 Laurie L. Molday
Laurie L. Molday Thomas Jefferies
Thomas Jefferies Robert S. Molday
Robert S. Molday- Department of Biochemistry and Molecular Biology, Center for Macular Research, University of British Columbia, Vancouver, BC, USA
Retinal degeneration 3 (RD3) is an evolutionarily conserved 23 kDa protein expressed in rod and cone photoreceptor cells. Mutations in the gene encoding RD3 resulting in unstable non-functional C-terminal truncated proteins are responsible for early onset photoreceptor degeneration in Leber Congenital Amaurosis 12 patients, the rd3 mice, and the rcd2 collies. Recent studies have shown that RD3 interacts with guanylate cyclases GC1 and GC2 in retinal cell extracts and HEK293 cells co-expressing GC and RD3. This interaction inhibits GC catalytic activity and promotes the exit of GC1 and GC2 from the endoplasmic reticulum and their trafficking to photoreceptor outer segments. Adeno-associated viral vector delivery of the normal RD3 gene to photoreceptors of the rd3 mouse restores GC1 and GC2 expression and outer segment localization and leads to the long-term recovery of visual function and photoreceptor cell survival. This review focuses on the genetic and biochemical studies that have provided insight into the role of RD3 in photoreceptor function and survival.
Introduction
Guanylate cyclase (GC) plays a key role in phototransduction by catalyzing the synthesis of cGMP. In the dark the basal catalytic activity of GC in photoreceptor outer segments (OS) is balanced by the basal activity of phosphodiesterase (PDE) to maintain cGMP at a level sufficient for maintaining a significant fraction of cGMP-gated channels in their open state. This allows the influx of Ca2+ and Na+ into the OS with the intracellular Ca2+ level reaching 250–500 nM (Gray-Keller and Detwiler, 1994; Woodruff et al., 2002). At this concentration, guanylate cyclase activating proteins (GCAPs) with bound Ca2+ maintain GC at its basal level (Gorczyca et al., 1994; Dizhoor et al., 1995; Polans et al., 1996). Following photoexcitation the Ca2+ level decreases to less than 25 nM through the combined closure of the cGMP-gated channels and continued efflux of Ca2+ by the Na/Ca-K exchanger (Woodruff et al., 2002). The reduced Ca2+ concentration triggers the exchange of Ca2+ for Mg2+ on GCAPs resulting in the activation GC (Peshenko and Dizhoor, 2006; Dizhoor et al., 2010). The increase in cGMP reopens the cGMP-gated channels and restores Ca2+ and cGMP to their dark state levels.
Vertebrate photoreceptors contain two membrane-bound GCs, generally called GC1 and GC2, that share a high degree of sequence identity (~49%) and structural organization (Shyjan et al., 1992; Lowe et al., 1995; Baehr et al., 2007; Karan et al., 2011). They consist of an N-terminal signal sequence followed by an extracellular domain, a transmembrane segment, a kinase homology domain, a dimerization domain, a cyclase catalytic domain, and a carboxyl-terminal extension. GC1 and GC2 exist as homodimers which localize to disk membranes of photoreceptor OS (Yang and Garbers, 1997; Ramamurthy et al., 2001). GC1 is present at relatively high concentrations in rod and cone OS, whereas GC2 is restricted to rod OS in mice and humans (Baehr et al., 2007; Azadi et al., 2010). The relative amounts of GC1 and GC2 vary with species with a GC1 to GC2 ratio of 4:1 in mouse (Peshenko et al., 2011b) and 30:1 in bovine photoreceptors (Helten et al., 2007; Kwok et al., 2008). The importance of GC1 and GC2 in the visual response and photoreceptor survival has been determined in knockout mice. In GC1 knockout mice, the cone photoresponse is absent and these cells undergo rapid degeneration (Baehr et al., 2007; Karan et al., 2010). Rod photoreceptors remain viable and functional due to the presence of GC2. In the GC1/GC2 double knockout mice, the rod and cone photoresponse is undetectable and both cell types undergo degeneration. The importance of GC1 in rod and cone function and survival is further highlighted by the finding that mutations in the GUCY2D gene encoding human GC1 cause Leber Congenital Amaurosis (LCA) Type 1 (LCA1), a severe early onset retinal dystrophy, and cone-rod dystrophy (Perrault et al., 1996; Kelsell et al., 1998; Karan et al., 2010).
Photoreceptor cells are highly polarized neurons with the OS segregated from the rest of the cell by a thin cilium. OS proteins must be efficiently transported from the endoplasmic reticulum (ER) in the inner segment through the cilium since OS turnover every 10 days through the phagocytosis of aged OS membranes by retinal pigment epithelial cells and the addition of new membrane at the base of the OS (Sung and Chuang, 2010). The molecular machinery which orchestrates the transport of proteins to the OS is highly complex involving vesicle budding, fusion, and motorized transport along microtubules and other cytoskeletal elements in the inner segment and cilium. Molecular-based studies indicate that the C-terminal targeting segment of several OS membrane proteins including rhodopsin and perpherin-2 play an essential role in vesicle trafficking through protein–protein interactions (Tam et al., 2000, 2004; Deretic et al., 2005; Chuang et al., 2007; Mazelova et al., 2009). The vesicle transport pathways, however, appear to be distinct for various OS membrane proteins (Fariss et al., 1997). Efforts to define a sequence required to transport GC to outer segments have been inconclusive (Karan et al., 2011). Recently, retinal degeneration 3 (RD3), a photoreceptor protein encoded by the gene associated with retinal degeneration in the rd3 mouse, rcd2 collie, and LCA12 patients has been shown to play a crucial role in the trafficking of GC1 and GC2 in photoreceptors (Azadi et al., 2010). In this review, we focus on the molecular properties of RD3 and its role in GC trafficking, catalytic activity, and photoreceptor degeneration.
Genetic and Protein Analysis of the RD3
The gene responsible for retinal degeneration in the rd3 mouse was first identified as an uncharacterized transcript in an in silico search of retina-specific transcripts and called C1orf36 for Chromosome 1 open reading frame 36 (Lavorgna et al., 2003). The gene now known as RD3 consists of 3 exons spanning the 5′ and 3′ untranslated region with the open reading frame comprised of part of exon 2 (amino acids 1–99) and exon 3 (100–195). RT-PCR and in situ hybridization confirmed the presence of RD3 in the retina with high expression in the photoreceptors.
The RD3 protein encoded by the RD3 gene is highly conserved across vertebrates with the human protein consisting of 195 amino acids and sharing 95% sequence identity with other primates, 86% with mouse and rat, 83% with bovine, 67% with chicken, and 50–60% with lower vertebrates including Danio rerio (Zebrafish) and Xenopus tropicalis (Western clawed frog). Computer algorithms indicate that RD3 lacks any known homology domains or transmembrane segments. RD3 is predicted to have a high α-helix content (~43%), no β-sheets and considerable disordered regions (Figure 1). There are four conserved stretches of predicted α-helices (H1–H4) with the first helix consisting of 34 amino acids and the last helix having 39 amino acids. Additional conserved features include a putative coil–coil region between amino acids 22–54 and several predicted phosphorylation sites.

FIGURE 1. Sequence alignment of RD3 proteins from various vertebrates. Sequences of human, mouse, chick, and Xenopus tropocalis RD3 were aligned using Clustal W multiple alignment program. Sequence identities relative to human RD3 are shown in yellow and * and similarities are indicated by (:). Helical predictions were based on the PSIPRED protein structure prediction program and coil–coil domain (enclosed in a rectangle) was predicted using Coils program found in the web-based ExPASy Bioinformatics Resource portal.
RD3 has been isolated from retinal homogenates and HEK293 cells and bacteria expressing the recombinant protein (Azadi et al., 2010; Peshenko et al., 2011a). On SDS gels RD3 migrates as a 23 kDa protein consistent with its amino acid sequence. Dynamic light scattering and gel filtration chromatography, however, indicate that purified RD3 in solution exists as a polydispersed oligomeric protein.
Truncation Mutations in RD3 Cause Photoreceptor Degeneration
The rd3 mouse was one of the first naturally occurring murine strains found to display early onset rod and cone degeneration (Chang et al., 1993). The rate of degeneration varies with the background strain (Linberg et al., 2005; Friedman et al., 2006; Danciger et al., 2008; Molday et al., 2013). The albino RBF/DnJ strain shows the fastest rate of degeneration with only a monolayer of photoreceptor nuclei present 8 weeks after birth. The pigmented In(5) 30RK/J strain displays the slowest rate with some photoreceptors still present after 30 weeks. The Rb(11.13)4Bnr/J strain is intermediate in its rate of degeneration. Rod-derived scotopic electroretinograms (ERG) are measurable in all strains at an early age prior to significant photoreceptor degeneration (~24–35 days postnatal), but a cone response has only been reported in the In(5) 30RK/J strain (Friedman et al., 2006; Molday et al., 2013). The mutation which causes photoreceptor degeneration in all strains was first reported by Friedman et al. (2006). They identified a homozygous C→T substitution (c.319C→T) in the Rd3 gene which causes a premature stop codon resulting in a protein lacking the last 99 amino acids.
An extensive human RD3 mutational screen was carried out on individuals with autosomal dominant and recessive retinal degenerative diseases. A homozygous G → A transition in the donor splice site at the end of exon 2 was found in two siblings from a family with LCA (Friedman et al., 2006). This mutation causes a premature stop codon following codon 99 of the RD3 gene. The RD3 gene was the twelfth gene associated with LCA and accordingly this subclass is called LCA12. Subsequent genetic screens revealed homozygous mutations in RD3 with severely truncated proteins in other LCA families (Preising et al., 2012; Perrault et al., 2013). World-wide genetic screens indicate that mutations in the RD3 gene causing LCA are rare. In addition to truncation mutations, missense mutations in RD3 have been observed in individuals with other retinopathies (Friedman et al., 2006). However, additional studies are needed to determine if these mutations are directly responsible for the disease.
A mutation in the canine Rd3 gene causes rod-cone dysplasia 2 (rcd2) in a strain of collies (Kukekova et al., 2009). An insertion in the gene leads to an alteration in the last 61 codons and a further extension of the open reading frame.
Biochemical studies indicate that the truncated RD3 proteins associated with photoreceptor degeneration in the rd3 mouse and LCA12 patients are highly unstable and non-functional (Friedman et al., 2006; Peshenko et al., 2011a). These studies support a crucial role of the C-terminal segment in the stability and function of RD3.
Interaction of RD3 with GC1 and GC2
RD3 was first detected in a mass-spectrometry-based proteomic analysis of bovine photoreceptor OS (Kwok et al., 2008). Monoclonal and polyclonal antibodies against RD3 confirmed the presence of the 23 kDa RD3 protein in mouse retinal extracts by western blotting (Azadi et al., 2010). These antibodies, however, proved to be problematic for immunolocalizing RD3 in photoreceptors as they failed to consistently label retinal cryosections above background levels due to either the inaccessibility of the epitopes or the low level of RD3 expression. One polyclonal antibody showed some immunoreactivity in outer and inner segment layers above control retinas, but in subsequent studies this observation could not be reproduced (Azadi et al., 2010). Additional studies are needed to definitively determine the subcellular localization of RD3 in photoreceptor cells.
The first insight into the possible role of RD3 in photoreceptors was obtained from co-immunoprecipitation experiments. A monoclonal antibody to murine RD3 coupled to Sepharose immunoprecipitated GC1 and GC2 together with RD3 from mouse retinal extracts (Azadi et al., 2010). The interaction of RD3 and GC1 was confirmed in co-expression and co-immunoprecipitation studies. When RD3 and GC1 were co-expressed in HEK293 cells, an anti-RD3 antibody co-precipitated GC1 with RD3 and in a reverse experiment an anti-GC1 antibody co-immunoprecipitated RD3 with GC1 confirming the direct interaction between these proteins (Azadi et al., 2010). Analysis of GC1 deletion mutants further indicated that the C-terminus of GC1 is required for binding of RD3.
RD3 is Important for the Expression and Localization of Guanylate Cyclases
To determine the significance of the RD3-GC interaction, the expression and localization of GC1 and GC2 in the rd3 mouse was compared with age-matched wild-type (WT) mice by western blotting and confocal microscopy (Azadi et al., 2010). GC1 and GC2 from WT mouse retina migrated as 120 and 125 kDa proteins on SDS gels, but were absent or present in reduced amounts in extracts from rd3 mice depending on the strain used for the analysis (Azadi et al., 2010; Molday et al., 2013). At a subcellular level, GC1 and GC2 was localized to the photoreceptor OS of WT mice with GC1 present in rod and cones and GC2 restricted to rod cells as reported previously (Karan et al., 2010). In contrast, GC1 and GC2 immunolabeling was absent in 21-day old Rb(11.13)4Bnr/J rd3 mice and mislocalized to the inner segment of the In(5)30RK/J strain (Azadi et al., 2010; Cheng and Molday, 2013; Molday et al., 2013). GCAP1 and GCAP2 were detected at a reduced level in rd3 mice and primarily confined to the inner segments as observed in GC1/GC2 knockout mice (Azadi et al., 2010; Karan et al., 2010). Other photoreceptor OS proteins including the cyclic GMP-gated channel, PDE6, peripherin-2, rhodopsin, and cone arrestin targeted normally to the OS of the rd3 mice. Gene expression observed by microarray analysis indicated that the expression levels of genes encoding RD3, GC1 and GC2 in 21 day old (Rb(11.13)4Bnr rd3 mice were similar to age-matched WT mice. Up-regulation of endothelin 2 (EDn2), glial fibrillary acid protein (Gfap), and complement component factor 1 (Cf1) and down-regulation of phosducin (Pdc), gap junction protein α5 (Gja5) and retinal G protein – coupled receptor (Rgr) genes, however, were observed for the rd3 retina (Cheng and Molday, 2013).
The expression and distribution of GC1 was also examined in the In(5)30RK/J strain of rd3 mice by immunofluorescence microscopy (Molday et al., 2013). GC1 was detected at reduced levels in this strain and confined primarily to the inner segments where it co-localized with the anti-KDEL ER marker. A small amount of GC1 was detected in the OS layer which may account for the attenuated scotopic and photopic ERGs observed in this strain. Similar mislocalization of GC in the rcd2 collie has been observed. When the normal murine Rd3 gene was delivered to photoreceptor cells of either strain of rd3 mice using adeno-associated virus (AAV), GC1 and GC2 expressed at levels comparable to that of WT mice and localized normally to the photoreceptor OS layer (Molday et al., 2013). Collectively, these studies indicate that RD3 is required for efficient expression and localization of GCs to the photoreceptor OS.
The effect of RD3 expression on the localization of GC was also examined in transfected culture cells (Azadi et al., 2010). GC1 primarily localized to the ER of transfected cells in the absence of RD3, whereas RD3 co-localized with Rab11 in endosomes. Co-expression of GC1 and RD3 resulted in the exit of GC1 from the ER and colocalization with RD3 in Rab11 containing vesicles. These studies are consistent with the observed effect of RD3 in promoting the exit of GC from the ER in photoreceptor cells.
RD3 Inhibits Guanylate Cyclase Activity
In addition to altering the expression level and trafficking of GC, RD3 inhibits its catalytic activity (Peshenko et al., 2011a). In the absence of GCAPs, RD3 expressed in either bacteria or HEK293 cells inhibited the basal and GCAP-activated GC catalytic activity at submicromolar concentrations, but did not alter the Ca2+ sensitivity of GCAPs. It remains to be determined if RD3 plays a significant role in modulating the activity of GCs during phototransduction. It is possible that RD3 only plays an important role in inhibiting cyclase activity during the trafficking of GCs within the inner segments. At present the quantity of RD3 in photoreceptors remains to be determined, but in in vitro measurements, RD3 at nanomolar concentrations competitively inhibited the activation of GC by GCAP (Peshenko et al., 2011a).
The effect of the RD3 mutation (F100ter) associated with LCA12 and other possible disease-linked missense mutations on the interaction of RD3 with GC was studied (Peshenko et al., 2011a). The F100ter and the G57V mutants were undetectable when expressed in HEK293 cells, but were expressed at significant levels in E. Coli. Other missense mutants (K130M, G35R, R68W, W6R/E23D) expressed at levels comparable to WT RD3 and bound GC1. The F100ter mutant had no effect of the cyclase activity, whereas the missense mutations inhibited GC activity to varying degrees. The W6R/E23D and G35R mutations inhibited the cyclase activity at a similar concentration as WT RD3, whereas the R68W, K130M and G57V were less effective. It remains to be determined if these missense mutations in RD3 alter the ability of RD3 to function in GC trafficking in photoreceptors.
Models for the Role of RD3 in Photoreceptors
Studies carried out to date suggest that RD3 serves two roles in photoreceptor cells. A primary function of RD3 is to facilitate the exit of GC from the ER and its trafficking to the photoreceptor cilium (Figure 2). RD3 may also be involved in the trafficking of GC within the cilium although this remains to be determined.
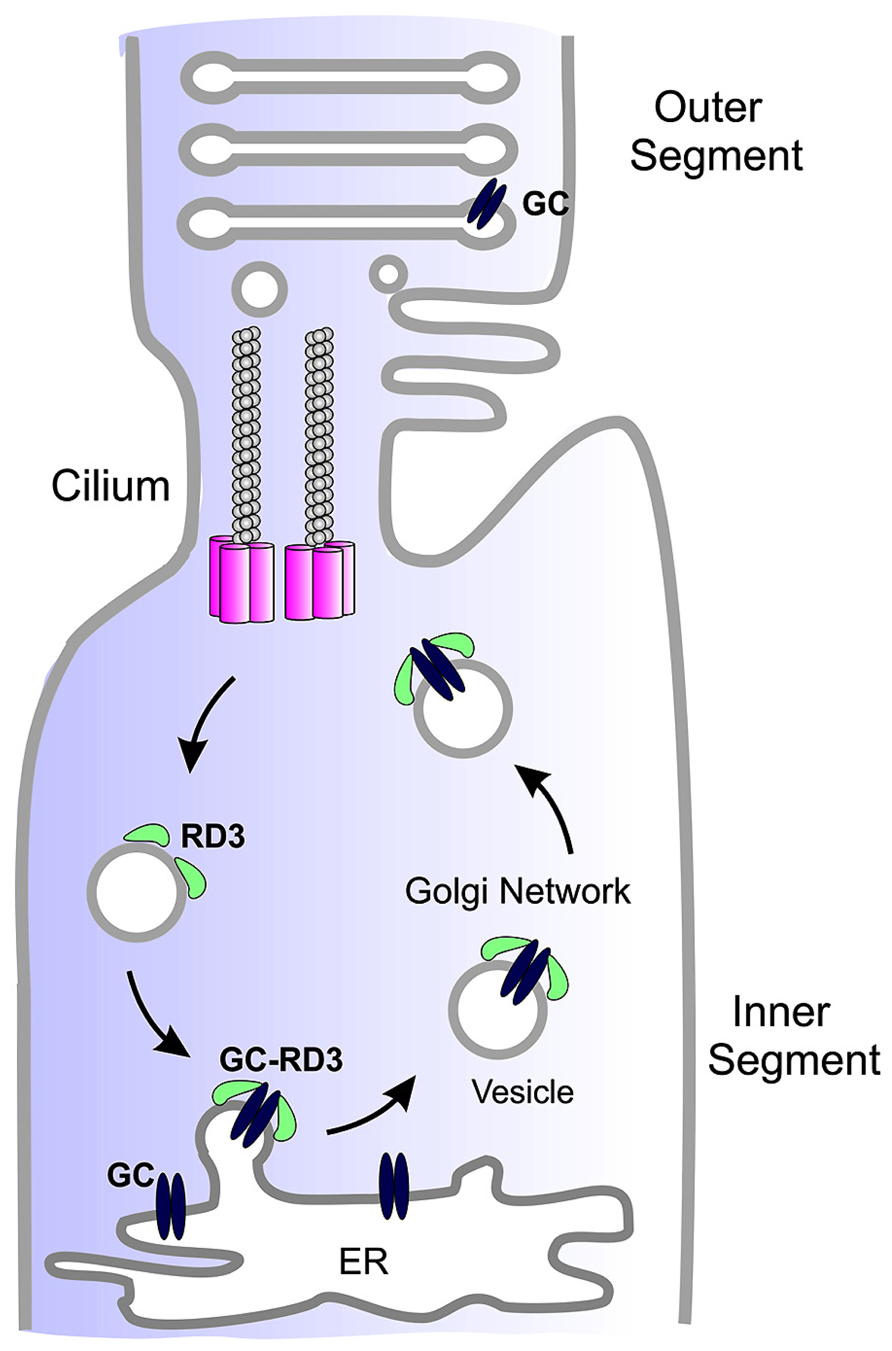
FIGURE 2. Simplified diagram showing the role of RD3 in GC trafficking. GC synthesized in the endoplasmic reticulum (ER) of photoreceptor inner segments binds to RD3 on the cytoplasmic side of the ER membrane. This interaction may induce membrane curvature required for the budding of GC-RD3 from the ER in the form of small vesicles. RD3 also inhibits the cyclase activity of GC. The RD3-GC complex together with other adaptor proteins is translocated through the Golgi to the base of the connecting cilium where GC perhaps in the absence of RD3 is transported through the cilium to the base of the outer segments where it is incorporated into disk membranes. At some point RD3 dissociates from GC possibly through phosphorylation or other post-translational modifications or protein–protein interaction. RD3 associated with endosomes is returned to the ER to initiate another cycle of RD3-mediated GC transport.
A second function of RD3 is to inhibit GC catalytic activity (Peshenko et al., 2011a). The inhibition of GC activity by RD3 may be crucial for insuring that cGMP is not produced in the inner segment of photoreceptors cells during the trafficking of GC to the outer segment. In the absence of RD3 inhibition, GC could either use up the supply of GTP required for the function of other proteins including small G-proteins such as Rabs required for protein vesicle trafficking or overproduction of cGMP in the inner segment could be toxic to photoreceptors. In this regard it is interesting to note that an early study reported that retinal extracts from the rcd2 collie showed a 10 times higher level of cGMP than control retinas during a 2–8 week period with only 25% reduction in PDE activity (Woodford et al., 1982).
Role of RD3 in Photoreceptor Cell Survival
The mechanism by which loss in RD3 causes photoreceptor degeneration remains to be determined. Here we discuss two possible mechanisms. The first mechanism is centered on the role of GC and cGMP in maintaining calcium homeostasis in photoreceptors. It is generally known that too high or too low Ca2+ levels can be toxic to cells (Fain, 2006). Calcium homeostasis in OS is maintained by the influx of Ca2+ through the cGMP-gated channels and the efflux through the Na/Ca-K exchanger. In the absence of GC in the OS, cGMP will not be produced resulting in permanent closure of cGMP-gated channels and a reduction in intracellular Ca2+ below a threshold required for photoreceptor survival. In this case the mechanism of photoreceptor degeneration in the rd3 mouse and LCA12 patients would be similar to photoreceptor degeneration in the GC1/GC2 knockout mouse and LCA1 patients. Direct comparison of the rate of photoreceptor degeneration in the rd3 mouse and GC1/GC2 knockout mouse is complicated by the large variation in the rate of degeneration observed for different strains of rd3 mice, although in general degeneration in the rd3 mouse appears to be more rapid. Comparison of LCA12 with LCA1 patients is complicated by the presence of functional GC2 in rod photoreceptors of LCA1 patients. However, limited clinical assessment of these patients suggests that LCA12 is more severe (Preising et al., 2012; Perrault et al., 2013).
A second mechanism involves the role of RD3 in the inhibition of GC activity (Peshenko et al., 2011a). In the absence of RD3, GC in photoreceptor inner segments is likely to be active in catalyzing the production of cGMP (Woodford et al., 1982). Elevated cGMP levels can be toxic through the unregulated activation of cGMP-dependent enzymes. High cGMP levels have been implicated in photoreceptor cell death in the rd1 and rd10 mice linked to mutations in the β-subunit of PDE (Bowes et al., 1990; Chang et al., 2007), mice deficient in the A3 subunit of the cone cGMP-gated channel (Xu et al., 2013), and transgenic mice harboring an Y99C mutation in GCAP1 (Olshevskaya et al., 2004). Both the loss in RD3 chaperone activity and inhibition of GC activity may contribute to photoreceptor degeneration.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grants from the National Institutes of Health (EY02422-33) and the Macular Vision Research Foundation.
References
Azadi, S., Molday, L. L., and Molday, R. S. (2010). RD3, the protein associated with Leber congenital amaurosis type 12, is required for guanylate cyclase trafficking in photoreceptor cells. Proc. Natl. Acad. Sci. U.S.A. 107, 21158–21163. doi: 10.1073/pnas.1010460107
Baehr, W., Karan, S., Maeda, T., Luo, D. G., Li, S., Bronson, J. D., et al. (2007). The function of guanylate cyclase 1 and guanylate cyclase 2 in rod and cone photoreceptors. J. Biol. Chem. 282, 8837–8847. doi: 10.1074/jbc.M610369200
Bowes, C., Li, T., Danciger, M., Baxter, L. C., Applebury, M. L., and Farber, D. B. (1990). Retinal degeneration in the rd mouse is caused by a defect in the beta subunit of rod cGMP-phosphodiesterase. Nature 347, 677–680. doi: 10.1038/347677a0
Chang, B., Hawes, N. L., Pardue, M. T., German, A. M., Hurd, R. E., Davisson, M. T., et al. (2007). Two mouse retinal degenerations caused by missense mutations in the beta-subunit of rod cGMP phosphodiesterase gene. Vision Res. 47, 624–633. doi: 10.1016/j.visres.2006.11.020
Chang, B., Heckenlively, J. R., Hawes, N. L., and Roderick, T. H. (1993). New mouse primary retinal degeneration (rd-3). Genomics 16, 45–49. doi: 10.1006/geno.1993.1138
Cheng, C. L., and Molday, R. S. (2013). Changes in gene expression associated with retinal degeneration in the rd3 mouse. Mol. Vis. 19, 955–969.
Chuang, J. Z., Zhao, Y., and Sung, C. H. (2007). SARA-regulated vesicular targeting underlies formation of the light-sensing organelle in mammalian rods. Cell 130, 535–547. doi: 10.1016/j.cell.2007.06.030
Danciger, M., Ogando, D., Yang, H., Matthes, M. T., Yu, N., Ahern, K., et al. (2008). Genetic modifiers of retinal degeneration in the rd3 mouse. Invest. Ophthalmol. Vis. Sci. 49, 2863–2869. doi: 10.1167/iovs.08-1715
Deretic, D., Williams, A. H., Ransom, N., Morel, V., Hargrave, P. A., and Arendt, A. (2005). Rhodopsin C terminus, the site of mutations causing retinal disease, regulates trafficking by binding to ADP-ribosylation factor 4 (ARF4). Proc. Natl. Acad. Sci. U.S.A. 102, 3301–3306. doi: 10.1073/pnas.0500095102
Dizhoor, A. M., Olshevskaya, E. V., Henzel, W. J., Wong, S. C., Stults, J. T., Ankoudinova, I., et al. (1995). Cloning, sequencing, and expression of a 24-kDa Ca(2+)-binding protein activating photoreceptor guanylyl cyclase. J. Biol. Chem. 270, 25200–25206. doi: 10.1074/jbc.270.42.25200
Dizhoor, A. M., Olshevskaya, E. V., and Peshenko, I. V. (2010). Mg2+/Ca2+ cation binding cycle of guanylyl cyclase activating proteins (GCAPs): role in regulation of photoreceptor guanylyl cyclase. Mol. Cell. Biochem. 334, 117–124. doi: 10.1007/s11010-009-0328-6
Fain, G. L. (2006). Why photoreceptors die (and why they don’t). Bioessays 28, 344–354. doi: 10.1002/bies.20382
Fariss, R. N., Molday, R. S., Fisher, S. K., and Matsumoto, B. (1997). Evidence from normal and degenerating photoreceptors that two outer segment integral membrane proteins have separate transport pathways. J. Comp. Neurol. 387, 148–156. doi: 10.1002/(SICI)1096-9861(19971013)387:1<148::AID-CNE12>3.0.CO;2-Q
Friedman, J. S., Chang, B., Kannabiran, C., Chakarova, C., Singh, H. P., Jalali, S., et al. (2006). Premature truncation of a novel protein, RD3, exhibiting subnuclear localization is associated with retinal degeneration. Am. J. Hum. Genet. 79, 1059–1070. doi: 10.1086/510021
Gorczyca, W. A., Gray-Keller, M. P., Detwiler, P. B., and Palczewski, K. (1994). Purification and physiological evaluation of a guanylate cyclase activating protein from retinal rods. Proc. Natl. Acad. Sci. U.S.A. 91, 4014–4018. doi: 10.1073/pnas.91.9.4014
Gray-Keller, M. P., and Detwiler, P. B. (1994). The calcium feedback signal in the phototransduction cascade of vertebrate rods. Neuron 13, 849–861. doi: 10.1016/0896-6273(94)90251-8
Helten, A., Saftel, W., and Koch, K. W. (2007). Expression level and activity profile of membrane bound guanylate cyclase type 2 in rod outer segments. J. Neurochem. 103, 1439–1446. doi: 10.1111/j.1471-4159.2007.04923.x
Karan, S., Frederick, J. M., and Baehr, W. (2010). Novel functions of photoreceptor guanylate cyclases revealed by targeted deletion. Mol. Cell. Biochem. 334, 141–155. doi: 10.1007/s11010-009-0322-z
Karan, S., Tam, B. M., Moritz, O. L., and Baehr, W. (2011). Targeting of mouse guanylate cyclase 1 (Gucy2e) to Xenopus laevis rod outer segments. Vision Res. 51, 2304–2311. doi: 10.1016/j.visres.2011.09.001
Kelsell, R. E., Gregory-Evans, K., Payne, A. M., Perrault, I., Kaplan, J., Yang, R. B., et al. (1998). Mutations in the retinal guanylate cyclase (RETGC-1) gene in dominant cone-rod dystrophy. Hum. Mol. Genet. 7, 1179–1184. doi: 10.1093/hmg/7.7.1179
Kukekova, A. V., Goldstein, O., Johnson, J. L., Richardson, M. A., Pearce-Kelling, S. E., Swaroop, A., et al. (2009). Canine RD3 mutation establishes rod-cone dysplasia type 2 (rcd2) as ortholog of human and murine rd3. Mamm. Genome 20, 109–123. doi: 10.1007/s00335-008-9163-4
Kwok, M. C., Holopainen, J. M., Molday, L. L., Foster, L. J., and Molday, R. S. (2008). Proteomics of photoreceptor outer segments identifies a subset of SNARE and Rab proteins implicated in membrane vesicle trafficking and fusion. Mol. Cell. Proteomics 7, 1053–1066. doi: 10.1074/mcp.M700571-MCP200
Lavorgna, G., Lestingi, M., Ziviello, C., Testa, F., Simonelli, F., Manitto, M. P., et al. (2003). Identification and characterization of C1orf36, a transcript highly expressed in photoreceptor cells, and mutation analysis in retinitis pigmentosa. Biochem. Biophys. Res. Commun. 308, 414–421. doi: 10.1016/S0006-291X(03)01410-4
Linberg, K. A., Fariss, R. N., Heckenlively, J. R., Farber, D. B., and Fisher, S. K. (2005). Morphological characterization of the retinal degeneration in three strains of mice carrying the rd-3 mutation. Vis. Neurosci. 22, 721–734. doi: 10.1017/S0952523805226044
Lowe, D. G., Dizhoor, A. M., Liu, K., Gu, Q., Spencer, M., Laura, R., et al. (1995). Cloning and expression of a second photoreceptor-specific membrane retina guanylyl cyclase (RetGC), RetGC-2. Proc. Natl. Acad. Sci. U.S.A. 92, 5535–5539. doi: 10.1073/pnas.92.12.5535
Mazelova, J., Astuto-Gribble, L., Inoue, H., Tam, B. M., Schonteich, E., Prekeris, R., et al. (2009). Ciliary targeting motif VxPx directs assembly of a trafficking module through Arf4. EMBO J. 28, 183–192. doi: 10.1038/emboj.2008.267
Molday, L. L., Djajadi, H., Yan, P., Szczygiel, L., Boye, S. L., Chiodo, V. A., et al. (2013). RD3 gene delivery restores guanylate cyclase localization and rescues photoreceptors in the Rd3 mouse model of Leber congenital amaurosis 12. Hum. Mol. Genet. 22, 3894–3905. doi: 10.1093/hmg/ddt244
Olshevskaya, E. V., Calvert, P. D., Woodruff, M. L., Peshenko, I. V., Savchenko, A. B., Makino, C. L., et al. (2004). The Y99C mutation in guanylyl cyclase-activating protein 1 increases intracellular Ca2+ and causes photoreceptor degeneration in transgenic mice. J. Neurosci. 24, 6078–6085. doi: 10.1523/JNEUROSCI.0963-04.2004
Perrault, I., Estrada-Cuzcano, A., Lopez, I., Kohl, S., Li, S., Testa, F., et al. (2013). Union makes strength: a worldwide collaborative genetic and clinical study to provide a comprehensive survey of RD3 mutations and delineate the associated phenotype. PLoS ONE 8:e51622. doi: 10.1371/journal.pone.0051622
Perrault, I., Rozet, J. M., Calvas, P., Gerber, S., Camuzat, A., Dollfus, H., et al. (1996). Retinal-specific guanylate cyclase gene mutations in Leber’s congenital amaurosis. Nat. Genet. 14, 461–464. doi: 10.1038/ng1296-461
Peshenko, I. V., and Dizhoor, A. M. (2006). Ca2+ and Mg2+ binding properties of GCAP-1. Evidence that Mg2+-bound form is the physiological activator of photoreceptor guanylyl cyclase. J. Biol. Chem. 281, 23830–23841. doi: 10.1074/jbc.M600257200
Peshenko, I. V., Olshevskaya, E. V., Azadi, S., Molday, L. L., Molday, R. S., and Dizhoor, A. M. (2011a). Retinal degeneration 3 (RD3) protein inhibits catalytic activity of retinal membrane guanylyl cyclase (RetGC) and its stimulation by activating proteins. Biochemistry 50, 9511–9519. doi: 10.1021/bi201342b
Peshenko, I. V., Olshevskaya, E. V., Savchenko, A. B., Karan, S., Palczewski, K., Baehr, W., et al. (2011b). Enzymatic properties and regulation of the native isozymes of retinal membrane guanylyl cyclase (RetGC) from mouse photoreceptors. Biochemistry 50, 5590–5600. doi: 10.1021/bi200491b
Polans, A., Baehr, W., and Palczewski, K. (1996). Turned on by Ca2+! The physiology and pathology of Ca(2+)-binding proteins in the retina. Trends Neurosci. 19, 547–554. doi: 10.1016/S0166-2236(96)10059-X
Preising, M. N., Hausotter-Will, N., Solbach, M. C., Friedburg, C., Ruschendorf, F., and Lorenz, B. (2012). Mutations in RD3 are associated with an extremely rare and severe form of early onset retinal dystrophy. Invest. Ophthalmol. Vis. Sci. 53, 3463–3472. doi: 10.1167/iovs.12-9519
Ramamurthy, V., Tucker, C., Wilkie, S. E., Daggett, V., Hunt, D. M., and Hurley, J. B. (2001). Interactions within the coiled-coil domain of RetGC-1 guanylyl cyclase are optimized for regulation rather than for high affinity. J. Biol. Chem. 276, 26218–26229. doi: 10.1074/jbc.M010495200
Shyjan, A. W., De Sauvage, F. J., Gillett, N. A., Goeddel, D. V., and Lowe, D. G. (1992). Molecular cloning of a retina-specific membrane guanylyl cyclase. Neuron 9, 727–737. doi: 10.1016/0896-6273(92)90035-C
Sung, C. H., and Chuang, J. Z. (2010). The cell biology of vision. J. Cell Biol. 190, 953–963. doi: 10.1083/jcb.201006020
Tam, B. M., Moritz, O. L., Hurd, L. B., and Papermaster, D. S. (2000). Identification of an outer segment targeting signal in the COOH terminus of rhodopsin using transgenic Xenopus laevis. J. Cell Biol. 151, 1369–1380. doi: 10.1083/jcb.151.7.1369
Tam, B. M., Moritz, O. L., and Papermaster, D. S. (2004). The C terminus of peripherin/rds participates in rod outer segment targeting and alignment of disk incisures. Mol. Biol. Cell 15, 2027–2037. doi: 10.1091/mbc.E03-09-0650
Woodford, B. J., Liu, Y., Fletcher, R. T., Chader, G. J., Farber, D. B., Santos-Anderson, R., et al. (1982). Cyclic nucleotide metabolism in inherited retinopathy in collies: a biochemical and histochemical study. Exp. Eye Res. 34, 703–714. doi: 10.1016/S0014-4835(82)80031-6
Woodruff, M. L., Sampath, A. P., Matthews, H. R., Krasnoperova, N. V., Lem, J., and Fain, G. L. (2002). Measurement of cytoplasmic calcium concentration in the rods of wild-type and transducin knock-out mice. J. Physiol. 542, 843–854. doi: 10.1113/jphysiol.2001.013987
Xu, J., Morris, L., Thapa, A., Ma, H., Michalakis, S., Biel, M., et al. (2013). cGMP accumulation causes photoreceptor degeneration in CNG channel deficiency: evidence of cGMP cytotoxicity independently of enhanced CNG channel function. J. Neurosci. 33, 14939–14948. doi: 10.1523/JNEUROSCI.0909-13.2013
Keywords: retinal degeneration 3, guanylate cyclase, photoreceptor cells, protein trafficking, photoreceptor degeneration, Leber congenital amaurosis, gene therapy
Citation: Molday LL, Jefferies T and Molday RS (2014) Insights into the role of RD3 in guanylate cyclase trafficking, photoreceptor degeneration, and Leber congenital amaurosis. Front. Mol. Neurosci. 7:44. doi: 10.3389/fnmol.2014.00044
Received: 28 February 2014; Accepted: 29 April 2014;
Published online: 26 May 2014.
Edited by:
Wolfgang Baehr, University of Utah, USAReviewed by:
John Oberdick, The Ohio State University, USAJohann H. Brandstätter, Friedrich-Alexander-Universität Erlangen-Nuremberg, Germany
Copyright © 2014 Molday, Jefferies and Molday. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Robert S. Molday, Department of Biochemistry and Molecular Biology, Center for Macular Research, University of British Columbia, 2350 Health Sciences Mall, Vancouver, BC V6T 1Z3, Canada e-mail:bW9sZGF5QG1haWwudWJjLmNh