 Zaira Ortega
Zaira Ortega Jose J. Lucas
Jose J. Lucas- 1Department of Molecular Biology, Centro de Biología Molecular “Severo Ochoa,” Consejo Superior de Investigaciones Científicas (CSIC), Universidad
- 2Autónoma de Madrid (UAM), Centro Investigación Biomédica en Red Enfermedades Neurodegenerativa (CIBERNED), Madrid, Spain
Huntington’s disease (HD) is a genetic autosomal dominant neurodegenerative disease caused by the expansion of a CAG repeat in the huntingtin (htt) gene. This triplet expansion encodes a polyglutamine stretch (polyQ) in the N-terminus of the high molecular weight (348-kDa) and ubiquitously expressed protein htt. Normal individuals have between 6 and 35 CAG triplets, while expansions longer than 40 repeats lead to HD. The onset and severity of the disease depend on the length of the polyQ tract: the longer the polyglutamine stretch (polyQ) is, the earlier the disease begins and the more severe the symptoms are. One of the main histopathological hallmarks of HD is the presence of intraneuronal proteinaceous inclusion bodies, whose prominent and invariant feature is the presence of ubiquitin (Ub); therefore, they can be detected with anti-ubiquitin and anti-proteasome antibodies. This, together with the observation that mutations in components of the ubiquitin–proteasome system (UPS) give rise to some neurodegenerative diseases, suggests that UPS impairment may be causative of HD. Even though the link between disrupted Ub homeostasis and protein aggregation to HD is undisputed, the functional significance of these correlations and their mechanistic implications remains unresolved. Moreover, there is no consistent evidence documenting an accompanying decrease in levels of free Ub or disruption of Ub pool dynamics in neurodegenerative disease or models thus suggesting that the Ub-conjugate accumulation may be benign and just underlie lesion in 26S function. In this chapter we will elaborate on the different studies that have been performed using different experimental approaches, in order to shed light to this matter.
Introduction
Huntington’s disease (HD) is a genetic autosomal dominant neurodegenerative disease (Wexler et al., 1987) that affects approximately 1 out of 10.000 individuals in most of the populations with European background (Harper, 1992). It shows symptoms in midlife and patients often die 15–20 years after the onset of the symptoms (Ambrose et al., 1994). Currently, there is no effective treatment to prevent or delay disease progression (Vonsattel and DiFiglia, 1998). HD patients suffer from motor dysfunction (chorea, rigidity, dystonia, and oculomotor dysfunction among others), cognitive decline also known as dementia (subcortical dementia, including affective and personality changes, and problems acquiring new knowledge), and psychopathological dysfunction (depression, suicide, and mania are the most frequent ones). Emotional and cognitive changes often precede motor dysfunction by several years (about 3 years). These symptoms are the result of the selective neurodegeneration that occurs preferentially in the striatum of the patients (Graveland et al., 1985; Gusella et al., 1993; Vonsattel and DiFiglia, 1998).
Huntington’s disease is included in a group of neurodegenerative diseases called proteinopathies [which include pathologies such as Alzheimer’s disease (AD) or Parkinson’s disease (PD)] due to the fact that aggregate-prone proteins cause all of them. The main histopathological hallmark of these diseases is the presence of aggregates constituted by the mutant or modified proteins: these inclusion bodies (IBs) can be predominantly cytosolic (such as in PD, and HD), intranuclear [for example, spinocerebellar ataxia type 1 (SCA1)], aggregated in the endoplasmic reticulum (as seen with neuroserpin mutations that cause familial encephalopathy with neuroserpin IBs) or extracellularly secreted (for example amyloid- β in AD). In HD, the mutated protein is the ubiquitously expressed protein huntingtin (htt) and the mutation consists of an expansion of a CAG repeat located in the 5′ terminus of the htt gene (HDCRG, 1993) which translates into a polyQ in the N’ terminus of the protein (Gusella et al., 1993; Locke et al., 1993). Normal individuals have between 6 and 35 CAG triplets, while expansions longer than 40 repeats lead to HD (Andrew et al., 1993; HDCRG, 1993). The onset and severity of the disease depend on the length of the polyQ tract, the longer the polyQ is, the earlier the disease begins and the more severe the symptoms are (Andrew et al., 1993; Snell et al., 1993). Apart from HD, there are eight additional hereditary diseases caused by CAG/polyQ expansion (Zoghbi and Orr, 2000; Ross, 2002), all of them are neurological diseases, despite the different nature of the proteins involved and their ubiquitous expression, suggesting a selective vulnerability of the neurons for polyQ (Figure 1). In all these diseases the triplet expansions are within the coding sequence of the gene, and they are always translated in the reading frame that produces a polyQ sequence. Moreover, the threshold for the expansion to become pathogenic is around 40 repeats in most of these diseases (Zoghbi and Orr, 2000) both in culture and in vivo. Interestingly, the threshold length for in vitro aggregation correlates with the pathogenic repeat length threshold (Scherzinger et al., 1997), thus suggesting that PolyQ aggregation is a key element in the pathogenesis.
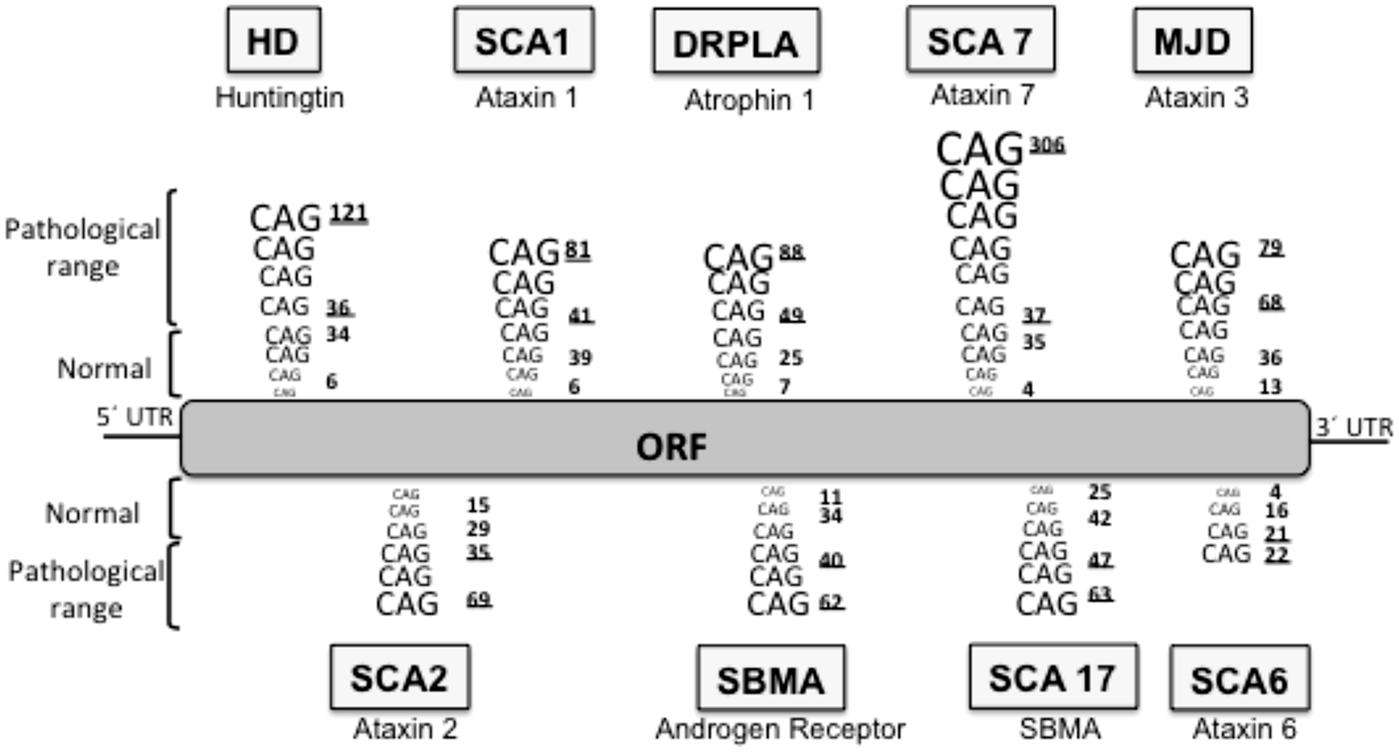
FIGURE 1. Neurodegenerative diseases caused by CAG/polyQ expansion. HD, Huntington’s disease; SCA1, spinocerebellar ataxia 1; DRPLA, dentatorubral-pallidoluysian atrophy; SCA7, spinocerebellar ataxia 7; MJD, Machado–Joseph dystrophy; SCA2, spinocerebellar ataxia 2; SBMA, spinobulbar muscular atrophy; SCA17, spinocerebellar ataxia 17; SCA6, spinocerebellar ataxia 6. The lower underlined number in each pathology represent the pathological threshold of the disease, the last underlined number represents the longest repetition analyzed. ORF, open reading frame.
Although the hemizygous loss of function of normal proteins in polyQ diseases, and particularly in HD, may contribute to some aspects of the pathologies, a toxic gain of function of the expanded polyQ is the most likely determinant of the disease. It can cause disease by conferring additional properties on the mutant gene product that may include hyperactivity of normal function and/or new toxic properties unrelated to normal function. Mice lacking one htt allele are essentially normal, although complete loss of htt causes embryonic lethality (Duyao et al., 1995; Nasir et al., 1995; Zeitlin et al., 1995). Humans with Wolf–Hirschhorn syndrome have hemizygous loss of the tip of chromosome 4p, which includes the HD gene, interestingly, these individuals do not show features of HD (Harper, 1996). Following these findings, a number of transgenic animal models have been made that express a mutant htt∗ transgene, comprising either the whole coding region, an amino-terminal fragment or simply isolated expanded polyQ repeats. Despite also having two normal htt orthologs, these animals recapitulate many of the features of the human disease (for a review, see Rubinsztein, 2002). Furthermore, expression of hypoxanthine phosphoribosyl transferase, a non-disease related protein that doesnot express polyQ, with expanded polyQ caused the mice to develop a progressive neurological disorder with clinical and pathological features reminiscent of HD, which implies that transferring the polyQ tract itself is sufficient to induce aggregation and disease (Ordway et al., 1997).
Protein Degradation Pathways
In cells, the efficient folding of new polypeptides and the efficient elimination of misfolded or damaged proteins is critical to the maintenance of protein homeostasis and cellular health. The presence of IBs in neurons in proteinopathies suggests a failure in the degradation pathways. Eukaryotic cells have two main routes for clearing misfolded or toxic proteins, the ubiquitin–proteasome system (UPS) and the autophagy-lysosome pathways. The UPS works both in the nucleus and in the cytoplasm and is responsible for the recycling and degradation of most of the short-lived and misfolded soluble proteins (Hershko and Ciechanover, 1998). On the contrary, the autophagy-lysosome pathway mainly degrades long-lived proteins and degenerated organelles, and requires the formation of double-membrane-bounded autophagosomes (Klionsky et al., 2003, 2008; Suzuki and Ohsumi, 2007) and it is thus restricted to the cytoplasm. Both pathways have been suggested to play a role in HD (Rubinsztein, 2006; Thompson et al., 2009), although recent studies suggest that the UPS is more important than autophagy for removing toxic- N-terminal htt∗ fragments (Li et al., 2010). If an impairment of the degradation pathway is the triggering step or a secondary effect in HD is still unclear. There are previous reviews on this matter (Valera et al., 2005; Ortega et al., 2007; Rubinsztein, 2007) and in this review we also discuss more recent reports on the status of the UPS in HD.
The Ubiquitin–Proteasome System
As one of the main routes of protein degradation, the UPS is involved in many cellular mechanisms in the nervous system such as neuronal plasticity, memory, and regulation of neurotransmission at pre- and post-synaptic sites, thus it plays a critical role in neuronal signaling (Krug et al., 1984; Fonseca et al., 2006; Karpova et al., 2006). It represents a major defense against misfolded proteins, particularly in post-mitotic neurons that are unable to divide to reduce their burden of damaged proteins. Despite being highly conserved across species, structurally and functionally distinct subunit compositions of the proteasome have been identified in different tissues (Glickman and Raveh, 2005; Drews et al., 2007; Tai et al., 2010). These variants have been attributed to alterations in ubiquitin (Ub) ligase activity, proteasome subunit composition, and tissue-specific proteasome-interacting proteins (Glickman and Raveh, 2005; Drews et al., 2007; Tai et al., 2010). In UPS degradation pathway there are two differentiated steps: (1) targeting of the protein for degradation and (2) substrate proteolysis in the proteasome. There are many molecules involved in these steps. In protein targeting for proteasomal degradation substrates must be covalently modified with Ub, which is conjugated through its carboxy terminus to form chains of four or more Ub molecules linked by lysines at residue 48 (Hershko and Ciechanover, 1998; Thrower et al., 2000; Pickart, 2001; Kuhlbrodt et al., 2005). This conjugation typically involves three types of enzyme: E1 (ubiquitin-activating enzyme) hydrolyses ATP and forms a thioester-linked conjugate between itself and Ub; E2 (ubiquitin-conjugating enzyme) receives Ub from E1 and forms a similar thioester intermediate with Ub; and E3 (ubiquitin-ligase) binds both E2 and the substrate, and transfers the Ub to the substrate (Figure 2; Hershko and Ciechanover, 1998; Pickart, 2001). Polyubiquitinated proteins are recognized and subsequently degraded by the 26S proteasome. This ATP-dependent proteolytic complex consists of a 20S core particle and one or two 19S regulatory particle(s). The barrel-shaped 20S complex is composed of four heptagonal rings where the proteolytic activities reside (Groll et al., 1997; DeMartino and Slaughter, 1999). The 19S regulatory particles are important for the recognition, unfolding, and translocation of ubiquitinated substrates into the 20S core subunit for degradation (Voges et al., 1999; Hartmann-Petersen et al., 2003). The polyUb chains are not degraded by the proteasome, deubiquitilating enzymes (DUBs) remove the chain from the substrates once they have been recognized by the 19S subunit of the proteasome and separate it into monomers ready to be reused (Kawakami et al., 1999).
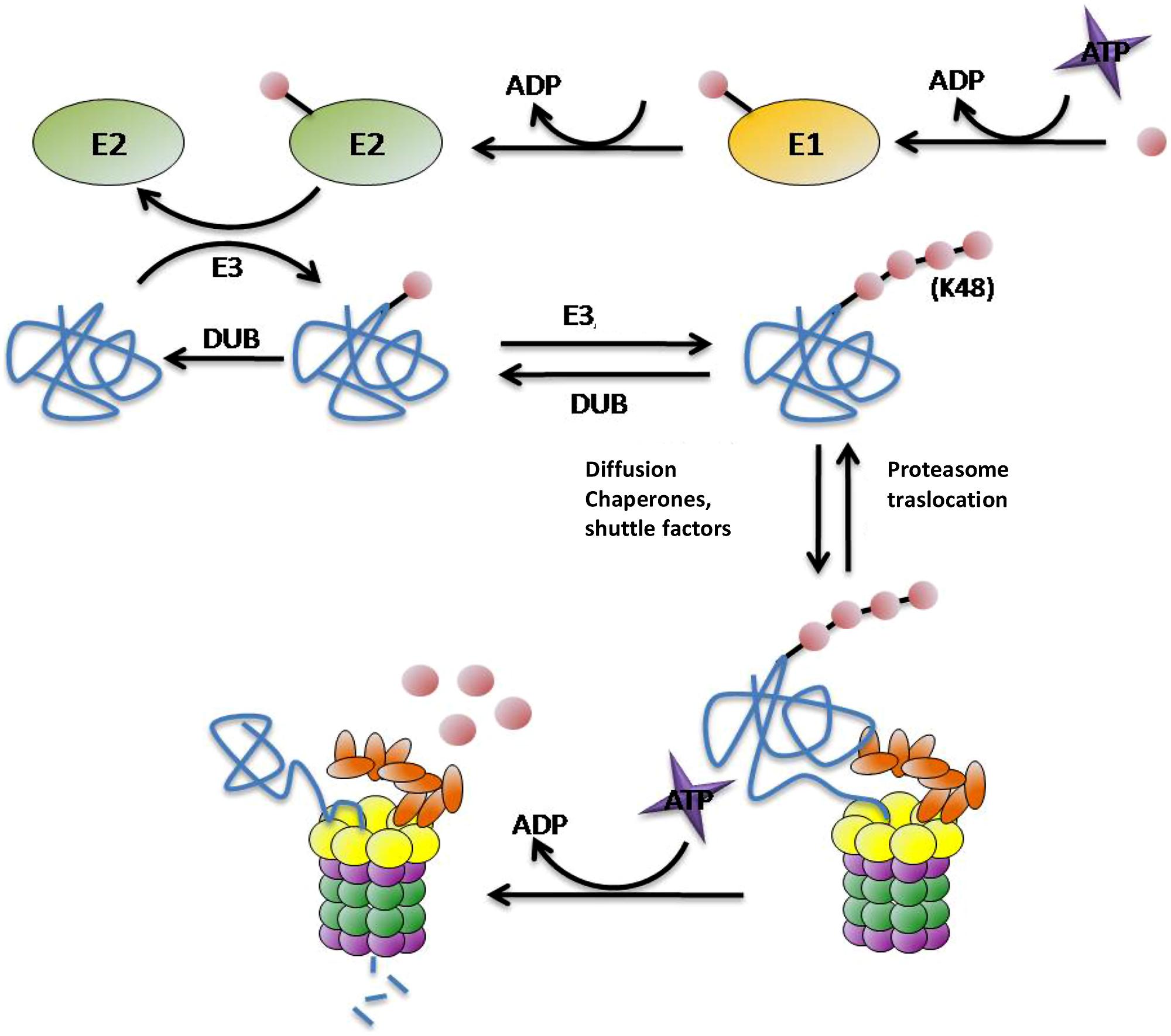
FIGURE 2. Protein targeting and degradation by the UPS. E1, E2, E3, and E4 enzymes are in charge for transferring the Ub molecules (pink circles) to the substrates (blue). This process requires energetic in term of ATP molecules. The proteasome is the protease responsible for the proteolysis of the substrates into small peptides. The polyubiquitin chain is not degraded by the proteasome, DUBs enzymes separate them from the substrate and divide them into monomers ready to be reused.
The UPS in HD
There is cellular and genetic evidence supporting the hypothesis of UPS impairment in neurodegenerative diseases. Regarding genetic evidence several neurodegenerative diseases have been described to be caused by mutations in different components at different levels of the labeling and degradation (ubiquitilation, deubiquitilation, and substrate delivery) of substrates by the UPS (Table 1). These pathologies suggest that primary genetic deficiencies of components of the UPS are sufficient to cause neurodegeneration. Regarding cellular evidence, the presence of IBs constituted by the mutant proteins has already been described both in human brain (DiFiglia et al., 1997) and in animal models of HD (Davies et al., 1997). In addition, complementary pharmacological data show that both in cellular (Waelter et al., 2001) and animal models (McNaught et al., 2004), the inhibition of the proteasome, by using specific inhibitors, produces an increase in aggregation of htt∗ that is critically dependent on the proteasomal activity and can cause parkinsonian features, including Lewy body-like aggregates (McNaught et al., 2004). Similarly, the reversal of aggregates that takes place in primary neurons from the HD inducible mouse model upon shutdown of htt∗ expression no longer takes place in the presence of proteasome inhibitors (Martin-Aparicio et al., 2001). These IBs are labeled with antibodies that recognize Ub and different proteasome subunits (in the 20S core and 19S caps; DiFiglia et al., 1997; Cummings et al., 1998; Goedert et al., 1998; Sherman and Goldberg, 2001; Díaz-Hernández et al., 2003; Schmitt, 2006), suggesting a direct (htt∗ protein) or indirect (proteins associated to htt∗) sequestration of the proteasome into the IBs. Besides, it was reported the spatial restriction of proteasomes within aggregates in fluorescence recovery after photobleaching (FRAP) experiments (Holmberg et al., 2004). However, recent results support that proteasomes are dynamically and reversibly recruited into IBs (Schipper-Krom et al., 2014) and that they remain catalytically active and accessible to substrates. This challenges the concept of proteasome sequestration and impairment in HD, and supports the absence of proteasome impairment in mouse models of HD. In line with this, an increase in proteasome activity in the insoluble cellular fractions of mtHtt-Q150 expressing neuronal cells has also been described (Jana et al., 2001).Taking all these data as a starting point, many experiments testing the capability of the IBs of directly inhibiting the proteasome have been conducted. It has been tested whether 26S activity is inhibited by IBs in HD by incubating 26S purified proteasomes with in vitro generated polyQ aggregates (Bennett et al., 2005). No impairment in any of the proteasome catalytic activities were detected what argues against a decreased proteasome activity. However, if ubiquitilation of the aggregates were important for its potential inhibitory interaction with 26S proteasomes, the previous experiments would not detect it. To overcome this limitation, similar experiments were performed with aggregates purified from mouse models and post-mortem human brains (Díaz-Hernández et al., 2006) instead of in vitro generated polyQ aggregates this approach in fact detected 26S activity impairment upon incubation with isolated microaggregates such as htt filaments although not when incubated with isolated IBs. To test the hypothesis in a more physiological environment, similar experiments were performed with HD mouse model brain extracts (Díaz-Hernández et al., 2003; Bowman et al., 2005; Bett et al., 2006). By this approach, not only there was no decrease in the catalytic activities, but also there was a selective increase in the chymotrypsin- and trypsin-like activities that were believed to be a result of a qualitative change in the subunit composition of the proteasomes (Díaz-Hernández et al., 2003, 2004a). These sets of experiment report that the UPS remained active in HD and argue against the postulated inhibition of proteasomes. Thus, opening the possibility of reinterpreting the meaning of the marked accumulation of PolyUb chains observed in the brains of these mouse models and in human HD patients (Bennett et al., 2007). On the other hand, when these experiments were performed on human post-mortem HD brains tissue, a decreased activity of the proteasome was reported (Seo et al., 2004) suggesting differences in the UPS but, due to inherent limitations associated to analysis of enzymatic activities in post-mortem tissue, it is difficult to conclude whether proteasome activity was really altered or not. In summary, it is not possible to draw a definite conclusion from all these studies due to the limitations associated to each of the employed techniques to monitor status or function of UPS components (degradation of small fluorogenic peptides, degradation of ubiquitylated proteins, detection of Ub-conjugates, etc.) and the differences in the analyzed systems or samples (in vitro incubation of proteasomes with PolyQ species at different degrees of aggregation and ubiquitylation vs. tissue homogenates with the latter being either fresh frozen animal model tissue or human frozen tissue with varying extents of post-mortem intervals).
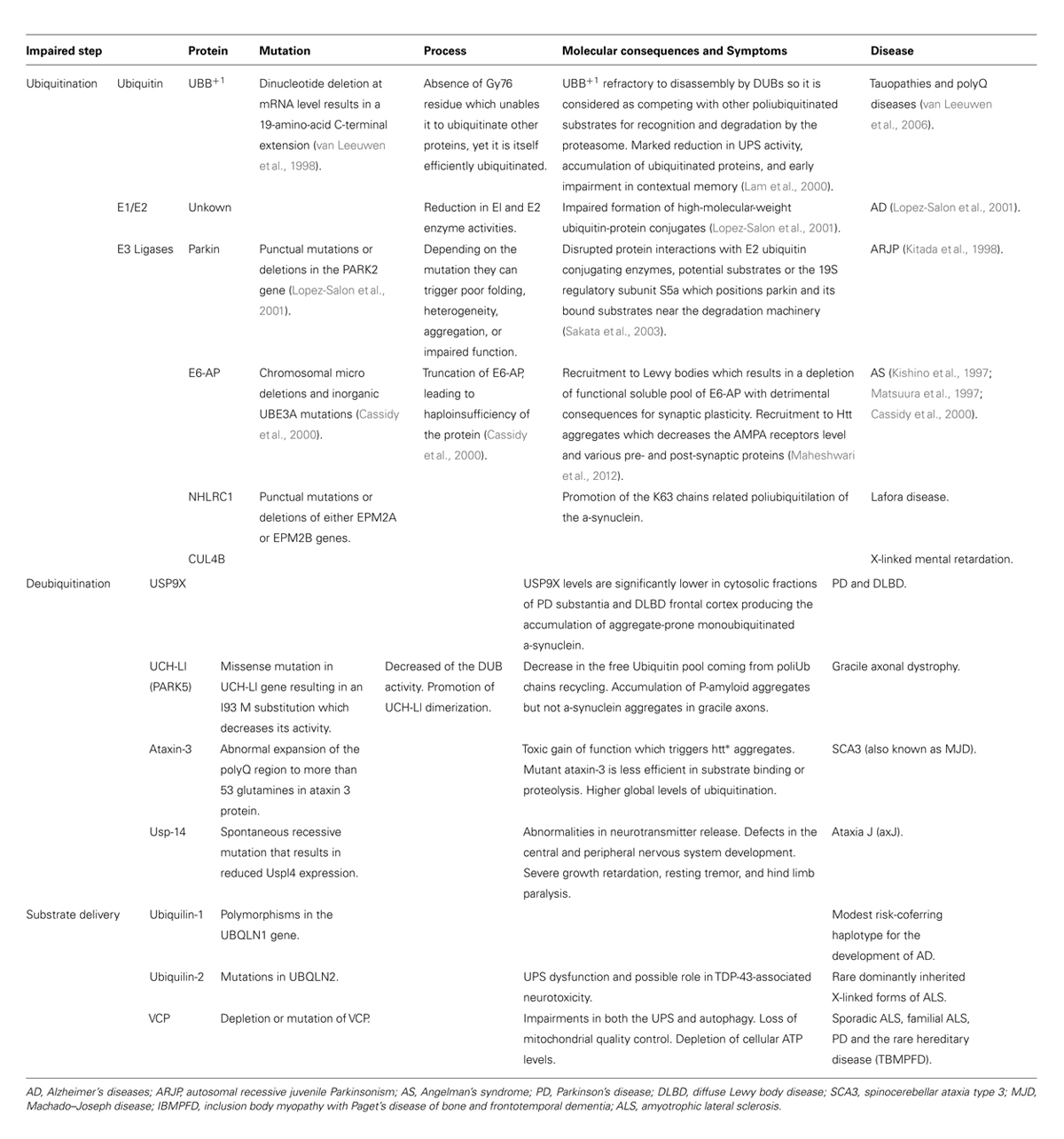
TABLE 1. Genetic evidence involving the UPS in neurodegenerative diseases.
In Konstantinova et al. (2008) the proteasome was described as dynamic structure in terms of its composition as 26S proteasome is constantly assembling and disassembling, and its 19S and 20S subunits are targets for a great number of post-translational modifications including phosphorylation and acetylation. 26S proteasomes would get assembled just to degrade the substrates and immediately disassembled again. These data led to hypothesize that IBs could interact with the disassembled 19S and 20S subunits preventing them from assembling again to degrade the substrates and retaining them in the IBs. Experiments testing the catalytic activities of the 26S proteasome and the 20S subunit alone in the presence of IBs purified from mouse models and post-mortem human brains (Díaz-Hernández et al., 2006) were performed. Experiments involving the 20S subunit were performed in the presence of sodium dodecyl sulfate (SDS) to facilitate the entrance of the substrate into the catalytic chamber however, as in the case of the 26S proteasome; they failed to detect enzymatic activity inhibition. These results confirm the previously obtained results supporting the absence of influence of IBs upon proteasome activity.
The above mentioned results were obtained from experimental approaches that assume that the proteasome degrades htt, however, the three endoproteolytic activities of the proteasome (trypsin-like, chymotrypsin-like and PGPH) cut the peptide bonds after basic, hydrophobic, or acid residues respectively (DeMartino and Slaughter, 1999) and glutamine does not really fit in any of these categories. This fact brought new hypothesis for a possible UPS impairment such as the possibility of htt∗ getting clogged in the channel of the 20S core subunit blocking access to other ubiquitinated substrates and therefore impairing the proteostasis of the cell. Several experiments have been performed in order to answer whether proteasome can degrade htt or not and results supporting both hypothesis can be found. Experiments performed with peptides containing polyQ tracts and incubated with purified eukaryotic proteasomes showed no digestion of the polyQ tract by the proteasome (Holmberg et al., 2004; Venkatraman et al., 2004), and more importantly, when degrading expanded polyQ-containing proteins, proteasomes might be generating the most toxic and aggregation-prone fragments. Such polyQ sequences (38–300Qs) exceed the lengths of normal proteasome products (2–25 residues) and a failure of theses fragments to exit the proteasome may interfere with proteasome function and it is known that expression of pure polyQ peptides is sufficient for aggregation (IB formation) and toxicity in cells (Yang et al., 2002; Raspe et al., 2009). However, more recent studies showed through quantitative flow cytometry and live-cell time-lapse imaging that N-htt—whether aggregated or not—does not choke or clog 26S and proposes that 26S activity is compromised only indirectly as a result of disrupted protein folding homeostasis (Hipp et al., 2012). As a matter of fact, UPS has been proved to be able to degrade htt∗-exon1 completely, including the expanded polyQ sequence (Michalik and Van Broeckhoven, 2004; Juenemann et al., 2013), although it has also been shown that the degradation signal that accompanies the polyQ tract is determinant to obtain this result (Juenemann et al., 2013).
There has always been a controversy regarding the pathogenicity of the IBs. Studies using transfected cells further suggested that toxicity might be induced by aggregates (Waelter et al., 2001). On the other hand, more recent observations support they hypothesis that aggregates are not pathogenic, or even that they might be protective (Saudou et al., 1998; Kopito, 2000; Arrasate et al., 2004; Bennett et al., 2005; Bowman et al., 2005) and that the pathogenic species could be the intermediate species that generate during IB formation. Htt∗ aggregation in mouse brain is not only an early event, but occurs rapidly (Gong et al., 2012). It has been found that IBs are present in the cortex of HD brains before any sign of degeneration can be detected, and many MSSNs in the striatum lack IBs despite the presence of significant neuronal loss (Gutekunst et al., 1999). Furthermore, there are also some transgenic mouse models of HD in which IBs appear only after symptoms onset (Menalled et al., 2003), and in transfected primary cultured neurons, their ability to build IBs protects them from the toxicity elicited by htt∗ (Arrasate et al., 2004). In Poirier et al. (2002) described in detail the process of IB formation. They are dynamic structures that require constant production of htt∗ to maintain them. If the influx of the mutated protein is inhibited, IBs disappear and the neurologic phenotype of the disease improves (Yamamoto et al., 2000; Díaz-Hernández et al., 2004b, 2005). IBs are not amorphous associations of N-terminal htt∗ fragments but highly organized structures. During the formation of an IB, several intermediate species are constituted and their organized interactions give rise to the IB. The most simple species, and therefore the one that appears earlier in the disease, is the N-terminal htt∗ fragments, also called monomers. These monomers carry the expanded polyQ that renders them highly prone to aggregate. Monomers associate to form globular assemblies with an average size of 4–5 nm called oligomers. These oligomers serve as nucleation seeds to form more complicated aggregation structures; they linearly associate to form protofibrils that have an undefined length. Finally, protofibrils assemble through polar zippers to obtain β-laminas called fibers. The unorganized assembly of these fibers gives rise to IBs. In order to confirm or discard the hypothesis that intermediate species as the pathogenic structures in HD, (Bennett et al.) tested the effect of in vitro-generated soluble htt∗ fragments (Bowman et al., 2005), highly aggregated fibrillar species or soluble oligomeric aggregates (Chen and Wetzel, 2001; Poirier et al., 2002) on the degradation of ubiquitin-dependent and ubiquitin-independent substrates by purified 26S proteasomes. No differences in proteasome activity were observed in any of the analyzed species, which argues against the notion that a direct interaction between 26S proteasomes and monomers or aggregates of expanded polyQ could result in decreased proteasome activity. However, as in those experiments performed with purified IBs the ubiquitilation process is not considered, if it were impaired it would not be detected. To overcome this limitation, Díaz-Hernández et al. (2006) tested the potential inhibition of the 26S and 20S proteasome by polyQ-containing filaments isolated from the brain of the Tet/HD94 inducible mouse model or from post-mortem HD human brain tissue. Filaments isolated from brain inhibited the endoproteolytic activities of the 26S proteasome. However, as above mentioned, when the same experiments were performed with the 20S subunit (in the presence of SDS to facilitate the entrance of the substrate into the catalytic chamber) no inhibition was detected. The selective inhibition of 26S proteasome but not of 20S subunit activities suggested a direct interaction of the ubiquitinated filaments and the 19S ubiquitin-interacting regulatory caps of the 26S proteasome. Interestingly, this interaction was confirmed by immunoelectron microscopy (Díaz-Hernández et al., 2006). These results advocate that fibrillar, and possibly also oligomeric, ubiquitinated polyQ aggregates have the potential to interfere with 26S proteasome through interacting with its 19S subunit but only when theses aggregates are not recruited into IBs. These results therefore strengthen the notion that IB formation may be protective, in this case, by neutralizing the inhibitory action of dispersed ubiquitynated polyQ smaller aggregates.
Short-Lived Fluorescent UPS Reporter Proteins
All the above mentioned experimental approaches focus their attention only on the proteasome activity without taking into account the complexity of the UPS. As shown by numerous genetic evidence, neurodegenerative diseases can be caused by alterations not only at the proteasome level but also at any other step that affects a substrate to be targeted to UPS degradation such as E1, E2, or E3 ubiquitin-ligating processes. In the human genome there are only 16 subtypes of E1 (Ardley and Robinson, 2005), which reflects the low specificity of this step as one E1 can recognize 100s of substrates. There are only 53 E2 coded in the human genome (Ardley and Robinson, 2005), this shows a higher specificity than E1 enzymes but still a single E2 can recognize several substrates. However, more than 527 E3 enzymes have been described (Ardley and Robinson, 2005), hence indicating that they are highly specialized to certain families of substrates. Taking into account the specificity of the pathogenic hallmarks of each neurodegenerative disease, if we are considering the possibility of finding UPS impairment in the ubiquitilating process, the best candidates would be the E3 ligases due to their substrate specificity. A new tool has been recently developed to test the implication of the ubiquitilating process in a very physiological environment. This tool consists of the use of degron-reporter proteins. These reporter proteins result from fusing a UPS degradation signal to a fluorescent protein that converts it into a reporter of UPS activity. These modified proteins have an extremely short half-life and will accumulate only in those cells where the UPS is not working efficiently thus offering cellular resolution (Dantuma et al., 2000; Stack et al., 2000; Neefjes and Dantuma, 2004). Many degradation signals can be attached to the protein and each one will undergo ubiquitilation through different combinations of E1-E2-E3 enzymes, as a consequence, if the combination is not the one affected in the disease, the impairment could be undetected.
The most frequently used degradation signals in HD models are CL1 degron and ubiquitin fusion degradation (UFD) signal. CL1 degron is a 16 amino acid sequence that easily destabilizes proteins by labeling them for ubiquitilation. CL1 degron has been used mainly in cellular models (Bence et al., 2001; Bennett et al., 2005) in which global impairment of the UPS, that results from an intrinsic property of N-terminal htt∗ and not from its sequestration into IBs, was detected. It should be noted though that CL1 is aggregation-prone so would also co-aggregate and increase in levels when the proteostasis system slows down, independent of proteasome function. Moreover, Wang et al used this reporter protein in a mouse model of HD, the R6/2 mouse model, and reported a synapse-specific loss of proteasome activity in R6/2 mice by measuring peptidase activity in isolated synaptosomes.
Ubiquitin fusion degradation signal is an N-terminal-linked Ub molecule that, on one hand has a G76V substitution that prevents removal of the Ub by DUBs and, on the other hand, serves as acceptor for polyUb chains. This degradation signal has been used both in cellular and animal HD models. To generate a mouse model to explore UPS dysfunction, the UFD signal was fused to the GFP protein and the transgene is under the control of the cytomegalovirus immediate-early enhancer and the chicken β-actin promoter so the mice show ubiquitous expression of the reporter (Dantuma et al., 2000). Experiments performed in double transgenic R6/2 ubiquitin-reporter mouse models reported that the UPS remained functionally active in HD and that an age-dependent decline in UPS activity was found to correlate with the age-related accumulation and aggregation of htt∗ in HD mouse brains (Maynard et al., 2009). These results appear to contradict the accumulation of polyUb chains in the brain of R6/2 mice and human HD patients; however, experiments with the inducible HD94 mouse model (Ortega et al., 2010) reconcile the data from cell models supporting polyQ-induced UPS impairment with the contradictory findings of no impairment in constitutive mouse models by showing that the expression of htt∗ does have the potential to induce UPS impairment in vivo in mouse models, thus fitting with previous observations in cell models expressing fluorescent reporter proteins. However, htt∗-induced UPS impairment in vivo is transient and, in good agreement with previous reports combining polyQ mouse models with the same or similar reporter mice, it is not detected with constitute htt∗ expression in adult mice. That the aggregate formation correlates with UPS recovery had also been reported in a cell model (Mitra et al., 2009), and Ortega et al. (2010) was able to demonstrate causality with the use of anti-aggregation compounds in a cell model and also in vivo in mouse models supporting the notion that formation of IBs has a beneficial effect by sequestering the smaller and more toxic species of htt∗.
UPS Impairment as Secondary Effect and its Therapeutic Implications
Most of the experiments conducted to elucidate the implication of the UPS in HD have been pursued considering htt∗ directly involved with the UPS. However, UPS impairment could be a consequence of the impairment of other metabolic pathways in which htt participates. N line with this, it has been suggested that UPS impairment might originate at the level of mitochondrial function/dysfunction. As an ATP-dependent process, the efficiency of ubiquitinated substrate degradation by the proteasome is linked to mitochondrial respiration. Htt∗ has been shown to interfere with mitochondria, leading to reduction in mitochondrial trafficking (Orr et al., 2008), and reduced ATP content has been detected in synaptosomes fractions prepared from the brains of HD knock-in mice (Orr et al., 2008; Wang et al., 2008) These findings suggest that mitochondrial dysfunction may contribute to UPS impairment in HD by depleting critical ATP levels. More recently, a theory of global proteostasis network dysfunction has been proposed in which a rising concentration of htt∗ causes delayed maturation of other cellular chaperon clients, promoting their ubiquitilation and proteasomal degradation (Hipp et al., 2012). This would trigger a competition between increasing numbers of ubiquitinated substrates that may result in UPS dysfunction, independent of any impairment in proteasome activity.
Regarding the therapeutic implications of the current knowledge of the UPS in HD, it seems reasonable to think that any agent that directly or indirectly increases proteolytic processing might be benefitial. While pharmacological inhibitors of the proteasome such as bortezomib are available and have reached the clinic for treatment of various types of cancer (Papandreou and Logothetis, 2004; Nawrocki et al., 2005; Richardson et al., 2006), no pharmacological activators of the proteasome are available. Interestingly, 36% of cancer patients treated with the proteasome inhibitor bortezomib develop peripheral neuropathy (Richardson et al., 2006), thus confirming the neurotoxicity decreased proteasome activity and strengthen the notion of a potential neuroprotective action of agents able to boost proteasome activity. Another possibility would be the use of pharmacological chaperones that bind to and stabilize the folded, functional form of a mutant protein or help to direct it to degradation or refolding pathways (Balch et al., 2008). In the meantime in the absence of proteasome activating drugs, any other agent able to diminish the load of unfolded proteins could be beneficial and, so far, related clinical trials for HD are based on compounds that might indirectly alleviate burden on proteasome by decreasing protein aggregation or by increasing degradation through other pathways like autophagy as is the case of trehalose and rapamycin (Sarkar and Rubinsztein, 2008).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the Spanish Ministry of Science MINECO (SAF2012-34177 grant to Jose J. Lucas), by CIBERNED and by an institutional grant from Fundación Ramón Areces.
References
Ambrose, C. M., Duyao, M. P., Barnes, G., Bates, G. P., Lin, C. S., Srinidhi, J.,et al. (1994). Structure and expression of the Huntington’s disease gene: evidence against simple inactivation due to an expanded CAG repeat. Somat. Cell Mol. Genet. 20, 27–38. doi: 10.1007/BF02257483
Andrew, S. E., Goldberg, Y. P., Kremer, B., Telenius, H., Theilmann, J., Adam, S.,et al. (1993). The relationship between trinucleotide (CAG) repeat length and clinical features of Huntington’s disease. Nat. Genet. 4, 398–403. doi: 10.1038/ng0893-398
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ardley, H. C., and Robinson, P. A. (2005). E3 ubiquitin ligases. Essays Biochem. 41, 15–30. doi: 10.1042/EB0410015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Arrasate, M., Mitra, S., Schweitzer, E. S., Segal, M. R., and Finkbeiner, S. (2004). Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 431, 805–810. doi: 10.1038/nature02998
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Balch, W. E., Morimoto, R. I., Dillin, A., and Kelly, J. W. (2008). Adapting proteostasis for disease intervention. Science 319, 916–919. doi: 10.1126/science.1141448
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bence, N. F., Sampat, R. M., and Kopito, R. R. (2001). Impairment of the ubiquitin-proteasome system by protein aggregation. Science 292, 1552–1555. doi: 10.1126/science.292.5521.1552
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bennett, E. J., Bence, N. F., Jayakumar, R., and Kopito, R. R. (2005). Global impairment of the ubiquitin-proteasome system by nuclear or cytoplasmic protein aggregates precedes inclusion body formation. Mol. Cell 17, 351–365. doi: 10.1016/j.molcel.2004.12.021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bennett, E. J., Shaler, T. A., Woodman, B., Ryu, K. Y., Zaitseva, T. S., Becker, C. H.,et al. (2007). Global changes to the ubiquitin system in Huntington’s disease. Nature 448, 704–708. doi: 10.1038/nature06022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bett, J. S., Goellner, G. M., Woodman, B., Pratt, G., Rechsteiner, M., and Bates, G. P. (2006). Proteasome impairment does not contribute to pathogenesis in R6/2 Huntington’s disease mice: exclusion of proteasome activator REGgamma as a therapeutic target. Hum. Mol. Genet. 15, 33–44. doi: 10.1093/hmg/ddi423
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bowman, A. B., Yoo, S. Y., Dantuma, N. P., and Zoghbi, H. Y. (2005). Neuronal dysfunction in a polyglutamine disease model occurs in the absence of ubiquitin-proteasome system impairment and inversely correlates with the degree of nuclear inclusion formation. Hum. Mol. Genet. 14, 679–691. doi: 10.1093/hmg/ddi064
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cassidy, S. B., Dykens, E., and Williams, C. A. (2000). Prader-Willi and Angelman syndromes: sister imprinted disorders. Am. J. Med. Genet. 97, 136–146. doi: 10.1002/1096-8628(200022)97:2<136::AID-AJMG5>3.0.CO;2-V
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, S., and Wetzel, R. (2001). Solubilization and disaggregation of polyglutamine peptides. Protein Sci. 10, 887–891. doi: 10.1110/ps.42301
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cummings, C. J., Mancini, M. A., Antalffy, B., DeFranco, D. B., Orr, H. T., and Zoghbi, H. Y. (1998). Chaperone suppression of aggregation and altered subcellular proteasome localization imply protein misfolding in SCA1. Nat. Genet. 19, 148–154. doi: 10.1038/502
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dantuma, N. P., Lindsten, K., Glas, R., Jellne, M., and Masucci, M. G. (2000). Short-lived green fluorescent proteins for quantifying ubiquitin/proteasome-dependent proteolysis in living cells. Nat. Biotechnol. 18, 538–543. doi: 10.1038/75406
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Davies, S. W., Turmaine, M., Cozens, B. A., DiFiglia, M., Sharp, A. H., Ross, C. A.,et al. (1997). Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell 90, 537–548. doi: 10.1016/S0092-8674(00)80513-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
DeMartino, G. N., and Slaughter, C. A. (1999). The proteasome, a novel protease regulated by multiple mechanisms. J. Biol. Chem. 274, 22123–22126. doi: 10.1074/jbc.274.32.22123
Díaz-Hernández, M., Hernández, F., Martín-Aparicio, E., Gómez-Ramos, P., Morán, M. A., Castño, J. G.,et al. (2003). Neuronal induction of the immunoproteasome in Huntington’s disease. J. Neurosci. 23, 11653–11661.
Díaz-Hernández, M., Martín-Aparicio, E., Avila, J., Hernández, F., and Lucas, J. J. (2004a). Enhanced induction of the immunoproteasome by interferon gamma in neurons expressing mutant Huntingtin. Neurotox. Res. 6, 463–468. doi: 10.1007/BF03033282
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Díaz-Hernández, M., Moreno-Herrero, F., Gómez-Ramos, P., Moran, M. A., Ferrer, I., Baro, A. M.,et al. (2004b). Biochemical, ultrastructural, and reversibility studies on huntingtin filaments isolated from mouse and human brain. J. Neurosci. 24, 9361–9371. doi: 10.1523/JNEUROSCI.2365-04.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Díaz-Hernández, M., Torres-Peraza, J., Salvatori-Abarca, A., Morán, M. A., Gómez-Ramos, P., Alberch, J.,et al. (2005). Full motor recovery despite striatal neuron loss and formation of irreversible amyloid-like inclusions in a conditional mouse model of Huntington’s disease. J. Neurosci. 25, 9773–9781. doi: 10.1523/JNEUROSCI.3183-05.2005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Díaz-Hernández, M., Valera, A. G., Morán, M. A., Gómez-Ramos, P., Alvarez-Castelao, B., Castaño, J. G.,et al. (2006). Inhibition of 26S proteasome activity by huntingtin filaments but not inclusion bodies isolated from mouse and human brain. J. Neurochem. 98, 1585–1596. doi: 10.1111/j.1471-4159.2006.03968.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
DiFiglia, M., Sapp, E., Chase, K. O., Davies, S. W., Bates, G. P., Vonsattel, J. P.,et al. (1997). Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 277, 1990–1993. doi: 10.1126/science.277.5334.1990
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Drews, O., Wildgruber, R., Zong, C., Sukop, U., Nissum, M., Weber, G.,et al. (2007). Mammalian proteasome subpopulations with distinct molecular compositions and proteolytic activities. Mol. Cell. Proteomics 6, 2021–2031. doi: 10.1074/mcp.M700187-MCP200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Duyao, M. P., Auerbach, A. B., Ryan, A., Persichetti, F., Barnes, G. T., McNeil, S. M.,et al. (1995). Inactivation of the mouse Huntington’s disease gene homolog Hdh. Science 269, 407–410. doi: 10.1126/science.7618107
Fonseca, R., Vabulas, R. M., Hartl, F. U., Bonhoeffer, T., and Nagerl, U. V. (2006). A balance of protein synthesis and proteasome-dependent degradation determines the maintenance of LTP. Neuron 52, 239–245. doi: 10.1016/j.neuron.2006.08.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Glickman, M. H., and Raveh, D. (2005). Proteasome plasticity. FEBS Lett. 579, 3214–3223. doi: 10.1016/j.febslet.2005.04.048
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Goedert, M., Spillantini, M. G., and Davies, S. W. (1998). Filamentous nerve cell inclusions in neurodegenerative diseases. Curr. Opin. Neurobiol. 8, 619–632. doi: 10.1016/S0959-4388(98)80090-1
Gong, B., Kielar, C., and Morton, A. J. (2012). Temporal separation of aggregation and ubiquitination during early inclusion formation in transgenic mice carrying the Huntington’s disease mutation. PLoS ONE 7:e41450. doi: 10.1371/journal.pone.0041450
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Graveland, G. A., Williams, R. S., and DiFiglia, M. (1985). Evidence for degenerative and regenerative changes in neostriatal spiny neurons in Huntington’s disease. Science 227, 770–773. doi: 10.1126/science.3155875
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Groll, M., Ditzel, L., Lowe, J., Stock, D., Bochtler, M., Bartunik, H. D.,et al. (1997). Structure of 20S proteasome from yeast at 2.4 A resolution. Nature 386, 463–471. doi: 10.1038/386463a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gusella, J. F., MacDonald, M. E., Ambrose, C. M., and Duyao, M. P. (1993). Molecular genetics of Huntington’s disease. Arch. Neurol. 50, 1157–1163. doi: 10.1001/archneur.1993.00540110037003
Gutekunst, C. A., Li, S. H., Yi, H., Mulroy, J. S., Kuemmerle, S., Jones, R.,et al. (1999). Nuclear and neuropil aggregates in Huntington’s disease: relationship to neuropathology. J. Neurosci. 19, 2522–2534.
Hartmann-Petersen, R., Seeger, M., and Gordon, C. (2003). Transferring substrates to the 26S proteasome. Trends Biochem. Sci. 28, 26–31. doi: 10.1016/S0968-0004(02)00002-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
HDCRG. (1993). A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell 72, 971–983. doi: 10.1016/0092-8674(93)90585-E
Hershko, A., and Ciechanover, A. (1998). The ubiquitin system. Annu. Rev. Biochem. 67, 425–479. doi: 10.1146/annurev.biochem.67.1.425
Hipp, M. S., Patel, C. N., Bersuker, K., Riley, B. E., Kaiser, S. E., Shaler, T. A.,et al. (2012). Indirect inhibition of 26S proteasome activity in a cellular model of Huntington’s disease. J. Cell Biol. 196, 573–587. doi: 10.1083/jcb.201110093
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Holmberg, C. I., Staniszewski, K. E., Mensah, K. N., Matouschek, A., and Morimoto, R. I. (2004). Inefficient degradation of truncated polyglutamine proteins by the proteasome. EMBO J. 23, 4307–4318. doi: 10.1038/sj.emboj.7600426
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jana, N. R., Zemskov, E. A., Wang, G., and Nukina, N. (2001). Altered proteasomal function due to the expression of polyglutamine-expanded truncated N-terminal huntingtin induces apoptosis by caspase activation through mitochondrial cytochrome c release. Hum. Mol. Genet. 10, 1049–1059. doi: 10.1093/hmg/10.10.1049
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Juenemann, K., Schipper-Krom, S., Wiemhoefer, A., Kloss, A., Sanz Sanz, A., and Reits, E. A. (2013). Expanded polyglutamine-containing N-terminal huntingtin fragments are entirely degraded by mammalian proteasomes. J. Biol. Chem. 288, 27068–27084. doi: 10.1074/jbc.M113.486076
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Karpova, A., Mikhaylova, M., Thomas, U., Knopfel, T., and Behnisch, T. (2006). Involvement of protein synthesis and degradation in long-term potentiation of Schaffer collateral CA1 synapses. J. Neurosci. 26, 4949–4955. doi: 10.1523/JNEUROSCI.4573-05.2006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kawakami, T., Suzuki, T., Baek, S. H., Chung, C. H., Kawasaki, H., Hirano, H.,et al. (1999). Isolation and characterization of cytosolic and membrane-bound deubiquitinylating enzymes from bovine brain. J. Biochem. 126, 612–623. doi: 10.1093/oxfordjournals.jbchem.a022493
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kishino, T., Lalande, M., and Wagstaff, J. (1997). UBE3A/E6-AP mutations cause Angelman syndrome. Nat. Genet. 15, 70–73. doi: 10.1038/ng0197-70
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kitada, T., Asakawa, S., Hattori, N., Matsumine, H., Yamamura, Y., Minoshima, S.,et al. (1998). Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392, 605–608. doi:10.1038/33416
Klionsky, D. J., Abeliovich, H., Agostinis, P., Agrawal, D. K., Aliev, G., Askew, D. S.,et al. (2008). Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 4, 151–175. doi: 10.4161/auto.5338
Klionsky, D. J., Cregg, J. M., Dunn, W. A. Jr., Emr, S. D., Sakai, Y., Sandoval, I. V.,et al. (2003). A unified nomenclature for yeast autophagy-related genes. Dev. Cell 5, 539–545. doi: 10.1016/S1534-5807(03)00296-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Konstantinova, I. M., Tsimokha, A. S., and Mittenberg, A. G. (2008). Role of proteasomes in cellular regulation. Int. Rev. Cell Mol. Biol. 267, 59–124. doi: 10.1016/S1937-6448(08)00602-3
Kopito, R. R. (2000). Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 10, 524–530. doi: 10.1016/S0962-8924(00)01852-3
Krug, M., Lossner, B., and Ott, T. (1984). Anisomycin blocks the late phase of long-term potentiation in the dentate gyrus of freely moving rats. Brain Res. Bull. 13, 39–42. doi: 10.1016/0361-9230(84)90005-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kuhlbrodt, K., Mouysset, J., and Hoppe, T. (2005). Orchestra for assembly and fate of polyubiquitin chains. Essays Biochem. 41, 1–14. doi: 10.1042/EB0410001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lam, Y. A., Pickart, C. M., Alban, A., Landon, M., Jamieson, C., Ramage, R.,et al. (2000). Inhibition of the ubiquitin-proteasome system in Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A. 97, 9902–9906. doi: 10.1073/pnas.170173897
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, X., Wang, C. E., Huang, S., Xu, X., Li, X. J., Li, H.,et al. (2010). Inhibiting the ubiquitin-proteasome system leads to preferential accumulation of toxic N-terminal mutant huntingtin fragments. Hum. Mol. Genet. 19, 2445–2455. doi: 10.1093/hmg/ddq127
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Locke, P. A., MacDonald, M. E., Srinidhi, J., Gilliam, T. C., Tanzi, R. E., Conneally, P. M.,et al. (1993). A genetic linkage map of the chromosome 4 short arm. Somat. Cell Mol. Genet. 19, 95–101. doi: 10.1007/BF01233958
Lopez-Salon, M., Alonso, M., Vianna, M. R., Viola, H., Mello e Souza, T., Izquierdo, I.,et al. (2001). The ubiquitin-proteasome cascade is required for mammalian long-term memory formation. Eur. J. Neurosci. 14, 1820–1826. doi: 10.1046/j.0953-816x.2001.01806.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Maheshwari, M., Samanta, A., Godavarthi, S. K., Mukherjee, R., and Jana, N. R. (2012). Dysfunction of the ubiquitin ligase Ube3a may be associated with synaptic pathophysiology in a mouse model of Huntington disease. J. Biol. Chem. 287, 29949–29957. doi: 10.1074/jbc.M112.371724
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Martin-Aparicio, E., Yamamoto, A., Hernandez, F., Hen, R., Avila, J., and Lucas, J. J. (2001). Proteasomal-dependent aggregate reversal and absence of cell death in a conditional mouse model of Huntington’s disease. J. Neurosci. 21, 8772–8781.
Matsuura, T., Sutcliffe, J. S., Fang, P., Galjaard, R. J., Jiang, Y. H., Benton, C. S.,et al. (1997). De novo truncating mutations in E6-AP ubiquitin-protein ligase gene (UBE3A) in Angelman syndrome. Nat. Genet. 15, 74–77. doi: 10.1038/ng0197-74
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Maynard, C. J., Bottcher, C., Ortega, Z., Smith, R., Florea, B. I., Diaz-Hernandez, M.,et al. (2009). Accumulation of ubiquitin conjugates in a polyglutamine disease model occurs without global ubiquitin/proteasome system impairment. Proc. Natl. Acad. Sci. U.S.A. 106, 13986–13991. doi: 10.1073/pnas.0906463106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
McNaught, K. S., Perl, D. P., Brownell, A. L., and Olanow, C. W. (2004). Systemic exposure to proteasome inhibitors causes a progressive model of Parkinson’s disease. Ann. Neurol. 56, 149–162. doi: 10.1002/ana.20186
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Menalled, L. B., Sison, J. D., Dragatsis, I., Zeitlin, S., and Chesselet, M. F. (2003). Time course of early motor and neuropathological anomalies in a knock-in mouse model of Huntington’s disease with 140 CAG repeats. J. Comp. Neurol. 465, 11–26. doi: 10.1002/cne.10776
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Michalik, A., and Van Broeckhoven, C. (2004). Proteasome degrades soluble expanded polyglutamine completely and efficiently. Neurobiol. Dis. 16, 202–211. doi: 10.1016/j.nbd.2003.12.020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mitra, S., Tsvetkov, A. S., and Finkbeiner, S. (2009). Single neuron ubiquitin-proteasome dynamics accompanying inclusion body formation in huntington disease. J. Biol. Chem. 284, 4398–4403. doi: 10.1074/jbc.M806269200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nasir, J., Floresco, S. B., O’Kusky, J. R., Diewert, V. M., Richman, J. M., Zeisler, J.,et al. (1995). Targeted disruption of the Huntington’s disease gene results in embryonic lethality and behavioral and morphological changes in heterozygotes. Cell 81, 811–823. doi: 10.1016/0092-8674(95)90542-1
Nawrocki, S. T., Carew, J. S., Pino, M. S., Highshaw, R. A., Dunner, K. Jr., Huang, P.,et al. (2005). Bortezomib sensitizes pancreatic cancer cells to endoplasmic reticulum stress-mediated apoptosis. Cancer Res. 65, 11658–11666. doi: 10.1158/0008-5472.CAN-05-2370
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Neefjes, J., and Dantuma, N. P. (2004). Fluorescent probes for proteolysis: tools for drug discovery. Nat. Rev. Drug Discov. 3, 58–69. doi: 10.1038/nrd1282
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ordway, J. M., Tallaksen-Greene, S., Gutekunst, C. A., Bernstein, E. M., Cearley, J. A., Wiener, H. W.,et al. (1997). Ectopically expressed CAG repeats cause intranuclear inclusions and a progressive late onset neurological phenotype in the mouse. Cell 91, 753–763. doi: 10.1016/S0092-8674(00)80464-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Orr, A. L., Li, S., Wang, C. E., Li, H., Wang, J., Rong, J.,et al. (2008). N-terminal mutant huntingtin associates with mitochondria and impairs mitochondrial trafficking. J. Neurosci. 28, 2783–2792. doi: 10.1523/JNEUROSCI.0106-08.2008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ortega, Z., Diaz-Hernandez, M., and Lucas, J. J. (2007). Is the ubiquitin-proteasome system impaired in Huntington’s disease? Cell. Mol. Life Sci. 64, 2245–2257. doi: 10.1007/s00018-007-7222-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ortega, Z., Diaz-Hernandez, M., Maynard, C. J., Hernandez, F., Dantuma, N. P., and Lucas, J. J. (2010). Acute polyglutamine expression in inducible mouse model unravels ubiquitin/proteasome system impairment and permanent recovery attributable to aggregate formation. J. Neurosci. 30, 3675–3688. doi: 10.1523/JNEUROSCI.5673-09.2010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Papandreou, C. N., Logothetis, C. J. (2004). Bortezomib as a potential treatment for prostate cancer. Cancer Res. 64, 5036–5043. doi: 10.1158/0008-5472.CAN-03-2707
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pickart, C. M. (2001). Mechanisms underlying ubiquitination. Annu. Rev. Biochem. 70, 503–533. doi: 10.1146/annurev.biochem.70.1.503
Poirier, M. A., Li, H., Macosko, J., Cai, S., Amzel, M., and Ross, C. A. (2002). Huntingtin spheroids and protofibrils as precursors in polyglutamine fibrilization. J. Biol. Chem. 277, 41032–41037. doi: 10.1074/jbc.M205809200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Raspe, M., Gillis, J., Krol, H., Krom, S., Bosch, K., van Veen, H.,et al. (2009). Mimicking proteasomal release of polyglutamine peptides initiates aggregation and toxicity. J. Cell Sci. 122, 3262–3271. doi: 10.1242/jcs.045567
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Richardson, P. G., Mitsiades, C., Hideshima, T., and Anderson, K. C. (2006). Bortezomib: proteasome inhibition as an effective anticancer therapy. Annu Rev. Med. 57, 33–47. doi: 10.1146/annurev.med.57.042905.122625
Ross, C. A. (2002). Polyglutamine pathogenesis: emergence of unifying mechanisms for Huntington’s disease and related disorders. Neuron 35, 819–822. doi: 10.1016/S0896-6273(02)00872-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rubinsztein, D. C. (2002). Lessons from animal models of Huntington’s disease. Trends Genet. 18, 202–209. doi: 10.1016/S0168-9525(01)02625-7
Rubinsztein, D. C. (2006). The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 443, 780–786. doi: 10.1038/nature05291
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rubinsztein, D. C. (2007). Autophagy induction rescues toxicity mediated by proteasome inhibition. Neuron 54, 854–856. doi: 10.1016/j.neuron.2007.06.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sakata, E., Yamaguchi, Y., Kurimoto, E., Kikuchi, J., Yokoyama, S., Yamada, S.,et al. (2003). Parkin binds the Rpn10 subunit of 26S proteasomes through its ubiquitin-like domain. EMBO Rep. 4, 301–306. doi: 10.1038/sj.embor.embor764
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sarkar, S., and Rubinsztein, D. C. (2008). Huntington’s disease: degradation of mutant huntingtin by autophagy. FEBS J. 275, 4263–4270. doi: 10.1111/j.1742-4658.2008.06562.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Saudou, F., Finkbeiner, S., Devys, D., and Greenberg, M. E. (1998). Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell 95, 55–66. doi: 10.1016/S0092-8674(00)81782-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Scherzinger, E., Lurz, R., Turmaine, M., Mangiarini, L., Hollenbach, B., Hasenbank, R.,et al. (1997). Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell 90, 549–558. doi: 10.1016/S0092-8674(00)80514-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schipper-Krom, S., Juenemann, K., Jansen, A. H., Wiemhoefer, A., van den Nieuwendijk, R., Smith, D. L.,et al. (2014). Dynamic recruitment of active proteasomes into polyglutamine initiated inclusion bodies. FEBS Lett. 588, 151–159. doi: 10.1016/j.febslet.2013.11.023
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schmitt, H. P. (2006). Protein ubiquitination, degradation and the proteasome in neuro-degenerative disorders: no clear evidence for a significant pathogenetic role of proteasome failure in Alzheimer’s disease and related disorders. Med. Hypotheses 67, 311–317. doi: 10.1016/j.mehy.2006.02.023
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Seo, H., Sonntag, K. C., and Isacson, O. (2004). Generalized brain and skin proteasome inhibition in Huntington’s disease. Ann. Neurol. 56, 319–328. doi: 10.1002/ana.20207
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sherman, M. Y., and Goldberg, A. L. (2001). Cellular defenses against unfolded proteins: a cell biologist thinks about neurodegenerative diseases. Neuron 29, 15–32. doi: 10.1016/S0896-6273(01)00177-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Snell, R. G., MacMillan, J. C., Cheadle, J. P., Fenton, I., Lazarou, L. P., Davies, P.,et al. (1993). Relationship between trinucleotide repeat expansion and phenotypic variation in Huntington’s disease. Nat. Genet. 4, 393–397. doi: 10.1038/ng0893-393
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Stack, J. H., Whitney, M., Rodems, S. M., and Pollok, B. A. (2000). A ubiquitin-based tagging system for controlled modulation of protein stability. Nat. Biotechnol. 18, 1298–1302. doi: 10.1038/82422
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Suzuki, K., and Ohsumi, Y. (2007). Molecular machinery of autophagosome formation in yeast, Saccharomyces cerevisiae. FEBS Lett. 581, 2156–2161. doi: 10.1016/j.febslet.2007.01.096
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tai, H. C., Besche, H., Goldberg, A. L., and Schuman, E. M. (2010). Characterization of the brain 26S proteasome and its interacting proteins. Front. Mol. Neurosci. 3:12. doi: 10.3389/fnmol.2010.00012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Thompson, L. M., Aiken, C. T., Kaltenbach, L. S., Agrawal, N., Illes, K., Khoshnan, A.,et al. (2009). IKK phosphorylates Huntingtin and targets it for degradation by the proteasome and lysosome. J. Cell Biol. 187, 1083–1099. doi: 10.1083/jcb.200909067
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Thrower, J. S., Hoffman, L., Rechsteiner, M., and Pickart, C. M. (2000). Recognition of the polyubiquitin proteolytic signal. EMBO J. 19, 94–102. doi: 10.1093/emboj/19.1.94
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Valera, A. G., Díaz-Hernández, M., Hernández, F., Ortega, Z., and Lucas, J. J. (2005). The ubiquitin-proteasome system in Huntington’s disease. Neuroscientist 11, 583–594. doi: 10.1177/1073858405280639
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
van Leeuwen, F. W., de Kleijn, D. P., van den Hurk, H. H., Neubauer, A., Sonnemans, M. A., Sluijs, J. A.,et al. (1998). Frameshift mutants of beta amyloid precursor protein and ubiquitin-B in Alzheimer’s and Down patients. Science 279, 242–247. doi: 10.1126/science.279.5348.242
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
van Leeuwen, F. W., Hol, E. M., and Fischer, D. F. (2006). Frameshift proteins in Alzheimer’s disease and in other conformational disorders: time for the ubiquitin-proteasome system. J. Alzheimers Dis. 9(3 Suppl), 319–325.
Venkatraman, P., Wetzel, R., Tanaka, M., Nukina, N., and Goldberg, A. L. (2004). Eukaryotic proteasomes cannot digest polyglutamine sequences and release them during degradation of polyglutamine-containing proteins. Mol. Cell 14, 95–104. doi: 10.1016/S1097-2765(04)00151-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Voges, D., Zwickl, P., and Baumeister, W. (1999). The 26S proteasome: a molecular machine designed for controlled proteolysis. Annu. Rev. Biochem. 68, 1015–1068. doi: 10.1146/annurev.biochem.68.1.1015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vonsattel, J. P., and DiFiglia, M. (1998). Huntington disease. J. Neuropathol. Exp. Neurol. 57, 369–384. doi: 10.1097/00005072-199805000-00001
Waelter, S., Boeddrich, A., Lurz, R., Scherzinger, E., Lueder, G., Lehrach, H.,et al. (2001). Accumulation of mutant huntingtin fragments in aggresome-like inclusion bodies as a result of insufficient protein degradation. Mol. Biol. Cell 12, 1393–1407. doi: 10.1091/mbc.12.5.1393
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, J., Wang, C. E., Orr, A., Tydlacka, S., Li, S. H., and Li, X. J. (2008). Impaired ubiquitin-proteasome system activity in the synapses of Huntington’s disease mice. J. Cell Biol. 180, 1177–1189. doi: 10.1083/jcb.200709080
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wexler, N. S., Young, A. B., Tanzi, R. E., Travers, H., Starosta-Rubinstein, S., Penney, J. B.,et al. (1987). Homozygotes for Huntington’s disease. Nature 326, 194–197. doi: 10.1038/326194a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yamamoto, A., Lucas, J. J., and Hen, R. (2000). Reversal of neuropathology and motor dysfunction in a conditional model of Huntington’s disease. Cell 101, 57–66. doi: 10.1016/S0092-8674(00)80623-6
Yang, W., Dunlap, J. R., Andrews, R. B., and Wetzel, R. (2002). Aggregated polyglutamine peptides delivered to nuclei are toxic to mammalian cells. Hum. Mol. Genet. 11, 2905–2917. doi: 10.1093/hmg/11.23.2905
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zeitlin, S., Liu, J. P., Chapman, D. L., Papaioannou, V. E., and Efstratiadis, A. (1995). Increased apoptosis and early embryonic lethality in mice nullizygous for the Huntington’s disease gene homologue. Nat. Genet. 11, 155–163. doi: 10.1038/ng1095-155
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: ubiquitin–proteasome system, Huntington’s disease, inclusion body, degron-fluorescent proteins, animal models
Citation: Ortega Z and Lucas JJ (2014) Ubiquitin–proteasome system involvement in Huntington’s disease. Front. Mol. Neurosci. 7:77. doi: 10.3389/fnmol.2014.00077
Received: 30 April 2014; Accepted: 10 September 2014;
Published online: 29 September 2014.
Edited by:
Fred Van Leeuwen, Maastricht University, NetherlandsReviewed by:
Stefano Sensi, University of California, Irvine, USAEric Reits, University of Amsterdam, Netherlands
Copyright © 2014 Ortega and Lucas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zaira Ortega, Laboratory 209, Department of Molecular Biology, Centro de Biología Molecular “Severo Ochoa,” Consejo Superior de Investigaciones Científicas (CSIC), Universidad Autónoma de Madrid (UAM), Centro Investigación Biomédica en Red Enfermedades Neurodegenerativa (CIBERNED), 28049 Madrid, Spain e-mail:WmFpcmFvcnRlZ2FsbG9yZW50ZUBnbWFpbC5jb20=