 Rajvinder Karda1
Rajvinder Karda1 Suzanne M. K. Buckley1Citra N. Mattar2Joanne Ng1
Suzanne M. K. Buckley1Citra N. Mattar2Joanne Ng1 Giulia Massaro3Michael P. Hughes3Manju A. Kurian4Julien Baruteau1Paul Gissen5Jerry K. Y. Chan2Chiara Bacchelli6
Giulia Massaro3Michael P. Hughes3Manju A. Kurian4Julien Baruteau1Paul Gissen5Jerry K. Y. Chan2Chiara Bacchelli6 Simon N. Waddington1,7
Simon N. Waddington1,7 Ahad A. Rahim3*
Ahad A. Rahim3*
- 1Gene Transfer Technology Group, UCL EGA Institute for Women’s Health, University College London, London, UK
- 2Experimental Fetal Medicine Group, Department of Obstetrics and Gynaecology, National University of Singapore, Singapore, Singapore
- 3Department of Pharmacology, UCL School of Pharmacy, University College London, London, UK
- 4Neurosciences Unit, UCL Institute of Child Health, University College London, London, UK
- 5Clinical and Molecular Genetics Unit, UCL Institute of Child Health, University College London, London, UK
- 6Centre for Translational Research – GOSgene, UCL Institute of Child Health, University College London, London, UK
- 7School of Pathology, University of the Witwatersrand, Johannesburg, South Africa
Neurodegenerative monogenic diseases often affect tissues and organs beyond the nervous system. An effective treatment would require a systemic approach. The intravenous administration of novel therapies is ideal but is hampered by the inability of such drugs to cross the blood–brain barrier (BBB) and precludes efficacy in the central nervous system. A number of these early lethal intractable diseases also present devastating irreversible pathology at birth or soon after. Therefore, any therapy would ideally be administered during the perinatal period to prevent, stop, or ameliorate disease progression. The concept of perinatal gene therapy has moved a step further toward being a feasible approach to treating such disorders. This has primarily been driven by the recent discoveries that particular serotypes of adeno-associated virus (AAV) gene delivery vectors have the ability to cross the BBB following intravenous administration. Furthermore, safety has been demonstrated after perinatal administration mice and non-human primates. This review focuses on the progress made in using AAV to achieve systemic transduction and what this means for developing perinatal gene therapy for early lethal neurodegenerative diseases.
Introduction
The development of intravenously administered therapies for neurodegenerative diseases is often impeded by the blood–brain barrier (BBB). The BBB isolates and protects the central nervous system (CNS) by restricting large or hydrophobic molecules such as viruses and large proteins from crossing into the CNS (Rubin and Staddon, 1999). Remarkable efficacy has been reported in clinical trials for neurodegenerative disorders using ex vivo hematopoietic stem cell gene therapy (Cartier et al., 2009; Biffi et al., 2013). While this approach is hugely promising for those diseases where secretion of a soluble protein from bone marrow derived microglia and therapeutic recapture from neighboring neural cells is possible, other conditions may not be amenable to this approach. Therefore, highly efficient and direct delivery of genes, ideally targeted to specific neural cells using cell type specific promoters, would be required. The adeno-associated virus (AAV) vector system has been subject to recent, intense, scrutiny due to: (1) high efficiency of neural cell transduction (Gray et al., 2011); (2) excellent safety profile in humans (Nakai et al., 2005); (3) low toxicity (Daya and Berns, 2008); and (4) lack of integration into the host genome, therefore imposing a lower risk of insertional oncogenesis associated with integrating vectors such as lentivirus (Themis et al., 2005; Howe et al., 2008). The post-mitotic status of neurons makes them a good target for non-integrating vectors. Thus, AAV is now a commonly used vector for clinical trials addressing neurological disease. This must be balanced with specific disadvantages associated with AAV vectors: (1) a limited packaging capacity of around 4.7 kb (Dong et al., 2010) and (2) the possibility of pre-existing humoral response to capsid protein because of prior wild-type AAV infections (Boutin et al., 2010).
Numerous AAV serotypes have been isolated from human and non-human primates (NHPs). Their tropisms following intravenous administration have been studied in mice and NHPs, these include AAV6, 6.2, 7, 8, 9, rh.8, rh.10, rh.39, and rh.43 (Towne et al., 2008; Zincarelli et al., 2008; Gray et al., 2011; Yang et al., 2014). Interestingly, AAV9 has been shown to cross the BBB in adult and neonatal animals following intravenous infusion of the virus (Zincarelli et al., 2008; Duque et al., 2009; Foust et al., 2009; Fu et al., 2011). Other AAV serotypes have since demonstrated the ability to cross the BBB in neonatal mice following intravenous administration with AAVrh.10 and AAVrh.8 both showing promise (Zhang et al., 2011).
An example of a lethal neurodegenerative monogenic disease with visceral involvement is neuronopathic Gaucher disease (GD). Patients exhibit hepatomegaly and splenomegaly in addition to the ultimately lethal brain pathology (Gupta et al., 2011). Enzyme replacement therapy (ERT) has been highly successful in treating the visceral pathology associated to GD (Cox, 2013) and other lysosomal storage disorders (Ortolano et al., 2014). However, the presence of the BBB has severely restricted recombinant enzyme from crossing into the CNS and rendering ERT ineffective in preventing death from neurodegeneration (Pastores et al., 1993; Wynn et al., 2009). Such systemic diseases will ideally require a systemic treatment. AAV serotypes that can be intravenously administered and cross the BBB raises hope that this approach may have clinical potential for treating such disorders. Furthermore, the prospect of administering gene therapy during the perinatal period is attractive for a number of reasons. Here we discuss the potential of AAV administered during the perinatal period to treat systemic disorders, with an emphasis upon neurodegenerative conditions, using a selection of studies from a rapidly growing field to highlight progress.
Perinatal Gene Delivery
Gene delivery to the developing fetus or neonate has been discussed extensively over the years (David and Peebles, 2008; Rahim et al., 2010; Buckley et al., 2011; Coutelle and Waddington, 2012; Mattar et al., 2012; Roybal et al., 2012). While this is yet to be realized in the clinic, the advantages that such an approach offers are appealing (1) A higher vector to cell ratio enhancing the efficiency of gene delivery, (2) A naïve immune system allows for the induction of ‘tolerance’ to vector-mediated expression of a protein that the body may not recognize as self, (3) Reduced chances of an immune response to a viral vector since the fetus is unlikely to have had previous viral infections, (4) Potentially enhanced access of vectors to progenitor and stem cell niches, and (5) The ability to prevent pathology from manifesting – arguably most important in the context of acute neurodegenerative disorders where susceptible post-mitotic neurons must be protected at the earliest possible moment to prevent irrevocable loss. The arguments in favor of in utero gene therapy must be balanced with those against such as (1) Is there an established method for early diagnosis for the disease in question? (2) If the fetus is diagnosed with an intractable genetic disorder then would the mother opt to terminate the pregnancy? We refer the reader to a separate elegant review by Coutelle and Ashcroft (2012) that discusses the ethical, legal and societal implications. (3) Fetal developmental and cell physiological abnormalities caused by overexpression of the therapeutic transgene. Realistically, gene therapy for the neonate will be developed first. Symptoms may be more obvious; further investigation by newborn screening programs or scans may facilitate and confirm diagnosis followed by administration of gene therapy – but in many cases of acute and aggressive neurodegenerative disorders that present during gestation, neonatal intervention could already be too late.
AAV in Fetal and Neonatal Rodents
Very few studies have examined systemic gene delivery via intravenous administration to the fetal rodent. This is probably due to the technical expertise required to perform this procedure. Rahim et al. (2011) described extensive reporter gene expression in mice following in utero intravenous injection of AAV9. This vector efficiently crossed the BBB and mediated global gene expression throughout the nervous system. More studies have examined gene delivery following intravenous administration of AAV into neonatal rodents. Foust et al. (2009) showed that intravenous injections of AAV9 carrying the GFP reporter gene into neonatal mice resulted in extensive transduction of neuronal cells within the spinal cord and the brain and within the heart and skeletal muscles. Similarly, Duque et al. (2009) showed efficient transduction of motor neurons within the spinal cords of adult mice and cats after intravenous injections of AAV9. Foust et al. (2009) further elaborated on their previous AAV9 study by rescuing an early lethal mouse model of spinal muscular atrophy (SMA). Remarkably, a single intravenous injection at birth of AAV9 carrying the SMN gene was sufficient to correct the neuromuscular electrophysiology and motor functions (Foust et al., 2010). In contrast, injection of therapeutic AAV9 vector at later time points (5 and 10 days after birth) produced partial correction or little effect, respectively. This highlights the critical importance of identifying the therapeutic window of opportunity that may be narrow and early in development. Towne et al. (2008) demonstrated motor neuronal transduction within the spinal cord and the brain stem through intravenous administration of AAV6 to adult mice. AAV8, 9, and rh.10 also achieved impressive nervous system transduction (Yang et al., 2014). This also supports the observations of impressive neuronal and glial transduction in a separate study using AAV9 in adult mice (Gray et al., 2011). Furthermore, a systematic comparison of various AAV serotypes in the neonatal mouse revealed that AAV9 was highly efficient in crossing the BBB, although AAVrh.10 also holds great promise and must be investigated further (Zhang et al., 2011). To date, given the larger body of evidence and data on systemic delivery, the review will focus on AAV9.
The timing of systemic administration of AAV9 is also important in the context of gene expression in particular neural cells. Foust et al. (2009) demonstrated that AAV9 carrying the GFP gene driven by the chicken beta actin hybrid promoter effectively transduces neurons when intravenously administered to neonatal mice. However, expression becomes preferentially astrocyte specific when administered to adult mice. Further analysis revealed that this ‘switch’ between expression in cell types occurs within the first 10 days of life (Foust et al., 2010). Rahim et al. (2011) reported that intravenous injection of AAV9 carrying the GFP gene driven by the CMV promoter to E15 fetal mice led to preferential neuronal expression and minimal astrocyte expression. When the same vector was administered to P1 neonatal mice, preferential astrocyte expression was observed. In some cases, expression throughout the nervous system may not be required and preferably targeted to a specific neuronal population. This refinement in targeted gene expression will require more work in identifying suitable promoters or microRNA sequences.
AAV in Fetal and Neonatal Non-Human Primates
Non-human primates are a valuable pre-clinical model given the significant difference between fetal and neonatal development and cell type expression in rodents and humans. Mattar et al. (2013) evaluated the ability of AAV9 to transduce the CNS through a single intravenous injection into a late gestation fetal macaque. The administration of the virus was conducted using an established ultrasound guided clinical protocol that is routinely used worldwide. Examination of administered animals at 6 and 14 days of age revealed significant neuron transduction in the CNS and the PNS, with a lower efficiency seen in astrocytes. Furthermore, significant transduction in the skeletal muscle, heart and liver was observed. This work supports the findings of Rahim et al. (2011) in fetal mice intravenously administered with AAV9. However, the potential differences between mouse and NHPs were highlighted by Bevan et al. (2011) who intravenously administered AAV9 carrying the GFP gene driven by the chicken beta actin hybrid promoter to NHPs at P1, P30, and P90. Monkeys that received vector at P1 showed mostly astrocyte and microglial targeting in the brain that is in contrast to mice that received the same vector at P1 and demonstrated predominantly neuronal transduction (Foust et al., 2010). Bevan et al. (2011) attribute this discrepancy to differences in the timing of gliogenesis between the two species. The same study also reported sustained expression in the musculature and liver of injected monkeys. Table 1 summarizes the key features described above. Intravenous injection of the vector used by Bevan et al. (2011) has also been administered to older 3–4 year old rhesus macaques (Bevan et al., 2011; Gray et al., 2011). 4-weeks after injection, analysis showed similar gene expression profiles – again predominantly glial in the brain with extensive transduction of the musculature and liver. Interestingly, monkeys that were serotype positive for AAV9 and had pre-existing circulating AAV9 neutralizing antibodies exhibited significantly reduced levels of transduction (Gray et al., 2011). This issue will be further discussed later but represents a strong argument for administering during the perinatal period where the subject is less likely to have been exposed to AAV infection and more likely to be serotype negative.
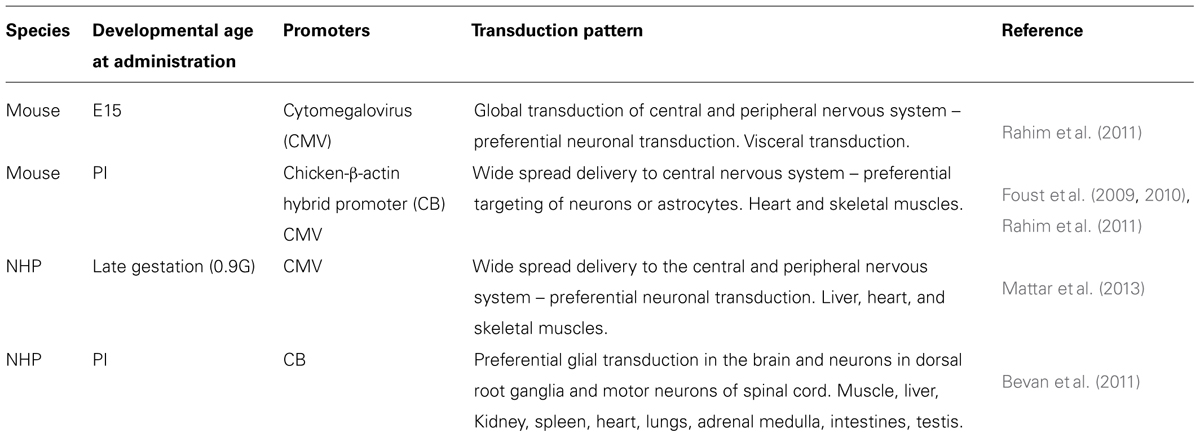
TABLE 1. Summary of intravenously administered AAV9-mediated gene delivery studies in perinatal mice and non-human primates.
Clinical Translation of Systemic AAV-Mediated Gene Therapy
Systemic delivery of AAV vectors in patients has already been demonstrated in a recent successful clinical trial using AAV8 to express the FIX gene to treat patients with the blood clotting disorder hemophilia B (Nathwani et al., 2011). AAV8 carrying the FIX gene driven by the LP1 liver specific promoter was administered, intravenously, to six patients with severe hemophilia B all over the age of 18 years old. Four of the six patients did not require prophylactic recombinant FIX. The other two patients that received a higher dose of vector either developed an AAV8-capsid specific T cell response or a small increase in liver enzyme levels – both symptoms were treated using glucocorticoids (Nathwani et al., 2011). While this study is in older patients, it provides an important proof-of-principle; high doses of AAV can be intravenously administered and tolerated without any life-threatening adverse effects. Furthermore, a Phase I clinical trial for the neuromuscular condition SMA is currently recruiting patients less than 9 months old (ClinicalTrials.gov Identifier: NCT02122952). Patients will receive an intravenous infusion of AAV9 carrying the therapeutic SMN gene driven by the CMV enhancer/chicken-beta-actin promoter. Interestingly, an inclusion criteria is onset of disease at birth to 6 months of age and patients are eligible to be included if they are 9 months or younger at time of infusion. The young age of the potential participants exemplifies the volition to intervene as early as possible and vindicates a strategy to treat diseases that may manifest at birth. This trial will also address potential logistical and immunological barriers that have been proposed to impede such an approach. It has been suggested that this approach for using AAV9 to treat neurodegenerative diseases would require large vector preparations to achieve the necessary therapeutic transduction levels in the CNS (Foust et al., 2009; Gray et al., 2011). Producing the amount of vector required for a human trial could be challenging with the current AAV production technology, although the baculoviral system (Cecchini et al., 2011) has been successfully used by uniQure to manufacture the first licensed gene therapy product in Europe using an AAV vector, Glybera (Yla-Herttuala, 2012).
Administering such a high titre may result in an adverse immune response, particularly in those patients that have low levels of pre-existing AAV antibodies, reducing the efficiency of therapy and increasing the chance of an adverse immune response (Boutin et al., 2010; Samaranch et al., 2012). Treating younger and, therefore, smaller patients may well overcome any issue of production and also pre-existing antibodies against AAV9 (although an exclusion criteria in the SMA trial is the presence of anti-AAV9 antibodies at a titre of >1:50 as determined by ELISA binding immunoassay). However, high levels of maternal alloantibodies against AAV could trigger an immunization response through maternal microchimerism that is thought to play a role in the development of the fetal immune system (Merianos et al., 2009; Jeanty et al., 2014).
A criticism of perinatal gene therapy is diagnosing a fetus or neonate early enough for therapeutic intervention. This is dependent upon diagnostic technologies and resources in any given country, e.g., the availability of prenatal screening and diagnosis programs. Screening programs even for rare neurological conditions have been established in communities that have a high prevalence such as Tay–Sachs and GD in the Ashkenazi Jewish populations (Eng et al., 1997). Advances in ultrasound technology have led to the ability to identify fetal abnormalities in utero. An example of this is Niemann–Pick disease type C where although the severity of disease and age of onset can be highly variable, fetal presentation in the form of splenomegaly, hepatomegaly, and ascites are detectable by ultrasound scan (Spiegel et al., 2009). The continuing development of next generation sequencing (NGS) over the past decade may ultimately hold the key to early or pre-term diagnosis. The ability to obtain fetal genetic information during pregnancy from the mother’s blood has been a long-term goal. Originally, studies focused on identification of such cells in the maternal circulation. Once cell-free fetal DNA (cffDNA) was identified (Lo et al., 1997), the focus directed toward ways of analyzing such DNA material for prenatal testing. Recently, non-invasive prenatal diagnosis (NIPD) utilizing cffDNA as early as 7 weeks’ gestation has been introduced into clinical practice as reviewed by Daley et al. (2014). This approach has been successfully used for fetal gender identification (Devaney et al., 2011), a highly accurate screening test for aneuploidies like trisomy 21 (Fan et al., 2008), other major trisomies (Norton et al., 2012), sex chromosomes abnormalities (Bianchi et al., 2012; Futch et al., 2013), and the diagnosis of some single gene disorders including cystic fibrosis, thalassaemia, and achondroplasia (Lench et al., 2013).
A more reliable, sensitive and cost effective method for NIPD in monogenic disorders is provided by NGS platforms. Now available in routine diagnostic laboratories, they can accurately quantify certain sequences in the maternal blood. An example was for the identification of Down Syndrome by whole genome sequencing by quantifying cffDNA sequences mapping to chromosome 21 (Fan et al., 2008). A more recent application of NGS in NIPD for b-thalassemia has been described (Lam et al., 2012).
Next generation sequencing presents many advantages over standard PCR-based genetic testing. The high throughput of NGS allows samples from different patients to be multiplexed in a single run. This combined with the ability to design assays with multiple mutations in multiple genes to be included in a single test are allowing a reduced cost per sample as well as a quick turn-around-time for reporting results. With the cost of NGS constantly decreasing and the imminent implementation of whole exome sequencing (which allows the simultaneous sequence of all coding exons) in diagnostic laboratories, NGS and NIPD will be increasingly used for the diagnosis of a range of monogenic disorders (Lench et al., 2013). Although the cost is still high and the time of data analysis long, whole genome sequencing could be implemented to profile the entire fetal genomes (Lo et al., 2010).
Concluding Remarks
Perinatal gene therapy has distinct advantages (Summarized in Table 2) and is essential in addressing those diseases that manifest during the neonatal stage or even in utero. This is particularly important in early lethal neurodegenerative disorders where vulnerable post-mitotic neurons must be rescued or risk irrecoverable loss. The presence of additional systemic pathology is a further complication. However, the potential of intravenously administered AAV to provide systemic gene therapy is highly attractive in treating such pleiotropic conditions. Of course, there are significant practical issues that need to be overcome for perinatal gene therapy to be realized in the clinic. However, we are moving in the right direction with advances in prenatal diagnostic technologies, clinical trials demonstrating that intravenously administered AAV vectors are safe and plans to take this further in young children with SMA. The ascendancy of gene therapy through a number of recent successful gene therapy trials (Gaspar et al., 2004; Bainbridge et al., 2008; Cartier et al., 2009; Nathwani et al., 2011; Aiuti et al., 2013; Biffi et al., 2013) and the approval of the first gene therapy product to be sold in Europe (Yla-Herttuala, 2012) has meant that the field in general is gathering momentum and is an opportune moment to bring systemic perinatal gene therapy into the limelight.

TABLE 2. Summary of using AAV in perinatal gene therapy and its clinical implication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work is supported by the UK Medical Research Council (G1000709), Gauchers Association, Niemann-Pick Disease Group, Niemann-Pick Research Foundation, and the US Neurodegeneration with Brain Iron Accumulation Disorders Association.
References
Aiuti, A., Biasco, L., Scaramuzza, S., Ferrua, F., Cicalese, M. P., Baricordi, C.,et al. (2013). Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott-Aldrich syndrome. Science 341:1233151. doi: 10.1126/science.1233151
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bainbridge, J. W., Smith, A. J., Barker, S. S., Robbie, S., Henderson, R., Balaggan, K.,et al. (2008). Effect of gene therapy on visual function in Leber’s congenital amaurosis. N. Engl. J. Med. 358, 2231–2239. doi: 10.1056/NEJMoa0802268
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bevan, A. K., Duque, S., Foust, K. D., Morales, P. R., Braun, L., Schmelzer, L.,et al. (2011). Systemic gene delivery in large species for targeting spinal cord, brain, and peripheral tissues for pediatric disorders. Mol. Ther. 19, 1971–1980. doi: 10.1038/mt.2011.157
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bianchi, D. W., Platt, L. D., Goldberg, J. D., Abuhamad, A. Z., Sehnert, A. J., Rava, R. P.,et al. (2012). Genome-wide fetal aneuploidy detection by maternal plasma DNA sequencing. Obstet. Gynecol. 119, 890–901. doi: 10.1097/AOG.0b013e31824fb482
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Biffi, A., Montini, E., Lorioli, L., Cesani, M., Fumagalli, F., Plati, T.,et al. (2013). Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science 341:1233158. doi: 10.1126/science.1233158
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Boutin, S., Monteilhet, V., Veron, P., Leborgne, C., Benveniste, O., Montus, M. F.,et al. (2010). Prevalence of serum IgG and neutralizing factors against adeno-associated virus (AAV) types 1, 2, 5, 6, 8, and 9 in the healthy population: implications for gene therapy using AAV vectors. Hum. Gene Ther. 21, 704–712. doi: 10.1089/hum.2009.182
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Buckley, S. M., Rahim, A. A., Chan, J. K., David, A. L., Peebles, D. M., Coutelle, C.,et al. (2011). Recent advances in fetal gene therapy. Ther. Deliv. 2, 461–469. doi: 10.4155/tde.11.17
Cartier, N., Hacein-Bey-Abina, S., Bartholomae, C. C., Veres, G., Schmidt, M., Kutschera, I.,et al. (2009). Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science 326, 818–823. doi: 10.1126/science.1171242
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cecchini, S., Virag, T., and Kotin, R. M. (2011). Reproducible high yields of recombinant adeno-associated virus produced using invertebrate cells in 0.02- to 200-liter cultures. Hum. Gene Ther. 22, 1021–1030. doi: 10.1089/hum.2010.250
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Coutelle, C., and Ashcroft, R. (2012). Risks, benefits and ethical, legal, and societal considerations for translation of prenatal gene therapy to human application. Methods Mol. Biol. 891, 371–387. doi: 10.1007/978-1-61779-873-3_17
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Coutelle, C., and Waddington, S. N. (2012). The concept of prenatal gene therapy. Methods Mol. Biol. 891, 1–7. doi: 10.1007/978-1-61779-873-3_1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cox, T. M. (2013). Competing for the treasure in exceptions. Am. J. Hematol. 88, 163–165. doi: 10.1002/ajh.23399
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Daley, R., Hill, M., and Chitty, L. S. (2014). Non-invasive prenatal diagnosis: progress and potential. Arch. Dis. Child Fetal Neonatal Ed. 99, 426–430. doi: 10.1136/archdischild-2013-304828
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
David, A. L., and Peebles, D. (2008). Gene therapy for the fetus: is there a future? Best Pract. Res. Clin. Obstet. Gynaecol. 22, 203–218. doi: 10.1016/j.bpobgyn.2007.08.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Daya, S., and Berns, K. I. (2008). Gene therapy using adeno-associated virus vectors. Clin. Microbiol. Rev. 21, 583–593. doi: 10.1128/CMR.00008-08
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Devaney, S. A., Palomaki, G. E., Scott, J. A., and Bianchi, D. W. (2011). Noninvasive fetal sex determination using cell-free fetal DNA: a systematic review and meta-analysis. JAMA 306, 627–636. doi: 10.1001/jama.2011.1114
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dong, B., Nakai, H., and Xiao, W. (2010). Characterization of genome integrity for oversized recombinant AAV vector. Mol. Ther. 18, 87–92. doi: 10.1038/mt.2009.258
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Duque, S., Joussemet, B., Riviere, C., Marais, T., Dubreil, L., Douar, A. M.,et al. (2009). Intravenous administration of self-complementary AAV9 enables transgene delivery to adult motor neurons. Mol. Ther. 17, 1187–1196. doi: 10.1038/mt.2009.71
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Eng, C. M., Schechter, C., Robinowitz, J., Fulop, G., Burgert, T., Levy, B.,et al. (1997). Prenatal genetic carrier testing using triple disease screening. JAMA 278, 1268–1272. doi: 10.1001/jama.1997.03550150072038
Fan, H. C., Blumenfeld, Y. J., Chitkara, U., Hudgins, L., and Quake, S. R. (2008). Noninvasive diagnosis of fetal aneuploidy by shotgun sequencing DNA from maternal blood. Proc. Natl. Acad. Sci. U.S.A. 105, 16266–16271. doi: 10.1073/pnas.0808319105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Foust, K. D., Nurre, E., Montgomery, C. L., Hernandez, A., Chan, C. M., and Kaspar, B. K. (2009). Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat. Biotechnol. 27, 59–65. doi: 10.1038/nbt.1515
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Foust, K. D., Wang, X., Mcgovern, V. L., Braun, L., Bevan, A. K., Haidet, A. M.,et al. (2010). Rescue of the spinal muscular atrophy phenotype in a mouse model by early postnatal delivery of SMN. Nat. Biotechnol. 28, 271–274. doi: 10.1038/nbt.1610
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fu, H., Dirosario, J., Killedar, S., Zaraspe, K., and Mccarty, D. M. (2011). Correction of neurological disease of mucopolysaccharidosis IIIB in adult mice by rAAV9 trans-blood-brain barrier gene delivery. Mol. Ther. 19, 1025–1033. doi: 10.1038/mt.2011.34
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Futch, T., Spinosa, J., Bhatt, S., De Feo, E., Rava, R. P., and Sehnert, A. J. (2013). Initial clinical laboratory experience in noninvasive prenatal testing for fetal aneuploidy from maternal plasma DNA samples. Prenat. Diagn. 33, 569–574. doi: 10.1002/pd.4123
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gaspar, H. B., Parsley, K. L., Howe, S., King, D., Gilmour, K. C., Sinclair, J.,et al. (2004). Gene therapy of X-linked severe combined immunodeficiency by use of a pseudotyped gammaretroviral vector. Lancet 364, 2181–2187. doi: 10.1016/S0140-6736(04)17590-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gray, S. J., Matagne, V., Bachaboina, L., Yadav, S., Ojeda, S. R., and Samulski, R. J. (2011). Preclinical differences of intravascular AAV9 delivery to neurons and glia: a comparative study of adult mice and nonhuman primates. Mol. Ther. 19, 1058–1069. doi: 10.1038/mt.2011.72
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gupta, N., Oppenheim, I. M., Kauvar, E. F., Tayebi, N., and Sidransky, E. (2011). Type 2 Gaucher disease: phenotypic variation and genotypic heterogeneity. Blood Cells Mol. Dis. 46, 75–84. doi: 10.1016/j.bcmd.2010.08.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Howe, S. J., Mansour, M. R., Schwarzwaelder, K., Bartholomae, C., Hubank, M., Kempski, H.,et al. (2008). Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J. Clin. Invest. 118, 3143–3150. doi: 10.1172/JCI35798
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jeanty, C., Derderian, S. C., and Mackenzie, T. C. (2014). Maternal-fetal cellular trafficking: clinical implications and consequences. Curr. Opin. Pediatr. 26, 377–382. doi: 10.1097/MOP.0000000000000087
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lam, K. W., Jiang, P., Liao, G. J., Chan, K. C., Leung, T. Y., Chiu, R. W.,et al. (2012). Noninvasive prenatal diagnosis of monogenic diseases by targeted massively parallel sequencing of maternal plasma: application to beta-thalassemia. Clin. Chem. 58, 1467–1475. doi: 10.1373/clinchem.2012.189589
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lench, N., Barrett, A., Fielding, S., Mckay, F., Hill, M., Jenkins, L.,et al. (2013). The clinical implementation of non-invasive prenatal diagnosis for single-gene disorders: challenges and progress made. Prenat. Diagn. 33, 555–562. doi: 10.1002/pd.4124
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lo, Y. M., Chan, K. C., Sun, H., Chen, E. Z., Jiang, P., Lun, F. M.,et al. (2010). Maternal plasma DNA sequencing reveals the genome-wide genetic and mutational profile of the fetus. Sci. Transl. Med. 2:61ra91. doi: 10.1126/scitranslmed.3001720
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lo, Y. M., Corbetta, N., Chamberlain, P. F., Rai, V., Sargent, I. L., Redman, C. W.,et al. (1997). Presence of fetal DNA in maternal plasma and serum. Lancet 350, 485–487. doi: 10.1016/S0140-6736(97)02174-0
Mattar, C. N., Waddington, S. N., Biswas, A., Davidoff, A. M., Choolani, M., Chan, J. K.,et al. (2012). The case for intrauterine gene therapy. Best Pract. Res. Clin. Obstet. Gynaecol. 26, 697–709. doi: 10.1016/j.bpobgyn.2012.06.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mattar, C. N., Waddington, S. N., Biswas, A., Johana, N., Ng, X. W., Fisk, A. S.,et al. (2013). Systemic delivery of scAAV9 in fetal macaques facilitates neuronal transduction of the central and peripheral nervous systems. Gene Ther. 20, 69–83. doi: 10.1038/gt.2011.216
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Merianos, D. J., Tiblad, E., Santore, M. T., Todorow, C. A., Laje, P., Endo, M.,et al. (2009). Maternal alloantibodies induce a postnatal immune response that limits engraftment following in utero hematopoietic cell transplantation in mice. J. Clin. Invest. 119, 2590–2600. doi: 10.1172/JCI38979
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nakai, H., Wu, X., Fuess, S., Storm, T. A., Munroe, D., Montini, E.,et al. (2005). Large-scale molecular characterization of adeno-associated virus vector integration in mouse liver. J. Virol. 79, 3606–3614. doi: 10.1128/JVI.79.6.3606-3614.2005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nathwani, A. C., Tuddenham, E. G., Rangarajan, S., Rosales, C., McIntosh, J., Linch, D. C.,et al. (2011). Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N. Engl. J. Med. 365, 2357–2365. doi: 10.1056/NEJMoa1108046
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Norton, M. E., Brar, H., Weiss, J., Karimi, A., Laurent, L. C., Caughey, A. B.,et al. (2012). Non-Invasive Chromosomal Evaluation (NICE) Study: results of a multicenter prospective cohort study for detection of fetal trisomy 21 and trisomy 18. Am. J. Obstet. Gynecol. 207, 137.e131–137.e138. doi: 10.1016/j.ajog.2012.05.021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ortolano, S., Vieitez, I., Navarro, C., and Spuch, C. (2014). Treatment of lysosomal storage diseases: recent patents and future strategies. Recent Pat. Endocr. Metab. Immune Drug Discov. 8, 9–25. doi: 10.2174/1872214808666140115111350
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pastores, G. M., Sibille, A. R., and Grabowski, G. A. (1993). Enzyme therapy in Gaucher disease type 1: dosage efficacy and adverse effects in 33 patients treated for 6 to 24 months. Blood 82, 408–416.
Rahim, A. A., Wong, A. M., Buckley, S. M., Chan, J. K., David, A. L., Cooper, J. D.,et al. (2010). In utero gene transfer to the mouse nervous system. Biochem. Soc. Trans. 38, 1489–1493. doi: 10.1042/BST0381489
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rahim, A. A., Wong, A. M., Hoefer, K., Buckley, S. M., Mattar, C. N., Cheng, S. H.,et al. (2011). Intravenous administration of AAV2/9 to the fetal and neonatal mouse leads to differential targeting of CNS cell types and extensive transduction of the nervous system. FASEB J. 25, 3505–3518. doi: 10.1096/fj.11-182311
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Roybal, J. L., Endo, M., Buckley, S. M., Herbert, B. R., Waddington, S. N., and Flake, A. W. (2012). Animal models for prenatal gene therapy: rodent models for prenatal gene therapy. Methods Mol. Biol. 891, 201–218. doi: 10.1007/978-1-61779-873-3_10
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rubin, L. L., and Staddon, J. M. (1999). The cell biology of the blood-brain barrier. Annu. Rev. Neurosci. 22, 11–28. doi: 10.1146/annurev.neuro.22.1.11
Samaranch, L., Salegio, E. A., San Sebastian, W., Kells, A. P., Foust, K. D., Bringas, J. R.,et al. (2012). Adeno-associated virus serotype 9 transduction in the central nervous system of nonhuman primates. Hum. Gene Ther. 23, 382–389. doi: 10.1089/hum.2011.200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Spiegel, R., Raas-Rothschild, A., Reish, O., Regev, M., Meiner, V., Bargal, R.,et al. (2009). The clinical spectrum of fetal Niemann-Pick type C. Am. J. Med. Genet. A 149A, 446–450. doi: 10.1002/ajmg.a.32642
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Themis, M., Waddington, S. N., Schmidt, M., Von Kalle, C., Wang, Y., Al-Allaf, F.,et al. (2005). Oncogenesis following delivery of a nonprimate lentiviral gene therapy vector to fetal and neonatal mice. Mol. Ther. 12, 763–771. doi: 10.1016/j.ymthe.2005.07.358
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Towne, C., Raoul, C., Schneider, B. L., and Aebischer, P. (2008). Systemic AAV6 delivery mediating RNA interference against SOD1: neuromuscular transduction does not alter disease progression in fALS mice. Mol. Ther. 16, 1018–1025. doi: 10.1038/mt.2008.73
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wynn, R. F., Wraith, J. E., Mercer, J., O’Meara, A., Tylee, K., Thornley, M.,et al. (2009). Improved metabolic correction in patients with lysosomal storage disease treated with hematopoietic stem cell transplant compared with enzyme replacement therapy. J. Pediatr. 154, 609–611. doi: 10.1016/j.jpeds.2008.11.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yang, B., Li, S., Wang, H., Guo, Y., Gessler, D. J., Cao, C.,et al. (2014). Global CNS transduction of adult mice by intravenously delivered rAAVrh.8 and rAAVrh.10 and nonhuman primates by rAAVrh.10. Mol. Ther. 22, 1299–1309. doi: 10.1038/mt.2014.68
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yla-Herttuala, S. (2012). Endgame: glybera finally recommended for approval as the first gene therapy drug in the European union. Mol. Ther. 20, 1831–1832. doi: 10.1038/mt.2012.194
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, H., Yang, B., Mu, X., Ahmed, S. S., Su, Q., He, R.,et al. (2011). Several rAAV vectors efficiently cross the blood-brain barrier and transduce neurons and astrocytes in the neonatal mouse central nervous system. Mol. Ther. 19, 1440–1448. doi: 10.1038/mt.2011.98
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zincarelli, C., Soltys, S., Rengo, G., and Rabinowitz, J. E. (2008). Analysis of AAV serotypes 1-9 mediated gene expression and tropism in mice after systemic injection. Mol. Ther. 16, 1073–1080. doi: 10.1038/mt.2008.76
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: adeno-associated virus, systemic delivery, perinatal, gene therapy, neurodegenerative disease, mouse models, non-human primates, metabolic disease
Citation: Karda R, Buckley SMK, Mattar CN, Ng J, Massaro G, Hughes MP, Kurian MA, Baruteau J, Gissen P, Chan JKY, Bacchelli C, Waddington SN and Rahim AA (2014) Perinatal systemic gene delivery using adeno-associated viral vectors. Front. Mol. Neurosci. 7:89. doi: 10.3389/fnmol.2014.00089
Received: 23 June 2014; Accepted: 29 October 2014;
Published online: 14 November 2014.
Edited by:
Deniz Dalkara, Université Pierre et Marie Curie, FranceReviewed by:
David J. Margolis, Rutgers University, USAMarie Reine Haddad, National Institutes of Health, USA (in collaboration with Stephen Gerard Kaler)
Stephen Gerard Kaler, National Institutes of Health, USA
Copyright © 2014 Karda, Buckley, Mattar, Ng, Massaro, Hughes, Kurian, Baruteau, Gissen, Chan, Bacchelli, Waddington and Rahim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ahad A. Rahim, Department of Pharmacology, UCL School of Pharmacy, University College London, 29-39 Brunswick Square, London WC1N 1AX, UK e-mail:YS5yYWhpbUB1Y2wuYWMudWs=