, A., Buck, C. R., Sehgal, A., Morgan, C., Mercer, E., et al. (1986). Expression and structure of the human NGF receptor. Cell 47, 545–554. doi: 10.1016/0092-8674(86)90619-7
Johnson, E. M. Jr., and Yip, H. K. (1985). Central nervous system and peripheral nerve growth factor provide trophic support critical to mature sensory neuronal survival. Nature 314, 751–752. doi: 10.1038/314751a0
Jullien, J., Guili, V., Derrington, E. A., Darlix, J. L., Reichardt, L. F., and Rudkin, B. B. (2003). Trafficking of TrkA-green fluorescent protein chimerae during nerve growth factor-induced differentiation. J. Biol. Chem. 278, 8706–8716. doi: 10.1074/jbc.M202401200
Jullien, J., Guili, V., Reichardt, L. F., and Rudkin, B. B. (2002). Molecular kinetics of nerve growth factor receptor trafficking and activation. J. Biol. Chem. 277, 38700–38708. doi: 10.1074/jbc.M202348200
Kamura, T., Sato, S., Haque, D., Liu, L., Kaelin, W. G. Jr., Conaway, R. C., et al. (1998). The Elongin BC complex interacts with the conserved SOCS-box motif present in members of the SOCS, ras, WD-40 repeat, and ankyrin repeat families. Genes Dev. 12, 3872–3881. doi: 10.1101/gad.12.24.3872
Kaplan, D. R., Hempstead, B. L., Martin-Zanca, D., Chao, M. V., and Parada, L. F. (1991b). The trk proto-oncogene product: a signal transducing receptor for nerve growth factor. Science 252, 554–558. doi: 10.1126/science.1850549
Kaplan, D. R., Martin-Zanca, D., and Parada, L. F. (1991a). Tyrosine phosphorylation and tyrosine kinase activity of the trk proto-oncogene product induced by NGF. Nature 350, 158–160. doi: 10.1038/350158a0
Kershaw, N. J., Murphy, J. M., Liau, N. P., Varghese, L. N., Laktyushin, A., Whitlock, E. L., et al. (2013). SOCS3 binds specific receptor-JAK complexes to control cytokine signaling by direct kinase inhibition. Nat. Struct. Mol. Biol. 20, 469–476. doi: 10.1038/nsmb.2519
Kiris, E., Wang, T., Yanpallewar, S., Dorsey, S. G., Becker, J., Bavari, S., et al. (2014). TrkA In vivo function is negatively regulated by ubiquitination. J. Neurosci. 34, 4090–4098. doi: 10.1523/JNEUROSCI.4294-13.2014
Klein, R., Jing, S. Q., Nanduri, V., O'Rourke, E., and Barbacid, M. (1991a). The trk proto-oncogene encodes a receptor for nerve growth factor. Cell 65, 189–197. doi: 10.1016/0092-8674(91)90419-Y
Klein, R., Nanduri, V., Jing, S. A., Lamballe, F., Tapley, P., Bryant, S., et al. (1991b). The trkB tyrosine protein kinase is a receptor for brain-derived neurotrophic factor and neurotrophin-3. Cell 66, 395–403. doi: 10.1016/0092-8674(91)90628-C
Komander, D., and Rape, M. (2012). The ubiquitin code. Annu. Rev. Biochem. 81, 203–229. doi: 10.1146/annurev-biochem-060310-170328
Krebs, D. L., Uren, R. T., Metcalf, D., Rakar, S., Zhang, J. G., Starr, R., et al. (2002). SOCS-6 binds to insulin receptor substrate 4, and mice lacking the SOCS-6 gene exhibit mild growth retardation. Mol. Cell Biol. 22, 4567–4578. doi: 10.1128/MCB.22.13.4567-4578.2002
Kremer, B. E., Adang, L. A., and Macara, I. G. (2007). Septins regulate actin organization and cell-cycle arrest through nuclear accumulation of NCK mediated by SOCS7. Cell 130, 837–850. doi: 10.1016/j.cell.2007.06.053
Laz, E. V., Sugathan, A., and Waxman, D. J. (2009). Dynamic in vivo binding of STAT5 to growth hormone-regulated genes in intact rat liver. Sex-specific binding at low- but not high-affinity STAT5 sites. Mol. Endocrinol. 23, 1242–1254. doi: 10.1210/me.2008-0449
Lee, F. S., Kim, A. H., Khursigara, G., and Chao, M. V. (2001). The uniqueness of being a neurotrophin receptor. Curr. Opin. Neurobiol. 11, 281–326. doi: 10.1016/S0959-4388(00)00209-9
Lee, S. H., Yun, S., Piao, Z. H., Jeong, M., Kim, D. O., Jung, H., et al. (2010). Suppressor of cytokine signaling 2 regulates IL-15-primed human NK cell function via control of phosphorylated Pyk2. J. Immunol. 185, 917–928. doi: 10.4049/jimmunol.1000784
Li, Z., Beutel, G., Rhein, M., Meyer, J., Koenecke, C., Neumann, T., et al. (2009). High-affinity neurotrophin receptors and ligands promote leukemogenesis. Blood 113, 2028–2037. doi: 10.1182/blood-2008-05-155200
Lou, X., Yano, H., Lee, F., Chao, M. V., and Farquhar, M. G. (2001). GIPC and GAIP form a complex with TrkA: a putative link between G protein and receptor tyrosine kinase pathways. Mol. Biol. Cell 12, 615–627. doi: 10.1091/mbc.12.3.615
Mahadeo, D., Kaplan, L., Chao, M. V., and Hempstead, B. L. (1994). High affinity nerve growth factor binding displays a faster rate of association than p140trk binding. Implications for multi-subunit polypeptide receptors. J. Biol. Chem. 269, 6884–6891.
Marsh, H. N., Dubreuil, C. I., Quevedo, C., Lee, A., Majdan, M., Walsh, G. S., et al. (2003). SHP-1 negatively regulates neuronal survival by functioning as a TrkA phosphatase. J. Cell Biol. 163, 999–1010. doi: 10.1083/jcb.200309036
McPherson, P. S., Kay, B. K., and Hussain, N. K. (2001). Signaling on the endocytic pathway. Traffic 2, 375–384. doi: 10.1034/j.1600-0854.2001.002006375.x
Metcalf, D., Greenhalgh, C. J., Viney, E., Willson, T. A., Starr, R., Nicola, N. A., et al. (2000). Gigantism in mice lacking suppressor of cytokine signalling-2. Nature 405, 1069–1073. doi: 10.1038/35016611
Miao, T., Wu, D., Zhang, Y., Bo, X., Subang, M. C., Wang, P., et al. (2006). Suppressor of cytokine signaling-3 suppresses the ability of activated signal transducer and activator of transcription-3 to stimulate neurite growth in rat primary sensory neurons. J. Neurosci. 26, 9512–9519. doi: 10.1523/JNEUROSCI.2160-06.2006
Miranda, C., Fumagalli, T., Anania, M. C., Vizioli, M. G., Pagliardini, S., Pierotti, M. A., et al. (2010). Role of STAT3 in in vitro transformation triggered by TRK oncogenes. PLoS ONE 5:e9446. doi: 10.1371/journal.pone.0009446
Naka, T., Narazaki, M., Hirata, M., Matsumoto, T., Minamoto, S., Aono, A., et al. (1997). Structure and function of a new STAT-induced STAT inhibitor. Nature 387, 924–929. doi: 10.1038/43219
Ng, Y. P., Cheung, Z. H., and Ip, N. Y. (2006). STAT3 as a downstream mediator of Trk signaling and functions. J. Biol. Chem. 281, 15636–15644. doi: 10.1074/jbc.M601863200
Obermeier, A., Halfter, H., Wiesmuller, K. H., Jung, G., Schlessinger, J., and Ullrich, A. (1993a). Tyrosine 785 is a major determinant of Trk—substrate interaction. EMBO J. 12, 933–941.
Obermeier, A., Lammers, R., Wiesmuller, K. H., Jung, G., Schlessinger, J., and Ullrich, A. (1993b). Identification of Trk binding sites for SHC and phosphatidylinositol 3′-kinase and formation of a multimeric signaling complex. J. Biol. Chem. 268, 22963–22966.
Okada, N., Wada, K., Goldsmith, B. A., and Koizumi, S. (1996). SHP-2 is involved in neurotrophin signaling. Biochem. Biophys. Res. Commun. 229, 607–611. doi: 10.1006/bbrc.1996.1851
Ong, S. H., Guy, G. R., Hadari, Y. R., Laks, S., Gotoh, N., Schlessinger, J., et al. (2000). FRS2 proteins recruit intracellular signaling pathways by binding to diverse targets on fibroblast growth factor and nerve growth factor receptors. Mol. Cell Biol. 20, 979–989. doi: 10.1128/MCB.20.3.979-989.2000
Ossipov, M. H. (2011). Growth factors and neuropathic pain. Curr. Pain Headache Rep. 15, 185–192. doi: 10.1007/s11916-011-0183-5
O'Sullivan, L. A., Liongue, C., Lewis, R. S., Stephenson, S. E., and Ward, A. C. (2007). Cytokine receptor signaling through the Jak-Stat-Socs pathway in disease. Mol. Immunol. 44, 2497–2506. doi: 10.1016/j.molimm.2006.11.025
Palmer, D. C., and Restifo, N. P. (2009). Suppressors of cytokine signaling (SOCS) in T cell differentiation, maturation, and function. Trends Immunol. 30, 592–602. doi: 10.1016/j.it.2009.09.009
Paravicini, U., Stoeckel, K., and Thoenen, H. (1975). Biological importance of retrograde axonal transport of nerve growth factor in adrenergic neurons. Brain Res. 84, 279–291. doi: 10.1016/0006-8993(75)90982-8
Piessevaux, J., De Ceuninck, L., Catteeuw, D., Peelman, F., and Tavernier, J. (2008b). Elongin B/C recruitment regulates substrate binding by CIS. J. Biol. Chem. 283, 21334–21346. doi: 10.1074/jbc.M803742200
Piessevaux, J., Lavens, D., Montoye, T., Wauman, J., Catteeuw, D., Vandekerckhove, J., et al. (2006). Functional cross-modulation between SOCS proteins can stimulate cytokine signaling. J. Biol. Chem. 281, 32953–32966. doi: 10.1074/jbc.M600776200
Piessevaux, J., Lavens, D., Peelman, F., and Tavernier, J. (2008a). The many faces of the SOCS box. Cytokine Growth Factor Rev. 19, 371–381. doi: 10.1016/j.cytogfr.2008.08.006
Polizzotto, M. N., Bartlett, P. F., and Turnley, A. M. (2000). Expression of “suppressor of cytokine signalling” (SOCS) genes in the developing and adult mouse nervous system. J. Comp. Neurol. 423, 348–358. doi: 10.1002/1096-9861(20000724)423:2<348::AID-CNE11>3.0.CO;2-W
Qin, H., Niyongere, S. A., Lee, S. J., Baker, B. J., and Benveniste, E. N. (2008). Expression and functional significance of SOCS-1 and SOCS-3 in astrocytes. J. Immunol. 181, 3167–3176. doi: 10.4049/jimmunol.181.5.3167
Radeke, M. J., Misko, T. P., Hsu, C., Herzenberg, L. A., and Shooter, E. M. (1987). Gene transfer and molecular cloning of the rat nerve growth factor receptor. Nature 325, 593–597. doi: 10.1038/325593a0
Ransome, M. I., Goldshmit, Y., Bartlett, P. F., Waters, M. J., and Turnley, A. M. (2004). Comparative analysis of CNS populations in knockout mice with altered growth hormone responsiveness. Eur. J. Neurosci. 19, 2069–2079. doi: 10.1111/j.0953-816X.2004.03308.x
Ransome, M. I., and Turnley, A. M. (2005). Analysis of neuronal subpopulations in mice over-expressing suppressor of cytokine signaling-2. Neuroscience 132, 673–687. doi: 10.1016/j.neuroscience.2004.12.041
Ransome, M. I., and Turnley, A. M. (2007). Systemically delivered Erythropoietin transiently enhances adult hippocampal neurogenesis. J. Neurochem. 102, 1953–1965. doi: 10.1111/j.1471-4159.2007.04684.x
Reichardt, L. F. (2006). Neurotrophin-regulated signalling pathways. Philos. Trans. R. Soc. Lond. B Biol. Sci. 361, 1545–1564. doi: 10.1098/rstb.2006.1894
Rezaee, F., Rellick, S. L., Piedimonte, G., Akers, S. M., O'Leary, H. A., Martin, K., et al. (2010). Neurotrophins regulate bone marrow stromal cell IL-6 expression through the MAPK pathway. PLoS ONE 5:e9690. doi: 10.1371/journal.pone.0009690
Rockow, S., Tang, J., Xiong, W., and Li, W. (1996). Nck inhibits NGF and basic FGF induced PC12 cell differentiation via mitogen-activated protein kinase-independent pathway. Oncogene 12, 2351–2359.
Rodriguez-Tebar, A., Dechant, G., and Barde, Y. A. (1990). Binding of brain-derived neurotrophic factor to the nerve growth factor receptor. Neuron 4, 487–492. doi: 10.1016/0896-6273(90)90107-Q
Rodriguez-Tebar, A., Dechant, G., Gotz, R., and Barde, Y. A. (1992). Binding of neurotrophin-3 to its neuronal receptors and interactions with nerve growth factor and brain-derived neurotrophic factor. EMBO J. 11, 917–922.
Romanelli, R. J., and Wood, T. L. (2008). Directing traffic in neural cells: determinants of receptor tyrosine kinase localization and cellular responses. J. Neurochem. 105, 2055–2068. doi: 10.1111/j.1471-4159.2008.05263.x
Rosario, M., Franke, R., Bednarski, C., and Birchmeier, W. (2007). The neurite outgrowth multiadaptor RhoGAP, NOMA-GAP, regulates neurite extension through SHP2 and Cdc42. J. Cell Biol. 178, 503–516. doi: 10.1083/jcb.200609146
Rusanescu, G., Yang, W., Bai, A., Neel, B. G., and Feig, L. A. (2005). Tyrosine phosphatase SHP-2 is a mediator of activity-dependent neuronal excitotoxicity. EMBO J. 24, 305–314. doi: 10.1038/sj.emboj.7600522
Sadowski, L., Pilecka, I., and Miaczynska, M. (2009). Signaling from endosomes: location makes a difference. Exp. Cell Res. 315, 1601–1609. doi: 10.1016/j.yexcr.2008.09.021
Sasaki, A., Yasukawa, H., Suzuki, A., Kamizono, S., Syoda, T., Kinjyo, I., et al. (1999). Cytokine-inducible SH2 protein-3 (CIS3/SOCS3) inhibits Janus tyrosine kinase by binding through the N-terminal kinase inhibitory region as well as SH2 domain. Genes Cells 4, 339–351. doi: 10.1046/j.1365-2443.1999.00263.x
Schneider, R., and Schweiger, M. (1991). A novel modular mosaic of cell adhesion motifs in the extracellular domains of the neurogenic trk and trkB tyrosine kinase receptors. Oncogene 6, 1807–1811.
Scott, H. J., Stebbing, M. J., Walters, C. E., McLenachan, S., Ransome, M. I., Nichols, N. R., et al. (2006). Differential effects of SOCS2 on neuronal differentiation and morphology. Brain Res. 1067, 138–145. doi: 10.1016/j.brainres.2005.10.032
Scuri, M., Samsell, L., and Piedimonte, G. (2010). The role of neurotrophins in inflammation and allergy. Inflamm. Allergy Drug Targets 9, 173–180. doi: 10.2174/187152810792231913
Shintani, T., and Noda, M. (2008). Protein tyrosine phosphatase receptor type Z dephosphorylates TrkA receptors and attenuates NGF-dependent neurite outgrowth of PC12 cells. J. Biochem. 144, 259–266. doi: 10.1093/jb/mvn064
Shoba, L., An, M. R., Frank, S. J., and Lowe, W. L. Jr. (1999). Developmental regulation of insulin-like growth factor-I and growth hormone receptor gene expression. Mol. Cell Endocrinol. 152, 125–136. doi: 10.1016/S0303-7207(99)00045-3
Skaper, S. D. (2012). The neurotrophin family of neurotrophic factors: an overview. Methods Mol. Biol. 846, 1–12. doi: 10.1007/978-1-61779-536-7_1
Snider, W. D. (1994). Functions of the neurotrophins during nervous system development: what the knockouts are teaching us. Cell 77, 627–638. doi: 10.1016/0092-8674(94)90048-5
Song, E. J., Hong, H. M., and Yoo, Y. S. (2009). Proteasome inhibition induces neurite outgrowth through posttranslational modification of TrkA receptor. Int. J. Biochem. Cell Biol. 41, 539–545. doi: 10.1016/j.biocel.2008.04.022
Stark, J. L., and Cross, A. H. (2006). Differential expression of suppressors of cytokine signaling-1 and -3 and related cytokines in central nervous system during remitting versus non-remitting forms of experimental autoimmune encephalomyelitis. Int. Immunol. 18, 347–353. doi: 10.1093/intimm/dxh373
Starr, R., Willson, T. A., Viney, E. M., Murray, L. J., Rayner, J. R., Jenkins, B. J., et al. (1997). A family of cytokine-inducible inhibitors of signalling. Nature 387, 917–921. doi: 10.1038/43206
Stephens, R. M., Loeb, D. M., Copeland, T. D., Pawson, T., Greene, L. A., and Kaplan, D. R. (1994). Trk receptors use redundant signal transduction pathways involving SHC and PLC-gamma 1 to mediate NGF responses. Neuron 12, 691–705. doi: 10.1016/0896-6273(94)90223-2
Takahashi, Y., Shimokawa, N., Esmaeili-Mahani, S., Morita, A., Masuda, H., Iwasaki, T., et al. (2011). Ligand-induced downregulation of TrkA is partly regulated through ubiquitination by Cbl. FEBS Lett. 585, 1741–1747. doi: 10.1016/j.febslet.2011.04.056
Tannahill, G. M., Elliott, J., Barry, A. C., Hibbert, L., Cacalano, N. A., and Johnston, J. A. (2005). SOCS2 can enhance interleukin-2 (IL-2) and IL-3 signaling by accelerating SOCS3 degradation. Mol. Cell Biol. 25, 9115–9126. doi: 10.1128/MCB.25.20.9115-9126.2005
Tollet-Egnell, P., Flores-Morales, A., Stavreus-Evers, A., Sahlin, L., and Norstedt, G. (1999). Growth hormone regulation of SOCS-2, SOCS-3, and CIS messenger ribonucleic acid expression in the rat. Endocrinology 140, 3693–3704.
Tsui-Pierchala, B. A., and Ginty, D. D. (1999). Characterization of an NGF-P-TrkA retrograde-signaling complex and age-dependent regulation of TrkA phosphorylation in sympathetic neurons. J. Neurosci. 19, 8207–8218.
Turnley, A. M., Faux, C. H., Rietze, R. L., Coonan, J. R., and Bartlett, P. F. (2002b). Suppressor of cytokine signaling 2 regulates neuronal differentiation by inhibiting growth hormone signaling. Nat. Neurosci. 5, 1155–1162. doi: 10.1038/nn954
Turnley, A. M., Starr, R., and Bartlett, P. F. (2001). SOCS1 regulates interferon-gamma mediated sensory neuron survival. Neuroreport 12, 3443–3445. doi: 10.1097/00001756-200111160-00013
Turnley, A. M., Starr, R., and Bartlett, P. F. (2002a). Failure of sensory neurons to express class I MHC is due to differential SOCS1 expression. J. Neuroimmunol. 123, 35–40. doi: 10.1016/S0165-5728(01)00480-5
Uren, R. T., Turbic, A., Wong, A. W., Klein, R., Murray, S. S., and Turnley, A. M. (2014). A novel role of suppressor of cytokine signaling-2 in the regulation of TrkA neurotrophin receptor biology. J. Neurochem. 129, 614–627. doi: 10.1111/jnc.12671
Vaegter, C. B., Jansen, P., Fjorback, A. W., Glerup, S., Skeldal, S., Kjolby, M., et al. (2011). Sortilin associates with Trk receptors to enhance anterograde transport and neurotrophin signaling. Nat. Neurosci. 14, 54–61. doi: 10.1038/nn.2689
Valdez, G., Akmentin, W., Philippidou, P., Kuruvilla, R., Ginty, D. D., and Halegoua, S. (2005). Pincher-mediated macroendocytosis underlies retrograde signaling by neurotrophin receptors. J. Neurosci. 25, 5236–5247. doi: 10.1523/JNEUROSCI.5104-04.2005
Valdez, G., Philippidou, P., Rosenbaum, J., Akmentin, W., Shao, Y., and Halegoua, S. (2007). Trk-signaling endosomes are generated by Rac-dependent macroendocytosis. Proc. Natl. Acad. Sci. U.S.A. 104, 12270–12275. doi: 10.1073/pnas.0702819104
Van Kanegan, M. J., and Strack, S. (2009). The protein phosphatase 2A regulatory subunits B'beta and B'delta mediate sustained TrkA neurotrophin receptor autophosphorylation and neuronal differentiation. Mol. Cell Biol. 29, 662–674. doi: 10.1128/MCB.01242-08
Varsano, T., Dong, M. Q., Niesman, I., Gacula, H., Lou, X., Ma, T., et al. (2006). GIPC is recruited by APPL to peripheral TrkA endosomes and regulates TrkA trafficking and signaling. Mol. Cell Biol. 26, 8942–8952. doi: 10.1128/MCB.00305-06
Vesterlund, M., Zadjali, F., Persson, T., Nielsen, M. L., Kessler, B. M., Norstedt, G., et al. (2011). The SOCS2 ubiquitin ligase complex regulates growth hormone receptor levels. PLoS ONE 6:e25358. doi: 10.1371/journal.pone.0025358
Wang, J., and Campbell, I. L. (2002). Cytokine signaling in the brain: putting a SOCS in it? J. Neurosci. Res. 67, 423–427. doi: 10.1002/jnr.10145
Watson, F. L., Heerssen, H. M., Moheban, D. B., Lin, M. Z., Sauvageot, C. M., Bhattacharyya, A., et al. (1999). Rapid nuclear responses to target-derived neurotrophins require retrograde transport of ligand-receptor complex. J. Neurosci. 19, 7889–7900.
Windisch, J. M., Auer, B., Marksteiner, R., Lang, M. E., and Schneider, R. (1995a). Specific neurotrophin binding to leucine-rich motif peptides of TrkA and TrkB. FEBS Lett. 374, 125–129. doi: 10.1016/0014-5793(95)01047-I
Windisch, J. M., Marksteiner, R., Lang, M. E., Auer, B., and Schneider, R. (1995b). Brain-derived neurotrophic factor, neurotrophin-3, and neurotrophin-4 bind to a single leucine-rich motif of TrkB. Biochemistry 34, 11256–11263. doi: 10.1021/bi00035a035
Wu, C., Cui, B., He, L., Chen, L., and Mobley, W. C. (2009). The coming of age of axonal neurotrophin signaling endosomes. J. Proteomics 72, 46–55. doi: 10.1016/j.jprot.2008.10.007
Yamamoto, S., Yoshino, I., Shimazaki, T., Murohashi, M., Hevner, R. F., Lax, I., et al. (2005). Essential role of Shp2-binding sites on FRS2alpha for corticogenesis and for FGF2-dependent proliferation of neural progenitor cells. Proc. Natl. Acad. Sci. U.S.A. 102, 15983–15988. doi: 10.1073/pnas.0507961102
Yamashita, T., Higuchi, H., and Tohyama, M. (2002). The p75 receptor transduces the signal from myelin-associated glycoprotein to Rho. J. Cell Biol. 157, 565–570. doi: 10.1083/jcb.200202010
Yamashita, T., Tucker, K. L., and Barde, Y. A. (1999). Neurotrophin binding to the p75 receptor modulates Rho activity and axonal outgrowth. Neuron 24, 585–593. doi: 10.1016/S0896-6273(00)81114-9
Yano, H., and Chao, M. V. (2005). Biochemical characterization of intracellular membranes bearing Trk neurotrophin receptors. Neurochem. Res. 30, 767–777. doi: 10.1007/s11064-005-6870-z
Yasukawa, H., Misawa, H., Sakamoto, H., Masuhara, M., Sasaki, A., Wakioka, T., et al. (1999). The JAK-binding protein JAB inhibits Janus tyrosine kinase activity through binding in the activation loop. EMBO J. 18, 1309–1320. doi: 10.1093/emboj/18.5.1309
Ye, H., Kuruvilla, R., Zweifel, L. S., and Ginty, D. D. (2003). Evidence in support of signaling endosome-based retrograde survival of sympathetic neurons. Neuron 39, 57–68. doi: 10.1016/S0896-6273(03)00266-6
Yoshimura, A., Ohkubo, T., Kiguchi, T., Jenkins, N. A., Gilbert, D. J., Copeland, N. G., et al. (1995). A novel cytokine-inducible gene CIS encodes an SH2-containing protein that binds to tyrosine-phosphorylated interleukin 3 and erythropoietin receptors. EMBO J. 14, 2816–2826.
Yu, T., Calvo, L., Anta, B., Lopez-Benito, S., Southon, E., Chao, M. V., et al. (2011). Regulation of trafficking of activated TrkA is critical for NGF-mediated functions. Traffic 12, 521–534. doi: 10.1111/j.1600-0854.2010.01156.x
Yuan, X. B., Jin, M., Xu, X., Song, Y. Q., Wu, C. P., Poo, M. M., et al. (2003). Signalling and crosstalk of Rho GTPases in mediating axon guidance. Nat. Cell Biol. 5, 38–45. doi: 10.1038/ncb895
Zagrebelsky, M., Holz, A., Dechant, G., Barde, Y. A., Bonhoeffer, T., and Korte, M. (2005). The p75 neurotrophin receptor negatively modulates dendrite complexity and spine density in hippocampal neurons. J. Neurosci. 25, 9989–9999. doi: 10.1523/JNEUROSCI.2492-05.2005
Zhang, J. G., Farley, A., Nicholson, S. E., Willson, T. A., Zugaro, L. M., Simpson, R. J., et al. (1999). The conserved SOCS box motif in suppressors of cytokine signaling binds to elongins B and C and may couple bound proteins to proteasomal degradation. Proc. Natl. Acad. Sci. U.S.A. 96, 2071–2076. doi: 10.1073/pnas.96.5.2071
Zhang, Y., Moheban, D. B., Conway, B. R., Bhattacharyya, A., and Segal, R. A. (2000). Cell surface Trk receptors mediate NGF-induced survival while internalized receptors regulate NGF-induced differentiation. J. Neurosci. 20, 5671–5678.
Zhou, Y., Xu, B. C., Maheshwari, H. G., He, L., Reed, M., Lozykowski, M., et al. (1997). A mammalian model for Laron syndrome produced by targeted disruption of the mouse growth hormone receptor/binding protein gene (the Laron mouse). Proc. Natl. Acad. Sci. U.S.A. 94, 13215–13220. doi: 10.1073/pnas.94.24.13215
Keywords: NGF, ubiquitin, signal transduction, Trk receptors, PC12 cells, DRG neurons, SOCS-2, neurite outgrowth
Citation: Uren RT and Turnley AM (2014) Regulation of neurotrophin receptor (Trk) signaling: suppressor of cytokine signaling 2 (SOCS2) is a new player. Front. Mol. Neurosci. 7:39. doi: 10.3389/fnmol.2014.00039
Received: 13 March 2014; Accepted: 21 April 2014;
Published online: 14 May 2014.
Edited by:
Andreas Vlachos, Goethe University Frankfurt, GermanyReviewed by:
Hansjürgen Volkmer, NMI an der Universität Tübingen, GermanyAsya Rolls, Technion, Israel Technology Institute, Israel
Copyright © 2014 Uren and Turnley. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ann M. Turnley, Department of Anatomy and Neuroscience, Melbourne Brain Centre, 30 Royal Parade, The University of Melbourne, Melbourne, VIC 3010, Australia e-mail:dHVybmxleUB1bmltZWxiLmVkdS5hdQ==
†Present address: Rachel T. Uren, The Walter and Eliza Hall Institute of Medical Research, Parkville, Australia