 Yasuhito Watanabe1
Yasuhito Watanabe1 Michaela K. Müller2,3
Michaela K. Müller2,3 Jakob von Engelhardt2,3
Jakob von Engelhardt2,3 Rolf Sprengel4
Rolf Sprengel4 Peter H. Seeburg4
Peter H. Seeburg4 Hannah Monyer1*
Hannah Monyer1*- 1Department of Clinical Neurobiology, University Hospital and German Cancer Research Center Heidelberg, Heidelberg, Germany
- 2Synaptic Signalling and Neurodegeneration, German Center for Neurodegenerative Diseases, Bonn, Germany
- 3Synaptic Signalling and Neurodegeneration, German Cancer Research Center Heidelberg, Heidelberg, Germany
- 4Department of Molecular Neurobiology, Max Planck Institute for Medical Research, Heidelberg, Germany
N-methyl-D-aspartate receptors (NMDARs) in all hippocampal areas play an essential role in distinct processes of memory formation as well as in sustaining cell survival of postnatally generated neurons in the dentate gyrus (DG). In contrast to the beneficial effects, over-activation of NMDARs has been implicated in many acute and chronic neurological diseases, reason why therapeutic approaches and clinical trials involving receptor blockade have been envisaged for decades. Here we employed genetically engineered mice to study the long-term effect of NMDAR ablation on selective hippocampal neuronal populations. Ablation of either GluN1 or GluN2B causes degeneration of the DG. The neuronal demise affects mature neurons specifically in the dorsal DG and is NMDAR subunit-dependent. Most importantly, the degenerative process exacerbates with increasing age of the animals. These results lead us to conclude that mature granule cells in the dorsal DG undergo neurodegeneration following NMDAR ablation in aged mouse. Thus, caution needs to be exerted when considering long-term administration of NMDAR antagonists for therapeutic purposes.
Introduction
The dentate gyrus (DG) in the hippocampus has been the focus of numerous studies for two main reasons. Firstly, the DG is a major station in the trisynaptic pathway that conveys input from the cortex to the hippocampus. DG granule cells excite CA3 neurons, which in turn project to CA1 neurons. All hippocampal subregions have been much investigated in the context of distinct forms of plasticity, learning, and memory (e.g., McHugh et al., 2007; Niewoehner et al., 2007; Gu et al., 2012; Nakashiba et al., 2012). Secondly, the DG is one of the two principal neurogenic niches in the postnatal brain, and has been therefore frequently studied in the context of neurogenesis (reviewed in Jessberger and Gage, 2014). Some studies addressed questions that link these two research areas. Thus, young DG granule cells support pattern separation, whereas mature granule cells facilitate pattern completion (Nakashiba et al., 2012). The DG comprises a dorsal and a ventral part that can be distinguished on anatomical grounds, and also based on distinct functions that the two areas support. Thus, the dorsal DG is involved in cognitive functions such as spatial memory, whilst the ventral DG supports brain functions related to stress, emotion, and affect (reviewed in Fanselow and Dong, 2010). Several decades of pharmacological and genetic research have taught us that N-methyl-D-aspartate receptors (NMDARs) are intricately linked to hippocampus-dependent memory (reviewed in Bannerman et al., 2014). Mouse genetics was fundamental to correlate subregions of the hippocampus, including the DG, with distinct functions supporting spatial memory (McHugh et al., 2007; Niewoehner et al., 2007). Thus, NMDARs in the DG are required for pattern separation (McHugh et al., 2007) and spatial working memory (Niewoehner et al., 2007). Furthermore, studies based on mouse mutants were instrumental in helping delineate the differential functions of NMDAR subunits. DG granule cells, like most neurons in the hippocampus, harbor NMDARs built from the obligatory GluN1 subunit (encoded by Grin1) and the developmentally regulated GluN2A and GluN2B subunits (encoded by Grin2a and Grin2b, respectively; Monyer et al., 1994; Rodenas-Ruano et al., 2012; Paoletti et al., 2013). NMDARs have been much discussed with respect to their function in postnatal neurogenesis. Thus, NMDARs in postnatally generated DG granule cells are a prerequisite for the survival of newborn neurons (Tashiro et al., 2006).
In mature neurons, over-activation or suppression of NMDARs are detrimental for cell function and survival. Thus, enhanced receptor activity as it occurs in many pathophysiological neurological diseases, e.g., stroke, epilepsy, induces excitotoxic cell death (reviewed in Choi, 1992; Paoletti et al., 2013). Conversely, pharmacological blockade of NMDARs for hours and days also has an adverse effect, as it induces cellular vacuolization in pyramidal neurons in many cortical brain areas (e.g., Ikonomidou et al., 1999). The effect of prolonged or even permanent NMDAR blockade on the survival of mature neurons has not yet been tested systematically. Moreover, a potential role of NMDARs for cell survival in aged animals has not been investigated. This is surprising, given that the NMDAR has been a key candidate when considering therapeutic strategies for stroke.
Here we took advantage of genetically modified mice and addressed the question whether the survival of mature and aged neurons depends on NMDARs. We demonstrate that in the hippocampus, mature neurons in the DG, but not in the CA1 region, require intact NMDAR function for survival.
Materials and Methods
Animals
Animals were housed and handled according to the respected animal welfare guidelines and rules of the Max Planck Society and the Germany government animal welfare office in Karlsruhe, Germany. The following genetically modified male and female C57Bl/6N mice were used in this study: DG and CA1 selective Grin1 knockout (Grin1ΔDGCA1) mice (Bannerman et al., 2012), DG selective Grin1 knockout (Grin1ΔDG, previously called NR1ΔDG) mice (Niewoehner et al., 2007), Grin2a knockout (Grin2a-/-, previously called homozygous GluR𝜀1 mutant) mice (Sakimura et al., 1995), and DG and CA1 selective Grin2b knockout (Grin2bΔDGCA1, previously called NR2B ΔHPC) mice (von Engelhardt et al., 2008).
Grin1ΔDGCA1 mice are homozygous for the floxed Grin1 and carry two transgenes, TgLC1 and TgCN12, which enable doxycycline-sensitive, Cre-mediated gene ablation in CA1 and DG excitatory neurons and piriform cortex in the adult brain by use of a CamKIIa/Grin2c hybrid promoter (Bannerman et al., 2012). Grin1ΔDG mice are homozygous for Grin1 and carry the transgenes TgLC1 and TgCN10-itTA, which are the same transgenes as in TgCN12, but yield differential expression in the two transgenic mouse lines. In Grin1ΔDG mice, Cre is specifically expressed in the DG granule cells and some CA1 pyramidal neurons (Niewoehner et al., 2007) which express calbindin (CB; data not shown). Grin2bΔDGCA1 mice are homozygous for the floxed Grin2b and carry TgLC1 and TgCN12. For the selective expression of Cre in Grin1ΔDGCA1, Grin1ΔDG and Grin2bΔDGCA1 mice, doxycycline was given via the drinking water to pregnant mice to suppress Cre expression of the offspring during embryonic development, and was withdrawn after birth (Bannerman et al., 2012). Doxycycline treatment was performed under the license 35-9185.81/G71/10 of the governmental council in the Karlsruhe, Germany. In Grin1ΔDGCA1 mice, Cre expression becomes detectable as early as postnatal day P28 (Bannerman et al., 2012). Control mice (i.e., mice with no Cre expression) to be compared with Grin1ΔDGCA1 and Grin1ΔDG mice were homozygous for floxed Grin1 and carried one or none of the transgenes. Control mice for the Grin2a knockout mice are wild-type mice. Control mice for Grin2bΔDGCA1 mice were homozygous for floxed Grin2b and carried one or none of the transgene. As control mice for Cre expressing mice (carrying only TgLC1 and TgCN12 but no floxed gene) we used either wild-type mice or carriers of one of the transgenes. Following mice are available at EMMA (https://www.infrafrontier.eu) with respective EMMA ID: Floxed Grin1 mice (EM: 09220), mice carrying TgLC1 and TgCN12 transgenes (EM: 09256), and mice carrying TgCN10-itTA transgene (EM: 09255). All animal experiments were performed according to the regulations of Heidelberg University/German Cancer Research Center/Max Planck Institute.
Immunohistochemistry
Mice were perfused with phosphate buffered saline (PBS, pH 7.4) followed by 4% paraformaldehyde (PFA). Brains were removed and postfixed with 2% PFA at 4°C overnight. Coronal sections were prepared at 50 μm using a vibratome. Sections were blocked and permeabilized with 5% bovine serum albumin (BSA) in PBS containing 0.5% Triton X-100, incubated with primary antibodies overnight at 4°C, then incubated with appropriate secondary antibodies. Primary and secondary antibodies were diluted in PBS containing 0.2% Triton X-100 before incubation. Sections were washed three times with PBS after every antibody incubation step. Sections were counterstained with DAPI (Invitrogen, Carlsbad, CA, USA) after the secondary antibody incubation step. For GluN1 staining, before blocking the sections, antigen retrieval was performed as follows. Sections were immersed in 10 mM sodium citrate (pH 6.0) and heated in a microwave oven for 5 min at 650 W twice with a 5 min break in between, kept for 20 min at room temperature, and washed twice with PBS. The following primary antibodies were used: rabbit active caspase-3 (catalog # AF835; 1:2000; polyclonal; R&D systems, Minneapolis, MN, USA), mouse CB D-28k (300; 1:2000; monoclonal; Swant, Switzerland), goat DCX (sc-8066; 1:500; polyclonal; Santa Cruz Biotechnology, Santa Cruz, CA, USA), goat Sox-2 (sc-17320; 1:500; polyclonal; Santa Cruz Biotechnology), mouse GFAP (G3893; 1:10000; monoclonal; Sigma–Aldrich, Saint Louis, MO, USA), rabbit S100β (721; 1:2000; polyclonal; Swant), rabbit NMDAR1 (for GluN1; AB9864R; 1:250, monoclonal; Millipore, Billerica, MA, USA), rabbit Cre [1:2000; polyclonal; a generous gift from Dr. Günther Schütz (Kellendonk et al., 1999)], mouse Mineralocorticoid receptor [6G1; 1:100; monoclonal; a generous gift from Dr. Elise P. Gomez-Sanchez (Gomez-Sanchez et al., 2006)].
Nissl Staining
Sections were mounted on microscope slides, air dried, immersed in 0.1% thionin, washed with water, dehydrated in 70, 95, and 100% ethanol for several minutes, cleared in xylene for 5 min twice. The slides were subsequently covered with Eukitt mounting medium, and mounted with coverslips.
Image Processing and Quantification
Fluorescent images were acquired using a LSM 700 confocal microscope (Zeiss, Oberkochen, Germany). A single focal plane picture of the whole DG area from one hemisphere was taken from each brain section using a tile scan function of the confocal microscope. Bright field images were acquired using a BX51W microscope (Olympus, Tokyo, Japan). All quantification analyses were done using ImageJ software. The DG thickness was measured from both dorsal/suprapyramidal blade and ventral/infrapyramidal blade. Care was taken to minimize variations in choosing sections in the rostrocaudal axis. Several, typically three, DG images from one animal were used for the analysis whenever possible. Measured values were averaged to create one representative value for each animal. To calculate DG thickness, a part of the DG with uniform thickness was selected as rectangular area of interest, and the area was then divided by the base length of the rectangular area. Dorsal DG thickness was calculated from the proximal areas to the crest where the two blades connect. Ventral DG thickness was calculated from the most ventral area close to the end of the blade. Degenerating cells were counted using the green channel after staining with antibody against active caspase-3. A cell was considered DCX-positive when the signal outlined the shape of a cell body with processes. To confirm that active capsase-3 (stained in green fluorescence) is expressed in CB-positive (stained in red fluorescence) cells, images of active caspase-3 positive cells were taken when there was no auto-fluorescence signal from degenerating cells in an unstained (far-red fluorescence) channel. Degenerating cells were counted as follows. The DG area to be examined was selected with selection tools. Putative degenerating cells were selected by MaxEntropy method of the autothreshold function, and signals bigger than 3 μm2 were counted using “Analyze particles” function. We confirmed that this counting method matches manual counting. This method enabled us to quantify degenerating cells more objectively than manual counting. Except for degenerating cells, all other fluorescently labeled cells were counted manually using cell counter function of ImageJ. Three to six images of the DG were used for counting fluorescent positive cells, and the obtained values were pooled to obtain one value for each mouse.
Statistical Analysis
Quantifications were carried out without referring to the genotype information. Differences or correlations between groups were examined using the statistical tests indicated in the figure legends. Statistical analyses were performed using R 3.1.3. Values were expressed as mean ± standard deviation unless otherwise mentioned in the figure legends. Values of p < 0.05 were considered statically significant (∗p < 0.05, ∗∗p < 0.005, ∗∗∗p < 0.0005).
Results
GluN1 Ablation Causes Progressive Degeneration of Granule Cells in the Dorsal DG
In 6 months old Grin1ΔDGCA1 mice, in which Grin1 is deleted in both the DG and CA1, we found a reduced thickness of the DG granule cell layer (Figures 1A,B,H). This phenotype could be readily detected by eye after DAPI staining, and was more pronounced in 18 months old mice (Figures 1C,D,H). In contrast, there was no apparent abnormality in the CA1 region in 18 months old Grin1ΔDGCA1 mice (Figures 1E,F,I). Expression of active caspase-3 in CB-positive granule cells was indicative of mature granule cell demise (Figure 1G). The alteration in the granule cell layer was restricted to the dorsal DG. In 18 months old mice, the ventral DG in Grin1ΔDGCA1 mice was comparable to that of controls (Supplementary Figures S1A–C). We confirmed GluN1 ablation in the CA1 region and in both the dorsal and ventral DG of 6 months old Grin1ΔDGCA1 mice by immunohistochemistry (Supplementary Figures S1D–G). Neuronal degeneration did not result from Cre expression itself, but was a consequence of Grin1 deletion via Cre-mediated recombination, as the DG was normal in Cre expressing mice with wild-type Grin1 alleles (Supplementary Figure S2). Thus, ablation of NMDARs in CA1 and DG causes selective degeneration of granule cells in the dorsal DG.
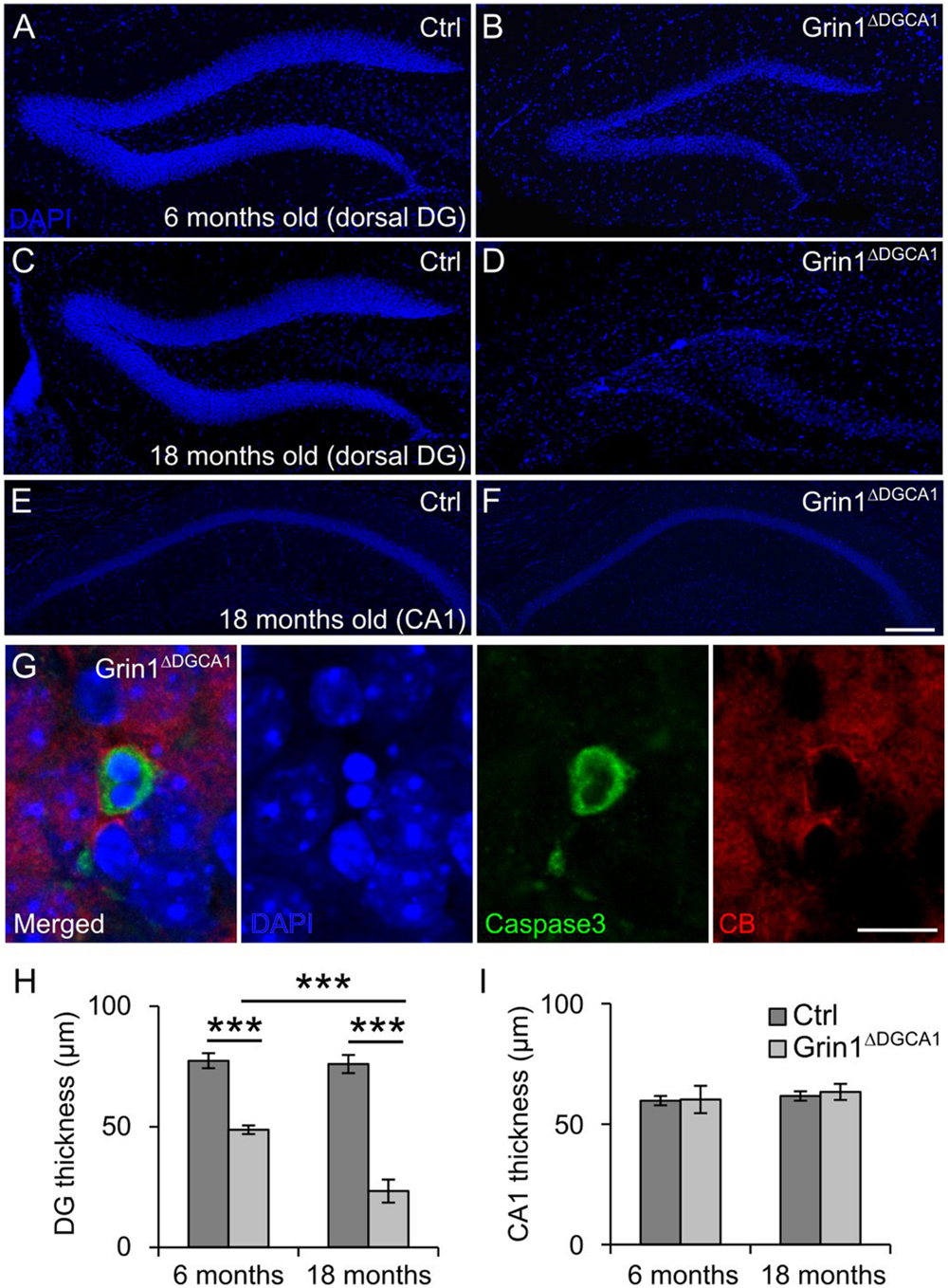
FIGURE 1. GluN1 deletion causes age-dependent neurodegeneration in the dorsal dentate gyrus (DG). (A) DAPI staining illustrating a section of a dorsal DG from a 6 months old control (Ctrl) mouse. (B) DAPI staining of a dorsal DG from a 6 months old Grin1ΔDGCA1 mouse. Note the thinning of the dentate granule cell layer. (C) DAPI staining illustrating a section of a dorsal DG from an 18 months old control mouse. (D) DAPI staining of a dorsal DG from an 18 months old Grin1ΔDGCA1 mouse. Note the thinning of the dentate granule cell layer following profound neurodegeneration. (E) DAPI staining illustrating a section of a dorsal CA1 from an 18 months old control mouse. (F) DAPI staining of a dorsal DG from an 18 months old Grin1ΔDGCA1 mouse. There is no difference between the two genotypes. (G) Representative image from the DG showing an apoptotic, i.e., caspase-3-positive, CB-positive neuron in a 6 months old Grin1ΔDGCA1 mouse. The section is counterstained with DAPI. (H) Quantitative evaluation of dorsal DG thickness in 6 and 18 months old control and Grin1ΔDGCA1 mice. The thickness of the DG was analyzed by two-factor ANOVA, followed by pairwise comparison test with p-value modification by Holm’s method, ∗∗∗p < 0.0005. The DG is thinner in Grin1ΔDGCA1 mice [F(1,16) = 662.39, p < 0.0005], and the difference increases with aging [F(1,16) = 59.92, p < 0.0005]. There is a significant interaction between genotype and age [F(1,16) = 53.99, p < 0.005]. (I) Quantitative evaluation of CA1 thickness in the dorsal hippocampus in 6 and 18 months old control and Grin1ΔDGCA1 mice. There is no difference in the thickness of the CA1 layer (two-factor ANOVA, no significant effect of genotype [F(1,16) = 0.594], no significant effect of age [F(1,16) = 2.793], and no significant interaction between age and genotype [F(1,16) = 0.168]. Scale bars, in (F), which applies to (A–E), and (G), 200 and 10 μm, respectively. Five and four mice for 6 months old control and Grin1ΔDGCA1, and six and five mice for 18 months old control and Grin1ΔDGCA1 were used, respectively.
Neuronal Degeneration in Grin1ΔDGCA1 Mice Affects Mature DG Granule Cells
To corroborate that NMDAR deletion causes cell death of mature DG granule cells, we compared the distribution of immature doublecortin (DCX) expressing cells (Figure 2A) and of degenerating cells visualized by active caspase-3 immunostaining (Figure 2B) within the DG granule cell layer (Figure 2C). If degeneration affected primarily immature DG granule cells, degenerating cells should be confined to the innermost rim of the DG granule cell layer that harbors DCX expressing neurons. This, however, was not the case. Degenerating cells were found throughout the entire DG granule cell layer (Figure 2D). We next examined whether there was a correlation between the number of immature and degenerating DG granule cells. Since the number of immature DG granule cells decreases with age, both populations would have to decrease in parallel, if degenerating cells were primarily immature DG granule cells. However, while the number of immature DG granule cells decreased with age both in control and Grin1ΔDGCA1 mice (Figure 2E), the number of degenerating cells was higher in Grin1ΔDGCA1 mice than in controls both at 6 and 18 months of age (Figure 2F). Furthermore, in Grin1ΔDGCA1 mice, there was no correlation between the number of immature DG granule cells and the number of degenerating cells, suggesting that degenerating cells were not immature cells (Figure 2G). Together these results indicate that degenerating DG granule cells are primarily mature DG granule cells.
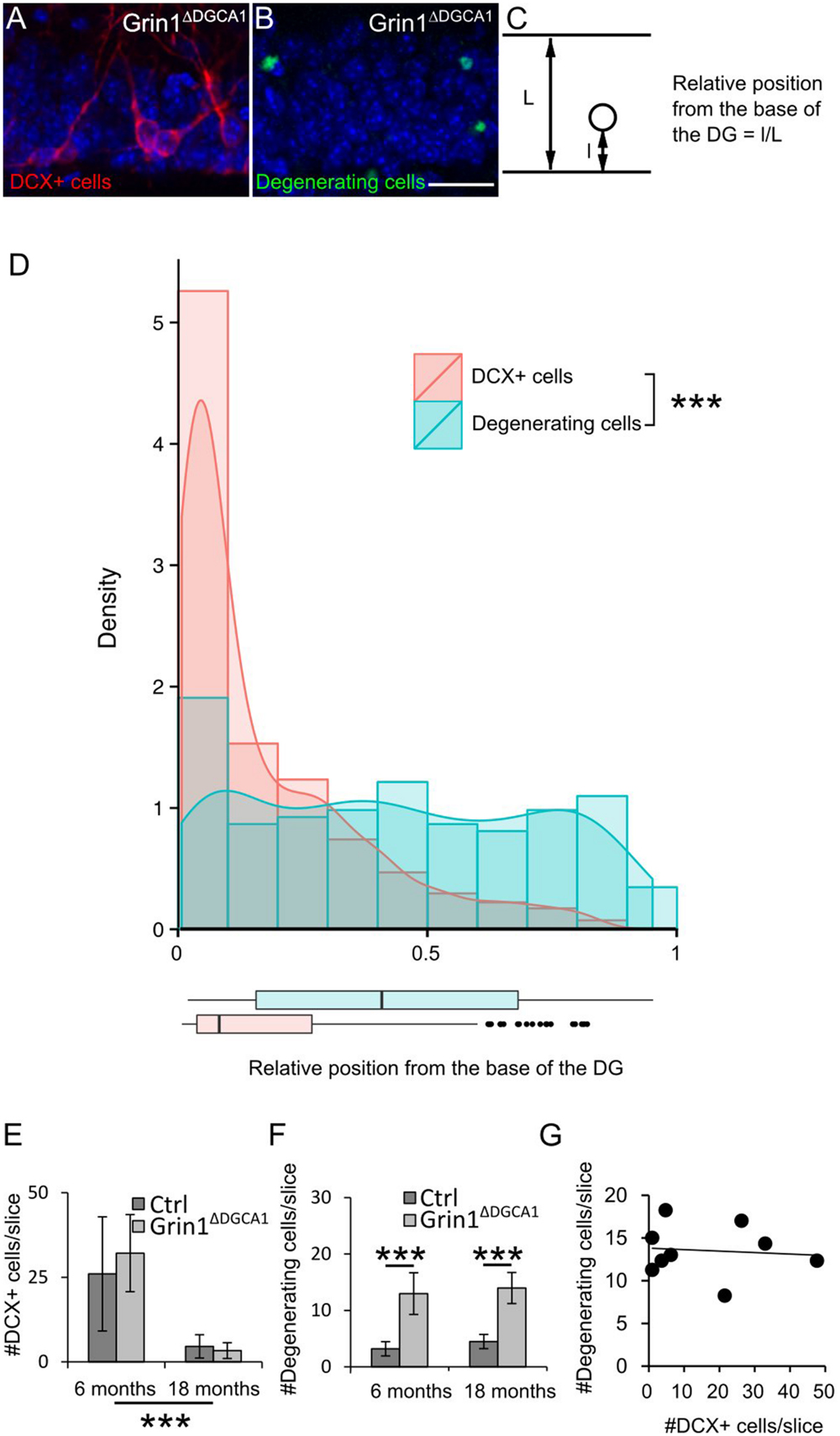
FIGURE 2. N-methyl-D-aspartate receptor (NMDAR) depletion in Grin1ΔDGCA1 mice causes age-dependent degeneration of mature DG granule cells. (A) DAPI stained section (blue) from a 6 months old Grin1ΔDGCA1 mouse brain showing DCX-positive (abbreviated DCX+ in the figure) neurons (red) located in the inner DG granule cell layer. (B) DAPI stained section (blue) from a 6 months old Grin1ΔDGCA1 mouse with degenerating neurons (green) dispersed throughout the layer. (C) Diagram indicating the parameters that were used to calculate the relative position of individual DG granule cells. (D) Histogram, density plot (curved line), and boxplot (bottom) of the relative position of DCX-positive cells (pink) and degenerating cells (blue) showing the differential distribution within the DG granule cell layer (significant difference in both Kolmogorov–Smirnov Tests and Mann–Whitney–Wilcoxon Test, ∗∗∗p < 0.0005). Whiskers extending from the boxes in the boxplot indicate the maximum and minimum value within the 1.5-fold of the interquartile range from the higher (75th percentile) and the lower quartile (25th percentile). Dots in the boxplot are outliers. This analysis comprised 405 DCX-positive cells and 173 degenerating cells. (E) The number of DCX-positive cells in the DG decreases with age both in control and Grin1ΔDGCA1 mice [two-factor ANOVA, F(1,16) = 30.090, p < 0.00005]. There is neither a significant difference between genotypes [F(1,16) = 0.653], nor a significant interaction between age and genotype [F(1,16) = 0.423]. (F) The number of degenerating neurons in the DG is higher in Grin1ΔDGCA1 mice [two-factor ANOVA, F(1,16) = 85.593, p < 0.00005, followed by post hoc analysis with Holm’s p-value modification, ∗∗∗p < 0.0005]. There is neither an effect of age [F(1,16) = 0.247, ns], nor a significant interaction between age and genotype [F(1,16) = 0.888]. (G) There is no correlation between the number of degenerating and newborn neurons in the DG of Grin1ΔDGCA1 mice (Spearman’s rank correlation coefficient test). The same animals were used as in Figure 1. Scale bar in (B), 25 μm.
NMDAR Dependent Survival is a Cell Type-Specific Property of Mature DG Granule Cells
In Grin1ΔDGCA1 mice, DG granule cell degeneration might be a complex phenotype that results from changed interactions between CA1 and DG network activities, and might reflect, at least in part, altered output activity from CA1 neurons, in which NMDARs were also deleted. To directly test whether the observed degeneration of mature DG granule cells following GluN1 depletion is indicative of a specific DG granule cell vulnerability, we investigated the effect of NMDAR depletion in genetically modified mice (Grin1ΔDG), in which the manipulation was restricted to the DG. Indeed, a reduction in the thickness of the dentate granule cell layer with degenerating cells was seen also in 6 months old Grin1ΔDG mice (Figures 3A–D,G,H), indicating that adult granule cells require NMDAR-mediated activity for their survival.
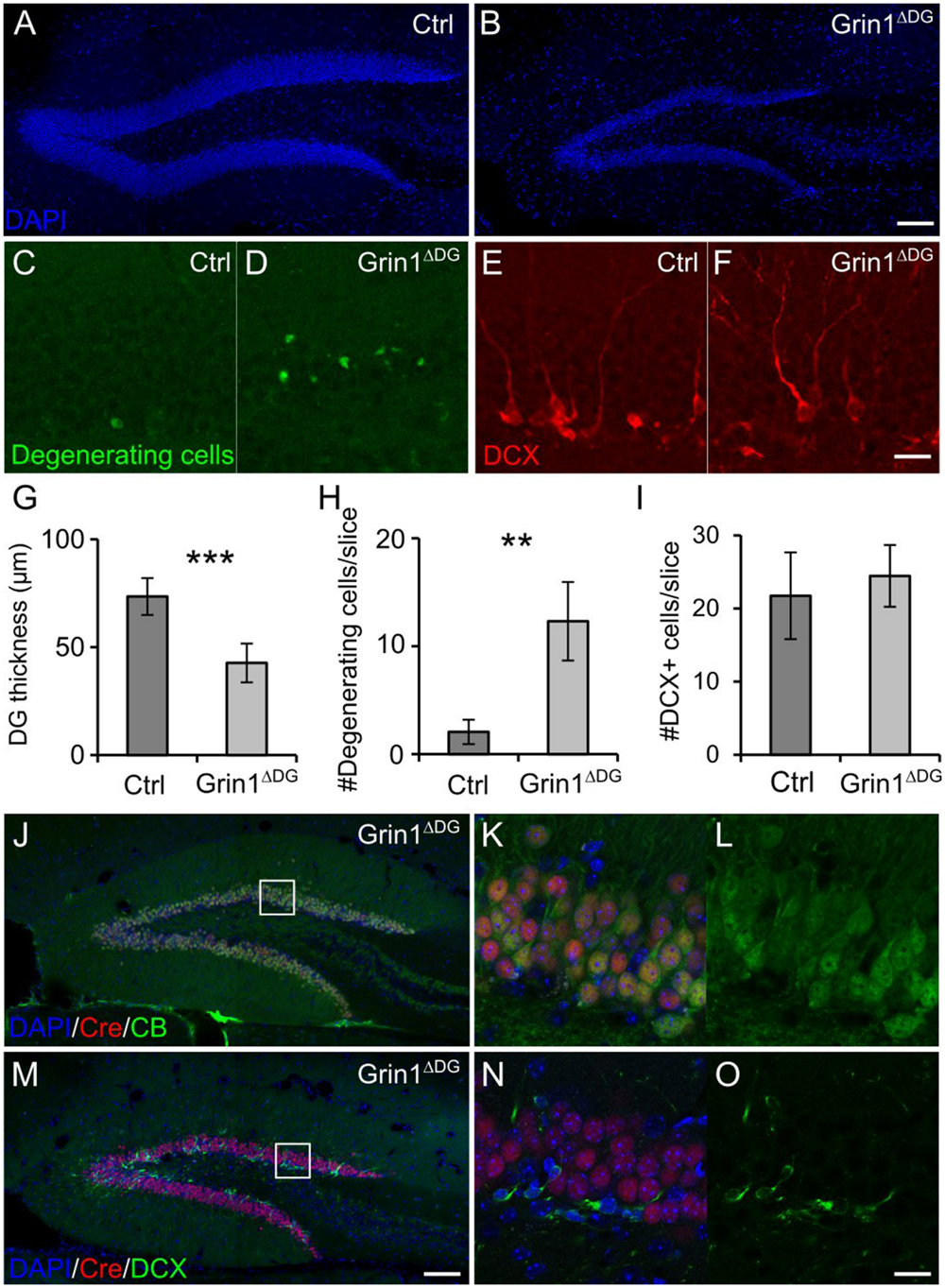
FIGURE 3. DG selective deletion of GluN1 in Grin1ΔDG recapitulates the phenotype observed in Grin1ΔDGCA1 mice. (A) DAPI stained section showing the DG of a 6 months old control mouse. (B) DAPI stained section from a 6 months old Grin1ΔDG mouse brain showing thinning of the DG. (C–F) Degenerating cells [green in (C,D)] and DCX-positive cells [red in (E,F)] in the DG of a 6 months old control and Grin1ΔDG mouse. (G) DG thickness is reduced in Grin1ΔDG mice (Welch’s t-test, ∗∗∗p < 0.0005). (H) The number of degenerating cells is increased in the DG of Grin1ΔDG mice (Welch’s t-test, ∗∗p < 0.005). (I) The number of DCX-positive immature cells is comparable between genotypes (Welch’s t-test). (J–O) In the DG of Grin1ΔDG mice, Cre (red) is expressed in CB-positive [green in (J–L)] mature neurons, but not in DCX-positive [green in (M–O)] immature neurons. Right panels (K,L,N,O) show a magnified view of the indicated area [white boxes in (J,M)]. Scale bars in (B,F,M,O), 100, 20, 100, and 20 μm, respectively. Five and seven mice were used for control and Grin1ΔDG, respectively.
Similarly to Grin1ΔDGCA1 mice, also in Grin1ΔDG mice the number of immature DG granule cells was not reduced (Figures 3E,F,I), suggesting that GluN1 depletion was restricted to mature DG granule cells. Hence we determined Cre expression in mature and immature DG granule cells, and found that indeed, Cre expression was detected in CB-positive, but not DCX-positive neurons both in Grin1ΔDG (Figures 3J–O) and in Grin1ΔDGCA1 mice (data not shown). Accordingly, GluN1 expression was present in immature DG granule cells (Supplementary Figures S3A–D). Together, these data provide evidence that GluN1 ablation was restricted to mature DG granule cells.
We wondered whether DG granule cell degeneration following NMDAR ablation was noticeable already at earlier developmental stages, and thus investigated 2 months old Grin1ΔDG mice. Despite of the GluN1 ablation, the number of immature and degenerating DG granule cells, and the overall thickness of the granule cell layer were comparable between control and mutant mice (Supplementary Figures S3E–I). It thus appears that the phenotype of DG granule cell degeneration following NMDAR ablation worsens in an age-dependent fashion.
Next we examined whether the genetic manipulation altered the number of stem cells in the DG of 6 months old Grin1ΔDG mice. The number of stem cells, i.e., GFAP/Sox2 positive cells, was not different between the two genotypes. The increased number of GFAP/Sox2/S100β positive cells in Grin1ΔDG mice (Supplementary Figure S4), reflects enhanced proliferation of astrocytes as often reported in brain areas with neurodegeneration. Hence NMDAR depletion in mature DG granule cells does not affect stem cell proliferation.
Survival of Mature DG Granule Cells Requires GluN2B Expression
Finally, to examine the composition of the NMDARs involved in the degeneration of DG granule cells, we investigated the survival of mature DG granule cells in mice with either GluN2A or GluN2B receptor ablation. To this end we took recourse to 18 months old Grin2a-/- and Grin2bΔDGCA1 mice. In Grin2a-/- mice, the overall appearance and thickness of the DG granule cell layer was not different from that in control mice (Figures 4A,B,E). In contrast, the DG of Grin2bΔDGCA1 mice was reminiscent of the above-described phenotype in Grin1ΔDGCA1 and Grin1ΔDG mice (Figures 4C,D,F). These data demonstrate that GluN2B-containing NMDARs support the survival of aged dorsal DG granule cells.
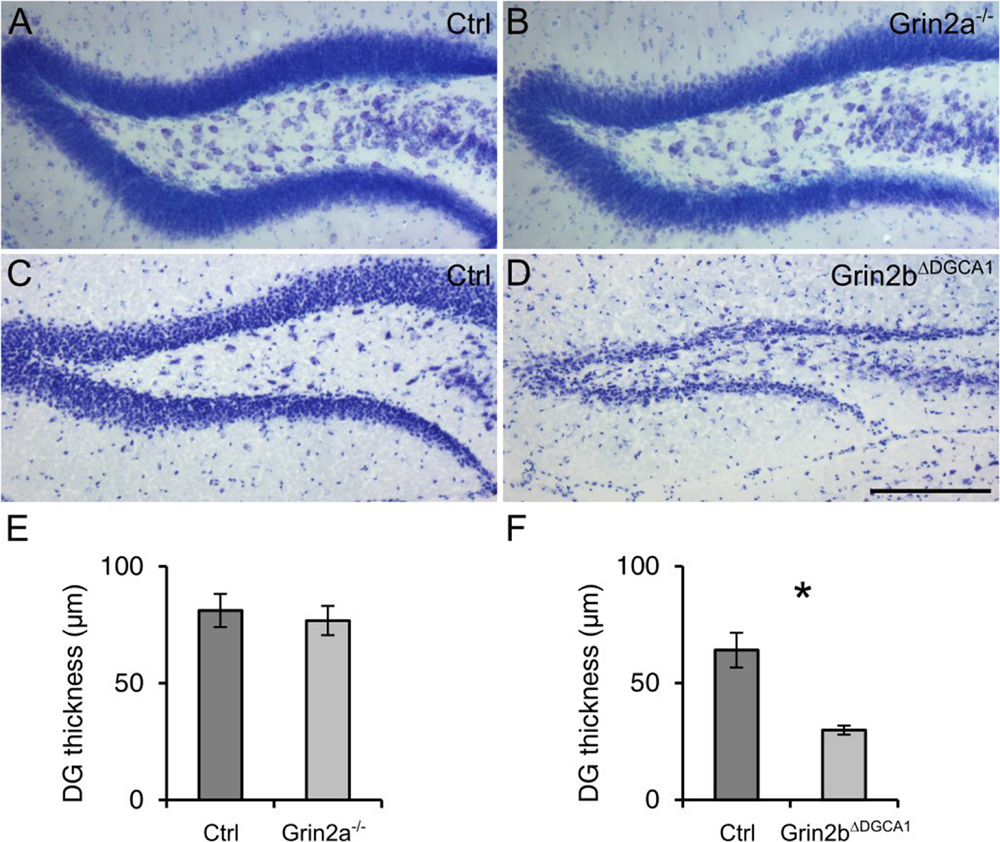
FIGURE 4. Absence of GluN2B-, but not GluN2A-containing NMDARs causes age-associated neurodegeneration of granule cells in the dorsal DG. (A) Nissl staining of the DG from an 18 months old control mouse. (B) Nissl staining of the DG from an 18 months old Grin2a-/- mouse. (C) Nissl staining of the DG from an 18 months old control mouse. (D) Nissl staining of the DG from an 18 months old control mouse (Grin2bΔDGCA1). (E) Quantitative evaluation of the granule cell layer thickness in the DG of control and Grin2a-/- mice (Student’s t-test, ns). (F) Quantitative evaluation of the granule cell layer thickness in the DG of control and Grin2bΔDGCA1 mice (Welch’s t-test, ∗p < 0.05). Scale bar in (D), 200 μm. Four and three mice for control and Grin2a-/-, and three and three mice for control and Grin2bΔDGCA1 were used, respectively.
Discussion
Here we demonstrate that NMDAR ablation causes neurodegeneration of the DG. This phenotype derives from the demise of mature DG granule cells and not from altered neurogenesis for the following reasons: First, enhanced caspase-3 expression and degenerating neurons were detected in the outer DG granule cell layer in the mutant mice. Second, the number of stem cells in the DG did not differ between control and mutant mice. Third, neurogenesis in the DG is known to decrease with age. Thus, if NMDAR ablation affected newborn cells, one would expect a progressive age-dependent decline of degenerating neurons. This, however, was not the case. Fourth, GluN1 was depleted primarily in mature DG granule cells as evidenced both by Cre expression and GluN1 immunohistochemistry in Grin1ΔDG mice. In addition, previous studies indicated that in several mouse mutants with reduced adult or absent adult neurogenesis the size or thickness of the DG granule cell layer was not visibly altered (Ansorg et al., 2012; Ouchi et al., 2013).
As evidenced in Grin1ΔDGCA1 mice, neurodegeneration following NMDAR ablation was region- and cell type-specific. Thus, dorsal but not ventral DG granule cell or CA1 pyramidal neurons were affected. A similar phenotype as the one detected in Grin1ΔDGCA1 and Grin1ΔDG mice was also reported in two other animal models of neurodegeneration. Thus, in rats, adrenalectomy causes degeneration of granule cells in the dorsal DG. The ventral DG is much less affected, and CA1 neurons or other hippocampal regions not at all (e.g., Sloviter et al., 1993). The pro-survival effect of corticosterone appears to be mediated by mineralocorticoid receptors (Gass et al., 2000). We do not think, however, that altered downstream signaling in mice with NMDAR ablation in DG granule cell involves signaling via mineralocorticoid receptors as their expression was not affected in our mutants (data not shown).
Notably, transgenic mice overexpressing Gsk3β also exhibit degeneration of granule cells preferentially in the dorsal DG (Fuster-Matanzo et al., 2011), and the alteration is accompanied by activated astrocytes (e.g., Fuster-Matanzo et al., 2011). Interestingly, Gsk3β is more active in the dorsal, whilst Akt, an inhibitor of Gsk3β, is more active in the ventral hippocampus (Fuster-Matanzo et al., 2011). Conversely, chronic application of lithium, an inhibitor of Gsk3β, ameliorates DG neurodegeneration induced by injection of amyloid-β fibrils (De Ferrari et al., 2003). Notably, there is evidence that β-catenin, a substrate of Gsk3β, directly interacts with the NMDAR (Al-Hallaq et al., 2007). Accordingly, GluN2B-containing NMDARs suppress apoptosis induced by Gsk3β overexpression in cultured neurons (Habas et al., 2006).
It was surprising that GluN2B, but not GluN2A, was required for the survival of DG granule cells, as several studies provided ample evidence that GluN2B-containing receptors mediate excitotoxic neuronal cell death (reviewed in Hardingham and Bading, 2010). There are nevertheless reports proposing that under certain conditions GluN2B activation provides also pro-survival signals (Habas et al., 2006).
Our findings have implications for therapeutic approaches in which chronic NMDAR antagonist treatment is envisaged. Thus, NMDAR antagonists have been considered for treatment of many neurological diseases, including cerebral ischemia, traumatic brain injury, pain, Alzheimer’s disease, Huntington’s disease, Parkinson’s disease, autism spectrum disorders, and depression (reviewed in Paoletti et al., 2013). In some cases, treatment appeared promising, at least in animal models (Paoletti et al., 2013; Patrizi et al., 2015). In humans, the NMDAR antagonist ketamine shows promising effects in patients with neuropathic pain and depression (reviewed in Niesters et al., 2014; Iadarola et al., 2015). Surveillance of the literature, however, indicates that in most studies the treatment was short, ranging from 1 day to few weeks. Importantly, ketamine, is also used for recreational purposes, and the longest period of drug intake exceeds 10 years (Liao et al., 2010). Chronic use of ketamine has long-lasting devastating effects on brain function, including working memory and episodic memory deficits (reviewed in Morgan and Curran, 2012). Based on our results, we propose that NMDAR antagonists, whilst potentially beneficial for acute treatment, bear the risk of causing DG degeneration following chronic administration, which would contribute to the worsening of cognitive functions, considering the crucial role of the DG in memory processes. It is not clear, however, to which extent the results that we obtained in mice following genetic receptor ablation can be transferred to humans, and in particular whether chronic NMDAR antagonist administration mimics the results reported here. Finally, the results warrant further investigations as to the downstream cellular signal cascade that eventually leads to the region-specific cell death. It is tempting to propose a putative involvement of Gsk3β, but so far this hypothesis remains speculative.
In summary, mature granule cells in the dorsal DG undergo neurodegeneration following NMDAR ablation in aged mice. The study provides evidence that caution must be exerted, especially in aged patients, when long-term administration of NMDAR antagonists is considered for therapeutic purposes. Identification of downstream pathways leading to neurodegeneration following NMDAR ablation is warranted to eventually establish safe clinical administration of NMDAR antagonists.
Author Contributions
YW and HM designed the experiments and wrote the manuscript. YW performed the experiments. YW and MM evaluated the data and performed the statistical analysis. All authors provided critical input for the generation of the final version of the manuscript. RS and PS provided the Grin1ΔDGCA1, Grin1ΔDG, and Grin2a-/- mice. All authors read and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank R. Hinz-Herkommer, I. Preugschat-Gumprecht, and U. Amtmann for technical assistance, M. Higuchi and A. Herb for animal management. This study was supported in part by a Postdoctoral Fellowship from the Uehara Memorial Foundation to YW.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fnmol.2015.00087
References
Al-Hallaq, R. A., Conrads, T. P., Veenstra, T. D., and Wenthold, R. J. (2007). NMDA di-heteromeric receptor populations and associated proteins in rat hippocampus. J. Neurosci. 27, 8334–8343. doi: 10.1523/JNEUROSCI.2155-07.2007
Ansorg, A., Witte, O. W., and Urbach, A. (2012). Age-dependent kinetics of dentate gyrus neurogenesis in the absence of cyclin D2. BMC Neurosci. 13:46. doi: 10.1186/1471-2202-13-46
Bannerman, D. M., Bus, T., Taylor, A., Sanderson, D. J., Schwarz, I., Jensen, V., et al. (2012). Dissecting spatial knowledge from spatial choice by hippocampal NMDA receptor deletion. Nat. Neurosci. 15, 1153–1159. doi: 10.1038/nn.3166
Bannerman, D. M., Sprengel, R., Sanderson, D. J., McHugh, S. B., Rawlins, J. N. P., Monyer, H., et al. (2014). Hippocampal synaptic plasticity, spatial memory and anxiety. Nat. Rev. Neurosci. 15, 181–192. doi: 10.1038/nrn3677
De Ferrari, G. V., Chacón, M. A., Barría, M. I., Garrido, J. L., Godoy, J. A., Olivares, G., et al. (2003). Activation of Wnt signaling rescues neurodegeneration and behavioral impairments induced by β-amyloid fibrils. Mol. Psychiatry 8, 195–208. doi: 10.1038/sj.mp.4001208
Fanselow, M. S., and Dong, H.-W. (2010). Are the dorsal and ventral hippocampus functionally distinct structures? Neuron 65, 7–19. doi: 10.1016/j.neuron.2009.11.031
Fuster-Matanzo, A., Llorens-Martín, M., de Barreda, E. G., Ávila, J., and Hernández, F. (2011). Different susceptibility to neurodegeneration of dorsal and ventral hippocampal dentate gyrus: a study with transgenic mice overexpressing GSK3β. PLoS ONE 6:e27262. doi: 10.1371/journal.pone.0027262
Gass, P., Kretz, O., Wolfer, D. P., Berger, S., Tronche, F., Reichardt, H. M., et al. (2000). Genetic disruption of mineralocorticoid receptor leads to impaired neurogenesis and granule cell degeneration in the hippocampus of adult mice. EMBO Rep. 1, 447–451. doi: 10.1093/embo-reports/kvd088
Gomez-Sanchez, C. E., de Rodriguez, A. F., Romero, D. G., Estess, J., Warden, M. P., Gomez-Sanchez, M. T., et al. (2006). Development of a panel of monoclonal antibodies against the mineralocorticoid receptor. Endocrinology 147, 1343–1348. doi: 10.1210/en.2005-0860
Gu, Y., Arruda-Carvalho, M., Wang, J., Janoschka, S. R., Josselyn, S. A., Frankland, P. W., et al. (2012). Optical controlling reveals time-dependent roles for adult-born dentate granule cells. Nat. Neurosci. 15, 1700–1706. doi: 10.1038/nn.3260
Habas, A., Kharebava, G., Szatmari, E., and Hetman, M. (2006). NMDA neuroprotection against a phosphatidylinositol-3 kinase inhibitor, LY294002 by NR2B-mediated suppression of glycogen synthase kinase-3β-induced apoptosis. J. Neurochem. 96, 335–348. doi: 10.1111/j.1471-4159.2005.03543.x
Hardingham, G. E., and Bading, H. (2010). Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat. Rev. Neurosci. 11, 682–696. doi: 10.1038/nrn2911
Iadarola, N. D., Niciu, M. J., Richards, E. M., Vande Voort, J. L., Ballard, E. D., Lundin, N. B., et al. (2015). Ketamine and other N-methyl-D-aspartate receptor antagonists in the treatment of depression: a perspective review. Ther. Adv. Chronic Dis. 6, 97–114. doi: 10.1177/2040622315579059
Ikonomidou, C., Bosch, F., Miksa, M., Bittigau, P., Vöckler, J., Dikranian, K., et al. (1999). Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science 283, 70–74. doi: 10.1126/science.283.5398.70
Jessberger, S., and Gage, F. H. (2014). Adult neurogenesis: bridging the gap between mice and humans. Trends Cell Biol. 24, 558–563. doi: 10.1016/j.tcb.2014.07.003
Kellendonk, C., Tronche, F., Casanova, E., Anlag, K., Opherk, C., and Schütz, G. (1999). Inducible site-specific recombination in the brain. J. Mol. Biol. 285, 175–182. doi: 10.1006/jmbi.1998.2307
Liao, Y., Tang, J., Ma, M., Wu, Z., Yang, M., Wang, X., et al. (2010). Frontal white matter abnormalities following chronic ketamine use: a diffusion tensor imaging study. Brain J. Neurol. 133, 2115–2122. doi: 10.1093/brain/awq131
McHugh, T. J., Jones, M. W., Quinn, J. J., Balthasar, N., Coppari, R., Elmquist, J. K., et al. (2007). Dentate gyrus NMDA receptors mediate rapid pattern separation in the hippocampal network. Science 317, 94–99. doi: 10.1126/science.1140263
Monyer, H., Burnashev, N., Laurie, D. J., Sakmann, B., and Seeburg, P. H. (1994). Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron 12, 529–540. doi: 10.1016/0896-6273(94)90210-0
Morgan, C. J. A., and Curran, H. V. (2012). Independent scientific committee on drugs. Ketamine use: a review. Addict. Abingdon Engl. 107, 27–38. doi: 10.1111/j.1360-0443.2011.03576.x
Nakashiba, T., Cushman, J. D., Pelkey, K. A., Renaudineau, S., Buhl, D. L., McHugh, T. J., et al. (2012). Young dentate granule cells mediate pattern separation, whereas old granule cells facilitate pattern completion. Cell 149, 188–201. doi: 10.1016/j.cell.2012.01.046
Niesters, M., Martini, C., and Dahan, A. (2014). Ketamine for chronic pain: risks and benefits. Br. J. Clin. Pharmacol. 77, 357–367. doi: 10.1111/bcp.12094
Niewoehner, B., Single, F. N., Hvalby,Ø, Jensen, V., Meyer zum Alten Borgloh, S., Seeburg, P. H., et al. (2007). Impaired spatial working memory but spared spatial reference memory following functional loss of NMDA receptors in the dentate gyrus. Eur. J. Neurosci. 25, 837–846. doi: 10.1111/j.1460-9568.2007.05312.x
Ouchi, Y., Banno, Y., Shimizu, Y., Ando, S., Hasegawa, H., Adachi, K., et al. (2013). Reduced adult hippocampal neurogenesis and working memory deficits in the Dgcr8-deficient mouse model of 22q11.2 deletion-associated schizophrenia can be rescued by IGF2. J. Neurosci. 33, 9408–9419. doi: 10.1523/JNEUROSCI.2700-12.2013
Paoletti, P., Bellone, C., and Zhou, Q. (2013). NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci. 14, 383–400. doi: 10.1038/nrn3504
Patrizi, A., Picard, N., Simon, A. J., Gunner, G., Centofante, E., Andrews, N. A., et al. (2015). Chronic administration of the N-methyl-D-aspartate receptor antagonist ketamine improves Rett syndrome phenotype. Biol. Psychiatry doi: 10.1016/j.biopsych.2015.08.018 [Epub ahead of print].
Rodenas-Ruano, A., Chavez, A. E., Cossio, M. J., Castillo, P. E., and Zukin, R. S. (2012). REST-dependent epigenetic remodeling promotes the developmental switch in synaptic NMDA receptors. Nat. Neurosci. 15, 1382–1390. doi: 10.1038/nn.3214
Sakimura, K., Kutsuwada, T., Ito, I., Manabe, T., Takayama, C., Kushiya, E., et al. (1995). Reduced hippocampal LTP and spatial learning in mice lacking NMDA receptor epsilon 1 subunit. Nature 373, 151–155. doi: 10.1038/373151a0
Sloviter, R. S., Sollas, A. L., Dean, E., and Neubort, S. (1993). Adrenalectomy-induced granule cell degeneration in the rat hippocampal dentate gyrus: characterization of an in vivo model of controlled neuronal death. J. Comp. Neurol. 330, 324–336. doi: 10.1002/cne.903300304
Tashiro, A., Sandler, V. M., Toni, N., Zhao, C., and Gage, F. H. (2006). NMDA-receptor-mediated, cell-specific integration of new neurons in adult dentate gyrus. Nature 442, 929–933. doi: 10.1038/nature05028
Keywords: NMDA receptors, aging, neurodegeneration, hippocampus, dentate gyrus, mouse
Citation: Watanabe Y, Müller MK, von Engelhardt J, Sprengel R, Seeburg PH and Monyer H (2016) Age-Dependent Degeneration of Mature Dentate Gyrus Granule Cells Following NMDA Receptor Ablation. Front. Mol. Neurosci. 8:87. doi: 10.3389/fnmol.2015.00087
Received: 06 November 2015; Accepted: 18 December 2015;
Published: 12 January 2016.
Edited by:
Robert J. Harvey, UCL School of Pharmacy, UKReviewed by:
Hansjürgen Volkmer, Universität Tübingen, GermanyHansen Wang, University of Toronto, Canada
Copyright © 2016 Watanabe, Müller, von Engelhardt, Sprengel, Seeburg and Monyer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hannah Monyer, aC5tb255ZXJAZGtmei1oZWlkZWxiZXJnLmRl