 Tianda Chen
Tianda Chen Qinwei Wu
Qinwei Wu Yang Zhang1,2
Yang Zhang1,2 Dai Zhang
Dai Zhang- 1Institute of Mental Health, Peking University Sixth Hospital, Beijing, China
- 2Key Laboratory of Mental Health, Ministry of Health & National Clinical Research Center for Mental Disorders, Peking University, Beijing, China
- 3Academy for Advanced Interdisciplinary Studies, Peking University, Beijing, China
- 4Peking-Tsinghua Center for Life Sciences, Peking University, Beijing, China
- 5PKU-IDG/McGovern Institute for Brain Research, Peking University, Beijing, China
Background: Transcription factor 4 (TCF4) is found to be associated with schizophrenia. TCF4 mutations also cause Pitt-Hopkins Syndrome, a neurodevelopmental disorder associated with severe mental retardation. However, the function of TCF4 during brain development remains unclear.
Results: Here, we report that Tcf4 is expressed in the developing cerebral cortex. In utero suppression of Tcf4 arrested neuronal migration, leading to accumulation of ectopic neurons in the intermediate zone. Knockdown of Tcf4 impaired leading process formation. Furthermore, Bone Morphogenetic Protein 7 (Bmp7) is upregulated in Tcf4-deficient neurons. In vivo gain of function and rescue experiments demonstrated that Bmp7 is the major downstream effector of Tcf4 required for neuronal migration.
Conclusion: Thus, we have uncovered a new Tcf4/Bmp7-dependent mechanism underlying neuronal migration, and provide insights into the pathogenesis of neurodevelopmental disorders.
Introduction
Transcription factor 4 (TCF4) (Gene ID: 6925) is a member of the E-protein family which encodes a highly homologous helix-turn-helix domains. It is expressed in the nervous system as well as the immune system (Einarson and Chao, 1995). Recently, genetic studies have demonstrated that defects in this gene are a cause of Pitt-Hopkins syndrome, a neurodevelopmental disease characterized by mental retardation, seizures, and hyperventilation (Amiel et al., 2007; Brockschmidt et al., 2007). Additional genetic evidence associates TCF4 with schizophrenia-relevant phenotypes (Blake et al., 2010; Li et al., 2010; Lennertz et al., 2011; Quednow et al., 2011; Aberg et al., 2013). TCF4 expression also differs in postmortem brains (Mudge et al., 2008) and blood from schizophrenia patients (Kurian et al., 2011).
Although TCF4 is strongly associated with several neuropsychiatric phenotypes, its role in brain development has not been studied in detail. Tcf4(-/-) mice have disrupted pontine nucleus development (Flora et al., 2007). Tcf4 upregulation enhances the expression of the cyclin-dependent kinase inhibitor gene p57(Kip2) and increases the number of cells in G1 phase among neuronal progenitors (Schmidt-Edelkraut et al., 2014). The major functional studies of TCF4 are in the immune system. TCF4 is essential for the development of B cell and plasmacytoid dendritic cell (PDC) differentiation (Zhuang et al., 1996; Nagasawa et al., 2008).
Here, we report that Tcf4 is expressed in developing mouse cortex. Tcf4-deficient neurons fail to grow leading process, resulting in impaired radial migration. In addition, we show that the expression of Bmp7 is increased in Tcf4-deficient neurons. We further demonstrate that Tcf4 regulate neuronal migration through Bmp7. Overall, our findings establish a new Tcf4/Bmp7 dependent mechanism underlying neuronal migration.
Results
Tcf4 is Expressed in the Developing Cerebral Cortex
As a first step in studying whether Tcf4 plays a role in cortical development, we examined the expression profile of Tcf4 in the developing mouse brain. Using in situ hybridization we found that Tcf4 mRNA was present in the subplate (SP) and proliferative zones of the cortical wall [the ventricular zone (VZ) and the subventricular zone (SVZ)] at E14 and in the cortical plate (CP) of the cerebral cortex and hippocampus at embryonic (E) day 17.5 and postnatal (P) day 0 (Figure 1A and Supplementary Figure S1). Immunofluorescent staining also shows that Tcf4 is highly expressed in the E17.5 mouse cortex (Figure 1B). Furthermore, we measured the abundance of Tcf4 in the developing cerebral cortex and found that high levels of Tcf4 protein were present in the neocortex from E14.5 to P0 (Figures 1C,D).
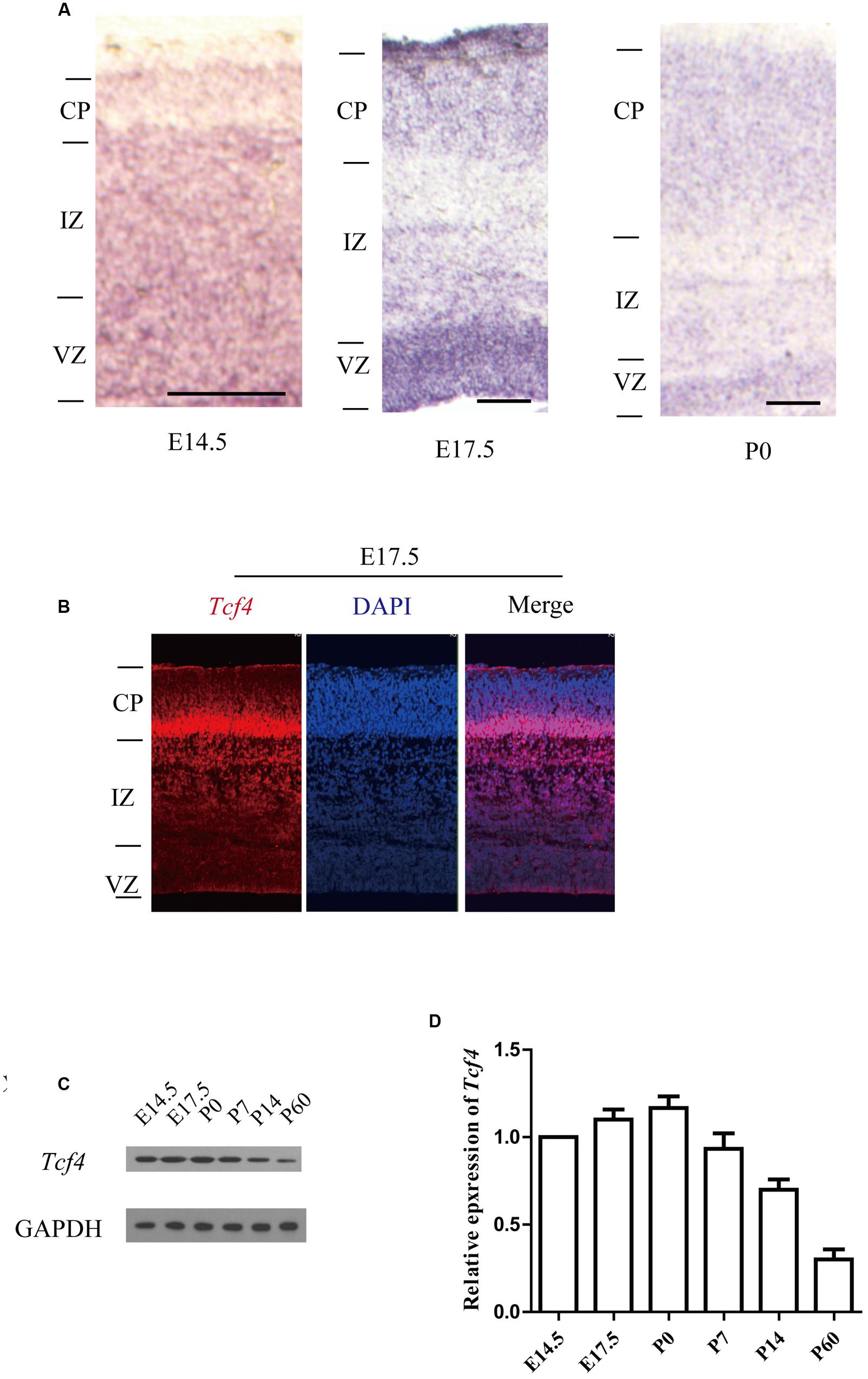
FIGURE 1. Tcf4 expression in developing cortical neurons. (A) In situ hybridization shows Tcf4 expression in the developing cerebral cortex of mice. (B) Immunofluorescent staining shows that Tcf4 is highly expressed in the E17.5 mouse cortex. (C,D) Immunoblotting reveals the Tcf4 protein levels in the cerebral cortex during development (n = 3). Scale bars, E14.5 is 100 μm, E17.5 and P0 are 200 μm. Data are shown as the mean ± SEM. one-way ANOVA, followed by an LSD post hoc test. CP, cortical plate; VZ, ventricular zone; IZ, intermediate zone.
Tcf4 is Essential for Neuronal Migration
Because Tcf4 is expressed in the developing cerebral cortex, we wondered whether Tcf4 may regulate neuronal migration. To test this possibility, in utero electroporation was used to cotransfect radial glial progenitors in the cerebral cortex of E14.5 mice with plasmids expressing Tcf4 shRNA and GFP as a marker. The distribution of GFP-positive cells was examined at P0. Approximately 80% of the Tcf4 shRNA-expressing neurons were in the IZ and VZ, whereas the control cortical neurons migrated into CP (Figures 2A,B). The shTcf4 1# specificity was demonstrated by the rescue of neuronal migration following co-electroporation of a plasmid-expressing Tcf4 mutant resistant to the shTcf4 1# sequences (Tcf4R) (Figures 2A–C). Furthermore, the western blot results showed that shTcf4 1# caused an 80% reduction of Tcf4 expression, whereas 2# caused a 75% reduction (Figures 2C,D). Thus, Tcf4 is essential for neuronal migration.
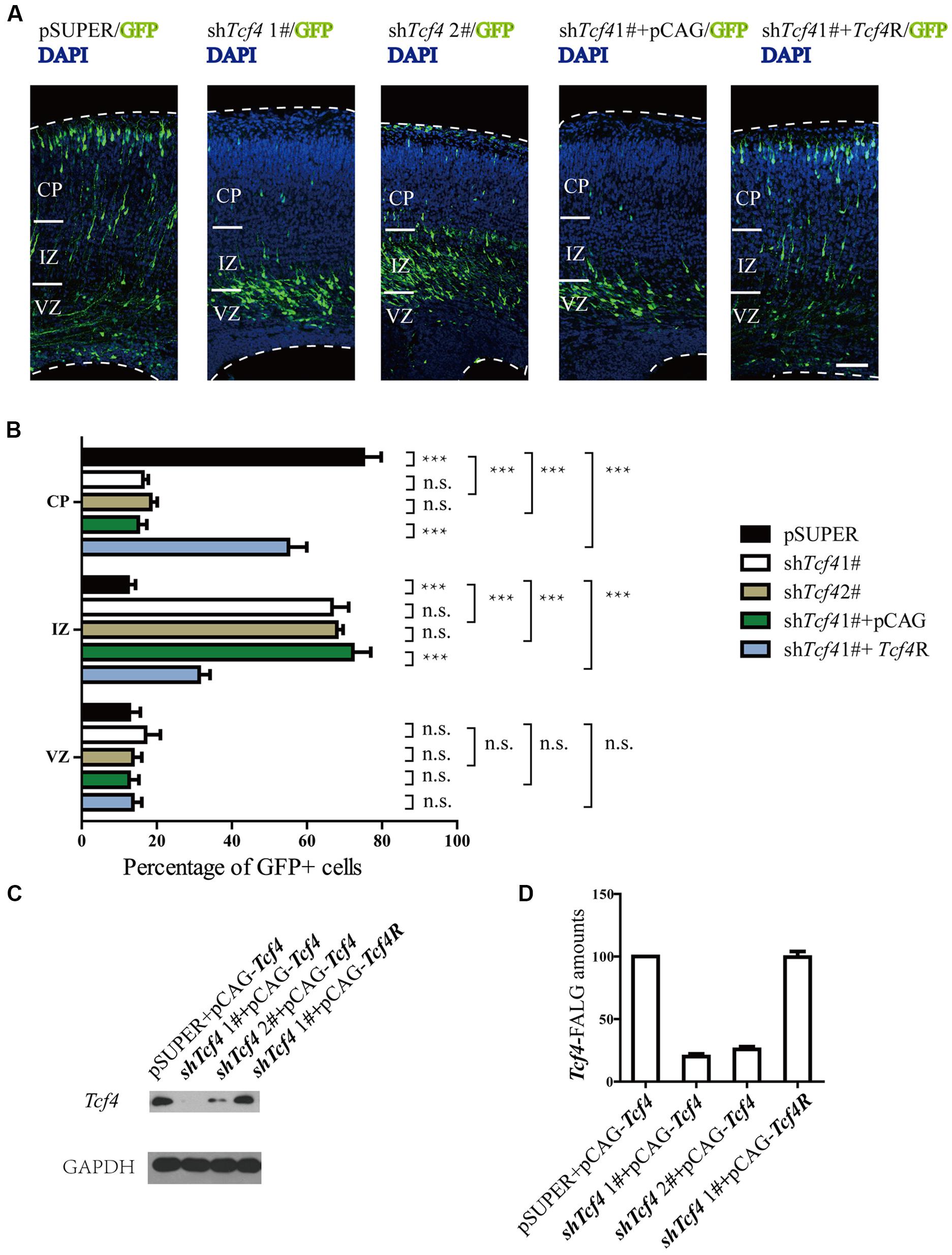
FIGURE 2. Tcf4 is essential for neuronal migration in developing cortex. (A,B) Results of in utero electroporation of E14.5 mouse embryos with GFP plasmids together with pSUPER vector (pSUPER) or pSUPER-based shRNAs targeting Tcf4 (shTcf4 1# and 2#). Knockdown of Tcf4 impairs radial migration. Overexpressed Tcf4R but not PCAG vector (E14.5-P0) can rescue the radial migration defect caused by in utero electroporation with shTcf4 (GFP-positive). Representative coronal brain sections at P0 were stained with antibodies to GFP (green), and counterstained with DAPI, a nuclear marker (blue). The distribution of GFP-positive neurons is quantified (>600 neurons from three mice). (C,D) Immunoblot analysis in HEK293T cells shows that Tcf4-specific shRNAs, shTcf4 1# and shTcf4 2#, are both sufficient to knock down Tcf4. Cells were co-transfected with Tcf4-FLAG and individual shRNA and total cell lysates were prepared for immunoblotting 48 h after transfection (n = 4). Scale bars, 100 μm. Data are shown as the mean ± SEM. n.s., not significant; ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001; one-way ANOVA, followed by an LSD post hoc test or two-way ANOVA followed by a Bonferroni post hoc test. VZ, ventricular zone; CP, cortical plate; IZ, intermediate zone.
Knockdown of Tcf4 Impairs Leading Process Formation
To explore the underlying mechanism by which Tcf4 controls radial migration, we analyzed the morphology of neurons in the IZ in detail. E14.5 mice were transfected with pSUPER and the short hairpin RNA against Tcf4 (shTcf4 1#) constructs into the developing mouse brain using in utero electroporation. We noticed that Tcf4 knockdown neurons exhibited impairments in leading processes formation at E17.5 with a large portion of cells (50.3%, compared to 12%in the control) lacking a healthy leading process (Figures 3A–C).
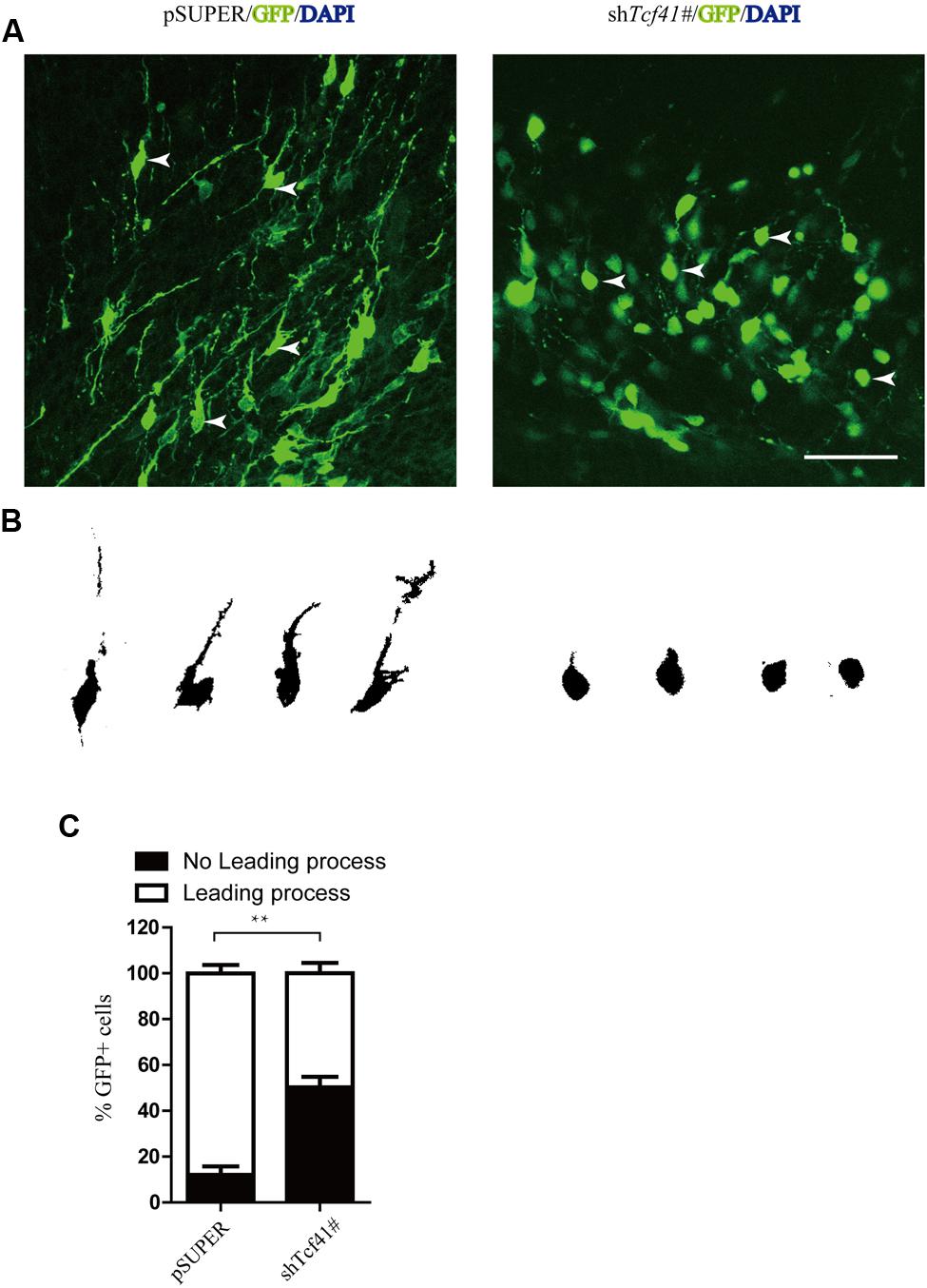
FIGURE 3. Knockdown of Tcf4 impairs leading process formation and neuronal differentiation. (A) Coronal sections around the IZ from brains electroporated at E14.5. Tcf4 shRNA inhibits the formation of leading processes at E17.5. Scale bar, 50 mm. (B) Tracings of representative transfected neurons from (A). (C) Quantification of the experiment shown in (A) (n = 3). Scale bars, 50 μm. Data are shown as the mean ± SEM. n.s., not significant; ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001; CP, cortical plate; VZ, ventricular zone; IZ, intermediate zone.
We also investigate whether Tcf4 knockdown might arrest neuronal migration by altering radial glia organization, cell division or cell survival. After 3 days of electroporation, we stained slices with Nestin to indicate radial glia organization, Ki67 to show cell division, and cleaved caspas3 to mark cell survival. There were no significant differences between control and Tcf4 knockdown slices (Supplementary Figure S2A–E). Tcf4 knockdown thus does not affect radial glia organization, cell division or cell survival.
Tcf4 Deficient Neurons Negatively Regulate Bmp7
Knockdown of human TCF4 is known to affect Bmp7 (Forrest et al., 2013). Here, we found that knockdown of Tcf4 upregulated Bmp7 expression in mouse progenitor cells (Figures 4C,D and Supplementary Figure S3). We also found that the Bmp7 expression profile in the developing mouse brain negatively correlates with Tcf4 (Figures 1B and 4A,B). We next identified a putative regulatory region of 1 kb upstream of the transcriptional start site of Bmp7 which contained six E-box (5′-CANNTG-3′) motifs that are known binding sites of Tcf4. Binding of Tcf4 to these motifs was tested by chromatin immunoprecipitation (ChIP), followed by quantitative real-time PCR using primer pairs (R1–R5) that specifically detected binding to these six motifs (R5 contains two E-box motifs). Figure 4F shows that Tcf4 binds on -201–49 bp of the Bmp7 promoter (R4 and R5). In addition, we found an enrichment of precipitated DNA of more than 5-fold using a Tcf4-specific antibody compared with an immunoglobulin G (IgG) control antibody (Figure 4E). Furthermore, this 1 kb regulatory sequence was tested for transcriptional activity by luciferase assays, yielding a repression of 50.5% (Figure 4G). This indicates that this sequence conveys functional repression through Tcf4 binding. Thus, Tcf4 binds to the Bmp7 promoter and negatively regulates Bmp7.
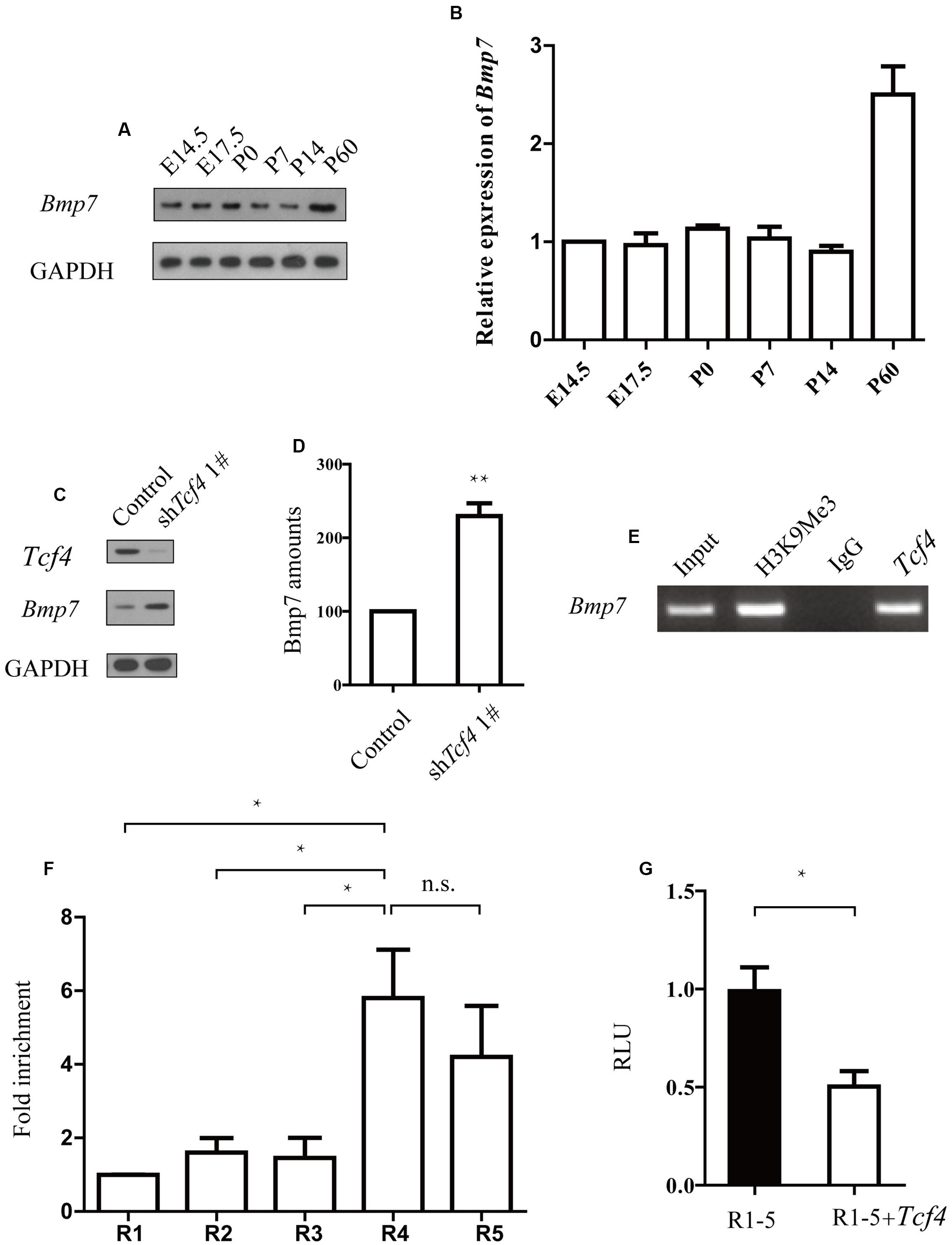
FIGURE 4. TCf4 binds to the Bmp7 promoter and negatively regulates Bmp7 expression. (A,B) Immunoblotting reveals Bmp7 protein levels in the cerebral cortex during development (n = 3). (C,D) Knockdown of Tcf4 increased Bmp7 levels in primary dissociated cortical neurons. Western blot analysis of the protein extracts from dissociated cortical neurons infected with control and Tcf4-shRNA lentivirus (n = 5). (E,F) ChIP analysis using a Tcf4 antibody and E14.5 cortical tissue detects Tcf4 binding on -201 bp to +49 bp of the Bmp7 promoter (R4 and R5) (n = 3). (G) Luciferase assays in HEK293 cells transfected with Tcf4 or a control vector show repression of luciferase activity of the R1–R5 reporter construct by Tcf4 (n = 3) 2 days after transfection. Data are shown as the mean ± SEM. n.s., not significant; ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001; Student’s t-test or one-way ANOVA, followed by an LSD post hoc test.
Bmp7 is an Important Downstream Effector of Tcf4 Required for Neuronal Migration
Electroporation of Bmp7 cDNA at E14.5 resulted in migration defects of cortical neurons similar to those observed in Tcf4-depleted neurons at E17.5 (Figures 5A,B). In addition, the proportion of neurons without leading processes was significantly increased upon overexpression of Bmp7 in comparison to a control vector in IZ (Figures 5C,D). Finally, we performed rescue experiments by introducing Bmp7 shRNA constructs into Tcf4-deficient cells. Figures 6A,B show that knockdown of Bmp7 partially rescued neuronal migration defects in Tcf4-deficient neurons. Altogether, our data provide evidence that Bmp7 is an important downstream effector of Tcf4, regulating radial migration of cortical neurons.
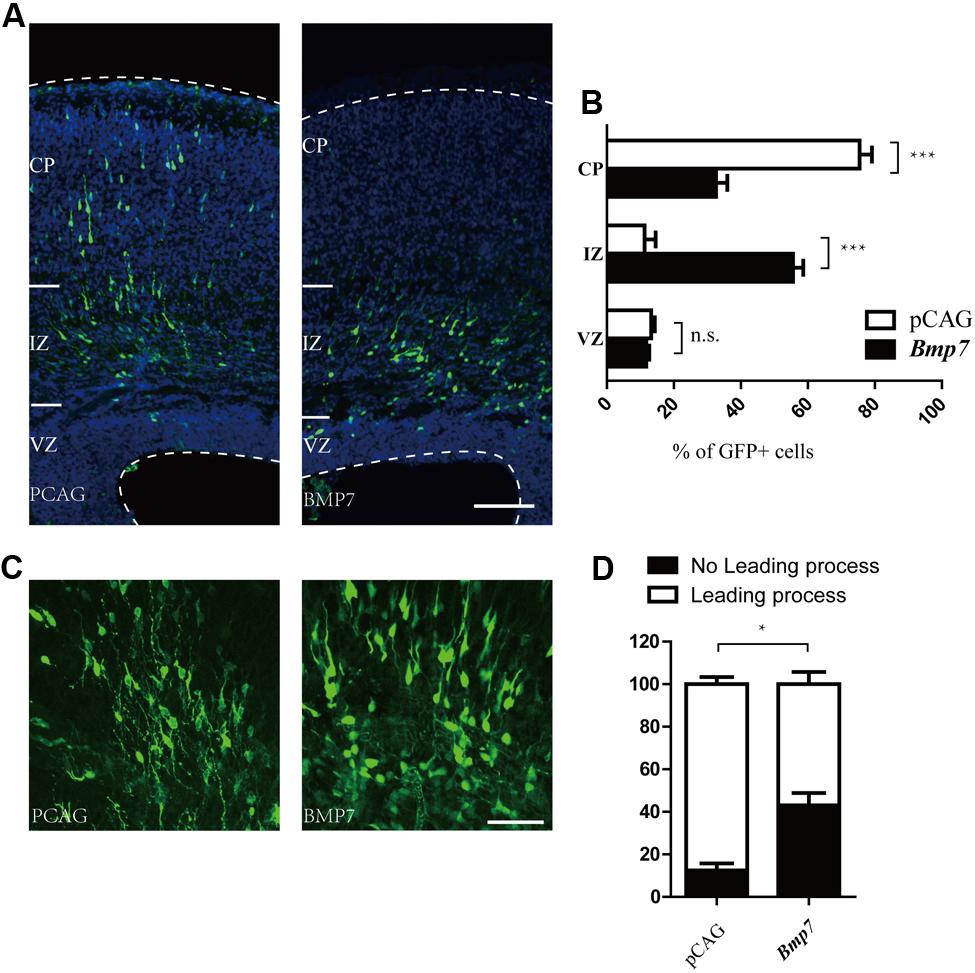
FIGURE 5. Bmp7 regulates neuronal migration to the cortical plate. (A,B) Results of in utero electroporation of E14.5 mouse embryos with GFP plasmids together with pCAG vector or pCAG-Bmp7. Representative coronal brain sections at E17.5 were stained with antibodies to GFP (green), and counterstained with DAPI, a nuclear marker (blue). The distribution of GFP-positive neurons is quantified (>600 neurons from three mice). Scale bar, 100 μm. (C) Coronal sections from brains electroporated at E14.5. Bmp7 overexpression inhibits the formation of leading processes at E17.5. Scale bar, 50 μm. (D) Quantification of the experiment shown in (C) (n = 3). Data are shown as the mean ± SEM. n.s., not significant; ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001; Student’s t-test or two-way ANOVA followed by a Bonferroni post hoc test. CP, cortical plate; VZ, ventricular zone; IZ, intermediate zone.
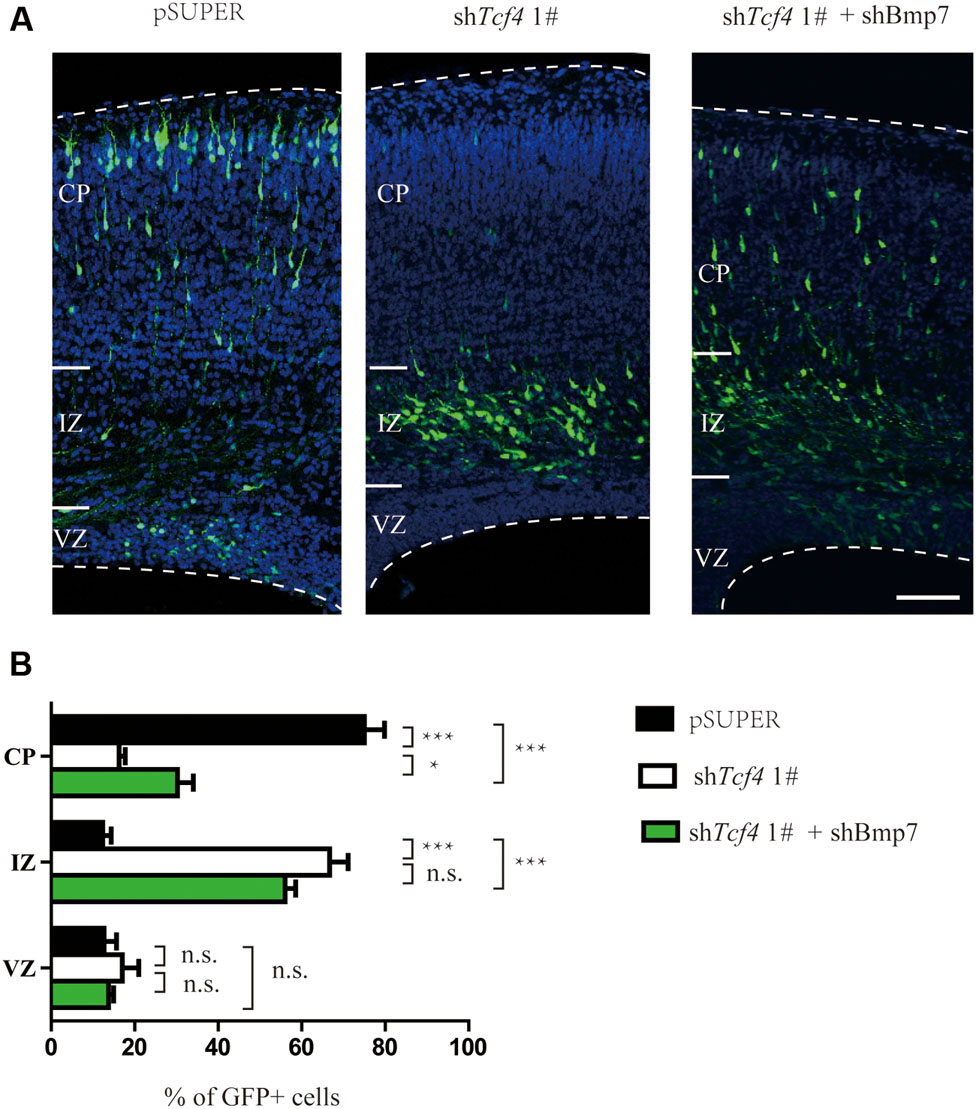
FIGURE 6. Bmp7 is an important downstream effector of Tcf4 required for neuronal migration. (A,B) Bmp7 shRNA can partially rescue the radial migration defect caused by in utero electroporation (E14.5-P0) with shTcf4 (GFP-positive). The number of GFP-positive neurons in each cortical zone is quantified (>600 neurons in three mice). Scale bar, 100 μm. Data are shown as the mean ± SEM. n.s., not significant; ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001; two-way ANOVA followed by a Bonferroni post hoc test. CP, cortical plate; VZ, ventricular zone; IZ, intermediate zone.
Discussion
Defects in neuronal cell migration in the cerebral cortex can lead to mental retardation, schizophrenia, and epilepsy (Kerjan and Gleeson, 2007; Walsh and Engle, 2010). The human TCF4 gene is implicated in susceptibility to schizophrenia and TCF4 haploinsufficiency is the cause of the Pitt-Hopkins mental retardation syndrome. However, the in vivo role of TCF4 in cortical development has remained unclear. Emerging evidence suggests that abnormalities in neuronal positioning are among the underlying causes contributing to the clinical symptoms of these diseases (Moffat et al., 2015). In support of this idea, we show that knockdown of Tcf4 impairs neuronal migration. The current demonstration opens a new avenue for research on the function of Tcf4.
Our data demonstrate that knockdown of Tcf4 impairs leading process growth. The precise mechanism of leading process is still unknown. The formation of proximal cytoplasmic dilation in the leading process (PCDLP) of migratory neocortical neurons is crucial for somal translocation and neuronal migration (Yang et al., 2012). Interestingly, a large portion of Tcf4 knock down cells lack the characteristic proximal cytoplasmic dilation presented in most of control neurons (Figure 3A). These data suggest that the loss of Tcf4 may affect PCDLP and thus impair leading process growth and neuronal migration.
Our data do not exclude the possibility that Tcf4 knockdown may affect other processes of neuronal migration, such as neuronal progenitor cell differentiation (Fischer et al., 2014). It is reported that Tcf4 interacts with Math1 to regulate differentiation of a specific subset of neuronal progenitors (Flora et al., 2007). Compatible with this, our data demonstrate that knockdown of Tcf4 impairs progenitor cell differentiation (Supplementary Figure S4). However, at E17.5, most of the Tcf4 depleted neurons leave VZ/SVZ (Supplementary Figure S5). Two days later, these neurons still had not migrated into the CP zone (data not shown). These data indicate that, at least for the neurons which have left VZ/SVZ, differentiation deficiency is not the reason why they cannot migrate into the CP zone.
Here, we show that Tcf4 knockdown upregulates Bmp7. We found that Tcf4 negatively regulates Bmp7, which conflicts with a previous report (Forrest et al., 2013). The reason might be the differences between 293T cells and mouse neurons (Segklia et al., 2012; Forrest et al., 2013). Our finding is consistent with previous research showing that BMP7 affects radial neuronal migration (Ortega and Alcantara, 2010). However, this does not exclude the possibility that additional downstream targets of TCF4 are involved in this process. It has been reported previously that knockdown of TCF4 affects multiple signaling pathways including NOTCH1 and NEUROG2 (Forrest et al., 2013). NOTCH1 has been shown to interact with Reelin signaling and regulate neuronal migration in the cerebral cortex (Hashimoto-Torii et al., 2008). Neurog2 has been found to control two waves of neuronal differentiation in the piriform cortex (Dixit et al., 2014). Thus, it remains to be addressed whether TCF4 transduces other signaling networks to regulate migratory behavior of neurons.
Materials and Methods
Plasmid Constructions
The shRNA target sequence of shTcf41# was 5′-GAACGGAGGATGGCCAATAAT-3′. shTcf42# was GGTCAAGATCTAGCAATAACG. α2-shRNA-2 was 5′-ACATATGCCAGTCCTGAAA-3′. Bmp7 was 5′-TCCATCTCCGTAGTATCCG-3′. shRNA sequences were designed and subcloned into pSUPER plasmid. The cDNA of mouse Tcf4 and shRNA-resistant constructs of mutants of Tcf4 were generated with the QuikChange mutagenesis kit (Stratagene) were cloned into pCAG-IRES-EGFP plasmid.
Cell Culture
For neurosphere cultures, single dissociated cortical progenitor cells (E12.5) were cultured in serum-free DMEM medium with 20 ng/mL FGF2 and EGF (Invitrogen) for 7 DIV. To subclone, neurospheres were collected and gently dissociated using Accutase (Gibco) for 20 min at 37°C. Cells were replated at 100 cells per ul for each condition. The differentiation media was made up of low glucose DMEM with penicillin-streptomycin-glutamine, 2% B27 supplement, and 1% fetal bovine serum (Invitrogen).
Real-Time PCR
Total mRNAs of the neocortex of E14.5, E17.5, P0, P7, P14, P28, and P60 mice were extracted with TRIzol reagent(Invitrogen). Super-Script II reverse transcriptase (Invitrogen) was used for reverse transcription to produce complementary DNAs (cDNAs). Real-time PCR was performed with the KAPA SYBR FAST qPCR Kits (Kapa Biosystems) and on a Roche LC96 apparatus. Primer pairs are, forward, 5′-TCTTCTCTCAGCCAACAGACAC-3′ and reverse, 5′-TTCAAGTCAGGGGAAGTTGC-3′.
In situ Hybridization
In situ hybridization on the brain sections was performed with digoxigenin-labeled antisense riboprobes. Full length cDNA of Tcf4 was amplified with PCR primers and cloned into pGEM-T vector (Promega) to generate antisense and sense probes for Tcf4. The digoxigenin-labeled antisense and sense riboprobes were synthesized by in vitro transcription with DIG RNA Labeling Mix (Roche). Mice were perfused with 4% paraformaldehyde (PFA) and fixed overnight in 4% PFA at 4°C. Fixed brains were cryoprotected overnight in 30% sucrose/ phosphate buffered saline (PBS) at 4°C and mounted in OCT compound and sectioned coronally (20 mm) with a cryostat (Leica). In situ hybridization was performed as described previously (Wu et al., 2012). Briefly, brain sections were hybridized for 18 h at 65°C. The hybridization signal was detected with an alkaline phosphatase-coupled antibody (1:2,000) against digoxigenin, as well as nitro blue tetrazolium and 5-bromo-4-chloro-3-indolyl phosphate as color reaction substrates.
Immunostaining
For immunohistochemistry, brain sections were washed in PBS for three times, and blocked in PBS supplemented with 5% BSA and 0.1% Triton X-100 for 30 min at room temperature. Thereafter, the brain sections were incubated with primary antibodies against Cux1 (1:50, Santa Cruz), Ki67 (1:1,000, Invitrogen), Cleaved Caspase-3 (1:1,000, Cell Signaling Technology), Nestin (1:200, Sigma), Pax6(1:100, Millipore), and GFP(1:1000, Molecular Probes), overnight at 4°C. After washing, sections were incubated with the correspondent secondary antibodies for 2 h at room temperature and then counterstained with DAPI (1:2,000, Sigma) for 15 min at room temperature before coverslipping.
Immunoblotting
Tissues dissected from the mouse brains and cultured cells were lysed in RIPA buffer [20 mM Tris-HCl (pH = 7.5), 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% NP-40, 1% sodium deoxycholate, 1 mM PMSF, 10 mg/ml aprotinin, 1 mg/ml pepstatin A and 1 mg/ml leupeptin]. The equivalent denatured samples were subjected to SDS-PAGE, transferred and probed with antibodies against Tcf4 (1:1,000, proteintech), GAPDH (1:4,000, Abcam). Bmp7 (1:1000, Proteintech), Smad1 (1:1000, Proteintech), phosphorylated Smad1 (1:1000, Abcam), Neurogenin2 (1:1000, Abcam), and FLAG (1:1000, sigma).
In utero Electroporation
E14.5 ICR-strain mice were used for in utero electroporation as described previously (Ip et al., 2012). Briefly, E14.5 pregnant mice were anesthetized with 0.7% pentobarbital sodium. The midline laparotomy was performed after the cleaning of the abdomen and the uterus were taken out. DNA plasmids (1–2 μl) of high concentration with 0.05% Fast Green (Sigma) were injected into the lateral ventricle through a polished micropipette. pSUPER shRNA was mixed with pCAG-IRES vector expressing GFP at 1:1 ratio (1 μg:1 μg) with a final concentration of 2 μg/μl. In rescue experiments, pSUPER shRNA was mixed with indicated pCAG-IRES expressing constructs at a 1:2 ratio (1 μg:2 μg) with a final concentration of 3 μg/μl. Square electric pulses were delivered at a rate of one pulse per second to embryos through the uterus by holding them with forceps-type electrodes, while the uterus was kept wet by dropping saline (prewarmed at 37°C) between the electrodes. Five electrical pulses (33V, 50 ms, 1s interval for mice) were applied across the uterine wall using electroporator (ECM-830 BTX). The uterine horns were then replaced in the abdominal cavity and the abdomen wall and skin were sutured using surgical needle and thread.
CHIP Assay
ChIPs were performed as previously described (Chen et al., 2012), using anti-H3K9Me3 (Abcam), anti-TCF4 (Abcam), anti-IgG (invitrogen), DNA released from the precipitated complexes was amplified by PCR using sequence-specific primers. Primer pairs used in Figure 5C are, Bmp7 forward GATCGGAAAGGGGTTTGTTG, reverse ACCCGAGGTCACTTGCTG; β-actin forward, AAATGCTGCACTGTGCGGCGAA, reverse TGCTCGCGGGCGGACGCGGTCTCGG.
Luciferase Assay
The 1 kb region in the promoter of Bmp7 was cloned into the PGL-3 basic Vector (Promega). This construct was transfected into HEK293 cells with and without pCAG-Tcf4 using Lipofectamine 2000 in accordance with the manufacturer’s instructions (Invitrogen). Renilla was co-transfected in each well as a transfection control. Supernatant from transfected cells was analyzed 48 h after transfection. Luciferase assays were performed using the Dual Luminesence Reporter Assay system (Promega) in accordance with the manufacturer’s instructions and BioTek’s Synergy H1. The values are reported as the mean ratio of luminescence intensity (RLU) of firefly over Renilla. Values were collected from three independent experiments performed with at least three replicates perexperiment.
Statistical Analysis
All date are presented as the mean ± SEM. Statistical significance was calculated using an unpaired Student’s t-test, one-way ANOVA, or two-way ANOVA. Differences were considered significant at p < 0.05. Quantification of neuronal migration was estimated by recording GFP positive neurons in distinct regions of the cerebral cortices (CP, IZ, and VZ). Two-way ANOVA followed by a Bonferroni post hoc test was used. More than 600 neurons GFP+ neurons from three brains were analyzed in each group. Quantification of morphology of neurons was estimated by recording percentage GFP posictive of neurons with or without leading process. Student’s t-test. More than 100 GFP+ neurons from three brains were examined in each group.
Ethics Statement
All methods were carried out in accordance with the approved guidelines.
All protocols was approved by the Animal Care and Use Committee of Peking University Health Science Center.
Author Contributions
TC and QW wrote the main manuscript text and prepared all figures. YZ, TL, and WY prepared plasmids. DZ supervised the work. All authors reviewed the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work is supported by grants from the National Natural Science Foundation of China (81401111) Beijing Natural Science Foundation (7144259) and Specialized Research Fund for the Doctoral Program of Higher Education (20130001120115).
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fnmol.2016.00094
FIGURE S1 |(A–C) In situ hybridization shows Tcf4 expression in the developing cerebral cortex of mice. Scale bars, 200 μm.
FIGURE S2 |(A–E) In utero electroporation at E14.5 with shTcf4 or pSUPER as indicated. The results were quantified by counting the number of stained cells in a constant area of each section, and averaged across sections from at least three different embryos for each antibody. Immunostaining at E17.5 for Nestin, cleaved Caspase 3 or Ki67.Scale bars, 100 μm. Data are shown as the mean ± SEM. n.s., not significant; *p < 0.05, **p < 0.01, and ***p < 0.001; CP, cortical plate; VZ, ventricular zone; IZ, intermediate zone.
FIGURE S3 |(A) Immunoblot analysis in HEK293T cells shows that Bmp7-specific shRNA, shBmp7 is sufficient to knockdown Bmp7. Cells were co-transfected with Bmp7-FLAG and individual shRNA and total cell lysates were prepared for immunoblotting 48 h after transfection. (B) Tcf4 knockdown increased Bmp7 protein levels in primary neuronal progenitor cells. Western blot analysis of the protein extracts from progenitor cells infected with control and Tcf4-shRNA lentivirus.
FIGURE S4 |(A,B) Immunocytochemistry for Nestin (red) on control and shTcf4 neurosphere cultures after 5 days under differentiation conditions. Scale bars, 100 μm. (B) Quantification of Nestin positive cells in (A). Data are shown as the mean ± SEM. n.s., not significant; *p < 0.05, **p < 0.01, and ***p < 0.001; Student’s t-test or one-way ANOVA, followed by an LSD post hoc test.
FIGURE S5 |(A,B) In utero electroporation at E14.5 with shTcf4 as indicated. Immunostaining at E17.5 for Pax6 or Tbr2 (markers for VZ). VZ, ventricular zone.
References
Aberg, K. A., Liu, Y., Bukszár, J., McClay, J. L., Khachane, A. N., Andreassen, O. A., et al. (2013). A comprehensive family-based replication study of schizophrenia genes. JAMA Psychiatry 70, 573–581. doi: 10.1001/jamapsychiatry.2013.288
Amiel, J., Rio, M., de, Pontual L, Redon, R., Malan, V., Boddaert, N., et al. (2007). Mutations in TCF4, encoding a class I basic helix-loop-helix transcription factor, are responsible for Pitt-Hopkins syndrome, a severe epileptic encephalopathy associated with autonomic dysfunction. Am. J. Hum. Genet. 80, 988–993. doi: 10.1086/515582
Blake, D. J., Forrest, M., Chapman, R. M., Tinsley, C. L., O’Donovan, M. C., and Owen, M. J. (2010). TCF4, schizophrenia, and Pitt-Hopkins Syndrome. Schizophr. Bull. 36, 443–447. doi: 10.1093/schbul/sbq035
Brockschmidt, A., Todt, U., Ryu, S., Hoischen, A., Landwehr, C., Birnbaum, S., et al. (2007). Severe mental retardation with breathing abnormalities (Pitt-Hopkins syndrome) is caused by haploinsufficiency of the neuronal bHLH transcription factor TCF4. Hum. Mol. Genet. 16, 1488–1494. doi: 10.1093/hmg/ddm099
Chen, T., Xue, L., Niu, J., Ma, L., Li, N., Cao, X., et al. (2012). The retinoblastoma protein selectively represses E2F1 targets via a TAAC DNA element during cellular senescence. J. Biol. Chem. 287, 37540–37541.
Dixit, R., Wilkinson, G., Cancino, G. I., Shaker, T., Adnani, L., Li, S., et al. (2014). Neurog1 and Neurog2 control two waves of neuronal differentiation in the piriform cortex. J. Neurosci. 34, 539–553. doi: 10.1523/JNEUROSCI.0614-13.2014
Einarson, M. B., and Chao, M. V. (1995). Regulation of Id1 and its association with basic helix-loop-helix proteins during nerve growth factor-induced differentiation of PC12 cells. Mol. Cell Biol. 15, 4175–4183. doi: 10.1128/MCB.15.8.4175
Fischer, B., Azim, K., Hurtado-Chong, A., Ramelli, S., Fernández, M., and Raineteau, O. (2014). E-proteins orchestrate the progression of neural stem cell differentiation in the postnatal forebrain. Neural Dev. 9:23. doi: 10.1186/1749-8104-9-23
Flora, A., Garcia, J. J., Thaller, C., and Zoghbi, H. Y. (2007). The E-protein Tcf4 interacts with Math1 to regulate differentiation of a specific subset of neuronal progenitors. Proc. Natl. Acad. Sci. U.S.A. 104, 15382–15387. doi: 10.1073/pnas.0707456104
Forrest, M. P., Waite, A. J., Martin-Rendon, E., and Blake, D. J. (2013). Knockdown of human TCF4 affects multiple signaling pathways involved in cell survival, epithelial to mesenchymal transition and neuronal differentiation. PLoS ONE 8:e73169. doi: 10.1371/journal.pone.0073169
Hashimoto-Torii, K., Torii, M., Sarkisian, M. R., Bartley, C. M., Shen, J., Radtke, F., et al. (2008). Interaction between Reelin and Notch signaling regulates neuronal migration in the cerebral cortex. Neuron 60, 273–284. doi: 10.1016/j.neuron.2008.09.026
Ip, J. P., Shi, L., Chen, Y., Itoh, Y., Fu, W.-Y., Betz, A., et al. (2012). alpha2-chimaerin controls neuronal migration and functioning of the cerebral cortex through CRMP-2. Nat. Neurosci. 15, 39–47. doi: 10.1038/nn.2972
Kerjan, G., and Gleeson, J. G. (2007). Genetic mechanisms underlying abnormal neuronal migration in classical lissencephaly. Trends Genet. 23, 623–630. doi: 10.1016/j.tig.2007.09.003
Kurian, S. M., Le-Niculescu, H., Patel, S. D., Bertram, D., Davis, J., Dike, C., et al. (2011). Identification of blood biomarkers for psychosis using convergent functional genomics. Mol. Psychiatry 16, 37–58. doi: 10.1038/mp.2009.117
Lennertz, L., Rujescu, D., Wagner, M., Frommann, I., Schulze-Rauschenbach, S., Schuhmacher, A., et al. (2011). Novel schizophrenia risk gene TCF4 influences verbal learning and memory functioning in schizophrenia patients. Neuropsychobiology 63, 131–136. doi: 10.1159/000317844
Li, T., Li, Z., Chen, P., Zhao, Q., Wang, T., Huang, K., et al. (2010). Common variants in major histocompatibility complex region and TCF4 gene are significantly associated with schizophrenia in Han Chinese. Biol. Psychiatry 68, 671–673. doi: 10.1016/j.biopsych.2010.06.014
Moffat, J. J., Ka, M., Jung, E. M., and Kim, W. Y. (2015). Genes and brain malformations associated with abnormal neuron positioning. Mol. Brain 8:72. doi: 10.1186/s13041-015-0164-4
Mudge, J., Miller, N. A., Khrebtukova, I., Lindquist, I. E., May, G. D., Huntley, J. J., et al. (2008). Genomic convergence analysis of schizophrenia: mRNA sequencing reveals altered synaptic vesicular transport in post-mortem cerebellum. PLoS ONE 3:e3625. doi: 10.1371/journal.pone.0003625
Nagasawa, M., Schmidlin, H., Hazekamp, M. G., Schotte, R., and Blom, B. (2008). Development of human plasmacytoid dendritic cells depends on the combined action of the basic helix-loop-helix factor E2-2 and the Ets factor Spi-B. Eur. J. Immunol. 38, 2389–2400. doi: 10.1002/eji.200838470
Ortega, J. A., and Alcantara, S. (2010). BDNF/MAPK/ERK-induced BMP7 expression in the developing cerebral cortex induces premature radial glia differentiation and impairs neuronal migration. Cereb. Cortex. 20, 2132–2144. doi: 10.1093/cercor/bhp275
Quednow, B. B., Ettinger, U., Mössner, R., Rujescu, D., Giegling, I., Collier, D. A., et al. (2011). The schizophrenia risk allele C of the TCF4 rs9960767 polymorphism disrupts sensorimotor gating in schizophrenia spectrum and healthy volunteers. J. Neurosci. 31, 6684–6691. doi: 10.1523/JNEUROSCI.0526-11.2011
Schmidt-Edelkraut, U., Daniel, G., Hoffmann, A., and Spengler, D. (2014). Zac1 regulates cell cycle arrest in neuronal progenitors via Tcf4. Mol. Cell Biol. 34, 1020–1030. doi: 10.1128/MCB.01195-13
Segklia, A., Seuntjens, E., Elkouris, M., Tsalavos, S., Stappers, E., Mitsiadis, T. A., et al. (2012). Bmp7 regulates the survival, proliferation, and neurogenic properties of neural progenitor cells during corticogenesis in the mouse. PLoS ONE 7:e34088. doi: 10.1371/journal.pone.0034088
Walsh, C. A., and Engle, E. C. (2010). Allelic diversity in human developmental neurogenetics: insights into biology and disease. Neuron 68, 245–253. doi: 10.1016/j.neuron.2010.09.042
Wu, Q. F., Yang, L., Li, S., Wang, Q., Yuan, X. B., Gao, X., et al. (2012). Fibroblast growth factor 13 is a microtubule-stabilizing protein regulating neuronal polarization and migration. Cell 149, 1549–1564. doi: 10.1016/j.cell.2012.04.046
Yang, T., Sun, Y., Zhang, F., Zhu, Y., Shi, L., Li, H., et al. (2012). POSH localizes activated Rac1 to control the formation of cytoplasmic dilation of the leading process and neuronal migration. Cell Rep. 2, 640–651. doi: 10.1016/j.celrep.2012.08.007
Keywords: schizophrenia, brain development, neuronal migration, TCF4, Bmp7
Citation: Chen T, Wu Q, Zhang Y, Lu T, Yue W and Zhang D (2016) Tcf4 Controls Neuronal Migration of the Cerebral Cortex through Regulation of Bmp7. Front. Mol. Neurosci. 9:94. doi: 10.3389/fnmol.2016.00094
Received: 24 May 2016; Accepted: 20 September 2016;
Published: 03 October 2016.
Edited by:
Robert W. Burgess, The Jackson Laboratory, USAReviewed by:
Christian Gonzalez-Billault, University of Chile, ChileHansen Wang, University of Toronto, Canada
Copyright © 2016 Chen, Wu, Zhang, Lu, Yue and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tianda Chen, dGlhbmRhY2hlbkBiam11LmVkdS5jbg== Dai Zhang, ZGFpemhhbmdAYmptdS5lZHUuY24=
†These authors have contributed equally to this work.