 Yili Wu
Yili Wu Xin Wang1,3
Xin Wang1,3 Gongying Li
Gongying Li- 1Department of Psychiatry, Jining Medical University, Jining, China
- 2Collaborative Innovation Center for Birth Defect Research and Transformation of Shandong Province, Jining Medical University, Jining, China
- 3Shandong Key Laboratory of Behavioral Medicine, Jining Medical University, Jining, China
- 4Neurobiology Institute, Jining Medical University, Jining, China
Stroke is one of the leading causes of death and disability worldwide, and ischemic stroke accounts for approximately 87% of cases. Improving post-stroke recovery is a major challenge in stroke treatment. Accumulated evidence indicates that the apelinergic system, consisting of apelin and apelin receptor (APLNR), is temporally dysregulated in ischemic stroke. Moreover, the apelinergic system plays a pivotal role in post-stroke recovery by inhibiting neuronal apoptosis and facilitating angiogenesis through various molecular pathways. In this review article, we summarize the temporal expression of apelin and APLNR in ischemic stroke and the mechanisms of their dysregulation. In addition, the protective role of the apelinergic system in ischemic stroke and the underlying mechanisms of its protective effects are discussed. Furthermore, critical issues in activating the apelinergic system as a potential therapy will also be discussed. The aim of this review article is to shed light on exploiting the activation of the apelinergic system to treat ischemic stroke.
Introduction
Stroke is one of the leading causes of death and disability worldwide and the majority of cases are ischemic stroke. To improve recovery and reduce disability, novel therapeutic approaches need to be developed, as current treatments are only limited to thrombolytic therapy within an extremely narrow time window (Craig and Housley, 2016). A growing body of evidence indicates that the apelinergic system, consisting of apelin and apelin receptor (APLNR), is implicated in ischemic stroke. First, Hata et al. (2007, 2011) showed that a single-nucleotide polymorphism (SNP, rs9943582) in APLNR is significantly associated with the increased risk of ischemic stroke in human. Moreover, the expression of apelin and APLNR is temporally dysregulated during different phases of ischemic stroke. Importantly, a number of studies showed that the apelinergic system plays a pivotal role in protecting cells from ischemic stroke-induced apoptosis and improving behavioral performance. The aforementioned evidence indicates that the apelinergic system might be a potential target for novel treatments. Therefore, we summarize the temporal expression of apelin and APLNR in ischemic stroke, the mechanisms of their dysregulation, the protective effect of the apelinergic system on ischemic stroke and the underlying mechanisms of its protective effect. In addition, we also discuss critical issues that may affect strategies for activating the apelinergic system as a potential therapy.
Overview of Ischemic Stroke
Epidemiology and Prevalence of Ischemic Stroke
Stroke, caused by the disturbance of blood supply to the brain, is the second leading cause of death and the third leading cause of disability worldwide (Wang et al., 2012). In the United States, stroke is the leading cause of long-term disability, including both physical and cognitive deficits. By 2030, the prevalence of stroke will increase by more than 20% over 2012 and the direct medical costs are projected to reach $184.1 billion, which is a great challenge to human society and healthcare system (Mozaffarian et al., 2016). The situation is much worse in China, where stroke is the leading cause of death (Wang H. et al., 2016). Ischemic stroke is caused by the blockage of an artery in the brain, accounting for approximately 87% of stroke cases, which contributes to the major portion of death and post-stroke disability in patients (Mozaffarian et al., 2016). In addition to acute brain damage, stroke significantly increases the risk of neurodegenerative diseases, such as Alzheimer’s disease (Wu et al., 2014). However, current treatments are only limited to thrombolytic therapy within an extremely narrow time window. Therefore, it is urgent to develop novel therapeutic approaches for stroke treatment.
Mechanisms of Cell Death in Ischemic Stroke
Depending on the severity of lack of blood supply, the ischemic territory consists of the ischemic core and penumbra, which are respectively characterized by acute and delayed cell death, necrosis and apoptosis (Lipton, 1999; Lo, 2008). Ischemic core, the center area of the ischemic territory, is caused by an acute blood reduction to less than 15%, while ischemic penumbra, the surrounding area of the core, has a higher blood supply. Necrosis occurs within minutes after stroke, thus, cells within the ischemic core cannot be rescued. However, cells in the penumbra experience less severe blood reduction and may undergo apoptosis (Lipton, 1999; Lo, 2008). Although cells in the penumbra are vulnerable, their death is salvageable by proper intervention, suggesting that preventing penumbra cell death is the key to improve the recovery from and the prognosis of ischemic stroke. During acute ischemia, the diffusion of toxic contents released from dead cells in the core region triggers apoptotic responses. In addition, ischemia/reperfusion injury contributes to the apoptosis of penumbral cells by a number of molecular mechanisms, such as oxidative stress, inflammatory response, endoplasmic reticulum (ER) stress and abnormal protein degradation (Moskowitz et al., 2010; Zhang et al., 2014b). Therefore, blocking the detrimental pathways has a therapeutic potential to reduce penumbra cell death and improve stroke recovery.
The Apelinergic System—Apelin and APLNR
Apelin
The apelin (APLN) gene, located on chromosome Xq25, spans 9698bp, consisting of three exons and two introns. It encodes the preproapelin of 77 amino acids. Various mature peptides are derived from the 55 amino-acid propeptide, ranging from 13–36 amino acids. The mature peptides are highly conserved between species. In particular, the C-terminal 23 amino acids are 100% homologous between human and rat, mouse or bovine. Tatemoto et al. (1998) first identified apelin-36 in vivo and predicted it can be further processed to apelin-17 and apelin-13 with 17 and 13 amino acids, respectively. In addition, the shorter C-terminal peptides, apelin-13 and its pyroglutamate-modified form Pyr-apelin-13, have higher activity compared with apelin-36 (Hosoya et al., 2000). Mesmin et al. (2011) identified 46 endogenous apelin peptides in bovine colostrums. Apelin-13, the most active isoform, has been widely studied in ischemia/reperfusion injury (Tao et al., 2011; Khaksari et al., 2012; Yang et al., 2014, 2015; Chen et al., 2015; Li et al., 2016). Although all apelin isoforms may function through the unique APLNR, their tissue specificity, binding affinity to APLNR and efficacy in APLNR recycling may lead to differential functions of isoforms. However, the proapelin processing and the proteases involved in the generation of apelin isoforms have received less investigation. Recently, Shin et al. (2013) showed that proprotein convertase subtilisin/kexin 3 (PCSK3) directly cleaves proapelin to generate apelin-13.
Apelin is expressed in both brain and peripheral tissues in human. In the brain, apelin is expressed in thalamus, frontal cortex and hippocampus, while it is also expressed in placenta, heart, lung and other peripheral tissues. The expression of apelin is controlled at both transcriptional and post-translational levels (Figure 1). First, multiple transcriptional factors are involved in the transcriptional regulation of apelin, such as Sp1 transcription factor (SP1), signal transducer and activator of transcription 3 (STAT3), hypoxia inducible factor 1 alpha (HIF-1α), upstream transcription factor 1/upstream transcription factor 2 (USF1/USF2) and activating transcription factor 4 (ATF4; Wang et al., 2006; Han et al., 2008; Lv et al., 2013; Jeong et al., 2014; He et al., 2015). Moreover, tumor necrosis factor alpha (TNF-α) increases the expression of APLN mRNA via phosphatidyl Inositol 3-kinase (PI3K) activation (Daviaud et al., 2006). In addition, apelin has a high turnover rate, with a half life of less than 8 min (Aydin et al., 2014; Juhl et al., 2016). Two identified proteases, angiotensin-converting enzyme 2 (ACE2) and metalloprotease (NEP), are involved in apelin proteolysis, which partially or fully inactivate the binding activity of apelin to its receptor and contribute to its degradation (Kalea and Batlle, 2010; McKinnie et al., 2016; Wang W. et al., 2016).
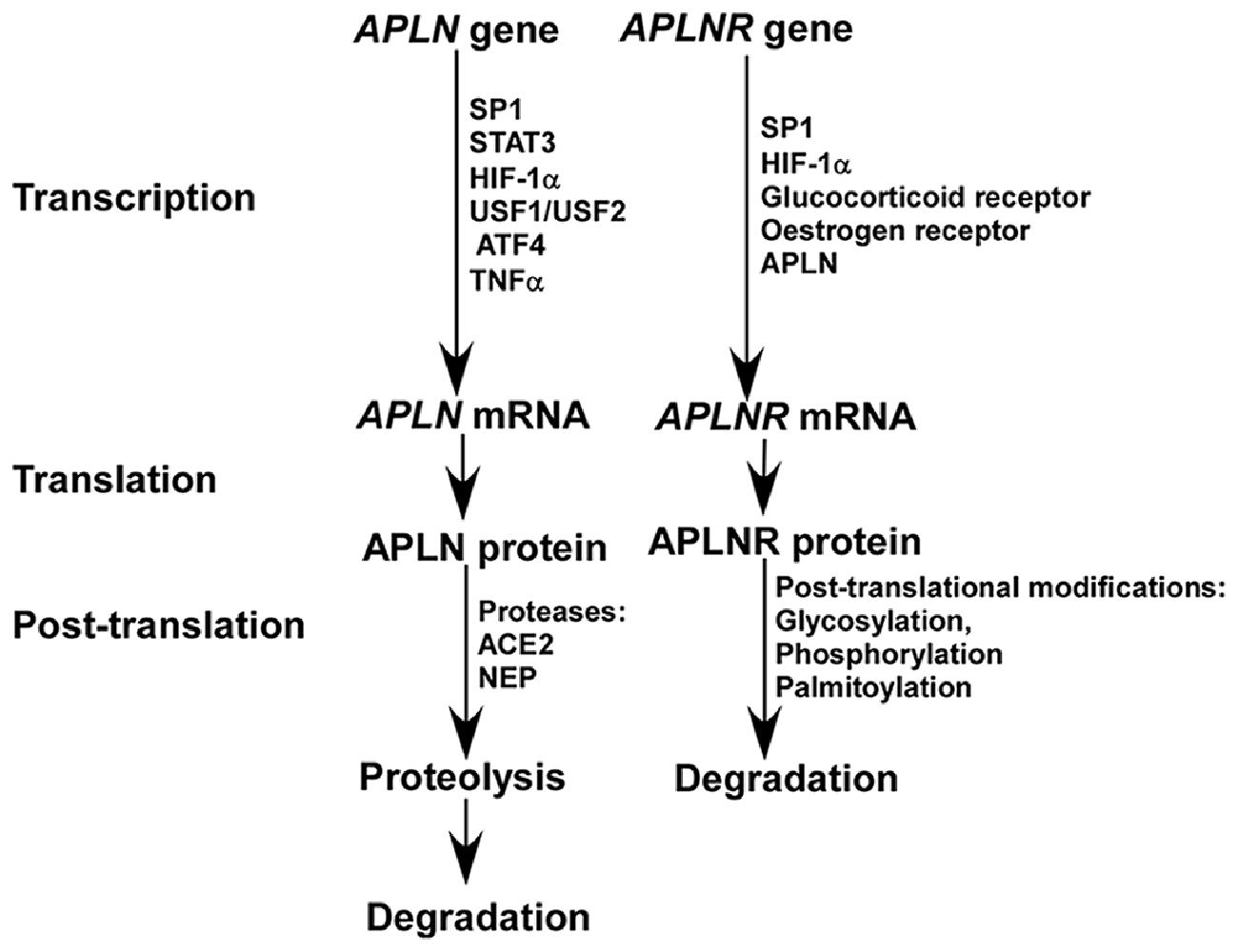
Figure 1. The expression of apelin and apelin receptor (APLNR) is differentially regulated at transcriptional and post-translational levels.
APLNR
APLNR is the unique receptor of apelin, while apelin has until recently been considered the only endogenous ligand of APLNR. Recently, Chng et al. (2013) reported that elabela, also named as apela or toddler, is another endogenous ligand of APLNR in zebrafish, which is essential for early cardiovascular development. However, the expression of elabela is restricted to pluripotent cells and kidney in human (Wang et al., 2015).
The APLNR gene is located on chromosome 11q12, spanning 3877bp genomic DNA without introns. Human APLNR, consisting of 380 amino acids, shares more than 90% amino acid sequence homology to mouse and rat APLNR. It is widely distributed in human tissues. In human brain, APLNR mRNA is highly expressed in caudate nucleus, corpus callosum and hippocampus (Matsumoto et al., 1996; Edinger et al., 1998; Medhurst et al., 2003). Recently, its expression is also detected in the cortex (Hansen et al., 2007). The aforementioned evidence suggests that APLNR has important functions in multiple brain regions, which has not been fully investigated. APLNR, a typical G protein coupled receptor (GPCR), mainly mediates signal transduction via Gα subunit (Gαi or Gαq) of heterotrimeric G protein. For example, Gαi contributes to PI3K/serine/threonine kinase (PI3K/AKT) activation and extracellular signal-regulated kinase (ERK) activation, while Gαq contributes to phospholipase C beta-protein kinase C (PLCβ-PKC) activation (Chapman et al., 2014). In addition, it may mediate signal transduction through a β-arrestin-dependent mechanism (Chapman et al., 2014). However, the exact role of the β-arrestin-dependent pathway in signal transduction remains elusive.
A number of transcription factors are implicated in the regulation of APLNR transcription, as multiple functional binding sites are located in the promoter region of the APLNR gene, including SP1, HIF-1α, glucocorticoid receptor and estrogen receptor (O’Carroll et al., 2006; Figure 1). Insulin is also involved in the regulation of APLNR expression (Dray et al., 2010). In addition, apelin deficiency reduces the level of APLNR mRNA (Wang et al., 2009). Hypoxia promotes both apelin and APLNR expression accompanying with HIF-1α induction, suggesting that upregulation of APLNR may be mediated by both apelin and/or HIF-1α (He et al., 2015). Moreover, APLNR undergoes multiple post-translational modifications, including glycosylation, phosphorylation and palmitoylation, which may contribute to the alteration of APLNR at the protein level by affecting its turnover rate (Figure 1; O’Carroll et al., 2013; Chen et al., 2014). For example, the phosphorylation status of regulator of calcineurin 1(RCAN1) has a significant effect on its degradation rate (Genescà et al., 2003).
Temporal Expression of Apelin/APLNR in Ischemic Stroke
Increased Expression of Apelin/APLNR During the Ischemic Phase of Ischemic Stroke
Ischemia/reperfusion injury occurs in both heart and brain, accompanied by many common pathophysiological alterations. Numerous in vitro and in vivo studies showed that the expression of apelin and APLNR is altered by ischemia/reperfusion injury via a number of mechanisms (Figure 2). First, hypoxia and glucose deprivation, the major consequences of ischemia, are implicated in the aberrant expression of apelin and APLNR. The expression of apelin and APLNR is increased under hypoxic conditions, which is mediated by the induction of HIF-1α (He et al., 2015). HIF-1α can functionally bind to hypoxia response elements (HREs) within the promoter region of the apelin and APLNR genes, leading to the upregulation of apelin and APLNR (Ronkainen et al., 2007; He et al., 2015). In addition, glucose deprivation upregulates apelin and APLNR expression at early stages (Zhang et al., 2009). Moreover, the increase of SP1 promotes apelin and APLNR expression in ischemic neurons at the early stage, which may be mediated by HIF-1α induction (Woo et al., 2012; O’Carroll et al., 2006; Yeh et al., 2011; Lv et al., 2013). Consistently, the expression of both apelin and APLNR is increased by ischemia in rat hearts (Atluri et al., 2007; Sheikh et al., 2008).
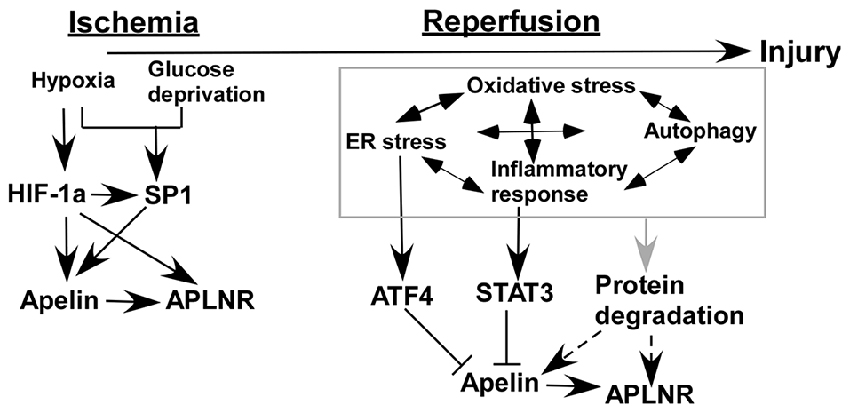
Figure 2. Temporal expression of apelin/APLNR in ischemic stroke. HIF-1α and SP1 transcription factor (SP1) contribute to the upregulation of apelin/APLNR during the ischemic phase, while multiple mechanisms are involved in the reduction of apelin/APLNR during the reperfusion phase.
Reduced Expression of Apelin/APLNR During the Reperfusion Phase of Ischemic Stroke
Recent studies indicate that the expression of apelin and APLNR may be temporally regulated in ischemic stroke. The expression of apelin and APLNR is only increased early after ischemia in heart (Rastaldo et al., 2011). Although glucose deprivation upregulates the expression of apelin and APLNR at early stages, it significantly reduces apelin and APLNR expression at later stages (Zhang et al., 2009). The later-stage reduction of apelin and APLNR may be associated with a plethora of mechanisms in ischemic stroke (Figure 2). For example, ER stress is a major mechanism contributing to ischemia/reperfusion injury in both heart and brain (Morimoto et al., 2007; Nakka et al., 2010; Tao et al., 2011; Xin et al., 2014). However, it is only activated at the reperfusion stage but not at the ischemic stage in both heart and brain (Morimoto et al., 2007; Nakka et al., 2010; Tao et al., 2011). ATF4, an important mediator of ER stress, significantly reduces apelin expression via a p38 mitogen-activated protein kinase (p38-MAPK) dependent pathway (Jeong et al., 2014), suggesting that ER stress may contribute to the reduction of apelin expression during the reperfusion phase (Figure 2). Subsequently, APLNR may be downregulated by apelin reduction (Wang et al., 2009). In addition, inflammatory responses may be implicated in the regulation of apelin and APLNR expression mediated by STAT3, as it directly regulates the promoter activity of the apelin gene (Han et al., 2008; Liang et al., 2016). Interestingly, STAT3 is only activated during the reperfusion phase but not the ischemic phase. The inhibition of STAT3 signaling dramatically improves the outcome of ischemic stroke, suggesting that STAT3 may contribute to the reduction of apelin expression during the reperfusion phase (Figure 2; Li and Zhang, 2003; Yu et al., 2013). Moreover, the interplay among oxidative stress, ER stress, autophagy and inflammatory responses may be implicated in the temporal expression of apelin and APLNR (Sheng et al., 2012; Mei et al., 2015).
Understanding the temporal expression of apelin/APLNR is critical for defining the timing of treatment with apelin or APLNR agonists. For example, Rastaldo et al. (2011) reported that apelin can only reduce infarct size of heart when it is administrated at the reperfusion phase. Therefore, the temporal expression of apelin/APLNR in ischemic stroke and the underlying mechanisms need to be further investigated, which is crucial for the therapeutic application of apelin or APLNR agonists to treat ischemic stroke.
Protective Effects of Apelin/APLNR in Ischemic Stroke and the Underlying Mechanisms
Apelin/APLNR Inhibits Cell Death in Ischemic Stroke
Accumulated evidence indicates that apelin/APLNR improves the recovery from ischemic stroke by inhibiting cell death and facilitating angiogenesis, which are mediated by the activation of PI3K/AKT and ERKs signaling pathways, respectively (Figure 3; Chuang et al., 2011; Tao et al., 2011; Gu et al., 2013; Wang et al., 2013; Zhu et al., 2013; Yang et al., 2014; Chen et al., 2015; Liu et al., 2015; Huang et al., 2016; Novakova et al., 2016; Zou et al., 2016). Oxidative stress is a major mechanism of ischemia/reperfusion injury, leading to cell death. Mitochondria-derived oxidative stress is involved in ischemia/reperfusion injury by causing an overload in reactive oxygen species (ROS) and the increased ROS further promotes mitochondria impairment, forming a vicious feedback loop. Blocking this vicious cycle by suppressing oxidative stress is a major mechanism of apelin/APLNR’s protective effects on ischemic stroke (Figure 3). First, apelin reduces ischemia/reperfusion injury-induced oxidative stress by increasing the activity of antioxidant enzymes in kidneys, such as superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GSH-Px) (Bircan et al., 2016). Than et al. (2014) showed that apelin promotes the expression of antioxidant enzymes via MAPK kinase/ERK pathway in adipocytes. Moreover, apelin analogs inhibit mitochondrial ROS generation in hearts (Pisarenko et al., 2015). The aforementioned evidence suggests that the protective effect apelin/APLNR on oxidative stress-induced cell death may be mediated by suppressing ROS generation and promoting ROS scavenging, which remains to be investigated.
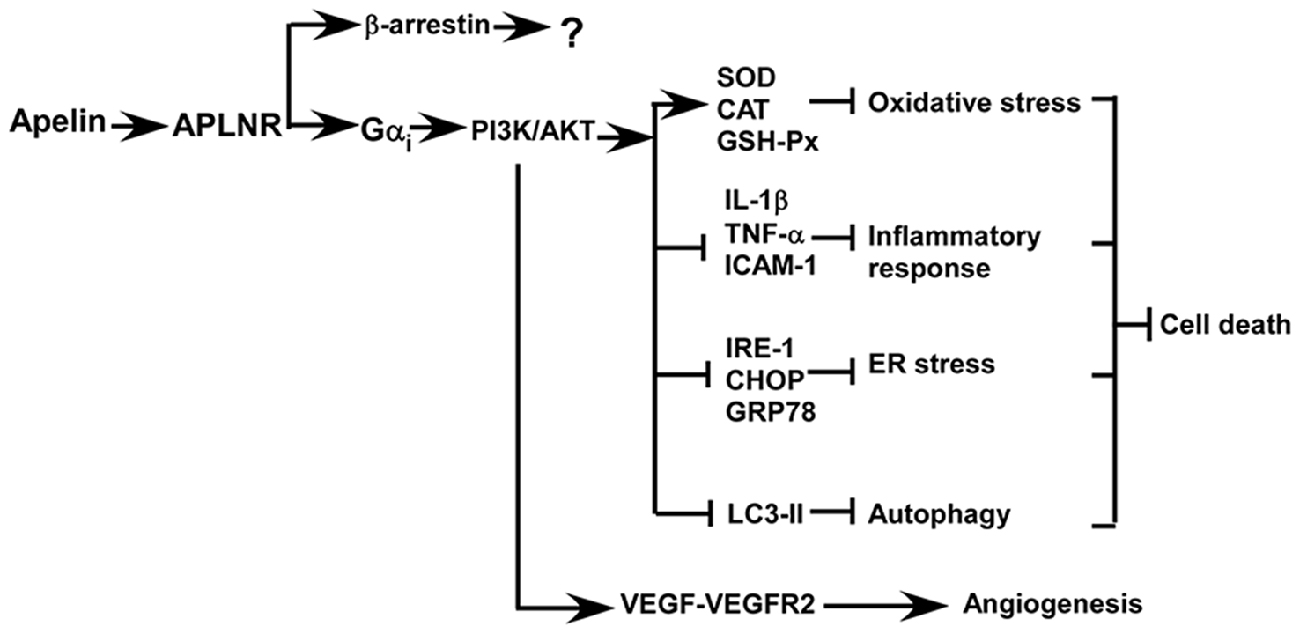
Figure 3. Apelin/APLNR improves the recovery from ischemic stroke by inhibiting cell death and facilitating angiogenesis via Gαi- dependent mechanisms, while the role of β–arrestin-dependent pathways in ischemic stroke is unknown.
Oxidative stress not only promotes cell death but also contributes to the induction of inflammatory responses, which is another mechanism contributing to cell death in ischemic stroke. Apelin inhibits inflammatory response via reducing the generation of inflammatory cytokines (Figure 3). For example, apelin significantly reduces the expression of inflammatory cytokines in rat brains, such as interleukin 1 beta (IL-1β), TNF-α and intercellular adhesion molecule 1(ICAM-1), and inhibits cell apoptosis in rats with ischemic stroke (Chen et al., 2015; Xin et al., 2015; Yan et al., 2015). Moreover, apelin markedly reduces microglial activation and their recruitment to the ischemic penumbra, which is associated with a decrease in cell death (Chen et al., 2015).
Apelin can protect cells from apoptosis by preventing ER stress, which is activated by ischemia/reperfusion injury in both brain and heart (Figure 3; Morimoto et al., 2007; Nakka et al., 2010; Tao et al., 2011; Xin et al., 2014). ER stress is only activated at the reperfusion stage but not activated at the ischemic stage, suggesting that administration of apelin during the reperfusion stage may be effective in protecting cells from ER stress-induced apoptosis (Morimoto et al., 2007; Nakka et al., 2010; Tao et al., 2011). For example, Nakka et al. (2010) showed that ER stress is activated around 3–6 h after reperfusion in rat brains. Consistently, Tao et al. (2011) showed that ER stress is activated 2 h after reperfusion in heart. Moreover, apelin significantly reduces ER stress and infarct size via a PI3K/AKT-dependent pathway (Tao et al., 2011). Importantly, apelin has a long-term effect on the inhibition of ER stress, beyond 24 h after reperfusion (Tao et al., 2011). However, the expression of apelin is downregulated by ER stress (Jeong et al., 2014). The aforementioned evidence indicates that apelin reduction and the activation of ER stress may form a vicious feedback loop during the reperfusion phase, similar to mitochondria-induced stress, which potentiates ER stress-induced apoptosis. Thus, apelin administration may block this vicious cycle to inhibit ER stress-induced apoptosis in ischemic stroke.
Growing evidence indicates that the protective effect of apelin on ischemia/reperfusion-induced apoptosis may be associated with its role in the regulation of autophagy (Figure 3). First, apelin is able to inhibit glucose deprivation-induced autophagy, contributing to the protective effect on cardiomyocytes. This effect is associated with the activation of PI3K/AKT/mechanistic target of rapamycin (mTOR) pathway (Jiao et al., 2013). In addition, apelin inhibits hypoxia-induced autophagy via activating PI3K/AKT/mTOR pathway in muscle cells (Zhang et al., 2014a). Moreover, apelin attenuates traumatic brain injury-induced damage by suppressing autophagy (Bao et al., 2015).
Apelin/APLNR Promotes Angiogenesis in Ischemic Stroke
Post-stroke angiogenesis has a beneficial effect on cell survival and stroke recovery. Apelin not only protects cells from death but also promotes angiogenesis against ischemic stroke and improves stroke recovery (Chen et al., 2015; Huang et al., 2016). Apelin facilitates angiogenesis and blood flow restoration, which is dependent on an increase in vascular endothelial growth factor- vascular endothelial growth factor receptor 2 (VEGF-VEGFR2) signaling. In addition, PI3K/AKT and ERKs signaling pathways play a crucial role in apelin/APLNR-induced angiogenesis in ischemic stroke, which may be mediated by VEGF-VEGFR2 (Wang et al., 2013; Chen et al., 2015; Liu et al., 2015; Novakova et al., 2016). Consistently, loss of apelin impairs the angiogenesis and functional recovery (Wang et al., 2013).
Conclusions and Perspectives
The aforementioned evidence suggests that activating apelin/APLNR system may be a potent approach for protecting against ischemic stroke and improving recovery following an ischemic event. However, several critical issues need to be well resolved. First, the temporal expression of apelin/ APLNR during ischemic stroke needs to be determined, as it is crucial for deciding the timing of treatment with apelin or APLNR agonists. Based on the alteration of apelin/APLNR expression in ischemia/reperfusion injury of heart, application of apelin after thrombolytic therapy may be an effective approach to protect penumbra cells from apoptosis. Moreover, modified apelin or APLNR agonists with longer half life need to be developed, as the half life of apelin is too short to be applied clinically. Finally, non-invasive drug delivery methods need to be further explored. As APLNR is widely expressed in peripheral tissues, systematic delivery may generate more side effects. Thus, apelin was administered through lateral ventricle injection in most in vivo studies. However, this approach is not suitable for clinical application. A recent study has applied intranasal delivery of apelin for stroke treatment, shedding a light on exploiting non-invasive apelin application to treat ischemic stroke (Chen et al., 2015).
Author Contributions
YW formulated the study, wrote the manuscript and designed the figures. XW, XZ, BC and GL wrote the manuscript. BB provided intellectual thoughts, revised the manuscript and was the project leader.
Funding
This work was supported by grants from the National Natural Science Foundation of China (81070961 to BB, 81571334 to GL, 81671276 to BC), the Jining science and technology development plan (2013jnwk75 to BB), Natural Science Foundation of Shandong Province (ZR2016HM30 to YW, ZR2011HM023 to GL, ZR2014HL040 to BC, ZR2010HL057 and ZR2012HL24 to BC), Science and Technology Project of Higher education of Shandong Province (J10LF01 to GL), the Development of Medical Science and Technology Project of Shandong Province (2011HZ011 to GL), Postgraduate Education Innovation Program of Shandong Province (SDYY15012 to GL) and Research Project of Teaching Reform in Undergraduate Colleges and Universities in Shandong Province (2015M049 to GL), Project of Shandong Province Higher Educational Science and Technology Program (J12LE10 to BC).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
ACE2, angiotensin-converting enzyme 2; AKT, serine/threonine kinase; APLN, apelin; APLNR, apelin receptor; ATF4, activating transcription factor 4; CAT, catalase; ER, endoplasmic reticulum; ERK, extracellular signal-regulated kinase; GSH-Px, glutathione peroxidase; HIF-1α, hypoxia inducible factor 1 alpha; HREs, hypoxia response elements; ICAM-1, intercellular adhesion molecule 1; IL-1β, interleukin 1 beta; mTOR, mechanistic target of rapamycin; NEP, metalloprotease; p38-MAPK, p38 mitogen-activated protein kinase; PCSK3, proprotein convertase subtilisin/kexin 3; PI3K, phosphatidyl Inositol 3-kinase; PLCβ-PKC, phospholipase C beta-protein kinase C; ROS, reactive oxygen species; SOD, superoxide dismutase; SP1, Sp1 transcription factor; STAT3, signal transducer and activator of transcription 3; TNF-α, tumor necrosis factor alpha; USF1/USF2, upstream transcription factor 1/upstream transcription factor 2; VEGF, vascular endothelial growth factor; VEGFR2, vascular endothelial growth factor receptor 2.
References
Atluri, P., Morine, K. J., Liao, G. P., Panlilio, C. M., Berry, M. F., Hsu, V. M., et al. (2007). Ischemic heart failure enhances endogenous myocardial apelin and APJ receptor expression. Cell Mol. Biol. Lett. 12, 127–138. doi: 10.2478/s11658-006-0058-7
Aydin, S., Eren, M. N., Sahin, I., and Aydin, S. (2014). The role of apelins in the physiology of the heart. Protein Pept. Lett. 21, 2–9. doi: 10.2174/09298665113209990083
Bao, H. J., Zhang, L., Han, W. C., and Dai, D. K. (2015). Apelin-13 attenuates traumatic brain injury-induced damage by suppressing autophagy. Neurochem. Res. 40, 89–97. doi: 10.1007/s11064-014-1469-x
Bircan, B., Çakir, M., Kirbag, S., and Gül, H. F. (2016). Effect of apelin hormone on renal ischemia/reperfusion induced oxidative damage in rats. Ren. Fail. 38, 1122–1128. doi: 10.1080/0886022x.2016.1184957
Chapman, N. A., Dupré, D. J., and Rainey, J. K. (2014). The apelin receptor: physiology, pathology, cell signalling and ligand modulation of a peptide-activated class A GPCR. Biochem. Cell Biol. 92, 431–440. doi: 10.1139/bcb-2014-0072
Chen, X., Bai, B., Tian, Y., Du, H., and Chen, J. (2014). Identification of serine 348 on the apelin receptor as a novel regulatory phosphorylation site in apelin-13-induced G protein-independent biased signaling. J. Biol. Chem. 289, 31173–31187. doi: 10.1074/jbc.m114.574020
Chen, D., Lee, J., Gu, X., Wei, L., and Yu, S. P. (2015). Intranasal delivery of apelin-13 is neuroprotective and promotes angiogenesis after ischemic stroke in mice. ASN Neuro. 7, 1–15. doi: 10.1177/1759091415605114
Chng, S. C., Ho, L., Tian, J., and Reversade, B. (2013). ELABELA: a hormone essential for heart development signals via the apelin receptor. Dev. Cell 27, 672–680. doi: 10.1016/j.devcel.2013.11.002
Chuang, D. M., Wang, Z., and Chiu, C. T. (2011). GSK-3 as a target for lithium-induced neuroprotection against excitotoxicity in neuronal cultures and animal models of ischemic stroke. Front. Mol. Neurosci. 4:15. doi: 10.3389/fnmol.2011.00015
Craig, A. J., and Housley, G. D. (2016). Evaluation of gene therapy as an intervention strategy to treat brain injury from stroke. Front. Mol. Neurosci. 9:34. doi: 10.3389/fnmol.2016.00034
Daviaud, D., Boucher, J., Gesta, S., Dray, C., Guigne, C., Quilliot, D., et al. (2006). TNFα up-regulates apelin expression in human and mouse adipose tissue. FASEB J. 20, 1528–1530. doi: 10.1096/fj.05-5243fje
Dray, C., Debard, C., Jager, J., Disse, E., Daviaud, D., Martin, P., et al. (2010). Apelin and APJ regulation in adipose tissue and skeletal muscle of type 2 diabetic mice and humans. Am. J. Physiol. Endocrinol. Metab. 298, E1161–E1169. doi: 10.1152/ajpendo.00598.2009
Edinger, A. L., Hoffman, T. L., Sharron, M., Lee, B., Yi, Y., Choe, W., et al. (1998). An orphan seven-transmembrane domain receptor expressed widely in the brain functions as a coreceptor for human immunodeficiency virus type 1 and simian immunodeficiency virus. J. Virol. 72, 7934–7940.
Genescà, L., Aubareda, A., Fuentes, J. J., Estivill, X., De La Luna, S., and Pérez-Riba, M. (2003). Phosphorylation of calcipressin 1 increases its ability to inhibit calcineurin and decreases calcipressin half-life. Biochem. J. 374, 567–575. doi: 10.1042/bj20030267
Gu, Q., Zhai, L., Feng, X., Chen, J., Miao, Z., Ren, L., et al. (2013). Apelin-36, a potent peptide, protects against ischemic brain injury by activating the PI3K/Akt pathway. Neurochem. Int. 63, 535–540. doi: 10.1016/j.neuint.2013.09.017
Han, S., Wang, G., Qi, X., Englander, E. W., and Greeley, G. H., Jr. (2008). Involvement of a Stat3 binding site in inflammation-induced enteric apelin expression. Am. J. Physiol. Gastrointest. Liver Physiol. 295, G1068–G1078. doi: 10.1152/ajpgi.90493.2008
Hansen, A., Chen, Y., Inman, J. M., Phan, Q. N., Qi, Z. Q., Xiang, C. C., et al. (2007). Sensitive and specific method for detecting G protein-coupled receptor mRNAs. Nat. Methods 4, 35–37. doi: 10.1038/nmeth977
Hata, J., Kubo, M., and Kiyohara, Y. (2011). Genome-wide association study for ischemic stroke based on the Hisayama study. Nihon Eiseigaku Zasshi 66, 47–52. doi: 10.1265/jjh.66.47
Hata, J., Matsuda, K., Ninomiya, T., Yonemoto, K., Matsushita, T., Ohnishi, Y., et al. (2007). Functional SNP in an Sp1-binding site of AGTRL1 gene is associated with susceptibility to brain infarction. Hum. Mol. Genet. 16, 630–639. doi: 10.1093/hmg/ddm005
He, L., Xu, J., Chen, L., and Li, L. (2015). Apelin/APJ signaling in hypoxia-related diseases. Clin. Chim. Acta 451, 191–198. doi: 10.1016/j.cca.2015.09.029
Hosoya, M., Kawamata, Y., Fukusumi, S., Fujii, R., Habata, Y., Hinuma, S., et al. (2000). Molecular and functional characteristics of APJ. Tissue distribution of mRNA and interaction with the endogenous ligand apelin. J. Biol. Chem. 275, 21061–21067. doi: 10.1074/jbc.m908417199
Huang, C., Dai, C., Gong, K., Zuo, H., and Chu, H. (2016). Apelin-13 protects neurovascular unit against ischemic injuries through the effects of vascular endothelial growth factor. Neuropeptides doi: 10.1016/j.npep.2016.08.006
Jeong, K., Oh, Y., Kim, S. J., Kim, H., Park, K. C., Kim, S. S., et al. (2014). Apelin is transcriptionally regulated by ER stress-induced ATF4 expression via a p38 MAPK-dependent pathway. Apoptosis 19, 1399–1410. doi: 10.1007/s10495-014-1013-0
Jiao, H., Zhang, Z., Ma, Q., Fu, W., and Liu, Z. (2013). Mechanism underlying the inhibitory effect of Apelin-13 on glucose deprivation-induced autophagy in rat cardiomyocytes. Exp. Ther. Med. 5, 797–802. doi: 10.3892/etm.2013.902
Juhl, C., Els-Heindl, S., Schönauer, R., Redlich, G., Haaf, E., Wunder, F., et al. (2016). Development of potent and metabolically stable APJ ligands with high therapeutic potential. ChemMedChem 11, 2378–2384. doi: 10.1002/cmdc.201600307
Kalea, A. Z., and Batlle, D. (2010). Apelin and ACE2 in cardiovascular disease. Curr. Opin. Investig. Drugs 11, 273–282.
Khaksari, M., Aboutaleb, N., Nasirinezhad, F., Vakili, A., and Madjd, Z. (2012). Apelin-13 protects the brain against ischemic reperfusion injury and cerebral edema in a transient model of focal cerebral ischemia. J. Mol. Neurosci. 48, 201–208. doi: 10.1007/s12031-012-9808-3
Li, E., Deng, H., Wang, B., Fu, W., You, Y., and Tian, S. (2016). Apelin-13 exerts antidepressant-like and recognition memory improving activities in stressed rats. Eur. Neuropsychopharmacol. 26, 420–430. doi: 10.1016/j.euroneuro.2016.01.007
Li, H. C., and Zhang, G. Y. (2003). Inhibitory effect of genistein on activation of STAT3 induced by brain ischemia/reperfusion in rat hippocampus. Acta Pharmacol. Sin. 24, 1131–1136.
Liang, Z., Wu, G., Fan, C., Xu, J., Jiang, S., Yan, X., et al. (2016). The emerging role of signal transducer and activator of transcription 3 in cerebral ischemic and hemorrhagic stroke. Prog. Neurobiol. 137, 1–16. doi: 10.1016/j.pneurobio.2015.11.001
Liu, Q., Hu, T., He, L., Huang, X., Tian, X., Zhang, H., et al. (2015). Genetic targeting of sprouting angiogenesis using Apln-CreER. Nat. Commun. 6:6020. doi: 10.1038/ncomms7020
Lo, E. H. (2008). A new penumbra: transitioning from injury into repair after stroke. Nat. Med. 14, 497–500. doi: 10.1038/nm1735
Lv, X. R., Zheng, B., Li, S. Y., Han, A. L., Wang, C., Shi, J. H., et al. (2013). Synthetic retinoid Am80 up-regulates apelin expression by promoting interaction of RARα with KLF5 and Sp1 in vascular smooth muscle cells. Biochem. J. 456, 35–46. doi: 10.1042/bj20130418
Matsumoto, M., Hidaka, K., Akiho, H., Tada, S., Okada, M., and Yamaguchi, T. (1996). Low stringency hybridization study of the dopamine D4 receptor revealed D4-like mRNA distribution of the orphan seven-transmembrane receptor, APJ, in human brain. Neurosci. Lett. 219, 119–122. doi: 10.1016/s0304-3940(96)13198-0
McKinnie, S. M., Fischer, C., Tran, K. M., Wang, W., Mosquera, F., Oudit, G. Y., et al. (2016). The metalloprotease neprilysin degrades and inactivates apelin peptides. ChemBioChem 17, 1495–1498. doi: 10.1002/cbic.201600244
Medhurst, A. D., Jennings, C. A., Robbins, M. J., Davis, R. P., Ellis, C., Winborn, K. Y., et al. (2003). Pharmacological and immunohistochemical characterization of the APJ receptor and its endogenous ligand apelin. J. Neurochem. 84, 1162–1172. doi: 10.1046/j.1471-4159.2003.01587.x
Mei, Y., Thompson, M. D., Cohen, R. A., and Tong, X. (2015). Autophagy and oxidative stress in cardiovascular diseases. Biochim. Biophys. Acta 1852, 243–251. doi: 10.1016/j.bbadis.2014.05.005
Mesmin, C., Fenaille, F., Becher, F., Tabet, J. C., and Ezan, E. (2011). Identification and characterization of apelin peptides in bovine colostrum and milk by liquid chromatography-mass spectrometry. J. Proteome. Res. 10, 5222–5231. doi: 10.1021/pr200725x
Morimoto, N., Oida, Y., Shimazawa, M., Miura, M., Kudo, T., Imaizumi, K., et al. (2007). Involvement of endoplasmic reticulum stress after middle cerebral artery occlusion in mice. Neuroscience 147, 957–967. doi: 10.1016/j.neuroscience.2007.04.017
Moskowitz, M. A., Lo, E. H., and Iadecola, C. (2010). The science of stroke: mechanisms in search of treatments. Neuron 67, 181–198. doi: 10.1016/j.neuron.2010.08.019
Mozaffarian, D., Benjamin, E. J., Go, A. S., Arnett, D. K., Blaha, M. J., Cushman, M., et al. (2016). Heart disease and stroke statistics-2016 update: a report from the American Heart Association. Circulation 133, e38–360. doi: 10.1161/CIR.0000000000000350
Nakka, V. P., Gusain, A., and Raghubir, R. (2010). Endoplasmic reticulum stress plays critical role in brain damage after cerebral ischemia/reperfusion in rats. Neurotox. Res. 17, 189–202. doi: 10.1007/s12640-009-9110-5
Novakova, V., Sandhu, G. S., Dragomir-Daescu, D., and Klabusay, M. (2016). Apelinergic system in endothelial cells and its role in angiogenesis in myocardial ischemia. Vascul. Pharmacol. 76, 1–10. doi: 10.1016/j.vph.2015.08.005
O’Carroll, A. M., Lolait, S. J., and Howell, G. M. (2006). Transcriptional regulation of the rat apelin receptor gene: promoter cloning and identification of an Sp1 site necessary for promoter activity. J. Mol. Endocrinol. 36, 221–235. doi: 10.1677/jme.1.01927
O’Carroll, A. M., Lolait, S. J., Harris, L. E., and Pope, G. R. (2013). The apelin receptor APJ: journey from an orphan to a multifaceted regulator of homeostasis. J. Endocrinol. 219, R13–R35. doi: 10.1530/joe-13-0227
Pisarenko, O., Shulzhenko, V., Studneva, I., Pelogeykina, Y., Timoshin, A., Anesia, R., et al. (2015). Structural apelin analogues: mitochondrial ROS inhibition and cardiometabolic protection in myocardial ischaemia reperfusion injury. Br. J. Pharmacol. 172, 2933–2945. doi: 10.1111/bph.13038
Rastaldo, R., Cappello, S., Folino, A., and Losano, G. (2011). Effect of apelin-apelin receptor system in postischaemic myocardial protection: a pharmacological postconditioning tool? Antioxid. Redox Signal. 14, 909–922. doi: 10.1089/ars.2010.3355
Ronkainen, V. P., Ronkainen, J. J., Hänninen, S. L., Leskinen, H., Ruas, J. L., Pereira, T., et al. (2007). Hypoxia inducible factor regulates the cardiac expression and secretion of apelin. FASEB J. 21, 1821–1830. doi: 10.1096/fj.06-7294com
Sheikh, A. Y., Chun, H. J., Glassford, A. J., Kundu, R. K., Kutschka, I., Ardigo, D., et al. (2008). In vivo genetic profiling and cellular localization of apelin reveals a hypoxia-sensitive, endothelial-centered pathway activated in ischemic heart failure. Am. J. Physiol. Heart Circ. Physiol. 294, H88–H98. doi: 10.1152/ajpheart.00935.2007
Sheng, R., Liu, X. Q., Zhang, L. S., Gao, B., Han, R., Wu, Y. Q., et al. (2012). Autophagy regulates endoplasmic reticulum stress in ischemic preconditioning. Autophagy 8, 310–325. doi: 10.4161/auto.18673
Shin, K., Pandey, A., Liu, X. Q., Anini, Y., and Rainey, J. K. (2013). Preferential apelin-13 production by the proprotein convertase PCSK3 is implicated in obesity. FEBS Open Bio 3, 328–333. doi: 10.1016/j.fob.2013.08.001
Tao, J., Zhu, W., Li, Y., Xin, P., Li, J., Liu, M., et al. (2011). Apelin-13 protects the heart against ischemia-reperfusion injury through inhibition of ER-dependent apoptotic pathways in a time-dependent fashion. Am. J. Physiol. Heart Circ. Physiol. 301, H1471–H1486. doi: 10.1152/ajpheart.00097.2011
Tatemoto, K., Hosoya, M., Habata, Y., Fujii, R., Kakegawa, T., Zou, M. X., et al. (1998). Isolation and characterization of a novel endogenous peptide ligand for the human APJ receptor. Biochem. Biophys. Res. Commun. 251, 471–476. doi: 10.1006/bbrc.1998.9489
Than, A., Zhang, X., Leow, M. K., Poh, C. L., Chong, S. K., and Chen, P. (2014). Apelin attenuates oxidative stress in human adipocytes. J. Biol. Chem. 289, 3763–3774. doi: 10.1074/jbc.m113.526210
Wang, G., Kundu, R., Han, S., Qi, X., Englander, E. W., Quertermous, T., et al. (2009). Ontogeny of apelin and its receptor in the rodent gastrointestinal tract. Regul. Pept. 158, 32–39. doi: 10.1016/j.regpep.2009.07.016
Wang, W., McKinnie, S. M., Farhan, M., Paul, M., McDonald, T., McLean, B., et al. (2016). Angiotensin-converting enzyme 2 metabolizes and partially inactivates pyr-apelin-13 and apelin-17: physiological effects in the cardiovascular system. Hypertension 68, 365–377. doi: 10.1161/HYPERTENSIONAHA.115.06892
Wang, W., McKinnie, S. M., Patel, V. B., Haddad, G., Wang, Z., Zhabyeyev, P., et al. (2013). Loss of Apelin exacerbates myocardial infarction adverse remodeling and ischemia-reperfusion injury: therapeutic potential of synthetic Apelin analogues. J. Am. Heart Assoc. 2:e000249. doi: 10.1161/JAHA.113.000249
Wang, H., Naghavi, M., Allen, C., Barber, R. M., Bhutta, Z. A., Carter, A., et al. (2016). Global, regional and national life expectancy, all-cause mortality and cause-specific mortality for 249 causes of death, 1980–2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet 388, 1459–1544. doi: 10.1016/s0140-6736(16)31012-1
Wang, G., Qi, X., Wei, W., Englander, E. W., and Greeley, G. H. Jr. (2006). Characterization of the 5′-regulatory regions of the rat and human apelin genes and regulation of breast apelin by USF. FASEB J. 20, 2639–2641. doi: 10.1096/fj.06-6315fje
Wang, R., Ying, Z., Zhao, J., Zhang, Y., Lu, H., Deng, Y., et al. (2012). Lys(203) and Lys(382) are essential for the proteasomal degradation of BACE1. Curr. Alzheimer Res. 9, 606–615. doi: 10.2174/156720512800618026
Wang, Z., Yu, D., Wang, M., Wang, Q., Kouznetsova, J., Yang, R., et al. (2015). Elabela-apelin receptor signaling pathway is functional in mammalian systems. Sci. Rep. 5:8170. doi: 10.1038/srep08170
Woo, S. K., Kwon, M. S., Geng, Z., Chen, Z., Ivanov, A., Bhatta, S., et al. (2012). Sequential activation of hypoxia-inducible factor 1 and specificity protein 1 is required for hypoxia-induced transcriptional stimulation of Abcc8. J. Cereb. Blood Flow Metab. 32, 525–536. doi: 10.1038/jcbfm.2011.159
Wu, Y., Xu, Q., and Song, W. (2014). Oxidative stress and Alzheimer’s disease. Systems Biology of Free Radicals and Antioxidants 2147–2174. doi: 10.1007/978-3-642-30018-9_197
Xin, Q., Cheng, B., Pan, Y., Liu, H., Yang, C., Chen, J., et al. (2015). Neuroprotective effects of apelin-13 on experimental ischemic stroke through suppression of inflammation. Peptides 63, 55–62. doi: 10.1016/j.peptides.2014.09.016
Xin, Q., Ji, B., Cheng, B., Wang, C., Liu, H., Chen, X., et al. (2014). Endoplasmic reticulum stress in cerebral ischemia. Neurochem. Int. 68, 18–27. doi: 10.1016/j.neuint.2014.02.001
Yan, X. G., Cheng, B. H., Wang, X., Ding, L. C., Liu, H. Q., Chen, J., et al. (2015). Lateral intracerebroventricular injection of Apelin-13 inhibits apoptosis after cerebral ischemia/reperfusion injury. Neural. Regen. Res. 10, 766–771. doi: 10.4103/1673-5374.157243
Yang, S., Li, H., Tang, L., Ge, G., Ma, J., Qiao, Z., et al. (2015). Apelin-13 protects the heart against ischemia-reperfusion injury through the RISK-GSK-3β-mPTP pathway. Arch. Med. Sci. 11, 1065–1073. doi: 10.5114/aoms.2015.54863
Yang, Y., Zhang, X., Cui, H., Zhang, C., Zhu, C., and Li, L. (2014). Apelin-13 protects the brain against ischemia/reperfusion injury through activating PI3K/Akt and ERK1/2 signaling pathways. Neurosci. Lett. 568, 44–49. doi: 10.1016/j.neulet.2014.03.037
Yeh, S. H., Yang, W. B., Gean, P. W., Hsu, C. Y., Tseng, J. T., Su, T. P., et al. (2011). Translational and transcriptional control of Sp1 against ischaemia through a hydrogen peroxide-activated internal ribosomal entry site pathway. Nucl. Acids Res. 39, 5412–5423. doi: 10.1093/nar/gkr161
Yu, L., Chen, C., Wang, L. F., Kuang, X., Liu, K., Zhang, H., et al. (2013). Neuroprotective effect of kaempferol glycosides against brain injury and neuroinflammation by inhibiting the activation of NF-kappaB and STAT3 in transient focal stroke. PLoS One 8:e55839. doi: 10.1371/journal.pone.0055839
Zhang, H., Gong, Y., Wang, Z., Jiang, L., Chen, R., Fan, X., et al. (2014a). Apelin inhibits the proliferation and migration of rat PASMCs via the activation of PI3K/Akt/mTOR signal and the inhibition of autophagy under hypoxia. J. Cell. Mol. Med. 18, 542–553. doi: 10.1111/jcmm.12208
Zhang, S., Zis, O., Ly, P. T., Wu, Y., Zhang, M., Cai, F., et al. (2014b). Down-regulation of MIF by NFκB under hypoxia accelerated neuronal loss during stroke. FASEB J. 28, 4394–4407. doi: 10.1096/fj.14-253625
Zhang, Z., Yu, B., and Tao, G. Z. (2009). Apelin protects against cardiomyocyte apoptosis induced by glucose deprivation. Chin. Med. J. 122, 2360–2365.
Zhu, Y. M., Wang, C. C., Chen, L., Qian, L. B., Ma, L. L., Yu, J., et al. (2013). Both PI3K/Akt and ERK1/2 pathways participate in the protection by dexmedetomidine against transient focal cerebral ischemia/reperfusion injury in rats. Brain Res. 1494, 1–8. doi: 10.1016/j.brainres.2012.11.047
Keywords: apelin, apelin receptor (APLNR), ischemic stroke, temporal expression, neuroprotection
Citation: Wu Y, Wang X, Zhou X, Cheng B, Li G and Bai B (2017) Temporal Expression of Apelin/Apelin Receptor in Ischemic Stroke and its Therapeutic Potential. Front. Mol. Neurosci. 10:1. doi: 10.3389/fnmol.2017.00001
Received: 24 October 2016; Accepted: 04 January 2017;
Published: 23 January 2017.
Edited by:
Detlev Boison, Legacy Health, USAReviewed by:
Samaneh Maysami, University of Manchester, UKPhilip Forsyth Copenhaver, Oregon Health & Science University, USA
Copyright © 2017 Wu, Wang, Zhou, Cheng, Li and Bai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution and reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yili Wu, eWlsaV93dTIwMDRAeWFob28uY2E=
Bo Bai, YmJhaUBtYWlsLmpubWMuZWR1LmNu