 Miguel Mompeán
Miguel Mompeán Marco Baralle
Marco Baralle Emanuele Buratti
Emanuele Buratti Douglas V. Laurents
Douglas V. Laurents- 1Instituto de Química Física “Rocasolano”, Consejo Superior de Investigaciones Científicas (CSIC), Madrid, Spain
- 2International Centre for Genetic Engineering and Biotechnology (ICGEB), Trieste, Italy
TDP-43 is an essential RNA-binding protein forming aggregates in almost all cases of sporadic amyotrophic lateral sclerosis (ALS) and many cases of frontotemporal lobar dementia (FTLD) and other neurodegenerative diseases. TDP-43 consists of a folded N-terminal domain with a singular structure, two RRM RNA-binding domains, and a long disordered C-terminal region which plays roles in functional RNA regulatory assemblies as well as pernicious aggregation. Evidence from pathological mutations and seeding experiments strongly suggest that TDP-43 aggregates are pathologically relevant through toxic gain-of-harmful-function and/or harmful loss-of-native-function mechanisms. Recent, but not early, microscopy studies and the ability of TDP-43 aggregates to resist harsh treatment and to seed new pathological aggregates in vitro and in cells strongly suggest that TDP-43 aggregates have a self-templating, amyloid-like structure. Based on the importance of the Gln/Asn-rich 341–367 residue segment for efficient aggregation of endogenous TDP-43 when presented as a 12X-repeat and extensive spectroscopic and computational experiments, we recently proposed that this segment adopts a beta-hairpin structure that assembles in a parallel with a beta-turn configuration to form an amyloid-like structure. Here, we propose that this conformer is stabilized by an especially strong class of hypercooperative hydrogen bonding unique to Gln and Asn sidechains. The clinical existence of this conformer is supported by very recent LC-MS/MS characterization of TDP-43 from ex vivo aggregates, which show that residues 341–367 were protected in vivo from Ser phosphorylation, Gln/Asn deamidation and Met oxidation. Its distinct pattern of SDS-PAGE bands allows us to link this conformer to the exceptionally stable seed of the Type A TDP-43 proteinopathy.
Introduction
Amyotrophic lateral sclerosis (ALS) is a rare, paralyzing mortal disease which involves the death of motor neurons, leading to progressive muscle atrophy and death (Kiernan et al., 2011). In 1991, ubiquitinated aggregates were detected in the motor neurons of patients affected by ALS (Leigh et al., 1991). In 2006, these aggregates were identified as ubiquitinated and polyphosphorylated TDP-43 (Arai et al., 2006; Neumann et al., 2006), an essential, well-conserved protein involved in RNA splicing, transport and translation regulation (Buratti and Baralle, 2012). C-terminal fragments of 35 kD and 25 kD were also observed. Soon afterwards, many mutations in TDP-43 genetically linked to ALS and frontotemporal lobar dementia (FTLD) were discovered, as recently reviewed (Buratti, 2016). These mutations and the observation of TDP-43 aggregates in the motor neurons of practically all ALS and many FTLD patients are evidence of TDP-43’s central role in this spectrum of neuromuscular/dementia pathologies (Kiernan et al., 2011). From the point of view of the protein architecture, it is fascinating that almost all of these mutations are located in the C-terminal region (Pesiridis et al., 2009). In concert with mutational, functional and dysfunctional results from in vitro and in vivo studies, an advanced knowledge of TDP-43’s structure is key to understand its roles in physiological and pathological processes, which are summarized in Figure 1A, that could eventually guide the development of prophylactics and treatments.
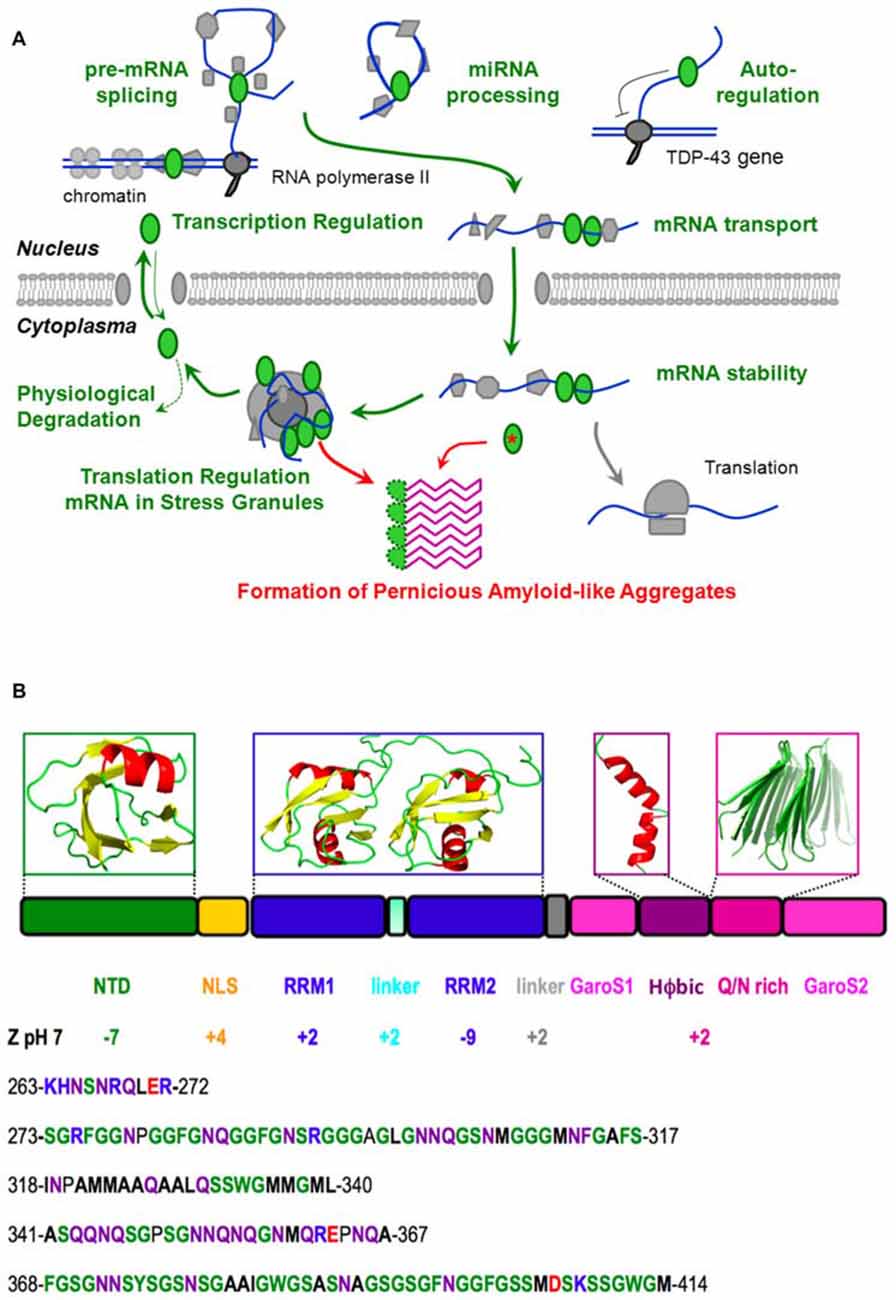
Figure 1. Native function, pathological aggregation and structure of TDP-43. (A) TDP-43 (green ellipses) performs vital physiological roles (green) in pre-mRNA splicing, microRNA processing, transcriptional regulation and post-transcriptional autoregulation of its own expression, mRNA transport, stability and translational regulation (through stress granule formation) before being degraded or shuttling back to the nucleus. However, the chance formation of aberrant conformers, which could be facilitated by mutation (red asterisk), aberrant cleavage, cytoplasmic mislocalization, aging and/or high local TDP-43 concentration in stress granules, may lead to the formation of a pathological amyloid-like aggregate. We propose that this aggregate consists of beta hairpins formed by residues 341–367 (purple) and is stabilized by hyper-stable inter-molecular hydrogen bonds. In this schematic diagram, DNA is represented by a double blue line, RNA is shown as a curved blue line and other proteins, the nuclear membrane and the ribosome are colored gray. (B) The domain composition and 3D structures of the N-terminal and RRM domains of TDP-43, as well as plausible, partially populated helical and quasi-amyloid conformers of the disordered C-terminal regions are shown. The net charge of the different domains, segments and the C-terminal region are indicated. The lower portion of the figure shows the sequence of the linker (residues 263–272), and the C-terminal subregions: G, S, aromatic-rich segment (GaroS1; 273–317), hydrophobic (318–340), Q/N-rich (341–367) and second GaroS region (368–414). Residues are colored blue (KHR), red (E, D), green (G, S, F, Y, W), purple (N, Q) or written in bold (aliphatics).
TDP-43 Contains Three Folded Domains and an Intrinsically Unfolded C-Terminal Region
Due to its intrinsic high-aggregation propensity, it has been impossible to solve the full structure of TDP-43. However, according to all the evidence gathered in recent years, TDP-43 consists of a folded N-terminal domain, two folded RRM domains and an intrinsically disordered C-terminal region (Figure 1B). The N-terminal domain has a singular tertiary structure, contains two negatively charged loops (Qin et al., 2014; Mompeán et al., 2016c) and is important for TDP-43’s physiological and pathological functions (Zhang et al., 2013; Budini et al., 2015; Sasaguri et al., 2016). In particular, the N-terminal domain is essential for the efficient capture of endogenous TDP-43 molecules into aggregates and induces a loss-of-function phenotype in cultured neuronal cells (Budini et al., 2015).
The RRM domains bind UG-rich segments of RNA and are essential for TDP-43’s physiological functions (Ayala et al., 2005; Lukavsky et al., 2013). Interestingly, both the N-terminal domain and the first RRM domain are stable (with TM’s close to 50°C; Kuo et al., 2009; Wang et al., 2013; Mompeán et al., 2016c), but the second RRM domain is much more stable (TM = >85°C; Kuo et al., 2009; Wang et al., 2013). Although the physiological relevance of these differences are unknown; here we speculatively propose that the second RRM domain’s high stability could aid the proper presentation of the nuclear export signal present within this domain (residues 238–250) or chaperone residues 246–255 which tend to form amyloid and hydrogels (Wang et al., 2013; Saini and Chauhan, 2014). Among the similarities and differences between the two domains involved in nucleic acids, RRM2 adopts an atypical RRM-fold with an additional beta-strand that could mediate RRM2-RRM2 protein interactions.
The preferred binding motif of these RRMs is represented by single-stranded UG-rich regions/repeats that are recognized with high affinity (Buratti and Baralle, 2011; Polymenidou et al., 2011; Tollervey et al., 2011). Both RRMs contribute to nucleic acid binding, with RRM1 specifically recognizing one GU repeat. RRM2 recognizes one UG repeat and binds to additional nucleotides nonspecifically. Thus, the emerging binding consensus sequence is 5′-N(N)GUGN(N)UGN-3′. A unique feature of the TDP-43—RNA complex is the reversed binding of the UG-rich RNA with the 5′end being bound to RRM1, rather than RRM2 (Lukavsky et al., 2013). This is in contrast to all other tandem RRMs characterized to date and might explain some peculiarities of TDP-43 functions. In fact, whilst TDP-43 binding to UG-rich RNA through RRM1 shows a very clear correlation between binding and inhibitory splicing function, this does not occur for RRM2 where alanine mutations in its recognition sites have only a small effect on TDP-43’s overall RNA binding affinity. Nevertheless, these interactions are functionally important and are required for the splicing function of TDP-43. Thus, the role of RRM2 could be to orient the UG-rich RNA for productive TDP-43 dimerization on pre-mRNA target sites.
Another remarkable feature of the second RRM domain is that it carries an overall charge of −9 at pH 7. In contrast, the first RRM domain has a net positive charge (Figure 1B). This difference might account in part for the second RRM domain’s weaker RNA binding as its negative charges could electrostatically repel longer RNA oligos. In this respect, it is interesting to note that the second RRM domain binds shorter RNA oligos better than longer ones (Kuo et al., 2009).
In contrast to the N-terminal and RRM domains, the C-terminal region is intrinsically disordered (Lim et al., 2016). It is remarkably poor in charged residues (Figure 1B), which is related to its meager solubility (Mompeán et al., 2016a). It can be divided into four sub-regions; namely a Gly, Aromatic, Ser-rich (GaroS) segment, a hydrophobic segment, a Gln/Asn (Q/N)-rich segment, and a second GaroS rich segment. The GaroS segments resemble the [G,S]Y[G,S] motifs of the intrinsically disordered region of “Fused in Sarcoma” (FUS), an RNA-binding protein that like TDP-43 is also aberrantly aggregated in ALS and FTLD pathologies. These motifs were proposed to endow TDP-43 and FUS with the ability to participate in RNA granules and form hydrogels (Kato et al., 2012). Although these segments do not resemble those of pathological amyloids, their ability to increase the local concentration of TDP-43 within RNA granules may boost the probability that the aberrant oligomers might form, along the lines of the model recently proposed for FUS (Patel et al., 2015), particularly in the context of some disease-related mutations (Lim et al., 2016).
These considerations raise several hypotheses for the origin of the pathology that involve a crucial role for interdomain contacts to eventually trigger the pathological aggregation process. In this respect, Zhang et al. (2013) have modeled the possible interaction between the N-terminal and C-terminal domains to regulate proper homodimerization of TDP-43 while Mackness et al. (2014) have investigated the different potential folding intermediates in the RRM1 and RRM2 domains. In addition, Wang et al. (2013) have shown that TDP-43 without the C-terminal region can form a homodimer primarily via N-terminal domain contacts.
Although the previously mentioned genetic evidence and these biochemical data strongly link TDP-43’s C-terminal region to pathology, many aspects of this issue still remain to be addressed. For example, there is still a very open debate regarding the amyloid vs. amorphous nature of these aggregates, their atomic level structure, and what interactions could stabilize them. In this Perspective, we shall put together diverse results from the literature to address these points and advance a working hypothesis that an amyloid-like conformer, composed of beta-hairpins formed by residues 341–357, is the relevant pernicious structure for TDP-43 Type A proteinopathies.
TDP-43 can Form Amyloid-Like Fibrils which Seem to be Pathologically Relevant
One important and long-standing issue is whether the TDP-43 aggregates seen in diseased tissues are amyloid-like or not.
First, it has to be acknowledged that early studies generally found that TDP-43 in cell inclusions from ex vivo tissues did not bind amyloid specific dyes like Thioflavin S or show much fibril formation when examined by immunostaining and optical microscopy (Cairns et al., 2007; Neumann et al., 2007). Furthermore, when TDP-43 is expressed in bacteria and purified from inclusion bodies (Capitini et al., 2014) or expressed in yeast (Johnson et al., 2008) no evidence for amyloid formation was detected. Moreover, TDP-43 produced from human cells or E. coli did not bind Thioflavin T or Congo Red, although it did form highly cytotoxic oligomers (Fang et al., 2014).
In contrast, several studies found that short peptides derived from TDP-43’s second RRM domain and C-terminal region adopt fibril structures that bind amyloid-specific dyes (Chen et al., 2010; Guo et al., 2011; Saini and Chauhan, 2011, 2014; Jiang et al., 2013; Mompeán et al., 2014; Sun et al., 2014; Zhu et al., 2014).
Finally, even middle-of-the-road positions have been described. For example, Wang et al. (2013) found that C-terminal truncated forms of TDP-43 can adopt well defined fibril structure which nonetheless does not bind amyloid specific dyes or antibodies.
Of course, in most cases these studies’ relevance for human pathology still needs to be demonstrated. It has to be taken in consideration, in fact, that TDP-43 is not naturally present in lower organisms and therefore its conformation could be altered by diverse in vivo environments. Likewise, the different context of isolated polypeptide segments vs. their presence within the complete protein has to be considered. Nonetheless, studies of ex vivo patient tissues using higher resolution immuno-gold labeling and electron microscopy did uncover solid evidence for amyloid-like fibrils formed by TDP-43 in ALS/FTLD affected neurons (Thorpe et al., 2008) as well as cells affected by other neurodegenerative diseases (Lin and Dickson, 2008). More recently, an optical microscopy study using Thioflavin S and optical microscopy has reported evidence for amyloid formation by TDP-43 in 30% of ALS patients (Robinson et al., 2013). Finally, upon treating tissues to remove background due to lipid autofluorescence, TDP-43 amyloid-like structures could be detected by Thioflavin S fluorescence in the relevant tissues of most patients with ALS or FTLD (Bigio et al., 2013). In conclusion, these results are good evidence that TDP-43 in patients can form amyloid-like fibrils that are pathologically relevant.
Hyperphosphorylation of TDP-43
One key hallmark of TDP-43 aggregates is hyperphosphorylation (Arai et al., 2006; Buratti and Baralle, 2012), and antibodies raised against specific phosphoserines enabled the identification of phosphorylated S409 and S410 as characteristic signatures of pathological TDP-43 aggregates (Neumann et al., 2009). These negatively charged phosphates seem to decrease the protein’s aggregation (Brady et al., 2011; Li et al., 2011) and phosphorylation appears to occur after aggregation in vivo as the cell attempts to solubilize and degrade the inclusions.
Additional phosphorylation sites were identified utilizing a set of specific antibodies (Hasegawa et al., 2008) and mass spectrometry (Kametani et al., 2009). Most phosphorylated residues (18 of 29) are in the C-terminal region, including S342, S347 and S350 in the Q/N-rich subregion. More recently, this laboratory has also analyzed TDP-43 aggregates from two ex vivo ALS brains (Kametani et al., 2016). Remarkably, they found 13 phosphorylation sites in one brain and 15 sites in the second. By comparing these results to those obtained using soluble TDP-43 (Kametani et al., 2009), it is clear that some serines; namely S342, S347, and S350, seem to be hidden from kinase and thus protected from phosphorylation in the context of the pathological inclusions. This observation is particularly important as it may give a clue about the location of the aggregation core.
The Formation of a Helix in the Hydrophobic C-Terminal Segment Could Modulate Toxic Aggregate Formation
The relevance of the hydrophobic and Q/N-rich segments to TDP-43 aggregation was demonstrated by Jiang et al. (2013) who observed that the C. elegans homolog of TDP-43, which lacks these segments, is much less prone to aggregation.
The hydrophobic stretch has a very weak tendency to form helix in aqueous solution, as evidenced by very small 1Hα, 13Cα conformational chemical shifts and a lack of 1HNi-1HNi+1,i+3,i+4 NOES (Mompeán et al., 2015); nonetheless, a significant amount of helix does form in the presence of TFE or DPC micelles (Jiang et al., 2013; Lim et al., 2016). These are important results as pathological amyloid formation is often characterized by a loss of native helix in favor of a gain in beta structure. In agreement with these findings, evidence for a helix to beta conformational conversion was found in this hydrophobic segment (Jiang et al., 2013).
Our own recent results (Budini et al., 2012; Mompeán et al., 2014, 2015) also point to the Q/N-rich segment as being very important for TDP-43 aggregation and amyloid formation. Moreover, the recent mass-spectrometry/proteolysis study of TDP-43 from ex vivo brains of two ALS patients provides insight into the relative importance of the hydrophobic and Q/N rich regions (Kametani et al., 2016). In this report, Kametani and co-workers found that residues 341–346 or 341–360 of the Q/N-rich segment were protected against proteolytic cleavage, deamidation and phosphorylation (in patient 1 and patient 2, respectively). Although more cases are needed to confirm whether this is indeed a general pathological feature, these data are consistent with these segments forming an highly stable amyloid-like structure, such as the one we have proposed (Mompeán et al., 2015), which protects them against cleavage and modification. In contrast, residues of the hydrophobic segment are heavily modified by Ser phosphorylation, Gln deamidation and Met oxidation, which suggests they are exposed in patient aggregates. This implies that the hydrophobic segment does not form an amyloid structure which would prevent these modifications.
The Q/N-Rich Segment of TDP-43’s C-Terminal Region can Quickly form a Stable, Amyloid-Like Structure
Regarding TDP-43 aggregation, the Q/N-rich segment is especially interesting because variants lacking it or substituting a polyAla segment do not aggregate (Budini et al., 2012). We have recently investigated this segment’s conformational transitions using CD/NMR spectroscopies and computational methods. Our studies have shown that the Q/N-rich segment is initially unfolded but over the course of several hours it converts into a structure rich in beta structure which binds thioflavin T (Mompeán et al., 2014). This segment, modeled as a beta hairpin, is stable in long molecular dynamics simulations. Additional studies, using conformational antibodies specific for amyloid, solid state NMR, X-ray diffraction and electron microscopic, confirmed the ability of the Q/N-rich segment of TDP-43 to adopt an amyloid-like structure, whose fibrils are broad and tend to associate laterally, like those of polyglutamine (Mompeán et al., 2015).
Using computational tools, we built up an amyloid-like structural model based on beta-hairpins composed of residues 341–357 of TDP-43 arranged in parallel and with a beta-turn topology. This arrangement was found to be stable in long MD simulations (Figure 2). By contrast, other configurations with consecutive beta hairpins arranged in an anti-parallel fashion or with beta-arc topology were unstable (not shown). Based on this model, the efficient aggregation of the 12X Q/N TDP-43 construct in vivo suggests that these 12 repetitions are likely to assemble into two stacked beta sheets, each containing six beta-strands or 12 beta strands that form the nascent, quasi-amyloid core. Residues 358–366 act as a linker to connect 341–357 beta-hairpins and allow proper cross-beta spine formation. This segment could connect the beta-hairpins within each sheet, and link the two sheets, allowing them to pack against each other (Figure 2B; Mompeán et al., 2015).
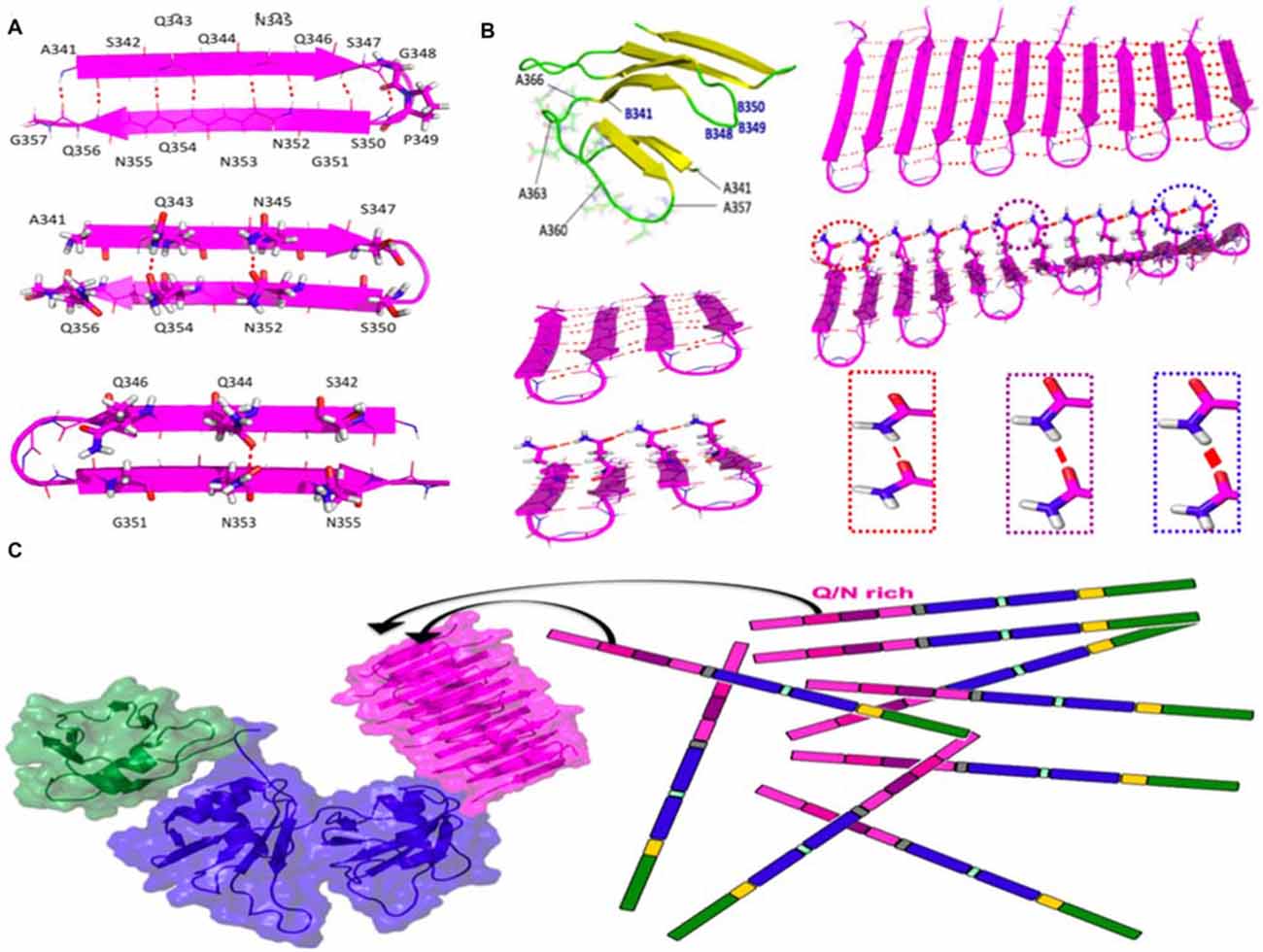
Figure 2. Structure and stabilizing H-bonds in the amyloid-like conformer of TDP-43 composed of residues 341–366. (A) Schematic diagram of the beta-hairpin adopted by residues 341–357 of TDP-43. (B) Residues 358–366 could act as a loop to link different beta-hairpins in the same beta-sheet or between different beta sheets (yellow strands and green loops). One of the two sheets of the 4X Q/N (4 hairpins) or of 12X Q/N (6 hairpins) are shown in magneta. Cooperative and hyperpolarization effects greatly fortify Q/N side chain H-bonds (red lines) and moderately strengthen backbone H-bonds (dashed red lines) in longer 12X Q/N assembles (right) but not the shorter 4X Q/N sheet (left). The thickness of the red lines reflects the strength of the H-bonds. (C) The formation of exceptionally stable H-bonds provides a free energy sink which drives the binding of endogenous TDP-43 molecules to the quasi-amyloid structure formed by the 12X Q/N TDP-43 construct. The color code of the domains matches that of the sequence diagram in Figure 1 and the structures were modeled according to Mompeán et al. (2014, 2015) for the Q/N segments (magenta), Mompeán et al. (2016c) for the NTD domain (green) and Lukavsky et al. (2013) for the RRM domains (blue).
Based on the apparent protection of residues 341–367 against Ser phosphorylation, Gln/Asn deamidation and Met oxidation in TDP-43 ex vivo aggregates, we therefore propose as a working hypothesis that our conformational model for this segment (Figure 2) may correspond to the pathological conformer present in the ex vivo aggregates of patient 2 described by Kametani et al. (2016). Indeed, based on the similarities of the characteristic bands in SDS-PAGE chromatograms previously described (Nonaka et al., 2013; Kametani et al., 2016), our conformer may represent the structural core of Type A TDP-43 proteinopathy, the most common class of TDP-43 maladies (Mackenzie et al., 2011). As TDP-43 Type A aggregates are remarkably more heat-stable as seeds than Type B or Type C aggregates (Nonaka et al., 2013), what remains to be addressed is the basis of this exceptional stability. Below, we present a model that can account for this.
Hyperpolarization-Mediated Stabilization of Gln and Asn Side Chain Hydrogen Bonds can Account for the Remarkable Stability of the Putative Amyloid Formed by the Q/N-Rich Segment of TDP-43
We have recently compared hydrogen bond cooperativity in a Q/N-rich amyloid, the paradigmatical GNNQQNY heptapeptide form the yeast prion Sup35, to the hydrophobic amyloid M35VGGVV40 from Aβ (Mompeán et al., 2016b). Whereas the backbone amide groups of both Q/N-rich and hydrophobic amyloids experience stabilization due to hyperpolarization, an additional and stronger type of hyperpolarization occurs in Asn and Gln side chain amides (Figure 2B). The strengthening due to hyperpolarization increases with the number of strands.
According to this working hypothesis, hydrogen bond hyperpolarization provides a very favorable free energy change for binding monomers to the quasi-amyloid structure formed by the Q/N-rich segment, thus permitting the TDP-43-12X Q/N construct to efficiently sponge up endogenous TDP-43 (Figure 2C). Hydrogen bond hypercooperativity can also explain how the Q/N-rich subregion enables TDP-43 to bind and co-aggregate with polyglutamine (Fuentealba et al., 2010). As a side note, this also provides a logical explanation for why TDP-43 12X Q/N repeats readily form a stable amyloid capable of sequestering endogenous TDP-43 whereas a stoichiometrically equivalent amount of Q/N repeats in the context of 2X Q/N or 4X Q/N constructs does not (Budini et al., 2012).
Recruitment of endogenous TDP-43 into aggregates within this cellular model can be tentatively attributed to two different processes. First, the tandem repetitions could form an amyloid fold that might sponge up and sequester nuclear TDP-43 through its Q/N segment. Second, heterodimerization via the NTDs of endogenous TDP-43 with TDP-43-12X Q/N would increase the local concentration of the Q/N rich segments and thereby favor their self-association (Romano et al., 2015). In the future, more complete structural models of TDP-43 could help determine whether these processes act antagonistically, independently or synergistically.
Recent studies strongly suggest that, once formed, pernicious conformers of TDP-43 could spread through the nervous system in a prion-like fashion (Nonaka et al., 2013; Smethurst et al., 2015). This issue will likely prove crucial for a better understanding of TDP-43 proteinopathies. In fact, the aggregates’ size, location, toxicity and their stability have been advanced as factors accounting for the efficiency of prion-like spreading by pernicious protein aggregates (Espargaró et al., 2016). In these conditions, the exceptional stability of our conformer/Type A could well be pertinent to provide a first structural framework for future studies aimed at further elucidating this issue.
Author Contributions
MM, MB, EB and DVL conceived the ideas, wrote the manuscript, critically revised it and approved the final version for publication. All four authors agree to be accountable for the contents.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by Grants SAF2013-49179-C2-2-R (DVL) and EU JPND AC14/00037 (DVL) and EU JPND RiModFTD, Italy, Ministero della Sanita’ (EB), and the Thierry Latran Foundation REHNPALS (EB).
References
Arai, T., Hasegawa, M., Akiyama, H., Ikeda, K., Nonaka, T., Mori, H., et al. (2006). TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 351, 602–611. doi: 10.1016/j.bbrc.2006.10.093
Ayala, Y. M., Pantano, S., D’Ambrogio, A., Buratti, E., Brindisi, A., Marchetti, C., et al. (2005). Human, Drosophila and C. elegans TDP-43: nucleic acid binding properties and splicing regulatory function. J. Mol. Biol. 348, 575–588. doi: 10.1016/j.jmb.2005.02.038
Bigio, E. H., Wu, J. Y., Deng, H.-X., Bit-Ivan, E. N., Mao, Q., Ganti, R., et al. (2013). Inclusions in frontotemporal lobar degeneration with TDP-43 proteinopathy (FTLD-TDP) and amyotrophic lateral sclerosis (ALS), but not FTLD with FUS proteinopathy (FTLD-FUS) have properties of amyloid. Acta Neuropathol. 125, 463–465. doi: 10.1007/s00401-013-1089-6
Brady, O. A., Meng, P., Zheng, Y., Mao, Y., and Hu, F. (2011). Regulation of TDP-43 aggregation by phosphorylation and p62/SQSTM1. J. Neurochem. 116, 248–259. doi: 10.1111/j.1471-4159.2010.07098.x
Budini, M., Buratti, E., Stuani, C., Guarnaccia, C., Romano, V., De Conti, L., et al. (2012). Cellular model of TAR DNA-binding protein 43 (TDP-43) aggregation based on its C-terminal Gln/Asn-rich region. J. Biol. Chem. 287, 7512–7525. doi: 10.1074/jbc.m111.288720
Budini, M., Romano, V., Quadri, Z., Buratti, E., and Baralle, F. E. (2015). TDP-43 loss of cellular function through aggregation requires additional structural determinants beyond its C-terminal Q/N prion-like domain. Hum. Mol. Genet. 24, 9–20. doi: 10.1093/hmg/ddu415
Buratti, E. (2016). Functional significance of TDP-43 mutations in disease. Adv. Genet. 91, 1–53. doi: 10.1016/bs.adgen.2015.07.001
Buratti, E., and Baralle, F. E. (2011). TDP-43: new aspects of autoregulation mechanisms in RNA binding proteins and their connection with human disease. FEBS J. 278, 3530–3538. doi: 10.1111/j.1742-4658.2011.08257.x
Buratti, E., and Baralle, F. E. (2012). TDP-43: gumming up neurons through protein-protein and protein-RNA interactions. Trends Biochem. Sci. 37, 237–247. doi: 10.1016/j.tibs.2012.03.003
Cairns, N. J., Neumann, M., Bigio, E. H., Holm, I. E., Troost, D., Hatanpaa, K. J., et al. (2007). TDP-43 in familial and sporadic frontotemporal lobar degeneration with ubiquitin inclusions. Am. J. Pathol. 171, 227–240. doi: 10.2353/ajpath.2007.070182
Capitini, C., Conti, S., Perni, M., Guidi, F., Cascella, R., De Poli, A., et al. (2014). TDP-43 inclusion bodies formed in bacteria are structurally amorphous, non-amyloid and inherently toxic to neuroblastoma cells. PLoS One 9:e86720. doi: 10.1371/journal.pone.0086720
Chen, A. K.-H., Lin, R. Y.-Y., Hsieh, E. Z.-J., Tu, P.-H., Chen, R. P.-Y., Liao, T.-Y., et al. (2010). Induction of amyloid fibrils by the C-terminal fragments of TDP-43 in amyotrophic lateral sclerosis. J. Am. Chem. Soc. 132, 1186–1187. doi: 10.1021/ja9066207
Espargaró, A., Busquets, M. A., Estelrich, J., and Sabate, R. (2016). Key points concerning amyloid infectivity and prion-like neuronal invasion. Front. Mol. Neurosci. 9:29. doi: 10.3389/fnmol.2016.00029
Fang, Y.-S., Tsai, K.-J., Chang, Y.-J., Kao, P., Woods, R., Kuo, P.-H., et al. (2014). Full-length TDP-43 forms toxic amyloid oligomers that are present in frontotemporal lobar dementia-TDP patients. Nat. Commun. 5:4824. doi: 10.1038/ncomms5824
Fuentealba, R. A., Udan, M., Bell, S., Wegorzewska, I., Shao, J., Diamond, M. I., et al. (2010). Interaction with polyglutamine aggregates reveals a Q/N-rich domain in TDP-43. J. Biol. Chem. 285, 26304–26314. doi: 10.1074/jbc.M110.125039
Guo, W., Chen, Y., Zhou, X., Kar, A., Ray, P., Chen, X., et al. (2011). An ALS-associated mutation affecting TDP-43 enhances protein aggregation, fibril formation and neurotoxicity. Nat. Struct. Mol. Biol. 18, 822–830. doi: 10.1038/nsmb.2053
Hasegawa, M., Arai, T., Nonaka, T., Kametani, F., Yoshida, M., Hashizume, Y., et al. (2008). Phosphorylated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann. Neurol. 64, 60–70. doi: 10.1002/ana.21425
Jiang, L.-L., Che, M.-X., Zhao, J., Zhou, C.-J., Xie, M.-Y., Li, H.-Y., et al. (2013). Structural transformation of the amyloidogenic core region of TDP-43 protein initiates its aggregation and cytoplasmic inclusion. J. Biol. Chem. 288, 19614–19624. doi: 10.1074/jbc.M113.463828
Johnson, B. S., McCaffery, S. M., Lindquist, S., and Gilter, A. D. (2008). A yeast TDP-43 protein pathology model: exploring the molecular determinants of TDP-43 aggregation and cellular toxicity. Proc. Natl. Acad. Sci. U S A 105, 6439–6444. doi: 10.1073/pnas.0802082105
Kametani, F., Nonaka, T., Suzuki, T., Arai, T., Dohmae, N., Akiyama, H., et al. (2009). Identification of casein kinase-1 phosphorylation sites on TDP-43. Biochem. Biophys. Res. Commun. 382, 405–409. doi: 10.1016/j.bbrc.2009.03.038
Kametani, F., Obi, T., Shishido, T., Akatsu, H., Murayama, S., Saito, Y., et al. (2016). Mass spectrometric analysis of accumulated TDP-43 in amyotrophic later sclerosis brains. Sci. Rep. 6:23281. doi: 10.1038/srep23281
Kato, M., Han, T. W., Xie, S., Shi, K., Du, X., Wu, L. C., et al. (2012). Cell-free formation of RNA granules: low complexity sequence domains form dynamic fibers within hydrogels. Cell 149, 753–767. doi: 10.1016/j.cell.2012.04.017
Kiernan, M. C., Vucic, S., Cheah, B. C., Turner, M. R., Eisen, A., Hardiman, O., et al. (2011). Amyotrophoic lateral sclerosis. Lancet 377, 942–955. doi: 10.1016/S0140-6736(10)61156-7
Kuo, P.-H., Doudeva, L. G., Wang, Y.-T., Shen, C.-K. J., and Yuan, H. S. (2009). Structural insights into TDP-43 in nucleic-acid binding and domain interactions. Nucleic Acids Res. 37, 1799–1808. doi: 10.1093/nar/gkp013
Leigh, P. N., Whitwell, H., Garofalo, O., Buller, J., Swash, M., Martin, J. E., et al. (1991). Ubiquitin-immunoreactive intraneuronal inclusions in amyotrophoic lateral sclerosis. Morphology, distribution and specificity. Brain 114, 775–788. doi: 10.1093/brain/114.2.775
Li, H.-Y., Yeh, P.-A., Chiu, H.-C., Tang, C.-Y., and Tu, B. P. (2011). Hyperphosphorylation as a defense mechanism to reduce TDP-43 aggregation. PLoS One 6:e23075. doi: 10.1371/journal.pone.0023075
Lim, L., Wei, Y., Lu, Y., and Song, J. (2016). ALS-causing mutations significantly perturb the self-assembly and interactions with nucleic acid of the intrinsically disordered prion-like domain of TDP-43. PLoS Biol. 14:e1002338. doi: 10.1371/journal.pbio.1002338
Lin, W. L., and Dickson, D. W. (2008). Ultrastructural localization of TDP-43 in filamentous neuronal inclusions in various neurodegenerative diseases. Acta Neuropathol. 116, 205–213. doi: 10.1007/s00401-008-0408-9
Lukavsky, P. J., Daujotyte, D., Tollervey, J. R., Ule, J., Stuani, C., Buratti, E., et al. (2013). Molecular basis of UG-rich RNA recognition by the human splicing factor TDP-43. Nat. Struct. Mol. Biol. 20, 1443–1449. doi: 10.1038/nsmb.2698
Mackenzie, I. R. A., Neumann, M., Baborie, A., Sampathu, D. M., Du Plessis, D., Jaros, E., et al. (2011). A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol. 122, 111–113. doi: 10.1007/s00401-011-0845-8
Mackness, B. C., Tran, M. T., McClain, S. P., Matthews, C. R., and Zitzewitz, J. A. (2014). Folding of the RNA recognition motif (RRM) domains of the amyotrophic lateral sclerosis (ALS)-linked protein TDP-43 reveal an intermediate state. J. Biol. Chem. 289, 8264–8276. doi: 10.1074/jbc.M113.542779
Mompeán, M., Buratti, E., Guarnaccia, C., Brito, R. M. M., Chakrabartty, A., Baralle, F. E., et al. (2014). Structural characterization of the minimal segment of TDP-43 competent for aggregation. Arch. Biochem. Biophys. 545, 53–62. doi: 10.1016/j.abb.2014.01.007
Mompeán, M., Chakrabartty, A., Buratti, E., and Laurents, D. V. (2016a). Electrostatic repulsion governs TDP-43 C-terminal domain aggregation. PLoS Biol. 14:e1002447. doi: 10.1371/journal.pbio.1002447
Mompeán, M., Nogales, A., Ezquerra, T. A., and Laurents, D. V. (2016b). Complex system assembly underlies a two-tiered model of highly delocalized electrons. J. Phys. Chem. Lett. 7, 1859–1864. doi: 10.1021/acs.jpclett.6b00699
Mompeán, M., Romano, V., Pantoja-Uceda, D., Stuani, C., Baralle, F. E., Buratti, E., et al. (2016c). The TDP-43 N-terminal domain structure at high resolution. FEBS J. 283, 1242–1260. doi: 10.1111/febs.13651
Mompeán, M., Hervás, R., Xu, Y., Tran, T. H., Guarnaccia, C., Buratti, E., et al. (2015). Structural evidence of amyloid fibril formation in the putative aggregation domain of TDP-43. J. Phys. Chem. Lett. 6, 2608–2615. doi: 10.1021/acs.jpclett.5b00918
Neumann, M., Kwong, L. K., Lee, E. B., Kremmer, E., Flatley, A., Xu, Y., et al. (2009). Phosphorylation of S409/S410 of TDP-43 is a consistent feature in all sporadic and familial forms of TDP-43 proteinpathies. Acta Neuropathol. 117, 137–149. doi: 10.1007/s00401-008-0477-9
Neumann, M., Kwong, L. K., Sampathu, D. M., Trojanowski, J. Q., and Lee, V. M.-Y. (2007). TDP-43 proteinopathy in frontotemporal lobar degeneration and amyotrophoic lateral sclerosis: protein misfolding diseases without amyloidosis. Arch. Neurol. 64, 1388–1394. doi: 10.1001/archneur.64.10.1388
Neumann, M., Sampathu, D. M., Kwong, L. K., Truax, A. C., Micsenyi, M. C., Chou, T. T., et al. (2006). Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133. doi: 10.1126/science.1134108
Nonaka, T., Masuda-Suzukake, M., Arai, T., Hasegawa, Y., Akatsu, H., Obi, T., et al. (2013). Prion-like properties of pathological TDP-43 aggregates from diseased brains. Cell Rep. 4, 124–134. doi: 10.1016/j.celrep.2013.06.007
Patel, A., Lee, H. O., Jawerth, L., Maharama, S., Jahnel, M., Hein, M. Y., et al. (2015). A liquid-to-solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell 162, 1066–1077. doi: 10.1016/j.cell.2015.07.047
Pesiridis, G. S., Lee, V. M.-Y., and Trojanowski, J. Q. (2009). Mutations in TDP-43 link glycine-rich domain function to amyotrophic lateral sclerosis. Hum. Mol. Genet. 18, R156–R162. doi: 10.1093/hmg/ddp303
Polymenidou, M., Lagier-Tourenne, C., Hutt, K. R., Huelga, S. C., Moran, J., Liang, T. Y., et al. (2011). Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat. Neurosci. 14, 459–468. doi: 10.1038/nn.2779
Qin, H., Lim, L.-Z., Wei, Y., and Song, J. (2014). TDP-43 N-terminus encodes a novel ubiquitin-like fold and its unfolded form in equilibrium that can be shifted by binding to ssDNA. Proc. Natl. Acad. Sci. U S A 111, 18619–18624. doi: 10.1073/pnas.1413994112
Robinson, J. L., Geser, F., Stieber, A., Umoh, M., Kwong, L. K., Van Deerlin, V. M., et al. (2013). TDP-43 skeins show properties of amyloid in a subset of ALS cases. Acta Neuropathol. 125, 121–131. doi: 10.1007/s00401-012-1055-8
Romano, V., Quadri, Z., Baralle, F. E., and Buratti, E. (2015). The structural integrity of TDP-43 N-terminus is required for efficient aggregate entrapment and consequent loss of protein function. Prion 9, 1–9. doi: 10.1080/19336896.2015.1011885
Saini, A., and Chauhan, V. S. (2011). Delineation of the core aggregation sequences of TDP-43 C-terminal fragment. Chembiochem 12, 2495–2501. doi: 10.1002/cbic.201100427
Saini, A., and Chauhan, V. S. (2014). Self-assembling properties of peptides derived from TDP-43 C-terminal fragment. Langmuir 30, 3845–3856. doi: 10.1021/la404710w
Sasaguri, H., Chew, J., Xu, Y. F., Gendron, T. F., Garrett, A., Lee, C. W., et al. (2016). The extreme N-terminus of TDP-43 mediates the cytoplasmic aggregation of TDP-43 and associated toxicity in vivo. Brain Res. 1647, 57–64. doi: 10.1016/j.brainres.2016.04.069
Smethurst, P., Sidle, K. C., and Hardy, J. (2015). Review: prion-like mechanisms of transactive response DNA binding protein of 43 kDa (TDP-43) in amyotrophic lateral sclerosis (ALS). Neuropathol. Appl. Neurobiol. 41, 578–597. doi: 10.1111/nan.12206
Sun, C.-S., Wang, C. Y.-H., Chen, B. P.-W., He, R.-Y., Liu, G. C.-H., Wang, C.-H., et al. (2014). The influence of pathological mutations and proline substitutions in TDP-43 glycine rich peptides on its amyloid properties and celular toxicity. PLoS One 9:e103644. doi: 10.1371/journal.pone.0103644
Thorpe, J. R., Tang, H., Atherton, J., and Cairns, N. J. (2008). Fine structural analysis of the neuronal inclusions of frontotemporal lobar degeneration with TDP-43 proteinpathy. J. Neural Transm. (Vienna) 115, 1661–1671. doi: 10.1007/s00702-008-0137-1
Tollervey, J. R., Curk, T., Rogelj, B., Briese, M., Cereda, M., Kayikci, M., et al. (2011). Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat. Neurosci. 14, 452–458. doi: 10.1038/nn.2778
Wang, Y.-T., Kuo, P.-H., Chiang, C.-H., Liang, J.-R., Chen, Y.-R., Wang, S., et al. (2013). The truncated C-terminal RNA recognition motif of TDP-43 protein plays a key role in forming proteinaceous aggregates. J. Biol. Chem. 288, 9049–9057. doi: 10.1074/jbc.M112.438564
Zhang, Y.-J., Caulfield, T., Xu, Y.-F., Gendron, T. F., Hubbard, J., Stetler, C., et al. (2013). The dual functions of the extreme N-terminus of TDP-43 in regulating its biological activity and inclusion formation. Hum. Mol. Genet. 22, 3112–3122. doi: 10.1093/hmg/ddt166
Keywords: pathological amyloids, transitive response DNA-bonding protein 43 kDa (TDP-43), hnRNPs, hyperstable H-bonds, amyotrophic lateral sclerosis (ALS), frontotemporal lobar degeneration, protein misfolding and disease
Citation: Mompeán M, Baralle M, Buratti E and Laurents DV (2016) An Amyloid-Like Pathological Conformation of TDP-43 Is Stabilized by Hypercooperative Hydrogen Bonds. Front. Mol. Neurosci. 9:125. doi: 10.3389/fnmol.2016.00125
Received: 19 July 2016; Accepted: 04 November 2016;
Published: 17 November 2016.
Edited by:
Lisbeth Birk Møller, Kennedy Center, DenmarkReviewed by:
Daniele Bottai, University of Milan, ItalyUtpal Das, University of California, San Diego, USA
Copyright © 2016 Mompeán, Baralle, Buratti and Laurents. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution and reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Douglas V. Laurents, ZGxhdXJlbnRzQGlxZnIuY3NpYy5lcw==