 Marcus O. W. Grimm
Marcus O. W. Grimm Janine Mett
Janine Mett Heike S. Grimm1
Heike S. Grimm1 Tobias Hartmann
Tobias Hartmann- 1Experimental Neurology, Saarland University, Homburg/Saar, Germany
- 2Neurodegeneration and Neurobiology, Saarland University, Homburg/Saar, Germany
- 3Deutsches Institut für DemenzPrävention (DIDP), Saarland University, Homburg/Saar, Germany
Extracellular neuritic plaques, composed of aggregated amyloid-β (Aβ) peptides, are one of the major histopathological hallmarks of Alzheimer’s disease (AD), a progressive, irreversible neurodegenerative disorder and the most common cause of dementia in the elderly. One of the most prominent risk factor for sporadic AD, carrying one or two aberrant copies of the apolipoprotein E (ApoE) ε4 alleles, closely links AD to lipids. Further, several lipid classes and fatty acids have been reported to be changed in the brain of AD-affected individuals. Interestingly, the observed lipid changes in the brain seem not only to be a consequence of the disease but also modulate Aβ generation. In line with these observations, protective lipids being able to decrease Aβ generation and also potential negative lipids in respect to AD were identified. Mechanistically, Aβ peptides are generated by sequential proteolytic processing of the amyloid precursor protein (APP) by β- and γ-secretase. The α-secretase appears to compete with β-secretase for the initial cleavage of APP, preventing Aβ production. All APP-cleaving secretases as well as APP are transmembrane proteins, further illustrating the impact of lipids on Aβ generation. Beside the pathological impact of Aβ, accumulating evidence suggests that Aβ and the APP intracellular domain (AICD) play an important role in regulating lipid homeostasis, either by direct effects or by affecting gene expression or protein stability of enzymes involved in the de novo synthesis of different lipid classes. This review summarizes the current literature addressing the complex bidirectional link between lipids and AD and APP processing including lipid alterations found in AD post mortem brains, lipids that alter APP processing and the physiological functions of Aβ and AICD in the regulation of several lipid metabolism pathways.
Alzheimer’S Disease
Worldwide currently there are more than 46 million people suffering from dementia and the number of affected individuals is estimated to double every 20 years. Alzheimer’s disease (AD) is a devastating neurodegenerative disorder, which is the most common cause of dementia in the elderly population. Clinically AD is characterized by a progressive loss of cognitive brain functions leading to memory dysfunction, impaired judgment, disorientation and finally to a total loss of memory and personality (Plassman et al., 2007; World Alzheimer Report, 2015). AD-patients typically die in average within 3–10 years after diagnosis due to secondary disorders (Zanetti et al., 2009). The clinical symptoms of AD might be caused by an extensive loss of synapses and neurons leading to a strong hippocampal and cortical atrophy (Scheff and Price, 1993; Gómez-Isla et al., 1996; Mouton et al., 1998; Dickerson et al., 2001). The characteristic neuropathological hallmarks of the disease are intracellular neurofibrillary tangles (NFTs) and extracellular localized amyloid plaques. While the NFTs are composed of the microtubuli-associated protein tau in a hyperphosphorylated state (Grundke-Iqbal et al., 1986a,b), the amyloid plaques are mainly built up of amyloid-β (Aβ) peptides. Aβ-peptides are hydrophobic, 38–43 amino acid long products generated by the sequential proteolytic processing of the amyloid precursor protein (APP; Glenner and Wong, 1984; Masters et al., 1985; Kang et al., 1987). The significant cerebral accumulation of Aβ, starting several years prior to the first symptoms, is respected to trigger the disease process (Glenner and Wong, 1984; Glenner, 1989; Hardy and Higgins, 1992; Hardy and Selkoe, 2002). Especially the accumulation of Aβ42 (indicating 42 amino acids), which is the major Aβ species found in neuritic plaques, is considered to initiate AD progression (Iwatsubo et al., 1994; Tamaoka et al., 1995). Due to the additional hydrophobic amino acids isoleucine and alanine Aβ42 has a higher tendency to aggregate compared to the more prevalent Aβ40 (indicating 40 amino acids; Jarrett et al., 1993). Increasing evidence suggests small oligomers of Aβ to represent the most toxic form of the peptide (Lambert et al., 1998; Lesné et al., 2006; Shankar et al., 2008). Several mechanisms are discussed to contribute to Aβ neurotoxicity, among them the induction of inflammatory processes, a disruption of calcium homeostasis and membrane integrity, cholinergic and mitochondrial dysfunction and increased oxidative stress (Grimm and Hartmann, 2012).
There are two forms of AD, the more common sporadic AD with a disease onset after the age of 65 (late onset AD, LOAD) and the genetically based form (familial AD, FAD) with an earlier manifestation of symptoms. The two variants are basically distinguishable from each other in clinical and neuropathological terms. Less than 5% of all AD-cases belong to FAD which is caused by mutations in the genes encoding for APP and the presenilins (PS) 1 and 2, proteins involved in proteolytic APP-processing (Levy et al., 1990; Goedert et al., 1994; Levy-Lahad et al., 1995; Sherrington et al., 1995; Tanzi, 2012). Besides aging, hypercholesterolemia, hypertension, atherosclerosis, homocysteinemia, diabetes mellitus and obesity are discussed as non-genetic risk factors for LOAD (Barnes and Yaffe, 2011; Polidori et al., 2012). The ε4 allele of the apolipoprotein E (ApoE) has been identified as the most important genetic risk factor for the sporadic form of the disease (Corder et al., 1993; Strittmatter et al., 1993).
As already mentioned, Aβ is generated by proteolytic processing of the precursor protein APP. APP is a ubiquitously expressed type I-transmembrane protein cycling between the plasma membrane and acidic intracellular compartments (Haass et al., 1992; Koo and Squazzo, 1994; Thinakaran and Koo, 2008). It consists of a large ectodomain, a single transmembrane domain and a short intracellular part. APP belongs to an evolutionary conserved protein family including the APP-like proteins 1 and 2 (APLP1, APLP2) in mammals. APP can be sequentially cleaved via two different pathways (Haass et al., 1992; Thinakaran and Koo, 2008; De Strooper, 2010; Figure 1). In the predominant non-amyloidogenic processing pathway the generation of Aβ is precluded. It is initiated by the α-secretase dependent cleavage of APP within the Aβ-domain shedding off the soluble ectodomain sAPPα and generating the membrane-anchored C-terminal fragment (CTF) C83 (indicating 83 amino acids). Members of the ADAM (a disintegrin and metalloprotease) protein family have been identified as catalytically active α-secretases with ADAM10 representing the physiologically relevant, constitutive α-secretase in neurons (Lammich et al., 1999; Kuhn et al., 2010). In contrast, the aspartyl protease β-site APP cleaving enzyme 1 (BACE1) initiates the amyloidogenic APP-processing pathway generating the membrane-spanning CTF C99 (indicating 99 amino acids) and releasing sAPPβ into the extracellular space (Vassar et al., 1999). The two alternative pathways differ in their subcellular localization: due to the acidic pH-optimum of BACE1 the amyloidogenic APP-processing is localized in acidic intracellular compartments, while non-amyloidogenic APP-processing mainly takes place at the cell surface (Parvathy et al., 1999; Grbovic et al., 2003; Carey et al., 2005). In both pathways the CTFs are subsequently processed by the γ-secretase complex, which consists of the proteins PS1 or PS2 as the catalytic core, Aph1 (anterior pharynx defective 1) a or b, PEN2 (presenilin enhancer 2) and nicastrin (Baulac et al., 2003; Edbauer et al., 2003; Kimberly et al., 2003). The γ-secretase possesses the unusual property to cleave its substrates within their transmembrane domains after shedding off the ectodomain, a process called regulated intramembrane proteolysis (RIP; Brown et al., 2000; Lichtenthaler et al., 2011). This catalytic activity leads to the generation of the non-toxic peptide p3 out of C83 and of Aβ out of C99 combined with the release of APP intracellular domain (AICD) into the cytosol in both processing pathways (Passer et al., 2000; Kakuda et al., 2006; Grimm and Hartmann, 2012). Due to multiple γ-secretase cleavage sites within the transmembrane domain of APP, the generated Aβ- and AICD-peptides can vary in length (Funamoto et al., 2004; Qi-Takahara et al., 2005; Kakuda et al., 2006). AICD is reported to translocate to the nucleus and to regulate the transcription of target genes, among them the genes encoding for APP, BACE1, the Aβ-degrading protease neprilysin (NEP) as well as several enzymes involved in lipid metabolism (Cao and Südhof, 2001; von Rotz et al., 2004; Grimm et al., 2011b,d, 2012c, 2013, 2015a).
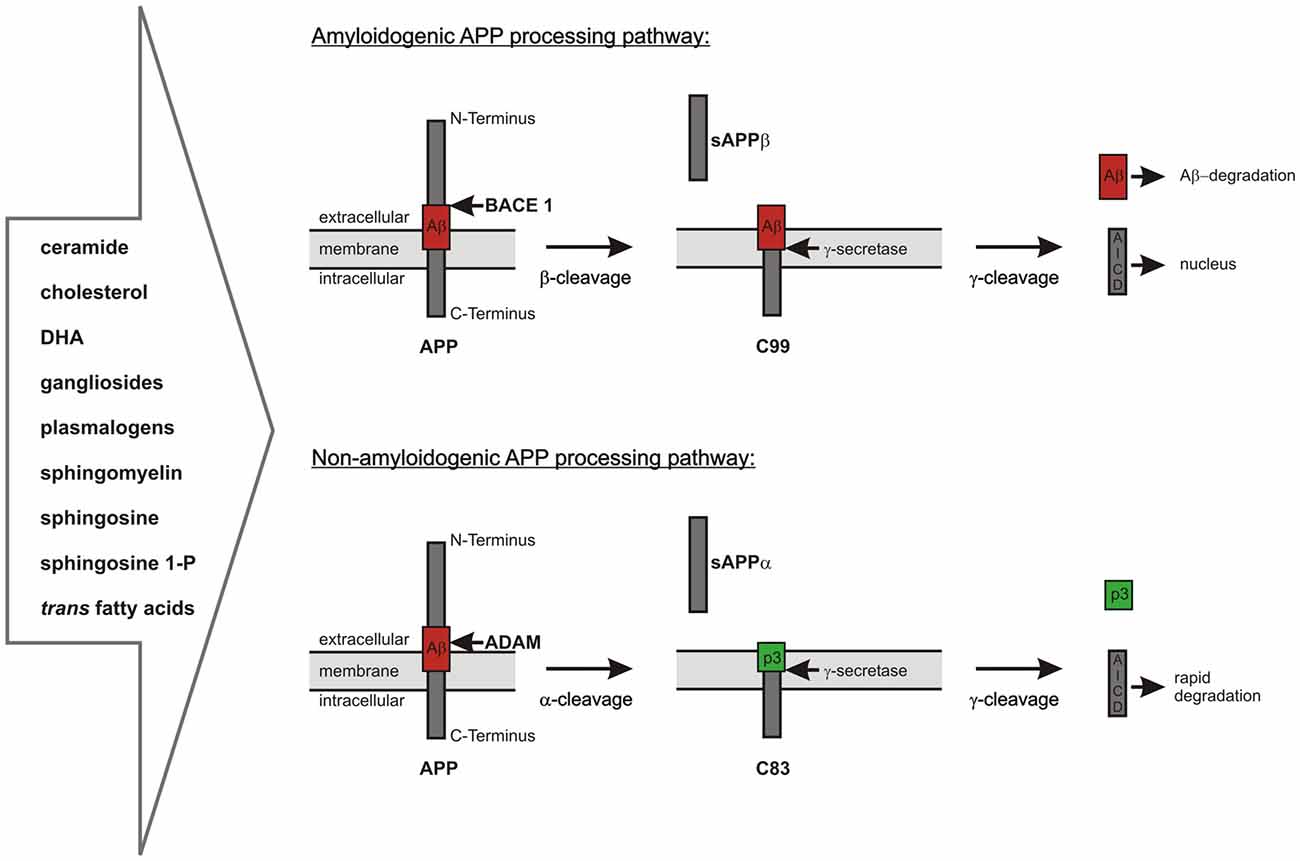
Figure 1. Overview of the two alternative amyloid precursor protein (APP) processing pathways, which are highly influenced by lipid homeostasis. Amyloidogenic pathway: APP is first cleaved by the β-secretase β-site APP cleaving enzyme 1 (BACE1) resulting in the release of sAPPβ and the generation of C99, which is further processed by the γ-secretase complex to amyloid-β (Aβ)-peptides and the APP intracellular domain (AICD). The neurotoxic Aβ-peptides can be cleared by different mechanisms including enzymatic degradation. The intracellular AICD-domain is known to translocate into the nucleus and to regulate the transcription of several target genes. Non-amyloidogenic pathway: APP is cleaved by the α-secretases belonging to the ADAM protein family within the Aβ-domain. This results in the release of sAPPα into the extracellular space and the formation of C83. C83 is further processed by the γ-secretase complex resulting in the release of the non-toxic peptide p3 into the extracellular space and of AICD into the cytosol. In contrast to the AICD generated by amyloidogenic APP processing the AICD derived from α-/γ-secretase-dependent APP processing is rapidly degraded in the cytosol and transcriptionally inactive.
Link Between Lipids and AD
A link between AD pathology and lipids was already observed more than a century ago by Alois Alzheimer, who described a higher occurrence of “adipose inclusions” or “lipoid granules” in post mortem AD-brain tissue as a third pathological hallmark of the disease (Foley, 2010). In the meantime the content of several lipid classes and fatty acids has been found to be altered in the brain of AD-patients. A physiological function of Aβ and AICD in the regulation of several lipid metabolism pathways has been reported, possibly explaining the altered cerebral content of some lipid species in AD-affected brain tissue. Inversely, APP-processing is strongly influenced by the surrounding lipid environment indicating a bidirectional link between APP-proteolysis and lipid metabolism (Grimm et al., 2012b; Mett et al., 2014).
The link between lipid homeostasis and AD-pathology is strengthened by the identification of the ApoEε4-allele as the most important genetic risk factor for LOAD. ApoE is a lipoprotein involved in the transport of cholesterol and other lipids in the central nervous system (CNS). In humans there are three different ApoEε alleles encoding for the isoforms ApoEε2, ApoEε3 and ApoEε4 (Weisgraber et al., 1981; Mahley et al., 1996; Holtzman et al., 2012). The ApoEε4-allele is associated with an increased AD-risk, earlier disease onset and enhanced cerebral plaque load (Corder et al., 1993; Kuusisto et al., 1994; Breitner et al., 1999; Tiraboschi et al., 2004). In contrast, ApoEε2-carriers have a reduced risk of developing AD (Corder et al., 1994). These associations might be explained by an isoform-dependent binding of ApoEε (ε2 > ε3 > ε4) to Aβ-peptides influencing the clearance and aggregation of the peptide (Ma et al., 1994; Deane et al., 2008; Castellano et al., 2011; Holtzman et al., 2012).
A strong impact of the surrounding lipid bilayer on APP-processing is given by the fact that APP as well as all secretases are transmembrane proteins and that γ-secretase dependent APP cleavage even takes place in the hydrophobic membrane environment. For example, the exact position of γ-secretase cleavage and hence the length of the generated Aβ-peptides depends on membrane thickness (Grziwa et al., 2003; Winkler et al., 2012). In addition, the membrane fluidity influences APP-processing. Increased membrane fluidity seems to stimulate the non-amyloidogenic APP-processing by reducing APP internalization (Kojro et al., 2001). In this context it is important to note that APP-processing is also influenced by the subcompartmentalization of the membrane. Lipid raft microdomains are compact, dynamic assemblies of membrane proteins enriched in cholesterol, gangliosides and other sphingolipids. They are detergent-resistent and strongly differ in their lipid composition from the surrounding non-raft domains. Implications of lipid rafts in the intracellular protein trafficking, protein-lipid and protein-protein interactions as well as transmembrane signaling have been reported (Brown and Rose, 1992; Lingwood and Simons, 2010). The generation of Aβ has been shown to mainly take place in lipid rafts due to the co-localization of APP with BACE1 and the γ-secretase complex within these membrane microdomains (Lee et al., 1998; Riddell et al., 2001; Ehehalt et al., 2003; Vetrivel et al., 2004). In contrast, the non-amyloidogenic APP-proteolysis seems to occur predominantly in non-raft regions (Ehehalt et al., 2003; Harris et al., 2009). All these details indicate that a modulation of the membrane lipid composition might provide the opportunity of influencing Aβ-generation.
In the following sections of this article, the impact of several lipids and fatty acids on Aβ-associated AD-pathology is reviewed as well as the regulation of the corresponding metabolic pathways by APP-processing.
The Impact of Cholesterol on AD
The brain is the most cholesterol-rich organ in the body (23 mg/g), it contains 23% of the total body sterol while only accounting for 2.1% of the total body weight (Dietschy and Turley, 2004). Within brain tissue cholesterol is mainly present in myelin sheaths and in the membranes of glial cells and neurons in its unesterified form. Due to the limited transport of cholesterol across the blood-brain barrier the cerebral cholesterol level is mainly dependent on de novo synthesis by oligodendrocytes, astrocytes and to a lesser extent by neurons. The conversion of 3-hydroxy-3-methylglutaryl-CoA to mevalonate catalyzed by the hydroxymethylglutaryl-CoA reductase (HMGCR), which is inhibited by statins, is the rate-controlling step in cholesterol biosynthesis (Martins et al., 2009; Di Paolo and Kim, 2011). The first evidence for a link between AD-pathogenesis and cholesterol metabolism was provided in 1994 by the observation that dietary cholesterol increases Aβ-production in rabbits (Sparks et al., 1994). Today there are many lines of evidence arguing for a connection between the pathology of AD and cholesterol homeostasis which are summarized below.
In several epidemiological studies elevated serum/plasma cholesterol contents have been identified as a risk factor for developing AD. Especially high serum cholesterol level in midlife are associated with a higher AD-risk (Pappolla et al., 2003; Solomon et al., 2009; Matsuzaki et al., 2011; Meng et al., 2014). Additionally, enhanced level of low-density lipoprotein (LDL) cholesterol and reduced level of high-density lipoprotein (HDL) cholesterol in serum correlate with the cerebral amyloid deposition in living human beings (Reed et al., 2014). In line, in human post mortem AD-brains cholesterol was found to be elevated and highly enriched in amyloid plaques (Cutler et al., 2004; Xiong et al., 2008; Panchal et al., 2010).
Most cell culture studies revealed that increasing cellular cholesterol level lead to an enhanced Aβ production whereas a depletion or reduction of cholesterol by e.g., cyclodextrin or statins shows the opposite effect (Simons et al., 1998; Fassbender et al., 2001; Maulik et al., 2013). The Aβ increasing property of cholesterol is based on a direct activation of β- and γ-secretase proteolytic activity (Kalvodova et al., 2005; Grimm et al., 2008; Osenkowski et al., 2008). Cholesterol is enriched in lipid raft membrane microdomains, in which amyloidogenic APP-processing mainly takes place. Thus modulating cellular cholesterol content inevitably affects membrane structure, membrane fluidity as well as cellular processes associated with lipid raft microdomains. Cholesterol depletion leads to the disruption of lipid rafts and therefore to a reduced association of APP, BACE1 and the components of the γ-secretase complex to lipid raft membrane microdomains, resulting in decreased amyloidogenic APP processing. Vice versa, an increase of cellular cholesterol leads to a higher lipid raft content of the membranes and hence to Aβ-overproduction (Simons et al., 1998; Hao et al., 2001; Hicks et al., 2012). High membrane cholesterol levels additionally promote APP endocytosis leading to enhanced Aβ-production in acidic intracellular compartments (Cossec et al., 2010). Conversely, APP is primarily localized at the cell surface in cholesterol-depleted cells leading to increased α-secretase-dependent non-amyloidogenic APP processing (Kojro et al., 2001). Beside the cholesterol-mediated effects on APP-proteolytic processing, cholesterol has been shown to promote Aβ-aggregation and -toxicity (Schneider et al., 2006; Ferrera et al., 2008; Abramov et al., 2011).
A strong correlation between hypercholesterolemia and enhanced Aβ level has also been observed in several animal models (Sparks et al., 1994; Refolo et al., 2000; Maulik et al., 2013). Inversely, a reduction of accumulated Aβ-peptides along with improved behavioral memory was achieved in animal models after administration of cholesterol-lowering drugs including statins (Fassbender et al., 2001; Refolo et al., 2001; Kurata et al., 2012). It should be noted, that there are also a few studies in which statins had no or oppositional effects on the cerebral Aβ-content in vivo (Park et al., 2003; Cibickova et al., 2009).
The impact of statins on AD has also been analyzed in observational studies and randomized controlled trials leading to inhomogeneous results. Statin intake is associated with a reduced incidence of AD or dementia in general in most, but not all of these studies (Wolozin et al., 2000, 2007; Rea et al., 2005; Arvanitakis et al., 2008; Haag et al., 2009). Especially the reduction of serum cholesterol level by the intake of statins in midlife might have a preventive effect towards the development of AD (Kivipelto et al., 2002; Pappolla et al., 2003; Shinohara et al., 2014). In strong contrast, most clinical trials failed to observe any benefit of statins in individuals already suffering from AD (Feldman et al., 2010; Sano et al., 2011; McGuinness et al., 2014), indicating cholesterol-lowering drugs to have rather a protective than a therapeutic potential in respect to AD.
Beside the described influence of cholesterol on APP-proteolysis, there is also an impact of APP-processing on cholesterol homeostasis. APP/APLP2- and PS1/PS2-deficient fibroblasts have a significantly increased cellular cholesterol content, which can be reversed by the supplementation of Aβ40-peptides. In line with this, enhanced cerebral cholesterol concentrations were found in APP- and PS-deficient mice (Grimm et al., 2005; Umeda et al., 2010). Analysis of the underlying mechanisms revealed that Aβ40 reduces cholesterol de novo synthesis by inhibiting HMGCR activity (Grimm et al., 2005).
Summary
The existence of a regulatory feedback cycle, in which Aβ-production is stimulated by cholesterol while cholesterol de novo synthesis is inhibited by high cellular Aβ40-concentrations is indicated.
Future Directions
The heterogeneous results of studies analyzing the impact of statins on the incidence of AD denote the existence of responders and non-responders. For the future it will be important to find biomarkes to identify patients that might profit from statins.
The Impact of Docosahexaenoic Acid (DHA) on AD
Docosahexaenoic acid (DHA, 22:6) is a polyunsaturated fatty acid (PUFA) naturally occurring in high amounts in marine food, especially in fish oil (Mann et al., 2010). It accounts for 30%–40% of all esterified fatty acids in neuronal plasma membrane phospholipids and for 8% of the brain dry weight, thus belonging together with α-linolenic acid (ALA, 18:3) and eicosapentaenoic acid (EPA, 20:5) to the most important ω3-fatty acids in the CNS (Lauritzen et al., 2001; Muskiet et al., 2006). As endogenous DHA-biosynthesis is highly limited in humans, the main part of this fatty acid is provided by dietary intake (Pawlosky et al., 2001). DHA is efficiently transported across the blood brain barrier (Ouellet et al., 2009; Nguyen et al., 2014) and rapidly incorporates into phospholipids of cellular membranes leading to increased membrane fluidity (Horrocks and Farooqui, 2004; Yang et al., 2011).
The DHA content is reported to be reduced in the serum/plasma of AD-patients as well as in certain regions of post mortem AD-brains (Söderberg et al., 1991; Conquer et al., 2000; Tully et al., 2003). Because of its six double-bonds DHA is very susceptible to lipid-peroxidation resulting in oxidative stress known to be involved in AD pathogenesis (Smith et al., 1994; Yatin et al., 1998; Fam et al., 2002; Cai et al., 2011). Indeed, the levels of PUFA oxidation products are elevated in AD-affected brains, indicating the reduced DHA content in these tissues to be caused by increased oxidative damage (Sayre et al., 1997; Markesbery and Lovell, 1998; Montine and Morrow, 2005; Grimm et al., 2016a).
Several epidemiological trials found the dietary intake of DHA or higher DHA serum/plasma levels to be associated with a reduced risk of developing AD indicating a potential of DHA in AD-prevention (Kalmijn et al., 1997; Barberger-Gateau et al., 2002; Morris et al., 2003b). However, other studies failed to find an association between PUFAs and AD-risk (Engelhart et al., 2002; Kröger et al., 2009; Jicha and Markesbery, 2010; Mett et al., 2014).
We and others analyzed the impact of DHA on APP-processing revealing the fatty acid to reduce Aβ-levels via pleiotropic mechanisms. DHA reduces β- and γ-secretase activity and stimulates α-secretase-dependent APP-cleavage. In addition to direct effects, the activities of γ- and β-secretase are reduced by DHA due to a PS1-displacement out of lipid rafts and a reduced BACE1 internalization. The stimulated α-secretase activity in presence of DHA is based on the enhanced gene expression and protein stability of ADAM17. Altogether these effects lead to a shift from amyloidogenic to non-amyloidogenic APP-processing and thus to reduced total Aβ-level. DHA additionally has cholesterol-lowering effects further inhibiting Aβ-production. It reduces cholesterol de novo synthesis via inhibition of HMGCR and disturbs lipid raft integrity by shifting cholesterol out of these membrane microdomains (Hashimoto et al., 2005a; Stillwell et al., 2005; Grimm et al., 2011c). Beside the described effects on APP-processing an impact of DHA on Aβ-degradation and -aggregation has also been reported. We recently observed a highly enhanced insulin-degrading enzyme (IDE)-dependent Aβ-degradation in neuroblastoma cells after the supplementation of DHA- and EPA-containing phosphatidylcholine (PC; Grimm et al., 2016b). Others reported an increased microglial phagocytosis of Aβ as well as a reduction of Aβ-fibrillation and Aβ-induced toxicity in the presence of DHA (Hossain et al., 2009; Hjorth et al., 2013).
A protective effect of dietary DHA with regard to cerebral Aβ-level and amyloid plaque load could be further confirmed in vivo in several animal models (Lim et al., 2005; Green et al., 2007; Perez et al., 2010). In line with this, higher cognitive performances were observed in AD-animal models after DHA supplementation (Hashimoto et al., 2002, 2005b; Calon et al., 2004). However, others failed to find any beneficial effect of DHA in AD transgenic mice (Arendash et al., 2007).
A possible therapeutic use of DHA regarding AD has been investigated in several clinical trials showing inconsistent results. Some studies revealed a beneficial effect of daily DHA treatment in patients with very mild cognitive dysfunctions (Freund-Levi et al., 2006; Kotani et al., 2006; Chiu et al., 2008). Others did not observe any influence of DHA on AD-biomarkers and cognitive decline in AD patients (Freund-Levi et al., 2009; Quinn et al., 2010). It should be mentioned, that oxidized DHA species and the lipid-peroxidation products of PUFAs are able to increase amyloidogenic APP-processing and hence Aβ-generation. In a recent study we demonstrated, that only 1% oxidized DHA reverts the positives effects of DHA on Aβ-production indicating that PUFAs have to be prevented from oxidation in nutritional approaches (Grimm et al., 2016a). In such approaches DHA often is combined with E-vitamins due to their high antioxidative properties acting as scavengers of radicals and peroxides (Kamal-Eldin and Appelqvist, 1996). However, we demonstrated in two recent studies that several tocopherol and tocotrienol species have beside their protective antioxidative properties the undesirable effect of increasing amyloidogenic APP processing and reducing the enzymatic degradation of Aβ-peptides (Grimm et al., 2015b, 2016c).
Summary
Despite the inhomogeneous results of clinical studies there are several epidemiological and molecular indications for a beneficial effect of DHA in preventing AD and possibly halting its progression, at least at very early disease stages. The fact that its oxidation products are able to reverse the beneficial effects of DHA might partially explain the divergent outcomes of clinical DHA studies and underlines the need to prevent DHA from oxidation in such trials.
Future Directions
Because of the controversial effects of several E-vitamins regarding the molecular mechanisms of AD, the identification of further molecules for the prevention of DHA from oxidative damage in nutritional approaches without side effects on APP processing might be valuable. Additionally, the combination of DHA with precursors/cofactors for membrane synthesis and synaptogenesis as for example uridine-monophosphate, choline and phospholipids might further strengthen its beneficial effects on cognition as demonstrated in a transgenic mouse model of AD (Koivisto et al., 2014).
The Impact of Trans Fatty Acids on AD
Trans fatty acids (TFAs) are unsaturated fatty acids, which are characterized by having at least one double-bond in trans-configuration. This means that the two hydrogen atoms are, in contrast to cis-configuration, localized on opposite sides of the double-bond. Because of their straighter shape compared to the cis-counterparts, TFAs have higher melting points and lead to a decreased fluidity of biological membranes (Roach et al., 2004; Ibrahim et al., 2005). TFAs in our diet arise from industrial procedures and to a lesser extent from biological processes in the digestive tract of ruminant animals. The key source of TFAs is commercially prepared food due to hydrogenation or thermal treatment of oils (Bhardwaj et al., 2011). Accumulation of these fatty acids in the body as well as incorporation in brain tissue has been reported indicating an impact of TFAs on cerebral biochemistry (Laryea et al., 1990; Teixeira et al., 2012).
Studies analyzing the relationship between TFAs and AD-risk or the progression of cognitive decline came to inconsistent results. A positive correlation between dietary TFA intake and AD-risk was found in one study while others reported the AD-risk not to be influenced by TFAs (Engelhart et al., 2002; Morris et al., 2003a). Similarly, some authors observed the TFA intake to result in a higher rate of cognitive decline in women with type 2 diabetes, in persons with high copper consumption and in older persons in general while others failed to find a relationship between TFA intake and cognitive decline in women (Morris et al., 2004, 2006; Devore et al., 2009; Naqvi et al., 2011; Okereke et al., 2012).
We investigated the effects of TFAs on APP-processing and Aβ-generation in neuroblastoma cells compared to their cis-counterparts. In presence of TFAs, we found a shift from non-amyloidogenic to amyloidogenic APP-processing accompanied by a significant increase in Aβ-production. TFA supplementation increases the activity of β- and γ-secretase due to direct effects and an enhanced gene expression of BACE1 and the γ-secretase complex components (Grimm et al., 2012a). The direct effect on γ-secretase activity was confirmed by others demonstrating the activity of purified γ-secretase to be stimulated by an increased trans/cis-ratio of supplemented fatty acids (Holmes et al., 2012). In contrast, non-amyloidogenic APP-processing is reduced in TFA-treated cells because of enhanced APP-internalization and a reduction in ADAM10 gene expression. Additionally, we found TFAs to stimulate Aβ-aggregation in vitro (Grimm et al., 2012a).
The impact of TFAs on cerebral Aβ-levels and cognition has also been investigated in vivo with less clear results. In a study by Phivilay et al. (2009), Aβ- and tau-pathology was unaltered in the brain tissue of an AD-mouse model after dietary supplementation of TFAs. Another study reported a declined spatial learning performance of mice fed with a TFA- and monosodium glutamate-rich diet (Collison et al., 2010).
As TFAs are reported to be linked to cholesterol and DHA homeostasis they might also affect APP-processing and Aβ-generation via indirect mechanisms. The dietary intake of TFA leads to an inauspicious enhanced ratio of LDL/HDL plasma cholesterol (Mensink and Katan, 1990; Judd et al., 1994), which might be associated with a higher AD-risk as described above. Furthermore, high TFA consumption was shown to modify the fatty acid profile of murine brain tissue with a reduction in DHA content. Nevertheless, in this study the cerebral Aβ-levels were unaltered as already mentioned (Phivilay et al., 2009).
Summary
Due to the dissimilar results of studies analyzing the impact of TFAs on AD-risk and Aβ-associated pathology in vivo, further trials are necessary to clarify the role of these fatty acids in AD-pathogenesis.
Future Directions
If the negative effects of TFA on AD-risk can be confirmed in vivo, a stronger reduction of TFA intake should be recommended, particularly because of the accumulation of these fatty acids in the human body over time and their incorporation into brain tissue (Laryea et al., 1990; Teixeira et al., 2012).
The Impact of Plasmalogens on AD
Plasmalogens (PL) are commonly occurring phospholipids accounting for 22% of the total phospholipid mass in human brain tissue. They are characterized by an enol ether double-bond at the sn1-position, which links an alkenyl chain to the glycerol backbone. At the sn2-position they are enriched in PUFAs including DHA and arachidonic acid (AA, 20:4). Phosphatidylethanolamine (PE) and PC are the most common polar head groups of PLs, which have a high susceptibility to oxidative stress due to their enol ether double-bond (Broniec et al., 2011; Braverman and Moser, 2012). PL-biosynthesis takes place in peroxisomes and the endoplasmic reticulum. The initial committed step reaction of PL de novo synthesis is catalyzed by the peroxisomal enzyme alkyl-dihydroxyacetonephosphate-synthase (AGPS; De Vet et al., 1999). PL level in the human body are mainly modulated by PL metabolism, but to a lesser extent also by the dietary consumption of PL-rich meat and fish (Blank et al., 1992).
While one study by Pettegrew et al. (2001) did not detect an AD-dependent alteration in the cerebral PL-content, we and others found a reduction of PE-PLs and PC-PLs in human post mortem AD-brains (Ginsberg et al., 1995; Han et al., 2001; Grimm et al., 2011a; Igarashi et al., 2011; Rothhaar et al., 2012). In line with this, a reduced PE-PL content was also observed in the serum and in erythrocyte membranes of AD-patients (Goodenowe et al., 2007; Oma et al., 2012).
The reduction of PL content in AD-affected brain tissue might be explained by enhanced PL degradation due to increased oxidative stress and a stimulated activity of phospholipases in presence of Aβ-peptides (Sanchez-Mejia et al., 2008). Additionally, we demonstrated PL biosynthesis to be regulated by APP-processing. Under physiological conditions AGPS gene expression and hence PL biosynthesis is upregulated by AICD. In contrast, under pathological conditions the Aβ-induced reactive oxidative species impair AGPS protein stability leading to a decreased PL de novo synthesis (Grimm et al., 2011d).
Because of the altered PL content in AD-brain tissue, we analyzed the impact of PLs on APP-processing. Our results demonstrate that PLs reduce γ-secretase activity in living cells as well as in purified membranes derived from neuroblastoma cells and murine brain tissue. Compared to the corresponding phospholipids lacking the enol ether, all tested PC-PL- and PE-PL-species independent of the bound fatty acid directly inhibited γ-secretase activity. In contrast, the activities of α- and β-secretase remained unchanged after PL-supplementation (Rothhaar et al., 2012). The direct inhibitory effect of PE-PLs on the γ-secretase complex has been recently confirmed by others (Onodera et al., 2015). Interestingly, in our study the addition of PLs to cellular membranes derived from human AD-brains also resulted in a decreased γ-secretase activity. This indicates the rebuilding of a normal PL level to have a positive impact in the pathologic situation of AD (Rothhaar et al., 2012). However, such ex vivo experiments have their clear limitations and further studies are necessary to analyze the in vivo relevance of PLs on APP-processing. In addition to Aβ-production, there is also an impact of PLs on the aggregation of Aβ-peptides. PE-PL has been reported to eliminate the neurotoxicity-associated Aβ-oligomerization phase while allowing fibril formation (Lee et al., 2011).
Summary
In the pathologic situation of AD a vicious cycle between PLs and Aβ-generation can be postulated: the accumulation of Aβ results in a reduced cerebral PL content stimulating γ-secretase activity and hence leading to a further increased Aβ-production.
Future Directions
In the future the in vivo-relevance of the effects of PLs on the generation of Aβ-peptides should be analyzed. An interesting human model for such trials could be cells derived from patients affected by Zellweger syndrome, which show deficient PL-levels due to a defective peroxisome assembly (Styger et al., 2002; Saitoh et al., 2009).
The Impact of Sphingolipids on AD
Sphingolipids are an inhomogeneous group of lipids characterized by a backbone consisting of the amino alcohol sphingosine. Sphingolipid biosynthesis is initiated by the serine palmitoyl-CoA transferase (SPT) catalyzing the condensation of palmitoyl-CoA and L-serine to 3-ketosphinganin, which is further metabolized to ceramide. Ceramide is the most important branching point within the sphingolipid metabolism pathways serving as precursor for the generation of sphingosine, sphingomyelin (SM) and more complex glycosphingolipids.
All sphingolipids are anchored in the membrane bilayer via their ceramide moiety, besides cholesterol they represent major components of lipid raft membrane microdomains (Posse de Chaves and Sipione, 2010). The first evidence for a role of sphingolipids in neurodegeneration came from the observation of lysosomal storage diseases, inherited disorders characterized by the lysosomal accumulation of different sphingolipids. These diseases are associated with early dementia and the development of AD-related Aβ- and tau-pathology (Tarasiuk et al., 2012). The link between sphingolipid metabolism and AD-pathogenesis is further strengthened by alterations of several sphingolipids in post mortem AD-brain tissue and their potential to modulate APP-processing and Aβ-aggregation summarized below. Additionally, SPT gene expression and hence total sphingolipid biosynthesis is downregulated by the APP-processing product AICD (Grimm et al., 2011b).
Ceramide
As already mentioned, ceramide is generated by de novo synthesis and by hydrolysis of various more complex sphingolipids. Ceramides are pro-apoptotic and neurotoxic signaling molecules, additionally participating in the regulation of cellular proliferation and differentiation (Dawson et al., 1998; Toman et al., 2002).
The ceramide level has been reported to be increased in different brain regions and in the cerebrospinal fluid (CSF) of AD-patients. As the increase in ceramide content is already present at the earliest clinical stages of AD, it might be speculated that it is involved in disease development (Han et al., 2002; Satoi et al., 2005; Katsel et al., 2007; He et al., 2010; Filippov et al., 2012). Such a relationship is supported by a 9-year-follow-up study reporting an association between elevated baseline serum ceramide levels and an enhanced risk for developing AD (Mielke et al., 2012).
As reported by Katsel et al. (2007) the accumulation of ceramide in AD-affected individuals might be explained by multiple gene expression abnormalities. The authors found an increased cerebral expression of genes involved in ceramide de novo synthesis along with a reduced expression of genes required for glycosphingolipid formation out of ceramide. Another explanation for the increased ceramide content in AD-brain tissue is the Aβ-mediated activation of sphingomyelinases (SMases) catalyzing the brake down of SM to ceramide. We and others found Aβ-peptides to directly stimulate neutral SMase (nSMase)-activity (Jana and Pahan, 2004; Lee et al., 2004; Grimm et al., 2005), a stimulation of acidic SMase (aSMase) by Aβ has also been observed (Malaplate-Armand et al., 2006). The resulting enhanced ceramide level is reported to be a mediator of Aβ-induced apoptosis. Besides a probable involvement in Aβ-induced cell death, ceramide also affects APP-cleavage. Accumulation of endogenous ceramide levels in cultured cells by the use of cell-permeable C6-ceramide or by nSMase treatment promotes amyloidogenic APP-processing. The resulting ceramide-induced enhanced Aβ biogenesis is caused by a post-translational stabilization of the β-secretase BACE1 due to elevated acetylation of the protein (Puglielli et al., 2003; Ko and Puglielli, 2009).
In their entirety these facts indicate the existence of a feed-forward cycle between ceramide and Aβ under the pathological conditions in AD-brain tissue: enhanced ceramide level lead to an increased Aβ-production resulting in the activation of SMases and hence in a further elevation of ceramide content, which stimulates Aβ-production and might be involved in the induction of apoptotic cell death.
Sphingomyelin
SM accounts for approximately 10% of mammalian cellular lipids and is highly enriched in myelin sheets. It is produced out of ceramide by the activity of SM-synthases, SMases catalyze the catabolic break down of SM back to ceramide.
The already mentioned increased ceramide content and the upregulation of SMases in post mortem AD brains (Katsel et al., 2007; He et al., 2010) suggests that SM concentrations might be reduced in these tissues. However, the results of studies analyzing the SM content in AD-affected brains are inhomogenous (Pettegrew et al., 2001; Cutler et al., 2004; Bandaru et al., 2009; He et al., 2010). In addition, SM level were found to be significantly increased in the CSF of individuals with prodromal AD while there was a slight, but not significant reduction of SM in the CSF of patients with mild and moderate AD (Kosicek et al., 2012). In an epidemiological study by Mielke et al. (2011) higher SM concentrations and an enhanced SM/ceramide-ratio in plasma was found to correlate with a decelerated disease progression among AD-patients.
In strong contrast to ceramide, SM was demonstrated to inhibit Aβ-production. Increasing SM content of cultured cells either by direct exposure or nSMase inhibition leads to a significant decrease of Aβ-peptides caused by an inhibition of γ-secretase dependent APP-processing. In this study we additionally identified the already mentioned direct stimulation of nSMase by Aβ42 (Grimm et al., 2005).
Accordingly, the Aβ-induced elevation of SMase-activity in AD-brain tissue results in an enhanced ceramide/SM-ratio. The increase in γ-secretase activity due to lowered SM-level in combination with the ceramide-dependent activation of β-secretase further promotes Aβ-production might result in a futile cycle.
Sphingosine and Sphingosine 1-Phosphate
Ceramidases catalyze the conversion of ceramide to sphingosine, which is phosphorylated by sphingosine kinase (SK) generating the anti-apoptotic and neuroprotective molecule sphingosine 1-phosphate (S1P). S1P has been demonstrated to induce cell survival and proliferation and to antagonize Aβ- and ceramide-induced cell death (Cuvillier et al., 1996; Gomez-Brouchet et al., 2007; Czubowicz and Strosznajder, 2014). In contrast, sphingosine seems to have a role in apoptosis, cooperatively or independently from ceramide signaling (Sweeney et al., 1998; Lepine et al., 2004).
In line with an increased acid ceramidase expression and activity, the sphingosine content has been found to be elevated in post mortem AD-brains (Huang et al., 2004; He et al., 2010). It should be mentioned, that there is also another study reporting a decreased acid ceramidase gene expression in AD-brain tissue (Katsel et al., 2007). In contrast, the cerebral S1P-content seems to be declined in AD-affected individuals and to negatively correlate with the level of Aβ and phosphorylated tau protein (He et al., 2010). In line with these observations, γ-secretase activity is reduced in cells devoid of S1P-lyase degrading intracellular S1P (Karaca et al., 2014). Contrariwise, S1P has been shown to increase the production of Aβ-peptides by directly stimulating β-secretase activity in another study (Takasugi et al., 2011). Therefore, further studies are necessary to clarify the role of sphingosine and S1P in APP-processing and AD-pathogenesis.
Sulfatides
Sulfatides are complex glycosphingolipids generated from ceramide by the addition of a galactose moiety and a sulfate group catalyzed by ceramide galactosyltransferase (CGT) and cerebrosidesulfotransferase (CST), respectively. They are highly enriched in myelin sheaths and mainly synthesized by oligodendrocytes.
Several studies reported the cerebral sulfatide content to be dramatically decreased in AD-patients compared to cognitive normal controls. These alterations were already observed in the earliest recognizable states of the disease (Han et al., 2002; Bandaru et al., 2009; Cheng et al., 2013). However, there are two other studies which failed to find a significant alteration in sulfatide content in AD-brain tissue (Cutler et al., 2004; Chan et al., 2012). CSF sulfatide level are also strongly reduced in AD-patients as reported by Han et al. (2003b) who suggested the sulfatide/phosphatidylinositol ratio in the CSF to be a potential AD-biomarker.
Interestingly, there seems to be a link between sulfatide homeostasis and ApoE: sulfatides are associated with ApoE-containing particles in the CSF and ApoE is involved in the modulation of cellular sulfatide content in an isoform-dependent manner. This possibly provides an explanation for the genetic association between ApoE and AD (Han, 2010). A role of ApoE in the regulation of cerebral sulfatide level has been demonstrated by Cheng et al. (2010). In this study the age-dependent decline in cortical sulfatide concentrations of APP transgenic mice was found to be totally abolished in ApoE-knockout animals. The sulfatide content in murine brain tissue was further demonstrated to be dependent on ApoE-genotype. In comparison to human ApoEε3 and wildtype ApoEε, the human ApoEε4-isoform is associated with a strong sulfatide depletion in the brain of transgenic mice (Han et al., 2003a). Additionally, sulfatides seem to be involved in ApoE-dependent Aβ-clearance. Treatment of cultured cells with sulfatides results in a strong reduction of Aβ-peptides in the culture media. The underlying mechanism was identified as a facilitated ApoE-mediated Aβ-clearance through an endocytotic pathway in response to elevated sulfatide levels (Zeng and Han, 2008).
Their robust depletion in post mortem AD-brain tissue and their potential to strongly reduce Aβ-levels in vitro indicate that sulfatides might be an attractive target in AD research. Further studies are necessary to investigate the role of this lipid class in the molecular mechanisms of the disease.
Gangliosides
Gangliosides, sialic acid containing glycosphingolipids, represent 6% of the total lipid content in brain. They are abundant in the luminal leaflet of cellular organelles and the outer leaflet of the plasma membrane, where they are localized in lipid raft microdomains. Important functions of gangliosides in the development, proliferation and differentiation of neuronal cells have been reported. The glycosylceramide synthase (GCS) catalyzes the first step of ganglioside biosynthesis by glycosylating ceramide. Dependent of the number of sialic acid residues gangliosides are classified into four catagories, the o-, a-, b- and c-series. In brain tissue the most common gangliosides are GM1, GD1a, GD1b and GT1b belonging to the a- and b-series. GM3 is the precursor of all a- and b-series gangliosides, which are segregated by the GD3-synthase (GD3S)-catalyzed addition of sialic acid to GM3 (Busam and Decker, 1986; Lahiri and Futerman, 2007; Yu et al., 2011).
In AD-brain tissue there is a reduction of total ganglioside content along with significant regional differences in the distribution of specific ganglioside species. In brains affected by FAD and LOAD the total ganglioside level is decreased in several brain regions (Kalanj et al., 1991; Svennerholm and Gottfries, 1994; Gottfries et al., 1996). Kracun et al. (1991) reported a reduction of all major brain gangliosides combined with an increase in the more simple GM2 and GM3 in the cortex of AD-patients. In line with this, the GM1 and GM2 level were found to be elevated in the lipid raft fraction derived from cortical regions of AD brains (Molander-Melin et al., 2005). In summary, in AD-affected brains complex gangliosides tend to decrease while there is an elevation of simple ganglioside species.
Interestingly, in post mortem AD-brains GM1 and GD1a have been found to be associated with Aβ-plaques forming GAβ-complexes exhibiting early pathological changes of AD. This indicates a role of these ganglioside species in Aβ-aggregation (Nishinaka et al., 1993; Yanagisawa et al., 1995). Indeed, GM1 induces a conformational transition of Aβ from random coil to β-sheet structure and triggers the formation of toxic Aβ-fibrils (Choo-Smith et al., 1997; Hayashi et al., 2004; Okada et al., 2007). Further studies demonstrated an accumulation and aggregation of Aβ in GM1-enriched lipid rafts leading to an increased cytotoxicity (Wakabayashi et al., 2005).
Besides Aβ-aggregation, APP-processing and hence Aβ-generation is also influenced by GM1 and other gangliosides. Direct administration of total ganglioside extract to purified γ-secretase leads to an enhanced enzyme activity and increases the ratio of generated Aβ42 to Aβ40 peptides (Holmes et al., 2012). In line, the inhibition of GCS and hence total ganglioside biosynthesis results in a significant reduction of Aβ-production in various cell lines. The addition of exogenous brain gangliosides reverses these effects indicating the reduction of total ganglioside biosynthesis to be beneficial in AD. In this study, the authors found glycosphingolipids to affect APP-processing via regulating the subcellular APP-transport in the secretory pathway (Tamboli et al., 2005). In our own study we demonstrated GCS gene expression to be regulated by PS and APP. Deficiency in these proteins or the inhibition of γ-secretase activity results in an increased GCS gene expression and hence in increased glycosylceramide and total ganglioside level in vitro and in vivo. We showed that GCS is upregulated in the brain tissue of an AD-mouse model and of patients suffering from LOAD. Accordingly, total ganglioside de novo synthesis is modulated by APP-processing and deregulated in the pathological situation of AD (Grimm et al., 2014).
The treatment of neuroblastoma cells with GM1 has been shown to stimulate Aβ-generation and to reduce the sAPPα level without affecting sAPPβ (Zha et al., 2004). In strong contrast to this, peripheral injections of GM1 reduce the cerebral Aβ-burden in an AD-mouse model, possibly due to the promotion of Aβ-degradation in the periphery (Matsuoka et al., 2003). In another study the impact of GD3S deficiency, which results in a loss of b-series gangliosides and an accumulation of GM3, GM1 and GD1a, on the cerebral Aβ-levels in an AD-mouse model has been analyzed. Compared to the control animals, the GD3S-depleted mice showed an almost completely eliminated Aβ-associated neuropathology and no cognitive decline (Bernardo et al., 2009). In line with this, we found the generation of Aβ in cultured cells to be reduced after GM3 supplementation while the addition of the GD3S-product GD3 stimulated Aβ-release. In this context it is important to mention that we also found a regulation of GD3S by APP-processing. The activity of GD3S is inhibited by a direct interaction of Aβ with GM3 leading to a reduced substrate availability and hence to an impaired conversion of GM3 to GD3. Additionally, the gene expression of GD3S is downregulated by AICD. These results indicate the existence of a regulatory feedback cycle, in which Aβ and AICD increase the GM3/GD3-ratio leading to a reduction of amyloidogenic APP-processing (Grimm et al., 2012c).
All these data indicate a strong link between ganglioside homeostasis and AD. As the single ganglioside species differ in their amyloidogenic potential, further studies are necessary to identify the most promising molecular target in ganglioside metabolism for developing therapeutic approaches regarding AD.
Summary
In post mortem AD-brain tissue there are alterations in the content of several sphingolipid species, which can be partially explained by an impact of Aβ and AICD on enzymes involved in sphingolipid homeostasis. Several sphingolipid classes have been shown to affect the proteolytic processing of APP and Aβ-clearance: ceramides, total gangliosides, GM1 and GD3 are associated with an increased Aβ-level while SM, sulfatides and GM3 have the opposite effect.
Future Directions
The fact that ceramide is associated with an increased amyloidogenic APP processing while an increase in SM-levels results in a decreased Aβ-generation indicates SMases to be interesting pharmacological targets regarding AD. Hence, the impact of SMase-inhibitors as for example fluoxetine, maprotiline or desipramine (Kölzer et al., 2004; Kornhuber et al., 2008) on the proteolytic processing of APP and on cognitive functions should be analyzed in suitable models. Another molecular target might be the GD3S, whose inhibition results in an enhanced GM3/GD3-ratio leading to a reduction in amyloidogenic APP proteolysis. In this context it should be mentioned, that mice lacking the GD3 synthase gene show abnormalities in the sciatic nerve and in peripheral nerve regeneration along with impaired neurogenesis and behavioral deficits (Ribeiro-Resende et al., 2014; Wang et al., 2014). This phenotype indicates that pharmacological interventions in ganglioside homeostasis might be associated with severe side effects.
Lipids as Potential Biomarkers for AD
Regarding therapeutic interventions for AD an early diagnosis of the disease and hence the identification of biomarkers, which can be used for the in vivo diagnosis prior to the first symptoms, is important. So, the identification of early AD-biomarkers with a high specificity and reliability is a central topic in AD research (Fiandaca et al., 2014). The lipid alterations connected to AD, which are partially detectable at the very early disease stages as described above, might have the potential to be used as biomarkers for early AD diagnosis by lipidomic approaches. For example, Mapstone et al. (2014) discovered a set of eight PC species and two acylcarnitines in the peripheral blood that predicts the development of mild cognitive impairment or AD within 2–3 years with an accuracy of more than 90%. However, further studies are needed to identify combinations of lipidomics-based biomarkers which can be used for the detection of preclinical AD with the required sensitivity and specificity.
Conclusion
In conclusion all these findings demonstrate a close link of APP, APP processing and AD to lipid homeostasis. It could be demonstrated that APP processing and especially AICD has a physiological function in in the regulation of several lipid metabolic pathways. Inversely, APP-processing is strongly dependent on the lipid microenvironment indicating a bidirectional link between APP-proteolysis and lipid metabolism. This results in tightly connected complex regulatory cycles (Figure 2). Under pathological situations such as AD, this balanced regulation might be disrupted leading to pathological alterations in lipid homeostasis and Aβ peptide overproduction, resulting in increased neurodegeneration.
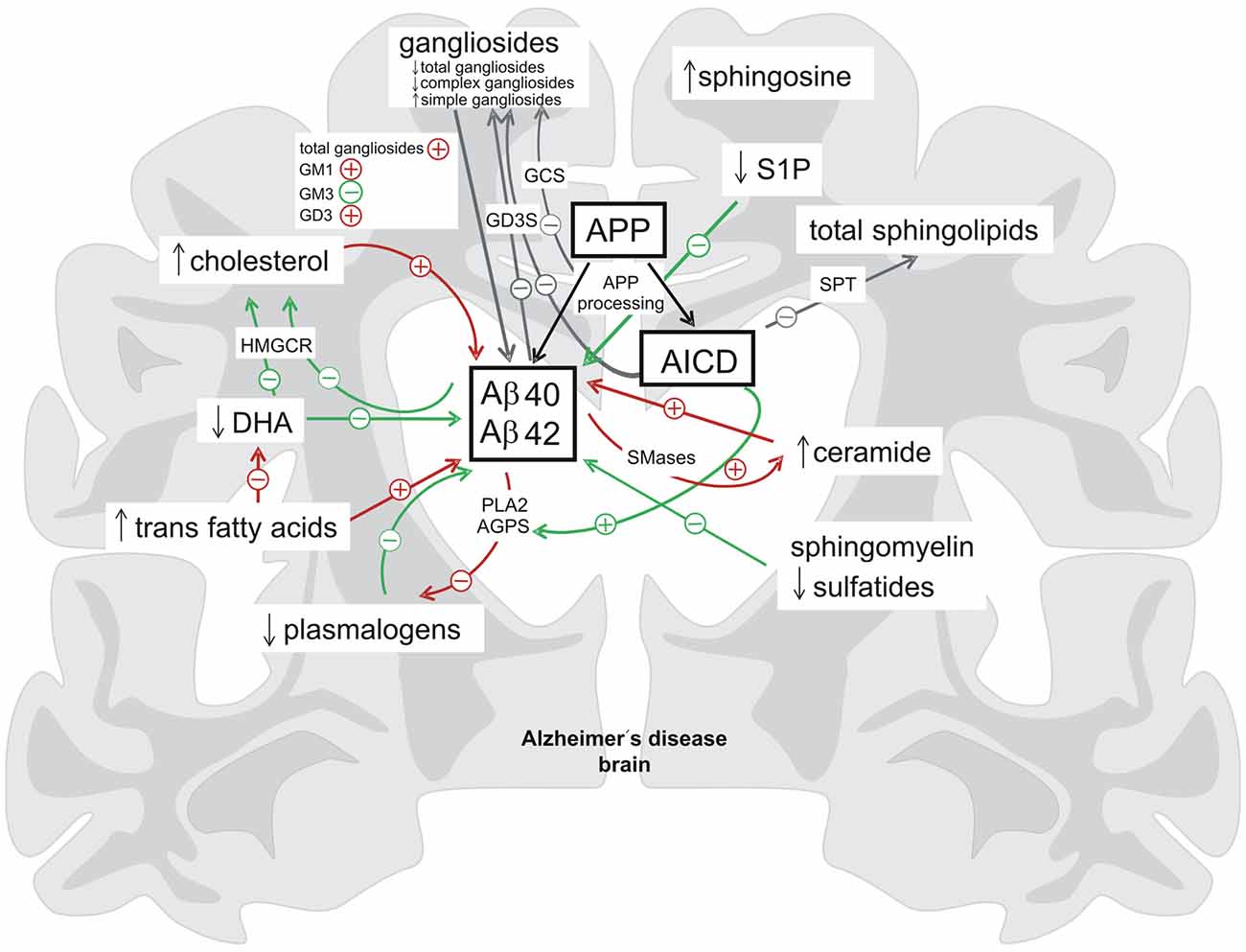
Figure 2. Summary of the bidirectional link between proteolytic processing of the APP and lipid homeostasis. In brain tissue affected by Alzheimer’s disease (AD) the levels of several lipid classes and fatty acids are altered (indicated by ↓ = decreased, ↑ = increased). Lipids and fatty acids have a strong impact on the cerebral Aβ-levels and there is also a regulation of lipid homeostasis by the APP-processing products Aβ and AICD (delineated by + = increasing effect, − = decreasing effect) indicating the existence of complex regulatory cycles between lipid homeostasis and proteolytic APP processing (green arrows = beneficial effects, red arrows = negative effects, gray arrows = neutral/unknown effects). AGPS, alkyl-dihydroxyacetonephosphate-synthase; DHA, docosahexaenoic acid; GCS, glycosylceramide synthase; GD3S, GD3-synthase; HMGCR, hydroxymethylglutaryl-CoA reductase; PLA2, phospholipase A2; S1P, sphingosine 1-phosphate; SMases, sphingomyelinases; SPT, the serine palmitoyl-CoA transferase.
Author Contributions
MOWG, JM, HSG and TH wrote the manuscript.
Funding
According to the author guidelines, funding for the research leading to these results were received from: the EU FP7 project LipiDiDiet, Grant Agreement No. 211696. Moreover funding for MOWG and TH was provided by Fundació la Maratò de TV3 and by JPND MindAD 1ED1508.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abramov, A. Y., Ionov, M., Pavlov, E., and Duchen, M. R. (2011). Membrane cholesterol content plays a key role in the neurotoxicity of β-amyloid: implications for Alzheimer’s disease. Aging Cell 10, 595–603. doi: 10.1111/j.1474-9726.2011.00685.x
Arendash, G. W., Jensen, M. T., Salem, N. Jr., Hussein, N., Cracchiolo, J., Dickson, A., et al. (2007). A diet high in omega-3 fatty acids does not improve or protect cognitive performance in Alzheimer’s transgenic mice. Neuroscience 149, 286–302. doi: 10.1016/j.neuroscience.2007.08.018
Arvanitakis, Z., Schneider, J. A., Wilson, R. S., Bienias, J. L., Kelly, J. F., Evans, D. A., et al. (2008). Statins, incident Alzheimer disease, change in cognitive function, and neuropathology. Neurology 70, 1795–1802. doi: 10.1212/01.wnl.0000288181.00826.63
Bandaru, V. V., Troncoso, J., Wheeler, D., Pletnikova, O., Wang, J., Conant, K., et al. (2009). ApoE4 disrupts sterol and sphingolipid metabolism in Alzheimer’s but not normal brain. Neurobiol. Aging 30, 591–599. doi: 10.1016/j.neurobiolaging.2007.07.024
Barberger-Gateau, P., Letenneur, L., Deschamps, V., Pérès, K., Dartigues, J. F., and Renaud, S. (2002). Fish, meat, and risk of dementia: cohort study. BMJ 325, 932–933. doi: 10.1136/bmj.325.7370.932
Barnes, D. E., and Yaffe, K. (2011). The projected effect of risk factor reduction on Alzheimer’s disease prevalence. Lancet Neurol. 10, 819–828. doi: 10.1016/S1474-4422(11)70072-2
Baulac, S., LaVoie, M. J., Kimberly, W. T., Strahle, J., Wolfe, M. S., Selkoe, D. J., et al. (2003). Functional γ-secretase complex assembly in Golgi/trans-Golgi network: interactions among presenilin, nicastrin, Aph1, Pen-2 and γ-secretase substrates. Neurobiol. Dis. 14, 194–204. doi: 10.1016/s0969-9961(03)00123-2
Bernardo, A., Harrison, F. E., McCord, M., Zhao, J., Bruchey, A., Davies, S. S., et al. (2009). Elimination of GD3 synthase improves memory and reduces amyloid-β plaque load in transgenic mice. Neurobiol. Aging 30, 1777–1791. doi: 10.1016/j.neurobiolaging.2007.12.022
Bhardwaj, S., Passi, S. J., and Misra, A. (2011). Overview of trans fatty acids: biochemistry and health effects. Diabetes Metab. Syndr. 5, 161–164. doi: 10.1016/j.dsx.2012.03.002
Blank, M. L., Cress, E. A., Smith, Z. L., and Snyder, F. (1992). Meats and fish consumed in the American diet contain substantial amounts of ether-linked phospholipids. J. Nutr. 122, 1656–1661.
Braverman, N. E., and Moser, A. B. (2012). Functions of plasmalogen lipids in health and disease. Biochim. Biophys. Acta 1822, 1442–1452. doi: 10.1016/j.bbadis.2012.05.008
Breitner, J. C., Wyse, B. W., Anthony, J. C., Welsh-Bohmer, K. A., Steffens, D. C., Norton, M. C., et al. (1999). APOE-ε4 count predicts age when prevalence of AD increases, then declines: the cache county study. Neurology 53, 321–331. doi: 10.1212/WNL.53.2.321
Broniec, A., Klosinski, R., Pawlak, A., Wrona-Krol, M., Thompson, D., and Sarna, T. (2011). Interactions of plasmalogens and their diacyl analogs with singlet oxygen in selected model systems. Free Radic. Biol. Med. 50, 892–898. doi: 10.1016/j.freeradbiomed.2011.01.002
Brown, D. A., and Rose, J. K. (1992). Sorting of GPI-anchored proteins to glycolipid-enriched membrane subdomains during transport to the apical cell surface. Cell 68, 533–544. doi: 10.1016/0092-8674(92)90189-j
Brown, M. S., Ye, J., Rawson, R. B., and Goldstein, J. L. (2000). Regulated intramembrane proteolysis: a control mechanism conserved from bacteria to humans. Cell 100, 391–398. doi: 10.1016/s0092-8674(00)80675-3
Busam, K., and Decker, K. (1986). Ganglioside biosynthesis in rat liver. Characterization of three sialyltransferases. Eur. J. Biochem. 160, 23–30. doi: 10.1111/j.1432-1033.1986.tb09934.x
Cai, Z., Zhao, B., and Ratka, A. (2011). Oxidative stress and β-amyloid protein in Alzheimer’s disease. Neuromolecular Med. 13, 223–250. doi: 10.1007/s12017-011-8155-9
Calon, F., Lim, G. P., Yang, F., Morihara, T., Teter, B., Ubeda, O., et al. (2004). Docosahexaenoic acid protects from dendritic pathology in an Alzheimer’s disease mouse model. Neuron 43, 633–645. doi: 10.1016/j.neuron.2004.08.013
Cao, X., and Südhof, T. C. (2001). A transcriptionally [correction of transcriptively] active complex of APP with Fe65 and histone acetyltransferase Tip60. Science 293, 115–120. doi: 10.1126/science.1058783
Carey, R. M., Balcz, B. A., Lopez-Coviella, I., and Slack, B. E. (2005). Inhibition of dynamin-dependent endocytosis increases shedding of the amyloid precursor protein ectodomain and reduces generation of amyloid β protein. BMC Cell Biol. 6:30. doi: 10.1186/1471-2121-6-30
Castellano, J. M., Kim, J., Stewart, F. R., Jiang, H., Demattos, R. B., Patterson, B. W., et al. (2011). Human apoE isoforms differentially regulate brain amyloid-β peptide clearance. Sci. Transl. Med. 3:89ra57. doi: 10.1126/scitranslmed.3002156
Chan, R. B., Oliveira, T. G., Cortes, E. P., Honig, L. S., Duff, K. E., Small, S. A., et al. (2012). Comparative lipidomic analysis of mouse and human brain with Alzheimer disease. J. Biol. Chem. 287, 2678–2688. doi: 10.1074/jbc.M111.274142
Cheng, H., Wang, M., Li, J.-L., Cairns, N. J., and Han, X. (2013). Specific changes of sulfatide levels in individuals with pre-clinical Alzheimer’s disease: an early event in disease pathogenesis. J. Neurochem. 127, 733–738. doi: 10.1111/jnc.12368
Cheng, H., Zhou, Y., Holtzman, D. M., and Han, X. (2010). Apolipoprotein E mediates sulfatide depletion in animal models of Alzheimer’s disease. Neurobiol. Aging 31, 1188–1196. doi: 10.1016/j.neurobiolaging.2008.07.020
Chiu, C. C., Su, K.-P., Cheng, T.-C., Liu, H.-C., Chang, C.-J., Dewey, M. E., et al. (2008). The effects of omega-3 fatty acids monotherapy in Alzheimer’s disease and mild cognitive impairment: a preliminary randomized double-blind placebo-controlled study. Prog. Neuropsychopharmacol. Biol. Psychiatry 32, 1538–1544. doi: 10.1016/j.pnpbp.2008.05.015
Choo-Smith, L.-P., Garzon-Rodriguez, W., Glabe, C. G., and Surewicz, W. K. (1997). Acceleration of amyloid fibril formation by specific binding of Aβ-(1–40) peptide to ganglioside-containing membrane vesicles. J. Biol. Chem. 272, 22987–22990. doi: 10.1074/jbc.272.37.22987
Cibickova, L., Hyspler, R., Micuda, S., Cibicek, N., Zivna, H., Jun, D., et al. (2009). The influence of simvastatin, atorvastatin and high-cholesterol diet on acetylcholinesterase activity, amyloid β and cholesterol synthesis in rat brain. Steroids 74, 13–19. doi: 10.1016/j.steroids.2008.08.007
Collison, K. S., Makhoul, N. J., Inglis, A., Al-Johi, M., Zaidi, M. Z., Maqbool, Z., et al. (2010). Dietary trans-fat combined with monosodium glutamate induces dyslipidemia and impairs spatial memory. Physiol. Behav. 99, 334–342. doi: 10.1016/j.physbeh.2009.11.010
Conquer, J. A., Tierney, M. C., Zecevic, J., Bettger, W. J., and Fisher, R. H. (2000). Fatty acid analysis of blood plasma of patients with Alzheimer’s disease, other types of dementia, and cognitive impairment. Lipids 35, 1305–1312. doi: 10.1007/s11745-000-0646-3
Corder, E. H., Saunders, A. M., Risch, N. J., Strittmatter, W. J., Schmechel, D. E., Gaskell, P. C. Jr., et al. (1994). Protective effect of apolipoprotein E type 2 allele for late onset alzheimer disease. Nat. Genet. 7, 180–184. doi: 10.1038/ng0694-180
Corder, E. H., Saunders, A. M., Strittmatter, W. J., Schmechel, D. E., Gaskell, P. C., Small, G. W., et al. (1993). Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261, 921–923. doi: 10.1126/science.8346443
Cossec, J. C., Simon, A., Marquer, C., Moldrich, R. X., Leterrier, C., Rossier, J., et al. (2010). Clathrin-dependent APP endocytosis and Aβ secretion are highly sensitive to the level of plasma membrane cholesterol. Biochim. Biophys. Acta 1801, 846–852. doi: 10.1016/j.bbalip.2010.05.010
Cutler, R. G., Kelly, J., Storie, K., Pedersen, W. A., Tammara, A., Hatanpaa, K., et al. (2004). Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer’s disease. Proc. Natl. Acad. Sci. U S A 101, 2070–2075. doi: 10.1073/pnas.0305799101
Cuvillier, O., Pirianov, G., Kleuser, B., Vanek, P. G., Coso, O. A., Gutkind, S., et al. (1996). Suppression of ceramide-mediated programmed cell death by sphingosine-1-phosphate. Nature 381, 800–803. doi: 10.1038/381800a0
Czubowicz, K., and Strosznajder, R. (2014). Ceramide in the molecular mechanisms of neuronal cell death. The role of sphingosine-1-phosphate. Mol. Neurobiol. 50, 26–37. doi: 10.1007/s12035-013-8606-4
Dawson, G., Goswami, R., Kilkus, J., Wiesner, D., and Dawson, S. (1998). The formation of ceramide from sphingomyelin is associated with cellular apoptosis. Acta Biochim. Pol. 45, 287–297.
Deane, R., Sagare, A., Hamm, K., Parisi, M., Lane, S., Finn, M. B., et al. (2008). apoE isoform-specific disruption of amyloid β peptide clearance from mouse brain. J. Clin. Invest. 118, 4002–4013. doi: 10.1172/JCI36663
De Strooper, B. (2010). Proteases and proteolysis in Alzheimer disease: a multifactorial view on the disease process. Physiol. Rev. 90, 465–494. doi: 10.1152/physrev.00023.2009
De Vet, E. C., Ijlst, L., Oostheim, W., Dekker, C., Moser, H. W., van Den Bosch, H., et al. (1999). Ether lipid biosynthesis: alkyl-dihydroxyacetonephosphate synthase protein deficiency leads to reduced dihydroxyacetonephosphate acyltransferase activities. J. Lipid Res. 40, 1998–2003.
Devore, E. E., Stampfer, M. J., Breteler, M. M., Rosner, B., Kang, J. H., Okereke, O., et al. (2009). Dietary fat intake and cognitive decline in women with type 2 diabetes. Diabetes Care 32, 635–640. doi: 10.2337/dc08-1741
Dickerson, B. C., Goncharova, I., Sullivan, M. P., Forchetti, C., Wilson, R. S., Bennett, D. A., et al. (2001). MRI-derived entorhinal and hippocampal atrophy in incipient and very mild Alzheimer’s disease. Neurobiol. Aging 22, 747–754. doi: 10.1016/s0197-4580(01)00271-8
Dietschy, J. M., and Turley, S. D. (2004). Thematic review series: brain Lipids. Cholesterol metabolism in the central nervous system during early development and in the mature animal. J. Lipid Res. 45, 1375–1397. doi: 10.1194/jlr.R400004-JLR200
Di Paolo, G., and Kim, T. W. (2011). Linking lipids to Alzheimer’s disease: cholesterol and beyond. Nat. Rev. Neurosci. 12, 284–296. doi: 10.1038/nrn3012
Edbauer, D., Winkler, E., Regula, J. T., Pesold, B., Steiner, H., and Haass, C. (2003). Reconstitution of γ-secretase activity. Nat. Cell Biol. 5, 486–488. doi: 10.1038/ncb960
Ehehalt, R., Keller, P., Haass, C., Thiele, C., and Simons, K. (2003). Amyloidogenic processing of the Alzheimer β-amyloid precursor protein depends on lipid rafts. J. Cell Biol. 160, 113–123. doi: 10.1083/jcb.200207113
Engelhart, M. J., Geerlings, M. I., Ruitenberg, A., Van Swieten, J. C., Hofman, A., Witteman, J. C., et al. (2002). Diet and risk of dementia: does fat matter? the rotterdam study. Neurology 59, 1915–1921. doi: 10.1212/01.wnl.0000038345.77753.46
Fam, S. S., Murphey, L. J., Terry, E. S., Zackert, W. E., Chen, Y., Gao, L., et al. (2002). Formation of highly reactive A-ring and J-ring isoprostane-like compounds (A4/J4-neuroprostanes) in vivo from docosahexaenoic acid. J. Biol. Chem. 277, 36076–36084. doi: 10.1074/jbc.m205638200
Fassbender, K., Simons, M., Bergmann, C., Stroick, M., Lutjohann, D., Keller, P., et al. (2001). Simvastatin strongly reduces levels of Alzheimer’s disease β -amyloid peptides Aβ 42 and Aβ 40 in vitro and in vivo. Proc. Natl. Acad. Sci. U S A 98, 5856–5861. doi: 10.1073/pnas.081620098
Feldman, H. H., Doody, R. S., Kivipelto, M., Sparks, D. L., Waters, D. D., Jones, R. W., et al. (2010). Randomized controlled trial of atorvastatin in mild to moderate Alzheimer disease: LEADe. Neurology 74, 956–964. doi: 10.1212/WNL.0b013e3181d6476a
Ferrera, P., Mercado-Gómez, O., Silva-Aguilar, M., Valverde, M., and Arias, C. (2008). Cholesterol potentiates β-amyloid-induced toxicity in human neuroblastoma cells: involvement of oxidative stress. Neurochem. Res. 33, 1509–1517. doi: 10.1007/s11064-008-9623-y
Fiandaca, M. S., Mapstone, M. E., Cheema, A. K., and Federoff, H. J. (2014). The critical need for defining preclinical biomarkers in Alzheimer’s disease. Alzheimers Dement. 10, S196–S212. doi: 10.1016/j.jalz.2014.04.015
Filippov, V., Song, M. A., Zhang, K., Vinters, H. V., Tung, S., Kirsch, W. M., et al. (2012). Increased ceramide in brains with Alzheimer’s and other neurodegenerative diseases. J. Alzheimers Dis. 29, 537–547. doi: 10.3233/JAD-2011-111202
Foley, P. (2010). Lipids in Alzheimer’s disease: a century-old story. Biochim. Biophys. Acta 1801, 750–753. doi: 10.1016/j.bbalip.2010.05.004
Freund-Levi, Y., Eriksdotter-Jönhagen, M., Cederholm, T., Basun, H., Faxén-Irving, G., Garlind, A., et al. (2006). ω-3 fatty acid treatment in 174 patients with mild to moderate Alzheimer disease: OmegAD study: a randomized double-blind trial. Arch. Neurol. 63, 1402–1408. doi: 10.1001/archneur.63.10.1402
Freund-Levi, Y., Hjorth, E., Lindberg, C., Cederholm, T., Faxen-Irving, G., Vedin, I., et al. (2009). Effects of omega-3 fatty acids on inflammatory markers in cerebrospinal fluid and plasma in Alzheimer’s disease: the OmegAD study. Dement. Geriatr. Cogn. Disord. 27, 481–490. doi: 10.1159/000218081
Funamoto, S., Morishima-Kawashima, M., Tanimura, Y., Hirotani, N., Saido, T. C., and Ihara, Y. (2004). Truncated carboxyl-terminal fragments of β-amyloid precursor protein are processed to amyloid β-proteins 40 and 42. Biochemistry 43, 13532–13540. doi: 10.1021/bi049399k
Ginsberg, L., Rafique, S., Xuereb, J. H., Rapoport, S. I., and Gershfeld, N. L. (1995). Disease and anatomic specificity of ethanolamine plasmalogen deficiency in Alzheimer’s disease brain. Brain Res. 698, 223–226. doi: 10.1016/0006-8993(95)00931-f
Glenner, G. G. (1989). Amyloid β protein and the basis for Alzheimer’s disease. Prog. Clin. Biol. Res. 317, 857–868.
Glenner, G. G., and Wong, C. W. (1984). Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 120, 885–890. doi: 10.1016/s0006-291x(84)80190-4
Goedert, M., Strittmatter, W. J., and Roses, A. D. (1994). Alzheimer’s disease. Risky apolipoprotein in brain. Nature 372, 45–46. doi: 10.1038/372045a0
Gomez-Brouchet, A., Pchejetski, D., Brizuela, L., Garcia, V., Altié, M. F., Maddelein, M. L., et al. (2007). Critical role for sphingosine kinase-1 in regulating survival of neuroblastoma cells exposed to amyloid-β peptide. Mol. Pharmacol. 72, 341–349. doi: 10.1124/mol.106.033738
Gómez-Isla, T., Price, J. L., McKeel, D. W. Jr., Morris, J. C., Growdon, J. H., and Hyman, B. T. (1996). Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer’s disease. J. Neurosci. 16, 4491–4500.
Goodenowe, D. B., Cook, L. L., Liu, J., Lu, Y., Jayasinghe, D. A., Ahiahonu, P. W., et al. (2007). Peripheral ethanolamine plasmalogen deficiency: a logical causative factor in Alzheimer’s disease and dementia. J. Lipid Res. 48, 2485–2498. doi: 10.1194/jlr.p700023-jlr200
Gottfries, C. G., Karlsson, I., and Svennerholm, L. (1996). Membrane components separate early-onset alzheimer’s disease from senile dementia of the Alzheimer type. Int. Psychogeriatr. 8, 365–372. doi: 10.1017/s1041610296002736
Grbovic, O. M., Mathews, P. M., Jiang, Y., Schmidt, S. D., Dinakar, R., Summers-Terio, N. B., et al. (2003). Rab5-stimulated up-regulation of the endocytic pathway increases intracellular β-cleaved amyloid precursor protein carboxyl-terminal fragment levels and Aβ production. J. Biol. Chem. 278, 31261–31268. doi: 10.1074/jbc.M304122200
Green, K. N., Martinez-Coria, H., Khashwji, H., Hall, E. B., Yurko-Mauro, K. A., Ellis, L., et al. (2007). Dietary docosahexaenoic acid and docosapentaenoic acid ameliorate amyloid-β and tau pathology via a mechanism involving presenilin 1 levels. J. Neurosci. 27, 4385–4395. doi: 10.1523/JNEUROSCI.0055-07.2007
Grimm, M. O. W., Grimm, H. S., Pätzold, A. J., Zinser, E. G., Halonen, R., Duering, M., et al. (2005). Regulation of cholesterol and sphingomyelin metabolism by amyloid-β and presenilin. Nat. Cell Biol. 7, 1118–1123. doi: 10.1038/ncb1313
Grimm, M. O. W., Grimm, H. S., Tomic, I., Beyreuther, K., Hartmann, T., and Bergmann, C. (2008). Independent inhibition of Alzheimer disease β- and γ-secretase cleavage by lowered cholesterol levels. J. Biol. Chem. 283, 11302–11311. doi: 10.1074/jbc.M801520200
Grimm, M. O. W., Grösgen, S., Riemenschneider, M., Tanila, H., Grimm, H. S., and Hartmann, T. (2011a). From brain to food: analysis of phosphatidylcholins, lyso-phosphatidylcholins and phosphatidylcholin-plasmalogens derivates in Alzheimer’s disease human post mortem brains and mice model via mass spectrometry. J. Chromatogr. A 1218, 7713–7722. doi: 10.1016/j.chroma.2011.07.073
Grimm, M. O. W., Grösgen, S., Rothhaar, T. L., Burg, V. K., Hundsdörfer, B., Haupenthal, V. J., et al. (2011b). Intracellular APP domain regulates serine-palmitoyl-CoA transferase expression and is affected in Alzheimer’s disease. Int. J. Alzheimers Dis. 2011:695413. doi: 10.4061/2011/695413
Grimm, M. O. W., Kuchenbecker, J., Grösgen, S., Burg, V. K., Hundsdörfer, B., Rothhaar, T. L., et al. (2011c). Docosahexaenoic acid reduces amyloid β production via multiple pleiotropic mechanisms. J. Biol. Chem. 286, 14028–14039. doi: 10.1074/jbc.M110.182329
Grimm, M. O. W., Kuchenbecker, J., Rothhaar, T. L., Grösgen, S., Hundsdörfer, B., Burg, V. K., et al. (2011d). Plasmalogen synthesis is regulated via alkyl-dihydroxyacetonephosphate-synthase by amyloid precursor protein processing and is affected in Alzheimer’s disease. J. Neurochem. 116, 916–925. doi: 10.1111/j.1471-4159.2010.07070.x
Grimm, M. O. W., and Hartmann, T. (2012). Recent understanding of the molecular mechanisms of Alzheimer’s disease. J. Addict. Res. Ther. S5:004. doi: 10.4172/2155-6105.s5-004
Grimm, M. O. W., Haupenthal, V. J., Mett, J., Stahlmann, C. P., Blümel, T., Mylonas, N. T., et al. (2016a). Oxidized docosahexaenoic acid species and lipid peroxidation products increase amyloidogenic amyloid precursor protein processing. Neurodegener. Dis. 16, 44–54. doi: 10.1159/000440839
Grimm, M. O. W., Mett, J., Stahlmann, C. P., Haupenthal, V. J., Blümel, T., Stötzel, H., et al. (2016b). Eicosapentaenoic acid and docosahexaenoic acid increase the degradation of amyloid-β by affecting insulin-degrading enzyme. Biochem. Cell Biol. 94, 534–542. doi: 10.1139/bcb-2015-0149
Grimm, M. O. W., Regner, L., Mett, J., Stahlmann, C. P., Schorr, P., Nelke, C., et al. (2016c). Tocotrienol affects oxidative stress, cholesterol homeostasis and the amyloidogenic pathway in neuroblastoma cells: consequences for Alzheimer’s disease. Int. J. Mol. Sci. 17:E1809. doi: 10.3390/ijms17111809
Grimm, M. O. W., Hundsdörfer, B., Grösgen, S., Mett, J., Zimmer, V. Z., Stahlmann, C. P., et al. (2014). PS dependent APP cleavage regulates glucosylceramide synthase and is affected in Alzheimer’s disease. Cell. Physiol. Biochem. 34, 92–110. doi: 10.1159/000362987
Grimm, M. O. W., Mett, J., Stahlmann, C. P., Grösgen, S., Haupenthal, V. J., Blümel, T., et al. (2015a). APP intracellular domain derived from amyloidogenic β- and γ-secretase cleavage regulates neprilysin expression. Front. Aging Neurosci. 7:77. doi: 10.3389/fnagi.2015.00077
Grimm, M. O. W., Stahlmann, C. P., Mett, J., Haupenthal, V. J., Zimmer, V. C., Lehmann, J., et al. (2015b). Vitamin E: curse or benefit in Alzheimer’s disease? A systematic investigation of the impact of α-, γ- and δ-tocopherol on Aβ generation and degradation in neuroblastoma cells. J. Nutr. Health Aging 19, 646–656. doi: 10.1007/s12603-015-0506-z
Grimm, M. O. W., Mett, J., Stahlmann, C. P., Haupenthal, V. J., Zimmer, V. C., and Hartmann, T. (2013). Neprilysin and Aβ clearance: impact of the APP intracellular domain in NEP regulation and implications in Alzheimer’s disease. Front. Aging Neurosci. 5:98. doi: 10.3389/fnagi.2013.00098
Grimm, M. O. W., Rothhaar, T. L., Grösgen, S., Burg, V. K., Hundsdörfer, B., Haupenthal, V. J., et al. (2012a). Trans fatty acids enhance amyloidogenic processing of the Alzheimer amyloid precursor protein (APP). J. Nutr. Biochem. 23, 1214–1223. doi: 10.1016/j.jnutbio.2011.06.015
Grimm, M. O. W., Rothhaar, T. L., and Hartmann, T. (2012b). The role of APP proteolytic processing in lipid metabolism. Exp. Brain Res. 217, 365–375. doi: 10.1007/s00221-011-2975-6
Grimm, M. O. W., Zinser, E. G., Grösgen, S., Hundsdorfer, B., Rothhaar, T. L., Burg, V. K., et al. (2012c). Amyloid precursor protein (APP) mediated regulation of ganglioside homeostasis linking Alzheimer’s disease pathology with ganglioside metabolism. PLoS One 7:e34095. doi: 10.1371/journal.pone.0034095
Grundke-Iqbal, I., Iqbal, K., Quinlan, M., Tung, Y. C., Zaidi, M. S., and Wisniewski, H. M. (1986a). Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J. Biol. Chem. 261, 6084–6089.
Grundke-Iqbal, I., Iqbal, K., Tung, Y. C., Quinlan, M., Wisniewski, H. M., and Binder, L. I. (1986b). Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc. Natl. Acad. Sci. U S A 83, 4913–4917. doi: 10.1073/pnas.83.13.4913
Grziwa, B., Grimm, M. O. W., Masters, C. L., Beyreuther, K., Hartmann, T., and Lichtenthaler, S. F. (2003). The transmembrane domain of the amyloid precursor protein in microsomal membranes is on both sides shorter than predicted. J. Biol. Chem. 278, 6803–6808. doi: 10.1074/jbc.M210047200
Haag, M. D., Hofman, A., Koudstaal, P. J., Stricker, B. H., and Breteler, M. M. (2009). Statins are associated with a reduced risk of Alzheimer disease regardless of lipophilicity. The rotterdam study. J. Neurol. Neurosurg. Psychiatry 80, 13–17. doi: 10.1136/jnnp.2008.150433
Haass, C., Koo, E. H., Mellon, A., Hung, A. Y., and Selkoe, D. J. (1992). Targeting of cell-surface β-amyloid precursor protein to lysosomes: alternative processing into amyloid-bearing fragments. Nature 357, 500–503. doi: 10.1038/357500a0
Han, X. (2010). The pathogenic implication of abnormal interaction between apolipoprotein E isoforms, amyloid-β peptides and sulfatides in Alzheimer’s disease. Mol. Neurobiol. 41, 97–106. doi: 10.1007/s12035-009-8092-x
Han, X., Cheng, H., Fryer, J. D., Fagan, A. M., and Holtzman, D. M. (2003a). Novel role for apolipoprotein E in the central nervous system. Modulation of sulfatide content. J. Biol. Chem. 278, 8043–8051. doi: 10.1074/jbc.M212340200
Han, X., Fagan, A. M., Cheng, H., Morris, J. C., Xiong, C., and Holtzman, D. M. (2003b). Cerebrospinal fluid sulfatide is decreased in subjects with incipient dementia. Ann. Neurol. 54, 115–119. doi: 10.1002/ana.10618
Han, X., Holtzman, D. M., and McKeel, D. W. Jr. (2001). Plasmalogen deficiency in early Alzheimer’s disease subjects and in animal models: molecular characterization using electrospray ionization mass spectrometry. J. Neurochem. 77, 1168–1180. doi: 10.1046/j.1471-4159.2001.00332.x
Han, X., Holtzman, D. M., McKeel, D. W. Jr., Kelley, J., and Morris, J. C. (2002). Substantial sulfatide deficiency and ceramide elevation in very early Alzheimer’s disease: potential role in disease pathogenesis. J. Neurochem. 82, 809–818. doi: 10.1046/j.1471-4159.2002.00997.x
Hao, M., Mukherjee, S., and Maxfield, F. R. (2001). Cholesterol depletion induces large scale domain segregation in living cell membranes. Proc. Natl. Acad. Sci. U S A 98, 13072–13077. doi: 10.1073/pnas.231377398
Hardy, J. A., and Higgins, G. A. (1992). Alzheimer’s disease: the amyloid cascade hypothesis. Science 256, 184–185. doi: 10.1126/science.1566067
Hardy, J., and Selkoe, D. J. (2002). The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297, 353–356. doi: 10.1126/science.1072994
Harris, B., Pereira, I., and Parkin, E. (2009). Targeting ADAM10 to lipid rafts in neuroblastoma SH-SY5Y cells impairs amyloidogenic processing of the amyloid precursor protein. Brain Res. 1296, 203–215. doi: 10.1016/j.brainres.2009.07.105
Hashimoto, M., Hossain, S., Agdul, H., and Shido, O. (2005a). Docosahexaenoic acid-induced amelioration on impairment of memory learning in amyloid β-infused rats relates to the decreases of amyloid β and cholesterol levels in detergent-insoluble membrane fractions. Biochim. Biophys. Acta 1738, 91–98. doi: 10.1016/j.bbalip.2005.11.011
Hashimoto, M., Tanabe, Y., Fujii, Y., Kikuta, T., Shibata, H., and Shido, O. (2005b). Chronic administration of docosahexaenoic acid ameliorates the impairment of spatial cognition learning ability in amyloid β-infused rats. J. Nutr. 135, 549–555.
Hashimoto, M., Hossain, S., Shimada, T., Sugioka, K., Yamasaki, H., Fujii, Y., et al. (2002). Docosahexaenoic acid provides protection from impairment of learning ability in Alzheimer’s disease model rats. J. Neurochem. 81, 1084–1091. doi: 10.1046/j.1471-4159.2002.00905.x
Hayashi, H., Kimura, N., Yamaguchi, H., Hasegawa, K., Yokoseki, T., Shibata, M., et al. (2004). A seed for Alzheimer amyloid in the brain. J. Neurosci. 24, 4894–4902. doi: 10.1523/JNEUROSCI.0861-04.2004
He, X., Huang, Y., Li, B., Gong, C. X., and Schuchman, E. H. (2010). Deregulation of sphingolipid metabolism in Alzheimer’s disease. Neurobiol. Aging 31, 398–408. doi: 10.1016/j.neurobiolaging.2008.05.010
Hicks, D. A., Nalivaeva, N. N., and Turner, A. J. (2012). Lipid rafts and Alzheimer’s disease: protein-lipid interactions and perturbation of signaling. Front. Physiol. 3:189. doi: 10.3389/fphys.2012.00189
Hjorth, E., Zhu, M., Toro, V. C., Vedin, I., Palmblad, J., Cederholm, T., et al. (2013). Omega-3 fatty acids enhance phagocytosis of Alzheimer’s disease-related amyloid-β42 by human microglia and decrease inflammatory markers. J. Alzheimers Dis. 35, 697–713. doi: 10.3233/JAD-130131
Holmes, O., Paturi, S., Ye, W., Wolfe, M. S., and Selkoe, D. J. (2012). Effects of membrane lipids on the activity and processivity of purified γ-secretase. Biochemistry 51, 3565–3575. doi: 10.1021/bi300303g
Holtzman, D. M., Herz, J., and Bu, G. (2012). Apolipoprotein E and apolipoprotein E receptors: normal biology and roles in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2:a006312. doi: 10.1101/cshperspect.a006312
Horrocks, L. A., and Farooqui, A. A. (2004). Docosahexaenoic acid in the diet: its importance in maintenance and restoration of neural membrane function. Prostaglandins Leukot. Essent. Fatty Acids 70, 361–372. doi: 10.1016/j.plefa.2003.12.011
Hossain, S., Hashimoto, M., Katakura, M., Miwa, K., Shimada, T., and Shido, O. (2009). Mechanism of docosahexaenoic acid-induced inhibition of in vitro Aβ1–42 fibrillation and Aβ1–42-induced toxicity in SH-S5Y5 cells. J. Neurochem. 111, 568–579. doi: 10.1111/j.1471-4159.2009.06336.x
Huang, Y., Tanimukai, H., Liu, F., Iqbal, K., Grundke-Iqbal, I., and Gong, C. X. (2004). Elevation of the level and activity of acid ceramidase in Alzheimer’s disease brain. Eur. J. Neurosci. 20, 3489–3497. doi: 10.1111/j.1460-9568.2004.03852.x
Ibrahim, A., Natrajan, S., and Ghafoorunissa, R. (2005). Dietary trans-fatty acids alter adipocyte plasma membrane fatty acid composition and insulin sensitivity in rats. Metabolism 54, 240–246. doi: 10.1016/j.metabol.2004.08.019
Igarashi, M., Ma, K., Gao, F., Kim, H. W., Rapoport, S. I., and Rao, J. S. (2011). Disturbed choline plasmalogen and phospholipid fatty acid concentrations in Alzheimer’s disease prefrontal cortex. J. Alzheimers Dis. 24, 507–517. doi: 10.3233/JAD-2011-101608
Iwatsubo, T., Odaka, A., Suzuki, N., Mizusawa, H., Nukina, N., and Ihara, Y. (1994). Visualization of Aβ42(43) and Aβ40 in senile plaques with end-specific Aβ monoclonals: evidence that an initially deposited species is Aβ42(43). Neuron 13, 45–53. doi: 10.1016/0896-6273(94)90458-8
Jana, A., and Pahan, K. (2004). Fibrillar amyloid-β peptides kill human primary neurons via NADPH oxidase-mediated activation of neutral sphingomyelinase. Implications for Alzheimer’s disease. J. Biol. Chem. 279, 51451–51459. doi: 10.1074/jbc.M404635200
Jarrett, J. T., Berger, E. P., and Lansbury, P. T. Jr. (1993). The carboxy terminus of the β amyloid protein is critical for the seeding of amyloid formation: implications for the pathogenesis of Alzheimer’s disease. Biochemistry 32, 4693–4697. doi: 10.1021/bi00069a001
Jicha, G. A., and Markesbery, W. R. (2010). Omega-3 fatty acids: potential role in the management of early Alzheimer’s disease. Clin. Interv. Aging 5, 45–61. doi: 10.2147/cia.s5231
Judd, J. T., Clevidence, B. A., Muesing, R. A., Wittes, J., Sunkin, M. E., and Podczasy, J. J. (1994). Dietary trans fatty acids: effects on plasma lipids and lipoproteins of healthy men and women. Am. J. Clin. Nutr. 59, 861–868.
Kakuda, N., Funamoto, S., Yagishita, S., Takami, M., Osawa, S., Dohmae, N., et al. (2006). Equimolar production of amyloid β-protein and amyloid precursor protein intracellular domain from β-carboxyl-terminal fragment by γ-secretase. J. Biol. Chem. 281, 14776–14786. doi: 10.1074/jbc.m513453200
Kalanj, S., Kracun, I., Rosner, H., and Cosović, C. (1991). Regional distribution of brain gangliosides in Alzheimer’s disease. Neurol. Croat. 40, 269–281.
Kalmijn, S., Launer, L. J., Ott, A., Witteman, J. C., Hofman, A., and Breteler, M. M. (1997). Dietary fat intake and the risk of incident dementia in the Rotterdam study. Ann. Neurol. 42, 776–782. doi: 10.1002/ana.410420514
Kalvodova, L., Kahya, N., Schwille, P., Ehehalt, R., Verkade, P., Drechsel, D., et al. (2005). Lipids as modulators of proteolytic activity of BACE: involvement of cholesterol, glycosphingolipids, and anionic phospholipids in vitro. J. Biol. Chem. 280, 36815–36823. doi: 10.1074/jbc.M504484200
Kamal-Eldin, A., and Appelqvist, L. A. (1996). The chemistry and antioxidant properties of tocopherols and tocotrienols. Lipids 31, 671–701. doi: 10.1007/bf02522884
Kang, J., Lemaire, H. G., Unterbeck, A., Salbaum, J. M., Masters, C. L., Grzeschik, K. H., et al. (1987). The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature 325, 733–736. doi: 10.1038/325733a0
Karaca, I., Tamboli, I. Y., Glebov, K., Richter, J., Fell, L. H., Grimm, M. O. W., et al. (2014). Deficiency of Sphingosine-1-phosphate lyase impairs lysosomal metabolism of the amyloid precursor protein. J. Biol. Chem. 289, 16761–16772. doi: 10.1074/jbc.m113.535500
Katsel, P., Li, C., and Haroutunian, V. (2007). Gene expression alterations in the sphingolipid metabolism pathways during progression of dementia and Alzheimer’s disease: a shift toward ceramide accumulation at the earliest recognizable stages of Alzheimer’s disease? Neurochem. Res. 32, 845–856. doi: 10.1007/s11064-007-9297-x
Kimberly, W. T., LaVoie, M. J., Ostaszewski, B. L., Ye, W., Wolfe, M. S., and Selkoe, D. J. (2003). γ-secretase is a membrane protein complex comprised of presenilin, nicastrin, Aph-1 and Pen-2. Proc. Natl. Acad. Sci. U S A 100, 6382–6387. doi: 10.1073/pnas.1037392100
Kivipelto, M., Helkala, E. L., Laakso, M. P., Hänninen, T., Hallikainen, M., Alhainen, K., et al. (2002). Apolipoprotein E ε4 allele, elevated midlife total cholesterol level, and high midlife systolic blood pressure are independent risk factors for late-life Alzheimer disease. Ann. Intern. Med. 137, 149–155. doi: 10.7326/0003-4819-137-3-200208060-00006
Ko, M. H., and Puglielli, L. (2009). Two endoplasmic reticulum (ER)/ER Golgi intermediate compartment-based lysine acetyltransferases post-translationally regulate BACE1 levels. J. Biol. Chem. 284, 2482–2492. doi: 10.1074/jbc.m804901200
Koivisto, H., Grimm, M. O. W., Rothhaar, T. L., Berkecz, R., Lütjohann, D. D., Giniatullina, R., et al. (2014). Special lipid-based diets alleviate cognitive deficits in the APPswe/PS1dE9 transgenic mouse model of Alzheimer’s disease independent of brain amyloid deposition. J. Nutr. Biochem. 25, 157–169. doi: 10.1016/j.jnutbio.2013.09.015
Kojro, E., Gimpl, G., Lammich, S., Marz, W., and Fahrenholz, F. (2001). Low cholesterol stimulates the nonamyloidogenic pathway by its effect on the α-secretase ADAM 10. Proc. Natl. Acad. Sci. U S A 98, 5815–5820. doi: 10.1073/pnas.081612998
Kölzer, M., Werth, N., and Sandhoff, K. (2004). Interactions of acid sphingomyelinase and lipid bilayers in the presence of the tricyclic antidepressant desipramine. FEBS Lett. 559, 96–98. doi: 10.1016/s0014-5793(04)00033-x
Koo, E. H., and Squazzo, S. L. (1994). Evidence that production and release of amyloid β-protein involves the endocytic pathway. J. Biol. Chem. 269, 17386–17389.
Kornhuber, J., Tripal, P., Reichel, M., Terfloth, L., Bleich, S., Wiltfang, J., et al. (2008). Identification of new functional inhibitors of acid sphingomyelinase using a structure-property-activity relation model. J. Med. Chem. 51, 219–237. doi: 10.1021/jm070524a
Kosicek, M., Zetterberg, H., Andreasen, N., Peter-Katalinic, J., and Hecimovic, S. (2012). Elevated cerebrospinal fluid sphingomyelin levels in prodromal Alzheimer’s disease. Neurosci. Lett. 516, 302–305. doi: 10.1016/j.neulet.2012.04.019
Kotani, S., Sakaguchi, E., Warashina, S., Matsukawa, N., Ishikura, Y., Kiso, Y., et al. (2006). Dietary supplementation of arachidonic and docosahexaenoic acids improves cognitive dysfunction. Neurosci. Res. 56, 159–164. doi: 10.1016/j.neures.2006.06.010
Kracun, I., Rosner, H., Drnovsek, V., Heffer-Lauc, M., Cosović, C., and Lauc, G. (1991). Human brain gangliosides in development, aging and disease. Int. J. Dev. Biol. 35, 289–295.
Kröger, E., Verreault, R., Carmichael, P. H., Lindsay, J., Julien, P., Dewailly, E., et al. (2009). Omega-3 fatty acids and risk of dementia: the canadian study of health and aging. Am. J. Clin. Nutr. 90, 184–192. doi: 10.3945/ajcn.2008.26987
Kuhn, P. H., Wang, H., Dislich, B., Colombo, A., Zeitschel, U., Ellwart, J. W., et al. (2010). ADAM10 is the physiologically relevant, constitutive α-secretase of the amyloid precursor protein in primary neurons. EMBO J. 29, 3020–3032. doi: 10.1038/emboj.2010.167
Kurata, T., Kawai, H., Miyazaki, K., Kozuki, M., Morimoto, N., Ohta, Y., et al. (2012). Statins have therapeutic potential for the treatment of Alzheimer’s disease, likely via protection of the neurovascular unit in the AD brain. J. Neurol. Sci. 322, 59–63. doi: 10.1016/j.jns.2012.06.011
Kuusisto, J., Koivisto, K., Kervinen, K., Mykkänen, L., Helkala, E. L., Vanhanen, M., et al. (1994). Association of apolipoprotein E phenotypes with late onset alzheimer’s disease: population based study. BMJ 309, 636–638. doi: 10.1136/bmj.309.6955.636
Lahiri, S., and Futerman, A. H. (2007). The metabolism and function of sphingolipids and glycosphingolipids. Cell. Mol. Life Sci. 64, 2270–2284. doi: 10.1007/s00018-007-7076-0
Lambert, M. P., Barlow, A. K., Chromy, B. A., Edwards, C., Freed, R., Liosatos, M., et al. (1998). Diffusible, nonfibrillar ligands derived from Aβ1–42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. U S A 95, 6448–6453. doi: 10.1073/pnas.95.11.6448
Lammich, S., Kojro, E., Postina, R., Gilbert, S., Pfeiffer, R., Jasionowski, M., et al. (1999). Constitutive and regulated α-secretase cleavage of Alzheimer’s amyloid precursor protein by a disintegrin metalloprotease. Proc. Natl. Acad. Sci. U S A 96, 3922–3927. doi: 10.1073/pnas.96.7.3922
Laryea, M., Cieslicki, P., Diekmann, E., and Wendel, U. (1990). Age-dependent fatty acid composition of erythrocyte membrane phospholipids in healthy children. Z. Ernahrungswiss. 29, 284–294. doi: 10.1007/bf02023085
Lauritzen, L., Hansen, H. S., Jørgensen, M. H., and Michaelsen, K. F. (2001). The essentiality of long chain n-3 fatty acids in relation to development and function of the brain and retina. Prog. Lipid Res. 40, 1–94. doi: 10.1016/s0163-7827(00)00017-5
Lee, J., Culyba, E. K., Powers, E. T., and Kelly, J. W. (2011). Amyloid-β forms fibrils by nucleated conformational conversion of oligomers. Nat. Chem. Biol. 7, 602–609. doi: 10.1038/nchembio.624
Lee, S. J., Liyanage, U., Bickel, P. E., Xia, W., Lansbury, P. T. Jr., and Kosik, K. S. (1998). A detergent-insoluble membrane compartment contains Aβ in vivo. Nat. Med. 4, 730–734. doi: 10.1038/nm0698-730
Lee, J. T., Xu, J., Lee, J. M., Ku, G., Han, X., Yang, D. I., et al. (2004). Amyloid-β peptide induces oligodendrocyte death by activating the neutral sphingomyelinase-ceramide pathway. J. Cell Biol. 164, 123–131. doi: 10.1083/jcb.200307017
Lepine, S., Lakatos, B., Courageot, M. P., Le Stunff, H., Sulpice, J. C., and Giraud, F. (2004). Sphingosine contributes to glucocorticoid-induced apoptosis of thymocytes independently of the mitochondrial pathway. J. Immunol. 173, 3783–3790. doi: 10.4049/jimmunol.173.6.3783
Lesné, S., Koh, M. T., Kotilinek, L., Kayed, R., Glabe, C. G., Yang, A., et al. (2006). A specific amyloid-β protein assembly in the brain impairs memory. Nature 440, 352–357. doi: 10.1038/nature04533
Levy, E., Carman, M. D., Fernandez-Madrid, I. J., Power, M. D., Lieberburg, I., van Duinen, S. G., et al. (1990). Mutation of the Alzheimer’s disease amyloid gene in hereditary cerebral hemorrhage, Dutch type. Science 248, 1124–1126. doi: 10.1126/science.2111584
Levy-Lahad, E., Wijsman, E. M., Nemens, E., Anderson, L., Goddard, K. A., Weber, J. L., et al. (1995). A familial Alzheimer’s disease locus on chromosome 1. Science 269, 970–973. doi: 10.1126/science.7638621
Lichtenthaler, S. F., Haass, C., and Steiner, H. (2011). Regulated intramembrane proteolysis–lessons from amyloid precursor protein processing. J. Neurochem. 117, 779–796. doi: 10.1111/j.1471-4159.2011.07248.x
Lim, G. P., Calon, F., Morihara, T., Yang, F., Teter, B., Ubeda, O., et al. (2005). A diet enriched with the omega-3 fatty acid docosahexaenoic acid reduces amyloid burden in an aged Alzheimer mouse model. J. Neurosci. 25, 3032–3040. doi: 10.1523/JNEUROSCI.4225-04.2005
Lingwood, D., and Simons, K. (2010). Lipid rafts as a membrane-organizing principle. Science 327, 46–50. doi: 10.1126/science.1174621
Ma, J., Yee, A., Brewer, H. B. Jr., Das, S., and Potter, H. (1994). Amyloid-associated proteins α 1-antichymotrypsin and apolipoprotein E promote assembly of Alzheimer β-protein into filaments. Nature 372, 92–94. doi: 10.1038/372092a0
Mahley, R. W., Nathan, B. P., and Pitas, R. E. (1996). Apolipoprotein E. Structure, function, and possible roles in Alzheimer’s disease. Ann. N Y Acad. Sci. 777, 139–145. doi: 10.1111/j.1749-6632.1996.tb34412.x
Malaplate-Armand, C., Florent-Béchard, S., Youssef, I., Koziel, V., Sponne, I., Kriem, B., et al. (2006). Soluble oligomers of amyloid-β peptide induce neuronal apoptosis by activating a cPLA2-dependent sphingomyelinase-ceramide pathway. Neurobiol. Dis. 23, 178–189. doi: 10.1016/j.nbd.2006.02.010
Mann, N. J., O’Connell, S. L., Baldwin, K. M., Singh, I., and Meyer, B. J. (2010). Effects of seal oil and tuna-fish oil on platelet parameters and plasma lipid levels in healthy subjects. Lipids 45, 669–681. doi: 10.1007/s11745-010-3450-z
Mapstone, M., Cheema, A. K., Fiandaca, M. S., Zhong, X., Mhyre, T. R., MacArthur, L. H., et al. (2014). Plasma phospholipids identify antecedent memory impairment in older adults. Nat. Med. 20, 415–418. doi: 10.1038/nm.3466
Markesbery, W. R., and Lovell, M. A. (1998). Four-hydroxynonenal, a product of lipid peroxidation, is increased in the brain in Alzheimer’s disease. Neurobiol. Aging 19, 33–36. doi: 10.1016/s0197-4580(98)00009-8
Martins, I. J., Berger, T., Sharman, M. J., Verdile, G., Fuller, S. J., and Martins, R. N. (2009). Cholesterol metabolism and transport in the pathogenesis of Alzheimer’s disease. J. Neurochem. 111, 1275–1308. doi: 10.1111/j.1471-4159.2009.06408.x
Masters, C. L., Simms, G., Weinman, N. A., Multhaup, G., McDonald, B. L., and Beyreuther, K. (1985). Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. U S A 82, 4245–4249. doi: 10.1073/pnas.82.12.4245
Matsuoka, Y., Saito, M., LaFrancois, J., Saito, M., Gaynor, K., Olm, V., et al. (2003). Novel therapeutic approach for the treatment of Alzheimer’s disease by peripheral administration of agents with an affinity to β-amyloid. J. Neurosci. 23, 29–33.
Matsuzaki, T., Sasaki, K., Hata, J., Hirakawa, Y., Fujimi, K., Ninomiya, T., et al. (2011). Association of Alzheimer disease pathology with abnormal lipid metabolism: the hisayama study. Neurology 77, 1068–1075. doi: 10.1212/WNL.0b013e31822e145d
Maulik, M., Westaway, D., Jhamandas, J. H., and Kar, S. (2013). Role of cholesterol in APP metabolism and its significance in Alzheimer’s disease pathogenesis. Mol. Neurobiol. 47, 37–63. doi: 10.1007/s12035-012-8337-y
McGuinness, B., Craig, D., Bullock, R., Malouf, R., and Passmore, P. (2014). Statins for the treatment of dementia. Cochrane Database Syst. Rev. 8:CD007514. doi: 10.1002/14651858.CD007514.pub3
Meng, X. F., Yu, J. T., Wang, H. F., Tan, M. S., Wang, C., Tan, C. C., et al. (2014). Midlife vascular risk factors and the risk of Alzheimer’s disease: a systematic review and meta-analysis. J. Alzheimers Dis. 42, 1295–1310. doi: 10.3233/JAD-140954
Mensink, R. P., and Katan, M. B. (1990). Effect of dietary trans fatty acids on high-density and low-density lipoprotein cholesterol levels in healthy subjects. N. Engl. J. Med. 323, 439–445. doi: 10.1056/NEJM199008163230703
Mett, J., Hartmann, T., and Grimm, M. O. W. (2014). “The effects of glycerophospholipids and fatty acids on APP processing: implications for Alzheimer’s disease,” in Handbook of Lipids in Human Function, eds R. Ross Watson and F. De Meester, (Amsterdam: Academic Press), 377–421.
Mielke, M. M., Bandaru, V. V., Haughey, N. J., Xia, J., Fried, L. P., Yasar, S., et al. (2012). Serum ceramides increase the risk of Alzheimer disease: the women’s health and aging study II. Neurology 79, 633–641. doi: 10.1212/WNL.0b013e318264e380
Mielke, M. M., Haughey, N. J., Bandaru, V. V., Weinberg, D. D., Darby, E., Zaidi, N., et al. (2011). Plasma sphingomyelins are associated with cognitive progression in Alzheimer’s disease. J. Alzheimers Dis. 27, 259–269. doi: 10.3233/JAD-2011-110405
Molander-Melin, M., Blennow, K., Bogdanovic, N., Dellheden, B., Månsson, J. E., and Fredman, P. (2005). Structural membrane alterations in Alzheimer brains found to be associated with regional disease development; increased density of gangliosides GM1 and GM2 and loss of cholesterol in detergent-resistant membrane domains. J. Neurochem. 92, 171–182. doi: 10.1111/j.1471-4159.2004.02849.x
Montine, T. J., and Morrow, J. D. (2005). Fatty acid oxidation in the pathogenesis of Alzheimer’s disease. Am. J. Pathol. 166, 1283–1289. doi: 10.1016/s0002-9440(10)62347-4
Morris, M. C., Evans, D. A., Bienias, J. L., Tangney, C. C., Bennett, D. A., Aggarwal, N., et al. (2003a). Dietary fats and the risk of incident Alzheimer disease. Arch. Neurol. 60, 194–200. doi: 10.1001/archneur.60.8.1072
Morris, M. C., Evans, D. A., Bienias, J. L., Tangney, C. C., Bennett, D. A., Wilson, R. S., et al. (2003b). Consumption of fish and n-3 fatty acids and risk of incident Alzheimer disease. Arch. Neurol. 60, 940–946. doi: 10.1001/archneur.60.7.940
Morris, M. C., Evans, D. A., Bienias, J. L., Tangney, C. C., and Wilson, R. S. (2004). Dietary fat intake and 6-year cognitive change in an older biracial community population. Neurology 62, 1573–1579. doi: 10.1212/01.wnl.0000123250.82849.b6
Morris, M. C., Evans, D. A., Tangney, C. C., Bienias, J. L., Schneider, J. A., Wilson, R. S., et al. (2006). Dietary copper and high saturated and trans fat intakes associated with cognitive decline. Arch. Neurol. 63, 1085–1088. doi: 10.1001/archneur.63.8.1085
Mouton, P. R., Martin, L. J., Calhoun, M. E., Dal Forno, G., and Price, D. L. (1998). Cognitive decline strongly correlates with cortical atrophy in Alzheimer’s dementia. Neurobiol. Aging 19, 371–377. doi: 10.1016/s0197-4580(98)00080-3
Muskiet, F. A., van Goor, S. A., Kuipers, R. S., Velzing-Aarts, F. V., Smit, E. N., Bouwstra, H., et al. (2006). Long-chain polyunsaturated fatty acids in maternal and infant nutrition. Prostaglandins Leukot. Essent. Fatty Acids 75, 135–144. doi: 10.1016/j.plefa.2006.05.010
Naqvi, A. Z., Harty, B., Mukamal, K. J., Stoddard, A. M., Vitolins, M., and Dunn, J. E. (2011). Monounsaturated, trans, and saturated Fatty acids and cognitive decline in women. J. Am. Geriatr. Soc. 59, 837–843. doi: 10.1111/j.1532-5415.2011.03402.x
Nguyen, L. N., Ma, D., Shui, G., Wong, P., Cazenave-Gassiot, A., Zhang, X., et al. (2014). Mfsd2a is a transporter for the essential omega-3 fatty acid docosahexaenoic acid. Nature 509, 503–506. doi: 10.1038/nature13241
Nishinaka, T., Iwata, D., Shimada, S., Kosaka, K., and Suzuki, Y. (1993). Anti-ganglioside GD1a monoclonal antibody recognizes senile plaques in the brains of patients with Alzheimer-type dementia. Neurosci. Res. 17, 171–176. doi: 10.1016/0168-0102(93)90093-6
Okada, T., Wakabayashi, M., Ikeda, K., and Matsuzaki, K. (2007). Formation of toxic fibrils of Alzheimer’s amyloid β-protein-(1–40) by monosialoganglioside GM1, a neuronal membrane component. J. Mol. Biol. 371, 481–489. doi: 10.1016/j.jmb.2007.05.069
Okereke, O. I., Rosner, B. A., Kim, D. H., Kang, J. H., Cook, N. R., Manson, J. E., et al. (2012). Dietary fat types and 4-year cognitive change in community-dwelling older women. Ann. Neurol. 72, 124–134. doi: 10.1002/ana.23593
Oma, S., Mawatari, S., Saito, K., Wakana, C., Tsuboi, Y., Yamada, T., et al. (2012). Changes in phospholipid composition of erythrocyte membrane in Alzheimer’s disease. Dement. Geriatr. Cogn. Dis. Extra 2, 298–303. doi: 10.1159/000341603
Onodera, T., Futai, E., Kan, E., Abe, N., Uchida, T., Kamio, Y., et al. (2015). Phosphatidylethanolamine plasmalogen enhances the inhibiting effect of phosphatidylethanolamine on γ-secretase activity. J. Biochem. 157, 301–309. doi: 10.1093/jb/mvu074
Osenkowski, P., Ye, W., Wang, R., Wolfe, M. S., and Selkoe, D. J. (2008). Direct and potent regulation of γ-secretase by its lipid microenvironment. J. Biol. Chem. 283, 22529–22540. doi: 10.1074/jbc.M801925200
Ouellet, M., Emond, V., Chen, C. T., Julien, C., Bourasset, F., Oddo, S., et al. (2009). Diffusion of docosahexaenoic and eicosapentaenoic acids through the blood-brain barrier: an in situ cerebral perfusion study. Neurochem. Int. 55, 476–482. doi: 10.1016/j.neuint.2009.04.018
Panchal, M., Loeper, J., Cossec, J. C., Perruchini, C., Lazar, A., Pompon, D., et al. (2010). Enrichment of cholesterol in microdissected Alzheimer’s disease senile plaques as assessed by mass spectrometry. J. Lipid Res. 51, 598–605. doi: 10.1194/jlr.M001859
Pappolla, M. A., Bryant-Thomas, T. K., Herbert, D., Pacheco, J., Fabra Garcia, M., Manjon, M., et al. (2003). Mild hypercholesterolemia is an early risk factor for the development of Alzheimer amyloid pathology. Neurology 61, 199–205. doi: 10.1212/01.wnl.0000070182.02537.84
Park, I. H., Hwang, E. M., Hong, H. S., Boo, J. H., Oh, S. S., Lee, J., et al. (2003). Lovastatin enhances Aβ production and senile plaque deposition in female Tg2576 mice. Neurobiol. Aging 24, 637–643. doi: 10.1016/s0197-4580(02)00155-0
Parvathy, S., Hussain, I., Karran, E. H., Turner, A. J., and Hooper, N. M. (1999). Cleavage of Alzheimer’s amyloid precursor protein by α-secretase occurs at the surface of neuronal cells. Biochemistry 38, 9728–9734. doi: 10.1021/bi9906827
Passer, B., Pellegrini, L., Russo, C., Siegel, R. M., Lenardo, M. J., Schettini, G., et al. (2000). Generation of an apoptotic intracellular peptide by γ-secretase cleavage of Alzheimer’s amyloid β protein precursor. J. Alzheimers Dis. 2, 289–301.
Pawlosky, R. J., Hibbeln, J. R., Novotny, J. A., and Salem, N. Jr. (2001). Physiological compartmental analysis of α-linolenic acid metabolism in adult humans. J. Lipid Res. 42, 1257–1265.
Perez, S. E., Berg, B. M., Moore, K. A., He, B., Counts, S. E., Fritz, J. J., et al. (2010). DHA diet reduces AD pathology in young APPswe/PS1 δ E9 transgenic mice: possible gender effects. J. Neurosci. Res. 88, 1026–1040. doi: 10.1002/jnr.22266
Pettegrew, J. W., Panchalingam, K., Hamilton, R. L., and McClure, R. J. (2001). Brain membrane phospholipid alterations in Alzheimer’s disease. Neurochem. Res. 26, 771–782. doi: 10.1023/A:1011603916962
Phivilay, A., Julien, C., Tremblay, C., Berthiaume, L., Julien, P., Giguère, Y., et al. (2009). High dietary consumption of trans fatty acids decreases brain docosahexaenoic acid but does not alter amyloid-β and tau pathologies in the 3xTg-AD model of Alzheimer’s disease. Neuroscience 159, 296–307. doi: 10.1016/j.neuroscience.2008.12.006
Plassman, B. L., Langa, K. M., Fisher, G. G., Heeringa, S. G., Weir, D. R., Ofstedal, M. B., et al. (2007). Prevalence of dementia in the United States: the aging, demographics, and memory study. Neuroepidemiology 29, 125–132. doi: 10.1159/000109998
Polidori, M. C., Pientka, L., and Mecocci, P. (2012). A review of the major vascular risk factors related to Alzheimer’s disease. J. Alzheimers Dis. 32, 521–530. doi: 10.3233/JAD-2012-120871
Posse de Chaves, E., and Sipione, S. (2010). Sphingolipids and gangliosides of the nervous system in membrane function and dysfunction. FEBS Lett. 584, 1748–1759. doi: 10.1016/j.febslet.2009.12.010
Puglielli, L., Ellis, B. C., Saunders, A. J., and Kovacs, D. M. (2003). Ceramide stabilizes β-site amyloid precursor protein-cleaving enzyme 1 and promotes amyloid β-peptide biogenesis. J. Biol. Chem. 278, 19777–19783. doi: 10.1074/jbc.M300466200
Qi-Takahara, Y., Morishima-Kawashima, M., Tanimura, Y., Dolios, G., Hirotani, N., Horikoshi, Y., et al. (2005). Longer forms of amyloid β protein: implications for the mechanism of intramembrane cleavage by γ-secretase. J. Neurosci. 25, 436–445. doi: 10.1523/JNEUROSCI.1575-04.2005
Quinn, J. F., Raman, R., Thomas, R. G., Yurko-Mauro, K., Nelson, E. B., Van Dyck, C., et al. (2010). Docosahexaenoic acid supplementation and cognitive decline in Alzheimer disease: a randomized trial. JAMA 304, 1903–1911. doi: 10.1001/jama.2010.1510
Rea, T. D., Breitner, J. C., Psaty, B. M., Fitzpatrick, A. L., Lopez, O. L., Newman, A. B., et al. (2005). Statin use and the risk of incident dementia: the cardiovascular health study. Arch. Neurol. 62, 1047–1051. doi: 10.1001/archneur.62.7.1047
Reed, B., Villeneuve, S., Mack, W., DeCarli, C., Chui, H. C., and Jagust, W. (2014). Associations between serum cholesterol levels and cerebral amyloidosis. JAMA Neurol. 71, 195–200. doi: 10.1001/jamaneurol.2013.5390
Refolo, L. M., Malester, B., LaFrancois, J., Bryant-Thomas, T., Wang, R., Tint, G. S., et al. (2000). Hypercholesterolemia accelerates the Alzheimer’s amyloid pathology in a transgenic mouse model. Neurobiol. Dis. 7, 321–331. doi: 10.1006/nbdi.2000.0304
Refolo, L. M., Pappolla, M. A., Lafrancois, J., Malester, B., Schmidt, S. D., Thomas-Bryant, T., et al. (2001). A cholesterol-lowering drug reduces β-amyloid pathology in a transgenic mouse model of Alzheimer’s disease. Neurobiol. Dis. 8, 890–899. doi: 10.1006/nbdi.2001.0422
Ribeiro-Resende, V. T., Araujo Gomes, T., de Lima, S., Nascimento-Lima, M., Bargas-Rega, M., Santiago, M. F., et al. (2014). Mice lacking GD3 synthase display morphological abnormalities in the sciatic nerve and neuronal disturbances during peripheral nerve regeneration. PLoS One 9:e108919. doi: 10.1371/journal.pone.0108919
Riddell, D. R., Christie, G., Hussain, I., and Dingwall, C. (2001). Compartmentalization of β-secretase (Asp2) into low-buoyant density, noncaveolar lipid rafts. Curr. Biol. 11, 1288–1293. doi: 10.1016/s0960-9822(01)00394-3
Roach, C., Feller, S. E., Ward, J. A., Shaikh, S. R., Zerouga, M., and Stillwell, W. (2004). Comparison of cis and trans fatty acid containing phosphatidylcholines on membrane properties. Biochemistry 43, 6344–6351. doi: 10.1021/bi049917r
Rothhaar, T. L., Grösgen, S., Haupenthal, V. J., Burg, V. K., Hundsdörfer, B., Mett, J., et al. (2012). Plasmalogens inhibit APP processing by directly affecting γ-secretase activity in Alzheimer’s disease. ScientificWorldJournal 2012:141240. doi: 10.1100/2012/141240
Saitoh, M., Itoh, M., Takashima, S., Mizuguchi, M., and Iwamori, M. (2009). Phosphatidyl ethanolamine with increased polyunsaturated fatty acids in compensation for plasmalogen defect in the Zellweger syndrome brain. Neurosci. Lett. 449, 164–167. doi: 10.1016/j.neulet.2008.11.004
Sanchez-Mejia, R. O., Newman, J. W., Toh, S., Yu, G. Q., Zhou, Y., Halabisky, B., et al. (2008). Phospholipase A2 reduction ameliorates cognitive deficits in a mouse model of Alzheimer’s disease. Nat. Neurosci. 11, 1311–1318. doi: 10.1038/nn.2213
Sano, M., Bell, K. L., Galasko, D., Galvin, J. E., Thomas, R. G., van Dyck, C. H., et al. (2011). A randomized, double-blind, placebo-controlled trial of simvastatin to treat Alzheimer disease. Neurology 77, 556–563. doi: 10.1212/WNL.0b013e318228bf11
Satoi, H., Tomimoto, H., Ohtani, R., Kitano, T., Kondo, T., Watanabe, M., et al. (2005). Astroglial expression of ceramide in Alzheimer’s disease brains: a role during neuronal apoptosis. Neuroscience 130, 657–666. doi: 10.1016/j.neuroscience.2004.08.056
Sayre, L. M., Zelasko, D. A., Harris, P. L., Perry, G., Salomon, R. G., and Smith, M. A. (1997). 4-Hydroxynonenal-derived advanced lipid peroxidation end products are increased in Alzheimer’s disease. J. Neurochem. 68, 2092–2097. doi: 10.1046/j.1471-4159.1997.68052092.x
Scheff, S. W., and Price, D. A. (1993). Synapse loss in the temporal lobe in Alzheimer’s disease. Ann. Neurol. 33, 190–199. doi: 10.1002/ana.410330209
Schneider, A., Schulz-Schaeffer, W., Hartmann, T., Schulz, J. B., and Simons, M. (2006). Cholesterol depletion reduces aggregation of amyloid-β peptide in hippocampal neurons. Neurobiol. Dis. 23, 573–577. doi: 10.1016/j.nbd.2006.04.015
Shankar, G. M., Li, S., Mehta, T. H., Garcia-Munoz, A., Shepardson, N. E., Smith, I., et al. (2008). Amyloid-β protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 14, 837–842. doi: 10.1038/nm1782
Sherrington, R., Rogaev, E. I., Liang, Y., Rogaeva, E. A., Levesque, G., Ikeda, M., et al. (1995). Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature 375, 754–760. doi: 10.1038/375754a0
Shinohara, M., Sato, N., Shimamura, M., Kurinami, H., Hamasaki, T., Chatterjee, A., et al. (2014). Possible modification of Alzheimer’s disease by statins in midlife: interactions with genetic and non-genetic risk factors. Front. Aging Neurosci. 6:71. doi: 10.3389/fnagi.2014.00071
Simons, M., Keller, P., De Strooper, B., Beyreuther, K., Dotti, C. G., and Simons, K. (1998). Cholesterol depletion inhibits the generation of β-amyloid in hippocampal neurons. Proc. Natl. Acad. Sci. U S A 95, 6460–6464. doi: 10.1073/pnas.95.11.6460
Smith, M. A., Kutty, R. K., Richey, P. L., Yan, S. D., Stern, D., Chader, G. J., et al. (1994). Heme oxygenase-1 is associated with the neurofibrillary pathology of Alzheimer’s disease. Am. J. Pathol. 145, 42–47.
Söderberg, M., Edlund, C., Kristensson, K., and Dallner, G. (1991). Fatty acid composition of brain phospholipids in aging and in Alzheimer’s disease. Lipids 26, 421–425. doi: 10.1007/bf02536067
Solomon, A., Kivipelto, M., Wolozin, B., Zhou, J., and Whitmer, R. A. (2009). Midlife serum cholesterol and increased risk of Alzheimer’s and vascular dementia three decades later. Dement. Geriatr. Cogn. Disord. 28, 75–80. doi: 10.1159/000231980
Sparks, D. L., Scheff, S. W., Hunsaker, J. C. III, Liu, H., Landers, T., and Gross, D. R. (1994). Induction of Alzheimer-like β-amyloid immunoreactivity in the brains of rabbits with dietary cholesterol. Exp. Neurol. 126, 88–94. doi: 10.1006/exnr.1994.1044
Stillwell, W., Shaikh, S. R., Zerouga, M., Siddiqui, R., and Wassall, S. R. (2005). Docosahexaenoic acid affects cell signaling by altering lipid rafts. Reprod. Nutr. Dev. 45, 559–579. doi: 10.1051/rnd:2005046
Strittmatter, W. J., Saunders, A. M., Schmechel, D., Pericak-Vance, M., Enghild, J., Salvesen, G. S., et al. (1993). Apolipoprotein E: high-avidity binding to β-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc. Natl. Acad. Sci. U S A 90, 1977–1981. doi: 10.1073/pnas.90.5.1977
Styger, R., Wiesmann, U. N., and Honegger, U. E. (2002). Plasmalogen content and β-adrenoceptor signalling in fibroblasts from patients with Zellweger syndrome. Effects of hexadecylglycerol. Biochim. Biophys. Acta 1585, 39–43. doi: 10.1016/s1388-1981(02)00320-7
Svennerholm, L., and Gottfries, C. G. (1994). Membrane lipids, selectively diminished in Alzheimer brains, suggest synapse loss as a primary event in early-onset form (type I) and demyelination in late-onset form (type II). J. Neurochem. 62, 1039–1047. doi: 10.1046/j.1471-4159.1994.62031039.x
Sweeney, E. A., Inokuchi, J., and Igarashi, Y. (1998). Inhibition of sphingolipid induced apoptosis by caspase inhibitors indicates that sphingosine acts in an earlier part of the apoptotic pathway than ceramide. FEBS Lett. 425, 61–65. doi: 10.1016/s0014-5793(98)00198-7
Takasugi, N., Sasaki, T., Suzuki, K., Osawa, S., Isshiki, H., Hori, Y., et al. (2011). BACE1 activity is modulated by cell-associated sphingosine-1-phosphate. J. Neurosci. 31, 6850–6857. doi: 10.1523/JNEUROSCI.6467-10.2011
Tamaoka, A., Sawamura, N., Odaka, A., Suzuki, N., Mizusawa, H., Shoji, S., et al. (1995). Amyloid β protein 1–42/43 (A β 1-42/43) in cerebellar diffuse plaques: enzyme-linked immunosorbent assay and immunocytochemical study. Brain Res. 679, 151–156. doi: 10.1016/0006-8993(95)00162-J
Tamboli, I. Y., Prager, K., Barth, E., Heneka, M., Sandhoff, K., and Walter, J. (2005). Inhibition of glycosphingolipid biosynthesis reduces secretion of the β-amyloid precursor protein and amyloid β-peptide. J. Biol. Chem. 280, 28110–28117. doi: 10.1074/jbc.M414525200
Tanzi, R. E. (2012). The genetics of Alzheimer disease. Cold Spring Harb. Perspect. Med. 2:a006296. doi: 10.1101/cshperspect.a006296
Tarasiuk, J., Kapica-Topczewska, K., Kulakowska, A., Halicka, D., Drozdowski, W., Kornhuber, J., et al. (2012). Increased concentration of the CSF Tau protein and its phosphorylated form in the late juvenile metachromatic leukodystrophy form: a case report. J. Neural Transm. 119, 759–762. doi: 10.1007/s00702-012-0826-7
Teixeira, A. M., Dias, V. T., Pase, C. S., Roversi, K., Boufleur, N., Barcelos, R. C., et al. (2012). Could dietary trans fatty acids induce movement disorders? Effects of exercise and its influence on Na+K+-ATPase and catalase activity in rat striatum. Behav. Brain Res. 226, 504–510. doi: 10.1016/j.bbr.2011.10.005
Thinakaran, G., and Koo, E. H. (2008). Amyloid precursor protein trafficking, processing, and function. J. Biol. Chem. 283, 29615–29619. doi: 10.1074/jbc.R800019200
Tiraboschi, P., Hansen, L. A., Masliah, E., Alford, M., Thal, L. J., and Corey-Bloom, J. (2004). Impact of APOE genotype on neuropathologic and neurochemical markers of Alzheimer disease. Neurology 62, 1977–1983. doi: 10.1212/01.wnl.0000128091.92139.0f
Toman, R. E., Movsesyan, V., Murthy, S. K., Milstien, S., Spiegel, S., and Faden, A. I. (2002). Ceramide-induced cell death in primary neuronal cultures: upregulation of ceramide levels during neuronal apoptosis. J. Neurosci. Res. 68, 323–330. doi: 10.1002/jnr.10190
Tully, A. M., Roche, H. M., Doyle, R., Fallon, C., Bruce, I., Lawlor, B., et al. (2003). Low serum cholesteryl ester-docosahexaenoic acid levels in Alzheimer’s disease: a case-control study. Br. J. Nutr. 89, 483–489. doi: 10.1079/BJN2002804
Umeda, T., Mori, H., Zheng, H., and Tomiyama, T. (2010). Regulation of cholesterol efflux by amyloid β secretion. J. Neurosci. Res. 88, 1985–1994. doi: 10.1002/jnr.22360
Vassar, R., Bennett, B. D., Babu-Khan, S., Kahn, S., Mendiaz, E. A., Denis, P., et al. (1999). β-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 286, 735–741. doi: 10.1126/science.286.5440.735
Vetrivel, K. S., Cheng, H., Lin, W., Sakurai, T., Li, T., Nukina, N., et al. (2004). Association of γ-secretase with lipid rafts in post-Golgi and endosome membranes. J. Biol. Chem. 279, 44945–44954. doi: 10.1074/jbc.m407986200
von Rotz, R. C., Kohli, B. M., Bosset, J., Meier, M., Suzuki, T., Nitsch, R. M., et al. (2004). The APP intracellular domain forms nuclear multiprotein complexes and regulates the transcription of its own precursor. J. Cell Sci. 117, 4435–4448. doi: 10.1242/jcs.01323
Wakabayashi, M., Okada, T., Kozutsumi, Y., and Matsuzaki, K. (2005). GM1 ganglioside-mediated accumulation of amyloid β-protein on cell membranes. Biochem. Biophys. Res. Commun. 328, 1019–1023. doi: 10.1016/j.bbrc.2005.01.060
Wang, J., Cheng, A., Wakade, C., and Yu, R. K. (2014). Ganglioside GD3 is required for neurogenesis and long-term maintenance of neural stem cells in the postnatal mouse brain. J. Neurosci. 34, 13790–13800. doi: 10.1523/JNEUROSCI.2275-14.2014
Weisgraber, K. H., Rall, S. C. Jr., and Mahley, R. W. (1981). Human E apoprotein heterogeneity. Cysteine-arginine interchanges in the amino acid sequence of the apo-E isoforms. J. Biol. Chem. 256, 9077–9083.
Winkler, E., Kamp, F., Scheuring, J., Ebke, A., Fukumori, A., and Steiner, H. (2012). Generation of Alzheimer disease-associated amyloid β42/43 peptide by γ-secretase can be inhibited directly by modulation of membrane thickness. J. Biol. Chem. 287, 21326–21334. doi: 10.1074/jbc.M112.356659
Wolozin, B., Kellman, W., Ruosseau, P., Celesia, G. G., and Siegel, G. (2000). Decreased prevalence of Alzheimer disease associated with 3-hydroxy-3-methyglutaryl coenzyme A reductase inhibitors. Arch. Neurol. 57, 1439–1443. doi: 10.1001/archneur.57.10.1439
Wolozin, B., Wang, S. W., Li, N. C., Lee, A., Lee, T. A., and Kazis, L. E. (2007). Simvastatin is associated with a reduced incidence of dementia and Parkinson’s disease. BMC Med. 5:20. doi: 10.1186/1741-7015-5-20
World Alzheimer Report (2015). Alzheimer’s Disease International. London: Alzheimer’s Disease International (ADI).
Xiong, H., Callaghan, D., Jones, A., Walker, D. G., Lue, L. F., Beach, T. G., et al. (2008). Cholesterol retention in Alzheimer’s brain is responsible for high β- and γ-secretase activities and Aβ production. Neurobiol. Dis. 29, 422–437. doi: 10.1016/j.nbd.2007.10.005
Yanagisawa, K., Odaka, A., Suzuki, N., and Ihara, Y. (1995). GM1 ganglioside-bound amyloid β-protein (Aβ): a possible form of preamyloid in Alzheimer’s disease. Nat. Med. 1, 1062–1066. doi: 10.1038/nm1095-1062
Yang, X., Sheng, W., Sun, G. Y., and Lee, J. C. (2011). Effects of fatty acid unsaturation numbers on membrane fluidity and α-secretase-dependent amyloid precursor protein processing. Neurochem. Int. 58, 321–329. doi: 10.1016/j.neuint.2010.12.004
Yatin, S. M., Aksenova, M., Aksenov, M., Markesbery, W. R., Aulick, T., and Butterfield, D. A. (1998). Temporal relations among amyloid β-peptide-induced free-radical oxidative stress, neuronal toxicity, and neuronal defensive responses. J. Mol. Neurosci. 11, 183–197. doi: 10.1385/jmn:11:3:183
Yu, R. K., Tsai, Y. T., Ariga, T., and Yanagisawa, M. (2011). Structures, biosynthesis and functions of gangliosides–an overview. J. Oleo Sci. 60, 537–544. doi: 10.5650/jos.60.537
Zanetti, O., Solerte, S. B., and Cantoni, F. (2009). Life expectancy in Alzheimer’s disease (AD). Arch. Gerontol. Geriatr. 49, 237–243. doi: 10.1016/j.archger.2009.09.035
Zeng, Y., and Han, X. (2008). Sulfatides facilitate apolipoprotein E-mediated amyloid-β peptide clearance through an endocytotic pathway. J. Neurochem. 106, 1275–1286. doi: 10.1111/j.1471-4159.2008.05481.x
Keywords: lipids, APP processing, AICD, Abeta cholesterol, sphingolipids, PUFA, sulfatides, gangliosides
Citation: Grimm MOW, Mett J, Grimm HS and Hartmann T (2017) APP Function and Lipids: A Bidirectional Link. Front. Mol. Neurosci. 10:63. doi: 10.3389/fnmol.2017.00063
Received: 31 October 2016; Accepted: 24 February 2017;
Published: 10 March 2017.
Edited by:
Thomas Deller, Goethe-University, GermanyReviewed by:
Anthony J. Turner, University of Leeds, UKLorena Perrone, Université Grenoble Alpes, France
Joachim Herz, University of Texas Southwestern Medical Center, USA
Copyright © 2017 Grimm, Mett, Grimm and Hartmann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution and reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marcus O. W. Grimm, bWFyY3VzLmdyaW1tQHVrcy5ldQ==
† These authors have contributed equally to this work.