 Rafael Linden
Rafael Linden- Laboratory of Neurogenesis, Institute of Biophysics, Federal University of Rio de Janeiro, Rio de Janeiro, Brazil
The prion glycoprotein (PrPC) is mostly located at the cell surface, tethered to the plasma membrane through a glycosyl-phosphatydil inositol (GPI) anchor. Misfolding of PrPC is associated with the transmissible spongiform encephalopathies (TSEs), whereas its normal conformer serves as a receptor for oligomers of the β-amyloid peptide, which play a major role in the pathogenesis of Alzheimer’s Disease (AD). PrPC is highly expressed in both the nervous and immune systems, as well as in other organs, but its functions are controversial. Extensive experimental work disclosed multiple physiological roles of PrPC at the molecular, cellular and systemic levels, affecting the homeostasis of copper, neuroprotection, stem cell renewal and memory mechanisms, among others. Often each such process has been heralded as the bona fide function of PrPC, despite restricted attention paid to a selected phenotypic trait, associated with either modulation of gene expression or to the engagement of PrPC with a single ligand. In contrast, the GPI-anchored prion protein was shown to bind several extracellular and transmembrane ligands, which are required to endow that protein with the ability to play various roles in transmembrane signal transduction. In addition, differing sets of those ligands are available in cell type- and context-dependent scenarios. To account for such properties, we proposed that PrPC serves as a dynamic platform for the assembly of signaling modules at the cell surface, with widespread consequences for both physiology and behavior. The current review advances the hypothesis that the biological function of the prion protein is that of a cell surface scaffold protein, based on the striking similarities of its functional properties with those of scaffold proteins involved in the organization of intracellular signal transduction pathways. Those properties are: the ability to recruit spatially restricted sets of binding molecules involved in specific signaling; mediation of the crosstalk of signaling pathways; reciprocal allosteric regulation with binding partners; compartmentalized responses; dependence of signaling properties upon posttranslational modification; and stoichiometric requirements and/or oligomerization-dependent impact on signaling. The scaffold concept may contribute to novel approaches to the development of effective treatments to hitherto incurable neurodegenerative diseases, through informed modulation of prion protein-ligand interactions.
Introduction
The prion protein, often referred to as cellular prion protein (PrPC) was discovered amid studies of transmissible spongiform encephalopathies (TSEs), such as Creutzfeldt-Jakob Disease, a low-prevalence, mostly sporadic, fatal and still incurable neurodegenerative disease (Takada and Geschwind, 2013). Since the 1980s evidence has accumulated that these conditions are associated with the misfolding, aggregation, replication and spread of abnormal conformers of PrPC, in line with the concept of a protein-only, infectious particle which originated the sobriquet prion for the anomalous conformer (Prusiner, 1984).
Whereas misfolding of PrPC is usually considered the major, if not indispensable requirement for neurodegeneration in TSEs, experimental work indicated that the normal PrPC conformer serves as a binding site for diffusible Aß peptide oligomers (AßO) in the course of Alzheimer’s Disease (AD; Um and Strittmatter, 2013; Laurén, 2014; Kostylev et al., 2015). The AßO are deemed the major toxic species associated with AD, and accumulate as a consequence of disregulated proteolytic cleavage of the amyloid precursor protein (APP; Lambert et al., 1998; Walsh and Selkoe, 2007; Ferreira and Klein, 2011).
The involvement of PrPC in both TSEs and AD renewed and amplified the interest in this protein, that holds important clues towards the understanding of the pathogenesis, as well as the discovery of novel therapies for both those neurodegenerative diseases. Progress in this direction, however, suffers from controversies over functional properties of the prion protein, the corruption and/or loss of which are likely relevant to both TSEs and AD. The current review compares functional properties of PrPC with those of scaffold proteins involved in the organization of intracellular signal transduction pathways, in support of the hypothesis that the biological function of the prion protein is that of a cell surface scaffold protein (Linden et al., 2008, 2012, 2017).
Association of the Prion Protein with Both Transmissible Spongiform Encephalopathies and Alzheimer’s Disease
The course of the various types of TSEs (also known as Prion Diseases, henceforth abbreviated PrDis) involves the progressive cooptation and misfolding of PrPC molecules from an initial template of abnormal prions (Colby and Prusiner, 2011). Knowledge is still fragmentary as to the kinetics of aggregation and progressive growth of prion oligomers, the ensuing compaction of protease-resistant, insoluble deposits of abnormal conformers of PrPC, as well as the conditions that lead to their occasional organization as amyloid proper (Morris et al., 2009; Eichner and Radford, 2011; Corsaro et al., 2012; Wang et al., 2016). Also the purported toxic species are a matter of debate (Bucciantini et al., 2002; Silveira et al., 2005; Guerrero-Muñoz et al., 2014), as are hypotheses concerning the requirement for ancillary pathogenic factors (Cordeiro and Silva, 2005; Manuelidis, 2013). That the presence of the prion protein is required for the course of PrDis was, however, made clear by early experiments, in which neither the spread of abnormal conformers, nor the pathological hallmarks of PrDis were found in the brains of PrPC-null mice infected with extracts of diseased tissue (Büeler et al., 1993).
On the other hand, experimental studies showed that PrPC may bind oligomers of Aß peptide (AßO) and mediate signal transduction induced by the latter (Laurén et al., 2009; Nygaard and Strittmatter, 2009; Chen et al., 2010; Barry et al., 2011; Bate and Williams, 2011a; Larson et al., 2012; Ganzinger et al., 2014; Laurén, 2014). Notably, however, the reputed role of the prion protein as a receptor for AßO is not exclusive (Balducci et al., 2010; Calella et al., 2010; Cissé et al., 2011; Forloni and Balducci, 2011). Several other molecules interact with AßO in both neurons and glial cells (Mucke and Selkoe, 2012; Kam et al., 2014; Ferreira et al., 2015; Yu and Ye, 2015). Importantly, the composition of the preparations of AßO employed in distinct experimental studies is quite variable (Mucke and Selkoe, 2012; Ferreira et al., 2015), and for example, whereas the Frizzled receptor preferentially binds oligomers of low molecular weight and/or monomeric Aß peptide, higher molecular weight oligomers bind the prion protein (Magdesian et al., 2008; Kostylev et al., 2015). This probably explains the multitude of putative neurotoxic AßO receptors, albeit selective oligomer-receptor interactions may legitimately represent the progressive effects of the variegated and evolving Aß peptide aggregates present in the brains of patients along the course of AD (Amieva et al., 2005; Mucke and Selkoe, 2012; Villemagne et al., 2013; Bernard et al., 2014; Alzheimer’s Association, 2015).
The Quest for the Function of the Prion Protein
The production of the first Prnp-null mouse in the 1990s (Büeler et al., 1992) triggered major advances in the field, as it allowed proof that PrPC was required for progression of PrDis in the mouse brain (Büeler et al., 1993). In turn, the report that those mice developed normally and showed no overt behavioral or immunological defects (Büeler et al., 1992), depreciated somewhat the search for functional properties of the normal conformer of PrPC. The acme of such dismissal may well be a bold proposal that PrPC has no function, and that its conserved amino acid sequence was naturally selected as a consequence of the deadly effects of mutations (Prcina and Kontsekova, 2011).
Still, the last 15 years witnessed growing interest in the functional properties of PrPC, based on analyses of mice devoid of its coding gene Prnp, transgenic animals harboring various mutated or partially-deleted forms of PrPC, or Prnp-overexpressing mice, as well as experimental cross-linking of PrPC with antibodies, engagement with binding peptides or glycosaminoglycans (GAGs), and interference with plasma membrane lipids, eventually accompanied by simultaneous activation of other membrane proteins (reviewed in Martins et al., 2002; Westergard et al., 2007; Linden et al., 2008, 2012; Málaga-Trillo and Sempou, 2009; Martin-Lannerée et al., 2014; Onodera et al., 2014).
It appears to be settled that the prion protein mediates mechanisms of neuroprotection (Martins et al., 2010; Biasini et al., 2012; Béland and Roucou, 2014). However, contributions of PrPC have been reported also in immune responses, energy metabolism, cancer, and stress conditions in general (Linden et al., 2008; Li et al., 2011; Mariante et al., 2012; Martin-Lannerée et al., 2014; Onodera et al., 2014; Bakkebø et al., 2015; Zeng et al., 2015). Often, each such demonstration was heralded as the bona fide physiological function of PrPC, claims of which range from the systemic level, such as the consolidation of memory, through cellular, such as cytoprotection, down to the subcellular level, such as the homeostasis of copper (Table 1). Nonetheless, the vast majority of the corresponding data, actually disclosed either circumstantial contributions to cellular or systemic processes, or phenotypes, in some cases specific to certain mouse strains, rather than the unraveling of an unambiguous function at the molecular level (Ashburner et al., 2000; Dessimoz and Škunca, 2017).

Table 1. Keywords to processes at the molecular, cellular and system levels, upon which presumptive functions have been ascribed to the prion protein.
It is therefore not surprising that current literature pictures the function of PrPC as “unknown”, “unresolved”, “uncertain”, “obscure”, “abstruse”, or “elusive”, among other demeaning terms. Indeed, some of the alleged functions coexist with their opposites. For example, despite substantial agreement that PrPC supports cytoprotection (Liang et al., 2006; Martins et al., 2010; Mehrpour and Codogno, 2010; Santos et al., 2015), proapoptotic effects have also been reported (Paitel et al., 2002; Solforosi et al., 2004; Zhang et al., 2006). Whereas the binding of PrPC to the co-chaperone hop/STI1 triggers neuroprotective signals (Zanata et al., 2002), and the expression of PrPC is associated with enhanced synaptic function (Robinson et al., 2014), binding of PrPC to AßO induces synaptotoxic signals (Nygaard and Strittmatter, 2009). Also, the prion protein reportedly stimulates the proliferation of stem cells (Steele et al., 2006; Santos et al., 2011), but may also shift the phenotype of human embryonic stem cells from self-renewal to differentiation (Lee and Baskakov, 2013).
Granted, methodological differences as well as distinct experimental preparations might explain such contradictory effects. However, the latter are also consistent with a strong cell type- and context-dependency in the behavior of PrPC (Linden et al., 2008; Steele et al., 2009). Such an abundance and variety of functional properties is even more striking considering that the vast majority of mature PrPC molecules are tethered to the outer leaflet of the plasma membrane through a glycosyl-phosphatydil inositol (GPI) anchor (Stahl et al., 1987), and therefore lack an intracellular domain capable of transferring signals from the extracellular environment to the intracellular milieu. Signal transfer involving the prion protein must therefore be conveyed by transmembrane molecules engaged either together with or through PrPC. Analysis of such molecular complexes is required to understand the roles of the prion protein in physiological context, as well as its multiple interventions in both health and disease.
Research on PrPC-binding partners was originally directed at the identification of a so-called “protein X”, participant in the conversion of PrPC into the scrapie form (Yehiely et al., 1997), or otherwise involved in the formation and propagation of prions (Caughey and Baron, 2006). Over the years a list of putative PrPC-binding partners grew out of various approaches (Schmitt-Ulms et al., 2004; Aguzzi et al., 2008; Linden et al., 2008). Several such interactions were validated through compelling biochemical and cell biological procedures, and in each individual case the results were interpreted as evidence for the respective authors’ view of the long sought fundamental function of PrPC. In contrast, a number of other putative ligands still lack rigorous confirmation or, often, are unlikely to pair with PrPC in physiological context due to incongruous topologies (Aguzzi et al., 2008; Linden et al., 2008). Nonetheless, even the current consensus around only a handful of strictly validated binding partners allows for the conclusion that PrPC is poised to participate in a variety of combinatorial, multiprotein complexes at the cell surface (Martins et al., 2002, 2010; Linden et al., 2008). The composition of such molecular arrangements is expected to depend on both cell type and context—the former determines the repertoire of binding partners available at the cell surface, whereas the latter modulates their stoichiometry and pattern of activation. The influence of both these factors is further enriched by the rapid and continuous trafficking of PrPC among distinct plasma membrane domains, and the repeated cycles of endocytosis and resurfacing prior to degradation of individual PrPC molecules (Harris, 2003; Prado et al., 2004).
To account for the abundance of cell- and context-dependent, PrPC-related roles and phenotypes, as well as the growing list of validated binding partners, we advanced the hypothesis that PrPC functions as a dynamic platform for the assembly of signaling modules at the cell surface, analogous to the scaffold proteins involved in the organization of intracellular signal transduction pathways (Linden et al., 2008, 2012). This theory is further discussed here, in light of three decades of studies that led to the robust characterization of intracellular scaffold proteins (Langeberg and Scott, 2015).
The Prion Protein as a Cell Surface Scaffold Protein
The current concept of a natural scaffold protein is that of an intracellular, multivalent molecule that binds several members of a signaling pathway leading to a higher order, spatially restricted ensemble, which optimizes downstream signal transfer (Morrison and Davis, 2003; Good et al., 2011; Langeberg and Scott, 2015). Early work suggested that the role of such proteins was limited to the holding of intracellular enzymes in close proximity (Faux and Scott, 1996), but subsequent studies uncovered remarkable structural and functional plasticity (Chen et al., 2005; Brennan et al., 2011; Pan et al., 2012; Smith and Scott, 2013; Barbar and Nyarko, 2014), which helps these molecular platforms regulate spatial, temporal and kinetic properties of signal transduction pathways (Pan et al., 2012). The following sections consider the parallels between the fundamental properties of the prion protein and those of intracellular scaffold proteins.
Assembly of Multicomponent Signaling Modules
The basic property of a scaffold protein was originally seen as the offer of a molecular architecture that organizes an intracellular signaling cascade, through the binding of several sequential members of a defined pathway (Pawson and Scott, 1997; Whitmarsh and Davis, 1998). Consistent with this fundamental property, defined sets of molecules among those known to interact with the prion protein compose functional assemblies with specific signaling properties (Figure 1).
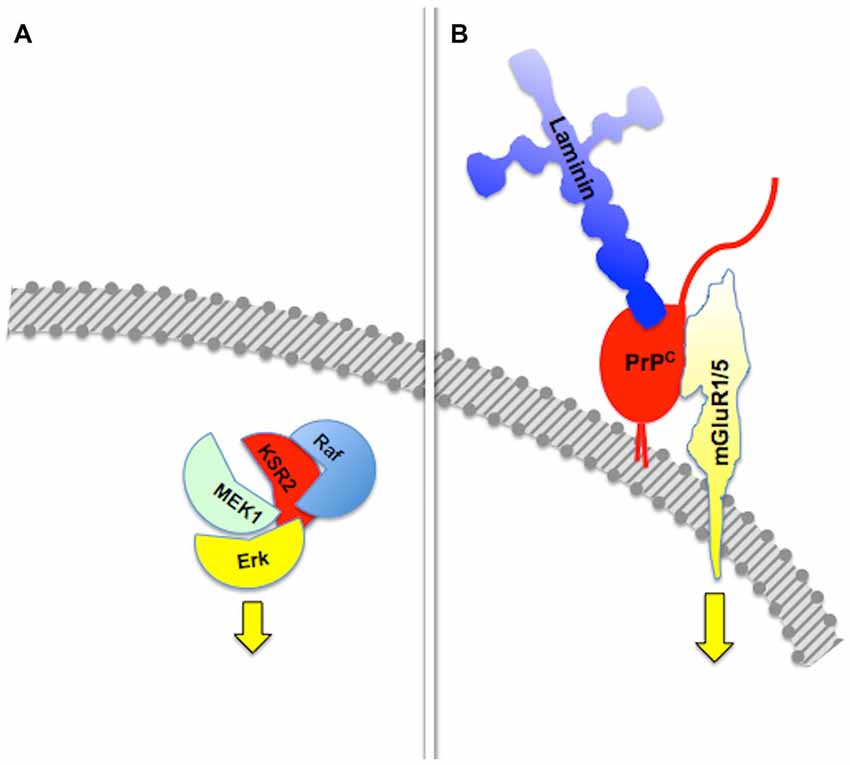
Figure 1. Assembly of multicomponent signaling modules. This and the following figures depict multiprotein signaling modules assembled around a scaffold protein (shown in red); the diagram to the left of the vertical bar portrays an intracellular signaling module organized by a consensual scaffold protein, and the scheme to the right represents a cell surface signaling module scaffolded by the prion protein. Yellow arrows indicate output signals from the scaffolded complex. Except where explicitly indicated, the form of each drawing or their juxtaposition indicate binding only, and do not imply either structural or spatial arrangements. (A) KSR2-scaffolded MAP kinase cascade based on Kolch (2005). (B) PrPC-scaffolded, mGluR1/5-mediated, laminin γ1-induced signaling module based on Beraldo et al. (2011).
Group I metabotropic receptors mGluR1 and mGluR5 belong to a subclass of receptors for the ubiquitous neurotransmitter glutamate (Ferraguti et al., 2008; Ribeiro et al., 2010). These receptors, originally identified as potential ligands of the prion protein in a PrPC-baited phage display screen and validated through biochemical experiments (Beraldo et al., 2011), are required to trigger intracellular phospholipase C (PLC)-mediated calcium signals induced in hippocampal neurons by the binding of PrPC to a peptide from the γ1 chain of the extracellular matrix protein Laminin (Ln-γ1; Graner et al., 2000; Beraldo et al., 2011). Signaling through this pathway induces neuritogenesis in both isolated hippocampal neurons and PC12 cells (Beraldo et al., 2011). In turn, the α7 type of nicotinic acetylcholine receptor (α7nAChR) was also shown to bind PrPC (Beraldo et al., 2010), and this interaction was required to trigger calcium influx, the activation of both protein kinase A and Erk, and trophic responses in isolated hippocampal neurons following the binding of the cochaperone hop/STI1 to PrPC (Zanata et al., 2002; Lopes et al., 2005; Beraldo et al., 2010).
Somewhat similar results were reported following experiments done with dorsal root ganglion (DRG) neurons (Santos et al., 2013), where both hop/STI1 and Ln-γ1 induced calcium responses and axon elongation. Here again, the responses triggered by Ln-γ1:PrPC interaction were mediated by mGluR1/5. Distinct from hippocampal neurons, however, signals triggered by hop/STI1:PrPC binding in DRG neurons were traced to the TRPC family of transient calcium receptor channels (Ramsey et al., 2006), rather than to α7nAChR (Santos et al., 2013). It is not known whether the latter result is due to direct PrPC:TRPC binding, or to an indirect cell surface interaction, but the differing results reported in neurons either from the central (CNS) or peripheral (PNS) nervous system (Beraldo et al., 2010; Santos et al., 2013) are consistent with the aforementioned cell- and context-dependence of PrPC-mediated signal transduction. Importantly, evidence was shown for DRG neurons, but not for hippocampal neurons, of synergism between the hop/STI-1:PrPC and Ln-γ1:PrPC effects, as well as of simultaneous occupation of binding sites in PrPC by both ligands (Santos et al., 2013), supporting the view that PrPC scaffolds multiple molecules at the cell surface, however depending on both cell type and context.
Crosstalk of Scaffolded Signaling Pathways
In contrast with the early idea of an exclusive intracellular scaffold protein for each defined set of signaling partners, subsequent work disclosed extensive crosstalk among scaffolded signaling networks (Pan et al., 2012). Thus, scaffold proteins such as β-arrestins or axin may each engage multiple signaling cascades (Luo and Lin, 2004; Dard and Peter, 2006). In turn, distinct scaffold proteins may organize the same set of signaling intermediates, such as the Raf-MEK-Erk kinase pathway (Pan et al., 2012; Witzel et al., 2012). In addition, scaffold proteins promote interactions of various signaling modules (Kolch, 2005; Dhanasekaran et al., 2007; Pan et al., 2012), and are subject to regulatory feedback control (Good et al., 2011; Witzel et al., 2012).
Certain signaling modules based on PrPC display similar features. Thus, PrPC binds several isoforms of the neural cell adhesion molecule (NCAM; Schmitt-Ulms et al., 2001; Slapšak et al., 2016), a cell surface-adhesion molecule of the immunoglobulin superfamily. NCAMs are widely expressed in many tissues and are especially abundant in the nervous system, where they mediate both neural histogenesis and plasticity through homophilic cell-cell interactions (Edelman, 1986; Rutishauser and Landmesser, 1996). Upon binding to PrPC, NCAM is recruited to lipid rafts, which facilitates interaction with the soluble Fyn protein kinase, thus leading to intracellular signaling (Santuccione et al., 2005). In addition, the extensive network of NCAM-binding cell adhesion molecules, proteoglycans and extracellular matrix molecules (Nielsen et al., 2010) adds an additional layer of complexity to PrPC-mediated signaling components containing NCAM. Interestingly, at least one NCAM binding partner, the cell adhesion molecule L1, also binds laminin (Hall et al., 1997), which forms a loop that may amplify the consequences of the PrPC-NCAM interaction. Thus, it is expected that the engagement of PrPC by binding to laminin entails cross-linked activation of multiple signaling pathways, through the concurring transfer of transmembrane signals through mGluR1/5, NCAM and L1 (Figure 2).
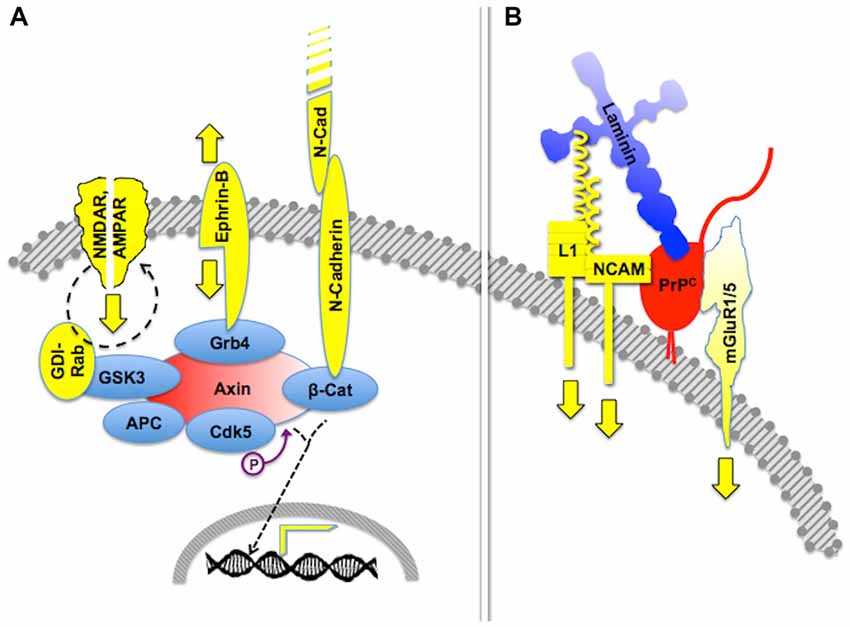
Figure 2. Crosstalk of multicomponent signaling modules. The drawings represent multiple signaling modules that may be organized by either an intracellular scaffold protein or the cell surface cellular prion protein PrPC. (A) Intracellular protein Axin functions as a major scaffold for components of synapses, which include interactive glutamate receptors of the NMDA and AMPA types, bidirectional signaling by ephrin-B, and adhesive interactions through N-cadherin, the latter of which modulates gene transcription through β-catenin. Based on Wei et al. (2010) and Chen et al. (2013). (B) Expected crosstalk of Laminin (Ln)-induced signals, transferred to the intracellular milieu through PrPC-scaffolded mGluR1/5-, NCAM- and L1-mediated pathways. Based on Nielsen et al. (2010) and Beraldo et al. (2011).
Importantly, the 37 kDa Laminin Receptor Precursor/Laminin Receptor (LRP/LR) has been identified as an additional binding partner of PrPC (Rieger et al., 1997). Two sets of cognate binding sites were identified in both partners, one of which required mediation of heparan sulfate proteoglycan, and included a heparin-binding site in PrPC (Hundt et al., 2001; Warner et al., 2002). Interestingly, the binding site in LRP/LR for both Laminin and PrPC is the same (Rieger et al., 1999), and the binding sites in PrPC for both LRP/LR and Laminin partially overlap (Linden et al., 2008), which implies an even more intricate arrangement of PrPC-mediated, laminin-induced signaling.
Allosteric Properties of Multiprotein Signaling Modules
Mechanisms of regulation of scaffold proteins and their clients include reciprocal allosteric changes (Pan et al., 2012; Langeberg and Scott, 2015). For example, the scaffold protein Kinase Suppressor of Ras (KSR), which regulates signal transduction through MAPK pathways (Witzel et al., 2012), not only allosterically modulates the activity of its client protein kinases (Langeberg and Scott, 2015), but its own kinase activity is unlocked upon binding to B-RAF, which facilitates downstream phosphorylation of MEK (Brennan et al., 2011; Hu et al., 2011).
Reciprocal allosteric effects have also been shown in experiments done with recombinant PrPC and some of its binding partners. Thus, the binding of the co-chaperone hop/STI1 to PrPC induced C-terminal compaction of the former, detected by modeling through small-angle X-ray spectroscopy (SAXS), as well as a slight loss of PrPC α-helical structure, involving at least the PrPC143–153 (H1) α-helix (Romano et al., 2009; Figure 3). The latter domain of PrPC contains binding sites for both LRP/LR and NCAM (Hundt et al., 2001; Santuccione et al., 2005), which raised the hypothesis that the prion protein may compute signaling triggered by multiple ligands. Interestingly, whereas a PrPC-binding hop/STI1 peptide mimicked the full hop/STI1 protein in the induction of several PrPC-mediated responses in neurons (Zanata et al., 2002; Lopes et al., 2005), the proliferative effect of hop/STI1 upon glioblastoma cells also depended on the hop/STI1:PrPC interaction, but was not induced by the peptide alone (Erlich et al., 2007; Linden et al., 2012). The latter effect is likely associated with the reciprocal allosteric modulation between hop/STI1 and PrPC, and a higher order interaction may involve one or more additional hop/STI1 partners (Linden et al., 2012).
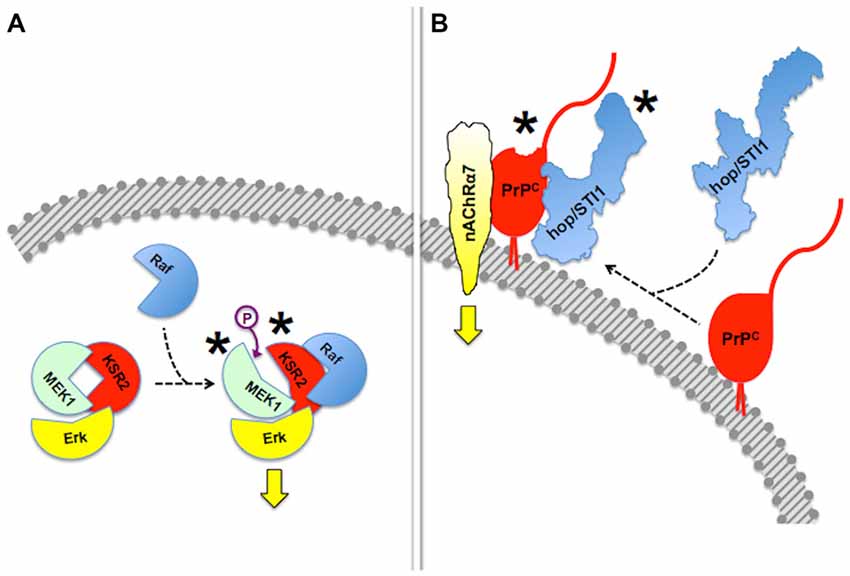
Figure 3. Allosteric properties of signaling modules. In this figure, asterisks indicate allosteric events. (A) Binding of Raf to scaffold protein KSR2 leads to structural rearrangement in the MEK1 kinase and ensuing activation of Erk. Based on Brennan et al. (2011) and Langeberg and Scott (2015). This simplified diagram does not include dimerization events also indicated by Brennan et al. (2011). (B) Reciprocal allosteric changes induced by the binding of hop/STI1 to PrPC. Compaction of hop/STI1 and structural remodeling within at least the PrPC143–153 α-helix may engage the transmembrane signaling proteins LRP/LR and NCAM, both of which bind that domain of PrPC, but the binding domains involved in the PrPC:nAChRα7 interaction are still unknown. Based on Romano et al. (2009) and Linden et al. (2012).
Also consistent with allosteric control of cell surface complexes, is the evidence that a variety of human TSE-related point mutations along the globular domain of PrPC both enhanced the binding of GAGs to the far N-terminal of PrPC, as well as unlocked a normally hidden GAG-binding site midway between differing mutations (Yin et al., 2007). These results may signify an impact on signaling properties of LRP/LR, since the interaction of this receptor with one of its target sites in PrPC depends on mediation by a heparan sulfate proteoglycan (Hundt et al., 2001). It should be noted that although PrPC mutations examined in this context were all disease-associated (Yin et al., 2007), several of those correspond to aminoacid residues located either within or close to the binding sites of functionally relevant PrPC ligands such as hop/STI1 (Zanata et al., 2002) or mGluR5 (Haas et al., 2014).
Furthermore, an antibody that targets residues in the α1 and α3 helices of the C-terminal globular domain of PrPC resulted in severe toxicity dependent on the latter’s N-terminal (Sonati et al., 2013). This study pointed to remarkable long-distance interactions along the full extent of PrPC. Interestingly, although the set of residues of PrPC that underwent chemical shifts detectable through nuclear magnetic resonance upon antibody binding did not include the N-terminal, they overlapped extensively with the domains involved in the interaction of the prion protein with both Laminin and NCAM (Gauczynski et al., 2001; Santuccione et al., 2005; Sonati et al., 2013). More recent work showed that an engineered GPI-anchored, N-terminal only PrPC molecule (PrPΔ141–225, dubbed FTgpi) mimicked the effect of the toxic antibody. Thus, FTgpi bound the endoplasmic reticulum (ER) chaperone Immunoglobulin heavy chain-Binding Protein/Glucose-Regulated Protein 78 (Bip/GRP78), and such binding was followed by sustained ER stress, reduced FTgpi protein/mRNA ratio due to rapid proteolysis, as well as activation of the Protein Kinase R-like ER Kinase (PERK), and cell death (Dametto et al., 2015). Differing, however, from these results, the previous study from the same group did not report any change in the content of full length PrPC upon binding of the toxic antibody (Sonati et al., 2013). Thus, it is not clear whether the toxicity of the latter engages the same mechanisms that link FTgpi with fatal ER stress, or alternatively, depend on interactions of the N-terminal of PrPC at the cell surface. Interestingly, other than its canonical location with the ER, Bip/GRP78 is also found both at the cell surface and in the extracellular medium upon cellular stress (Delpino and Castelli, 2002; Corrigall et al., 2004; Marín-Briggiler et al., 2010; Panayi and Corrigall, 2014; Tsai et al., 2015), therefore potentially subject to scaffolding by an allosterically activated N-terminal domain of PrPC.
Compartmentalization of Scaffolded Signaling Modules
Besides allosteric modulation, the activities of intracellular scaffold proteins are subject to robust regulation by several other mechanisms (Morrison and Davis, 2003; Dard and Peter, 2006). Distribution to selected subcellular domains is required for the spatial and temporal restriction of the activity of signaling modules, as exemplified by the nucleocytoplasmic shuttling of both the yeast Ste5p and mammalian β-arrestin (Mahanty et al., 1999; Wang et al., 2003), or the tethering of KSR to the plasma membrane (Zhou et al., 2002; Ory and Morrison, 2004; Koveal et al., 2012).
Effects induced by hop/STI1:PrPC interaction provide an example of compartmentalization of PrPC-mediated signaling (Figure 4). The binding of hop/STI1 to PrPC in CNS neurons engages the cAMP-PKA, as well as the Erk signaling pathways (Chiarini et al., 2002; Zanata et al., 2002). Both responses were blocked either by α-bungarotoxin, a specific inhibitor of the α7 nicotinic cholinergic receptor, or by the removal of extracellular calcium, which together with evidence of the binding of PrPC to α7nAChR, implicated this membrane receptor in the PrPC-mediated cell responses to hop/STI1 (Beraldo et al., 2010). Nevertheless, the activation of Erk induced by hop/STI1 was abolished by prevention of PrPC endocytosis, while that of the cAMP-PKA pathway persisted (Americo et al., 2007; Caetano et al., 2008). It is likely that the latter results reflect changes in PrPC-ligand interactions as the prion protein moves along distinct compartments. One hypothesis to explain these results stems from the changing physicochemical environment along the endocytic pathway. Thus, the concentration of calcium collected via pinocytosis to the lumen of endosomes undergoes an initial rapid decrease compared with the extracellular medium, due to both acidification and the activity of various transporters (Gerasimenko et al., 1998). This strongly affects the binding of several plasma membrane receptors to their cognate ligands (Andersen and Moestrup, 2014). Also, the progressive acidification along the endocytic pathway is, by itself, expected to promote changes in structure and thermodynamic stability of the prion protein (Gu et al., 2003; Zahn, 2003; Chiang et al., 2008; Biljan et al., 2012; Kovač et al., 2016), which in combination with changes in luminal calcium, may modulate the binding of PrPC ligands. Further work directed at the characterization of the dynamic behavior of PrPC ligands, in particular in response to Ca2+ levels, is therefore warranted to clarify mechanisms that regulate the compartmentalization of PrPC-mediated signal transduction.
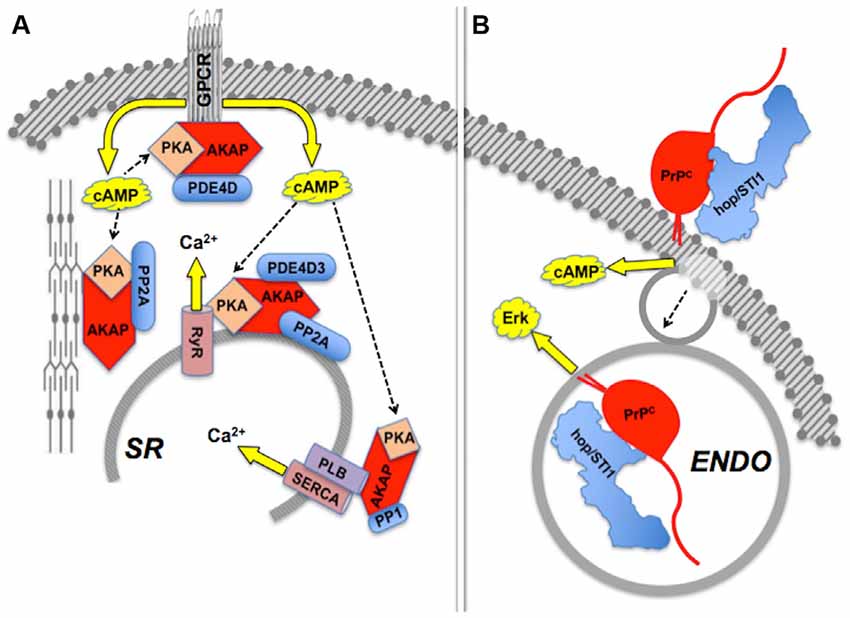
Figure 4. Compartmentalization of scaffolded signals. (A) Sets of AKAP-scaffolded client proteins located in distinct compartments lead to distinct intracellular signals mediated by cAMP-PKA activity or calcium fluxes. Modified from Fu et al. (2013). (B) Binding of hop/STI1 to PrPC leads to endocytosis-independent signaling through cAMP, and endocytosis-dependent Erk signaling. The PrPC-binding clients involved in each case are yet to be determined. Based on Americo et al. (2007) and Caetano et al. (2008).
In addition to the PrPC-ligand interactions in cis described above, other studies of both soluble PrPC and its fragments also typify compartmentalized signaling. Thus, various soluble recombinant forms of PrPC bound to and underwent partial LRP/LR-dependent internalization (Gauczynski et al., 2001), protected human neurons from Bax-mediated apoptosis (Bounhar et al., 2001), induced neurite outgrowth and/or synaptogenesis in cultured cerebellar and hippocampal neurons (Chen et al., 2003; Kanaani et al., 2005), and activated monocytes (Krebs et al., 2006; Jeon et al., 2013) and natural killer cells (Seong et al., 2015). Such effects are contingent upon the activation of a variety of intracellular signaling molecules, including PI3-kinase, Erk, cAMP/PKA or PKC (for review see Linden et al., 2008). Although those results were obtained with recombinant PrPC, they are consistent with physiological effects of PrPC in trans, either through the release of PrP-containing microvesicles (Porto-Carreiro et al., 2005; Robertson et al., 2006; Vella et al., 2008; Wang et al., 2011; Hajj et al., 2013; Ritchie et al., 2013; Berrone et al., 2015; Guo et al., 2015), or as soluble fragments derived from endoproteolysis of PrPC (Béland et al., 2012; Roucou, 2014).
An extended view of PrPC-based interactions in trans includes the recently disclosed role of the prion protein upon myelin homeostasis, through the specific interaction of its N-terminal flexible tail with the Adhesion G protein-coupled receptor Gpr126 (Adgrg6; Küffer et al., 2016 and see below). Also recently, evidence was shown that a recombinant, soluble PrPC promoted growth cone (GC) motility and extension of neurites, through in trans interactions that depend on cell surface PrPC as well as NCAM, both of which are recruited to common sites at the GC plasma membrane, and involve the activation of several downstream signaling pathways (Amin et al., 2016). The latter are analogous to effects triggered by other extracellular ligands of the prion protein, and suggest a physiological role of either soluble or microvesicle-associated PrPC upon neurite outgrowth. A notable requirement for the reported effect in trans was the integrity of the soluble PrPC molecule (Amin et al., 2016), which is consistent with long-range allosteric interactions throughout the full extent of the prion protein (Yao et al., 2003; Yin et al., 2007; Christen et al., 2009; Thakur et al., 2011; Sonati et al., 2013; Spevacek et al., 2013). It is, however, not yet known whether the effects of the recombinant PrPC in physiological context may require its location at the surface of extracellular microvesicles.
Posttranslational Regulation of Scaffolding Properties
Functional regulation of intracellular scaffold proteins also relies upon phosphorylation (Ory and Morrison, 2004; Good et al., 2011; Tacchelly-Benites et al., 2013; Langeberg and Scott, 2015) or ubiquitination (Shenoy et al., 2001). Neither has been so far described for the prion protein, but other posttranslational modifications of native PrPC molecules affect signaling properties.
The GPI anchor was reported as required for the formation of cell-surface PrPC dimers, which in turn were needed for PrPC-mediated protection from cellular stress (Rambold et al., 2008). Consistent with a previous theoretical model (Warwicker, 2000), the short internal hydrophobic domain PrPC113–133 was identified as the dimerization domain (Rambold et al., 2008; Figure 5). This finding strengthens the notion that posttranslational modifications impart PrPC properties relevant for signal transduction. The GPI anchor is also critical for the trafficking of PrPC along distinct plasma membrane domains (Harris, 2003; Prado et al., 2004), which underlies the above-mentioned dependence of downstream signals on endocytosis of PrPC, and in particular for the targeting of the prion protein to lipid rafts (Morris et al., 2006; Taylor and Hooper, 2006; Puig et al., 2014). The latter explains, for example, the recruitment of NCAM towards the preferential location of its intracellular signaling partner, the soluble Fyn kinase (Santuccione et al., 2005), as well as the association of PrPC with reggie/flotillins (Stuermer and Plattner, 2005), which drives both downstream MAP kinase and calcium signals (Stuermer et al., 2004). Recent work also attributes to the GPI anchor an important role in PrPC processing and the shedding of bioactive fragments (Puig et al., 2014).
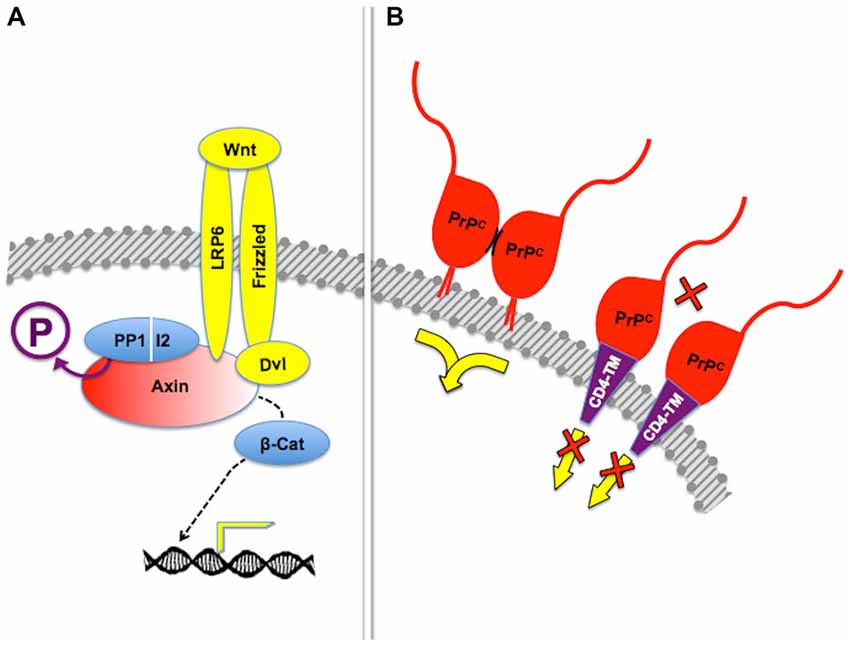
Figure 5. Posttranslational regulation of scaffolding properties. (A) Dephosphorylation of the scaffold protein Axin by the PP1-I2 phosphatase complex is required for β-catenin-induced gene expression in response to Frizzled-mediated Wnt signaling. Adapted from Tacchelly-Benites et al. (2013). (B) Tetethering of PrPC through the glycosyl-phosphatydil inositol (GPI) anchor, but not through an heterologous transmembrane domain, was needed for protection of neuroblastoma cell lines from cell death induced by the excitotoxin kainate. In this context, it was shown that stress protection depended on dimerization of PrPC (short, curved black lines), but additional molecules required for the transfer of cytoprotective signals have not been identified. Based on Rambold et al. (2008).
Furthermore, the composition of the GPI anchor was shown to regulate both the lipid content of membrane microdomains and the localization of PrPC therein (Bate and Williams, 2011b; Bate et al., 2016), with concurring changes in synaptotoxic signaling triggered by cross-linking of PrPC molecules with either AßO or antibodies, and mediated by phospholipase A2 (PLA2; West et al., 2015; Bate et al., 2016). Interestingly, PLA2 has also been identified as a mediator of the release of the APP ectodomain induced by activation of the PrPC ligand mGluR1 (Nitsch et al., 1997). These data are consistent with an important, selective role of the GPI anchor upon the interaction of PrPC with its client proteins.
Another critical postranslational modification of PrPC is the N-linked glycosylation of either one or both target asparagine residues (Turk et al., 1988; Rudd et al., 2002; Lawson et al., 2005). Comparative analysis of PrPC either from the brain or from peripheral blood mononuclear cells (PBMC), showed that the relative content of the unglycosylated and glycosylated forms differed between the two samples (Li et al., 2001). Distinct patterns of sialylation have also been described for PrPC from either brain or spleen (Baskakov and Katorcha, 2016), and possible roles of sialylation upon functional properties of PrPC were discussed (Baskakov and Katorcha, 2016). Other studies indicated that posttranslational modifications produce a collection of differing glycosylated forms of PrPC (Pan et al., 2002), which vary across distinct brain regions (Kuczius et al., 2007b) and change with aging (Goh et al., 2007). Heterogeneous glycosylation is likely to impart selectivity of ligand binding, as suggested both by differential binding to antibodies (Li et al., 2001; Kuczius et al., 2007a) and metal ions (Moudjou et al., 2007), as well as by the differing outcomes of peripheral inoculation of TSE upon experimental prion disease in mice expressing distinct glycosylated forms of PrPC (Cancellotti et al., 2010). Indeed, a coimmunoprecipitation experiment in our lab suggested that interaction of PrPC with the purinergic receptor P2X4R depends on the pattern of glycosylation of PrPC (Carneiro et al., 2016).
Stoichiometry and Oligomerization in Scaffold-Client Signaling Modules
In early attempts to model the behavior of scaffold proteins, attention was focused on their binding selectivity and their ensuing ability to spatially concentrate sequential components of intracellular signaling modules (Eungdamrong and Iyengar, 2004). Such studies gradually evolved to the matter of stoichiometry of multiprotein assemblies, and the effects of varying concentrations of either the scaffold or their client proteins (Bray and Lay, 1997; Bray, 1998; Levchenko et al., 2000; Heinrich et al., 2002; Locasale et al., 2007; Kholodenko et al., 2010). Pertinent to the matter of stoichiometry, the effects of certain scaffold proteins upon signaling efficacy were traced to their oligomerization (Yablonski et al., 1996; Elion, 2001; Ren et al., 2005; Chen et al., 2008; Gold et al., 2011; Abel et al., 2015; Liu et al., 2016).
A major challenge to a deeper understanding of the scaffolding function is, however, the scarcity of data regarding both kinetic parameters and relative concentrations of signal transducers in confined intracellular domains, which are required for the full understanding of signaling dynamics (Langeberg and Scott, 2015). Still, the stoichiometries of certain scaffold-client complexes have been unraveled. For example, the scaffold protein AKAP79 forms a 2:2:2:2 complex with its client proteins calmodulin, calcineurin and a PKA regulatory subunit (Gold et al., 2011), whereas AKAP18γ forms a 1:2 complex with a PKA regulatory subunit (Smith et al., 2013), and the NOD-like receptor NLRP3 was proposed to form a multimeric, equimolar inflammasome with Caspase-1 through the adaptor protein Apoptosis-associated Speck-like protein containing a CARD (ASC; Lechtenberg et al., 2014). An especially complex case is the postsynaptic density (PSD), which contains large numbers of proteins, including neurotransmitter receptors, adaptor and effector proteins organized in aggregates visible through conventional transmission electron microscopy (Harris and Weinberg, 2012). Importantly, certain differences were reported among the relative concentrations of PSD components in differing areas of the CNS (Cheng et al., 2006; Sheng and Hoogenraad, 2007; Lowenthal et al., 2015; Patrizio and Specht, 2016).
So far, growing interest in ligands of the prion protein has yet to lead to direct analysis of the stoichiometry of the PrPC-based signaling modules, and this is, at this time, the least understood among the features discussed herein with respect to intracellular scaffold proteins. Nevertheless, many studies have compared either the phenotypes of mice, or the properties of cells harboring differing contents of PrPC. For example, by comparing Prnp-KO, WT and Prnp-overexpressing mice subject to ischemic injury to the brain, it was reported both that PrPC accumulates at the penumbra of hypoxic damage, and that lack of PrPC is associated with aggravated ischemic injury (McLennan et al., 2004; Weise et al., 2004, 2006; Spudich et al., 2005; Mitsios et al., 2007). Transduction of the Prnp gene carried by a recombinant viral vector improved neurological behavior and reduced the volume of cerebral infarction in a rat model of cerebral ischemia (Shyu et al., 2005).
The results above suggest a dose-dependent neuroprotective effect of PrPC against hypoxic-ischemic insults, but its mechanisms are unclear. Enhanced sensitivity to ischemic damage in the absence of PrPC was originally attributed to an impairment of the antiapoptotic phosphatidylinositol 3-kinase/Akt pathway, resulting in enhanced postischemic activation of caspase-3 (Weise et al., 2006). However, mice harboring an increased content of PrPC displayed significantly smaller infarct volumes than wild type, accompanied by a reduction in early postischemic Erk1/2 phosphorylation, whereas no difference was detected in postischemic phosphorylation of Akt (Weise et al., 2008). Recently, the same group reported an increased content of lactate dehydrogenase (LDH), as well as evidence of physical interaction of LDH with PrPC, and suggested that LDH may mediate PrPC-dependent neuroprotection under low oxygen conditions, although the apparent physical LDH:PrPC interaction was localized to the cytoplasm (Ramljak et al., 2015). Still, in those reports no cell surface partners of PrPC have been associated with the altered intracellular signals, which preclude further consideration of stoichiometry.
Other studies showed that hop/STI1 haploinsufficient mice were more vulnerable to ischemic insult and their astrocytes secreted lower amounts of the cochaperone than wildtype. Significantly, PrPC mediated prevention of ischemic insult by extracellular hop/STI1 (Beraldo et al., 2013). Since neurotrophic signals induced by hop/STI1:PrPC interaction in central neurons depend on α7nAChR (Beraldo et al., 2010), and the latter has been implicated in neuronal resistance induced by either nicotine or melatonin against hypoxia (Hejmadi et al., 2003; Parada et al., 2014), the hop/STI1:PrPC:α7nAChR signaling complex may be a major player in neuroprotection against ischemic insults. Interestingly, examples of sexually dimorphic, ischemic brain injury mediated by both hormonal and non hormonal mechanisms (Liu et al., 2009; Herson and Hurn, 2010; Manwani and McCullough, 2011; Fairbanks et al., 2012; Herson et al., 2013; Zuo et al., 2013; Sanches et al., 2015) include the sensitivity of hippocampal neurons to ischemia in PrPC-null mice (Sakurai-Yamashita et al., 2005), and evidence has been reported of both sexually-dimorphic α-bungarotoxin binding (Arimatsu et al., 1981; Arimatsu and Seto, 1982) as well as changed content of α7nAChR following prenatal stress (Schulz et al., 2013). These data warrant a critical examination of the stoichiometry of hop/STI1:PrPC:α7nAChR complexes in the context of sensitivity to ischemic insults, especially in view of the variegated homo- and/or hetero-multimeric, cholinergic receptors that may assembled around α7nAChR subunits, as indicated by experimental work with various cell types (Bertrand et al., 2015; Wu et al., 2016).
In contrast with the reports above of an unimodal dose-response relationship between cell responses and the content of PrPC, differing results were reported as to the sensitivity to ischemic damage of transgenic Prnp-overexpressing mice (Spudich et al., 2005; Weise et al., 2006), and several experimental models failed to conform to a regular dose-dependent effect among mice harboring variable amounts of PrPC (Coulpier et al., 2006; Jouvin-Marche et al., 2006; Terra-Granado et al., 2007; Lobão-Soares et al., 2008; Steele et al., 2009; Rial et al., 2012; Alfaidy et al., 2013). In several such cases, it appears that either up- or downregulation of PrPC may induce cellular dysfunction, and again the effect depends on both cell type and context. For example, the recently described dose-response curve of the neuritogenic effect of a soluble recombinant PrPC upon GCs of hippocampal neurons was clearly biphasic (Amin et al., 2016). Results as such strengthen the need for studies of the stoichiometry of PrPC-ligand complexes.
In a distinct experimental setting, we showed that the expression of the the Prnp gene, as well as the content of PrPC at the cell surface of mouse neutrophils, are selectively augmented by both inflammatory and behavioral stress, as a response mediated by a combination of serum TGFβ and glucocorticoid (Mariante et al., 2012). The increased content of PrPC endowed neutrophils with enhanced peroxide-dependent cytotoxicity toward endothelial cells (Figure 6), the mechanism of which is currently unknown. Studies of the stoichiometry of PrPC-dependent signaling complexes in immune cells may thus contribute to the understanding of neurodegenerative events (Beckman and Linden, 2016), in particular those mediated by neutrophils which have recently been implicated in the pathogenesis of AD (Zenaro et al., 2015).
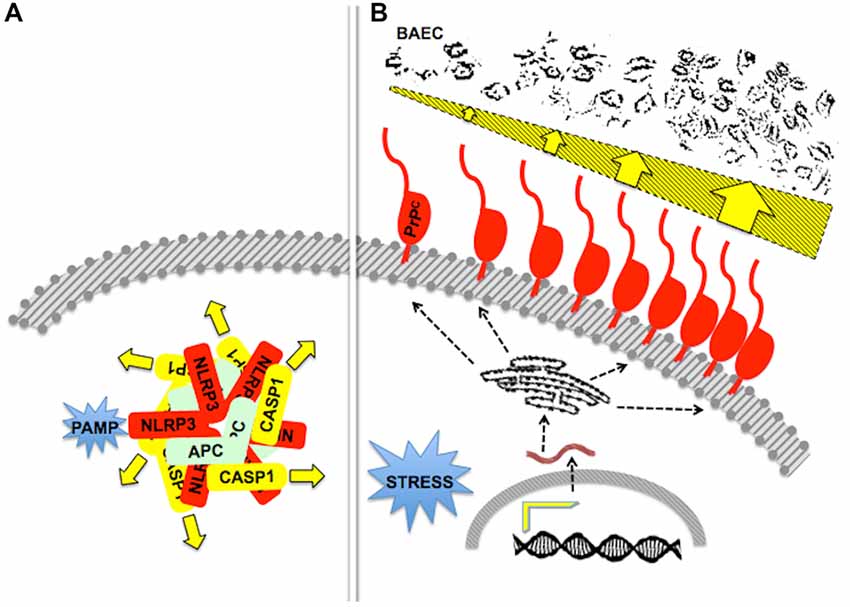
Figure 6. Stoichiometry and/or oligomerization in scaffold-client signaling modules. (A) A diagram of the pathogen-associated molecular pattern (PAMP)-activated, NLRP3-scaffolded inflammasome, formed by an equimolar associationof the latter with ASC and caspase-1. The drawing depicts a pentameric arrangement, but the actual stoichiometry is still unknown. Adapted from Lechtenberg et al. (2014). (B) The drawing represents the effect of an increased content of PrPC upon the killing of primary bovine aortic endothelial cells (BAEC) by neutrophils. Either inflammatory of behavioral stress induced increased gene expression and a higher content of PrPC at the surface of mouse neutrophils, which was associated with increased neutrophil cytotoxicity towards BAEC (dark profiles at the top). Adapted from Mariante et al. (2012). Effectors of cytotoxicity are still unknown.
In parallel, several lines of evidence indicate that clustering or oligomerization of native PrPC affect normal signal transduction. Various cellular responses were induced by cross-linking of PrPC with antibodies (Mouillet-Richard et al., 2000; Hugel et al., 2004; Solforosi et al., 2004; Pantera et al., 2009; Tomasi, 2010; Shi et al., 2013), as for example, the association of PrPC with microdomain-forming reggie/flotillin proteins, followed by recruitment of other transmembrane proteins and soluble intracellular protein kinases, leading to downstream signaling (Stuermer et al., 2004).
Native prion protein may likewise form dimers (Priola et al., 1995; Meyer et al., 2000), and in contrast with an earlier report that a recombinant protein failed to exhibit in vitro monomer-dimer equilibrium (Meyer et al., 2000), recombinant PrPC was shown to dimerize in solution at room temperature and upon crystalization, through domain swapping and rearrangement of disulfide bonds (Knaus et al., 2001). Copper ions at substoichiometric concentrations also induced self-association of PrPC molecules in vitro, without detectable conformational changes in the globular domain (Wells et al., 2006). Furthermore, adding to the above mentioned stress-protection effect of dimeric PrPC in cis (Rambold et al., 2008), homophilic interaction of PrPC molecules in trans were shown to impart cell adhesion properties particularly important for embryonic development in zebrafish (Málaga-Trillo et al., 2009).
Due to the evidence that the infectivity of abnormal conformers of the prion protein is associated with aggregation (Silveira et al., 2005), functional characterization of PrPC oligomers have usually been limited to their putative role as the basic components of pathogenic prions (Masel et al., 2005; Pan et al., 2005; Zhang et al., 2007; Gerber et al., 2008; Kaimann et al., 2008; Lee et al., 2010; Hafner-Bratkovič and Jerala, 2011; Hafner-Bratkovič et al., 2011; Apostol et al., 2013; Huang et al., 2013; Yu et al., 2016). Nonetheless, there is growing interest in physiological consequences of PrPC dimerization, such as their trafficking to the cell surface, endoproteolysis and shedding of soluble fragments with cytoprotective activity (Yusa et al., 2012; Roucou, 2014), all of which may be subject to stoichiometry-dependent multicomponent assemblies of PrPC and its ligands. This subject clearly needs further attention to allow better understanding of PrPC-dependent cell signaling and its consequences upon physiology and behavior.
Corruption of Prion Protein-Mediated Signaling and the Scaffold Hypothesis in Neuropathology
Mutations and polymorphisms in several members of the AKAP family of intracellular scaffold proteins, such as AKAP12, Ezrin and Merlin have been linked to hyperplastic syndromes and cancer (Poppinga et al., 2014; Han et al., 2015; Petrilli and Fernández-Valle, 2016), while other family members, such as Myospryn and AKAP9, have been associated with skeletal muscle and cardiovascular diseases (Tsoupri and Capetanaki, 2013; Diviani et al., 2016). In particular, a targeted mutation analysis has linked the Long-QT Syndrome (LQTS) to a single missense mutation in AKAP9, which disrupts its binding to a slowly activating cardiac potassium channel (IKs), thus preventing proper cAMP-dependent regulation of the latter, and leading to delayed repolarization of the ventricular action potential (Chen et al., 2007). The latter is a compelling example of the specific requirement of the scaffold-client interaction for maintaining a defined physiological condition. Robust, albeit less precise, genotype-phenotype correlations were inferred for other intracellular scaffold proteins and provisionally traced to scaffold-client interactions, such as the association of severe obesity with rare variants of KSR2, a member of the KSR family (Pearce et al., 2013), and that of certain transcripts of the dystrophin gene with cognitive impairment in a subset of muscular distrophy patients (Daoud et al., 2009; Desguerre et al., 2009; Taylor et al., 2010; Constantin, 2014; Molza et al., 2015). Further work is, however, warranted to reach a similar mechanistic understanding of scaffold corruption associated with mutations in either KSR or Dystrophin, as is the case of the AKAP9:IKs interaction associated with LQTS.
An analogous hypothesis of scaffold corruption applies to PrPC. Thus, the group I metabotropic glutamate receptor mGluR5 reportedly cooperates with PrPC for both AßO binding and toxicity Um and Strittmatter, 2013; Hu et al., 2014). AßO induced cell-surface clustering of PrPC (Caetano et al., 2011), while an mGluR5-selective negative allosteric modulator had a protective effect against both cognitive loss and the accumulation of neuropathological Aß oligomers and plaques in a transgenic AD mouse model (Hamilton et al., 2016). These results are consistent with the evidence for a pathogenic role of the PrPC:mGluR5 interaction, which may be linked to disruption of PrPC:mGluR5 stoichiometry. In addition, recent studies showed that the co-chaperone hop/STI1 has protective effects upon AßO toxicity, through direct interaction with the PrPC-α7nAChR complex (Ostapchenko et al., 2013), and evidence has been reported of a crosstalk between intracellular signaling induced by either AβO or the Ln-γ1 peptide through the PrPC-mGluR5 complex in both primary neuron cultures and cell lines (Beraldo et al., 2016). These data implicate at least two extracellular and two transmembrane ligands of PrPC in a cell-surface complex involved in both neurodegenerative and neuroprotective signaling associated with AD.
On the other hand, the recently disclosed interaction in trans of the N-terminal flexible tail of PrPC with the Adhesion G protein-coupled receptor Gpr126 was shown to favor myelination of peripheral axons through an increase in the levels of cAMP in Schwann cells, which likely explains the demyelinating polyneuropathy that affects aging PrPC-null mice (Küffer et al., 2016). Whereas possible roles of other PrPC-interacting molecules have not been examined, it is noteworthy that another known ligand of Gpr126 likewise involved in myelin homeostasis is Laminin-211 (Petersen et al., 2015), which bears the PrPC-interacting Laminin γ1 chain (Graner et al., 2000; Beraldo et al., 2011). These data raise the hypothesis of the operation of a signaling complex involving PrPC-laminin 211 binding in cis, and both PrPC- and Laminin 211-Gpr126 in trans, in both the physiological control of peripheral nerve myelination and in demyelination conditions.
The prevailing view that TSEs are caused by an exclusive gain-of-toxic function of the scrapie form of the prion protein, has often been challenged by an alternative view that loss-of-function of PrPC is likely to play a role in such diseases (for review see Leighton and Allison, 2016). The latter has historically been dismissed due to the lack of major neurological signs in PrPC-null mice (Büeler et al., 1992). Even the evidence of preclinical downregulation of PrPC in several disease models was discussed basically as a possible neuroprotective event, on the grounds that it would provide less substrate for conformational conversion and thus for disease progress (Mays et al., 2014). In fact, gain- and loss-of-function components are not mutually exclusive, and the abundant evidence for neuroprotective effects of PrPC (Zamponi and Stys, 2009; Martins et al., 2010; Biasini et al., 2012; Onodera et al., 2014; Zeng et al., 2015) concurs with the hypothesis that the early and robust loss of PrPC may be involved in the pathogenesis of TSEs. In turn, whereas PrPC has been identified as a pathogenic receptor for AβO in models of AD, binding of the prion protein to hop/STI1 also mediates neuroprotection against AβO neurotoxicity (Ostapchenko et al., 2013), which reinforce the interest in physiological properties of PrPC.
Conclusion and Further Directions
So far, the reported physiological roles of PrPC cannot be reduced to any intrinsic function beyond its ability to bind other molecules required to either overcome the lack of a transmembrane domain in the dominant form of PrPC, or to bridge in trans cell-cell interaction. Still, most attempts at understanding the Janus-faced behavior of the prion protein in various circumstances have led investigators to concentrate on effects of either the engagement or ablation of PrPC, or else to address single PrPC partners. This has usually led to equating the elusive physiological function of PrPC to its role in a particular process or phenotype. Contrary to such a restricted approach, current evidence supports the hypothesis that the functional properties of PrPC are based on its ability to serve as a hub for a large variety of multicomponent signaling modules, with widespread consequences for both physiology and pathology.
The data reviewed above highlight a striking resemblance of both the behavior of PrPC and that of intracellular signaling scaffold proteins. Similar to the latter, the prion protein displays the following properties (Figures 1–6): (a) ability to recruit spatially restricted sets of binding molecules involved in specific signaling; (b) mediation of the crosstalk of signaling pathways; (c) reciprocal allosteric regulation with its partners; (d) compartmentalized responses; (e) dependence of signaling properties upon posttranslational modification; and (f) stoichiometric requirements and/or oligomerization-dependent impact on PrPC-dependent effects. These features, added to the widely recognized pleiotropism of PrPC, are consistent with our view that the prion protein functions as a scaffold protein, which helps the assembly of various cell type- and context-specific, multicomponent signaling modules at the cell surface (Linden et al., 2008, 2012, 2017).
The recognition of PrPC as a scaffold protein appears to be the closest to philosophical concepts of biological function, which imply an unambiguous, unconditional, generalized property of a biological unit (Cummins, 1975; Griffiths, 1993; Diaz-Herrera, 2006; Seringhaus and Gerstein, 2008). Rather than concentrating on any selected, individual binding partner of PrPC, such a concept recommends a wider, systemic approach to the variety of signaling modules scaffolded by the prion protein in either physiological or pathophysiological contexts. In view of the failure of several clinical trials directed at either the TSEs or AD (Stewart et al., 2008; Gauthier et al., 2016), this approach may help devise a novel rationale to the development of effective therapeutic options for such refractory neurodegenerative conditions.
Author Contributions
RL is the sole author and fully responsible for this article.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Funding for experimental work, as well as various fellowships for members of the author’s lab have been provided by the Brazilian National Research Council (Conselho Nacional de Desenvolvimento Científico e Tecnológico, CNPq), the Rio de Janeiro State Research Foundation (Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro, FAPERJ), and occasionally by collaborative grants funded by the São Paulo State Research Foundation (Fundação de Amparo à Pesquisa do Estado de São Paulo, FAPESP). I am indebted to my colleagues Vilma R. Martins, Marco A.M. Prado, Ivan Izquierdo, Yraima Cordeiro, Luciana B. Chiarini, Luis Mauricio T.R. Lima and to the late Professor Ricardo Brentani, who shared with me many years of collaboration and exchange of ideas, as well as to the students and post-docs who carried the experimental work and contributed to the shaping of the concept expressed in this article. Special thanks to Yraima Cordeiro for critical reading of the manuscript.
References
Abel, A. M., Schuldt, K. M., Rajasekaran, K., Hwang, D., Riese, M. J., Rao, S., et al. (2015). IQGAP1: insights into the function of a molecular puppeteer. Mol. Immunol. 65, 336–349. doi: 10.1016/j.molimm.2015.02.012
Aguzzi, A., Baumann, F., and Bremer, J. (2008). The prion’s elusive reason for being. Annu. Rev. Neurosci. 31, 439–477. doi: 10.1146/annurev.neuro.31.060407.125620
Alfaidy, N., Chauvet, S., Donadio-Andrei, S., Salomon, A., Saoudi, Y., Richaud, P., et al. (2013). Prion protein expression and functional importance in developmental angiogenesis: role in oxidative stress and copper homeostasis. Antioxid. Redox Signal. 18, 400–411. doi: 10.1089/ars.2012.4637
Alzheimer’s Association. (2015). 2015 Alzheimer’s disease facts and figures. Alzheimers Dement. 11, 332–384. doi: 10.1016/j.jalz.2015.02.003
Americo, T. A., Chiarini, L. B., and Linden, R. (2007). Signaling induced by hop/STI-1 depends on endocytosis. Biochem. Biophys. Res. Commun. 358, 620–625. doi: 10.1016/j.bbrc.2007.04.202
Amieva, H., Jacqmin-Gadda, H., Orgogozo, J. M., Le Carret, N., Helmer, C., Letenneur, L., et al. (2005). The 9 year cognitive decline before dementia of the Alzheimer type: a prospective population-based study. Brain 128, 1093–1101. doi: 10.1093/brain/awh451
Amin, L., Nguyen, X. T., Rolle, I. G., D’Este, E., Giachin, G., Tran, T. H., et al. (2016). Characterization of prion protein function by focal neurite stimulation. J. Cell Sci. 129, 3878–3891. doi: 10.1242/jcs.183137
Andersen, C. B., and Moestrup, S. K. (2014). How calcium makes endocytic receptors attractive. Trends Biochem. Sci. 39, 82–90. doi: 10.1016/j.tibs.2013.12.003
Apostol, M. I., Perry, K., and Surewicz, W. K. (2013). Crystal structure of a human prion protein fragment reveals a motif for oligomer formation. J. Am. Chem. Soc. 135, 10202–10205. doi: 10.1021/ja403001q
Arimatsu, Y., and Seto, A. (1982). Ontogeny of sexual difference in α-bungarotoxin binding capacity in the mouse amygdala. Brain Res. 234, 27–39. doi: 10.1016/0006-8993(82)90470-x
Arimatsu, Y., Seto, A., and Amano, T. (1981). Sexual dimorphism in α-bungarotoxin binding capacity in the mouse amygdala. Brain Res. 213, 432–437. doi: 10.1016/0006-8993(81)90249-3
Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D., Butler, H., Cherry, J. M., et al. (2000). Gene ontology: tool for the unification of biology. The gene ontology consortium. Nat. Genet. 25, 25–29. doi: 10.1038/75556
Bakkebø, M. K., Mouillet-Richard, S., Espenes, A., Goldmann, W., Tatzelt, J., and Tranulis, M. A. (2015). The cellular prion protein: a player in immunological quiescence. Front. Immunol. 6:450. doi: 10.3389/fimmu.2015.00450
Balducci, C., Beeg, M., Stravalaci, M., Bastone, A., Sclip, A., Biasini, E., et al. (2010). Synthetic amyloid-β oligomers impair long-term memory independently of cellular prion protein. Proc. Natl. Acad. Sci. U S A 107, 2295–2300. doi: 10.1073/pnas.0911829107
Barbar, E., and Nyarko, A. (2014). NMR characterization of self-association domains promoted by interactions with LC8 hub protein. Comput. Struct. Biotechnol. J. 9:e201402003. doi: 10.5936/csbj.201402003
Barry, A. E., Klyubin, I., Mc Donald, J. M., Mably, A. J., Farrell, M. A., Scott, M., et al. (2011). Alzheimer’s disease brain-derived amyloid-β-mediated inhibition of LTP in vivo is prevented by immunotargeting cellular prion protein. J. Neurosci. 31, 7259–7263. doi: 10.1523/JNEUROSCI.6500-10.2011
Baskakov, I. V., and Katorcha, E. (2016). Multifaceted role of sialylation in prion diseases. Front. Neurosci. 10:358. doi: 10.3389/fnins.2016.00358
Bate, C., Nolan, W., and Williams, A. (2016). Sialic acid on the glycosylphosphatidylinositol anchor regulates PrP-mediated cell signaling and prion formation. J. Biol. Chem. 291, 160–170. doi: 10.1074/jbc.M115.672394
Bate, C., and Williams, A. (2011a). Amyloid-β-induced synapse damage is mediated via cross-linkage of the cellular prion protein. J. Biol. Chem. 286, 37955–37963. doi: 10.1074/jbc.M111.248724
Bate, C., and Williams, A. (2011b). Monoacylated cellular prion protein modifies cell membranes, inhibits cell signaling, and reduces prion formation. J. Biol. Chem. 286, 8752–8758. doi: 10.1074/jbc.M110.186833
Beckman, D., and Linden, R. (2016). A roadmap for investigating the role of the prion protein in depression associated with neurodegenerative disease. Prion 10, 131–142. doi: 10.1080/19336896.2016.1152437
Béland, M., Motard, J., Barbarin, A., and Roucou, X. (2012). PrPC homodimerization stimulates the production of PrPC cleaved fragments PrPN1 and PrPC1. J. Neurosci. 32, 13255–13263. doi: 10.1523/JNEUROSCI.2236-12.2012
Béland, M., and Roucou, X. (2014). Taking advantage of physiological proteolytic processing of the prion protein for a therapeutic perspective in prion and Alzheimer diseases. Prion 8, 106–110. doi: 10.4161/pri.27438
Beraldo, F. H., Arantes, C. P., Santos, T. G., Machado, C. F., Roffe, M., Hajj, G. N., et al. (2011). Metabotropic glutamate receptors transduce signals for neurite outgrowth after binding of the prion protein to laminin γ1 chain. FASEB J. 25, 265–279. doi: 10.1096/fj.10-161653
Beraldo, F. H., Arantes, C. P., Santos, T. G., Queiroz, N. G., Young, K., Rylett, R. J., et al. (2010). Role of α7 nicotinic acetylcholine receptor in calcium signaling induced by prion protein interaction with stress-inducible protein 1. J. Biol. Chem. 285, 36542–36550. doi: 10.1074/jbc.M110.157263
Beraldo, F. H., Ostapchenko, V. G., Caetano, F. A., Guimaraes, A. L., Ferretti, G. D., Daude, N., et al. (2016). Regulation of Amyloid β oligomer binding to neurons and neurotoxicity by the complex prion protein/mGluR5. J. Biol. Chem. 291, 21945–21955. doi: 10.1074/jbc.M116.738286
Beraldo, F. H., Soares, I. N., Goncalves, D. F., Fan, J., Thomas, A. A., Santos, T. G., et al. (2013). Stress-inducible phosphoprotein 1 has unique cochaperone activity during development and regulates cellular response to ischemia via the prion protein. FASEB J. 27, 3594–3607. doi: 10.1096/fj.13-232280
Bernard, C., Helmer, C., Dilharreguy, B., Amieva, H., Auriacombe, S., Dartigues, J. F., et al. (2014). Time course of brain volume changes in the preclinical phase of Alzheimer’s disease. Alzheimers Dement. 10, 143.e1–151.e1. doi: 10.1016/j.jalz.2013.08.279
Berrone, E., Corona, C., Mazza, M., Vallino Costassa, E., Faro Lo, M., Properzi, F., et al. (2015). Detection of cellular prion protein in exosome derived from ovine plasma. J. Gen. Virol. 96, 3698–3702. doi: 10.1099/jgv.0.000291
Bertrand, D., Lee, L. C.-H., Flood, D., Marger, F., and Donnelly-Roberts, D. (2015). Therapeutic potential of α7 nicotinic acetylcholine receptors. Pharmacol. Rev. 67, 1025–1073. doi: 10.1124/pr.113.008581
Biasini, E., Turnbaugh, J. A., Unterberger, U., and Harris, D. A. (2012). Prion protein at the crossroads of physiology and disease. Trends Neurosci. 35, 92–103. doi: 10.1016/j.tins.2011.10.002
Biljan, I., Ilc, G., Giachin, G., Plavec, J., and Legname, G. (2012). Structural rearrangements at physiological pH: nuclear magnetic resonance insights from the V210I human prion protein mutant. Biochemistry 51, 7465–7474. doi: 10.1021/bi3009856
Bounhar, Y., Zhang, Y., Goodyer, C. G., and LeBlanc, A. (2001). Prion protein protects human neurons against Bax-mediated apoptosis. J. Biol. Chem. 276, 39145–39149. doi: 10.1074/jbc.c100443200
Bray, D. (1998). Signaling complexes: biophysical constraints on intracellular communication. Annu. Rev. Biophys. Biomol. Struct. 27, 59–75. doi: 10.1146/annurev.biophys.27.1.59
Bray, D., and Lay, S. (1997). Computer-based analysis of the binding steps in protein complex formation. Proc. Natl. Acad. Sci. U S A 94, 13493–13498. doi: 10.1073/pnas.94.25.13493
Brennan, D. F., Dar, A. C., Hertz, N. T., Chao, W. C., Burlingame, A. L., Shokat, K. M., et al. (2011). A Raf-induced allosteric transition of KSR stimulates phosphorylation of MEK. Nature 472, 366–369. doi: 10.1038/nature09860
Bucciantini, M., Giannoni, E., Chiti, F., Baroni, F., Formigli, L., Zurdo, J., et al. (2002). Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 416, 507–511. doi: 10.1038/416507a
Büeler, H., Aguzzi, A., Sailer, A., Greiner, R.-A., Autenried, P., Aguet, M., et al. (1993). Mice devoid of PrP are resistant to scrapie. Cell 73, 1339–1347. doi: 10.1016/0092-8674(93)90360-3
Büeler, H., Fischer, M., Lang, Y., Bluethmann, H., Lipp, H. P., DeArmond, S. J., et al. (1992). Normal development and behaviour of mice lacking the neuronal cell-surface PrP protein. Nature 356, 577–582. doi: 10.1038/356577a0
Caetano, F. A., Beraldo, F. H., Hajj, G. N. M., Guimaraes, A. L., Jürgensen, S., Wasilewska-Sampaio, A. P., et al. (2011). Amyloid-β oligomers increase the localization of prion protein at the cell surface. J. Neurochem. 117, 538–553. doi: 10.1111/j.1471-4159.2011.07225.x
Caetano, F. A., Lopes, M. H., Hajj, G. N., Machado, C. F., Pinto Arantes, C., Magalhães, A. C., et al. (2008). Endocytosis of prion protein is required for ERK1/2 signaling induced by stress-inducible protein 1. J. Neurosci. 28, 6691–6702. doi: 10.1523/JNEUROSCI.1701-08.2008
Calella, A. M., Farinelli, M., Nuvolone, M., Mirante, O., Moos, R., Falsig, J., et al. (2010). Prion protein and Aβ-related synaptic toxicity impairment. EMBO Mol. Med. 2, 306–314. doi: 10.1002/emmm.201000082
Cancellotti, E., Bradford, B. M., Tuzi, N. L., Hickey, R. D., Brown, D., Brown, K. L., et al. (2010). Glycosylation of PrPC determines timing of neuroinvasion and targeting in the brain following transmissible spongiform encephalopathy infection by a peripheral route. J. Virol. 84, 3464–3475. doi: 10.1128/JVI.02374-09
Carneiro, M. V., Americo, T. A., Guimarães, M. Z., and Linden, R. (2016). The prion protein selectively binds to and modulates the content of purinergic receptor P2X4R. Biochem. Biophys. Res. Commun. 472, 293–298. doi: 10.1016/j.bbrc.2016.02.122
Caughey, B., and Baron, G. S. (2006). Prions and their partners in crime. Nature 443, 803–810. doi: 10.1038/nature05294
Chen, Y., Fu, A. K., and Ip, N. Y. (2013). Axin: an emerging key scaffold at the synapse. IUBMB Life 65, 685–691. doi: 10.1002/iub.1184
Chen, L., Kurokawa, J., and Kass, R. S. (2005). Phosphorylation of the A-kinase-anchoring protein Yotiao contributes to protein kinase A regulation of a heart potassium channel. J. Biol. Chem. 280, 31347–31352. doi: 10.1074/jbc.m505191200
Chen, C., Lewis, R. E., and White, M. A. (2008). IMP modulates KSR1-dependent multivalent complex formation to specify ERK1/2 pathway activation and response thresholds. J. Biol. Chem. 283, 12789–12796. doi: 10.1074/jbc.M709305200
Chen, S., Mangé, A., Dong, L., Lehmann, S., and Schachner, M. (2003). Prion protein as trans-interacting partner for neurons is involved in neurite outgrowth and neuronal survival. Mol. Cell. Neurosci. 22, 227–233. doi: 10.1016/s1044-7431(02)00014-3
Chen, L., Marquardt, M. L., Tester, D. J., Sampson, K. J., Ackerman, M. J., and Kass, R. S. (2007). Mutation of an A-kinase-anchoring protein causes long-QT syndrome. Proc. Natl. Acad. Sci. U S A 104, 20990–20995. doi: 10.1073/pnas.0710527105
Chen, S., Yadav, S. P., and Surewicz, W. K. (2010). Interaction between human prion protein and amyloid-β (Aβ) oligomers: role OF N-terminal residues. J. Biol. Chem. 285, 26377–26383. doi: 10.1074/jbc.m110.145516
Cheng, D., Hoogenraad, C. C., Rush, J., Ramm, E., Schlager, M. A., Duong, D. M., et al. (2006). Relative and absolute quantification of postsynaptic density proteome isolated from rat forebrain and cerebellum. Mol. Cell. Proteomics 5, 1158–1170. doi: 10.1074/mcp.d500009-mcp200
Chiang, Y. W., Otoshima, Y., Watanabe, Y., Inanami, O., and Shimoyama, Y. (2008). Dynamics and local ordering of spin-labeled prion protein: an ESR simulation study of a highly PH-sensitive site. J. Biomol. Struct. Dyn. 26, 355–366. doi: 10.1080/07391102.2008.10507250
Chiarini, L. B., Freitas, A. R., Zanata, S. M., Brentani, R. R., Martins, V. R., and Linden, R. (2002). Cellular prion protein transduces neuroprotective signals. EMBO J. 21, 3317–3326. doi: 10.1093/emboj/cdf324
Christen, B., Hornemann, S., Damberger, F. F., and Wüthrich, K. (2009). Prion protein NMR structure from tammar wallaby (Macropus eugenii) shows that the β2-α2 loop is modulated by long-range sequence effects. J. Mol. Biol. 389, 833–845. doi: 10.1016/j.jmb.2009.04.040
Cissé, M., Sanchez, P. E., Kim, D. H., Ho, K., Yu, G.-Q., and Mucke, L. (2011). Ablation of cellular prion protein does not ameliorate abnormal neural network activity or cognitive dysfunction in the J20 line of human amyloid precursor protein transgenic mice. J. Neurosci. 31, 10427–10431. doi: 10.1523/JNEUROSCI.1459-11.2011
Colby, D. W., and Prusiner, S. B. (2011). Prions. Cold Spring Harb. Perspect. Biol. 3:a006833. doi: 10.1101/cshperspect.a006833
Constantin, B. (2014). Dystrophin complex functions as a scaffold for signalling proteins. Biochim. Biophys. Acta 1838, 635–642. doi: 10.1016/j.bbamem.2013.08.023
Cordeiro, Y., and Silva, J. L. (2005). The hypothesis of the catalytic action of nucleic acid on the conversion of prion protein. Protein Pept. Lett. 12, 251–255. doi: 10.2174/0929866053587138
Corrigall, V. M., Bodman-Smith, M. D., Brunst, M., Cornell, H., and Panayi, G. S. (2004). Inhibition of antigen-presenting cell function and stimulation of human peripheral blood mononuclear cells to express an antiinflammatory cytokine profile by the stress protein BiP: relevance to the treatment of inflammatory arthritis. Arthritis Rheum. 50, 1164–1171. doi: 10.1002/art.20134
Corsaro, A., Thellung, S., Villa, V., Nizzari, M., and Florio, T. (2012). Role of prion protein aggregation in neurotoxicity. Int. J. Mol. Sci. 13, 8648–8669. doi: 10.3390/ijms13078648
Coulpier, M., Messiaen, S., Boucreaux, D., and Eloit, M. (2006). Axotomy-induced motoneuron death is delayed in mice overexpressing PrPc. Neuroscience 141, 1827–1834. doi: 10.1016/j.neuroscience.2006.05.037
Dametto, P., Lakkaraju, A. K., Bridel, C., Villiger, L., O’Connor, T., Herrmann, U. S., et al. (2015). Neurodegeneration and unfolded-protein response in mice expressing a membrane-tethered flexible tail of PrP. PLoS One 10:e0117412. doi: 10.1371/journal.pone.0117412
Daoud, F., Angeard, N., Demerre, B., Martie, I., Benyaou, R., Leturcq, F., et al. (2009). Analysis of Dp71 contribution in the severity of mental retardation through comparison of Duchenne and Becker patients differing by mutation consequences on Dp71 expression. Hum. Mol. Genet. 18, 3779–3794. doi: 10.1093/hmg/ddp320
Dard, N., and Peter, M. (2006). Scaffold proteins in MAP kinase signaling: more than simple passive activating platforms. Bioessays 28, 146–156. doi: 10.1002/bies.20351
Delpino, A., and Castelli, M. (2002). The 78 kDa glucose-regulated protein (GRP78/BIP) is expressed on the cell membrane, is released into cell culture medium and is also present in human peripheral circulation. Biosci. Rep. 22, 407–420. doi: 10.1023/A:1020966008615
Desguerre, I., Christov, C., Mayer, M., Zeller, R., Becane, H. M., Bastuji-Garin, S., et al. (2009). Clinical heterogeneity of duchenne muscular dystrophy (DMD): definition of sub-phenotypes and predictive criteria by long-term follow-up. PLoS One 4:e4347. doi: 10.1371/journal.pone.0004347
Dhanasekaran, D. N., Kashef, K., Lee, C. M., Xu, H., and Reddy, E. P. (2007). Scaffold proteins of MAP-kinase modules. Oncogene 26, 3185–3202. doi: 10.1038/sj.onc.1210411
Diaz-Herrera, P. (2006). “What is a biological function?” in Formal Ontology in Information Systems, eds B. Bennett and C. Fellbaum (Netherlands: IOS Press), 128–140.
Diviani, D., Reggi, E., Arambasic, M., Caso, S., and Maric, D. (2016). Emerging roles of A-kinase anchoring proteins in cardiovascular pathophysiology. Biochim. Biophys. Acta 1863, 1926–1936. doi: 10.1016/j.bbamcr.2015.11.024
Edelman, G. M. (1986). Cell adhesion molecules in neural histogenesis. Annu. Rev. Physiol. 48, 417–430. doi: 10.1146/annurev.physiol.48.1.417
Eichner, T., and Radford, S. E. (2011). A diversity of assembly mechanisms of a generic amyloid fold. Mol. Cell 43, 8–18. doi: 10.1016/j.molcel.2011.05.012
Erlich, R. B., Kahn, S. A., Lima, F. R., Muras, A. G., Martins, R. A., Linden, R., et al. (2007). STI1 promotes glioma proliferation through MAPK and PI3K pathways. Glia 55, 1690–1698. doi: 10.1002/glia.20579
Eungdamrong, N. J., and Iyengar, R. (2004). Computational approaches for modeling regulatory cellular networks. Trends Cell Biol. 14, 661–669. doi: 10.1016/j.tcb.2004.10.007
Fairbanks, S. L., Young, J. M., Nelson, J. W., Davis, C. M., Koerner, I. P., and Alkayed, N. J. (2012). Mechanism of the sex difference in neuronal ischemic cell death. Neuroscience 219, 183–191. doi: 10.1016/j.neuroscience.2012.05.048
Faux, M. C., and Scott, J. D. (1996). Molecular glue: kinase anchoring and scaffold proteins. Cell 85, 9–12. doi: 10.1016/s0092-8674(00)81075-2
Ferraguti, F., Crepaldi, L., and Nicoletti, F. (2008). Metabotropic glutamate 1 receptor: current concepts and perspectives. Pharmacol. Rev. 60, 536–581. doi: 10.1124/pr.108.000166
Ferreira, S. T., and Klein, W. L. (2011). The Aβ oligomer hypothesis for synapse failure and memory loss in Alzheimer’s disease. Neurobiol. Learn. Mem. 96, 529–543. doi: 10.1016/j.nlm.2011.08.003
Ferreira, S. T., Lourenco, M. V., Oliveira, M. M., and De Felice, F. G. (2015). Soluble amyloid-β oligomers as synaptotoxins leading to cognitive impairment in Alzheimer’s disease. Front. Cell. Neurosci. 9:191. doi: 10.3389/fncel.2015.00191
Forloni, G., and Balducci, C. (2011). β-amyloid oligomers and prion protein: fatal attraction? Prion 5, 10–15. doi: 10.4161/pri.5.1.14367
Fu, Q., Chen, X., and Xiang, Y. K. (2013). Compartmentalization of β-adrenergic signals in cardiomyocytes. Trends Cardiovasc. Med. 23, 250–256. doi: 10.1016/j.tcm.2013.02.001
Ganzinger, K. A., Narayan, P., Qamar, S. S., Weimann, L., Ranasinghe, R. T., Aguzzi, A., et al. (2014). Single-molecule imaging reveals that small amyloid-β1–42 oligomers interact with the cellular prion protein (PrPC). Chembiochem 15, 2515–2521. doi: 10.1002/cbic.201402377
Gauczynski, S., Peyrin, J. M., Haïk, S., Leucht, C., Hundt, C., Rieger, R., et al. (2001). The 37-kDa/67-kDa laminin receptor acts as the cell-surface receptor for the cellular prion protein. EMBO J. 20, 5863–5875. doi: 10.1093/emboj/20.21.5863
Gauthier, S., Albert, M., Fox, N., Goedert, M., Kivipelto, M., Mestre-Ferrandiz, J., et al. (2016). Why has therapy development for dementia failed in the last two decades? Alzheimers Dement. 12, 60–64. doi: 10.1016/j.jalz.2015.12.003
Gerasimenko, J. V., Tepikin, A. V., Petersen, O. H., and Gerasimenko, O. V. (1998). Calcium uptake via endocytosis with rapid release from acidifying endosomes. Curr. Biol. 8, 1335–1338. doi: 10.1016/s0960-9822(07)00565-9
Gerber, R., Voitchovsky, K., Mitchel, C., Tahiri-Alaoui, A., Ryan, J. F., Hore, P. J., et al. (2008). Inter-oligomer interactions of the human prion protein are modulated by the polymorphism at codon 129. J. Mol. Biol. 381, 212–220. doi: 10.1016/j.jmb.2008.05.057
Goh, A. X., Li, C., Sy, M. S., and Wong, B. S. (2007). Altered prion protein glycosylation in the aging mouse brain. J. Neurochem. 100, 841–854. doi: 10.1111/j.1471-4159.2006.04268.x
Gold, M. G., Stengel, F., Nygren, P. J., Weisbrod, C. R., Bruce, J. E., Robinson, C. V., et al. (2011). Architecture and dynamics of an A-kinase anchoring protein 79 (AKAP79) signaling complex. Proc. Natl. Acad. Sci. U S A 108, 6426–6431. doi: 10.1073/pnas.1014400108
Good, M. C., Zalatan, J. G., and Lim, W. A. (2011). Scaffold proteins: hubs for controlling the flow of cellular information. Science 332, 680–686. doi: 10.1126/science.1198701
Graner, E., Mercadante, A. F., Zanata, S. M., Forlenza, O. V., Cabral, A. L., Veiga, S. S., et al. (2000). Cellular prion protein binds laminin and mediates neuritogenesis. Brain Res. Mol. Brain Res. 76, 85–92. doi: 10.1016/s0169-328x(99)00334-4
Griffiths, P. E. (1993). Functional analysis and proper function. Brit. J. Phil. Sci. 44, 409–422. doi: 10.1093/bjps/44.3.409
Gu, W., Wang, T., Zhu, J., Shi, Y., and Liu, H. (2003). Molecular dynamics simulation of the unfolding of the human prion protein domain under low pH and high temperature conditions. Biophys. Chem. 104, 79–94. doi: 10.1016/s0301-4622(02)00340-x
Guerrero-Muñoz, M. J., Castillo-Carranza, D. L., and Kayed, R. (2014). Therapeutic approaches against common structural features of toxic oligomers shared by multiple amyloidogenic proteins. Biochem. Pharmacol. 88, 468–478. doi: 10.1016/j.bcp.2013.12.023
Guo, B. B., Bellingham, S. A., and Hill, A. F. (2015). The neutral sphingomyelinase pathway regulates packaging of the prion protein into exosomes. J. Biol. Chem. 290, 3455–3467. doi: 10.1074/jbc.M114.605253
Haas, L. T., Kostylev, M. A., and Strittmatter, S. M. (2014). Therapeutic molecules and endogenous ligands regulate the interaction between brain cellular prion protein (PrPC) and metabotropic glutamate receptor 5 (mGluR5). J. Biol. Chem. 289, 28460–28477. doi: 10.1074/jbc.M114.584342
Hafner-Bratkovič, I., Bester, R., Pristovsek, P., Gaedtke, L., Veranic, P., Gaspersic, J., et al. (2011). Globular domain of the prion protein needs to be unlocked by domain swapping to support prion protein conversion. J. Biol. Chem. 286, 12149–12156. doi: 10.1074/jbc.M110.213926
Hafner-Bratkovič, I., and Jerala, R. (2011). Disulfide mapping reveals the domain swapping as the crucial process of the structural conversion of prion protein. Prion 5, 56–59. doi: 10.4161/pri.5.2.16232
Hajj, G. N., Arantes, C. P., Dias, M. V., Roffé, M., Costa-Silva, B., Lopes, M. H., et al. (2013). The unconventional secretion of stress-inducible protein 1 by a heterogeneous population of extracellular vesicles. Cell. Mol. Life Sci. 70, 3211–3227. doi: 10.1007/s00018-013-1328-y
Hall, H., Carbonetto, S., and Schachner, M. (1997). L1/HNK-1 carbohydrate- and β 1 integrin-dependent neural cell adhesion to laminin-1. J. Neurochem. 68, 544–553. doi: 10.1046/j.1471-4159.1997.68020544.x
Hamilton, A., Vasefi, M., Tuin Vander, C., McQuaid, R. J., Anisman, H., and Ferguson, S. S. (2016). Chronic pharmacological mGluR5 inhibition prevents cognitive impairment and reduces pathogenesis in an Alzheimer disease mouse model. Cell Rep. 15, 1859–1865. doi: 10.1016/j.celrep.2016.04.077
Han, B., Poppinga, W. J., and Schmidt, M. (2015). Scaffolding during the cell cycle by A-kinase anchoring proteins. Pflugers Arch. 467, 2401–2411. doi: 10.1007/s00424-015-1718-0
Harris, D. A. (2003). Trafficking, turnover and membrane topology of PrP. Br. Med. Bull. 66, 71–85. doi: 10.1093/bmb/66.1.71
Harris, K. M., and Weinberg, R. J. (2012). Ultrastructure of synapses in the mammalian brain. Cold Spring Harb. Perspect. Biol. 4:a005587. doi: 10.1101/cshperspect.a005587
Heinrich, R., Neel, B. G., and Rapoport, T. A. (2002). Mathematical models of protein kinase signal transduction. Mol. Cell 9, 957–970. doi: 10.1016/s1097-2765(02)00528-2
Hejmadi, M. V., Dajas-Bailador, F., Barns, S. M., Jones, B., and Wonnacott, S. (2003). Neuroprotection by nicotine against hypoxia-induced apoptosis in cortical cultures involves activation of multiple nicotinic acetylcholine receptor subtypes. Mol. Cell. Neurosci. 24, 779–786. doi: 10.1016/s1044-7431(03)00244-6
Herson, P. S., and Hurn, P. D. (2010). Gender and the injured brain. Prog. Brain Res. 186, 177–187. doi: 10.1016/B978-0-444-53630-3.00012-9
Herson, P. S., Palmateer, J., and Hurn, P. D. (2013). Biological sex and mechanisms of ischemic brain injury. Transl. Stroke Res. 4, 413–419. doi: 10.1007/s12975-012-0238-x
Hu, N. W., Nicoll, A. J., Zhang, D., Mably, A. J., O’Malley, T., Purro, S. A., et al. (2014). mGlu5 receptors and cellular prion protein mediate amyloid-β-facilitated synaptic long-term depression in vivo. Nat. Commun. 5:3374. doi: 10.1038/ncomms4374
Hu, J., Yu, H., Kornev, A. P., Zhao, J., Filbert, E. L., Taylor, S. S., et al. (2011). Mutation that blocks ATP binding creates a pseudokinase stabilizing the scaffolding function of kinase suppressor of Ras, CRAF and BRAF. Proc. Natl. Acad. Sci. U S A 108, 6067–6072. doi: 10.1073/pnas.1102554108
Huang, P., Lian, F., Wen, Y., Guo, C., and Lin, D. (2013). Prion protein oligomer and its neurotoxicity. Acta Biochim. Biophys. Sin. 45, 442–451. doi: 10.1093/abbs/gmt037
Hugel, B., Martínez, M. C., Kunzelmann, C., Blättler, T., Aguzzi, A., and Freyssinet, J. M. (2004). Modulation of signal transduction through the cellular prion protein is linked to its incorporation in lipid rafts. Cell. Mol. Life Sci. 61, 2998–3007. doi: 10.1007/s00018-004-4318-2
Hundt, C., Peyrin, J. M., Haïk, S., Gauczynski, S., Leucht, C., Rieger, R., et al. (2001). Identification of interaction domains of the prion protein with its 37-kDa/67-kDa laminin receptor. EMBO J. 20, 5876–5886. doi: 10.1093/emboj/20.21.5876
Jeon, J. W., Park, B. C., Jung, J. G., Jang, Y. S., Shin, E. C., and Park, Y. W. (2013). The soluble form of the cellular prion protein enhances phagocytic activity and cytokine production by human monocytes via activation of ERK and NF-κB. Immune Netw. 13, 148–156. doi: 10.4110/in.2013.13.4.148
Jouvin-Marche, E., Attuil-Audenis, V., Aude-Garcia, C., Rachidi, W., Zabel, M., Podevin-Dimster, V., et al. (2006). Overexpression of cellular prion protein induces an antioxidant environment altering T cell development in the thymus. J. Immunol. 176, 3490–3497. doi: 10.4049/jimmunol.176.6.3490
Kaimann, T., Metzger, S., Kuhlmann, K., Brandt, B., Birkmann, E., Höltje, H. D., et al. (2008). Molecular model of an α-helical prion protein dimer and its monomeric subunits as derived from chemical cross-linking and molecular modeling calculations. J. Mol. Biol. 376, 582–596. doi: 10.1016/j.jmb.2007.11.035
Kam, T. I., Gwon, Y., and Jung, Y. K. (2014). Amyloid β receptors responsible for neurotoxicity and cellular defects in Alzheimer’s disease. Cell. Mol. Life Sci. 71, 4803–4813. doi: 10.1007/s00018-014-1706-0
Kanaani, J., Prusiner, S. B., Diacovo, J., Baekkeskov, S., and Legname, G. (2005). Recombinant prion protein induces rapid polarization and development of synapses in embryonic rat hippocampal neurons in vitro. J. Neurochem. 95, 1373–1386. doi: 10.1111/j.1471-4159.2005.03469.x
Kholodenko, B. N., Hancock, J. F., and Kolch, W. (2010). Signalling ballet in space and time. Nat. Rev. Mol. Cell Biol. 11, 414–426. doi: 10.1038/nrm2901
Knaus, K. J., Morillas, M., Swietnicki, W., Malone, M., Surewicz, W. K., and Yee, V. C. (2001). Crystal structure of the human prion protein reveals a mechanism for oligomerization. Nat. Struct. Biol. 8, 770–774. doi: 10.1038/nsb0901-770
Kolch, W. (2005). Coordinating ERK/MAPK signalling through scaffolds and inhibitors. Nat. Rev. Mol. Cell Biol. 6, 827–837. doi: 10.1038/nrm1743
Kostylev, M. A., Kaufman, A. C., Nygaard, H. B., Patel, P., Haas, L. T., Gunther, E. C., et al. (2015). Prion-protein-interacting amyloid-β oligomers of high molecular weight are tightly correlated with memory impairment in multiple Alzheimer mouse models. J. Biol. Chem. 290, 17415–17438. doi: 10.1074/jbc.m115.643577
Kovač, V., Hafner-Bratkovič, I., and Čurin Šerbec, V. (2016). Anchorless forms of prion protein - Impact of truncation on structure destabilization and prion protein conversion. Biochem. Biophys. Res. Commun. 481, 1–6. doi: 10.1016/j.bbrc.2016.11.036
Koveal, D., Schuh-Nuhfer, N., Ritt, D., Page, R., Morrison, D. K., and Peti, W. (2012). A CC-SAM, for coiled coil-sterile α motif, domain targets the scaffold KSR-1 to specific sites in the plasma membrane. Sci. Signal. 5:ra94. doi: 10.1126/scisignal.2003289
Krebs, B., Dorner-Ciossek, C., Schmalzbauer, R., Vassallo, N., Herms, J., and Kretzschmar, H. A. (2006). Prion protein induced signaling cascades in monocytes. Biochem. Biophys. Res. Commun. 340, 13–22. doi: 10.1016/j.bbrc.2005.11.158
Kuczius, T., Grassi, J., Karch, H., and Groschup, M. H. (2007a). Binding of N- and C-terminal anti-prion protein antibodies generates distinct phenotypes of cellular prion proteins (PrPC) obtained from human, sheep, cattle and mouse. FEBS J. 274, 1492–1502. doi: 10.1111/j.1742-4658.2007.05691.x
Kuczius, T., Koch, R., Keyvani, K., Karch, H., Grassi, J., and Groschup, M. H. (2007b). Regional and phenotype heterogeneity of cellular prion proteins in the human brain. Eur. J. Neurosci. 25, 2649–2655. doi: 10.1111/j.1460-9568.2007.05518.x
Küffer, A., Lakkaraju, A. K., Mogha, A., Petersen, S. C., Airich, K., Doucerain, C., et al. (2016). The prion protein is an agonistic ligand of the G protein-coupled receptor Adgrg6. Nature 536, 464–468. doi: 10.1038/nature19312
Lambert, M. P., Barlow, A. K., Chromy, B. A., Edwards, C., Freed, R., Liosatos, M., et al. (1998). Diffusible, nonfibrillar ligands derived from Aβ1–42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. U S A 95, 6448–6453. doi: 10.1073/pnas.95.11.6448
Langeberg, L. K., and Scott, J. D. (2015). Signalling scaffolds and local organization of cellular behaviour. Nat. Rev. Mol. Cell Biol. 16, 232–244. doi: 10.1038/nrm3966
Larson, M., Sherman, M. A., Amar, F., Nuvolone, M., Schneider, J. A., Bennett, D. A., et al. (2012). The complex PrPc-fyn couples human oligomeric Aβ with pathological tau changes in Alzheimer’s disease. J. Neurosci. 32, 16857–16871. doi: 10.1523/jneurosci.1858-12.2012
Laurén, J. (2014). Cellular prion protein as a therapeutic target in Alzheimer’s disease. J. Alzheimers Dis. 38, 227–244. doi: 10.3233/JAD-130950
Laurén, J., Gimbel, D. A., Nygaard, H. B., Gilbert, J. W., and Strittmatter, S. M. (2009). Cellular prion protein mediates impairment of synaptic plasticity by amyloid-β oligomers. Nature 457, 1128–1132. doi: 10.1038/nature07761
Lawson, V. A., Collins, S. J., Masters, C. L., and Hill, A. F. (2005). Prion protein glycosylation. J. Neurochem. 93, 793–801. doi: 10.1111/j.1471-4159.2005.03104.x
Lechtenberg, B. C., Mace, P. D., and Riedl, S. J. (2014). Structural mechanisms in NLR inflammasome signaling. Curr. Opin. Struct. Biol. 29, 17–25. doi: 10.1016/j.sbi.2014.08.011
Lee, S., Antony, L., Hartmann, R., Knaus, K. J., Surewicz, K., Surewicz, W. K., et al. (2010). Conformational diversity in prion protein variants influences intermolecular β-sheet formation. EMBO J. 29, 251–262. doi: 10.1038/emboj.2009.333
Lee, Y. J., and Baskakov, I. V. (2013). The cellular form of the prion protein is involved in controlling cell cycle dynamics, self-renewal and the fate of human embryonic stem cell differentiation. J. Neurochem. 124, 310–322. doi: 10.1111/j.1471-4159.2012.07913.x
Leighton, P. L., and Allison, W. T. (2016). Protein misfolding in prion and prion-like diseases: reconsidering a required role for protein loss-of-function. J. Alzheimers Dis. 54, 3–29. doi: 10.3233/JAD-160361
Levchenko, A., Bruck, J., and Sternberg, P. W. (2000). Scaffold proteins may biphasically affect the levels of mitogen-activated protein kinase signaling and reduce its threshold properties. Proc. Natl. Acad. Sci. U S A 97, 5818–5823. doi: 10.1073/pnas.97.11.5818
Li, R., Liu, D., Zanusso, G., Liu, T., Fayen, J. D., Huang, J. H., et al. (2001). The expression and potential function of cellular prion protein in human lymphocytes. Cell. Immunol. 207, 49–58. doi: 10.1006/cimm.2000.1751
Li, Q. Q., Sun, Y. P., Ruan, C. P., Xu, X. Y., Ge, J. H., He, J., et al. (2011). Cellular prion protein promotes glucose uptake through the Fyn-HIF-2α-Glut1 pathway to support colorectal cancer cell survival. Cancer Sci. 102, 400–406. doi: 10.1111/j.1349-7006.2010.01811.x
Liang, J., Pan, Y. L., Ning, X. X., Sun, L. J., Lan, M., Hong, L., et al. (2006). Overexpression of PrPC and its antiapoptosis function in gastric cancer. Tumour Biol. 27, 84–91. doi: 10.1159/000092488
Linden, R., Cordeiro, Y., and Lima, L. M. (2012). Allosteric function and dysfunction of the prion protein. Cell. Mol. Life Sci. 69, 1105–1124. doi: 10.1007/s00018-011-0847-7
Linden, R., Martins, V. R., and Prado, M. A. (2017). “Prion protein,” in Encyclopedia of Signaling Molecules, 2nd Edn. ed. S. Choi (New York, NY: Springer Science), in press.
Linden, R., Martins, V. R., Prado, M. A., Cammarota, M., Izquierdo, I., and Brentani, R. R. (2008). Physiology of the prion protein. Physiol. Rev. 88, 673–728. doi: 10.1152/physrev.00007.2007
Liu, M., Dziennis, S., Hurn, P. D., and Alkayed, N. J. (2009). Mechanisms of gender-linked ischemic brain injury. Restor. Neurol. Neurosci. 27, 163–179. doi: 10.3233/RNN-2009-0467
Liu, J., Kurella, V. B., LeCour, L., Vanagunas, T., and Worthylake, D. K. (2016). The IQGAP1 N-terminus forms dimers and the dimer interface is required for binding F-actin and calcium-bound calmodulin. Biochemistry 55, 6433–6444. doi: 10.1021/acs.biochem.6b00745
Lobão-Soares, B., Walz, R., Prediger, R. D., Freitas, R. L., Calvo, F., Bianchin, M. M., et al. (2008). Cellular prion protein modulates defensive attention and innate fear-induced behaviour evoked in transgenic mice submitted to an agonistic encounter with the tropical coral snake Oxyrhopus guibei. Behav. Brain Res. 194, 129–137. doi: 10.1016/j.bbr.2008.06.006
Locasale, J. W., Shaw, A. S., and Chakraborty, A. K. (2007). Scaffold proteins confer diverse regulatory properties to protein kinase cascades. Proc. Natl. Acad. Sci. U S A 104, 13307–13312. doi: 10.1073/pnas.0706311104
Lopes, M. H., Hajj, G. N. M., Muras, A. G., Mancini, G. L., Castro, R. M. P. S., Ribeiro, K. C. B., et al. (2005). Interaction of cellular prion and stress-inducible protein 1 promotes neuritogenesis and neuroprotection by distinct signaling pathways. J. Neurosci. 25, 11330–11339. doi: 10.1523/JNEUROSCI.2313-05.2005
Lowenthal, M. S., Markey, S. P., and Dosemeci, A. (2015). Quantitative mass spectrometry measurements reveal stoichiometry of principal postsynaptic density proteins. J. Proteome Res. 14, 2528–2538. doi: 10.1021/acs.jproteome.5b00109
Luo, W., and Lin, S. C. (2004). Axin: a master scaffold for multiple signaling pathways. Neurosignals 13, 99–113. doi: 10.1159/000076563
Magdesian, M. H., Carvalho, M. M., Mendes, F. A., Saraiva, L. M., Juliano, M. A., Juliano, L., et al. (2008). Amyloid-β binds to the extracellular cysteine-rich domain of Frizzled and inhibits Wnt/β-catenin signaling. J. Biol. Chem. 283, 9359–9368. doi: 10.1074/jbc.M707108200
Mahanty, S. K., Wang, Y., Farley, F. W., and Elion, E. A. (1999). Nuclear shuttling of yeast scaffold Ste5 is required for its recruitment to the plasma membrane and activation of the mating MAPK cascade. Cell 98, 501–512. doi: 10.1016/s0092-8674(00)81978-9
Málaga-Trillo, E., and Sempou, E. (2009). PrPs: proteins with a purpose: lessons from the zebrafish. Prion 3, 129–133. doi: 10.4161/pri.3.3.9651
Málaga-Trillo, E., Solis, G. P., Schrock, Y., Geiss, C., Luncz, L., Thomanetz, V., et al. (2009). Regulation of embryonic cell adhesion by the prion protein. PLoS Biol. 7:e55. doi: 10.1371/journal.pbio.1000055
Manuelidis, L. (2013). Infectious particles, stress, and induced prion amyloids: a unifying perspective. Virulence 4, 373–383. doi: 10.4161/viru.24838
Manwani, B., and McCullough, L. D. (2011). Sexual dimorphism in ischemic stroke: lessons from the laboratory. Womens Health 7, 319–339. doi: 10.2217/whe.11.22
Mariante, R. M., Nóbrega, A., Martins, R. A., Areal, R. B., Bellio, M., and Linden, R. (2012). Neuroimmunoendocrine regulation of the prion protein in neutrophils. J. Biol. Chem. 287, 35506–35515. doi: 10.1074/jbc.M112.394924
Marín-Briggiler, C. I., González-Echeverría, M. F., Munuce, M. J., Ghersevich, S., Caille, A. M., Hellman, U., et al. (2010). Glucose-regulated protein 78 (Grp78/BiP) is secreted by human oviduct epithelial cells and the recombinant protein modulates sperm-zona pellucida binding. Fertil. Steril. 93, 1574–1584. doi: 10.1016/j.fertnstert.2008.12.132
Martin-Lannerée, S., Hirsch, T. Z., Hernandez-Rapp, J., Halliez, S., Vilotte, J. L., Launay, J. M., et al. (2014). PrPC from stem cells to cancer. Front. Cell Dev. Biol. 2:55. doi: 10.3389/fcell.2014.00055
Martins, V. R., Beraldo, F. H., Hajj, G. N., Lopes, M. H., Lee, K. S., Prado, M. A., et al. (2010). Prion protein: orchestrating neurotrophic activities. Curr. Issues Mol. Biol. 12, 63–86. doi: 10.21775/cimb.012.063
Martins, V. R., Linden, R., Prado, M. A., Walz, R., Sakamoto, A. C., Izquierdo, I., et al. (2002). Cellular prion protein: on the road for functions. FEBS Lett. 512, 25–28. doi: 10.1016/s0014-5793(02)02291-3
Masel, J., Genoud, N., and Aguzzi, A. (2005). Efficient inhibition of prion replication by PrP-Fc2 suggests that the prion is a PrPSc oligomer. J. Mol. Biol. 345, 1243–1251. doi: 10.1016/j.jmb.2004.10.088
Mays, C. E., Kim, C., Haldiman, T., van der Merwe, J., Lau, A., Yang, J., et al. (2014). Prion disease tempo determined by host-dependent substrate reduction. J. Clin. Invest. 124, 847–858. doi: 10.1172/JCI72241
McLennan, N. F., Brennan, P. M., McNeill, A., Davies, I., Fotheringham, A., Rennison, K. A., et al. (2004). Prion protein accumulation and neuroprotection in hypoxic brain damage. Am. J. Pathol. 165, 227–235. doi: 10.1016/S0002-9440(10)63291-9
Mehrpour, M., and Codogno, P. (2010). Prion protein: from physiology to cancer biology. Cancer Lett. 290, 1–23. doi: 10.1016/j.canlet.2009.07.009
Meyer, R. K., Lustig, A., Oesch, B., Fatzer, R., Zurbriggen, A., and Vandevelde, M. (2000). A monomer-dimer equilibrium of a cellular prion protein (PrPC) not observed with recombinant PrP. J. Biol. Chem. 275, 38081–38087. doi: 10.1074/jbc.M007114200
Mitsios, N., Saka, M., Krupinski, J., Pennucci, R., Sanfeliu, C., Turu Miguel, M., et al. (2007). Cellular prion protein is increased in the plasma and peri-infarcted brain tissue after acute stroke. J. Neurosci. Res. 85, 602–611. doi: 10.1002/jnr.21142
Molza, A. E., Mangat, K., Le Rumeur, E., Hubert, J. F., Menhart, N., and Delalande, O. (2015). Structural basis of neuronal nitric-oxide synthase interaction with dystrophin repeats 16 and 17. J. Biol. Chem. 290, 29531–29541. doi: 10.1074/jbc.M115.680660
Morris, R. J., Parkyn, C. J., and Jen, A. (2006). Traffic of prion protein between different compartments on the neuronal surface, and the propagation of prion disease. FEBS Lett. 580, 5565–5571. doi: 10.1016/j.febslet.2006.07.053
Morris, A. M., Watzky, M. A., and Finke, R. G. (2009). Protein aggregation kinetics, mechanism, and curve-fitting: a review of the literature. Biochim. Biophys. Acta 1794, 375–397. doi: 10.1016/j.bbapap.2008.10.016
Morrison, D. K., and Davis, R. J. (2003). Regulation of MAP kinase signaling modules by scaffold proteins in mammals. Annu. Rev. Cell Dev. Biol. 19, 91–118. doi: 10.1146/annurev.cellbio.19.111401.091942
Moudjou, M., Bernard, J., Sabuncu, E., Langevin, C., and Laude, H. (2007). Glycan chains modulate prion protein binding to immobilized metal ions. Neurochem. Int. 50, 689–695. doi: 10.1016/j.neuint.2007.01.001
Mouillet-Richard, S., Ermonval, M., Chebassier, C., Laplanche, J. L., Lehmann, S., Launay, J. M., et al. (2000). Signal transduction through prion protein. Science 289, 1925–1928. doi: 10.1126/science.289.5486.1925
Mucke, L., and Selkoe, D. J. (2012). Neurotoxicity of amyloid β-protein: synaptic and network dysfunction. Cold Spring Harb. Perspect. Med. 2:a006338. doi: 10.1101/cshperspect.a006338
Nielsen, J., Kulahin, N., and Walmod, P. S. (2010). Extracellular protein interactions mediated by the neural cell adhesion molecule, NCAM: heterophilic interactions between NCAM and cell adhesion molecules, extracellular matrix proteins, and viruses. Adv. Exp. Med. Biol. 663, 23–53. doi: 10.1007/978-1-4419-1170-4_2
Nitsch, R. M., Deng, A., Wurtman, R. J., and Growdon, J. H. (1997). Metabotropic glutamate receptor subtype mGluR1α stimulates the secretion of the amyloid β-protein precursor ectodomain. J. Neurochem. 69, 704–712. doi: 10.1046/j.1471-4159.1997.69020704.x
Nygaard, H. B., and Strittmatter, S. M. (2009). Cellular prion protein mediates the toxicity of β-amyloid oligomers: implications for Alzheimer disease. Arch. Neurol. 66, 1325–1328. doi: 10.1001/archneurol.2009.223
Onodera, T., Sakudo, A., Tsubone, H., and Itohara, S. (2014). Review of studies that have used knockout mice to assess normal function of prion protein under immunological or pathophysiological stress. Microbiol. Immunol. 58, 361–374. doi: 10.1111/1348-0421.12162
Ory, S., and Morrison, D. K. (2004). Signal transduction: implications for Ras-dependent ERK signaling. Curr. Biol. 14, R277–R278. doi: 10.1016/j.cub.2004.03.023
Ostapchenko, V. G., Beraldo, F. H., Mohammad, A. H., Xie, Y. F., Hirata, P. H., Magalhaes, A. C., et al. (2013). The prion protein ligand, stress-inducible phosphoprotein 1, regulates amyloid-β oligomer toxicity. J. Neurosci. 33, 16552–16564. doi: 10.1523/JNEUROSCI.3214-13.2013
Paitel, E., Alves da Costa, C., Vilette, D., Grassi, J., and Checler, F. (2002). Overexpression of PrPc triggers caspase 3 activation: potentiation by proteasome inhibitors and blockade by anti-PrP antibodies. J. Neurochem. 83, 1208–1214. doi: 10.1046/j.1471-4159.2002.01234.x
Pan, T., Chang, B., Wong, P., Li, C., Li, R., Kang, S. C., et al. (2005). An aggregation-specific enzyme-linked immunosorbent assay: detection of conformational differences between recombinant PrP protein dimers and PrPSc aggregates. J. Virol. 79, 12355–12364. doi: 10.1128/jvi.79.19.12355-12364.2005
Pan, T., Li, R., Wong, B. S., Liu, T., Gambetti, P., and Sy, M. S. (2002). Heterogeneity of normal prion protein in two- dimensional immunoblot: presence of various glycosylated and truncated forms. J. Neurochem. 81, 1092–1101. doi: 10.1046/j.1471-4159.2002.00909.x
Pan, C. Q., Sudol, M., Sheetz, M., and Low, B. C. (2012). Modularity and functional plasticity of scaffold proteins as p(l)acemakers in cell signaling. Cell. Signal. 24, 2143–2165. doi: 10.1016/j.cellsig.2012.06.002
Panayi, G. S., and Corrigall, V. M. (2014). Immunoglobulin heavy-chain-binding protein (BiP): a stress protein that has the potential to be a novel therapy for rheumatoid arthritis. Biochem. Soc. Trans. 42, 1752–1755. doi: 10.1042/BST20140230
Pantera, B., Bini, C., Cirri, P., Paoli, P., Camici, G., Manao, G., et al. (2009). PrPc activation induces neurite outgrowth and differentiation in PC12 cells: role for caveolin-1 in the signal transduction pathway. J. Neurochem. 110, 194–207. doi: 10.1111/j.1471-4159.2009.06123.x
Parada, E., Buendia, I., León, R., Negredo, P., Romero, A., Cuadrado, A., et al. (2014). Neuroprotective effect of melatonin against ischemia is partially mediated by α-7 nicotinic receptor modulation and HO-1 overexpression. J. Pineal Res. 56, 204–212. doi: 10.1111/jpi.12113
Patrizio, A., and Specht, C. G. (2016). Counting numbers of synaptic proteins: absolute quantification and single molecule imaging techniques. Neurophotonics 3:041805. doi: 10.1117/1.NPh.3.4.041805
Pawson, T., and Scott, J. D. (1997). Signaling through scaffold, anchoring, and adaptor proteins. Science 278, 2075–2080. doi: 10.1126/science.278.5346.2075
Pearce, L. R., Atanassova, N., Banton, M. C., Bottomley, B., van der Klaauw, A. A., Revelli, J. P., et al. (2013). KSR2 mutations are associated with obesity, insulin resistance, and impaired cellular fuel oxidation. Cell 155, 765–777. doi: 10.1016/j.cell.2013.09.058
Petersen, S. C., Luo, R., Liebscher, I., Giera, S., Jeong, S. J., Mogha, A., et al. (2015). The adhesion GPCR GPR126 has distinct, domain-dependent functions in Schwann cell development mediated by interaction with laminin-211. Neuron 85, 755–769. doi: 10.1016/j.neuron.2014.12.057
Petrilli, A. M., and Fernández-Valle, C. (2016). Role of Merlin/NF2 inactivation in tumor biology. Oncogene 35, 537–548. doi: 10.1038/onc.2015.125
Poppinga, W. J., Muñoz-Llancao, P., González-Billault, C., and Schmidt, M. (2014). A-kinase anchoring proteins: cAMP compartmentalization in neurodegenerative and obstructive pulmonary diseases. Br. J. Pharmacol. 171, 5603–5623. doi: 10.1111/bph.12882
Porto-Carreiro, I., Février, B., Paquet, S., Vilette, D., and Raposo, G. (2005). Prions and exosomes: from PrPc trafficking to PrPsc propagation. Blood Cells Mol. Dis. 35, 143–148. doi: 10.1016/j.bcmd.2005.06.013
Prado, M. A., Alves-Silva, J., Magalhães, A. C., Prado, V. F., Linden, R., Martins, V. R., et al. (2004). PrPc on the road: trafficking of the cellular prion protein. J. Neurochem. 88, 769–781. doi: 10.1046/j.1471-4159.2003.02199.x
Prcina, M., and Kontsekova, E. (2011). Has prion protein important physiological function? Med. Hypotheses 76, 567–569. doi: 10.1016/j.mehy.2011.01.002
Priola, S. A., Caughey, B., Wehrly, K., and Chesebro, B. (1995). A 60-kDa prion protein (PrP) with properties of both the normal and scrapie-associated forms of PrP. J. Biol. Chem. 270, 3299–3305. doi: 10.1074/jbc.270.7.3299
Puig, B., Altmeppen, H., and Glatzel, M. (2014). The GPI-anchoring of PrP: implications in sorting and pathogenesis. Prion 8, 11–18. doi: 10.4161/pri.27892
Rambold, A. S., Müller, V., Ron, U., Ben-Tal, N., Winklhofer, K. F., and Tatzelt, J. (2008). Stress-protective signalling of prion protein is corrupted by scrapie prions. EMBO J. 27, 1974–1984. doi: 10.1038/emboj.2008.122
Ramljak, S., Schmitz, M., Zafar, S., Wrede, A., Schenkel, S., Asif, A. R., et al. (2015). Cellular prion protein directly interacts with and enhances lactate dehydrogenase expression under hypoxic conditions. Exp. Neurol. 271, 155–167. doi: 10.1016/j.expneurol.2015.04.025
Ramsey, I. S., Delling, M., and Clapham, D. E. (2006). An introduction to TRP channels. Annu. Rev. Physiol. 68, 619–647. doi: 10.1146/annurev.physiol.68.040204.100431
Ren, J. G., Li, Z., Crimmins, D. L., and Sacks, D. B. (2005). Self-association of IQGAP1: characterization and functional sequelae. J. Biol. Chem. 280, 34548–34557. doi: 10.1074/jbc.m507321200
Rial, D., Piermartiri, T. C., Duarte, F. S., Tasca, C. I., Walz, R., and Prediger, R. D. (2012). Overexpression of cellular prion protein (PrPC) prevents cognitive dysfunction and apoptotic neuronal cell death induced by amyloid-β (Aβ1−40) administration in mice. Neuroscience 215, 79–89. doi: 10.1016/j.neuroscience.2012.04.034
Ribeiro, F. M., Paquet, M., Cregan, S. P., and Ferguson, S. S. (2010). Group I metabotropic glutamate receptor signalling and its implication in neurological disease. CNS Neurol. Disord. Drug Targets 9, 574–595. doi: 10.2174/187152710793361612
Rieger, R., Edenhofer, F., Lasmézas, C. I., and Weiss, S. (1997). The human 37-kDa laminin receptor precursor interacts with the prion protein in eukaryotic cells. Nat. Med. 3, 1383–1388. doi: 10.1038/nm1297-1383
Rieger, R., Lasmézas, C. I., and Weiss, S. (1999). Role of the 37 kDa laminin receptor precursor in the life cycle of prions. Transfus. Clin. Biol. 6, 7–16. doi: 10.1016/s1246-7820(99)80006-8
Ritchie, A. J., Crawford, D. M., Ferguson, D. J., Burthem, J., and Roberts, D. J. (2013). Normal prion protein is expressed on exosomes isolated from human plasma. Br. J. Haematol. 163, 678–680. doi: 10.1111/bjh.12543
Robertson, C., Booth, S. A., Beniac, D. R., Coulthart, M. B., Booth, T. F., and McNicol, A. (2006). Cellular prion protein is released on exosomes from activated platelets. Blood 107, 3907–3911. doi: 10.1182/blood-2005-02-0802
Robinson, S. W., Nugent, M. L., Dinsdale, D., and Steinert, J. R. (2014). Prion protein facilitates synaptic vesicle release by enhancing release probability. Hum. Mol. Genet. 23, 4581–4596. doi: 10.1093/hmg/ddu171
Romano, S. A., Cordeiro, Y., Lima, L. M., Lopes, M. H., Silva, J. L., Foguel, D., et al. (2009). Reciprocal remodeling upon binding of the prion protein to its signaling partner hop/STI1. FASEB J. 23, 4308–4316. doi: 10.1096/fj.09-138974
Roucou, X. (2014). Regulation of PrPC signaling and processing by dimerization. Front. Cell Dev. Biol. 2:57. doi: 10.3389/fcell.2014.00057
Rudd, P. M., Merry, A. H., Wormald, M. R., and Dwek, R. A. (2002). Glycosylation and prion protein. Curr. Opin. Struct. Biol. 12, 578–586. doi: 10.1016/s0959-440x(02)00377-9
Rutishauser, U., and Landmesser, L. (1996). Polysialic acid in the vertebrate nervous system: a promoter of plasticity in cell-cell interactions. Trends Neurosci. 19, 422–427. doi: 10.1016/0166-2236(96)10041-2
Sakurai-Yamashita, Y., Sakaguchi, S., Yoshikawa, D., Okimura, N., Masuda, Y., Katamine, S., et al. (2005). Female-specific neuroprotection against transient brain ischemia observed in mice devoid of prion protein is abolished by ectopic expression of prion protein-like protein. Neuroscience 136, 281–287. doi: 10.1016/j.neuroscience.2005.06.095
Sanches, E. F., Arteni, N., Nicola, F., Aristimunha, D., and Netto, C. A. (2015). Sexual dimorphism and brain lateralization impact behavioral and histological outcomes following hypoxia-ischemia in P3 and P7 rats. Neuroscience 290, 581–593. doi: 10.1016/j.neuroscience.2014.12.074
Santos, T. G., Beraldo, F. H., Hajj, G. N., Lopes, M. H., Roffe, M., Lupinacci, F. C., et al. (2013). Laminin-γ1 chain and stress inducible protein 1 synergistically mediate PrPC-dependent axonal growth via Ca2+ mobilization in dorsal root ganglia neurons. J. Neurochem. 124, 210–223. doi: 10.1111/jnc.12091
Santos, T. G., Lopes, M. H., and Martins, V. R. (2015). Targeting prion protein interactions in cancer. Prion 9, 165–173. doi: 10.1080/19336896.2015.1027855
Santos, T. G., Silva, I. R., Costa-Silva, B., Lepique, A. P., Martins, V. R., and Lopes, M. H. (2011). Enhanced neural progenitor/stem cells self-renewal via the interaction of stress-inducible protein 1 with the prion protein. Stem Cells 29, 1126–1136. doi: 10.1002/stem.664
Santuccione, A., Sytnyk, V., Leshchyns’ka, I., and Schachner, I. (2005). Prion protein recruits its neuronal receptor NCAM to lipid rafts to activate p59fyn and to enhance neurite outgrowth. J. Cell Biol. 169, 341–354. doi: 10.1083/jcb.200409127
Schmitt-Ulms, G., Legname, G., Baldwin, M. A., Ball, H. L., Bradon, N., Bosque, P. J., et al. (2001). Binding of neural cell adhesion molecules (N-CAMs) to the cellular prion protein. J. Mol. Biol. 314, 1209–1225. doi: 10.1006/jmbi.2000.5183
Schmitt-Ulms, G., Hansen, K., Liu, J., Cowdrey, C., Yang, J., DeArmond, S. J., et al. (2004). Time-controlled transcardiac perfusion cross-linking for the study of protein interactions in complex tissues. Nat. Biotechnol. 22, 724–731. doi: 10.1038/nbt969
Schulz, K. M., Andrud, K. M., Burke, M. B., Pearson, J. N., Kreisler, A. D., Stevens, K. E., et al. (2013). The effects of prenatal stress on α4 β2 and α7 hippocampal nicotinic acetylcholine receptor levels in adult offspring. Dev. Neurobiol. 73, 806–814. doi: 10.1002/dneu.22097
Seong, Y. J., Sung, P. S., Jang, Y. S., Choi, Y. J., Park, B. C., Park, S. H., et al. (2015). Activation of human natural killer cells by the soluble form of cellular prion protein. Biochem. Biophys. Res. Commun. 464, 512–518. doi: 10.1016/j.bbrc.2015.06.172
Seringhaus, M., and Gerstein, M. (2008). Genomics confounds gene classification. Amer. Sci. 96, 466–473. doi: 10.1511/2008.75.466
Sheng, M., and Hoogenraad, C. C. (2007). The postsynaptic architecture of excitatory synapses: a more quantitative view. Annu. Rev. Biochem. 76, 823–847. doi: 10.1146/annurev.biochem.76.060805.160029
Shenoy, S. K., McDonald, P. H., Kohout, T. A., and Lefkowitz, R. J. (2001). Regulation of receptor fate by ubiquitination of activated β2-adrenergic receptor and β-arrestin. Science 294, 1307–1313. doi: 10.1126/science.1063866
Shi, Q., Jing, Y. Y., Wang, S. B., Chen, C., Sun, H., Xu, Y., et al. (2013). PrP octarepeats region determined the interaction with caveolin-1 and phosphorylation of caveolin-1 and Fyn. Med. Microbiol. Immunol. 202, 215–227. doi: 10.1007/s00430-012-0284-8
Shyu, W. C., Lin, S. Z., Chiang, M. F., Ding, D. C., Li, K. W., Chen, S. F., et al. (2005). Overexpression of PrPC by adenovirus-mediated gene targeting reduces ischemic injury in a stroke rat model. J. Neurosci. 25, 8967–8977. doi: 10.1523/JNEUROSCI.1115-05.2005
Silveira, J. R., Raymond, G. J., Hughson, A. G., Race, R. E., Sim, V. L., Hayes, S. F., et al. (2005). The most infectious prion protein particles. Nature 437, 257–261. doi: 10.1038/nature03989
Slapšak, U., Salzano, G., Amin, L., Abskharon, R. N., Ilc, G., Zupančič, B., et al. (2016). The N-terminus of the prion protein mediates functional interactions with NCAM fibronectin domain. J. Biol. Chem. 291, 21857–21868. doi: 10.1074/jbc.M116.743435
Smith, F. D., Reichow, S. L., Esseltine, J. L., Shi, D., Langeberg, L. K., Scott, J. D., et al. (2013). Intrinsic disorder within an AKAP-protein kinase A complex guides local substrate phosphorylation. Elife 2:e01319. doi: 10.7554/eLife.01319
Smith, F. D., and Scott, J. D. (2013). Scaffolding proteins: not such innocent bystanders. Curr. Biol. 23, R515–R517. doi: 10.1016/j.cub.2013.05.002
Solforosi, L., Criado, J. R., McGavern, D. B., Wirz, S., Sánchez-Alavez, M., Sugama, S., et al. (2004). Cross-linking cellular prion protein triggers neuronal apoptosis in vivo. Science 303, 1514–1516. doi: 10.1126/science.1094273
Sonati, T., Reimann, R. R., Falsig, J., Baral, P. K., O’Connor, T., Hornemann, S., et al. (2013). The toxicity of antiprion antibodies is mediated by the flexible tail of the prion protein. Nature 501, 102–106. doi: 10.1038/nature12402
Spevacek, A. R., Evans, E. G., Miller, J. L., Meyer, H. C., Pelton, J. G., and Millhauser, G. L. (2013). Zinc drives a tertiary fold in the prion protein with familial disease mutation sites at the interface. Structure 21, 236–246. doi: 10.1016/j.str.2012.12.002
Spudich, A., Frigg, R., Kilic, E., Kilic, U., Oesch, B., Raeber, A., et al. (2005). Aggravation of ischemic brain injury by prion protein deficiency: role of ERK-1/-2 and STAT-1. Neurobiol. Dis. 20, 442–449. doi: 10.1016/j.nbd.2005.04.002
Stahl, N., Borchelt, D. R., Hsiao, K., and Prusiner, S. B. (1987). Scrapie prion protein contains a phosphatidylinositol glycolipid. Cell 51, 229–240. doi: 10.1016/0092-8674(87)90150-4
Steele, A. D., Emsley, J. G., Ozdinler, P. H., Lindquist, S., and Macklis, J. D. (2006). Prion protein (PrPc) positively regulates neural precursor proliferation during developmental and adult mammalian neurogenesis. Proc. Natl. Acad. Sci. U S A 103, 3416–3421. doi: 10.1073/pnas.0511290103
Steele, A. D., Zhou, Z., Jackson, W. S., Zhu, C., Auluck, P., Moskowitz, M. A., et al. (2009). Context dependent neuroprotective properties of prion protein (PrP). Prion 3, 240–249. doi: 10.4161/pri.3.4.10135
Stewart, L. A., Rydzewska, L. H., Keogh, G. F., and Knight, R. S. (2008). Systematic review of therapeutic interventions in human prion disease. Neurology 70, 1272–1281. doi: 10.1212/01.WNL.0000308955.25760.c2
Stuermer, C. A., Langhorst, M. F., Wiechers, M. F., Legler, D. F., Von Hanwehr, S. H., Guse, A. H., et al. (2004). PrPc capping in T cells promotes its association with the lipid raft proteins reggie-1 and reggie-2 and leads to signal transduction. FASEB J. 18, 1731–1733. doi: 10.1096/fj.04-2150fje
Stuermer, C. A., and Plattner, H. (2005). The ‘lipid raft’ microdomain proteins reggie-1 and reggie-2 (flotillins) are scaffolds for protein interaction and signalling. Biochem. Soc. Symp. 72, 109–118. doi: 10.1042/bss0720109
Tacchelly-Benites, O., Wang, Z., Yang, E., Lee, E., and Ahmed, Y. (2013). Toggling a conformational switch in Wnt/β-catenin signaling: regulation of Axin phosphorylation. The phosphorylation state of Axin controls its scaffold function in two Wnt pathway protein complexes. Bioessays 35, 1063–1070. doi: 10.1002/bies.201300101
Takada, L. T., and Geschwind, M. D. (2013). Prion diseases. Semin. Neurol. 33, 348–356. doi: 10.1055/s-0033-1359314
Taylor, P. J., Betts, G. A., Maroulis, S., Gilissen, C., Pedersen, R. L., Mowat, D. R., et al. (2010). Dystrophin gene mutation location and the risk of cognitive impairment in Duchenne muscular dystrophy. PLoS One 5:e8803. doi: 10.1371/journal.pone.0008803
Taylor, D. R., and Hooper, N. M. (2006). The prion protein and lipid rafts. Mol. Membr. Biol. 23, 89–99. doi: 10.1080/09687860500449994
Terra-Granado, E., Berbert, L. R., de Meis, J., Nomizo, R., Martins, V. R., Savino, W., et al. (2007). Is there a role for cellular prion protein in intrathymic T cell differentiation and migration? Neuroimmunomodulation 14, 213–219. doi: 10.1159/000110649
Thakur, A. K., Srivastava, A. K., Srinivas, V., Chary, K. V., and Rao, C. M. (2011). Copper alters aggregation behavior of prion protein and induces novel interactions between its N- and C-terminal regions. J. Biol. Chem. 286, 38533–38545. doi: 10.1074/jbc.M111.265645
Tomasi, V. (2010). Signal transduction in neurons: effects of cellular prion protein on fyn kinase and ERK1/2 kinase. Immun. Ageing 7:S5. doi: 10.1186/1742-4933-7-s1-s5
Tsai, Y. L., Zhang, Y., Tseng, C. C., Stanciauskas, R., Pinaud, F., and Lee, A. S. (2015). Characterization and mechanism of stress-induced translocation of 78-kilodalton glucose-regulated protein (GRP78) to the cell surface. J. Biol. Chem. 290, 8049–8064. doi: 10.1074/jbc.M114.618736
Tsoupri, E., and Capetanaki, Y. (2013). Muyospryn: a multifunctional desmin-associated protein. Histochem. Cell Biol. 140, 55–63. doi: 10.1007/s00418-013-1103-z
Turk, E., Teplow, D. B., Hood, L. E., and Prusiner, S. B. (1988). Purification and properties of the cellular and scrapie hamster prion proteins. Eur. J. Biochem. 176, 21–30. doi: 10.1111/j.1432-1033.1988.tb14246.x
Um, J. W., and Strittmatter, S. M. (2013). Amyloid-β induced signaling by cellular prion protein and Fyn kinase in Alzheimer disease. Prion 7, 37–41. doi: 10.4161/pri.22212
Vella, L. J., Greenwood, D. L., Cappai, R., Scheerlinck, J. P., and Hill, A. F. (2008). Enrichment of prion protein in exosomes derived from ovine cerebral spinal fluid. Vet. Immunol. Immunopathol. 124, 385–393. doi: 10.1016/j.vetimm.2008.04.002
Villemagne, V. L., Burnham, S., Bourgeat, P., Brown, B., Ellis, K. A., Salvado, O., et al. (2013). Amyloid β deposition, neurodegeneration and cognitive decline in sporadic Alzheimer’s disease: a prospective cohort study. Lancet Neurol. 12, 357–367. doi: 10.1016/S1474-4422(13)70044-9
Walsh, D. M., and Selkoe, D. J. (2007). A β oligomers—a decade of discovery. J. Neurochem. 101, 1172–1184. doi: 10.1111/j.1471-4159.2006.04426.x
Wang, B., Lou, Z., Zhang, H., and Xu, B. (2016). Effect of the electrostatic surface potential on the oligomerization of full-length human recombinant prion protein at single-molecule level. J. Chem. Phys. 144:114701. doi: 10.1063/1.4943878
Wang, P., Wu, Y., Ge, X., Ma, L., and Pei, G. (2003). Subcellular localization of β-arrestins is determined by their intact N domain and the nuclear export signal at the C terminus. J. Biol. Chem. 278, 11648–11653. doi: 10.1074/jbc.M208109200
Wang, G. H., Zhou, X. M., Bai, Y., Yin, X. M., Yang, L. F., and Zhao, D. (2011). Hsp70 binds to PrPC in the process of PrPC release via exosomes from THP-1 monocytes. Cell Biol. Int. 35, 553–558. doi: 10.1042/CBI20090391
Warner, R. G., Hundt, C., Weiss, S., and Turnbull, J. E. (2002). Identification of the heparan sulfate binding sites in the cellular prion protein. J. Biol. Chem. 277, 18421–18430. doi: 10.1074/jbc.M110406200
Warwicker, J. (2000). Modeling a prion protein dimer: predictions for fibril formation. Biochem. Biophys. Res. Commun. 278, 646–652. doi: 10.1006/bbrc.2000.3829
Wei, J., Liu, W., and Yan, Z. (2010). Regulation of AMPA receptor trafficking and function by glycogen synthase kinase 3. J. Biol. Chem. 285, 26369–26376. doi: 10.1074/jbc.M110.121376
Weise, J., Crome, O., Sandau, R., Schulz-Schaeffer, W., Bahr, M., and Zerr, I. (2004). Upregulation of cellular prion protein (PrPc) after focal cerebral ischemia and influence of lesion severity. Neurosci. Lett. 372, 146–150. doi: 10.1016/j.neulet.2004.09.030
Weise, J., Doeppner, T. R., Muller, T., Wrede, A., Schulz-Schaeffer, W., Zerr, I., et al. (2008). Overexpression of cellular prion protein alters postischemic Erk1/2 phosphorylation but not Akt phosphorylation and protects against focal cerebral ischemia. Restor. Neurol. Neurosci. 26, 57–64.
Weise, J., Sandau, R., Schwarting, S., Crome, O., Wrede, A., Schulz-Schaeffer, W., et al. (2006). Deletion of cellular prion protein results in reduced Akt activation, enhanced postischemic caspase-3 activation and exacerbation of ischemic brain injury. Stroke 37, 1296–1300. doi: 10.1161/01.STR.0000217262.03192.d4
Wells, M. A., Jackson, G. S., Jones, S., Hosszu, L. L., Craven, C. J., Clarke, A. R., et al. (2006). A reassessment of copper(II) binding in the full-length prion protein. Biochem. J. 399, 435–444. doi: 10.1042/bj20060458
West, E., Osborne, C., Nolan, W., and Bate, C. (2015). Monoacylated cellular prion proteins reduce amyloid-β-induced activation of cytoplasmic phospholipase A2 and synapse damage. Biology (Basel) 4, 367–382. doi: 10.3390/biology4020367
Westergard, L., Christensen, H. M., and Harris, D. A. (2007). The cellular prion protein (PrPC): its physiological function and role in disease. Biochim. Biophys. Acta 1772, 629–644. doi: 10.1016/j.bbadis.2007.02.011
Whitmarsh, A. J., and Davis, R. J. (1998). Structural organization of MAP-kinase signaling modules by scaffold proteins in yeast and mammals. Trends Biochem. Sci. 23, 481–485. doi: 10.1016/s0968-0004(98)01309-7
Witzel, F., Maddison, L., and Bluthgen, N. (2012). How scaffolds shape MAPK signaling: what we know and opportunities for systems approaches. Front. Physiol. 3:475. doi: 10.3389/fphys.2012.00475
Wu, J., Liu, Q., Tang, P., Mikkelsen, J. D., Shen, J., Whiteaker, P., et al. (2016). Heteromeric α7β2 nicotinic acetylcholine receptors in the brain. Trends Pharmacol. Sci. 37, 562–574. doi: 10.1016/j.tips.2016.03.005
Yablonski, D., Marbach, I., and Levitzki, A. (1996). Dimerization of Ste5, a mitogen-activated protein kinase cascade scaffold protein, is required for signal transduction. Proc. Natl. Acad. Sci. U S A 93, 13864–13869. doi: 10.1073/pnas.93.24.13864
Yao, Y., Ren, J., and Jones, I. M. (2003). Amino terminal interaction in the prion protein identified using fusion to green fluorescent protein. J. Neurochem. 87, 1057–1065. doi: 10.1046/j.1471-4159.2003.02039.x
Yehiely, F., Bamborough, P., Da Costa, M., Perry, B. J., Thinakaran, G., Cohen, F. E., et al. (1997). Identification of candidate proteins binding to prion protein. Neurobiol. Dis. 3, 339–355. doi: 10.1006/nbdi.1997.0130
Yin, S., Pham, N., Yu, S., Li, C., Wong, P., Chang, B., et al. (2007). Human prion proteins with pathogenic mutations share common conformational changes resulting in enhanced binding to glycosaminoglycans. Proc. Natl. Acad. Sci. U S A 104, 7546–7551. doi: 10.1073/pnas.0610827104
Yu, Z., Huang, P., Yu, Y., Zheng, Z., Huang, Z., Guo, C., et al. (2016). Unique properties of the rabbit prion protein oligomer. PLoS One 11:e0160874. doi: 10.1371/journal.pone.0160874
Yu, Y., and Ye, R. D. (2015). Microglial Aβ receptors in Alzheimer’s disease. Cell. Mol. Neurobiol. 35, 71–83. doi: 10.1007/s10571-014-0101-6
Yusa, S., Oliveira-Martins, J. B., Sugita-Konishi, Y., and Kikuchi, Y. (2012). Cellular prion protein: from physiology to pathology. Viruses 4, 3109–3131. doi: 10.3390/v4113109
Zahn, R. (2003). The octapeptide repeats in mammalian prion protein constitute a pH-dependent folding and aggregation site. J. Mol. Biol. 334, 477–488. doi: 10.1016/j.jmb.2003.09.048
Zamponi, G. W., and Stys, P. K. (2009). Role of prions in neuroprotection and neurodegeneration: a mechanism involving glutamate receptors? Prion 3, 187–189. doi: 10.4161/pri.3.4.9549
Zanata, S. M., Lopes, M. H., Mercadante, A. F., Hajj, G. N., Chiarini, L. B., Nomizo, R., et al. (2002). Stress-inducible protein 1 is a cell surface ligand for cellular prion that triggers neuroprotection. EMBO J. 21, 3307–3316. doi: 10.1093/emboj/cdf325
Zenaro, E., Pietronigro, E., Bianca Della, V., Piacentino, G., Marongiu, L., Budui, S., et al. (2015). Neutrophils promote Alzheimer’s disease-like pathology and cognitive decline via LFA-1 integrin. Nat. Med. 21, 880–886. doi: 10.1038/nm.3913
Zeng, L., Zou, W., and Wang, G. (2015). Cellular prion protein (PrPC) and its role in stress responses. Int. J. Clin. Exp. Med. 8, 8042–8050.
Zhang, Z., Chen, H., Bai, H., and Lai, L. (2007). Molecular dynamics simulations on the oligomer-formation process of the GNNQQNY peptide from yeast prion protein Sup35. Biophys. J. 93, 1484–1492. doi: 10.1529/biophysj.106.100537
Zhang, Y., Qin, K., Wang, J., Hung, T., and Zhao, R. Y. (2006). Dividing roles of prion protein in staurosporine-mediated apoptosis. Biochem. Biophys. Res. Commun. 349, 759–768. doi: 10.1016/j.bbrc.2006.08.116
Zhou, M., Horita, D. A., Waugh, D. S., Byrd, R. A., and Morrison, D. K. (2002). Solution structure and functional analysis of the cysteine-rich C1 domain of kinase suppressor of Ras (KSR). J. Mol. Biol. 315, 435–446. doi: 10.1006/jmbi.2001.5263
Keywords: prion protein, neurodegeneration, prion diseases, Alzheimer disease, signal transduction, cell surface, scaffold proteins, signal corruption
Citation: Linden R (2017) The Biological Function of the Prion Protein: A Cell Surface Scaffold of Signaling Modules. Front. Mol. Neurosci. 10:77. doi: 10.3389/fnmol.2017.00077
Received: 26 December 2016; Accepted: 06 March 2017;
Published: 20 March 2017.
Edited by:
Rameshwar K. Sharma, Salus University, USAReviewed by:
Anuradha Ratnaparkhi, Agharkar Research Institute, IndiaHomira Behbahani, Karolinska Institutet, Sweden
Copyright © 2017 Linden. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution and reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rafael Linden, cmxpbmRlbkBiaW9mLnVmcmouYnI=