 Veronica Merelo1
Veronica Merelo1 Dante Durand1Adam R. Lescallette2,3Kent E. Vrana2
Dante Durand1Adam R. Lescallette2,3Kent E. Vrana2 L. Elliot Hong4
L. Elliot Hong4 Mohammad Ali Faghihi5
Mohammad Ali Faghihi5 Alfredo Bellon2,3*
Alfredo Bellon2,3*- 1Department of Psychiatry and Behavioral Sciences, University of Miami, Miller School of Medicine, Miami, FL, USA
- 2Penn State Hershey Medical Center, Department of Pharmacology, Hershey, PA, USA
- 3Penn State Hershey Medical Center, Department of Psychiatry, Hershey, PA, USA
- 4Maryland Psychiatric Research Center, Department of Psychiatry, University of Maryland School of Medicine, Baltimore, MD, USA
- 5Center for Therapeutic Innovation, Department of Psychiatry and Behavioral Sciences University of Miami, Miller School of Medicine, Miami, FL, USA
Several lines of evidence indicate that schizophrenia has a strong genetic component. But the exact nature and functional role of this genetic component in the pathophysiology of this mental illness remains a mystery. Long non-coding RNAs (lncRNAs) are a recently discovered family of molecules that regulate gene transcription through a variety of means. Consequently, lncRNAs could help us bring together apparent unrelated findings in schizophrenia; namely, genomic deficiencies on one side and neuroimaging, as well as postmortem results on the other. In fact, the most consistent finding in schizophrenia is decreased brain size together with enlarged ventricles. This anomaly appears to originate from shorter and less ramified dendrites and axons. But a decrease in neuronal arborizations cannot explain the complex pathophysiology of this psychotic disorder; however, dynamic changes in neuronal structure present throughout life could. It is well recognized that the structure of developing neurons is extremely plastic. This structural plasticity was thought to stop with brain development. However, breakthrough discoveries have shown that neuronal structure retains some degree of plasticity throughout life. What the neuroscientific field is still trying to understand is how these dynamic changes are regulated and lncRNAs represent promising candidates to fill this knowledge gap. Here, we present evidence that associates specific lncRNAs with schizophrenia. We then discuss the potential role of lncRNAs in neurostructural dynamics. Finally, we explain how dynamic neurostructural modifications present throughout life could, in theory, reconcile apparent unrelated findings in schizophrenia.
Introduction
Several lines of evidence indicate that schizophrenia has a strong genetic component, including; heritability estimates of 80–85% (Cardno et al., 1999), high rates of monozygotic concordance (Cardno et al., 1999; Cardno and Gottesman, 2000) and its association with several discrete genomic regions (Sullivan et al., 2012; Ripke et al., 2013). But the role of this genetics in the pathophysiology of schizophrenia remains a mystery. Recently discovered regulatory mechanisms of gene transcription could help us bring together apparent unrelated findings in schizophrenia namely genomic deficiencies on one side and neuroimaging findings, as well as postmortem results on the other. Long non-coding RNAs (lncRNAs) are an important part of these new regulatory mechanisms as they affect gene transcription through a variety of means such as acting as Natural antisense transcripts (NATs; Magistri et al., 2012) or via dosage compensation of the XX or XY chromosomes (Moran et al., 2012). LncRNAs also regulate gene expression by a mechanism known as genomic imprinting and through its interaction with paraspeckles (Mattick et al., 2009; Wang and Chang, 2011; Moran et al., 2012). Potential lncRNA implications throughout cell biology are enormous and neurons are not excluded. Numerous lncRNAs are expressed in the central nervous system (CNS) where they participate in essential processes for normal brain development (Fenoglio et al., 2013). This is of particular importance as schizophrenia is generally considered a neurodevelopmental disorder (Marenco and Weinberger, 2000; Rapoport et al., 2005). It is therefore not surprising that there is emerging evidence of lncRNAs association with this mental illness given these molecules play crucial roles in brain development. What remains to be determined is confirmatory evidence of these findings and more importantly, which cellular process they regulate. To explore such a complex question, we have to rely on commonalities in findings from patients with schizophrenia.
Among the most consistent findings in schizophrenia are ventricular enlargement and decreased brain volume (Van Horn and McManus, 1992; Wright et al., 2000; Woods et al., 2005). How these findings come about remains under intense research, however; neuropathological studies have shown that neurons from patients with schizophrenia present shorter and less ramified dendrites and axons (Van Horn and McManus, 1992; Wright et al., 2000; Woods et al., 2005; Bellon et al., 2011). Even neuronal-like cells differentiated from skin fibroblasts exhibit shorter extensions (Brennand et al., 2011), while glial cells also from psychotic individuals exhibit abnormal arborizations (Rajkowska et al., 2002). A discreet shrinkage of dendrites and axons present throughout the brain could lead to smaller brains and enlarged ventricles, but it does not explain the complex pathophysiology of schizophrenia. However, dynamic changes in neuronal structure that are present throughout life could provide an explanation.
It is well recognized that the structure of developing neurons is extremely plastic undergoing constant extension and retraction of neuronal arborizations (Tavosanis, 2012; Urbanska et al., 2012). This structural plasticity was once thought to stop with brain development. Nonetheless, breakthrough discoveries in neurosciences have shown that the old dogma of a static neuronal structure in adulthood is no longer applicable. Instead, it is now known that neuronal structure remains plastic throughout life (Holtmaat and Svoboda, 2009; Nacher et al., 2013). Adult neurons change their shape even at baseline when they are not being stimulated by environmental cues (Mizrahi and Katz, 2003; De Paola et al., 2006; Galimberti et al., 2006; Lee et al., 2006; Nishiyama et al., 2007; Chow et al., 2009; Marik et al., 2010; Chen et al., 2011). In the presence of stimuli such as learning, these structural modifications become even more dramatic (Holtmaat and Svoboda, 2009; Caroni et al., 2012). Stress also causes drastic changes in neuronal shape including dendritic atrophy in the prefrontal cortex and hippocampus, whereas in the amygdala it promotes dendritic growth (Radley and Morrison, 2005). The adult brain’s ability to retain a degree of structural plasticity is evident. What the neuroscientific field is still trying to understand is how these dynamic changes are regulated and lncRNAs are promising candidates. Therefore, the goals of this review are to: (1) briefly present how lncRNAs exert their multiple regulatory mechanisms; (2) determine if there is evidence associating lncRNAs to schizophrenia; and (3) explore if genes regulated by lncRNAs linked to schizophrenia impact neuronal structure. We then discuss the potential role of lncRNAs in neurostructural dynamics and how dynamic structural modifications occurring throughout life could, in theory, reconcile apparent unrelated findings in schizophrenia.
Methods
A systematic review of the literature was conducted via MEDLINE and Google Scholar using search terms such as lncRNAs, neuronal structure, dendrite, axons, dendritic spines, axonal boutons, structural plasticity, neuronal differentiation and schizophrenia. The search was limited to studies published in English. Regarding the association of lncRNAs with schizophrenia, only studies involving patients with schizophrenia were included; any articles associating lncRNAs with schizophrenia through animal models were excluded.
Long Non-Coding RNAs Mechanisms of Action
Seventy-to-seventy five percent of the eukaryotic genome encodes transcripts that do not have an obvious potential to encode proteins and are popularly called non-coding RNA (ncRNA; Consortium, 2012; Pennisi, 2012). For many years, the role of ncRNAs remained a mystery. Only recently, significant progress has been made in understanding how ncRNAs participate in a number of essential cellular processes. NcRNAs are broadly classified into long and small ncRNA depending upon the length of the transcript. Small ncRNAs (less than 200 bp in length) include micro RNAs (miRNAs), piwi-interacting RNAs (piRNAs), endogenous small-interfering RNAs (siRNAs) and small nucleolar RNAs (snoRNAs), and have all been extensively reviewed in the literature (Carthew and Sontheimer, 2009; Ghildiyal and Zamore, 2009; Malone and Hannon, 2009; Magistri et al., 2012). On the other hand, lncRNAs are defined as transcripts longer than 200 bp. Similar to messenger RNAs (mRNA) they are RNA polymerase II transcripts that are capped, spliced and polyadenylated, yet do not function as templates for protein synthesis (Mercer et al., 2009; Ponting et al., 2009). LncRNA genes maintain features common to protein-coding genes such as promoter regions, intron-exon boundaries and alternative splicing, but lack extended translational open reading frames (ORFs). Moreover, they are mainly nuclear localized, less polyadenylated and more tissue-specific than protein-coding genes (Barry, 2014). The number of total lncRNAs is in the range of 20,000 transcripts. The crucial task is to determine whether all these transcripts are functional or not. So far, over 200 lncRNAs have been studied and many of these show evidence of functionality, at least in vitro (Moran et al., 2012).
Intense research during the past few years has revealed that the main function of lncRNAs is to regulate gene expression. This regulation is executed through a variety of complex mechanisms that include: acting as NATs, as well as via dosage compensation, genomic imprinting and through nuclear organization (Mattick et al., 2009; Mercer et al., 2009; Wang and Chang, 2011). In the following sections, we will briefly review each of these regulatory processes to highlight the importance of lncRNAs to maintain cellular homeostasis.
NATs are a well characterized type of lncRNA which arise from the opposite strand of protein-coding or non-protein-coding genes (Faghihi and Wahlestedt, 2009). They can regulate mRNA expression at the level of transcription via competition for regulatory factors, or through physically hindering the progress of transcription, either topologically or by being transcribed themselves (Munroe and Zhu, 2006). It is also possible for NATs to edit or activate cellular siRNA-related pathways that lead to degradation of homologous transcripts ultimately eliciting gene silencing (Kumar and Carmichael, 1998; Munroe and Zhu, 2006). Consequently, NATs can potentially regulate any step in RNA processing including translation, polyadenylation, splicing, transport or even degradation (Munroe and Zhu, 2006). NATs can also bind to epigenetic enzymes and act as scaffold to form active or repressive chromatin modifying complexes (Magistri et al., 2012). DISC-2, Evf2 and BDNF-AS are examples of NATs that have been associated with several neurodegenerative, neurodevelopment and psychiatric disorders like schizophrenia, bipolar disorder and autism (Millar et al., 2004; Williams et al., 2009; Velmeshev et al., 2013). NATs have also been documented for the Fragile X Mental Retardation gene (FMR1). Although Fragile X Syndrome (FXS) is considered a monogenetic disorder, there is evidence that supports an alternative model in which other ncRNAs contribute to FXS pathogenesis and the phenotypic variations observed among patients (Ladd et al., 2007; Khalil et al., 2008).
Dosage compensation is a hypothetical genetic regulatory mechanism that operates to equalize the phenotypic expression of characteristics determined by genes on the X chromosome so that they are equally expressed in humans carrying the XY chromosomes or the XX combination. For instance, Xist—perhaps the best studied lncRNA—is responsible for the initiation of X chromosome inactivation (Xi) in female somatic cells (Augui et al., 2011; Pontier and Gribnau, 2011). Xist silences hundreds of genes on the inactive X chromosome (Zhao et al., 2008) and this process is essential for normal brain development (Qureshi and Mehler, 2010).
LncRNAs also regulate gene expression through a mechanism known as genomic imprinting. The expression of imprinted genes depends on their parental origin and the level of differential expression of the two alleles of an imprinted gene can vary from one imprinted gene to another (Moran et al., 2012). Some lncRNAs can exert their function by recruiting epigenetic factors such as PCR2 and G9a, in order to control the imprinted expression of neighboring coding genes (Nagano et al., 2008; Zhao et al., 2008). Other lncRNAs such as Kcna1ot1, regulate genomic imprinting by interacting with repressive chromatin modifying complexes that ultimately suppress paternally inherited genes (Kanduri et al., 2006; Pandey et al., 2008). Of note, a number of authors have proposed that mental and neurological disorders such as schizophrenia and autism are often influenced by deregulation of the imprinting processes (Francks et al., 2003; Schulze et al., 2004; Schanen, 2006).
A recently discovered mechanism by which lncRNAs regulate gene expression is through their interaction with paraspeckles. Paraspeckles are membraneless subnuclear bodies (Mao et al., 2011b) that participate in nuclear organization, a phenomenon linked to genome maintenance and to the control of gene expression (Bond and Fox, 2009). These subnuclear bodies seem to regulate gene expression post-transcriptionally by retaining hyper-edited mRNAs in the nucleus (Prasanth et al., 2005; Bond and Fox, 2009; Chen and Carmichael, 2009; Clemson et al., 2009). It is noteworthy that formation and maintenance of paraspeckles requires the lncRNA named NEAT1 that localizes exclusively to paraspeckles (Clemson et al., 2009; Sunwoo et al., 2009; Mao et al., 2011a; Shevtsov and Dundr, 2011). NEAT1 has been found to be upregulated in neurological diseases such as Huntington’s disease (Johnson, 2012). Furthermore, analyses of 633 human spinal motor neurons in six cases of amyotrophic lateral sclerosis (ALS) showed that NEAT1 was upregulated during the early stage of ALS pathogenesis (Nishimoto et al., 2013).
It is clear from the preceding short overview that lncRNAs regulate gene expression through a number of complex genetic mechanisms. Deregulation of these complex genetic processes can lead to mental illnesses such as schizophrenia and therefore lncRNAs are promising candidates to explain the strong, but still uncharacterized, genetic component of this psychotic disorder. In the following section, we present data connecting lncRNAs with schizophrenia.
Long Non-Coding RNAs (lncRNAs) and Schizophrenia
It is widely accepted that schizophrenia is a neurodevelopmental disorder (Marenco and Weinberger, 2000; Rapoport et al., 2005). It is also well recognized that schizophrenia is a severe multifactorial condition with a complex genetic component; therefore it is not surprising that previously unrecognized non-coding RNAs could participate in its pathophysiology. The lncRNAs currently associated with this mental illness are DISC-2, Gomafu, EVF-2 and BDNF-AS (Table 1). Their degree of association with schizophrenia varies from direct in cases where the actual lncRNA is abnormal in patients suffering this psychotic disorder to indirect in cases where lncRNAs regulate proteins or mRNAs known to be deficient in schizophrenia. In the following paragraphs we will discuss each of those lncRNAs with direct, as well as indirect, association with schizophrenia. We will specify the strength of the evidence linking each specific lncRNA with this psychotic disorder.

Table 1. Long non-coding RNAs associated with schizophrenia.
DISC-2
In the early 2000s, a balanced translocation (1:11) (q42.1:q14.3) that segregates with schizophrenia and related disorders was found in a large Scottish family with a high loading for major mental illnesses (Millar et al., 2000; Blackwood et al., 2001). Long-term follow-up of this family has provided information about 87 family members, of whom 37 carry the translocation. Twenty nine individuals carrying the translocation, for whom psychiatric assessment was possible, have psychiatric illnesses, seven have a diagnosis of schizophrenia, one has bipolar disorder and 10 subjects present recurrent major depressive disorder. Thus, 18 of 29 translocation carriers are diagnosed with major psychiatric disorders whereas none of the 38 non-translocation carriers have any mental illness (Blackwood et al., 2001). This translocation affects the Disrupted-in-Schizophrenia (DISC) locus and results in a defective DISC-1 protein that lacks a carboxy-terminal (Kamiya et al., 2005; Sachs et al., 2005). Other association studies have strengthened the relationship between the DISC locus and schizophrenia. For instance, several studies support the evidence that variations at the DISC locus predispose individuals to schizophrenia, schizoaffective disorder and bipolar illness. In Hodgkinson et al. (2004) conducted a case-control study of a North American white population, they analyzed the DISC locus, describing multiple haplotypes contained within four haplotype blocks extending between exon 1 and exon 9 that are associated with schizophrenia, schizoaffective disorder, and bipolar disorder. Later, in Sachs et al. (2005) sequenced portions of DISC-1 in 28 unrelated probands with schizophrenia and six unrelated probands with schizoaffective disorder. They detected a 4 bp deletion at the extreme 3’ end of exon 12 in a proband with schizophrenia. The mutation was also present in a sibling with schizophrenia, a sibling with schizoaffective disorder, and the unaffected father. These findings support the possibility that mutations in the DISC-1 gene can increase the risk for schizophrenia and related disorders.
Expression of DISC-1 is widespread, developmentally-regulated, and high in regions of the brain that are implicated in schizophrenia, most notably the hippocampus (Millar et al., 2004). In fact, one study, using western blot analysis in postmortem brain samples, detected one major band of 70–75 kDa, which was increased in the hippocampus of patients with schizophrenia when compared to controls (Lipska et al., 2006). As human brain tissue is not easily accessible; lymphoblastoid cell lines generated directly from patients with this psychotic disorder have been used to obtain data on gene expression. In cell lines derived from members of the t(1:11) family, DISC-1 gene expression was decreased at both the mRNA and protein level in translocation carriers compared to normal karyotype controls (Millar et al., 2005).
The role of DISC-1 for proper brain function and development is not entirely understood, but is thought to be involved in neuronal migration, synaptogenesis, and glutamatergic transmission (Miyoshi et al., 2003; Morris et al., 2003; Ishizuka et al., 2006; Porteous et al., 2006). It was also recently discovered that DISC-1 can regulate dopamine signaling by interacting with activating transcription factor 4 (ATF4) and then repressing phosphodiesterase 4D (PDE4D; Soda et al., 2013). In addition, several studies have directly related DISC-1 to neurostructural dynamics, as will be presented later in this manuscript (Miyoshi et al., 2003; Ozeki et al., 2003; Kamiya et al., 2005).
While significant information is available about DISC-1, less is known about DISC-2. DISC-2 is located on the opposite strand to DISC-1 and is transcribed in a distal to proximal orientation, thus representing a NAT that lacks any significant coding potential and partially overlaps DISC-1 (Millar et al., 2000). As a NAT, it is believed to function as a negative regulator of its protein-coding counterpart, DISC-1 (Millar et al., 2000; Taylor et al., 2003; Chubb et al., 2008). In Millar et al. (2000) cloned and sequenced the breakpoints of the 1:11(q42.1:a14.3) translocation linked to schizophrenia and identified both genes DISC-1 and DISC-2. They suggested that DISC-2 presents an attractive mechanism by which DISC-1 expression may be regulated and proposed that alteration of DISC-1 and/or DISC-2 activity, by truncation and/or by abnormal regulation of expression, is causally linked to the psychiatric illnesses present in translocation carriers. Later, a systematic review compiling information about the DISC locus and mental disorders also proposed that disruption of DISC-2 might have a role in the pathophysiology of schizophrenia through dysregulation of DISC-1 expression (Chubb et al., 2008).
Although there is no direct evidence linking the DISC-2 gene to schizophrenia there are strong data supporting an association between the DISC locus and this mental illness. It must be kept in mind that most association studies do not discriminate between DISC-2 and DISC-1 in terms of positive linkage and association (Chubb et al., 2008); furthermore, many studies report markers that are within the DISC-2 gene region (Ekelund et al., 2001, 2004; Hodgkinson et al., 2004; Cannon et al., 2005). Perhaps most importantly, as mentioned above, the DISC-2 gene is also disrupted by the t(1:11) translocation (Millar et al., 2000). Consequently, there is no direct evidence yet, linking DISC-2 with schizophrenia, but its role within the DISC locus makes this association very likely.
Gomafu
Gomafu—also known as myocardial infarction-associated transcript (MIAT) was originally identified in 2004. It was found in a specific set of neurons in the mouse retina and was described as a noncoding RNA (Blackshaw et al., 2004). In Sone et al. (2007) established Gomafu as a member of a novel family of noncoding RNAs that constitutes a cell-type-specific component of the nuclear matrix. Gomafu is located in a novel nuclear compartment, which is enriched in pre-mRNA splicing factors, where it specifically interacts with splicing factor 1 (SF1; Tsuiji et al., 2011). Indeed, Sone et al. (2007), coined the name “Gomafu” to reflect the Japanese word for the speckled pattern in which it is distributed throughout the nucleoplasm.
Gomafu is widely and abundantly expressed in the nervous system throughout development and its expression continues into adulthood (Sone et al., 2007; Tsuiji et al., 2011). In adult mice, Gomafu is found in CA1 pyramidal neurons of the hippocampus and large cortical neurons in the hindbrain (Sone et al., 2007). This family of noncoding RNA participates in retinal development (Blackshaw et al., 2004; Rapicavoli and Blackshaw, 2009; Rapicavoli et al., 2010), brain development (Mercer et al., 2010), and post-mitotic neuronal function (Sone et al., 2007; Mercer et al., 2008). Gomafu became clinically relevant, in 2006, when Albertson et al. (2006) found a transcript assumed to encode a hypothetical protein highly increased in postmortem brains of human drug abusers. Subsequent bioinformatic analyses revealed that this transcript did not encode a protein and, instead, it corresponded to Gomafu (MIAT) (Ishii et al., 2006). The same team later demonstrated that Gomafu is upregulated in the nucleus accumbens of human heroin users and suggested that dysregulation of Gomafu can influence behavior (Albertson et al., 2006; Michelhaugh et al., 2011).
Despite Gomafu’s location in a chromosomal region linked to schizophrenia (22q12.1) (Takahashi et al., 2003), it was not until 2013 that this long noncoding RNA was associated with this mental illness. An Australian team found Gomafu to be downregulated in the cortical gray matter from the superior temporal gyrus of patients with schizophrenia (Barry et al., 2014). They also demonstrated that this lncRNA regulates nuclear splicing factors that ultimately affect schizophrenia related genes such as DISC-1 and ErbB4 (Barry et al., 2014). In addition, Gomafu binds to QKI, a protein itself implicated in schizophrenia (Barry et al., 2014). The QKI gene codes for a family of alternative spliced gene products (QKI-5 kb, QKI-6 kb, QKI-7 kb and QKI-7 kb-B) that regulate myelination by Schwann cells and oligodendrocytes (McCullumsmith et al., 2007). All four QKI splicing variants are expressed in the frontal cortex of human brain and the relative mRNA expression levels of the QKI splice-variant QKI-7 kb is down-regulated in schizophrenic patients (Aberg et al., 2006). There are consequently direct and indirect data associating Gomafu with this psychotic disorder.
Evf2
Evf2 is a NAT of the DLx5/DLx6 gene (Magistri et al., 2012). Evf2 was discovered in the developing mouse forebrain and it is transcribed from the ultra-conserved Dlx5/6 region encoding the homeodomain transcription factors DLx5 and DLx6 (Feng et al., 2006). Dlx homeobox genes products play a crucial role in migration and differentiation of the subpallial precursor cells that give rise to various subtypes of gamma-aminobutiric acid (GABA)-expressing neurons of the forebrain, including local-circuit cortical interneurons (Poitras et al., 2010). Interneurons play a vital role in modulating the activity of the cerebral cortex and they rely on the enzyme glutamic acid decarboxylase 67 (GAD67) for the synthesis of GABA (Addington et al., 2005), the major inhibitory neurotransmitter in the brain. In Bond et al. (2009) demonstrated that Evf2 plays a critical role in regulating genes involved in the development of interneurons. This research team showed that loss of Evf2 results in imbalanced gene expression, leading to decreased GABAergic interneurons in early postnatal hippocampus and dentate gyrus (Bond et al., 2009). They also discovered that, in the developing ventral forebrain, Evf2 recruits Dlx and MECP2 transcription factors to key DNA regulatory elements in the DLx 5/6 intergenic region to control DLx5, DLx6 and GAD67 expression (Bond et al., 2009).
There is no direct evidence showing abnormal levels of Evf2 in schizophrenia. However, there are data demonstrating that GAD67 and DLx1 are defective in these patients and both GAD67 and DLx1 are directly regulated by Evf2 as mentioned earlier (Bond et al., 2009). Numerous independent research teams have found GAD67 expression to be decreased in various brain regions of patients with schizophrenia (Akbarian et al., 1995; Guidotti et al., 2000; Woo et al., 2004). Moreover, recently, Joshi et al. demonstrated for the first time that DLx1 and GAD67 mRNA levels are reduced in the orbitofrontal gray matter of individuals with this mental illness (Joshi et al., 2012). Thus, there is an indirect relationship between EVf2 and schizophrenia and more specifically between Evf2, the development of GABAergic interneurons, and this psychotic disorder.
BDNF-AS
BDNF antisense (BDNF-AS) has been described as an endogenous non-coding antisense RNA transcribed from the human BDNF gene locus (Pruunsild et al., 2007). High levels of BDNF-AS are present in the brain, kidney, spinal cord and testis and are generated as a result of alternative splicing (Pruunsild et al., 2007). Alternatively spliced isoform diversity is common to many eukaryotic organisms and is widely used in the nervous system (Lipscombe, 2005). It is important to note that both BDNF and BDNF-AS share a common sense-antisense overlapping region (Liu et al., 2006; Pruunsild et al., 2007). More importantly, it has been demonstrated that this NAT regulates the expression of BDNF mRNA and protein in vivo and in vitro (Modarresi et al., 2012). In fact, knockdown of BDNF-AS increases BDNF mRNA levels and considerably increases the expression of BDNF protein (Modarresi et al., 2012).
BDNF belongs to a class of secreted growth factors that are essential for supporting neuronal growth, survival, synaptic plasticity and it is also involved in learning and memory neuregulin can elicit neurite outgrowth throughk (Kang and Schuman, 1995; Figurov et al., 1996; Yamada et al., 2002). While BDNF-AS has not, to our knowledge, been studied in schizophrenia, BDNF gene and protein have been largely associated with this psychotic disorder (Bellon et al., 2011). An important reduction of BDNF mRNA was observed in the prefrontal cortex and the hippocampus of patients with chronic schizophrenia (Favalli et al., 2012). In addition, a similar reduction of BDNF mRNA was observed in the dentate gyrus and hippocampus of affected individuals (Thompson Ray et al., 2011). Hence, there is only indirect evidence of BDNF-AS association with schizophrenia, as only BDNF has been studied in this context.
Long Non-Coding RNAs, Schizophrenia and Neuronal Structure
It is well established that neurons are structurally plastic throughout development (Tavosanis, 2012; Urbanska et al., 2012). But it only recently was demonstrated that neurons retain a certain degree of structural plasticity in adulthood (Mizrahi and Katz, 2003; De Paola et al., 2006; Galimberti et al., 2006; Lee et al., 2006; Nishiyama et al., 2007; Chow et al., 2009; Holtmaat and Svoboda, 2009; Marik et al., 2010; Chen et al., 2011; Nacher et al., 2013). Moreover, this plasticity becomes even more evident during learning (Holtmaat and Svoboda, 2009; Caroni et al., 2012). The presence of a dynamic neuronal structure throughout life could help reconcile a number of apparent unrelated findings in schizophrenia as will be presented later in this manuscript. One of the factors to be established is which molecules regulate these dynamic changes in neuronal shape. LncRNAs represent promising candidates as they are prominent regulators of gene expression and they are highly active during brain development. As a first step in associating lncRNAs with variations in neuronal architecture, here we review if those genes regulated by lncRNAs linked to schizophrenia can alter neuronal structure.
DISC-1 has been consistently associated with neurostructural dynamics (Miyoshi et al., 2003; Ozeki et al., 2003; Kamiya et al., 2005). It has been shown that transfection of mutant DISC-1 into PC12 cells results in shorter neurites (Ozeki et al., 2003), while decreasing the expression of DISC-1 by RNA interference, inhibits neurite outgrowth in the same type of cells (Kamiya et al., 2005). Furthermore, in utero gene-transfer techniques in rodents have shown that DISC-1 loss induces shorter dendrites that are randomly oriented in the cerebral cortex (Ozeki et al., 2003). The mechanism of action for DISC-1 as a transformer of neuronal structure remains a work in progress. What is currently known is that DISC-1 interacts with FEZ-1 to promote cytoskeletal dynamics that lead to the extension of neuronal processes (Bloom and Horvitz, 1997; Kuroda et al., 1999). As we previously mentioned, it seems likely that DISC-2 regulates the expression of DISC-1 (Figure 1) at the DISC locus (Millar et al., 2000; Chubb et al., 2008). The fact that DISC-1 and its antisense partner DISC-2 overlap in a specific gene region and that both molecules are extensively expressed in the brain (Millar et al., 2000) during complex developmental processes and also throughout higher level cognitive tasks, suggests that there is at least some degree of interaction between DISC-1 and DISC-2 (Chubb et al., 2008).
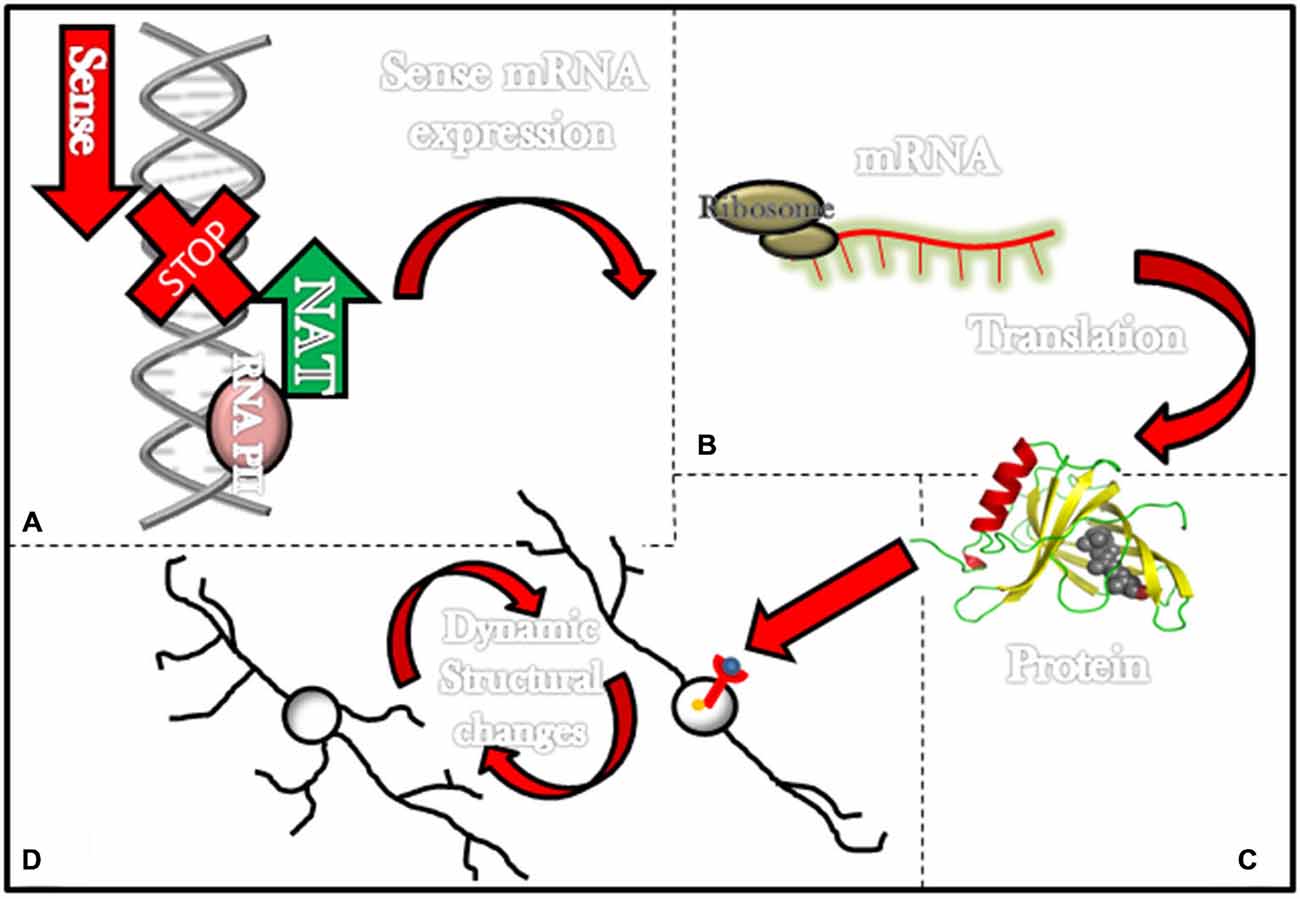
Figure 1. Long non-coding RNAs (ncRNAs) associated with schizophrenia and its effects in the neuronal structure. Most lncRNAs associated with schizophrenia are natural antisense transcripts (NATs; i.e., DISC-2, Evf2 and BDNF-AS). This figure illustrates in the upper left corner (A) a NAT (green arrow) being transcribed and consequently stopping transcription for genes that would have been transcribed in the normal transcription sense (red arrow). (B) In some instances transcription of NATs prevents the formation of mRNAs and (C) further translation of these mRNAs into proteins such as DISC-1 and BDNF. On the lower left corner (D) a diagram exemplifies how proteins such as BDNF can activate membrane receptors and then elicit dynamic structural changes in neurons.
Gomafu was recently implicated as another lncRNA involved in regulating the expression of DISC-1 and ErbB4. Overexpression of Gomafu in stem cells significantly decreases the expression of both DISC-1 and ErbB4 and their alternatively spliced variants (Barry, 2014). Conversely, knocking down Gomafu increases the expression of both DISC-1 and ErbB4 splice variants, but not their predominant, mature transcripts (Barry, 2014). Interestingly, these results almost exactly match the aberrant splicing pattern seen in post-mortem studies of patients with schizophrenia (Law et al., 2007; Nakata et al., 2009). Therefore, DISC-1 effects on neuronal structure could be regulated by either Gomafu or DISC-2.
As mentioned above, Gomafu also regulates the expression of ErbB4, a protein that has been associated with schizophrenia. Postmortem studies in patients with this mental illness showed altered levels of neuregulin-1 (NRG1) and its receptor, ErbB4, in prefrontal cortex (Chong et al., 2008). ErbB4 activation by neuregulin can elicit neurite outgrowth through two different enzymes—kinase (MAPK) and protein kinase C (PKC; Vaskovsky et al., 2000). The down-stream cascade of MAPK and PKC appears to be activation of Cdc42 which leads to fillopodium formation (Shigeta et al., 2003). Activation of Rac, also mediated by PKC, results in actin polymerization that leads to the formation of lamellipodium (Pilpel and Segal, 2004). Both fillopodium and lamellipodium are actin structures that participate in neurostructural rearrangements (Bellon, 2007). Another example of the role of ErbB4 in structural plasticity comes from a study published in 2008 that demonstrated that depletion of ErbB4 decreases the number of primary neurites in hippocampal cultures, whereas stimulation of ErbB4 using a soluble form of NRG1 results in increased dendritic arborizations via activation of the tyrosine kinase domain of ErbB4 and the phosphoinositide 3-kinase pathway (Krivosheya et al., 2008). Hence, there is clear evidence indicating that Gomafu regulates two proteins capable of transforming neuronal structure namely DISC-1 and ErbB4.
Another lncRNA, Evf2, affects neuronal shape through its association with Dlx genes and by regulating the expression of GAD67. In Plavicki et al. (2012) characterized the distal-less (dll) gene in the invertebrate nervous system; they used this single invertebrate gene as a homolog of the Dlx genes in vertebrates, proposing that the existence of a single dll gene in Drosophila has the potential to provide insight into how disruptions of Dlx function result in aberrant neuronal development in vertebrates. Altering the expression of the Dlx homolog, distal-less, disrupts normal dendrite and axon development in the olfactory system (Plavicki et al., 2012). The authors suggest that dll could serve as a pathfinding molecule in this brain area (Plavicki et al., 2012). Similar results are observed in rodents. Mice with a null allele of the Dlx5 gene do not form axons that would normally connect the olfactory epithelium with the olfactory bulb (Long et al., 2003). This lack of axonal extension appears to be due to the absence of Dlx5 chemoattractant effects (Levi et al., 2003).
The influence of GAD67 on neuronal structure is of a different nature. If the enzymatic activity of GAD67 is diminished during development, either by pharmacological or genetic means, then pruning of synapses decreases (Nakayama et al., 2012). This means that elimination of the GABA synthesizing enzyme GAD67 results in more prominent arborizations. Therefore, by controlling the expression of Dlx genes as well as the enzyme GAD67, Evf2 can transform neuronal structure.
Finally, it is widely accepted that BDNF promotes neurostructural dynamics during development and adult life (Bellon et al., 2011). For instance, mutant juvenile mice in which BDNF levels are modestly increased display higher numbers of first-order dendrites, total dendritic length, and total branch points in dentate gyrus granule cells (Tolwani et al., 2002; Bellon et al., 2011). Similarly, the introduction of BDNF to hippocampal slices elicits an increase in the number of spines and augments apical dendritic length in pyramidal cells (Alonso et al., 2004; Bellon et al., 2011). Also, BDNF triggers dendritic spine maturation during the transition between adolescence to adulthood (An et al., 2008). Most important to our current topic is a recent association between BDNF, BDNF-AS and changes in neuronal structure. In 2012, it was found that increasing endogenous BDNF by knocking down BDNF-AS transcript results in neurite outgrowth and maturation in human and mouse cell lines (Modarresi et al., 2012). This information directly associates BDNF-AS with modifications in neuronal architecture (Figure 1).
In summary, there are data suggesting lncRNAs, associated with schizophrenia, regulate proteins that can transform neuronal shape (Figure 1). For some, such as BDNF-AS, the evidence is strong while for others, such as Gomafu and Evf2, the evidence is indirect, and the data currently available on DISC-2 remain weak.
Discussion
After 50 years of intense research, it is now clear that a single gene or protein cannot explain such a complex illness as schizophrenia. We currently know that there are numerous genes, proteins, mRNAs, miRNAs and lncRNAs that have been associated with this mental disorder. The growing variety of molecules associated with schizophrenia could involve numerous cellular mechanisms including: cell migration, cell survival, neurogenesis, and many others. Consequently, one of the current challenges is to establish if there is a single cellular process that can consolidate all these molecules (and their functions) and, at the same time, explain the most consistent findings in schizophrenia.
We propose that a core deficit in schizophrenia is the establishment and maintenance of neuronal structure as it is now evident that the old dogma of a structural static brain is no longer valid. Recent in vivo evidence, using two-photon microscopy, has demonstrated that neurons remain structurally plastic throughout life (De Paola et al., 2006; Galimberti et al., 2006; Lee et al., 2006; Nishiyama et al., 2007; Chow et al., 2009; Holtmaat and Svoboda, 2009; Marik et al., 2010; Chen et al., 2011; Nacher et al., 2013) and not only during development as was originally thought. This structural plasticity involves, in addition to constant turnover of dendritic spines and axonal boutons (Holtmaat and Svoboda, 2009; Caroni et al., 2012), extension and retraction of dendritic and axonal shafts (De Paola et al., 2006; Galimberti et al., 2006; Lee et al., 2006; Nishiyama et al., 2007; Chow et al., 2009; Marik et al., 2010; Chen et al., 2011). Transformations in neuronal shape occur even when brain activity is at baseline without any particular environmental stimuli (De Paola et al., 2006; Galimberti et al., 2006; Lee et al., 2006; Nishiyama et al., 2007; Chow et al., 2009; Marik et al., 2010; Chen et al., 2011) and becomes more prominent during complex cognitive tasks such as learning (Holtmaat and Svoboda, 2009; Caroni et al., 2012). The relevance of these dynamic changes to the physiology of the brain is also apparent under stressful circumstances. Chronic stress elicits dendritic atrophy in the prefrontal cortex and hippocampus, whereas it promotes dendritic growth in the amygdala (Radley and Morrison, 2005). If the stressful event is removed, atrophic dendrites re-establish their original structure (Radley et al., 2005), which clearly illustrates how structurally plastic neurons can be. Structural plasticity is not limited to neurons. Astrocytes, after only minutes of brain stimulation, can extend processes in order to regulate synaptic transmission (Perez-Alvarez et al., 2014) and to promote excitatory synapse stability (Bernardinelli et al., 2014). Therefore, it is now obvious that continuous structural changes are part of normal brain functioning during development, as well as in adult life.
If schizophrenia is framed within the context of an ever changing neuronal shape, a number of apparent unrelated and inconsistent findings could be theoretically reconciled. For instance, it is widely accepted that schizophrenia is a developmental disorder in which exposure to famine or infection during pregnancy increases the risk for acquiring this mental disorder (MacDonald and Schulz, 2009). It has also been recognized that migrants to northern latitudes are at higher risk for developing this illness (MacDonald and Schulz, 2009). Similarly, lifetime cannabis use augments the probability of developing schizophrenia (MacDonald and Schulz, 2009). While these findings appear to be unrelated, all these risks factors may ultimately impact neuronal structure. Postmortem reports of malnourished infants have evidenced shorter and less ramified dendrites (Cordero et al., 1993; Benitez-Bribiesca et al., 1999). Similar structural deficits are observed in offspring when pregnant rodents undergo viral infection and these deficits persist even when the offspring have reached adulthood (Fatemi et al., 2002). Migration to northern latitudes is associated with vitamin D deficiency (Dealberto, 2013) and this vitamin maintains proteins directly involved in neurostructural dynamics (Féron et al., 2005). In fact, vitamin D can directly elicit growth of neuronal extensions in hippocampal neurons (Brown et al., 2003). Similarly, chronic cannabis exposure leads to altered dendritic structure in rats (Lawston et al., 2000). Unfortunately, data linking neuronal structure with all these risk factors for schizophrenia directly in patients’ cells is still lacking.
There is also information about the schizophrenia course and its clinical picture that could be explained by a defect in neurostructural dynamics. Neuroimaging studies strongly suggest that overt psychosis becomes evident while the last major pruning of brain connections is taking place (Thompson et al., 2001; Vidal et al., 2006; Sun et al., 2009a,b). This pruning normally occurs in late adolescence and early adulthood (Huttenlocher, 1990; Giedd et al., 1999; Gogtay et al., 2004; Lenroot and Giedd, 2006) when schizophrenia is most commonly diagnosed. It is possible that the removal of more connections in schizophrenia is a consequence of malformed dendrites and axons. But within this framework, how can we explain prodromal symptoms? There is no clear explanation for the origin of these symptoms; however, it is well recognized that during brain development and until adolescence the brain generates more connections than needed and only through different pruning episodes is the proper connectivity pattern finally acquired. This over-connectivity present until adolescence seems to protect patients from overt psychosis, but not from motor coordination anomalies, social difficulties and cognitive deficits (Marenco and Weinberger, 2000). These symptoms could result from neurons’ inability to constantly adjust their structure in response to external stimuli. Studies in rodents have demonstrated that learning involves drastic transformations in neuronal shape (Holtmaat and Svoboda, 2009; Caroni et al., 2012) while, in humans, different neuroimaging techniques have shown structural brain modifications after learning and training (Draganski et al., 2004, 2006; Sagi et al., 2012). One of the most consistent prodromal symptoms in schizophrenia is cognitive difficulties (Marenco and Weinberger, 2000). Consequently, a deficit in neurostructural dynamics could in theory explain the cognitive symptoms present not only during the prodromal phase but also throughout the illness course. Yet, there is still no information about brain structural deficits during learning in patients with schizophrenia.
The most consistent finding in schizophrenia, namely decreased brain volume and enlarged ventricles (Van Horn and McManus, 1992; Gur et al., 1994; Wright et al., 2000; Woods et al., 2005) can also stem from a deficit in neuronal structure. Postmortem studies have shown shorter and less ramified neuronal extensions in numerous brain areas including prefrontal cortex (Benes et al., 1986, 1991; Selemon et al., 1995, 1998; Kalus et al., 2000, 2002; Pierri et al., 2001, 2003; Broadbelt et al., 2002; Black et al., 2004), occipital cortex (Selemon et al., 1995), motor cortex (Benes et al., 1986), anterior cingulate cortex (Benes et al., 1986; Kalus et al., 2000, 2002; Chana et al., 2003), auditory cortex (Sweet et al., 2009), entorhinal cortex (Arnold et al., 1995), insular cortex (Pennington et al., 2008), subiculum (Arnold et al., 1995; Rosoklija et al., 2000), hippocampal subfields CA1, 2, 3 and 4 (Benes et al., 1991; Arnold et al., 1995; Zaidel et al., 1997; Jonsson et al., 1999; Kolomeets et al., 2005), cerebellar vermis (Tran et al., 1998), mediodorsal thalamus (Byne et al., 2002) and pulvinar (Byne et al., 2002). Even glial cells and neuronal-like cells derived from induced pluripotent stem cells have shown abnormal arborizations (Rajkowska et al., 2002). This broad range of affected brain regions strongly suggests that the entire brain is compromised.
The clinical picture of patients with schizophrenia supports this hypothesis of generalized brain dysfunction. Schizophrenia is commonly associated with psychotic symptoms; however, patients also experience negative symptoms such as lack of motivation and alogia, as well as cognitive difficulties such as attention problems and memory deficits. Less recognized, but also well documented in the scientific literature, are soft neurologic signs (Hui et al., 2009), oculomotor anomalies (Picard et al., 2008), disequilibrium (Picard et al., 2008), olfactory impediments (Turetsky et al., 2009) and the spontaneous appearance of abnormal movements even in the absence of antipsychotics (Walther and Strik, 2012). This wide variety of signs and symptoms is unlikely to originate from a single brain region or even from an isolated circuit. Instead, it could be explained by a generalized defect in neuronal structure. Within this framework, brain areas that contain neurons with more complex arborizations such as the hippocampus and the prefrontal cortex would be more affected, whereas brain regions carrying neurons with simple branches, like the olfactory bulb, would be less impaired. This proposition is consistent with the fact that the hippocampus and the prefrontal cortex are commonly associated with schizophrenia.
Based on this information, we believe there are a number of research lines suggesting a potential association between defective neurostructural dynamics and schizophrenia. One essential aspect is establishing which molecules regulate these constant changes in neuronal shape and lncRNAs are promising candidates. First, because the group of lncRNAs associated with schizophrenia is highly active during development when neuronal structure is more plastic and it is also during this life stage when schizophrenia seems to originate (Marenco and Weinberger, 2000; Rapoport et al., 2005). In addition, there is information that at least Gomafu remains active during adulthood (Albertson et al., 2006). Gomafu’s pattern of activity correlates with what is currently known about neuronal structural plasticity which is robust during development and then less active during adulthood.
A second important factor that suggests a potential association between lncRNAs, neurostructural dynamics and schizophrenia is that either lncRNAs (Table 1), or the genes they regulate, are active in numerous brain regions including: prefrontal cortex (Favalli et al., 2012), temporal gyrus (Barry et al., 2014) hippocampus, dentate gyrus (Bond et al., 2009; Thompson Ray et al., 2011; Favalli et al., 2012), nucleus accumbens (Albertson et al., 2006; Michelhaugh et al., 2011) and olfactory bulb (Long et al., 2003). This lncRNA’s capacity to act in several brain areas is in line with the clinical picture of schizophrenia and with anatomopathological results indicating there is broad brain involvement in this mental illness as was presented earlier in the discussion.
In addition, emerging information links known risk factors for schizophrenia such as malnutrition, infection during pregnancy and vitamin D deficiency with lncRNAs. For instance, the lncRNA growth arrest-specific 5 (Gas5) is implicated in certain metabolic pathways during starvation (Kino et al., 2010; Osborne-Majnik et al., 2013). It was also recently discovered that lncRNAs regulating responses to infections during pregnancy are involved with obstetric complications (Wang et al., 2014). Moreover, just last year it was revealed that vitamin D signaling can regulate the expression of certain lncRNAs (Jiang and Bikle, 2014). However, the role of these particular lncRNAs in the pathophysiology of schizophrenia needs to be further studied.
The strongest indication yet available about the possibility of lncRNAs controlling neurostructural dynamics and their potential involvement with schizophrenia comes from BDNF-AS and Gomafu. Data coming directly from patients have consistently associated BDNF with schizophrenia (Bellon et al., 2011; Thompson Ray et al., 2011; Favalli et al., 2012), while in vitro studies have shown that increasing endogenous BDNF by knocking down BDNF-AS transcript results in neurostructural rearrangements (Modarresi et al., 2012). But BDNF-AS has not been directly associated with schizophrenia and consequently its plausible role as a regulator of neurostructural dynamics in this mental illness has yet to be determined. In contrast, Gomafu has been directly linked with schizophrenia. This lncRNA is downregulated in the cortical gray matter of patients with this psychotic disorder (Barry et al., 2014). Moreover, Gomafu regulates genes also associated with schizophrenia (Barry et al., 2014) such as DISC-1 (Millar et al., 2000, 2005; Blackwood et al., 2001; Hodgkinson et al., 2004; Sachs et al., 2005; Lipska et al., 2006) and ErbB4 (Chong et al., 2008; Barry et al., 2014) and these genes have been shown to be directly involved in neurostructural dynamics (Bloom and Horvitz, 1997; Kuroda et al., 1999; Vaskovsky et al., 2000; Miyoshi et al., 2003; Ozeki et al., 2003; Kamiya et al., 2005; Krivosheya et al., 2008). Nonetheless, a direct deficit in neurostructural dynamics caused by either Gomafu, DISC-1 or ErbB4 in patients with schizophrenia has not been established.
In conclusion, there could be an association between schizophrenia, lncRNAs and neurostructural dynamics, but direct evidence is lacking. However, research on lncRNAs is rapidly evolving. There is tantalizing evidence that lncRNAs could help clarify the still enigmatic genetic contribution to schizophrenia. This could be particularly true if schizophrenia is approached within the context of dynamic changes in neuronal structure that start during development and continue throughout life.
Author Contributions
Author VM managed the literature searches and wrote the first draft of the manuscript. Author DD contributed to the discussion and second draft of the manuscript. Author KEV worked on the second draft of the manuscript. Author LEH contributed to the discussion and second draft of the manuscript. Author MAF contributed in the design as well as first and second draft. Author ARL contributed in parts of the design and development of the final manuscript. Author AB designed, developed and corrected all versions of this manuscript. All authors have approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was financially supported by Penn State College of Medicine and by Penn State Hershey Medical Center. No other source of funding was involved in this work.
References
Aberg, K., Saetre, P., Lindholm, E., Ekholm, B., Pettersson, U., Adolfsson, R., et al. (2006). Human QKI, a new candidate gene for schizophrenia involved in myelination. Am. J. Med. Genet. B Neuropsychiatr. Genet. 141, 84–90. doi: 10.1002/ajmg.b.30243
Addington, A. M., Gornick, M., Duckworth, J., Sporn, A., Gogtay, N., Bobb, A., et al. (2005). GAD1 (2q31.1), which encodes glutamic acid decarboxylase (GAD67), is associated with childhood-onset schizophrenia and cortical gray matter volume loss. Mol. Psychiatry 10, 581–588. doi: 10.1038/sj.mp.4001599
Akbarian, S., Kim, J. J., Potkin, S. G., Hagman, J. O., Tafazzoli, A., Bunney, W. E. Jr., et al. (1995). Gene expression for glutamic acid decarboxylase is reduced without loss of neurons in prefrontal cortex of schizophrenics. Arch. Gen. Psychiatry 52, 258–266. doi: 10.1001/archpsyc.1995.03950160008002
Albertson, D. N., Schmidt, C. J., Kapatos, G., and Bannon, M. J. (2006). Distinctive profiles of gene expression in the human nucleus accumbens associated with cocaine and heroin abuse. Neuropsychopharmacology 31, 2304–2312. doi: 10.1038/sj.npp.1301089
Alonso, M., Medina, J. H., and Pozzo-Miller, L. (2004). ERK1/2 activation is necessary for BDNF to increase dendritic spine density in hippocampal CA1 pyramidal neurons. Learn. Mem. 11, 172–178. doi: 10.1101/lm.67804
An, J. J., Gharami, K., Liao, G. Y., Woo, N. H., Lau, A. G., Vanevski, F., et al. (2008). Distinct role of long 3’ UTR BDNF mRNA in spine morphology and synaptic plasticity in hippocampal neurons. Cell 134, 175–187. doi: 10.1016/j.cell.2008.05.045
Arnold, S. E., Franz, B. R., Gur, R. C., Gur, R. E., Shapiro, R. M., Moberg, P. J., et al. (1995). Smaller neuron size in schizophrenia in hippocampal subfields that mediate cortical-hippocampal interactions. Am. J. Psychiatry 152, 738–748. doi: 10.1176/ajp.152.5.738
Augui, S., Nora, E. P., and Heard, E. (2011). Regulation of X-chromosome inactivation by the X-inactivation centre. Nat. Rev. Genet. 12, 429–442. doi: 10.1038/nrg2987
Barry, G. (2014). Integrating the roles of long and small non-coding RNA in brain function and disease. Mol. Psychiatry 19, 410–416. doi: 10.1038/mp.2013.196
Barry, G., Briggs, J. A., Vanichkina, D. P., Poth, E. M., Beveridge, N. J., Ratnu, V. S., et al. (2014). The long non-coding RNA Gomafu is acutely regulated in response to neuronal activation and involved in schizophrenia-associated alternative splicing. Mol. Psychiatry 19, 486–494. doi: 10.1038/mp.2013.45
Bellon, A. (2007). New genes associated with schizophrenia in neurite formation: a review of cell culture experiments. Mol. Psychiatry 12, 620–629. doi: 10.1038/sj.mp.4001985
Bellon, A., Krebs, M. O., and Jay, T. M. (2011). Factoring neurotrophins into a neurite-based pathophysiological model of schizophrenia. Prog. Neurobiol. 94, 77–90. doi: 10.1016/j.pneurobio.2011.04.003
Benes, F. M., Davidson, J., and Bird, E. D. (1986). Quantitative cytoarchitectural studies of the cerebral cortex of schizophrenics. Arch. Gen. Psychiatry 43, 31–35. doi: 10.1001/archpsyc.1986.01800010033004
Benes, F. M., McSparren, J., Bird, E. D., SanGiovanni, J. P., and Vincent, S. L. (1991). Deficits in small interneurons in prefrontal and cingulate cortices of schizophrenic and schizoaffective patients. Arch. Gen. Psychiatry 48, 996–1001. doi: 10.1001/archpsyc.1991.01810350036005
Benitez-Bribiesca, L., De la Rosa-Alvarez, I., and Mansilla-Olivares, A. (1999). Dendritic spine pathology in infants with severe protein-calorie malnutrition. Pediatrics 104:e21. doi: 10.1542/peds.104.2.e21
Bernardinelli, Y., Randall, J., Janett, E., Nikonenko, I., König, S., Jones, E. V., et al. (2014). Activity-dependent structural plasticity of perisynaptic astrocytic domains promotes excitatory synapse stability. Curr. Biol. 24, 1679–1688. doi: 10.1016/j.cub.2014.06.025
Black, J. E., Kodish, I. M., Grossman, A. W., Klintsova, A. Y., Orlovskaya, D., Vostrikov, V., et al. (2004). Pathology of layer V pyramidal neurons in the prefrontal cortex of patients with schizophrenia. Am. J. Psychiatry 161, 742–744. doi: 10.1176/appi.ajp.161.4.742
Blackshaw, S., Harpavat, S., Trimarchi, J., Cai, L., Huang, H., Kuo, W. P., et al. (2004). Genomic analysis of mouse retinal development. PLoS Biol. 2:E247. doi: 10.1371/journal.pbio.0020247
Blackwood, D. H., Fordyce, A., Walker, M. T., St Clair, D. M., Porteous, D. J., and Muir, W. J. (2001). Schizophrenia and affective disorders–cosegregation with a translocation at chromosome 1q42 that directly disrupts brain-expressed genes: clinical and P300 findings in a family. Am J Hum Genet 69, 428–433. doi: 10.1086/321969
Bloom, L., and Horvitz, H. R. (1997). The Caenorhabditis elegans gene unc-76 and its human homologs define a new gene family involved in axonal outgrowth and fasciculation. Proc. Natl. Acad. Sci. U S A 94, 3414–3419. doi: 10.1073/pnas.94.7.3414
Bond, A. M., Vangompel, M. J., Sametsky, E. A., Clark, M. F., Savage, J. C., Disterhoft, J. F., et al. (2009). Balanced gene regulation by an embryonic brain ncRNA is critical for adult hippocampal GABA circuitry. Nat. Neurosci. 12, 1020–1027. doi: 10.1038/nn.2371
Bond, C. S., and Fox, A. H. (2009). Paraspeckles: nuclear bodies built on long noncoding RNA. J. Cell. Biol. 186, 637–644. doi: 10.1083/jcb.200906113
Brennand, K. J., Simone, A., Jou, J., Gelboin-Burkhart, C., Tran, N., Sangar, S., et al. (2011). Modelling schizophrenia using human induced pluripotent stem cells. Nature 473, 221–225. doi: 10.1038/nature09915
Broadbelt, K., Byne, W., and Jones, L. B. (2002). Evidence for a decrease in basilar dendrites of pyramidal cells in schizophrenic medial prefrontal cortex. Schizophr. Res. 58, 75–81. doi: 10.1016/s0920-9964(02)00201-3
Brown, J., Bianco, J. I., McGrath, J. J., and Eyles, D. W. (2003). 1,25-dihydroxyvitamin D3 induces nerve growth factor, promotes neurite outgrowth and inhibits mitosis in embryonic rat hippocampal neurons. Neurosci. Lett. 343, 139–143. doi: 10.1016/s0304-3940(03)00303-3
Byne, W., Buchsbaum, M. S., Mattiace, L. A., Hazlett, E. A., Kemether, E., Elhakem, S. L., et al. (2002). Postmortem assessment of thalamic nuclear volumes in subjects with schizophrenia. Am. J. Psychiatry 159, 59–65. doi: 10.1176/appi.ajp.159.1.59
Cannon, T. D., Hennah, W., Van Erp, T. G., Thompson, P. M., Lonnqvist, J., Huttunen, M., et al. (2005). Association of DISC1/TRAX haplotypes with schizophrenia, reduced prefrontal gray matter and impaired short- and long-term memory. Arch. Gen. Psychiatry 62, 1205–1213. doi: 10.1001/archpsyc.62.11.1205
Cardno, A. G., and Gottesman, I. I. (2000). Twin studies of schizophrenia: from bow-and-arrow concordances to star wars Mx and functional genomics. Am. J. Med. Genet. 97, 12–17. doi: 10.1002/(sici)1096-8628(200021)97:1<12::aid-ajmg3>3.0.co;2-u
Cardno, A. G., Marshall, E. J., Coid, B., Macdonald, A. M., Ribchester, T. R., Davies, N. J., et al. (1999). Heritability estimates for psychotic disorders: the maudsley twin psychosis series. Arch. Gen. Psychiatry 56, 162–168. doi: 10.1001/archpsyc.56.2.162
Caroni, P., Donato, F., and Muller, D. (2012). Structural plasticity upon learning: regulation and functions. Nat. Rev. Neurosci. 13, 478–490. doi: 10.1038/nrn3258
Carthew, R. W., and Sontheimer, E. J. (2009). Origins and mechanisms of miRNAs and siRNAs. Cell 136, 642–655. doi: 10.1016/j.cell.2009.01.035
Chana, G., Landau, S., Beasley, C., Everall, I. P., and Cotter, D. (2003). Two-dimensional assessment of cytoarchitecture in the anterior cingulate cortex in major depressive disorder, bipolar disorder and schizophrenia: evidence for decreased neuronal somal size and increased neuronal density. Biol. Psychiatry 53, 1086–1098. doi: 10.1016/s0006-3223(03)00114-8
Chen, L. L., and Carmichael, G. G. (2009). Altered nuclear retention of mRNAs containing inverted repeats in human embryonic stem cells: functional role of a nuclear noncoding RNA. Mol. Cell 35, 467–478. doi: 10.1016/j.molcel.2009.06.027
Chen, J. L., Flanders, G. H., Lee, W. C., Lin, W. C., and Nedivi, E. (2011). Inhibitory dendrite dynamics as a general feature of the adult cortical microcircuit. J. Neurosci. 31, 12437–12443. doi: 10.1523/JNEUROSCI.0420-11.2011
Chong, V. Z., Thompson, M., Beltaifa, S., Webster, M. J., Law, A. J., and Weickert, C. S. (2008). Elevated neuregulin-1 and ErbB4 protein in the prefrontal cortex of schizophrenic patients. Schizophr. Res. 100, 270–280. doi: 10.1016/j.schres.2007.12.474
Chow, D. K., Groszer, M., Pribadi, M., Machniki, M., Carmichael, S. T., Liu, X., et al. (2009). Laminar and compartmental regulation of dendritic growth in mature cortex. Nat. Neurosci. 12, 116–118. doi: 10.1038/nn.2255
Chubb, J. E., Bradshaw, N. J., Soares, D. C., Porteous, D. J., and Millar, J. K. (2008). The DISC locus in psychiatric illness. Mol. Psychiatry 13, 36–64. doi: 10.1038/sj.mp.4002106
Clemson, C. M., Hutchinson, J. N., Sara, S. A., Ensminger, A. W., Fox, A. H., Chess, A., et al. (2009). An architectural role for a nuclear noncoding RNA: NEAT1 RNA is essential for the structure of paraspeckles. Mol. Cell 33, 717–726. doi: 10.1016/j.molcel.2009.01.026
Consortium, E. P. (2012). An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74. doi: 10.1038/nature11247
Cordero, M. E., D’Acuna, E., Benveniste, S., Prado, R., Nunez, J. A., and Colombo, M. (1993). Dendritic development in neocortex of infants with early postnatal life undernutrition. Pediatr. Neurol. 9, 457–464. doi: 10.1016/0887-8994(93)90025-8
Dealberto, M. J. (2013). Clinical symptoms of psychotic episodes and 25-hydroxy vitamin D serum levels in black first-generation immigrants. Acta Psychiatr. Scand. 128, 475–487. doi: 10.1111/acps.12086
De Paola, V., Holtmaat, A., Knott, G., Song, S., Wilbrecht, L., Caroni, P., et al. (2006). Cell type-specific structural plasticity of axonal branches and boutons in the adult neocortex. Neuron 49, 861–875. doi: 10.1016/j.neuron.2006.02.017
Draganski, B., Gaser, C., Busch, V., Schuierer, G., Bogdahn, U., and May, A. (2004). Neuroplasticity: changes in grey matter induced by training. Nature 427, 311–312. doi: 10.1038/427311a
Draganski, B., Gaser, C., Kempermann, G., Kuhn, H. G., Winkler, J., Büchel, C., et al. (2006). Temporal and spatial dynamics of brain structure changes during extensive learning. J. Neurosci. 26, 6314–6317. doi: 10.1523/jneurosci.4628-05.2006
Ekelund, J., Hennah, W., Hiekkalinna, T., Parker, A., Meyer, J., Lonnqvist, J., et al. (2004). Replication of 1q42 linkage in finnish schizophrenia pedigrees. Mol. Psychiatry 9, 1037–1041. doi: 10.1038/sj.mp.4001536
Ekelund, J., Hovatta, I., Parker, A., Paunio, T., Varilo, T., Martin, R., et al. (2001). Chromosome 1 loci in Finnish schizophrenia families. Hum. Mol. Genet. 10, 1611–1617. doi: 10.1093/hmg/10.15.1611
Faghihi, M. A., and Wahlestedt, C. (2009). Regulatory roles of natural antisense transcripts. Nat. Rev. Mol. Cell Biol. 10, 637–643. doi: 10.1038/nrm2738
Fatemi, S. H., Earle, J., Kanodia, R., Kist, D., Emamian, E. S., Patterson, P. H., et al. (2002). Prenatal viral infection leads to pyramidal cell atrophy and macrocephaly in adulthood: implications for genesis of autism and schizophrenia. Cell Mol. Neurobiol. 22, 25–33. doi: 10.1023/A:1015337611258
Favalli, G., Li, J., Belmonte-de-Abreu, P., Wong, A. H., and Daskalakis, Z. J. (2012). The role of BDNF in the pathophysiology and treatment of schizophrenia. J. Psychiatr. Res. 46, 1–11. doi: 10.1016/j.jpsychires.2011.09.022
Feng, J., Bi, C., Clark, B. S., Mady, R., Shah, P., and Kohtz, J. D. (2006). The Evf-2 noncoding RNA is transcribed from the Dlx-5/6 ultraconserved region and functions as a Dlx-2 transcriptional coactivator. Genes Dev. 20, 1470–1484. doi: 10.1101/gad.1416106
Fenoglio, C., Ridolfi, E., Galimberti, D., and Scarpini, E. (2013). An emerging role for long non-coding RNA dysregulation in neurological disorders. Int. J. Mol. Sci. 14, 20427–20442. doi: 10.3390/ijms141020427
Féron, F., Burne, T. H., Brown, J., Smith, E., Mcgrath, J. J., Mackay-Sim, A., et al. (2005). Developmental vitamin D3 deficiency alters the adult rat brain. Brain Res. Bull. 65, 141–148. doi: 10.1016/j.brainresbull.2004.12.007
Figurov, A., Pozzo-Miller, L. D., Olafsson, P., Wang, T., and Lu, B. (1996). Regulation of synaptic responses to high-frequency stimulation and LTP by neurotrophins in the hippocampus. Nature 381, 706–709. doi: 10.1038/381706a0
Francks, C., Delisi, L. E., Shaw, S. H., Fisher, S. E., Richardson, A. J., Stein, J. F., et al. (2003). Parent-of-origin effects on handedness and schizophrenia susceptibility on chromosome 2p12–q11. Hum. Mol. Genet. 12, 3225–3230. doi: 10.1093/hmg/ddg362
Galimberti, I., Gogolla, N., Alberi, S., Santos, A. F., Muller, D., and Caroni, P. (2006). Long-term rearrangements of hippocampal mossy fiber terminal connectivity in the adult regulated by experience. Neuron 50, 749–763. doi: 10.1016/j.neuron.2006.04.026
Ghildiyal, M., and Zamore, P. D. (2009). Small silencing RNAs: an expanding universe. Nat. Rev. Genet. 10, 94–108. doi: 10.1038/nrg2504
Giedd, J. N., Blumenthal, J., Jeffries, N. O., Castellanos, F. X., Liu, H., Zijdenbos, A., et al. (1999). Brain development during childhood and adolescence: a longitudinal MRI study. Nat. Neurosci. 2, 861–863. doi: 10.1038/13158
Gogtay, N., Giedd, J. N., Lusk, L., Hayashi, K. M., Greenstein, D., Vaituzis, A. C., et al. (2004). Dynamic mapping of human cortical development during childhood through early adulthood. Proc. Natl. Acad. Sci. U S A 101, 8174–8179. doi: 10.1073/pnas.0402680101
Guidotti, A., Auta, J., Davis, J. M., Di-Giorgi-Gerevini, V., Dwivedi, Y., Grayson, D. R., et al. (2000). Decrease in reelin and glutamic acid decarboxylase67 (GAD67) expression in schizophrenia and bipolar disorder: a postmortem brain study. Arch. Gen. Psychiatry 57, 1061–1069. doi: 10.1001/archpsyc.57.11.1061
Gur, R. E., Mozley, P. D., Shtasel, D. L., Cannon, T. D., Gallacher, F., Turetsky, B., et al. (1994). Clinical subtypes of schizophrenia: differences in brain and CSF volume. Am. J. Psychiatry 151, 343–350. doi: 10.1176/ajp.151.3.343
Hodgkinson, C. A., Goldman, D., Jaeger, J., Persaud, S., Kane, J. M., Lipsky, R. H., et al. (2004). Disrupted in schizophrenia 1 (DISC1): association with schizophrenia, schizoaffective disorder and bipolar disorder. Am. J. Hum. Genet. 75, 862–872. doi: 10.1086/425586
Holtmaat, A., and Svoboda, K. (2009). Experience-dependent structural synaptic plasticity in the mammalian brain. Nat. Rev. Neurosci. 10, 647–658. doi: 10.1038/nrn2699
Hui, C. L., Wong, G. H., Chiu, C. P., Lam, M. M., and Chen, E. Y. (2009). Potential endophenotype for schizophrenia: neurological soft signs. Ann. Acad. Med. Singapore 38, 408–506.
Huttenlocher, P. R. (1990). Morphometric study of human cerebral cortex development. Neuropsychologia 28, 517–527. doi: 10.1016/0028-3932(90)90031-I
Ishii, N., Ozaki, K., Sato, H., Mizuno, H., Saito, S., Takahashi, A., et al. (2006). Identification of a novel non-coding RNA, MIAT, that confers risk of myocardial infarction. J. Hum. Genet. 51, 1087–1099. doi: 10.1007/s10038-006-0070-9
Ishizuka, K., Paek, M., Kamiya, A., and Sawa, A. (2006). A review of Disrupted-In-Schizophrenia-1 (DISC1): neurodevelopment, cognition and mental conditions. Biol. Psychiatry 59, 1189–1197. doi: 10.1016/j.biopsych.2006.03.065
Jiang, Y. J., and Bikle, D. D. (2014). LncRNA: a new player in 1alpha, 25(OH)(2) vitamin D(3) /VDR protection against skin cancer formation. Exp. Dermatol. 23, 147–150. doi: 10.1111/exd.12341
Johnson, R. (2012). Long non-coding RNAs in huntington’s disease neurodegeneration. Neurobiol. Dis. 46, 245–254. doi: 10.1016/j.nbd.2011.12.006
Jonsson, S. A., Luts, A., Guldberg-Kjaer, N., and Ohman, R. (1999). Pyramidal neuron size in the hippocampus of schizophrenics correlates with total cell count and degree of cell disarray. Eur. Arch. Psychiatry Clin. Neurosci. 249, 169–173. doi: 10.1007/s004060050083
Joshi, D., Fung, S. J., Rothwell, A., and Weickert, C. S. (2012). Higher gamma-aminobutyric acid neuron density in the white matter of orbital frontal cortex in schizophrenia. Biol. Psychiatry 72, 725–733. doi: 10.1016/j.biopsych.2012.06.021
Kalus, P., Bondzio, J., Federspiel, A., Muller, T. J., and Zuschratter, W. (2002). Cell-type specific alterations of cortical interneurons in schizophrenic patients. Neuroreport 13, 713–717. doi: 10.1097/00001756-200204160-00035
Kalus, P., Muller, T. J., Zuschratter, W., and Senitz, D. (2000). The dendritic architecture of prefrontal pyramidal neurons in schizophrenic patients. Neuroreport 11, 3621–3625. doi: 10.1097/00001756-200011090-00044
Kamiya, A., Kubo, K., Tomoda, T., Takaki, M., Youn, R., Ozeki, Y., et al. (2005). A schizophrenia-associated mutation of DISC1 perturbs cerebral cortex development. Nat. Cell Biol. 7, 1167–1178. doi: 10.1038/ncb1328
Kanduri, C., Thakur, N., and Pandey, R. R. (2006). The length of the transcript encoded from the Kcnq1ot1 antisense promoter determines the degree of silencing. EMBO J. 25, 2096–2106. doi: 10.1038/sj.emboj.7601090
Kang, H., and Schuman, E. M. (1995). Long-lasting neurotrophin-induced enhancement of synaptic transmission in the adult hippocampus. Science 267, 1658–1662. doi: 10.1126/science.7886457
Khalil, A. M., Faghihi, M. A., Modarresi, F., Brothers, S. P., and Wahlestedt, C. (2008). A novel RNA transcript with antiapoptotic function is silenced in fragile X syndrome. PLoS One 3:e1486. doi: 10.1371/journal.pone.0001486
Kino, T., Hurt, D. E., Ichijo, T., Nader, N., and Chrousos, G. P. (2010). Noncoding RNA gas5 is a growth arrest- and starvation-associated repressor of the glucocorticoid receptor. Sci. Signal. 3:ra8. doi: 10.1126/scisignal.2000568
Kolomeets, N. S., Orlovskaya, D. D., Rachmanova, V. I., and Uranova, N. A. (2005). Ultrastructural alterations in hippocampal mossy fiber synapses in schizophrenia: a postmortem morphometric study. Synapse 57, 47–55. doi: 10.1002/syn.20153
Krivosheya, D., Tapia, L., Levinson, J. N., Huang, K., Kang, Y., Hines, R., et al. (2008). ErbB4-neuregulin signaling modulates synapse development and dendritic arborization through distinct mechanisms. J. Biol. Chem. 283, 32944–32956. doi: 10.1074/jbc.m800073200
Kumar, M., and Carmichael, G. G. (1998). Antisense RNA: function and fate of duplex RNA in cells of higher eukaryotes. Microbiol. Mol. Biol. Rev. 62, 1415–1434.
Kuroda, S., Nakagawa, N., Tokunaga, C., Tatematsu, K., and Tanizawa, K. (1999). Mammalian homologue of the Caenorhabditis elegans UNC-76 protein involved in axonal outgrowth is a protein kinase C zeta-interacting protein. J. Cell Biol. 144, 403–411. doi: 10.1083/jcb.144.3.403
Ladd, P. D., Smith, L. E., Rabaia, N. A., Moore, J. M., Georges, S. A., Hansen, R. S., et al. (2007). An antisense transcript spanning the CGG repeat region of FMR1 is upregulated in premutation carriers but silenced in full mutation individuals. Hum. Mol. Genet. 16, 3174–3187. doi: 10.1093/hmg/ddm293
Law, A. J., Kleinman, J. E., Weinberger, D. R., and Weickert, C. S. (2007). Disease-associated intronic variants in the ErbB4 gene are related to altered ErbB4 splice-variant expression in the brain in schizophrenia. Hum. Mol. Genet. 16, 129–141. doi: 10.1093/hmg/ddl449
Lawston, J., Borella, A., Robinson, J. K., and Whitaker-Azmitia, P. M. (2000). Changes in hippocampal morphology following chronic treatment with the synthetic cannabinoid WIN 55,212–2. Brain Res. 877, 407–410. doi: 10.1016/s0006-8993(00)02739-6
Lee, W. C., Huang, H., Feng, G., Sanes, J. R., Brown, E. N., So, P. T., et al. (2006). Dynamic remodeling of dendritic arbors in GABAergic interneurons of adult visual cortex. PLoS Biol. 4:e29. doi: 10.1371/journal.pbio.0040126
Lenroot, R. K., and Giedd, J. N. (2006). Brain development in children and adolescents: insights from anatomical magnetic resonance imaging. Neurosci. Biobehav. Rev. 30, 718–729. doi: 10.1016/j.neubiorev.2006.06.001
Levi, G., Puche, A. C., Mantero, S., Barbieri, O., Trombino, S., Paleari, L., et al. (2003). The Dlx5 homeodomain gene is essential for olfactory development and connectivity in the mouse. Mol. Cell. Neurosci. 22, 530–543. doi: 10.1016/s1044-7431(02)00041-6
Lipscombe, D. (2005). Neuronal proteins custom designed by alternative splicing. Curr. Opin. Neurobiol. 15, 358–363. doi: 10.1016/j.conb.2005.04.002
Lipska, B. K., Peters, T., Hyde, T. M., Halim, N., Horowitz, C., Mitkus, S., et al. (2006). Expression of DISC1 binding partners is reduced in schizophrenia and associated with DISC1 SNPs. Hum. Mol. Genet. 15, 1245–1258. doi: 10.1093/hmg/ddl040
Liu, Q. R., Lu, L., Zhu, X. G., Gong, J. P., Shaham, Y., and Uhl, G. R. (2006). Rodent BDNF genes, novel promoters, novel splice variants and regulation by cocaine. Brain Res. 1067, 1–12. doi: 10.1016/j.brainres.2005.10.004
Long, J. E., Garel, S., Depew, M. J., Tobet, S., and Rubenstein, J. L. (2003). DLX5 regulates development of peripheral and central components of the olfactory system. J. Neurosci. 23, 568–578.
MacDonald, A. W., and Schulz, S. C. (2009). What we know: findings that every theory of schizophrenia should explain. Schizophr. Bull. 35, 493–508. doi: 10.1093/schbul/sbp017
Magistri, M., Faghihi, M. A., St Laurent, G., IIIrd, and Wahlestedt, C. (2012). Regulation of chromatin structure by long noncoding RNAs: focus on natural antisense transcripts. Trends Genet. 28, 389–396. doi: 10.1016/j.tig.2012.03.013
Malone, C. D., and Hannon, G. J. (2009). Small RNAs as guardians of the genome. Cell 136, 656–668. doi: 10.1016/j.cell.2009.01.045
Mao, Y. S., Sunwoo, H., Zhang, B., and Spector, D. L. (2011a). Direct visualization of the co-transcriptional assembly of a nuclear body by noncoding RNAs. Nat. Cell Biol. 13, 95–101. doi: 10.1038/ncb2140
Mao, Y. S., Zhang, B., and Spector, D. L. (2011b). Biogenesis and function of nuclear bodies. Trends Genet. 27, 295–306. doi: 10.1016/j.tig.2011.05.006
Marenco, S., and Weinberger, D. R. (2000). The neurodevelopmental hypothesis of schizophrenia: following a trail of evidence from cradle to grave. Dev. Psychopathol. 12, 501–527. doi: 10.1017/s0954579400003138
Marik, S. A., Yamahachi, H., McManus, J. N., Szabo, G., and Gilbert, C. D. (2010). Axonal dynamics of excitatory and inhibitory neurons in somatosensory cortex. PLoS Biol. 8:e1000395. doi: 10.1371/journal.pbio.1000395
Mattick, J. S., Amaral, P. P., Dinger, M. E., Mercer, T. R., and Mehler, M. F. (2009). RNA regulation of epigenetic processes. Bioessays 31, 51–59. doi: 10.1002/bies.080099
McCullumsmith, R. E., Gupta, D., Beneyto, M., Kreger, E., Haroutunian, V., Davis, K. L., et al. (2007). Expression of transcripts for myelination-related genes in the anterior cingulate cortex in schizophrenia. Schizophr. Res. 90, 15–27. doi: 10.1016/j.schres.2006.11.017
Mercer, T. R., Dinger, M. E., and Mattick, J. S. (2009). Long non-coding RNAs: insights into functions. Nat. Rev. Genet. 10, 155–159. doi: 10.1038/nrg2521
Mercer, T. R., Dinger, M. E., Sunkin, S. M., Mehler, M. F., and Mattick, J. S. (2008). Specific expression of long noncoding RNAs in the mouse brain. Proc. Natl. Acad. Sci. U S A 105, 716–721. doi: 10.1073/pnas.0706729105
Mercer, T. R., Qureshi, I. A., Gokhan, S., Dinger, M. E., Li, G., Mattick, J. S., et al. (2010). Long noncoding RNAs in neuronal-glial fate specification and oligodendrocyte lineage maturation. BMC Neurosci. 11:14. doi: 10.1186/1471-2202-11-14
Michelhaugh, S. K., Lipovich, L., Blythe, J., Jia, H., Kapatos, G., and Bannon, M. J. (2011). Mining affymetrix microarray data for long non-coding RNAs: altered expression in the nucleus accumbens of heroin abusers. J. Neurochem. 116, 459–466. doi: 10.1111/j.1471-4159.2010.07126.x
Millar, J. K., James, R., Brandon, N. J., and Thomson, P. A. (2004). DISC1 and DISC2: discovering and dissecting molecular mechanisms underlying psychiatric illness. Ann. Med. 36, 367–378. doi: 10.1080/07853890410033603
Millar, J. K., Pickard, B. S., Mackie, S., James, R., Christie, S., Buchanan, S. R., et al. (2005). DISC1 and PDE4B are interacting genetic factors in schizophrenia that regulate cAMP signaling. Science 310, 1187–1191. doi: 10.1126/science.1112915
Millar, J. K., Wilson-Annan, J. C., Anderson, S., Christie, S., Taylor, M. S., Semple, C. A., et al. (2000). Disruption of two novel genes by a translocation co-segregating with schizophrenia. Hum. Mol. Genet. 9, 1415–1423. doi: 10.1093/hmg/9.9.1415
Miyoshi, K., Honda, A., Baba, K., Taniguchi, M., Oono, K., Fujita, T., et al. (2003). Disrupted-In-Schizophrenia 1, a candidate gene for schizophrenia, participates in neurite outgrowth. Mol. Psychiatry 8, 685–694. doi: 10.1038/sj.mp.4001352
Mizrahi, A., and Katz, L. C. (2003). Dendritic stability in the adult olfactory bulb. Nat. Neurosci. 6, 1201–1207. doi: 10.1038/nn1133
Modarresi, F., Faghihi, M. A., Lopez-Toledano, M. A., Fatemi, R. P., Magistri, M., Brothers, S. P., et al. (2012). Inhibition of natural antisense transcripts in vivo results in gene-specific transcriptional upregulation. Nat. Biotechnol. 30, 453–459. doi: 10.1038/nbt.2158
Moran, V. A., Perera, R. J., and Khalil, A. M. (2012). Emerging functional and mechanistic paradigms of mammalian long non-coding RNAs. Nucleic Acids Res. 40, 6391–6400. doi: 10.1093/nar/gks296
Morris, J. A., Kandpal, G., Ma, L., and Austin, C. P. (2003). DISC1 (Disrupted-In-Schizophrenia 1) is a centrosome-associated protein that interacts with MAP1A, MIPT3, ATF4/5 and NUDEL: regulation and loss of interaction with mutation. Hum. Mol. Genet. 12, 1591–1608. doi: 10.1093/hmg/ddg162
Munroe, S. H., and Zhu, J. (2006). Overlapping transcripts, double-stranded RNA and antisense regulation: a genomic perspective. Cell. Mol. Life Sci. 63, 2102–2118. doi: 10.1007/s00018-006-6070-2
Nacher, J., Guirado, R., and Castillo-Gómez, E. (2013). Structural plasticity of interneurons in the adult brain: role of PSA-NCAM and implications for psychiatric disorders. Neurochem. Res. 38, 1122–1133. doi: 10.1007/s11064-013-0977-4
Nagano, T., Mitchell, J. A., Sanz, L. A., Pauler, F. M., Ferguson-Smith, A. C., Feil, R., et al. (2008). The Air noncoding RNA epigenetically silences transcription by targeting G9a to chromatin. Science 322, 1717–1720. doi: 10.1126/science.1163802
Nakata, K., Lipska, B. K., Hyde, T. M., Ye, T., Newburn, E. N., Morita, Y., et al. (2009). DISC1 splice variants are upregulated in schizophrenia and associated with risk polymorphisms. Proc. Natl. Acad. Sci. U S A 106, 15873–15878. doi: 10.1073/pnas.0903413106
Nakayama, H., Miyazaki, T., Kitamura, K., Hashimoto, K., Yanagawa, Y., Obata, K., et al. (2012). GABAergic inhibition regulates developmental synapse elimination in the cerebellum. Neuron 74, 384–396. doi: 10.1016/j.neuron.2012.02.032
Nishimoto, Y., Nakagawa, S., Hirose, T., Okano, H. J., Takao, M., Shibata, S., et al. (2013). The long non-coding RNA nuclear-enriched abundant transcript 1_2 induces paraspeckle formation in the motor neuron during the early phase of amyotrophic lateral sclerosis. Mol. Brain 6:31. doi: 10.1186/1756-6606-6-31
Nishiyama, H., Fukaya, M., Watanabe, M., and Linden, D. J. (2007). Axonal motility and its modulation by activity are branch-type specific in the intact adult cerebellum. Neuron 56, 472–487. doi: 10.1016/j.neuron.2007.09.010
Osborne-Majnik, A., Fu, Q., and Lane, R. H. (2013). Epigenetic mechanisms in fetal origins of health and disease. Clin. Obstet. Gynecol. 56, 622–632. doi: 10.1097/grf.0b013e31829cb99a
Ozeki, Y., Tomoda, T., Kleiderlein, J., Kamiya, A., Bord, L., Fujii, K., et al. (2003). Disrupted-in-Schizophrenia-1 (DISC-1): mutant truncation prevents binding to NudE-like (NUDEL) and inhibits neurite outgrowth. Proc. Natl. Acad. Sci. U S A 100, 289–294. doi: 10.1073/pnas.0136913100
Pandey, R. R., Mondal, T., Mohammad, F., Enroth, S., Redrup, L., Komorowski, J., et al. (2008). Kcnq1ot1 antisense noncoding RNA mediates lineage-specific transcriptional silencing through chromatin-level regulation. Mol. Cell 32, 232–246. doi: 10.1016/j.molcel.2008.08.022
Pennington, K., Dicker, P., Hudson, L., and Cotter, D. R. (2008). Evidence for reduced neuronal somal size within the insular cortex in schizophrenia, but not in affective disorders. Schizophr. Res. 106, 164–171. doi: 10.1016/j.schres.2008.08.022
Pennisi, E. (2012). Genomics. ENCODE project writes eulogy for junk DNA. Science 337, 1159–1161. doi: 10.1126/science.337.6099.1159
Perez-Alvarez, A., Navarrete, M., Covelo, A., Martin, E. D., and Araque, A. (2014). Structural and functional plasticity of astrocyte processes and dendritic spine interactions. J. Neurosci. 34, 12738–12744. doi: 10.1523/JNEUROSCI.2401-14.2014
Picard, H., Amado, I., Mouchet-Mages, S., Olié, J. P., and Krebs, M. O. (2008). The role of the cerebellum in schizophrenia: an update of clinical, cognitive and functional evidences. Schizophr. Bull. 34, 155–172. doi: 10.1093/schbul/sbm049
Pierri, J. N., Volk, C. L., Auh, S., Sampson, A., and Lewis, D. A. (2001). Decreased somal size of deep layer 3 pyramidal neurons in the prefrontal cortex of subjects with schizophrenia. Arch. Gen. Psychiatry 58, 466–473. doi: 10.1001/archpsyc.58.5.466
Pierri, J. N., Volk, C. L., Auh, S., Sampson, A., and Lewis, D. A. (2003). Somal size of prefrontal cortical pyramidal neurons in schizophrenia: differential effects across neuronal subpopulations. Biol. Psychiatry 54, 111–120. doi: 10.1016/s0006-3223(03)00294-4
Pilpel, Y., and Segal, M. (2004). Activation of PKC induces rapid morphological plasticity in dendrites of hippocampal neurons via Rac and Rho-dependent mechanisms. Eur. J. Neurosci. 19, 3151–3164. doi: 10.1111/j.0953-816x.2004.03380.x
Plavicki, J., Mader, S., Pueschel, E., Peebles, P., and Boekhoff-Falk, G. (2012). Homeobox gene distal-less is required for neuronal differentiation and neurite outgrowth in the drosophila olfactory system. Proc. Natl. Acad. Sci. U S A 109, 1578–1583. doi: 10.1073/pnas.1016741109
Poitras, L., Yu, M., Lesage-Pelletier, C., Macdonald, R. B., Gagné, J. P., Hatch, G., et al. (2010). An SNP in an ultraconserved regulatory element affects Dlx5/Dlx6 regulation in the forebrain. Development 137, 3089–3097. doi: 10.1242/dev.051052
Pontier, D. B., and Gribnau, J. (2011). Xist regulation and function explored. Hum. Genet. 130, 223–236. doi: 10.1007/s00439-011-1008-7
Ponting, C. P., Oliver, P. L., and Reik, W. (2009). Evolution and functions of long noncoding RNAs. Cell 136, 629–641. doi: 10.1016/j.cell.2009.02.006
Porteous, D. J., Thomson, P., Brandon, N. J., and Millar, J. K. (2006). The genetics and biology of DISC1–an emerging role in psychosis and cognition. Biol. Psychiatry 60, 123–131. doi: 10.1016/j.biopsych.2006.04.008
Prasanth, K. V., Prasanth, S. G., Xuan, Z., Hearn, S., Freier, S. M., Bennett, C. F., et al. (2005). Regulating gene expression through RNA nuclear retention. Cell 123, 249–263. doi: 10.1016/j.cell.2005.08.033
Pruunsild, P., Kazantseva, A., Aid, T., Palm, K., and Timmusk, T. (2007). Dissecting the human BDNF locus: bidirectional transcription, complex splicing and multiple promoters. Genomics 90, 397–406. doi: 10.1016/j.ygeno.2007.05.004
Qureshi, I. A., and Mehler, M. F. (2010). Genetic and epigenetic underpinnings of sex differences in the brain and in neurological and psychiatric disease susceptibility. Prog. Brain Res. 186, 77–95. doi: 10.1016/b978-0-444-53630-3.00006-3
Radley, J. J., and Morrison, J. H. (2005). Repeated stress and structural plasticity in the brain. Ageing Res. Rev. 4, 271–287. doi: 10.1016/j.arr.2005.03.004
Radley, J. J., Rocher, A. B., Janssen, W. G., Hof, P. R., McEwen, B. S., and Morrison, J. H. (2005). Reversibility of apical dendritic retraction in the rat medial prefrontal cortex following repeated stress. Exp. Neurol. 196, 199–203. doi: 10.1016/j.expneurol.2005.07.008
Rajkowska, G., Miguel-Hidalgo, J. J., Makkos, Z., Meltzer, H., Overholser, J., and Stockmeier, C. (2002). Layer-specific reductions in GFAP-reactive astroglia in the dorsolateral prefrontal cortex in schizophrenia. Schizophr. Res. 57, 127–138. doi: 10.1016/s0920-9964(02)00339-0
Rapicavoli, N. A., and Blackshaw, S. (2009). New meaning in the message: noncoding RNAs and their role in retinal development. Dev. Dyn. 238, 2103–2114. doi: 10.1002/dvdy.21844
Rapicavoli, N. A., Poth, E. M., and Blackshaw, S. (2010). The long noncoding RNA RNCR2 directs mouse retinal cell specification. BMC Dev. Biol. 10:49. doi: 10.1186/1471-213x-10-49
Rapoport, J. L., Addington, A. M., Frangou, S., and Psych, M. R. (2005). The neurodevelopmental model of schizophrenia: update 2005. Mol. Psychiatry 10, 434–449. doi: 10.1038/sj.mp.4001642
Ripke, S., O’Dushlaine, C., Chambert, K., Moran, J. L., Kähler, A. K., Akterin, S., et al. (2013). Genome-wide association analysis identifies 13 new risk loci for schizophrenia. Nat. Genet. 45, 1150–1159. doi: 10.1038/ng.2742
Rosoklija, G., Toomayan, G., Ellis, S. P., Keilp, J., Mann, J. J., Latov, N., et al. (2000). Structural abnormalities of subicular dendrites in subjects with schizophrenia and mood disorders: preliminary findings. Arch. Gen. Psychiatry 57, 349–356. doi: 10.1001/archpsyc.57.4.349
Sachs, N. A., Sawa, A., Holmes, S. E., Ross, C. A., Delisi, L. E., and Margolis, R. L. (2005). A frameshift mutation in disrupted in schizophrenia 1 in an american family with schizophrenia and schizoaffective disorder. Mol. Psychiatry 10, 758–764. doi: 10.1038/sj.mp.4001667
Sagi, Y., Tavor, I., Hofstetter, S., Tzur-Moryosef, S., Blumenfeld-Katzir, T., and Assaf, Y. (2012). Learning in the fast lane: new insights into neuroplasticity. Neuron 73, 1195–1203. doi: 10.1016/j.neuron.2012.01.025
Schanen, N. C. (2006). Epigenetics of autism spectrum disorders. Hum. Mol. Genet. 15, R138–R150. doi: 10.1093/hmg/ddl213
Schulze, T. G., Buervenich, S., Badner, J. A., Steele, C. J., Detera-Wadleigh, S. D., Dick, D., et al. (2004). Loci on chromosomes 6q and 6p interact to increase susceptibility to bipolar affective disorder in the national institute of mental health genetics initiative pedigrees. Biol. Psychiatry 56, 18–23. doi: 10.1016/j.biopsych.2004.04.004
Selemon, L. D., Rajkowska, G., and Goldman-Rakic, P. S. (1995). Abnormally high neuronal density in the schizophrenic cortex. A morphometric analysis of prefrontal area 9 and occipital area 17. Arch. Gen. Psychiatry 52, 805–818; discussion 819–820. doi: 10.1001/archpsyc.1995.03950220015005
Selemon, L. D., Rajkowska, G., and Goldman-Rakic, P. S. (1998). Elevated neuronal density in prefrontal area 46 in brains from schizophrenic patients: application of a three-dimensional, stereologic counting method. J. Comp. Neurol. 392, 402–412. doi: 10.1002/(sici)1096-9861(19980316)392:3<402::aid-cne9>3.0.co;2-5
Shevtsov, S. P., and Dundr, M. (2011). Nucleation of nuclear bodies by RNA. Nat. Cell Biol. 13, 167–173. doi: 10.1038/ncb2157
Shigeta, M., Sanzen, N., Ozawa, M., Gu, J., Hasegawa, H., and Sekiguchi, K. (2003). CD151 regulates epithelial cell-cell adhesion through PKC- and Cdc42-dependent actin cytoskeletal reorganization. J. Cell Biol. 163, 165–176. doi: 10.1083/jcb.200301075
Soda, T., Frank, C., Ishizuka, K., Baccarella, A., Park, Y.-U., Flood, Z., et al. (2013). DISC1-ATF4 transcriptional repression complex: dual regulation of the cAMP-PDE4 cascade by DISC1. Mol. Psychiatry 18, 898–908. doi: 10.1038/mp.2013.38
Sone, M., Hayashi, T., Tarui, H., Agata, K., Takeichi, M., and Nakagawa, S. (2007). The mRNA-like noncoding RNA Gomafu constitutes a novel nuclear domain in a subset of neurons. J. Cell Sci. 120, 2498–2506. doi: 10.1242/jcs.009357
Sullivan, P. F., Daly, M. J., and O’Donovan, M. (2012). Genetic architectures of psychiatric disorders: the emerging picture and its implications. Nat. Rev. Genet. 13, 537–551. doi: 10.1038/nrg3240
Sun, D., Phillips, L., Velakoulis, D., Yung, A., McGorry, P. D., Wood, S. J., et al. (2009a). Progressive brain structural changes mapped as psychosis develops in ’at risk’ individuals. Schizophr. Res. 108, 85–92. doi: 10.1016/j.schres.2008.11.026
Sun, D., Stuart, G. W., Jenkinson, M., Wood, S. J., McGorry, P. D., Velakoulis, D., et al. (2009b). Brain surface contraction mapped in first-episode schizophrenia: a longitudinal magnetic resonance imaging study. Mol. Psychiatry 14, 976–986. doi: 10.1038/mp.2008.34
Sunwoo, H., Dinger, M. E., Wilusz, J. E., Amaral, P. P., Mattick, J. S., and Spector, D. L. (2009). MEN epsilon/beta nuclear-retained non-coding RNAs are up-regulated upon muscle differentiation and are essential components of paraspeckles. Genome Res. 19, 347–359. doi: 10.1101/gr.087775.108
Sweet, R. A., Henteleff, R. A., Zhang, W., Sampson, A. R., and Lewis, D. A. (2009). Reduced dendritic spine density in auditory cortex of subjects with schizophrenia. Neuropsychopharmacology 34, 374–389. doi: 10.1038/npp.2008.67
Takahashi, S., Ohtsuki, T., Yu, S. Y., Tanabe, E., Yara, K., Kamioka, M., et al. (2003). Significant linkage to chromosome 22q for exploratory eye movement dysfunction in schizophrenia. Am. J. Med. Genet. B Neuropsychiatr. Genet. 123, 27–32. doi: 10.1002/ajmg.b.10046
Tavosanis, G. (2012). Dendritic structural plasticity. Dev. Neurobiol. 72, 73–86. doi: 10.1002/dneu.20951
Taylor, M. S., Devon, R. S., Millar, J. K., and Porteous, D. J. (2003). Evolutionary constraints on the disrupted in schizophrenia locus. Genomics 81, 67–77. doi: 10.1016/s0888-7543(02)00026-5
Thompson Ray, M., Weickert, C. S., Wyatt, E., and Webster, M. J. (2011). Decreased BDNF, trkB-TK+ and GAD67 mRNA expression in the hippocampus of individuals with schizophrenia and mood disorders. J. Psychiatry Neurosci. 36, 195–203. doi: 10.1503/jpn.100048
Thompson, P. M., Vidal, C., Giedd, J. N., Gochman, P., Blumenthal, J., Nicolson, R., et al. (2001). Mapping adolescent brain change reveals dynamic wave of accelerated gray matter loss in very early-onset schizophrenia. Proc. Natl. Acad. Sci. U S A 98, 11650–11655. doi: 10.1073/pnas.201243998
Tolwani, R. J., Buckmaster, P. S., Varma, S., Cosgaya, J. M., Wu, Y., Suri, C., et al. (2002). BDNF overexpression increases dendrite complexity in hippocampal dentate gyrus. Neuroscience 114, 795–805. doi: 10.1016/s0306-4522(02)00301-9
Tran, K. D., Smutzer, G. S., Doty, R. L., and Arnold, S. E. (1998). Reduced Purkinje cell size in the cerebellar vermis of elderly patients with schizophrenia. Am. J. Psychiatry 155, 1288–1290. doi: 10.1176/ajp.155.9.1288
Tsuiji, H., Yoshimoto, R., Hasegawa, Y., Furuno, M., Yoshida, M., and Nakagawa, S. (2011). Competition between a noncoding exon and introns: gomafu contains tandem UACUAAC repeats and associates with splicing factor-1. Genes Cells 16, 479–490. doi: 10.1111/j.1365-2443.2011.01502.x
Turetsky, B. I., Hahn, C. G., Borgmann-Winter, K., and Moberg, P. J. (2009). Scents and nonsense: olfactory dysfunction in schizophrenia. Schizophr. Bull. 35, 1117–1131. doi: 10.1093/schbul/sbp111
Urbanska, M., Swiech, L., and Jaworski, J. (2012). Developmental plasticity of the dendritic compartment: focus on the cytoskeleton. Adv. Exp. Med. Biol. 970, 265–284. doi: 10.1007/978-3-7091-0932-8_12
Van Horn, J. D., and McManus, I. C. (1992. Ventricular enlargement in schizophrenia. A meta-analysis of studies of the ventricle: brain ratio (VBR). Br. J. Psychiatry 160, 687–697. doi: 10.1192/bjp.160.5.687
Vaskovsky, A., Lupowitz, Z., Erlich, S., and Pinkas-Kramarski, R. (2000). ErbB-4 activation promotes neurite outgrowth in PC12 cells. J. Neurochem. 74, 979–987. doi: 10.1046/j.1471-4159.2000.0740979.x
Velmeshev, D., Magistri, M., and Faghihi, M. A. (2013). Expression of non-protein-coding antisense RNAs in genomic regions related to autism spectrum disorders. Mol. Autism 4:32. doi: 10.1186/2040-2392-4-32
Vidal, C. N., Rapoport, J. L., Hayashi, K. M., Geaga, J. A., Sui, Y., McLemore, L. E., et al. (2006). Dynamically spreading frontal and cingulate deficits mapped in adolescents with schizophrenia. Arch. Gen. Psychiatry 63, 25–34. doi: 10.1001/archpsyc.63.1.25
Walther, S., and Strik, W. (2012). Motor symptoms and schizophrenia. Neuropsychobiology 66, 77–92. doi: 10.1159/000339456
Wang, H., Cao, Q., Ge, J., Liu, C., Ma, Y., Meng, Y., et al. (2014). LncRNA-regulated infection and inflammation pathways associated with pregnancy loss: genome wide differential expression of lncRNAs in early spontaneous abortion. Am. J. Reprod .Immunol. 72, 359–375. doi: 10.1111/aji.12275
Wang, K. C., and Chang, H. Y. (2011). Molecular mechanisms of long noncoding RNAs. Mol. Cell 43, 904–914. doi: 10.1016/j.molcel.2011.08.018
Williams, J. M., Beck, T. F., Pearson, D. M., Proud, M. B., Cheung, S. W., and Scott, D. A. (2009). A 1q42 deletion involving DISC1, DISC2 and TSNAX in an autism spectrum disorder. Am. J. Med. Genet. A 149, 1758–1762. doi: 10.1002/ajmg.a.32941
Woo, T. U., Walsh, J. P., and Benes, F. M. (2004). Density of glutamic acid decarboxylase 67 messenger RNA-containing neurons that express the N-methyl-D-aspartate receptor subunit NR2A in the anterior cingulate cortex in schizophrenia and bipolar disorder. Arch. Gen. Psychiatry 61, 649–657. doi: 10.1001/archpsyc.61.7.649
Woods, B. T., Ward, K. E., and Johnson, E. H. (2005). Meta-analysis of the time-course of brain volume reduction in schizophrenia: implications for pathogenesis and early treatment. Schizophr. Res. 73, 221–228. doi: 10.1016/j.schres.2004.05.014
Wright, I. C., Rabe-Hesketh, S., Woodruff, P. W., David, A. S., Murray, R. M., and Bullmore, E. T. (2000). Meta-analysis of regional brain volumes in schizophrenia. Am. J. Psychiatry 157, 16–25. doi: 10.1176/ajp.157.1.16
Yamada, K., Mizuno, M., and Nabeshima, T. (2002). Role for brain-derived neurotrophic factor in learning and memory. Life Sci. 70, 735–744. doi: 10.1016/s0024-3205(01)01461-8
Zaidel, D. W., Esiri, M. M., and Harrison, P. J. (1997). Size, shape and orientation of neurons in the left and right hippocampus: investigation of normal asymmetries and alterations in schizophrenia. Am. J. Psychiatry 154, 812–818. doi: 10.1176/ajp.154.6.812
Keywords: neurite formation, brain development, dendrites, axons, dendritic spines, brain structure, epigenetics, gene
Citation: Merelo V, Durand D, Lescallette AR, Vrana KE, Hong LE, Faghihi MA and Bellon A (2015) Associating schizophrenia, long non-coding RNAs and neurostructural dynamics. Front. Mol. Neurosci. 8:57. doi: 10.3389/fnmol.2015.00057
Received: 17 July 2015; Accepted: 10 September 2015;
Published: 30 September 2015.
Edited by:
Nicola Maggio, The Chaim Sheba Medical Center, IsraelReviewed by:
Michael J. Schmeisser, Ulm University, GermanySang H. Lee, Medical College of Wisconsin, USA
Copyright © 2015 Merelo, Durand, Lescallette, Vrana, Hong, Faghihi and Bellon. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution and reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alfredo Bellon, Penn State Hershey Medical Center, Department of Psychiatry, 500 University Drive, Hershey, PA 17033, USA, YWJlbGxvbkBobWMucHN1LmVkdQ==; YWxmcmVkb2JlbGxvbkB5YWhvby5jb20=