 Shannon E. Boye
Shannon E. Boye- Department of Ophthalmology, University of Florida, Gainesville, FL, USA
Vertebrate species possess two retinal guanylate cyclases (retGC1 and retGC2) and at least two guanylate cyclase activating proteins (GCAPs), GCAP1 and GCAP2. GCAPs function as Ca2+ sensors that regulate the activity of guanylate cyclases. Together, these proteins regulate cGMP and Ca2+ levels within the outer segments of rod and cone photoreceptors. Mutations in GUCY2D, the gene that encodes retGC1, are a leading cause of the most severe form of early onset retinal dystrophy, Leber congenital amaurosis (LCA1). These mutations, which reduce or abolish the ability of retGC1 to replenish cGMP in photoreceptors, are thought to lead to the biochemical equivalent of chronic light exposure in these cells. In spite of this, the majority of LCA1 patients retain normal photoreceptor laminar architecture aside from foveal cone outer segment abnormalities, suggesting they may be good candidates for gene replacement therapy. Work began in the 1980s to characterize multiple animal models of retGC1 deficiency. 34 years later, all models have been used in proof of concept gene replacement studies toward the goal of developing a therapy to treat GUCY2D-LCA1. Here we use the results of these studies as well as those of recent clinical studies to address specific questions relating to clinical application of a gene therapy for treatment of LCA1.
Introduction
The ability to process light into an electrochemical signal depends on the precise regulation of levels of cGMP and Ca2+ within the outer segments of rod and cone photoreceptor cells. In the absence of light stimulation, intracellular cGMP and Ca2+ levels are high and a continuous flow of Na+ and Ca2+ ions through cGMP-gated channels and Na+/Ca2+ exchangers keeps the cell in a depolarized state. Activation of rhodopsin or cone opsins in rod and cone outer segments, respectively, leads to activation of transducin that in turn activates cGMP phosphodiesterase (PDE) leading to a reduction in the concentration of cGMP in the outer segment. Reduction of cGMP leads to closure of cGMP-gated channels, reduced Na+/Ca2+ influx and hyperpolarization of the cell (Pugh et al., 1997). Recovery of light stimulated photoreceptors to the dark state is hastened by the decrease in intracellular Ca2+ that results from continued expulsion of Ca2+ by Na+/Ca2+-K+ exchangers. Calcium has several roles in photoreceptor cells, one of which is to regulate the activity of retinal guanylate cyclases, retGC1 and retGC2 (Koch and Stryer, 1988). It does so by binding to guanylate cyclase activating proteins (GCAPs), a family of calcium-binding proteins that regulate the activity of retGCs (Polans et al., 1996). Two variants of guanylate cyclases (retGC1 and retGC2) and at least two variants of GCAPs (GCAP1 and GCAP2) are present in the outer segments of vertebrate photoreceptors (Shyjan et al., 1992; Margulis et al., 1993; Dizhoor et al., 1994; Goraczniak et al., 1994, 1997; Palczewski et al., 1994; Lowe et al., 1995; Yang et al., 1995; Imanishi et al., 2004). In the dark, high levels of intracellular Ca2+ promote its binding to GCAP and inhibit the ability of this protein to activate retGCs. Reduction of free, intracellular Ca2+ following light stimulation leads to a decrease in the amount of Ca2+ bound to GCAP in exchange for Mg2+, which allows this protein to activate retGCs (Peshenko and Dizhoor, 2006; Figure 1). In vivo, retGC1 is the preferred target of GCAP1 (Olshevskaya et al., 2012). In vitro, some studies show that GCAP1 and GCAP2 can activate both retGCs, albeit with slightly different affinities and sensitivity to Ca2+ (Hwang et al., 2003; Peshenko and Dizhoor, 2004; Peshenko et al., 2011) while another shows that GCAP1 cannot activate retGC2 (Haeseleer et al., 1999).
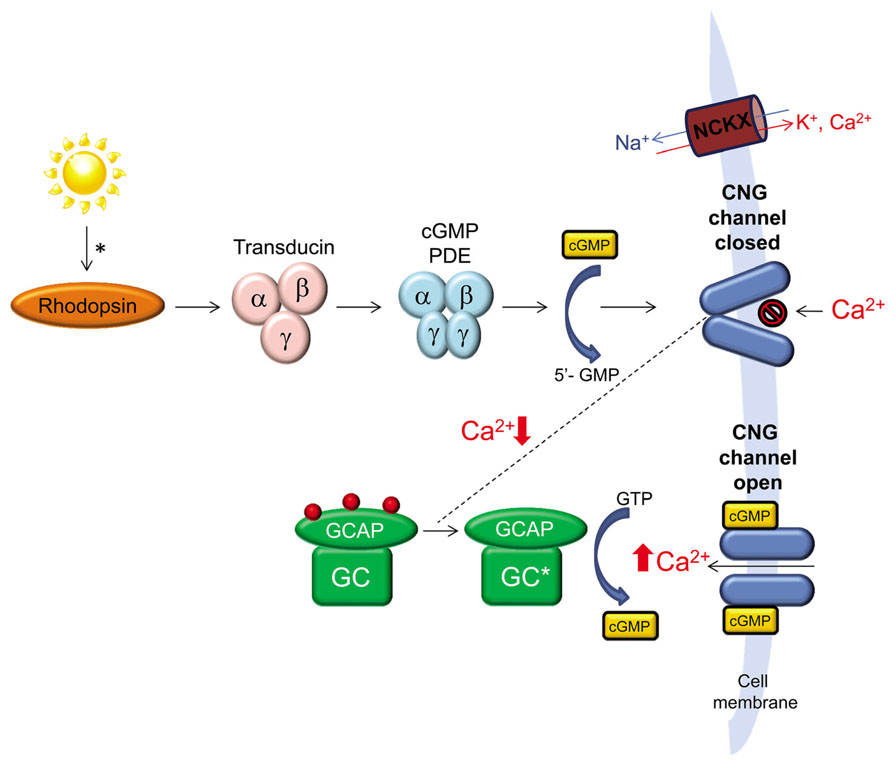
FIGURE 1. The role of retinal guanylate cyclase (retGC) in phototransduction. A photon of light activates rhodopsin initiating a cascade of events, the end result of which is reduction of cGMP, closure of cGMP-gated channels and reduction of intracellular Ca2+ in photoreceptor outer segments. Continued expulsion of Ca2+ by Na+/Ca2+-K+ exchangers (NCKX) results in activation of retGC by GCAP. retGC replenishes intracellular cGMP which reopens cGMP-gated channels allowing for Ca2+ influx and a return of the photoreceptor to its dark-adapted state.
Mutations in GUCY2D, the gene that encodes retGC1, are associated with the severe, early onset, autosomal recessive disorder Leber congenital amaurosis-1 (LCA1; Perrault et al., 1996, 2000). LCA1 causing mutations are found throughout GUCY2D, can alter the enzyme’s structure or stability, affect transport of other peripheral membrane associated protein, and are frequently null (Karan et al., 2010; Jacobson et al., 2013). Without fully functional retGC1 to replenish intracellular cGMP, as is the case in LCA1, cGMP-gated cation channels will remain closed as they are in the light stimulated state. Hence, mutations in GUCY2D are thought to induce the biochemical equivalent of chronic light exposure in photoreceptors. LCA1 patients routinely present with severely reduced visual acuities, attenuated or extinguished electroretinogram (ERG), nystagmus, digito-ocular signs, and apparently normal fundus appearance (Perrault et al., 1999a, 2000; Hanein et al., 2004; Yzer et al., 2006; den Hollander et al., 2008). While early reports indicated retinal degeneration was associated with this form of LCA (Milam et al., 2003; Porto et al., 2003), later studies revealed preservation of retinal laminar architecture in LCA1 patients (Simonelli et al., 2007; Pasadhika et al., 2010; Jacobson et al., 2013).
Preservation of retinal structure despite profound visual disturbance suggests that LCA1 patients would be good candidates for gene replacement therapy. Before initiating such clinical trials there are major questions that need to be answered. Here we stepwise address these questions by reviewing findings of proof of concept studies utilizing different animal models of retGC1 deficiency (results summarized in Table 1). Additionally, we provide an update on efforts to clinically apply a gene therapy for LCA1.
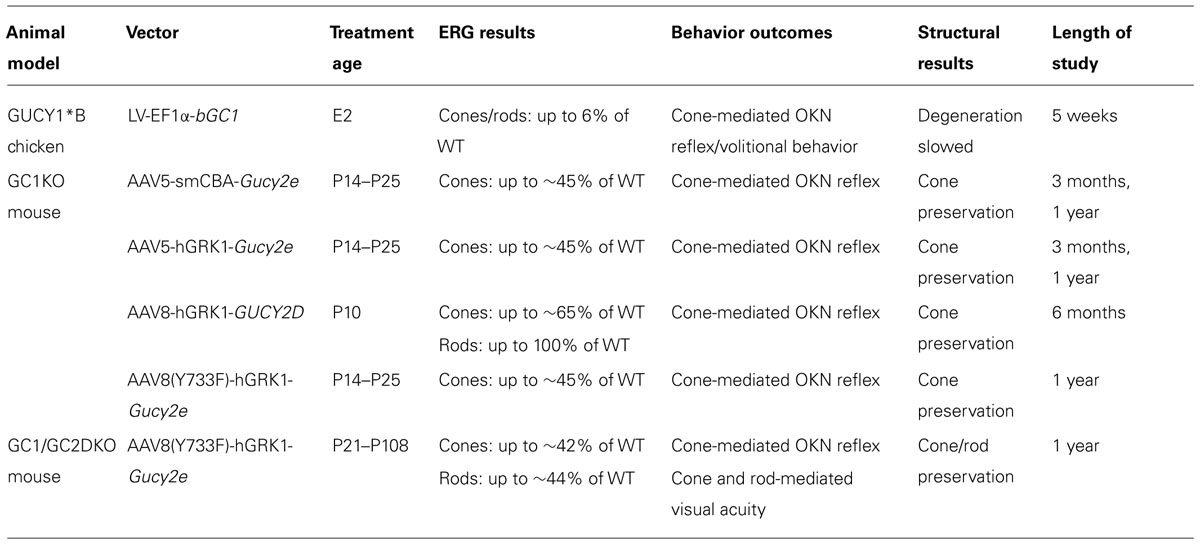
TABLE 1. Summary of proof of concept experiments.
Will GUCY2D Gene Replacement Restore Retinal Function in Vivo?
The first animal model used to investigate gene replacement for retGC1 was the retinal degeneration (rd) chicken, also referred to as the GUCY1*B chicken. This cone- dominant, avian model carries a null, naturally occurring deletion/insertion mutation in the gene encoding retGC1 (Semple-Rowland et al., 1998). As a consequence of retGC1 deficiency, photoreceptors of post-hatch day 1 (P1) GUCY1*B chickens have only 10–20% cGMP relative to that found in age-matched, wild type controls and affected chickens are blind at hatch. They have unrecordable ERGs and lack optokinetic and volitional visual behavior (Ulshafer et al., 1984; Williams et al., 2006). These biochemical and visual disturbances occur prior to photoreceptor loss which doesnot begin until 1 week post-hatch (Cheng et al., 1980; Ulshafer et al., 1984) and proceeds in a central to peripheral fashion with cones completely lost by 3.5 months and rods lost by 8 months (Ulshafer and Allen, 1985a,b).
Due to the technical difficulty associated with performing subretinal injections in chicken, an in ovo treatment paradigm was developed. For its ability to stably transduce retinal progenitor cells (Miyoshi et al., 1997), HIV-1-based lentivirus (LV) was used to deliver a cDNA encoding bovine retGC1 under the control of an elongation factor 1 alpha (EF1α) promoter to the neural tube of developing GUCY1*B embryos (Williams et al., 2006). Bovine retGC1 (bGC1) was chosen for its verified activity in the presence of chicken GCAPs in vitro (Williams et al., 2006). At 1 month post-hatch, ERG testing under both dark and light-adapted conditions revealed that LV-EF1α-bGC1 treatment produced modest increases in photoreceptor-mediated a- wave amplitudes in treated chickens (~6% of wild type). These results were the first demonstration that retGC1 gene replacement could restore retinal function. ERG improvements were associated with a restoration of visually guided behavior, as assessed by optokinetic reflex testing (Wallman and Velez, 1985) and volitional visually guided behavior tests (Williams et al., 2006). In addition, a slowing of retinal degeneration was observed (Williams et al., 2006). Taken together, these results were exciting proof of concept that gene replacement therapy could be effective for treatment of LCA1. However, there were multiple limitations associated with this study that needed to be overcome. First, the therapeutic effect was transient, with ERG and behavioral responses disappearing after the latest time points analyzed (5 weeks post-hatch) and retinal degeneration continuing unabated (Williams et al., 2006; Verrier et al., 2011). Second, multiple facets of the therapeutic strategy lacked clinical translatability- (1) Therapeutic retGC1 was delivered embryonically, a currently untenable task in patients and also requiring in-utero genotyping. (2) An integrating, lentiviral vector was used. While proven effective for transducing retinal precursors, LV has demonstrated little to no ability to transduce post-mitotic photoreceptors (Miyoshi et al., 1997; Bainbridge et al., 2001; Pang et al., 2006; Lipinski et al., 2014). (3) Data was obtained in a non-mammalian model of retGC1 deficiency. Together, this highlighted the need to test therapy using more clinically relevant animal models and vector platform.
Will Post-Natally Delivered retGC1 Restore Retinal Function in a Mammalian Model of retGC1 Deficiency?
The first mammalian model retGC1 deficiency to be described was the guanylate cyclase-1 knockout (GC1KO) mouse (Yang et al., 1999). Insertion of a neomycin resistance cassette in exon 5 of Gucy2e (the murine homolog of GUCY2D) resulted in truncation of retGC1, rendering it null (Yang et al., 1999). Cone-mediated ERGs are unrecordable by 5 weeks but cones are not completely degenerated in this mouse until ~6 months of age (Yang et al., 1999; Coleman et al., 2004). Rods, on the other hand, do not degenerate and maintain variable levels of function (~30–50% of WT; Yang et al., 1999) which is owed to the presence of retGC2 in those cells (Baehr et al., 2007). GCAP1 and GCAP2 transcripts and GCAP1 expression are down-regulated (Coleman et al., 2004) and light-induced translocation of cone arrestin is disrupted as a result of retGC1 deficiency in this model (Coleman and Semple-Rowland, 2005). The temporal dissociation between loss of cone function and structure in GC1KO mice, provides a window of therapy for gene replacement.
For its safety profile and proven ability to transduce post-mitotic photoreceptors following subretinal injection of murine retina (Yang et al., 2002), a serotype 5 adeno associated virus (AAV) was chosen to deliver bovine retGC1 (the same cDNA delivered to the GUCY1*B chicken) under the control of either the ubiquitous, small CBA (smCBA) or photoreceptor-specific, murine opsin (MOPS) promoter to the GC1KO mouse (Haire et al., 2006). Post-natal day 21 (P21) treatment with AAV5-bGC1 failed to improve retinal function (ERG) in treated mice. However, AAV-mediated retGC1 expression was restricted to photoreceptor outer segments and restoration of light-induced cone arrestin translocation, a biochemical correlate of functionality in these cells, was observed at 4 weeks post-treatment (Haire et al., 2006), suggesting that the light signaling cascade in GC1KO cones had been at least partially reset following treatment.
Investigators speculated that the species non-specific nature of the delivered transgene may have dampened the therapeutic outcome. Indeed, subretinal delivery of murine Gucy2e via an AAV5 vector containing either the ubiquitous smCBA or photoreceptor-specific human rhodopsin kinase (hGRK1) promoter led to robust improvements (~45% of WT) in cone-mediated ERGs (Boye et al., 2010). These improvements were stable over the course of this initial study (at least 3 months post-treatment) and provided the first evidence that an AAV-based vector could restore retinal function to a mammalian model of retGC1 deficiency (Boye et al., 2010). Later studies would go on to show that stable restoration of cone function (ERG) is achievable over the long term (Boye et al., 2011; Mihelec et al., 2011). Subretinally delivered AAV8 containing the hGRK1 promoter and human GUCY2D cDNA stably restored cone function (~65% of WT) for at least 6 months (Mihelec et al., 2011). In the longest follow up reported to date, subretinally delivered AAV5 or capsid mutant AAV8(Y733F) containing either the smCBA or hGRK1 promoter and murine Gucy2e cDNA stably restored cone function (~45% of WT) for at least 1 year post-treatment (Boye et al., 2011). A notable difference between these two studies was the level of cone function achieved (65 vs. 45% of WT). Mihelec et al. (2011) subretinally delivered AAV-GUCY2D to GC1KO mice at P10 (an age prior to natural eye opening) whereas Boye et al. (2011) delivered AAV-Gucy2e between P14–P25. Earlier intervention was likely more effective at combating the chronic effects of hyperpolarization that GC1KO cones endure upon light stimulation. Regardless of these differences, the robust and stable functional improvements in cone function following AAV-retGC1 treatment (including with a clinically relevant human cDNA) in GC1KO mice laid the groundwork for development of an AAV-based treatment for LCA1.
As a follow up to these studies, cone-mediated function was also evaluated in the retGC1/retGC2 double knockout (GCDKO mouse following subretinal delivery of AAV8(Y733F)-hGRK1-Gucy2e (Boye et al., 2013). Treatment of this model, in which cones and rods are functionally silent and progressively degenerate, also resulted in stable and robust improvements in cone function for at least 1 year post-treatment (Boye et al., 2013). As in all GC1KO studies, AAV-mediated retGC1 expression was restricted to the outer segments of rods and cones in treated GCDKO mice. The GCDKO mouse and gene replacement experiments in this model will be discussed in more detail below.
Does Restoration of Cone Function Translate into Useful Vision?
Adeno associated virus-mediated expression of retGC1 in photoreceptors of GC1KO mice effectively reset the phototransduction cascade in cones, allowing them to send electrochemical signals to downstream neurons which were detected via full field ERG electrodes (Boye et al., 2010, 2011; Mihelec et al., 2011). However, the existence of functional neural circuits within the retina does not ensure that electrochemical signals are properly relayed to higher order processing centers in the brain and experienced as “vision.” Even if the biochemical defect within photoreceptors is corrected via restoration of retGC1, it is unclear what role amblyopia (the inability of the retina to send visual information to the brain) will play on the treatment outcome. To gain insight into this question, multiple visually guided behavior tests were used to further characterize therapy in AAV-treated GC1KO mice.
Two tests, each of which evaluates the integrity of different visual processing centers of the brain, were used to determine whether useful cone vision could be restored to treated mice. A virtual optokinetic system was used to evaluate the integrity of subcortical retinal efferents (Douglas et al., 2005). While untreated GC1KO mice lack cone-mediated behavior, significant improvements in cone-mediated spatial frequency thresholds and contrast sensitivities were seen in GC1KO mice treated with AAV5-hGRK1-Gucy2e, AAV5-smCBA-Gucy2e, AAV8(Y733F)-hGRK1-Gucy2e or AAV8-hGRK1-GUCY2D (Boye et al., 2010, 2011; Mihelec et al., 2011) with behavior resembling that of normal, sighted congenic mice. Improvements in cone-mediated behavior were observed out to 4 months post-treatment (Mihelec et al., 2011), the latest time point evaluated in this mouse model. Analysis of cone-mediated behavior was also evaluated in GCDKO mice via Morris Water Maze (under photopic conditions), a test used to analyze cortically driven visual behavior (Boye et al., 2013). AAV8(Y733F)-hGRK1-Gucy2e treated mice exhibited significantly reduced escape latencies (the amount of time required to escape water bath via a platform demarcated with a flag) relative to untreated controls. Importantly, behavior of treated mice was not significantly different from age-matched WT controls. Notably, these results were obtained in 1 year old GCDKO mice treated 11 months prior, highlighting that retGC1 supplementation confers useful cone-mediated vision to mammalian models of LCA1 over the long term. This is significant as humans rely heavily on their cone-mediated vision for activities of daily living. It is also interesting to note that while cone-mediated ERG improvements in both GC1KO and GCDKO controls ranged between 45 and 65% that of WT controls, cone-mediated behavior was restored to “normal” in AAV-treated mice supporting previous work showing that behavior is a more sensitive indicator of therapy (Williams and Jacobs, 2007). This was also shown to be the case in LV-retGC1 treated GUCY1*B chickens whose modest ERG responses corresponded, albeit transiently, to robust visually guided behavior (Williams et al., 2006).
How do Rod Photoreceptors Respond to Treatment?
Prior to the most comprehensive clinical characterization of LCA1 patients published to date (Jacobson et al., 2013), multiple reports had described that both rod/cone function and rod/cone structure were compromised (Perrault et al., 1999a,b; Milam et al., 2003; Porto et al., 2003; den Hollander et al., 2008; Chung and Traboulsi, 2009; Jacobson et al., 2013). It was clear that, despite the presence of retGC2 in these cells (Lowe et al., 1995), rod photoreceptors of LCA1 patients are negatively impacted by retGC1 deficiency. Thus, the specific effects of post-natal gene replacement therapy on this photoreceptor subclass needed to be analyzed. Prior studies were conducted in a model that lacks rod degeneration and retains variable levels of rod function throughout its life, the GC1KO mouse. For these reasons, it was not possible to address whether gene replacement would prevent degeneration of rods or definitively restore their function, respectively. Significant improvements in rod-mediated ERGs were only obtained when vector was delivered prior to natural eye opening (P10; Mihelec et al., 2011), but it is difficult to ascertain whether these gains are meaningful because GC1KO mice exhibit normal rod-mediated visual behavior. It wasnot until generation of the GCDKO mouse that it became possible to ask these questions (Baehr et al., 2007).
Deletion of both retGC1 and retGC2 in the GCDKO mouse (Baehr et al., 2007) renders both cones and rods functionally silent (ERGs are unrecordable) and degenerative (Baehr et al., 2007). Several cone proteins (GCAP1, GCAP2, cone opsins, cone transducin, cone PDE, GRK1) are downregulated/mislocalized) and PDE6 is absent from rods (Baehr et al., 2007) in this model. By 2 months of age, overall outer segment length is reduced to 30–50% of normal (Baehr et al., 2007). Appreciable thinning of the outer nuclear layer (ONL) is apparent by ~3.5 months of age and by 6 months, only 3–4 photoreceptor nuclei remain (Boye et al., 2013).
For its success in the studies described above, AAV8(Y733)-hGRK1 was chosen to subretinally deliver Gucy2e to cohorts of GCDKO mice of various age groups (P18, P21–P25, P37–P49, and P108). ERG revealed that, like the cone results described above, rod function was also stably restored to ~40% of normal (at least 1 year post-treatment; Boye et al., 2013). Morris Water Maze testing revealed that these functional gains translated to useful rod-mediated vision, with AAV-retGC1- treated GCDKO mice performing the task at speeds that did not differ significantly from age-matched WT controls (Boye et al., 2013). As in the treated GC1KO mice, WT-like behavior was achievable in GCDKO mice that exhibited only partial ERG recovery. Optical coherence tomography (OCT) was used to monitor the rate of photoreceptor degeneration (predominantly rods) in treated vs. untreated mice. By 7–12 months post-treatment, there was significant structural preservation in all treated GCDKOs, barring those treated at the latest time point (P108), although retinas in these mice still exhibited greater preservation than controls (Boye et al., 2013). Importantly, the rate of photoreceptor cell loss in GCDKO mice treated as late as P49 was no different from that of age-matched WT controls. GCAP1 and GCAP2 were expressed at WT-like levels at 1 year post-injection and cone opsins were present only in treated cones (Boye et al., 2013). Taken together, this study showed for the first time that, along with the rescue effects seen on cones, AAV-mediated retGC1 expression can restore rod photoreceptor function and rod-mediated visual behavior and preserve rod structure over the long term in an animal model of LCA1.
What is the Functional Efficiency of AAV- Delivered retGC1 Enzyme?
In considering clinical application of a gene therapy, an important question becomes “how efficient is the vector-derived, recombinant gene product relative to the native protein?” The ability to assess this in vivo provides a powerful bioassay which can be used to establish dose-response relationships and equivalencies for batches of clinical vectors. Guanylate cyclase activity assays are performed on dark-adapted retinas to determine how much cGMP is produced by retGCs in the presence of a [α-32P]GTP substrate (Olshevskaya et al., 2004; Peshenko et al., 2011). This assay doesnot discriminate between the activity of retGC1 and retGC2. Therefore, isolating the functional efficiency of exogenous retGC1 requires delivery to photoreceptors that lack both endogenous retGC1 and retGC2, such as those in the GCDKO mouse. Boye et al. (2013) compared retGC activity in retinas of GCDKO mice that were treated subretinally between P30–P60 with AAV8(Y733F)-hGRK1-Gucy2e, relative to untreated GCDKO or WT controls and those that received a serotype-matched control vector expressing GFP. Maximal retGC activity in AAV-Gucy2e treated retinas was ~63% of normal. The known contribution to total cyclase activity by retGC2 in this assay is between 20 and 28% (Peshenko et al., 2011). In addition, the area of retGC1 expression in treated retinas is known to be restricted to the subretinal injection bleb (Timmers et al., 2001; Cideciyan et al., 2008). “Good” subretinal injections typically result in ~80–90% detachment meaning not all photoreceptors expressed the enzyme. Taking these two factors into account, the level of retGC1 activity suggested its near-complete restoration in the area exposed to vector. When the maximal activity of AAV-retGC1 was normalized to that seen in WT retinas, it was determined that the calcium sensitivity of the exogenous and endogenous enzymes were identical (Boye et al., 2013). Moving forward, this assay may be used to evaluate the relative potency of clinical vectors.
What are the Consequences of retGC1 Deficiency on Human Rod and Cone Photoreceptor Structure and Function?
While there is general consensus that LCA1 is associated with severely attenuated or ablated ERG (Perrault et al., 1999b, 2000; Hanein et al., 2004; Yzer et al., 2006; den Hollander et al., 2008), reports on the extent of photoreceptor degeneration associated with this form of LCA have been conflicting (Milam et al., 2003; Porto et al., 2003; Simonelli et al., 2007; Pasadhika et al., 2010). The most thorough clinical characterization of LCA1 performed to date focused on a cohort of patients ranging in age from 6 months to 37 years with different mutations in GUCY2D and has provided new insight on the effects of retGC1 deficiency on retinal structure and function (Jacobson et al., 2013).
Similar to previous reports Simonelli et al. (2007), Pasadhika et al. (2010), Jacobson et al. (2013) found that LCA1 patients retained normal retinal laminar architecture aside from foveal cone outer segment abnormalities and, in some cases, foveal cone loss. Within rod-dominant retina, ONL thickness and outer segment lengths were normal in all patients (Jacobson et al., 2013). Unlike previous reports, Jacobson et al. (2013) found that LCA1 patients can retain substantial rod function. Full field sensitivity testing (FST) revealed all patients effectively used their rods to detect blue stimuli, albeit at reduced sensitivities. These psychophysical findings were supported by ERG, pupillometry and mobility testing, all of which revealed variable levels of retained rod function that did not correlate with the patient’s age (Jacobson et al., 2013). In contrast, the majority of patients lacked cone sensitivity (FST) which correlated to severely reduced visual acuity and a lack of color perception. FST, microperimetry and mobility tests revealed a small subset had detectable, but reduced cone function with central fixation and some color perception. None of the patients had recordable cone-mediated ERGs (Jacobson et al., 2013).
To better understand these differential cone phenotypes, Jacobson et al. (2013) investigated the activity of patient-specific retGC1 mutants in vitro. Their activity was assayed by transfecting cDNA for each form, along with GCAPs in HEK293 cells under appropriate ionic conditions and quantifying the amount of cGMP produced relative to that achieved with wild type retGC1. Of the patient-specific mutants tested, some exhibited reduced catalytic activity (4–5 fold lower than WT) while others demonstrated no loss of function (Jacobson et al., 2013). Not surprisingly, those mutants that retained biochemical activity were expressed by LCA1 patients with measurable cone function. It is expected that those with normal activity in vitro may exhibit defective folding, expression or impaired trafficking to photoreceptor outer segments in situ. Investigations are currently underway to understand the fate of these mutants in vivo via AAV-mediated expression in GCDKO mice. retGC1 mutants that exhibited no activity in vitro corresponded to patients with profound loss of cone function (Jacobson et al., 2013).
What form Might a Clinical Trial for GUCY2D-LCA1 Take?
A few important choices must be made prior to clinical application of a gene replacement therapy for LCA1. These include the injection route and location, AAV serotype and promoter, and therapeutic endpoints. Route and location of injection should be dictated by the calculated risk(s)/benefit. The structural integrity of the patient’s retina and locus of existing vision must be considered. Evidence suggests GUCY2D-LCA1 is unlike any form of LCA studied in detail to date (Jacobson et al., 1998, 2003, 2005, 2007a,b, 2009b, 2011; Cideciyan et al., 2007). Their hallmark retinal preservation suggests that LCA1 patients may be good candidates for subretinal injection of AAV-GUCY2D (Simonelli et al., 2007; Pasadhika et al., 2010; Jacobson et al., 2013). Because retGC1 deficiency leads to profound visual impairment, stemming primarily from cones, the central retina should be the treatment target. In the small subset of patients that exhibit foveal cone losses, para- or peri-foveal areas may be targeted in the hopes that, like some RPE65-LCA2 patients, they will develop an eccentric locus of fixation (Cideciyan et al., 2009).
AAV5 and AAV8-based vectors were used in the aforementioned studies for developing a treatment for LCA1. These vectors have proven utility for photoreceptor-targeted therapy following subretinal injection in multiple mouse and dog models of inherited retinal disease (Min et al., 2005; Alexander et al., 2007; Boye et al., 2010, 2013; Gorbatyuk et al., 2010; Komaromy et al., 2010; Mao et al., 2011; Pang et al., 2011, 2012; Yao et al., 2011; Beltran et al., 2012; Petit et al., 2012; Lheriteau et al., 2014). Because AAV transduction profiles and the activity of promoters can vary across species, the final decision should be dictated by the serotype and promoter combination’s behavior in a species most closely related to man, non-human primate (NHP). In LCA1, photoreceptors of the central retina are the treatment target. Therefore, serotype and promoter combinations that effectively transduce cone photoreceptors in NHP are required. Transduction of both AAV5 and AAV8 vectors have recently been described in subretinally injected NHP (Boye et al., 2012; Vandenberghe et al., 2013). Only partial cone transduction was achieved following subretinal delivery of elevated doses of AAV8 containing the ubiquitous CMV promoter driving GFP (1011 vg delivered; Vandenberghe et al., 2011, 2013). At a lower dose (1010 vg delivered), AAV8-CMV-GFP failed to transduce foveal, parafoveal or perifoveal cones (Vandenberghe et al., 2011). In contrast, foveal, parafoveal, perifoveal and peripheral cones of NHP (as well as rods) were all effectively transduced following subretinal delivery of an AAV5 vector containing the photoreceptor-specific hGRK1 promoter (1010 vg delivered; Boye et al., 2012). This mirrors earlier findings in rodent that hGRK1 has exclusive activity in cones and rods (Khani et al., 2007; Tan et al., 2009; Boye et al., 2010, 2011; Pawlyk et al., 2010; Sun et al., 2010). Taken together, these results support the use of subretinally delivered AAV5-hGRK1-GUCY2D in a clinical setting.
Because LCA1 patients exhibit profound cone-mediated visual impairment and variable levels of rod-mediated vision, typical measures of vision must be supplemented with assays that are more suited to this severe disease. Most patients have nystagmus, making full field ERG recordings difficult. Additionally, because many LCA1 patients have unrecordable ERGs to begin with, it would be impossible to measure any negative treatment-associated changes with this assay. Tests which control for nystagmus such as microperimetry can be used to assess cone-mediated central vision (Cideciyan et al., 2012). Tests such as chromatic FST which was used in the most recent clinical characterization (Jacobson et al., 2013) and in the clinical trial for RPE65-LCA2 (Jacobson et al., 2012) could be used to measure localized improvements in rod function. The limits of variability for this assay have already been defined in patients with various retinal degenerations, including LCA (Roman et al., 2005; Roman et al., 2007; Jacobson et al., 2009a). OCT should be performed to monitor the safety of injections performed under the fovea or para/peri- fovea, with emphasis placed on the former given the slow return of cone outer segment structure observed in LCA2 patients treated subfoveally (Jacobson et al., 2012). The potential effects of amblyopia must also be considered. It is possible that AAV-mediated expression of wild type retGC1 in cones of LCA1 patients will reset their signaling cascade allowing for electrochemical signals to propagate through the retina, but that these signals will not be effectively relayed to the brain. Magnetic resonance imaging studies could be used to confirm the integrity of higher order visual processing pathways prior to treatment. Fully understanding how the brain of an LCA1 patient will accommodate GUCY2D gene replacement and whether this will differ depending the patient’s mutation and/or level of cone preservation will only be accomplished through a clinical trial.
Conclusion
Information gleaned from proof of concept experiments in multiple animal models of retGC1 deficiency have laid the groundwork for development of an AAV-based treatment for GUCY2D-LCA1 (Haire et al., 2006; Williams et al., 2006; Boye et al., 2010, 2011; Mihelec et al., 2011). Thorough clinical characterization of patients has provided new insights into the pathophysiology of this severe early onset inherited retinal disease, pointing to the central retina as the target for treatment and highlighting the importance of selecting appropriate outcome measures by which to score therapeutic efficacy in a clinical trial (Jacobson et al., 2013). Studies in NHP have revealed an optimal AAV serotype (AAV5) and promoter (hGRK1) combination for delivery of therapeutic transgene to all cone subclasses and rods (Boye et al., 2012). The majority of genes (13/19) known to account for some form of LCA, including GUCY2D, encode photoreceptor specific proteins (den Hollander et al., 2008; Wang et al., 2009; Estrada-Cuzcano et al., 2011; Sergouniotis et al., 2011; Abu-Safieh et al., 2013). In fact, inherited retinal diseases as a whole are caused primarily by defects in proteins expressed by photoreceptors (Wright et al., 2010), a cell type yet to be targeted by AAV in a clinical setting. Thus, developing a treatment for GUCY2D-LCA1 will serve as a framework for other photoreceptor-targeted gene replacement strategies.
Conflict of Interest Statement
Shannon E. Boye is a co-inventor on US patent # 61/327,521 which covers some aspects of the material discussed within.
Acknowledgment
I would like to thank Sanford L. Boye for his thoughtful critique of this review.
References
Abu-Safieh, L., Alrashed, M., Anazi, S., Alkuraya, H., Khan, A. O., Al-Owain, M., et al. (2013). Autozygome-guided exome sequencing in retinal dystrophy patients reveals pathogenetic mutations and novel candidate disease genes. Genome Res. 23, 236–247. doi: 10.1101/gr.144105.112
Alexander, J. J., Umino, Y., Everhart, D., Chang, B., Min, S. H., Li, Q., et al. (2007). Restoration of cone vision in a mouse model of achromatopsia. Nat. Med. 13, 685–687. doi: 10.1038/nm1596
Baehr, W., Karan, S., Maeda, T., Luo, D. G., Li, S., Bronson, J. D., et al. (2007). The function of guanylate cyclase 1 (GC1) and guanylate cyclase 2 (GC2) in rod and cone photoreceptors. J. Biol. Chem. 282, 8837–8847. doi: 10.1074/jbc.M610369200
Bainbridge, J. W., Stephens, C., Parsley, K., Demaison, C., Halfyard, A., Thrasher, A. J., et al. (2001). In vivo gene transfer to the mouse eye using an HIV-based lentiviral vector; efficient long-term transduction of corneal endothelium and retinal pigment epithelium. Gene Ther. 8, 1665–1668. doi: 10.1038/sj.gt.3301574
Beltran, W. A., Cideciyan, A. V., Lewin, A. S., Iwabe, S., Khanna, H., Sumaroka, A., et al. (2012). Gene therapy rescues photoreceptor blindness in dogs and paves the way for treating human X-linked retinitis pigmentosa. Proc. Natl. Acad. Sci. U.S.A. 109, 2132–2137. doi: 10.1073/pnas.1118847109
Boye, S. E., Alexander, J. J., Boye, S. L., Witherspoon, C. D., Sandefer, K. J., Conlon, T. J., et al. (2012). The human rhodopsin kinase promoter in an AAV5 vector confers rod- and cone-specific expression in the primate retina. Hum. Gene Ther. 23, 1101–1115. doi: 10.1089/hum.2012.125
Boye, S. E., Boye, S. L., Pang, J., Ryals, R., Everhart, D., Umino, Y., et al. (2010). Functional and behavioral restoration of vision by gene therapy in the guanylate cyclase-1 (GC1) knockout mouse. PLoS ONE 5:e11306. doi: 10.1371/journal.pone.0011306
Boye, S. L., Conlon, T., Erger, K., Ryals, R., Neeley, A., Cossette, T., et al. (2011). Long-term preservation of cone photoreceptors and restoration of cone function by gene therapy in the guanylate cyclase-1 knockout (GC1KO) mouse. Invest. Ophthalmol. Vis. Sci. 52, 7098–7108. doi: 10.1167/iovs.11-7867
Boye, S. L., Peshenko, I. V., Huang, W. C., Min, S. H., McDoom, I., Kay, C. N., et al. (2013). AAV-mediated gene therapy in the guanylate cyclase (RetGC1/RetGC2) double knockout mouse model of Leber congenital amaurosis. Hum. Gene Ther. 24, 189–202. doi: 10.1089/hum.2012.193
Cheng, K. M., Shoffner, R. N., Gelatt, K. N., Gum, G. G., Otis, J. S., and Bitgood, J. J. (1980). An autosomal recessive blind mutant in the chicken. Poult. Sci. 59, 2179–2181. doi: 10.3382/ps.0592179
Chung, D. C., and Traboulsi, E. I. (2009). Leber congenital amaurosis: clinical correlations with genotypes, gene therapy trials update, and future directions. J. AAPOS 13, 587–592. doi: 10.1016/j.jaapos.2009.10.004
Cideciyan, A. V., Aleman, T. S., Boye, S. L., Schwartz, S. B., Kaushal, S., Roman, A. J., et al. (2008). Human gene therapy for RPE65 isomerase deficiency activates the retinoid cycle of vision but with slow rod kinetics. Proc. Natl. Acad. Sci. U.S.A. 105, 15112–15117. doi: 10.1073/pnas.0807027105
Cideciyan, A. V., Aleman, T. S., Jacobson, S. G., Khanna, H., Sumaroka, A., Aguirre, G. K., et al. (2007). Centrosomal-ciliary gene CEP290/NPHP6 mutations result in blindness with unexpected sparing of photoreceptors and visual brain: implications for therapy of Leber congenital amaurosis. Hum. Mutat. 28, 1074–1083. doi: 10.1002/humu.20565
Cideciyan, A. V., Hauswirth, W. W., Aleman, T. S., Kaushal, S., Schwartz, S. B., Boye, S. L., et al. (2009). Human RPE65 gene therapy for Leber congenital amaurosis: persistence of early visual improvements and safety at 1 year. Hum. Gene Ther. 20, 999–1004. doi: 10.1089/hum.2009.086
Cideciyan, A. V., Swider, M., Aleman, T. S., Feuer, W. J., Schwartz, S. B., Russell, R. C., et al. (2012). Macular function in macular degenerations: repeatability of microperimetry as a potential outcome measure for ABCA4-associated retinopathy trials. Invest. Ophthalmol. Vis. Sci. 53, 841–852. doi: 10.1167/iovs.11-8415
Coleman, J. E., and Semple-Rowland, S. L. (2005). GC1 deletion prevents light-dependent arrestin translocation in mouse cone photoreceptor cells. Invest. Ophthalmol. Vis. Sci. 46, 12–16. doi: 10.1167/iovs.04-0691
Coleman, J. E., Zhang, Y., Brown, G. A., and Semple-Rowland, S. L. (2004). Cone cell survival and downregulation of GCAP1 protein in the retinas of GC1 knockout mice. Invest. Ophthalmol. Vis. Sci. 45, 3397–3403. doi: 10.1167/iovs.04-0392
den Hollander, A. I., Roepman, R., Koenekoop, R. K., and Cremers, F. P. (2008). Leber congenital amaurosis: genes, proteins and disease mechanisms. Prog. Retin. Eye Res. 27, 391–419. doi: 10.1016/j.preteyeres.2008.05.003
Dizhoor, A. M., Lowe, D. G., Olshevskaya, E. V., Laura, R. P., and Hurley, J. B. (1994). The human photoreceptor membrane guanylyl cyclase, RetGC, is present in outer segments and is regulated by calcium and a soluble activator. Neuron 12, 1345–1352. doi: 10.1016/0896-6273(94)90449-9
Douglas, R. M., Alam, N. M., Silver, B. D., McGill, T. J., Tschetter, W. W., and Prusky, G. T. (2005). Independent visual threshold measurements in the two eyes of freely moving rats and mice using a virtual-reality optokinetic system. Vis. Neurosci. 22, 677–684. doi: 10.1017/S0952523805225166
Estrada-Cuzcano, A., Koenekoop, R. K., Coppieters, F., Kohl, S., Lopez, I., Collin, R. W., et al. (2011). IQCB1 mutations in patients with leber congenital amaurosis. Invest. Ophthalmol. Vis. Sci. 52, 834–839. doi: 10.1167/iovs.10-5221
Goraczniak, R., Duda, T., and Sharma, R. K. (1997). Structural and functional characterization of a second subfamily member of the calcium-modulated bovine rod outer segment membrane guanylate cyclase, ROS-GC2. Biochem. Biophys. Res. Commun. 234, 666–670. doi: 10.1006/bbrc.1997.6579
Goraczniak, R. M., Duda, T., Sitaramayya, A., and Sharma, R. K. (1994). Structural and functional characterization of the rod outer segment membrane guanylate cyclase. Biochem. J. 302(Pt 2), 455–461.
Gorbatyuk, M. S., Knox, T., LaVail, M. M., Gorbatyuk, O. S., Noorwez, S. M., Hauswirth, W. W., et al. (2010). Restoration of visual function in P23H rhodopsin transgenic rats by gene delivery of BiP/Grp78. Proc. Natl. Acad. Sci. U.S.A. 107, 5961–5966. doi: 10.1073/pnas.0911991107
Haeseleer, F., Sokal, I., Li, N., Pettenati, M., Rao, N., Bronson, D., et al. (1999). Molecular characterization of a third member of the guanylyl cyclase-activating protein subfamily. J. Biol. Chem. 274, 6526–6535. doi: 10.1074/jbc.274.10.6526
Haire, S. E., Pang, J., Boye, S. L., Sokal, I., Craft, C. M., Palczewski, K., et al. (2006). Light-driven cone arrestin translocation in cones of postnatal guanylate cyclase-1 knockout mouse retina treated with AAV-GC1. Invest. Ophthalmol. Vis. Sci. 47, 3745–3753. doi: 10.1167/iovs.06-0086
Hanein, S., Perrault, I., Gerber, S., Tanguy, G., Barbet, F., Ducroq, D., et al. (2004). Leber congenital amaurosis: comprehensive survey of the genetic heterogeneity, refinement of the clinical definition, and genotype-phenotype correlations as a strategy for molecular diagnosis. Hum. Mutat. 23, 306–317. doi: 10.1002/humu.20010
Hwang, J. Y., Lange, C., Helten, A., Hoppner-Heitmann, D., Duda, T., Sharma, R. K., et al. (2003). Regulatory modes of rod outer segment membrane guanylate cyclase differ in catalytic efficiency and Ca(2+)-sensitivity. Eur. J. Biochem. 270, 3814–3821. doi: 10.1046/j.1432-1033.2003.03770.x
Imanishi, Y., Yang, L., Sokal, I., Filipek, S., Palczewski, K., and Baehr, W. (2004). Diversity of guanylate cyclase-activating proteins (GCAPs) in teleost fish: characterization of three novel GCAPs (GCAP4, GCAP5, GCAP7) from zebrafish (Danio rerio) and prediction of eight GCAPs (GCAP1-8) in pufferfish (Fugu rubripes). J. Mol. Evol. 59, 204–217. doi: 10.1007/s00239-004-2614-y
Jacobson, S. G., Aleman, T. S., Cideciyan, A. V., Roman, A. J., Sumaroka, A., Windsor, E. A., et al. (2009a). Defining the residual vision in leber congenital amaurosis caused by RPE65 mutations. Invest. Ophthalmol. Vis. Sci. 50, 2368–2375. doi: 10.1167/iovs.08-2696
Jacobson, S. G., Aleman, T. S., Cideciyan, A. V., Sumaroka, A., Schwartz, S. B., Windsor, E. A., et al. (2009b). Leber congenital amaurosis caused by Lebercilin (LCA5) mutation: retained photoreceptors adjacent to retinal disorganization. Mol. Vis. 15, 1098–1106.
Jacobson, S. G., Aleman, T. S., Cideciyan, A. V., Sumaroka, A., Schwartz, S. B., Windsor, E. A., et al. (2005). Identifying photoreceptors in blind eyes caused by RPE65 mutations: prerequisite for human gene therapy success. Proc. Natl. Acad. Sci. U.S.A. 102, 6177–6182. doi: 10.1073/pnas.0500646102
Jacobson, S. G., Cideciyan, A. V., Aleman, T. S., Pianta, M. J., Sumaroka, A., Schwartz, S. B., et al. (2003). Crumbs homolog 1 (CRB1) mutations result in a thick human retina with abnormal lamination. Hum. Mol. Genet. 12, 1073–1078. doi: 10.1093/hmg/ddg117
Jacobson, S. G., Cideciyan, A. V., Aleman, T. S., Sumaroka, A., Roman, A. J., Swider, M., et al. (2011). Human retinal disease from AIPL1 gene mutations: foveal cone loss with minimal macular photoreceptors and rod function remaining. Invest. Ophthalmol. Vis. Sci. 52, 70–79. doi: 10.1167/iovs.10-6127
Jacobson, S. G., Cideciyan, A. V., Aleman, T. S., Sumaroka, A., Schwartz, S. B., Roman, A. J., et al. (2007a). Leber’s congenital amaurosis caused by an RPGRIP1 mutation shows treatment potential. Ophthalmology 114, 895–898. doi: 10.1016/j.ophtha.2006.10.028
Jacobson, S. G., Cideciyan, A. V., Aleman, T. S., Sumaroka, A., Schwartz, S. B., Windsor, E. A., et al. (2007b). RDH12 and RPE65, visual cycle genes causing leber congenital amaurosis, differ in disease expression. Invest. Ophthalmol. Vis. Sci. 48, 332–338. doi: 10.1167/iovs.06-0599
Jacobson, S. G., Cideciyan, A. V., Huang, Y., Hanna, D. B., Freund, C. L., Affatigato, L. M., et al. (1998). Retinal degenerations with truncation mutations in the cone-rod homeobox (CRX) gene. Invest. Ophthalmol. Vis. Sci. 39, 2417–2426.
Jacobson, S. G., Cideciyan, A. V., Peshenko, I. V., Sumaroka, A., Olshevskaya, E. V., Cao, L., et al. (2013). Determining consequences of retinal membrane guanylyl cyclase (RetGC1) deficiency in human Leber congenital amaurosis en route to therapy: residual cone-photoreceptor vision correlates with biochemical properties of the mutants. Hum. Mol. Genet. 22, 168–183. doi: 10.1093/hmg/dds421
Jacobson, S. G., Cideciyan, A. V., Ratnakaram, R., Heon, E., Schwartz, S. B., Roman, A. J., et al. (2012). Gene therapy for leber congenital amaurosis caused by RPE65 mutations: safety and efficacy in 15 children and adults followed up to 3 years. Arch. Ophthalmol. 130, 9–24. doi: 10.1001/archophthalmol.2011.298
Karan, S., Frederick, J. M., and Baehr, W. (2010). Novel functions of photoreceptor guanylate cyclases revealed by targeted deletion. Mol. Cell. Biochem. 334, 141–155. doi: 10.1007/s11010-009-0322-z
Khani, S. C., Pawlyk, B. S., Bulgakov, O. V., Kasperek, E., Young, J. E., Adamian, M., et al. (2007). AAV-mediated expression targeting of rod and cone photoreceptors with a human rhodopsin kinase promoter. Invest. Ophthalmol. Vis. Sci. 48, 3954–3961. doi: 10.1167/iovs.07-0257
Koch, K. W., and Stryer, L. (1988). Highly cooperative feedback control of retinal rod guanylate cyclase by calcium ions. Nature 334, 64–66. doi: 10.1038/334064a0
Komaromy, A. M., Alexander, J. J., Rowlan, J. S., Garcia, M. M., Chiodo, V. A., Kaya, A., et al. (2010). Gene therapy rescues cone function in congenital achromatopsia. Hum. Mol. Genet. 19, 2581–2593. doi: 10.1093/hmg/ddq136
Lheriteau, E., Petit, L., Weber, M., Le Meur, G., Deschamps, J.-Y., Libeau, L., et al. (2014). Successful gene therapy in the RPGRIP1-deficient dog: a large model of cone-rod dystrophy. Mol. Ther. 22, 265–277. doi: 10.1038/mt.2013.232
Lipinski, D. M., Barnard, A. R., Charbel Issa, P., Singh, M. S., De Silva, S. R., Trabalza, A., et al. (2014). Vesicular stomatitis virus glycoprotein- and venezuelan equine encephalitis virus-derived glycoprotein-pseudotyped lentivirus vectors differentially transduce corneal endothelium, trabecular meshwork, and human photoreceptors. Hum. Gene Ther. 25, 50–62. doi: 10.1089/hum.2013.009
Lowe, D. G., Dizhoor, A. M., Liu, K., Gu, Q., Spencer, M. R., Laura, L., et al. (1995). Cloning and expression of a second photoreceptor-specific membrane retina guanylyl cyclase (RetGC), RetGC-2. Proc. Natl. Acad. Sci. U.S.A. 92, 5535–5539. doi: 10.1073/pnas.92.12.5535
Mao, H., James, T. Jr., Schwein, A., Shabashvili, A. E., Hauswirth, W. W., Gorbatyuk, M. S., et al. (2011). AAV delivery of wild-type rhodopsin preserves retinal function in a mouse model of autosomal dominant retinitis pigmentosa. Hum. Gene Ther. 22, 567–575. doi: 10.1089/hum.2010.140
Margulis, A., Goraczniak, R. M., Duda, T., Sharma, R. K., and Sitaramayya, A. (1993). Structural and biochemical identity of retinal rod outer segment membrane guanylate cyclase. Biochem. Biophys. Res. Commun. 194, 855–861. doi: 10.1006/bbrc.1993.1900
Mihelec, M., Pearson, R. A., Robbie, S. J., Buch, P. K., Azam, S. A., Bainbridge, J. W., et al. (2011). Long-term preservation of cones and improvement in visual function following gene therapy in a mouse model of leber congenital amaurosis caused by guanylate cyclase-1 deficiency. Hum. Gene Ther. 22, 1179–1190. doi: 10.1089/hum.2011.069
Milam, A. H., Barakat, M. R., Gupta, N., Rose, L., Aleman, T. S., Pianta, M. J., et al. (2003). Clinicopathologic effects of mutant GUCY2D in Leber congenital amaurosis. Ophthalmology 110, 549–558. doi: 10.1016/S0161-6420(02)01757-8
Min, S. H., Molday, L. L., Seeliger, M. W., Dinculescu, A., Timmers, A. M., Janssen, A., et al. (2005). Prolonged recovery of retinal structure/function after gene therapy in an Rs1h-deficient mouse model of x-linked juvenile retinoschisis. Mol. Ther. 12, 644–651. doi: 10.1016/j.ymthe.2005.06.002
Miyoshi, H., Takahashi, M., Gage, F. H., and Verma, I. M. (1997). Stable and efficient gene transfer into the retina using an HIV-based lentiviral vector. Proc. Natl. Acad. Sci. U.S.A. 94, 10319–10323. doi: 10.1073/pnas.94.19.10319
Olshevskaya, E. V., Calvert, P. D., Woodruff, M. L., Peshenko, I. V., Savchenko, A. B., Makino, C. L., et al. (2004). The Y99C mutation in guanylyl cyclase-activating protein 1 increases intracellular Ca2+ and causes photoreceptor degeneration in transgenic mice. J. Neurosci. 24, 6078–6085. doi: 10.1523/JNEUROSCI.0963-04.2004
Olshevskaya, E. V., Peshenko, I. V., Savchenko, A. B., and Dizhoor, A. M. (2012). Retinal guanylyl cyclase isozyme 1 is the preferential in vivo target for constitutively active GCAP1 mutants causing congenital degeneration of photoreceptors. J. Neurosci. 32, 7208–7217. doi: 10.1523/JNEUROSCI.0976-12.2012
Palczewski, K., Subbaraya, I., Gorczyca, W. A., Helekar, B. S., Ruiz, C. C., Ohguro, H., et al. (1994). Molecular cloning and characterization of retinal photoreceptor guanylyl cyclase-activating protein. Neuron 13, 395–404. doi: 10.1016/0896-6273(94)90355-7
Pang, J., Cheng, M., Haire, S. E., Barker, E., Planelles, V., and Blanks, J. C. (2006). Efficiency of lentiviral transduction during development in normal and rd mice. Mol. Vis. 12, 756–767.
Pang, J. J., Dai, X., Boye, S. E., Barone, I., Boye, S. L., Mao, S., et al. (2011). Long-term retinal function and structure rescue using capsid mutant AAV8 vector in the rd10 mouse, a model of recessive retinitis pigmentosa. Mol. Ther. 19, 234–242. doi: 10.1038/mt.2010.273
Pang, J. J., Deng, W. T., Dai, X., Lei, B., Everhart, D., Umino, Y., et al. (2012). AAV-mediated cone rescue in a naturally occurring mouse model of CNGA3-achromatopsia. PLoS ONE 7:e35250. doi: 10.1371/journal.pone.0035250
Pasadhika, S., Fishman, G. A., Stone, E. M., Lindeman, M., Zelkha, R., Lopez, I., et al. (2010). Differential macular morphology in patients with RPE65-, CEP290-, GUCY2D-, and AIPL1-related Leber congenital amaurosis. Invest. Ophthalmol. Vis. Sci. 51, 2608–2614. doi: 10.1167/iovs.09-3734
Pawlyk, B. S., Bulgakov, O. V., Liu, X., Xu, X., Adamian, M., Sun, X., et al. (2010). Replacement gene therapy with a human RPGRIP1 sequence slows photoreceptor degeneration in a murine model of Leber congenital amaurosis. Hum. Gene Ther. 21, 993–1004. doi: 10.1089/hum.2009.218
Perrault, I., Rozet, J. M., Calvas, P., Gerber, S., Camuzat, A., Dollfus, H., et al. (1996). Retinal-specific guanylate cyclase gene mutations in Leber’s congenital amaurosis. Nat. Genet. 14, 461–464. doi: 10.1038/ng1296-461
Perrault, I., Rozet, J. M., Gerber, S., Ghazi, I., Ducroq, D., Souied, E., et al. (2000). Spectrum of retGC1 mutations in Leber’s congenital amaurosis. Eur. J. Hum. Genet. 8, 578–582. doi: 10.1038/sj.ejhg.5200503
Perrault, I., Rozet, J. M., Gerber, S., Ghazi, I., Leowski, C., Ducroq, D., et al. (1999a). Leber congenital amaurosis. Mol. Genet. Metab. 68, 200–208. doi: 10.1006/mgme.1999.2906
Perrault, I., Rozet, J. M., Ghazi, I., Leowski, C., Bonnemaison, M., Gerber, S., et al. (1999b). Different functional outcome of RetGC1 and RPE65 gene mutations in Leber congenital amaurosis. Am. J. Hum. Genet. 64, 1225–1228. doi: 10.1086/302335
Peshenko, I. V., and Dizhoor, A. M. (2004). Guanylyl cyclase-activating proteins (GCAPs) are Ca2+/Mg2+ sensors: implications for photoreceptor guanylyl cyclase (RetGC) regulation in mammalian photoreceptors. J. Biol. Chem. 279, 16903–16906. doi: 10.1074/jbc.C400065200
Peshenko, I. V., and Dizhoor, A. M. (2006). Ca2+ and Mg2+ binding properties of GCAP-1. Evidence that Mg2+-bound form is the physiological activator of photoreceptor guanylyl cyclase. J. Biol. Chem. 281, 23830–23841. doi: 10.1074/jbc.M600257200
Peshenko, I. V., Olshevskaya, E. V., Savchenko, A. B., Karan, S., Palczewski, K., Baehr, W., et al. (2011). Enzymatic properties and regulation of the native isozymes of retinal membrane guanylyl cyclase (RetGC) from mouse photoreceptors. Biochemistry 50, 5590–5600. doi: 10.1021/bi200491b
Petit, L., Lhériteau, E., Weber, M., Le Meur, G., Deschamps, J.-Y., Provost, N., et al. (2012). Restoration of vision in the pde6beta-deficient dog, a large animal model of rod-cone dystrophy. Mol. Ther. 20, 2019–2030. doi: 10.1038/mt.2012.134
Polans, A., Baehr, W., and Palczewski, K. (1996). Turned on by Ca2+! The physiology and pathology of Ca(2+)-binding proteins in the retina. Trends Neurosci. 19, 547–554. doi: 10.1016/S0166-2236(96)10059-X
Porto, F. B., Perrault, I., Hicks, D., Rozet, J. M., Hanoteau, N., Hanein, S., et al. (2003). Prenatal human ocular degeneration occurs in Leber’s Congenital Amaurosis (LCA1 and 2). Adv. Exp. Med. Biol. 533, 59–68. doi: 10.1007/978-1-4615-0067-4_8
Pugh, E. N. Jr., Duda, T., Sitaramayya, A., and Sharma, R. K. (1997). Photoreceptor guanylate cyclases: a review. Biosci. Rep. 17, 429–473. doi: 10.1023/A:1027365520442
Roman, A. J., Cideciyan, A. V., Aleman, T. S., and Jacobson, S. G. (2007). Full-field stimulus testing (FST) to quantify visual perception in severely blind candidates for treatment trials. Physiol. Meas. 28, N51–N56. doi: 10.1088/0967-3334/28/8/N02
Roman, A. J., Schwartz, S. B., Aleman, T. S., Cideciyan, A. V., Chico, J. D., Windsor, E. A., et al. (2005). Quantifying rod photoreceptor-mediated vision in retinal degenerations: dark-adapted thresholds as outcome measures. Exp. Eye Res. 80, 259–272. doi: 10.1016/j.exer.2004.09.008
Semple-Rowland, S. L., Lee, N. R., Van Hooser, J. P., Palczewski, K., and Baehr, W. (1998). A null mutation in the photoreceptor guanylate cyclase gene causes the retinal degeneration chicken phenotype. Proc. Natl. Acad. Sci. U.S.A. 95, 1271–1276. doi: 10.1073/pnas.95.3.1271
Sergouniotis, P. I., Davidson, A. E., Mackay, D. S., Li, Z., Yang, X., Plagnol, V., et al. (2011). Recessive mutations in KCNJ13, encoding an inwardly rectifying potassium channel subunit, cause leber congenital amaurosis. Am. J. Hum. Genet. 89, 183–190. doi: 10.1016/j.ajhg.2011.06.002
Shyjan, A. W., de Sauvage, F. J., Gillett, N. A., Goeddel, D. V., and Lowe, D. G. (1992). Molecular cloning of a retina-specific membrane guanylyl cyclase. Neuron 9, 727–737. doi: 10.1016/0896-6273(92)90035-C
Simonelli, F., Ziviello, C., Testa, F., Rossi, S., Fazzi, E., Bianchi, P. E., et al. (2007). Clinical and molecular genetics of Leber’s congenital amaurosis: a multicenter study of Italian patients. Invest. Ophthalmol. Vis. Sci. 48, 4284–4290. doi: 10.1167/iovs.07-0068
Sun, X., Pawlyk, B., Xu, X., Liu, X., Bulgakov, O. V., Adamian, M., et al. (2010). Gene therapy with a promoter targeting both rods and cones rescues retinal degeneration caused by AIPL1 mutations. Gene Ther. 17, 117–131. doi: 10.1038/gt.2009.104
Tan, M. H., Smith, A. J., Pawlyk, B., Xu, X., Liu, X., Bainbridge, J. B., et al. (2009). Gene therapy for retinitis pigmentosa and Leber congenital amaurosis caused by defects in AIPL1: effective rescue of mouse models of partial and complete Aipl1 deficiency using AAV2/2 and AAV2/8 vectors. Hum. Mol. Genet. 18, 2099–2114. doi: 10.1093/hmg/ddp133
Timmers, A. M., Zhang, H., Squitieri, A., and Gonzalez-Pola, C. (2001). Subretinal injections in rodent eyes: effects on electrophysiology and histology of rat retina. Mol. Vis. 7, 131–137.
Ulshafer, R. J., and Allen, C. B. (1985a). Hereditary retinal degeneration in the Rhode Island Red chicken: ultrastructural analysis. Exp. Eye Res. 40, 865–877. doi: 10.1016/0014-4835(85)90131-9
Ulshafer, R. J., and Allen, C. B. (1985b). Ultrastructural changes in the retinal pigment epithelium of congenitally blind chickens. Curr. Eye Res. 4, 1009–1021. doi: 10.3109/02713688509003346
Ulshafer, R. J., Allen, C., Dawson, W. W., and Wolf, E. D. (1984). Hereditary retinal degeneration in the Rhode Island Red chicken. I. Histology and ERG. Exp. Eye Res. 39, 125–135. doi: 10.1016/0014-4835(84)90003-4
Vandenberghe, L. H., Bell, P., Maguire, A. M., Cearley, C. N., Xiao, R., Calcedo, R., et al. (2011). Dosage thresholds for AAV2 and AAV8 photoreceptor gene therapy in monkey. Sci. Transl. Med. 3:88ra54. doi: 10.1126/scitranslmed.3002103
Vandenberghe, L. H., Bell, P., Maguire, A. M., Xiao, R., Hopkins, T. B., Grant, R., et al. (2013). AAV9 targets cone photoreceptors in the nonhuman primate retina. PLoS ONE 8:e53463. doi: 10.1371/journal.pone.0053463
Verrier, J. D., Madorsky, I., Coggin, W. E., Geesey, M., Hochman, M., Walling, E., et al. (2011). Bicistronic lentiviruses containing a viral 2A cleavage sequence reliably co-express two proteins and restore vision to an animal model of LCA1. PLoS ONE 6:e20553. doi: 10.1371/journal.pone.0020553
Wallman, J., and Velez, J. (1985). Directional asymmetries of optokinetic nystagmus: developmental changes and relation to the accessory optic system and to the vestibular system. J. Neurosci. 5, 317–329.
Wang, H., den Hollander, A. I., Moayedi, Y., Abulimiti, A., Li, Y., Collin, R. W., et al. (2009). Mutations in SPATA7 cause Leber congenital amaurosis and juvenile retinitis pigmentosa. Am. J. Hum. Genet. 84, 380–387. doi: 10.1016/j.ajhg.2009.02.005
Williams, G. A., and Jacobs, G. H. (2007). Cone-based vision in the aging mouse. Vision Res. 47, 2037–2046. doi: 10.1016/j.visres.2007.03.023
Williams, M. L., Coleman, J. E., Haire, S. E., Aleman, T. S., Cideciyan, A. V., Sokal, I., et al. (2006). Lentiviral expression of retinal guanylate cyclase-1 (RetGC1) restores vision in an avian model of childhood blindness. PLoS Med. 3:e30201. doi: 10.1371/journal.pmed.0030201
Wright, A. F., Chakarova, C. F., Abd El-Aziz, M. M., and Bhattacharya, S. S. (2010). Photoreceptor degeneration: genetic and mechanistic dissection of a complex trait. Nat. Rev. Genet. 11, 273–284. doi: 10.1038/nrg2717
Yang, G. S., Schmidt, M., Yan, Z., Lindbloom, J. D., Harding, T. C., Donahue, B. A., et al. (2002). Virus-mediated transduction of murine retina with adeno-associated virus: effects of viral capsid and genome size. J. Virol. 76, 7651–7660. doi: 10.1128/JVI.76.15.7651-7660.2002
Yang, R. B., Foster, D. C., Garbers, D. L., and Fulle, H. J. (1995). Two membrane forms of guanylyl cyclase found in the eye. Proc. Natl. Acad. Sci. U.S.A. 92, 602–606. doi: 10.1073/pnas.92.2.602
Yang, R. B., Robinson, S. W., Xiong, W. H., Yau, K. W., Birch, D. G., and Garbers, D. L. (1999). Disruption of a retinal guanylyl cyclase gene leads to cone-specific dystrophy and paradoxical rod behavior. J. Neurosci. 19, 5889–5897.
Yao, J., Feathers, K. L., Khanna, H., Thompson, D., Tsilfidis, C., Hauswirth, W. W., et al. (2011). XIAP therapy increases survival of transplanted rod precursors in a degenerating host retina. Invest. Ophthalmol. Vis. Sci. 52, 1567–1572. doi: 10.1167/iovs.10-5998
Keywords: LCA1, Leber congenital amaurosis, GUCY2D, retGC1, GC1, guanylate cyclase, retinal gene therapy, AAV
Citation: Boye SE (2014) Insights gained from gene therapy in animal models of retGC1 deficiency. Front. Mol. Neurosci. 7:43. doi: 10.3389/fnmol.2014.00043
Received: 30 January 2014; Accepted: 26 April 2014;
Published online: 14 May 2014.
Edited by:
Wolfgang Baehr, University of Utah, USAReviewed by:
Wolfgang Baehr, University of Utah, USAIzhak Michaelevski, Tel Aviv University, Israel
Copyright © 2014 Boye. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shannon E. Boye, Department of Ophthalmology, University of Florida, P.O. Box 100284, Gainesville, FL 32610, USA e-mail:c2hhbm5vbi5ib3llQGV5ZS51ZmwuZWR1