 Netta Shemesh
Netta Shemesh Nadav Shai
Nadav Shai Lana Meshnik
Lana Meshnik Anat Ben-Zvi
Anat Ben-Zvi- Department of Life Sciences, The National Institute for Biotechnology in the Negev, Ben-Gurion University of the Negev, Beer Sheva, Israel
Caenorhabditis elegans somatic protein homeostasis (proteostasis) is actively remodeled at the onset of reproduction. This proteostatic collapse is regulated cell-nonautonomously by signals from the reproductive system that transmit the commitment to reproduction to somatic cells. Here, we asked whether the link between the reproductive system and somatic proteostasis could be uncoupled by activating downstream effectors in the gonadal longevity cascade. Specifically, we examined whether over-expression of lipl-4 (lipl-4(oe)), a target gene of the gonadal longevity pathway, or increase in arachidonic acid (AA) levels, associated with lipl-4(oe), modulated proteostasis and reproduction. We found that lipl-4(oe) rescued somatic proteostasis and postponed the onset of aggregation and toxicity in C. elegans models of polyglutamine (polyQ) diseases. However, lipl-4(oe) also disrupted fatty acid transport into developing oocytes and reduced reproductive success. In contrast, diet supplementation of AA recapitulated lipl-4(oe)-mediated proteostasis enhancement in wild type animals but did not affect the reproductive system. Thus, the gonadal longevity pathway mediates a trade-off between somatic maintenance and reproduction, in part by regulating the expression of genes, such as lipl-4, with inverse effects on somatic maintenance and reproduction. We propose that AA could uncouple such germline to soma crosstalk, with beneficial implications protein misfolding diseases.
Introduction
Aggregates or aggregation intermediates are strongly associated with the etiology of many late-onset neurodegenerative diseases, including Huntington’s disease, amyotrophic lateral sclerosis, Alzheimer’s disease and Parkinson’s disease (Davies et al., 1997; Nussbaum and Polymeropoulos, 1997; Johnston et al., 2000; Glabe and Kayed, 2006; Labbadia and Morimoto, 2015a). In Huntington’s disease, for example, the expansion of polyglutamine (polyQ) repeats is suggested to be the underlying cause of protein misfolding and gain-of-function toxicity (Orr and Zoghbi, 2007). Expression of mutant Huntingtin containing expanded polyQ or even the expanded glutamine tract alone is sufficient to cause cellular dysfunction in various animal models (Zoghbi and Botas, 2002; Sherman and Muchowski, 2003; Voisine and Hart, 2004). Sequestration of misfolded proteins into aggregates is also thought to be part of the cellular defense response against the accumulation of misfolded proteins (Cohen et al., 2006; Tyedmers et al., 2010).
Because chronic expression of misfolded proteins could interfere with and compete for cellular quality control machineries, it was suggested that disruption of protein homeostasis (proteostasis) could be a primary cause for cellular dysfunction and death in neurodegenerative diseases (Gidalevitz et al., 2006; Yerbury et al., 2016). Indeed, expression of aggregation-prone proteins in various model systems was shown to interfere with cellular proteostasis, including disruption of the clearance and folding machineries through competition with other proteins substrates (Suhr et al., 2001; Kim et al., 2002; Venkatraman et al., 2004; Bennett et al., 2007; Bilen and Bonini, 2007; Kitamura et al., 2014). This, in turn, can cause instability of the cellular proteome, affecting the folding of unrelated proteins (Gidalevitz et al., 2006, 2009; Olzscha et al., 2011; Eremenko et al., 2013; Yu et al., 2014; Klabonski et al., 2016). Moreover, changes in chaperone expression levels can disrupt cellular proteostasis and induce an accumulation of damaged protein (Blair et al., 2013; Guisbert et al., 2013; van Oosten-Hawle et al., 2013; Frumkin et al., 2014; Bar-Lavan et al., 2016; Lechler et al., 2017). Thus, maintaining proteostatic capacity is critical for protecting cells from the protein damage associated with protein misfolding diseases.
The challenge to cellular proteostasis is exacerbated in aged individuals as proteostasis maintenance and effective stress response activation decline with age (Taylor and Dillin, 2011; Shai et al., 2014; Labbadia and Morimoto, 2015a). In Caenorhabditis elegans, proteostatic capacity was shown to decline sharply following the onset of reproduction, thereby accelerating the accumulation of polyQ aggregation and toxicity (Ben-Zvi et al., 2009; Liu et al., 2011; Taylor and Dillin, 2013; Labbadia and Morimoto, 2015b; Walther et al., 2015). This decline was, in part, linked to remodeling of the chromatin accessibility of stress gene promoters (Labbadia and Morimoto, 2015b; Merkwirth et al., 2016; Tian et al., 2016). Proteostasis remodeling can be negated by the actions of the gonadal longevity pathway (Lapierre et al., 2011; Vilchez et al., 2012; Shemesh et al., 2013; Shai et al., 2014; Labbadia and Morimoto, 2015b). Signals from the reproductive system can regulate somatic proteostasis in response to inhibition of germline stem cell (GSC) proliferation by activating several transcription factors, including DAF-16/FOXO, SKN-1/Nrf and HSF-1, that are required for proteostasis maintenance during adulthood, as well as for extended lifespan (Hsin and Kenyon, 1999; Libina et al., 2003; Berman and Kenyon, 2006; Antebi, 2012; Shemesh et al., 2013; Steinbaugh et al., 2015; Wang et al., 2017). Thus, the gonadal longevity pathway could determine the investment in somatic maintenance in response to reproduction competence, making the soma available for the demands of reproduction (Kirkwood, 2005; Antebi, 2012; Shai et al., 2014). Given that this trade-off is a regulated switch (Shemesh et al., 2013; Labbadia and Morimoto, 2015b), we asked whether it is possible to uncouple somatic maintenance from reproduction and improve proteostasis without impacting fecundity. We reasoned that over-expression of genes down-regulated by the gonadal longevity pathway would alleviate the protein damage associated with age-dependent neurodegenerative diseases without affecting reproduction.
Inhibition of germline proliferation activates DAF-16 that, in turn, induces the expression of a large set of genes. One of the genes up-regulated by DAF-16 is the lysosomal acid lipase-encoding lipl-4 (Wang et al., 2008; Lapierre et al., 2011; McCormick et al., 2012; Folick et al., 2015; Figure 1A). LIPL-4 itself modulates C. elegans lifespan, with its function resulting in the activation of the nuclear hormone receptors NHR-49 and NHR-80 and in the induced expression of autophagy/lipolysis-related genes that modulate the fatty acid metabolism required for lipl-4-dependent lifespan extension (Goudeau et al., 2011; Lapierre et al., 2011; Ratnappan et al., 2014; Folick et al., 2015). Moreover, LIPL-4 over-expression results in the enrichment of long-chain fatty acids, including oleoylethanolamide (OEA), ω-6 arachidonic acid (AA) and dihomo-γ-linolenic acid (DGLA) and the ω-3 fatty acid eicosapentaenoic acid (EPA). Diet supplementation of OEA activates NHR-49 and NHR-80, while both DGLA and AA activate autophagy, extending lifespan (O’Rourke et al., 2013; Folick et al., 2015; Figure 1A). Given that lipl-4 is sufficient and required for lifespan extension (Wang et al., 2008), we asked whether over-expression of LIPL-4 could uncouple proteostasis from reproduction and delay the onset of protein aggregation and toxicity. We found that LIPL-4 modulated the proteostatic switch upon transition to adulthood, resulting in a delay in the onset of aggregation and toxicity in C. elegans models of polyQ diseases. However, over-expression of LIPL-4 negatively impacted fatty acid mobilization to the developing oocytes and disrupted reproduction. Surprisingly, diet supplementation of AA improved proteostasis without disrupting reproduction. AA supplementation could, therefore, uncouple somatic maintenance from reproduction, thereby mimicking the beneficial effects of inhibiting germline proliferation on somatic proteostasis without imposing a cost on reproduction.
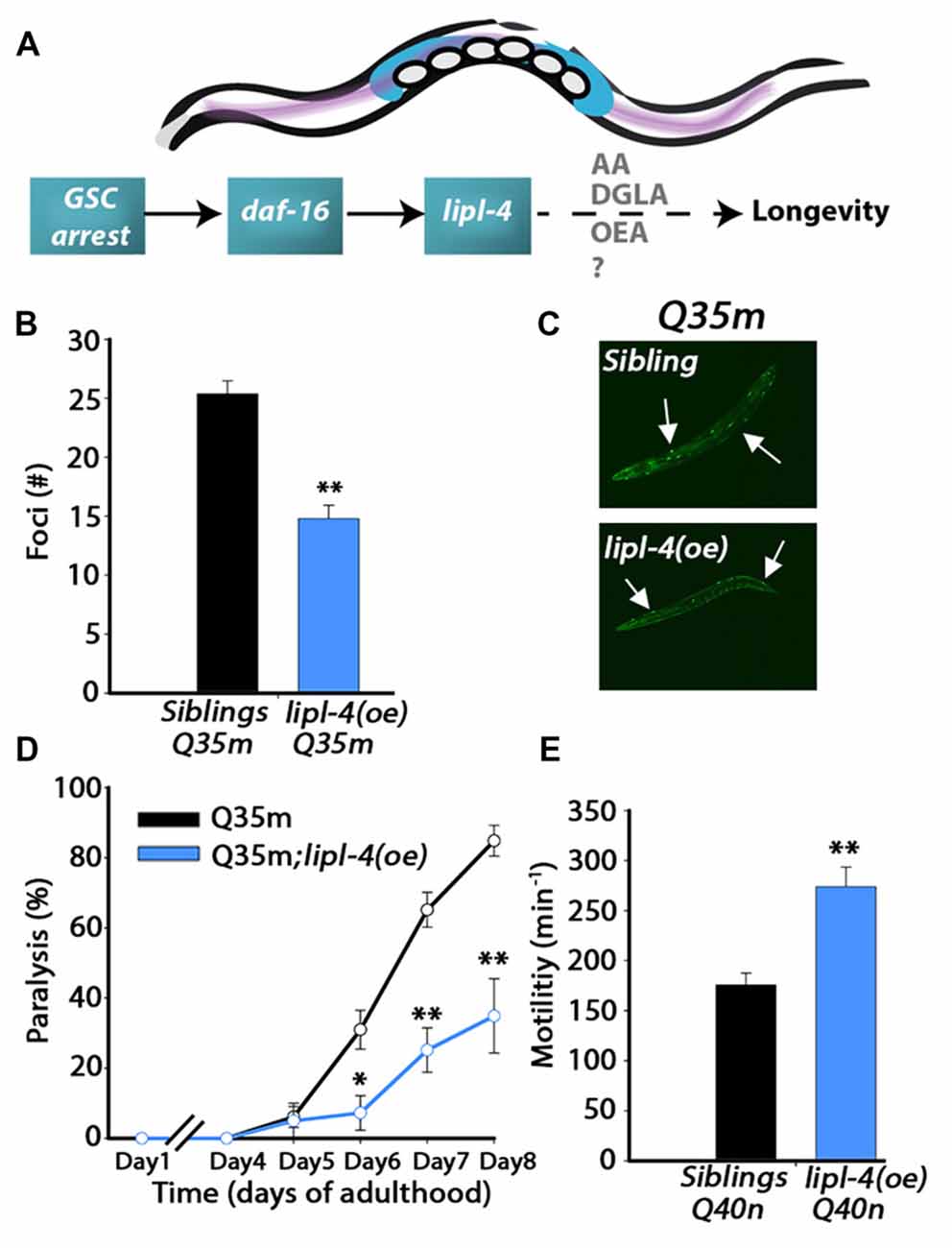
Figure 1. Over-expression of lipl-4 postpones the onset of polyglutamine (polyQ) aggregation and toxicity. (A) Schematic drawing of the gonadal longevity cascade regulating lipl-4 when germline stem cells (GSCs) are arrested in C. elegans. (B) The number of bright foci scored on day 2 of adulthood in age-synchronized Q35m;lipl-4(oe) animals and their siblings (n > 70). (C) Representative images of age-synchronized Q35m;lipl-4(oe) animals and their siblings on day 2 of adulthood. Arrows indicate foci. (D) Motility was scored in age-synchronized Q35m;lipl-4(oe) animals and their siblings by determining the percentage of paralyzed animals. (E) Motility was scored in age-synchronized Q40n;lipl-4(oe) animals and their siblings by counting the number of body bends per minute on day 2 of adulthood. Data was compared to age-matched sibling animals examined under the same condition. *Denotes P < 0.05, **denotes P < 0.01.
Materials and Methods
Nematodes and Growth Conditions
Nematodes were grown on nematode growth medium (NGM) plates seeded with the Escherichia coli OP50-1 strain. Unless otherwise stated, 30–80 embryos, laid at 15°C, were transferred to fresh plates and grown at 25°C for the duration of an experiment. The first day of adulthood (day 1) was set at 50 h after temperature shift, before the onset of egg-laying. Animals were moved every 1–2 days during the reproductive period to avoid progeny contamination. Heat shock-treated animals were discarded after scoring.
Statistical Analysis
Experiments were repeated at least three times and >15 animals per experimental condition were scored. Data are presented as means ± SEM. P values were calculated using the Wilcoxon Mann-Whitney rank sum test to compare two independent populations. *Denotes P < 0.05, **denotes P < 0.01.
Foci Quantification
Age-synchronized animals expressing punc-54::Q35::yellow fluorescent protein (YFP) (Q35m) were examined using a Leica M165 FC fluorescent stereoscope with a YFP filter and the number of bright foci was counted.
Paralysis Assay
A total of 15–30 age-synchronized animals were used for each assay. Animals were grown at 25°C for the duration of the experiment. Animals were moved every day, and paralyzed animals were scored by monitoring their movement 5 min after being transferred to a new plate. Animals that did not move were scored as paralyzed (Karady et al., 2013).
Motility Assay
A total of 15–30 age-synchronized animals were used for each assay. Day 2 adults were moved into M9 buffer and thrashing rates were measured by counting body bends for 15 s. One body bend was defined as a change in the direction of bending at mid-body. Values are presented as bends per minute.
Stiff Body Paralysis Assay
Animals expressing mutant unc-52(ts) were grown at 25°C until day 1 of adulthood, when they were shifted to 15°C and paralysis was scored.
Thermo-Resistance Assay
Animals were picked at the indicated ages and transferred to a 24-well plate containing heat shock buffer (100 mM Tris-HCl, pH 7.4, 17 mM NaCl and 1% cholesterol supplemented with bacteria). These animals were then subjected to a 37°C heat shock for 6 h. Heat shock buffer was supplemented with SYTOX orange (Invitrogene), and animal survival was scored by monitoring dye uptake, using a Leica M165 FC fluorescent stereoscope with a TXR filter. Fluorescent animals were scored as dead (Karady et al., 2013).
Heat Shock Treatment
A total of 30–60 age-synchronized animals grown at 25°C were used for each assay. Plates were sealed and placed in a 37°C bath for 90 min. Animals were frozen or fixed immediately following stress.
RNA Levels
Twenty animals were collected per condition. RNA was extracted using the TRIzol reagent (Invitrogene). For cDNA synthesis, mRNA was reverse-transcribed using the iScript cDNA Synthesis Kit (Bio-Rad). Quantitative PCR was performed on a C1000 Thermal Cycler (Bio-Rad) with KAPA SYBER FAST (KAPA BIOSYSTEMS; Shemesh et al., 2013).
Progeny Quantification
Individual age-synchronized animals of all tested condition (in parallel) were allowed to lay eggs on fresh plates at 24–25°C. Animals were moved every 24 h during the first 5 days of adulthood (i.e., past the reproduction span) and the number of offspring was scored 48–72 h later. The progeny of >25 animals per genotype were scored.
Oil-Red-O Staining
Animals were fixed and stained as previously described (O’Rourke et al., 2009) and subsequently mounted and imaged using a Leica DMIL microscope with a 10× 1.0 objective.
DAPI Staining
Gonad were dissected as in Colaiácovo et al. (2003). Gonads were fixed and stained as previously described (Karady et al., 2013) and subsequently mounted and imaged using an Olympus Fluoview FV1000 confocal microscope through a 60× 1.0 numerical aperture objective with a 405-nm line for excitation.
Yolk Levels
Age-synchronized animals expressing pwIs98(YP170::tdimer2) were fixed as in Karady et al. (2013) and imaged using a Leica M165 FC fluorescent stereoscope with a TXR filter. Pictures were analyzed using imageJ software (NIH).
Diet Supplementation of Fatty Acids
AA (50 μM dissolved in NP40) and control plates (containing NP40) were prepared as previously described (Deline et al., 2013). A total of 30–80 embryos were transferred to fresh plates and grown at 25°C for the duration of an experiment.
Results
lipl-4 Over-Expression Postponed the Onset of PolyQ-Associated Toxicity
Extended polyQ stretches (>35Q) fused to fluorescent proteins have been used in C. elegans as models for polyQ-associated toxicity (Morley et al., 2002; Brignull et al., 2006). To ask whether lipl-4 over-expression can modulate polyQ aggregation and toxicity, we first crossed animals expressing YFP fused to 35 repeats of glutamine in body wall muscle (Q35m; Morley et al., 2002) with animals expressing lipl-4 under the regulation of its own promoter as an extra chromosomal array, lipl-4(oe). We then monitored protein aggregation and toxicity and compared the same properties with non-transgenic siblings of lipl-4(oe) animals (siblings). By day 2 of adulthood, animals expressing Q35m in the lipl-4(oe) background showed 40% less visible foci than did their siblings expressing Q35m alone (Figures 1B,C and Supplementary Figure S1). When we examined motility as a measure of Q35m toxicity, we found that the onset of Q35m-mediated paralysis was delayed in lipl-4(oe) animals. By day 8 of adulthood, only 35 ± 11% of lipl-4(oe) animals were paralyzed, as compared to 85 ± 4% of their siblings (Figure 1D). Similar results were observed when we crossed animals expressing cyan fluorescent protein fused to 40 repeats of glutamine in neurons (Q40n; Brignull et al., 2006) with animals over-expressing lipl-4. The motility of Q40n:lipl-4(oe) animals, as measured by thrashing rate (body bends per min), was ~1.6-fold faster than that of their siblings (Figure 1E). Thus, lipl-4 over-expression modulated the onset and progression of protein aggregation and toxicity in C. elegans polyQ disease models.
lipl-4 Over-Expression Was Sufficient to Maintain Somatic Proteostasis
To extend our observations to other aspects of proteostatic function, we next analyzed the impact of lipl-4(oe) on the folding of metastable proteins. The ability of C. elegans to maintain metastable proteins is dependent on the cellular folding capacity. This ability becomes highly restricted early in adulthood, resulting in age-dependent misfolding (Ben-Zvi et al., 2009). A well-established protein folding reporter is the product of a temperature-sensitive mutation in the gene encoding perlecan, unc-52(e669, su250; unc-52(ts)), that causes an age-dependent disruption of muscle organization and motility (Ben-Zvi et al., 2009; Shemesh et al., 2013; Feldman et al., 2014). We, therefore, crossed unc-52(ts) mutant animals with animals expressing lipl-4(oe) and compared the motility of unc-52(ts);lipl-4(oe) with that of their siblings. We found that 79 ± 3% of the unc-52(ts);lipl-4(oe) mutant animals were motile on day 4 of adulthood, as opposing to only 12 ± 5% of the unc-52(ts) siblings, suggesting that lipl-4(oe) can improve proteostasis in adulthood (Figure 2A).
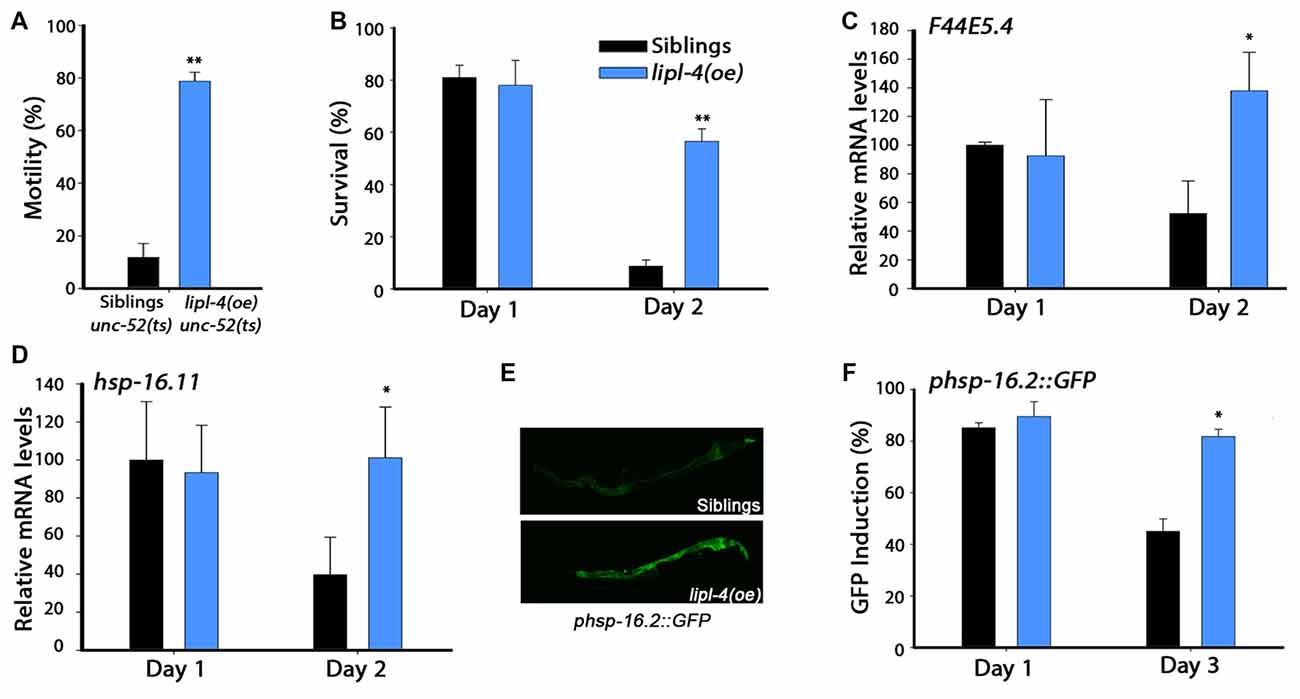
Figure 2. Over-expression of lipl-4 maintains proteostasis in adulthood. (A) Stiff-body paralysis was scored for age-synchronized lipl-4(oe) animals and their siblings on day 4 of adulthood. (B) Thermo-resistance was examined in age-synchronized lipl-4(oe) animals and their siblings. Animals were subjected to heat shock (6 h at 37°C) at the indicated times and survival was assayed. (C,D) Quantification of F44E5.4 (C) and hsp-16.11 (D) mRNA levels from age-synchronized lipl-4(oe) animals and their siblings following heat shock (90 min at 37°C). The data presented are normalized to treated siblings on day 1 of adulthood. (E) Representative images of age-synchronized lipl-4(oe) animals and their siblings expressing phsp-16.2::green fluorescent protein (GFP) that were subjected to heat shock (90 min at 37°C) on day 3 of adulthood. (F) Heat shock gene induction was examined in age-synchronized lipl-4(oe) animals and their siblings expressing phsp-16.2::GFP. Animals were subjected to heat shock (90 min at 37°C) and the percentage of animals expressing GFP was scored. Data was compared to age-matched sibling animals examined under the same condition. *Denotes P < 0.05, **denotes P < 0.01.
Cellular stress responses, such as the heat shock response, are diminished following the onset of reproduction in C. elegans. This is reflected in the sharp decline in the ability of animals to survive various stresses early in adulthood (Ben-Zvi et al., 2009; Shemesh et al., 2013; Labbadia and Morimoto, 2015b). While non-transgenic young adults survived well (81 ± 5%) when challenged by heat shock on day 1 of adulthood, only 9 ± 2.5% survived when first challenged on day 2 of adulthood. In contrast, the survival rates of lipl-4(oe) animals remained high, namely 78 ± 9.5% and 56.5 ± 4.7% on days 1 and 2 of adulthood, respectively (Figure 2B). Improved survival rates were also maintained later in life (Supplementary Figure S2A). To determine whether the increased thermo-resistance observed for lipl-4(oe) animals is associated with the ability to activate the heat shock response, we compared the stress-dependent induction of heat shock genes, whose expression depends on HSF-1 (F44E5.4 and hsp-70) or on HSF-1 and DAF-16 (hsp-16.11 and hsp-16.2). When animals were challenged by heat shock on day 1 of adulthood, F44E5.4, hsp-70, hsp-16.11 and hsp-16.2 mRNA expression levels were strongly induced in both lipl-4(oe) animals and their sibling. In contrast, when animals were heat shocked on day 2 of adulthood, the induced mRNA levels of sibling were 40%–60% lower than in lipl-4(oe) animals (Figures 2C,D, Supplementary Figures S2B,C). A transcriptional reporter of hsp-16.2, a reporter that regulates green fluorescent protein (GFP) expression in a stress-dependent manner, showed similar behavior. When animals were challenged by heat shock on day 1 of adulthood, strong GFP fluorescence was detected in intestinal cells of both lipl-4(oe) animals and their sibling (90 ± 6% and 85 ± 2%, respectively). While the percentage of animals showing high-induction levels of GFP following a heat shock on day 3 of adulthood was maintained for lipl-4(oe), this was not the case for sibling animals (82 ± 3% and 45 ± 5%, respectively; Figures 2E,F). These data suggest that lipl-4 over-expression is sufficient to modulate stress survival and stress response activation in somatic tissues after the onset of reproduction.
lipl-4-Dependent Rescue of Proteostasis Required the Reproductive System
In wild type animals, lipl-4 mRNA levels decline upon transition to adulthood (Supplementary Figure S3A). lipl-4 is regulated by DAF-16 as part of the gonadal longevity pathway, one of several transcription factors that are activated upon inhibition of GSCs proliferation (Hsin and Kenyon, 1999; Wang et al., 2008; Antebi, 2012; McCormick et al., 2012). Accordingly, lipl-4 mRNA levels were strongly induced (7-fold) in glp-1(e2141) (glp-1) germline proliferation mutant animals (Supplementary Figure S3B; Wang et al., 2008; McCormick et al., 2012). We, therefore, asked if down-regulation of lipl-4 is sufficient to impact germline-dependent rescue of proteostasis. For this, we examined whether lipl-4(RNAi) can affect Q35m-associated toxicity and heat shock activation of glp-1 mutant animals during adulthood. Q35m;glp-1 animals show reduced aggregation and reduced paralysis during adulthood (Shemesh et al., 2013). However, Q35m;glp-1 paralysis was induced 3.5-fold when treated with lipl-4(RNAi), as compared to Q35m;glp-1 animals treated with an empty vector (EV) control (Figure 3A). Likewise, glp-1 animals that were treated with lipl-4(RNAi) lost their ability to induce an effective heat shock response. Induction of hsp-16.2-dependent GFP on day 3 of adulthood was reduced 3-fold in lipl-4(RNAi)-treated glp-1 animals, as compared to glp-1 animals treated with an EV control (Figure 3B). We next ask whether over-expression of lipl-4 could further enhance proteostasis in glp-1 animals. glp-1 mutant animals were crossed with lipl-4(oe) animals and subjected to heat shock. Heat shock survival rates of glp-1;lipl-4(oe) animals and their siblings were unaffected by lipl-4(oe) (87.5 ± 4.8% vs. 89.7 ± 4%, Figure 3C). Thus, LIPL-4 is required for proteostatic remodeling downstream of the longevity reproductive pathway.
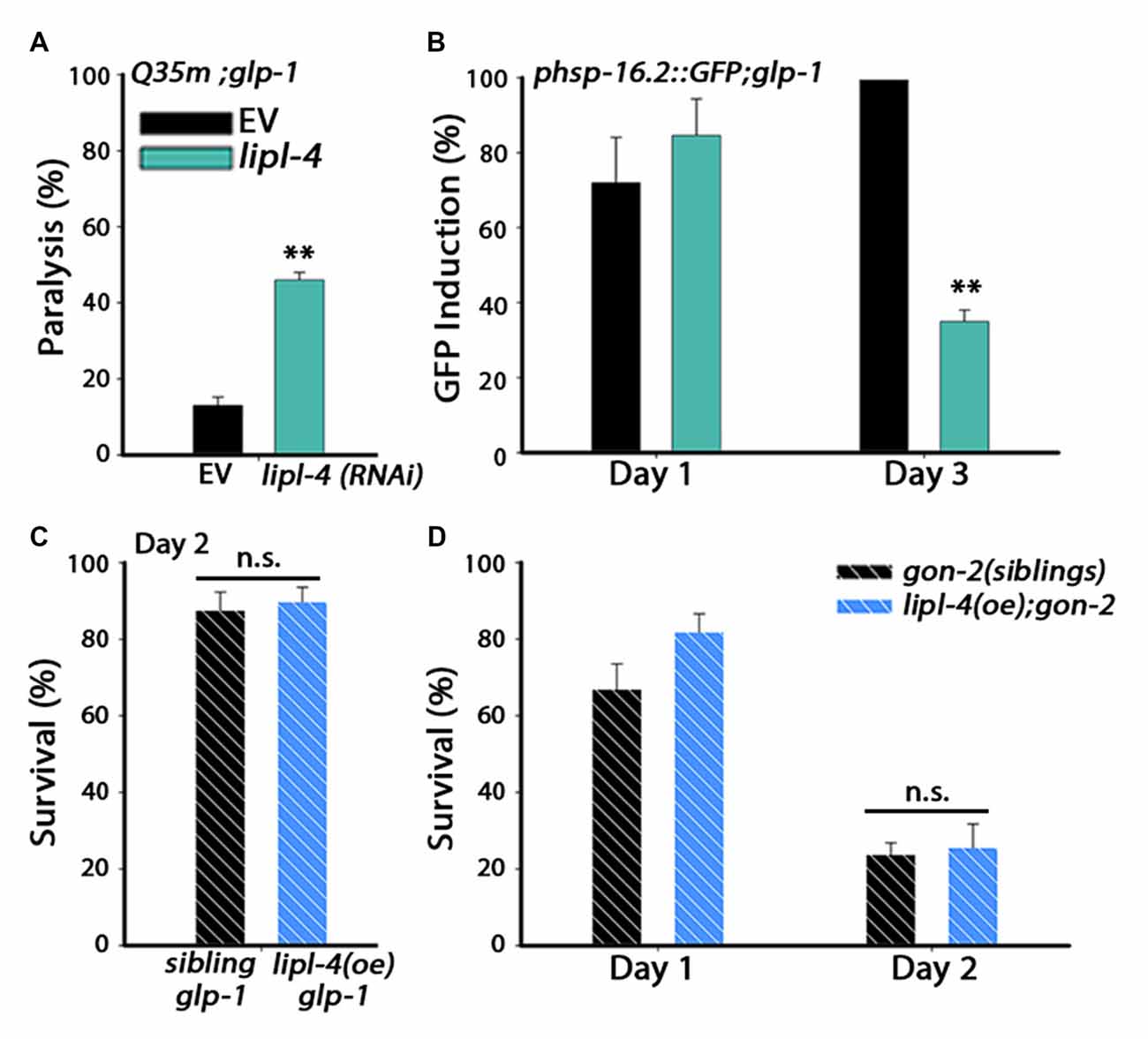
Figure 3. lipl-4-dependent rescue of somatic proteostasis requires the somatic gonad. (A) Motility was scored on day 6 of adulthood. Age-synchronized glp-1 animals expressing Q35m were treated with lipl-4 or empty vector (EV) control RNAi and the percentage of paralyzed animals was determined. (B) Heat shock gene induction was examined. Age-synchronized glp-1 animals expressing phsp-16.2::GFP were treated with lipl-4 or EV control RNAi and subjected to heat shock (90 min at 37°C) on day 1 and day 3 of adulthood. The percentage of animals showing a fluorescent signal in the gut was scored. (C) Thermo-resistance was examined in age-synchronized glp-1;lipl-4(oe) animals and their siblings. Animals were subjected to heat shock (6 h at 37°C) on day 2 of adulthood and survival was assayed. (D) Thermo-resistance was examined in age-synchronized gon-2;lipl-4(oe) animals and their siblings. Animals were subjected to heat shock (6 h at 37°C) on day 1 and day 2 of adulthoods and survival was assayed. For (A,B) data was compared to EV control. For (C,D) data was compared to age-matched sibling animals examined under the same condition. (n.s.) no statistical significance, **denotes P < 0.01.
LIPL-4 is expressed in the intestine, where it regulates DAF-16 function (Wang et al., 2008; Lapierre et al., 2011; Antebi, 2012; Folick et al., 2015). To determine whether the effects of lipl-4 on somatic proteostasis require the reproductive system, gonadogenesis-defective gon-2(q388), (gon-2) mutant animals were crossed with lipl-4(oe)-expressing animals and their ability to survive stress in adulthood was examined. Although lipl-4(oe) animals maintained high heat shock survival rates (Figure 2B), lipl-4(oe);gon-2 animals lost the ability to mount an effective heat shock response by day 2 of adulthood and their survival rates sharply declined from 82 ± 7% to 25.5 ± 6% (Figure 3D), similar to what was seen with the wild type (Figure 2B) and non-transgenic gon-2 animals (70 ± 7% to 24 ± 7%, Figure 3D). These data indicate that lipl-4-dependent rescue of somatic proteostasis required the gonad.
Over-Expression of lipl-4 Disrupted Reproduction
We established that lipl-4(oe) could modulate proteostasis and alleviate the protein damage associated with age-dependent misfolding, and that such rescue required the gonad. We next asked whether there is lipl-4(oe)-dependent impact on reproduction. lipl-4(oe) animals were smaller than their siblings but were not developmentally delayed. Therefore, we first examined the effect of lipl-4(oe) on fecundity by determining the number of progeny lipl-4(oe) and their siblings produced. Notably, lipl-4(oe) had ~40% less offspring than did their siblings (127 ± 16 and 211 ± 12 offspring, respectively; p < 0.0001; Figure 4A). This strong reduction in progeny number was not due to arrest of germline proliferation, since lipl-4(oe) and their siblings contained proliferating germ cells (Supplementary Figure S4).
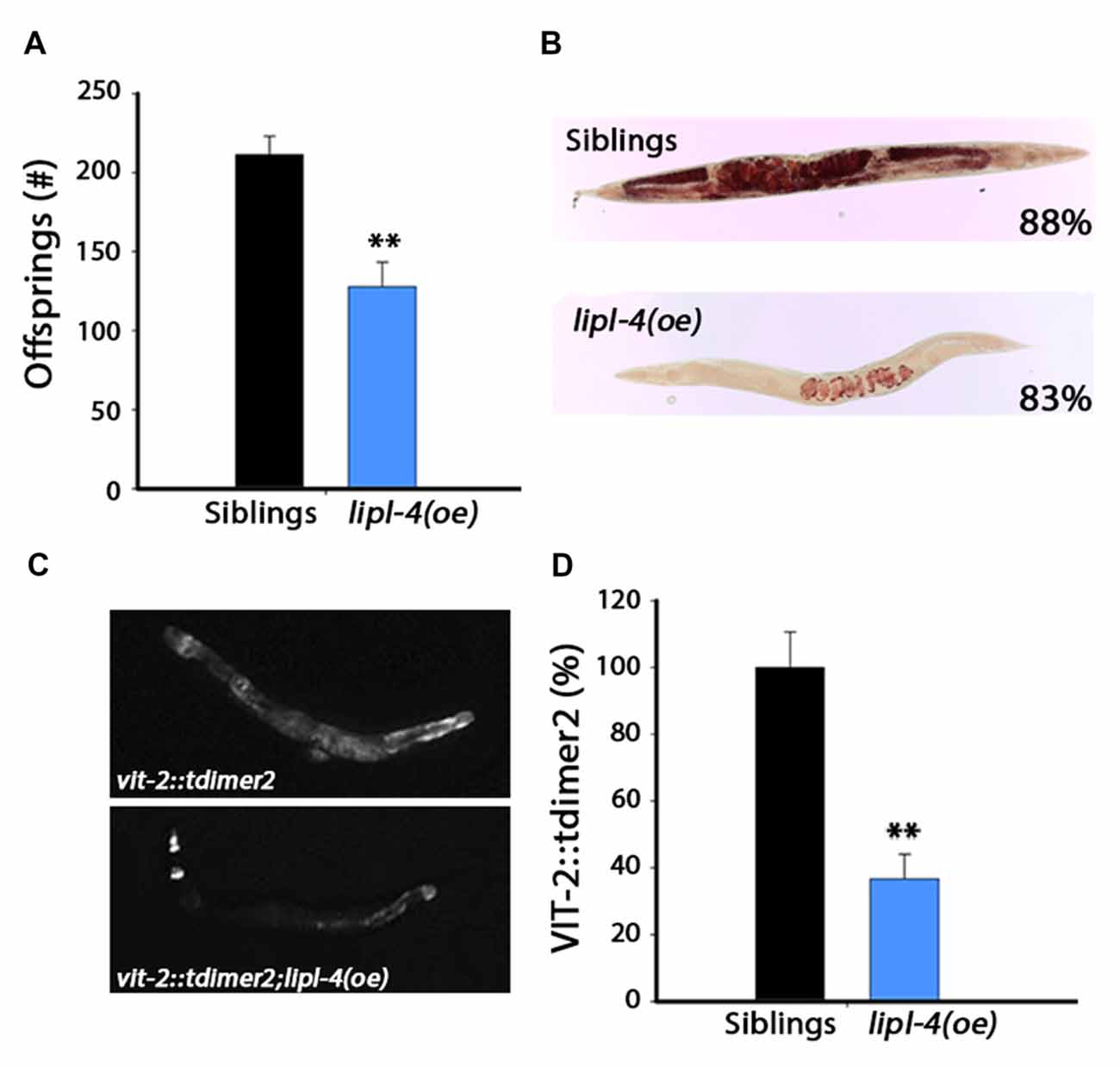
Figure 4. Over-expression of lipl-4 disrupts reproduction. (A) Progeny numbers were scored in age-synchronized lipl-4(oe) animals and their siblings. (B) Representative images of total fat stores. Age-synchronized lipl-4(oe) animals and their siblings were collected on day 2 of adulthood and total fat stores were analyzed using Oil-Red-O staining. Values indicate the percentage of animals showing the presented staining pattern (n > 18). (C) Representative images of age-synchronized lipl-4(oe) animals and their siblings carrying vit-2::tdimer2 on day 2 of adulthood. (D) VIT-2::tdimer2 levels were quantified from images using the ImageJ software and normalized to siblings levels. Data was compared to age-matched sibling animals examined under the same condition. **Denotes P < 0.01.
The gonadal longevity cascade modulates fatty acids metabolism and affects the levels of various fatty acids and their distribution in the nematode body (Wang et al., 2008; Goudeau et al., 2011; Lapierre et al., 2011; McCormick et al., 2012; Lapierre et al., 2013; Ratnappan et al., 2014; Steinbaugh et al., 2015). For example, when germline proliferation is inhibited, fatty acids accumulate in the body cavity, where they activate the transcription of genes required for fatty acid mobilization (Lynn et al., 2015; Steinbaugh et al., 2015). To ask how lipl-4(oe) could negatively impact reproduction, we first monitored total fat stores in lipl-4(oe) animals and their siblings using Oil-Red-O staining. Most of the lipl-4(oe) animals (83%) showed a strong reduction in lipid levels, with lipids being mostly located in the developing oocytes (Figure 4B and Supplementary Figure S5).
Levels of vitellogenic proteins are also affected by reproductive signals (DePina et al., 2011). Moreover, over-expression of vitellogenic proteins was previously shown to reduce lipl-4 levels (Seah et al., 2016). To further elucidate the effect of LIPL-4 on reproduction, we next examined transport of lipids from the intestine to the germline by monitoring the levels and localization of the vitellogenic protein VIT-2 tagged with tdimer2 (VIT-2::tdimer2). Coinciding with diminished fat stores, VIT-2 levels were reduced by ~60% in lipl-4(oe) animals, as compared to their siblings, on the day 2 of adulthood (Figures 4C,D). Fatty acid uptake by oocytes was, therefore, disrupted in lipl-4(oe) animals, which could, in turn, impact progeny production. Thus, while enhanced lipl-4 levels improved somatic proteostasis, they were detrimental to reproduction.
AA Uncouples Somatic Proteostasis from the Reproductive System
Over-expression of lipl-4 did not uncouple the trade-off between somatic maintenance and reproduction. Metabolite and lipid profiling of lipl-4(oe) identified accumulation of several long-chain fatty acids, including OEA, EPA, DGLA and AA (O’Rourke et al., 2013; Folick et al., 2015). While DGLA causes GSC death and complete sterility, EPA does not affect fertility (Watts and Browse, 2006). We, therefore, asked whether modulating lipl-4(oe) downstream products could potentially improve proteostasis without disrupting reproduction. We first examined whether diet supplementation of OEA, EPA or AA could regulate heat shock survival, as a simple readout of proteostasis. Embryos were transferred to fatty acid-supplemented or control plates (containing NP40) and their survival was examined on day 2 of adulthood. Diet supplementation of OEA or EPA had no significant effect on animal survival following heat shock on day 2 of adulthood (Supplementary Figure S6A). In contrast, when non-transgenic siblings were supplemented with AA, heat shock survival rates were significantly improved on day 2 of adulthood, as compared to siblings grown on control plates (56 + 5% and 25 + 3%, respectively; Figure 5A). Diet supplementation of AA to lipl-4(oe) animals did not further improve survival, as compared to lipl-4(oe) animals grown on control plates (65 + 4% and 63 ± 4%, respectively). Likewise, the percentage of sibling animals showing induction of hsp-16.2-dependent GFP upon heat shock was increased in animals supplemented with AA, as compared to controls. However, diet supplementation of AA to lipl-4(oe) animals did not further increase hsp-16.2-dependent GFP expression following heat shock (Supplementary Figure S6B). This was not due to AA-dependent increases in lipl-4 levels, as diet supplementation of AA did not affect lipl-4 mRNA levels (Supplementary Figure S7). These data thus support a role for AA in proteostasis remodeling.
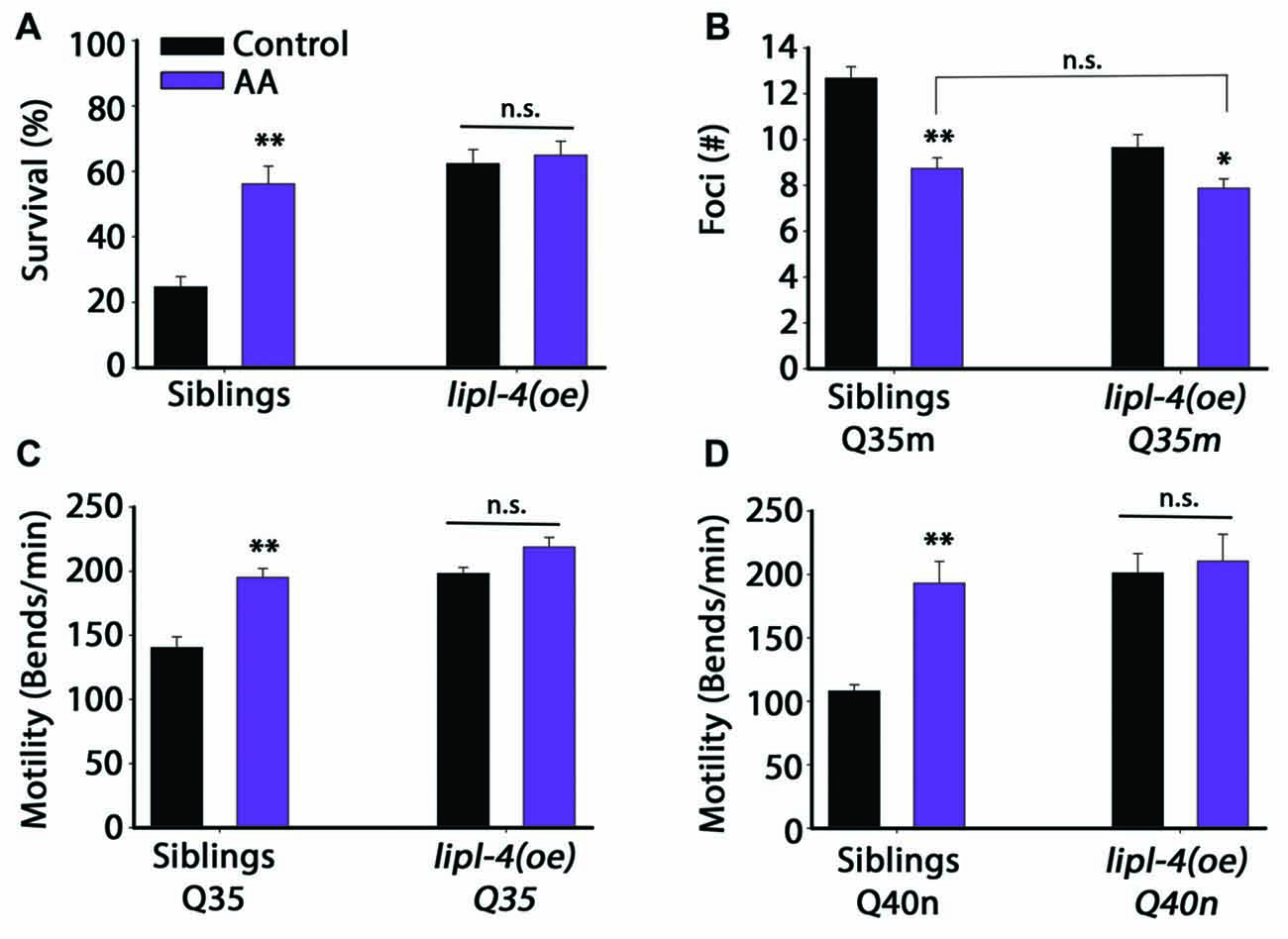
Figure 5. Diet supplementation of arachidonic acid (AA) could mimic the effects of lipl-4(oe) on proteostasis. (A) Thermo-resistance was examined in age-synchronized lipl-4(oe) animals and their siblings. Animals were grown on control (NP40) or AA-supplemented nematode growth medium (NGM) plates, subjected to heat shock (6 h at 37°C) and survival was assayed on day 2 of adulthood. (B) The number of bright foci was scored in age-synchronized Q35m;lipl-4(oe) animals and their siblings (n > 40). Animals were grown on control (NP40) or AA-supplemented NGM plates and the number of foci was counted on day 2 of adulthood. (C,D) Motility was scored in age-synchronized Q35m;lipl-4(oe), Q40n;lipl-4(oe) animals and their siblings. Animals were grown on control (NP40) or AA-supplemented NGM plates and the number of body bends per minute was scored on day 2 of adulthood. Data was compared to control plates (NP40). (n.s.) no statistical significance, *denotes P < 0.05, **denotes P < 0.01.
We then tested whether diet supplementation of AA could affect Q35m aggregation. A ~30% reduction in foci number was observed in Q35m siblings supplemented with AA. Whereas only a slight reduction in foci formation was observed in Q35m;lipl-4(oe) animals supplemented with AA and this reduction was not significantly different from Q35m siblings supplemented with AA (Figure 5B). Similar behavior was observed for Q35m- and Q40n-associated toxicity. Q35m;lipl-4(oe), Q40n;lipl-4(oe) animals and their siblings were grown on control or AA-supplemented plates and motility was monitored. Diet supplementation of AA to siblings expressing Q35m or Q40n improved their thrashing rates by 1.4- and 1.8-fold, respectively, compared to animals grown on control plates. In contrast, diet supplementation of AA did not further improve the motility of Q35m;lipl-4(oe) or Q40n;lipl-4(oe) animals (Figures 5C,D). Thus, diet supplementation with AA mimics the effects of lipl-4(oe) on proteostasis, suggesting that the increased AA level, observed in lipl-4(oe) animals is sufficient to remodel proteostasis in adulthood, likely via agents downstream of LIPL-4.
Given that AA mimics the effects of lipl-4(oe) on somatic proteostasis, we next asked whether AA supplementation has a negative impact on the reproductive system, similar to lipl-4(oe). For this, we examined the effect of AA on progeny production by lipl-4(oe) animals and their siblings. A previous study showed that only accumulation of high levels (>20% of their lipids) of AA affected fertility (Watts and Browse, 2006). We, therefore, asked whether diet supplementation of AA at a concentration that improved proteostasis (0.05 mM AA) affected reproduction. AA diet supplementation had no significant effect on the number of progeny produced by lipl-4(oe) siblings. Specifically, animals grown on control plates produced 210 ± 14 offspring, while AA-supplemented animals produced 196 ± 13 offspring. Likewise, lipl-4(oe) animals grown on control or AA produced similar numbers of offspring (160 ± 12 and 145 ± 10, respectively; Figure 6A). Moreover, diet supplementation of AA to lipl-4(oe) animals or their siblings did not affect fatty acid levels or localization, as monitored by Oil-Red-O staining or VIT-2 levels. Most siblings grown on either AA-supplemented or control plates (88%) showed normal fatty acid stores, while lipl-4(oe) animals showed low fatty acid levels localized in oocytes, regardless of diet (Figures 4B, 6B). Moreover, AA supplementation to wild type animals did not affect VIT-2 levels (Supplementary Figure S8). These data indicate that diet supplementation of AA can uncouple somatic proteostasis from the reproductive system.
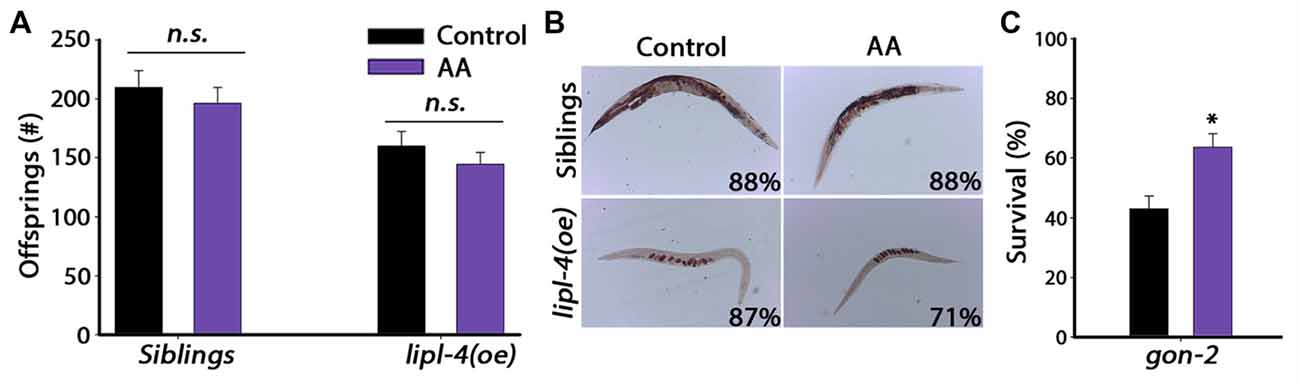
Figure 6. Reproduction is unaffected by diet supplementation of AA. (A) Progeny numbers were scored in age-synchronized lipl-4(oe) animals and their siblings grown on control (NP40) or AA-supplemented NGM plates. (B) Representative images of total fat stores. Age-synchronized lipl-4(oe) animals and their siblings were grown on control (NP40) or AA-supplemented NGM plates and collected on day 2 of adulthood. Total fat stores were analyzed using Oil-Red-O staining. Values indicate the percentage of animals showing the presented staining pattern (n > 14). (C) Thermo-resistance was examined in age-synchronized gon-2 animals. Animals grown on control (NP40) or AA-supplemented NGM plates were subjected to heat shock (6 h at 37°C) on day 2 of adulthood and survival was assayed. Data was compared to control plates (NP40). (n.s.) no statistical significance, *denotes P < 0.05.
Any benefits derived from AA supplementation in the soma came with no apparent cost to reproduction. We, therefore, asked whether AA-dependent rescue of proteostasis required the reproductive system. To test this directly, we examined the effect of AA supplementation on gonad-less animals by monitoring heat shock survival of gon-2 animals grown on control or AA-supplemented plates. While the survival of gon-2 animals grown on control plates was low on day 2 of adulthood (43 ± 4%; Shemesh et al., 2013), survival rates of gon-2 animals grown on AA-supplemented plates were surprisingly high (63.7 ± 4.4%, Figure 6C), similar to AA-supplemented siblings (Figure 5A). Thus, as opposed to lipl-4, the impact of AA on somatic proteostasis does not require the reproductive system. Rather, a small increase in AA levels was sufficient to improved proteostasis in adulthood with no apparent cost to reproduction.
Discussion
Complex signals control the proteostatic switch employed at the onset of reproduction to connect somatic maintenance to the reproductive system (Antebi, 2012). In wild type animals, the onset of egg laying is linked to a sharp decline in the ability to activate stress responses and maintain proteostasis mediated by signals from the gonad (Ben-Zvi et al., 2009; Liu et al., 2011; Taylor and Dillin, 2013; Labbadia and Morimoto, 2015b; Walther et al., 2015). The inhibition of germline proliferation, however, activates the gonadal longevity pathway, resulting in enhanced somatic proteostasis (Shemesh et al., 2013). Here, we showed that this proteostatic switch can be activated upon over-expression of LIPL-4 or AA enrichment, both of which act downstream of the gonadal longevity cascade (Wang et al., 2008; Lapierre et al., 2011; O’Rourke et al., 2013). We propose that the observed decline in lipl-4 levels upon transition to adulthood is associated with reduced proteostatic capacity but with improved progeny production (Figure 7A). Over-expression of lipl-4 thus restores somatic proteostasis, yet disrupts fatty acid accumulation in growing oocytes and thus, progeny production (Figure 7B). Because AA diet supplementation resulted in improved somatic proteostasis but did not affect or require the reproductive system, we suggest that the adverse effects of LIPL-4 function on reproduction can be bypassed and that its beneficial effects on somatic proteostasis can be retained by increased AA levels (Figure 7C). The somatic proteostasis rescue mediated by AA could thus delay the onset of toxicity and aggregation in C. elegans models of polyQ diseases without cost to reproduction.
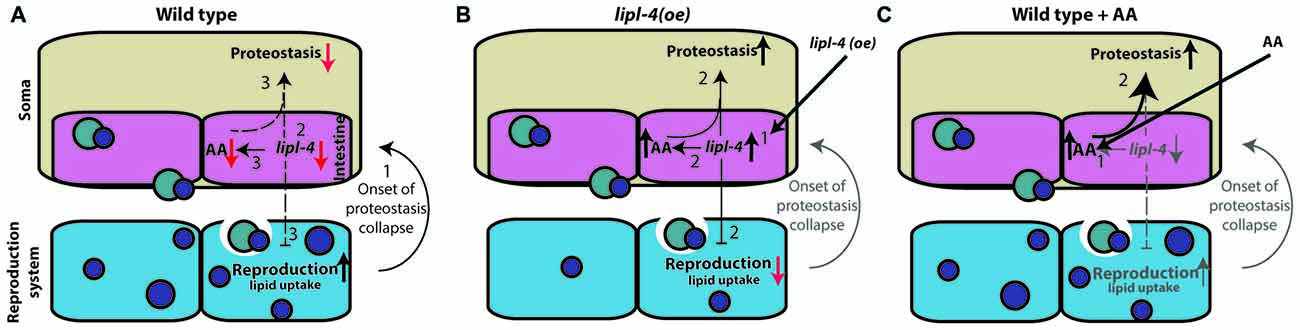
Figure 7. Signals from the reproductive system regulate somatic proteostasis. (A) The onset of reproduction regulates proteostasis cell-nonautonomously (1). This activates the gonadal longevity pathway that down regulates lipl-4 expression at the onset to reproduction (2). When embryo production begins proteostasis sharply declines. This is associated with decreased lipl-4 and AA levels (3). (B) Over-expression of lipl-4 (1) restores somatic proteostasis and enhances AA levels but also disrupts fatty acid mobilization to developing oocytes and thus disrupt reproduction (2). (C) Diet supplementation of AA (1) restores somatic proteostasis (2), like lipl-4 over-expression, but did not disrupt fatty acid mobilization or reproduction.
LIPL-4 has Inverse Effects on Reproduction and Somatic Maintenance
The antagonistic pleiotropy theory suggests that genes that have beneficial effects early in life could be selected for, even if they have detrimental effects later in life (Williams, 1957). Given how LIPL-4 has inverse effects on progeny production and proteostasis, we propose that lipl-4 represents such a gene, as elevated levels of lipl-4, although beneficial to the organism later in life, reduce reproductive success. The actions of LIPL-4 resulted in a trade-off between somatic maintenance and reproduction (Kirkwood, 2005). However, LIPL-4 function activates several different pathways that differ in terms of their effects on somatic proteostasis and reproduction. For example, lipl-4(oe) results in elevated levels of OEA that, in turn, activated NHR-49 and NHR-80. OEA did not affect proteostasis. However, NHR-49 and NHR-80 inversely regulated lipl-4 and vitellogenic gene expression (Seah et al., 2016; Figure 4), suggesting that OEA could regulate lipid resource allocation from yolk-bound lipoprotein to intestinal lipid droplets. In contrast, elevated levels of AA did not affect lipid distribution (Figure 6B) but rather were shown to induce autophagy in C. elegans and mammalian cells and extend C. elegans lifespan (O’Rourke et al., 2013). Moreover, lipl-4(oe) also resulted in a significant increase in DGLA (Folick et al., 2015). While DGLA, like AA, can induce autophagy and increase lifespan (O’Rourke et al., 2013), diet supplementation of DGLA resulted in a complete loss of germ cells and sterility, mediated by DGLA-derived epoxides (Watts and Browse, 2006; Deline et al., 2015). Finally, EPA did not affect proteostasis, autophagy, lifespan or reproduction in C. elegans (Watts and Browse, 2006; O’Rourke et al., 2013). Thus, the pleiotropic effects of LIPL-4 on reproduction and somatic maintenance are due to activation of different downstream pathways. Targeting metabolites that affect proteostasis but that have no or only mild effects on reproduction could artificially uncouple the two processes.
AA-Modulated Proteostasis Collapse
How does LIPL-4 impact proteostasis? Diet supplementation of AA did not affect lipid distribution (Figure 6B) but was sufficient to recapitulate the impact of lipl-4(oe) on proteostasis (Figure 5 and Supplementary Figure S6B). Thus, although redistribution of lipids could activate several transcriptional programs that affect stress response pathways (Steinbaugh et al., 2015), it is less likely that lipid redistribution impacts somatic proteostasis. Accumulation of polyunsaturated fatty acids, such AA and DGLA, induce autophagy. Such induction itself could enhance proteostasis, for example, by removal of protein aggregates (Vilchez et al., 2014). However, AA could potentially activate other somatic maintenance pathways. For instance, exposure of HeLa cells to AA was shown to activate HSF1 and induce the expression of heat shock genes (Jurivich et al., 1994; Horikawa and Sakamoto, 2009; Balogh et al., 2013). Moreover, AA could be further metabolized to eicosanoids, including various prostaglandins that could also induce the heat shock response and affect the expression of heat shock genes in human cells (Amici et al., 1992; Balogh et al., 2013). Thus, AA supplementation has the potential to modulate proteostasis, a function that could also be conserved in humans. Determining when AA is required for proteostasis remodeling might help us elucidate the function this molecule serves.
Regulation of the proteostatic switch has a strong impact on the maintenance of somatic tissues (Shai et al., 2014). Given that this switch can impact the onset of protein aggregation, our findings could have consequences for the treatment of neurodegenerative diseases. The question, however, remains as to whether this proteostatic switch is conserved. While it is clear that proteostasis is limited in adults across species, currently there are only indications that the proteostatic switch itself is conserved (Shai et al., 2014; Labbadia and Morimoto, 2015b). These include the impact of the reproductive system on aging in flies and mice and the conservation of many of the players in these signaling pathways, including LIPL-4 (Flatt et al., 2008; Mason et al., 2009; Shai et al., 2014; Folick et al., 2015; Labbadia and Morimoto, 2015b). By finding ways to uncouple proteostatic collapse from the reproductive system, we might be able to target multiple age-dependent protein misfolding diseases with different etiologies but with similar underlying biology.
Author Contributions
NShemesh and AB-Z designed the experiments and wrote the manuscript. NShemesh, NShai, LM and RK performed the experiments, analyzed the data and interpreted the results. NShemesh, NShai, LM, RK and AB-Z revised the text.
Funding
This research was supported by a grant from the Legacy Heritage Biomedical Science Partnership Program of the Israel Science Foundation (grant No. 804/13; https://www.isf.org.il/#/) and by a grant from the Binational Science Foundation (grant No. 2009187; http://www.bsf.org.il). NShemesh was supported by Kreitman Negev scholarship and a Kreitman short-term post-doctoral scholarship.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fnmol.2017.00101/full#supplementary-material
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Some nematode strains used in this work were provided by the Caenorhabditis Genetics Center, which is funded by the NIH National Center for Research Resources (NCRR).
References
Amici, C., Sistonen, L., Santoro, M. G., and Morimoto, R. I. (1992). Antiproliferative prostaglandins activate heat shock transcription factor. Proc. Natl. Acad. Sci. U S A 89, 6227–6231. doi: 10.1073/pnas.89.14.6227
Antebi, A. (2012). Regulation of longevity by the reproductive system. Exp. Gerontol. 48, 596–602. doi: 10.1016/j.exger.2012.09.009
Balogh, G., Péter, M., Glatz, A., Gombos, I., Török, Z., Horváth, I., et al. (2013). Key role of lipids in heat stress management. FEBS Lett. 587, 1970–1980. doi: 10.1016/j.febslet.2013.05.016
Bar-Lavan, Y., Shemesh, N., Dror, S., Ofir, R., Yeger-Lotem, E., and Ben-Zvi, A. (2016). A differentiation transcription factor establishes muscle-specific proteostasis in Caenorhabditis elegans. PLoS Genet. 12:e1006531. doi: 10.1371/journal.pgen.1006531
Bennett, E. J., Shaler, T. A., Woodman, B., Ryu, K. Y., Zaitseva, T. S., Becker, C. H., et al. (2007). Global changes to the ubiquitin system in Huntington’s disease. Nature 448, 704–708. doi: 10.1038/nature06022
Ben-Zvi, A., Miller, E. A., and Morimoto, R. I. (2009). Collapse of proteostasis represents an early molecular event in Caenorhabditis elegans aging. Proc. Natl. Acad. Sci. U S A 106, 14914–14919. doi: 10.1073/pnas.0902882106
Berman, J. R., and Kenyon, C. (2006). Germ-cell loss extends C. elegans life span through regulation of DAF-16 by kri-1 and lipophilic-hormone signaling. Cell 124, 1055–1068. doi: 10.1016/j.cell.2006.01.039
Bilen, J., and Bonini, N. M. (2007). Genome-wide screen for modifiers of ataxin-3 neurodegeneration in Drosophila. PLoS Genet. 3, 1950–1964. doi: 10.1371/journal.pgen.0030177
Blair, L. J., Nordhues, B. A., Hill, S. E., Scaglione, K. M., O’Leary, J. C. III., Fontaine, S. N., et al. (2013). Accelerated neurodegeneration through chaperone-mediated oligomerization of tau. J. Clin. Invest. 123, 4158–4169. doi: 10.1172/jci69003
Brignull, H. R., Moore, F. E., Tang, S. J., and Morimoto, R. I. (2006). Polyglutamine proteins at the pathogenic threshold display neuron-specific aggregation in a pan-neuronal Caenorhabditis elegans model. J. Neurosci. 26, 7597–7606. doi: 10.1523/JNEUROSCI.0990-06.2006
Cohen, E., Bieschke, J., Perciavalle, R. M., Kelly, J. W., and Dillin, A. (2006). Opposing activities protect against age-onset proteotoxicity. Science 313, 1604–1610. doi: 10.1126/science.1124646
Colaiácovo, M. P., MacQueen, A. J., Martinez-Perez, E., McDonald, K., Adamo, A., La Volpe, A., et al. (2003). Synaptonemal complex assembly in C. elegans is dispensable for loading strand-exchange proteins but critical for proper completion of recombination. Dev. Cell 5, 463–474. doi: 10.1016/s1534-5807(03)00232-6
Davies, S. W., Turmaine, M., Cozens, B. A., DiFiglia, M., Sharp, A. H., Ross, C. A., et al. (1997). Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell 90, 537–548. doi: 10.1016/s0092-8674(00)80513-9
Deline, M., Keller, J., Rothe, M., Schunck, W. H., Menzel, R., and Watts, J. L. (2015). Epoxides derived from dietary dihomo-gamma-linolenic acid induce germ cell death in C. elegans. Sci. Rep. 5:15417. doi: 10.1038/srep15417
Deline, M. L., Vrablik, T. L., and Watts, J. L. (2013). Dietary supplementation of polyunsaturated fatty acids in Caenorhabditis elegans. J. Vis. Exp. 81:e50879. doi: 10.3791/50879
DePina, A. S., Iser, W. B., Park, S.-S., Maudsley, S., Wilson, M. A., and Wolkow, C. A. (2011). Regulation of Caenorhabditis elegans vitellogenesis by DAF-2/IIS through separable transcriptional and posttranscriptional mechanisms. BMC Physiol. 11:11. doi: 10.1186/1472-6793-11-11
Eremenko, E., Ben-Zvi, A., Morozova-Roche, L. A., and Raveh, D. (2013). Aggregation of human S100A8 and S100A9 amyloidogenic proteins perturbs proteostasis in a yeast model. PLoS One 8:e58218. doi: 10.1371/journal.pone.0058218
Feldman, N., Kosolapov, L., and Ben-Zvi, A. (2014). Fluorodeoxyuridine improves Caenorhabditis elegans proteostasis independent of reproduction onset. PLoS One 9:e85964. doi: 10.1371/journal.pone.0085964
Flatt, T., Min, K. J., D’Alterio, C., Villa-Cuesta, E., Cumbers, J., Lehmann, R., et al. (2008). Drosophila germ-line modulation of insulin signaling and lifespan. Proc. Natl. Acad. Sci. U S A 105, 6368–6373. doi: 10.1073/pnas.0709128105
Folick, A., Oakley, H. D., Yu, Y., Armstrong, E. H., Kumari, M., Sanor, L., et al. (2015). Aging. Lysosomal signaling molecules regulate longevity in Caenorhabditis elegans. Science 347, 83–86. doi: 10.1126/science.1258857
Frumkin, A., Dror, S., Pokrzywa, W., Bar-Lavan, Y., Karady, I., Hoppe, T., et al. (2014). Challenging muscle homeostasis uncovers novel chaperone interactions in Caenorhabditis elegans. Front. Mol. Biosci. 1:21. doi: 10.3389/fmolb.2014.00021
Gidalevitz, T., Ben-Zvi, A., Ho, K. H., Brignull, H. R., and Morimoto, R. I. (2006). Progressive disruption of cellular protein folding in models of polyglutamine diseases. Science 311, 1471–1474. doi: 10.1126/science.1124514
Gidalevitz, T., Krupinski, T., Garcia, S. M., and Morimoto, R. I. (2009). Destabilizing protein polymorphisms in the genetic background direct phenotypic expression of mutant SOD1 toxicity. PLoS Genet. 5:e1000399. doi: 10.1371/journal.pgen.1000399
Glabe, C. G., and Kayed, R. (2006). Common structure and toxic function of amyloid oligomers implies a common mechanism of pathogenesis. Neurology 66, S74–S78. doi: 10.1212/01.wnl.0000192103.24796.42
Goudeau, J., Bellemin, S., Toselli-Mollereau, E., Shamalnasab, M., Chen, Y., and Aguilaniu, H. (2011). Fatty acid desaturation links germ cell loss to longevity through NHR-80/HNF4 in C. elegans. PLoS Biol. 9:e1000599. doi: 10.1371/journal.pbio.1000599
Guisbert, E., Czyz, D. M., Richter, K., McMullen, P. D., and Morimoto, R. I. (2013). Identification of a tissue-selective heat shock response regulatory network. PLoS Genet. 9:e1003466. doi: 10.1371/journal.pgen.1003466
Horikawa, M., and Sakamoto, K. (2009). Fatty-acid metabolism is involved in stress-resistance mechanisms of Caenorhabditis elegans. Biochem. Biophys. Res. Commun. 390, 1402–1407. doi: 10.1016/j.bbrc.2009.11.006
Hsin, H., and Kenyon, C. (1999). Signals from the reproductive system regulate the lifespan of C. elegans. Nature 399, 362–366. doi: 10.1038/20694
Johnston, J. A., Dalton, M. J., Gurney, M. E., and Kopito, R. R. (2000). Formation of high molecular weight complexes of mutant Cu, Zn-superoxide dismutase in a mouse model for familial amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. U S A 97, 12571–12576. doi: 10.1073/pnas.220417997
Jurivich, D. A., Sistonen, L., Sarge, K. D., and Morimoto, R. I. (1994). Arachidonate is a potent modulator of human heat shock gene transcription. Proc. Natl. Acad. Sci. U S A 91, 2280–2284. doi: 10.1073/pnas.91.6.2280
Karady, I., Frumkin, A., Dror, S., Shemesh, N., Shai, N., and Ben-Zvi, A. (2013). Using Caenorhabditis elegans as a model system to study protein homeostasis in a multicellular organism. J. Vis. Exp. 82:e50840. doi: 10.3791/50840
Kim, S., Nollen, E. A., Kitagawa, K., Bindokas, V. P., and Morimoto, R. I. (2002). Polyglutamine protein aggregates are dynamic. Nat. Cell Biol. 4, 826–831. doi: 10.1038/ncb863
Kirkwood, T. B. (2005). Understanding the odd science of aging. Cell 120, 437–447. doi: 10.1016/j.cell.2005.01.027
Kitamura, A., Inada, N., Kubota, H., Matsumoto, G., Kinjo, M., Morimoto, R. I., et al. (2014). Dysregulation of the proteasome increases the toxicity of ALS-linked mutant SOD1. Genes Cells 19, 209–224. doi: 10.1111/gtc.12125
Klabonski, L., Zha, J., Senthilkumar, L., and Gidalevitz, T. (2016). A bystander mechanism explains the specific phenotype of a broadly expressed misfolded protein. PLoS Genet. 12:e1006450. doi: 10.1371/journal.pgen.1006450
Labbadia, J., and Morimoto, R. I. (2015a). The biology of proteostasis in aging and disease. Annu. Rev. Biochem. 84, 435–464. doi: 10.1146/annurev-biochem-060614-033955
Labbadia, J., and Morimoto, R. I. (2015b). Repression of the heat shock response is a programmed event at the onset of reproduction. Mol. Cell 59, 639–650. doi: 10.1016/j.molcel.2015.06.027
Lapierre, L. R., De Magalhaes Filho, C. D., McQuary, P. R., Chu, C. C., Visvikis, O., Chang, J. T., et al. (2013). The TFEB orthologue HLH-30 regulates autophagy and modulates longevity in Caenorhabditis elegans. Nat. Commun. 4:2267. doi: 10.1038/ncomms3267
Lapierre, L. R., Gelino, S., Meléndez, A., and Hansen, M. (2011). Autophagy and lipid metabolism coordinately modulate life span in germline-less C. elegans. Curr. Biol. 21, 1507–1514. doi: 10.1016/j.cub.2011.07.042
Lechler, M. C., Crawford, E. D., Groh, N., Widmaier, K., Jung, R., Kirstein, J., et al. (2017). Reduced insulin/IGF-1 signaling restores the dynamic properties of key stress granule proteins during aging. Cell Rep. 18, 454–467. doi: 10.1016/j.celrep.2016.12.033
Libina, N., Berman, J. R., and Kenyon, C. (2003). Tissue-specific activities of C. elegans DAF-16 in the regulation of lifespan. Cell 115, 489–502. doi: 10.1016/s0092-8674(03)00889-4
Liu, G., Rogers, J., Murphy, C. T., and Rongo, C. (2011). EGF signalling activates the ubiquitin proteasome system to modulate C. elegans lifespan. EMBO J. 30, 2990–3003. doi: 10.1038/emboj.2011.195
Lynn, D. A., Dalton, H. M., Sowa, J. N., Wang, M. C., Soukas, A. A., and Curran, S. P. (2015). Omega-3 and -6 fatty acids allocate somatic and germline lipids to ensure fitness during nutrient and oxidative stress in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U S A 112, 15378–15383. doi: 10.1073/pnas.1514012112
Mason, J. B., Cargill, S. L., Anderson, G. B., and Carey, J. R. (2009). Transplantation of young ovaries to old mice increased life span in transplant recipients. J. Gerontol. A Biol. Sci. Med. Sci. 64, 1207–1211. doi: 10.1093/gerona/glp134
McCormick, M., Chen, K., Ramaswamy, P., and Kenyon, C. (2012). New genes that extend Caenorhabditis elegans’ lifespan in response to reproductive signals. Aging Cell 11, 192–202. doi: 10.1111/j.1474-9726.2011.00768.x
Merkwirth, C., Jovaisaite, V., Durieux, J., Matilainen, O., Jordan, S. D., Quiros, P. M., et al. (2016). Two conserved histone demethylases regulate mitochondrial stress-induced longevity. Cell 165, 1209–1223. doi: 10.1016/j.cell.2016.04.012
Morley, J. F., Brignull, H. R., Weyers, J. J., and Morimoto, R. I. (2002). The threshold for polyglutamine-expansion protein aggregation and cellular toxicity is dynamic and influenced by aging in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U S A 99, 10417–10422. doi: 10.1073/pnas.152161099
Nussbaum, R. L., and Polymeropoulos, M. H. (1997). Genetics of Parkinson’s disease. Hum. Mol. Genet. 6, 1687–1691. doi: 10.1093/hmg/6.10.1687
Olzscha, H., Schermann, S. M., Woerner, A. C., Pinkert, S., Hecht, M. H., Tartaglia, G. G., et al. (2011). Amyloid-like aggregates sequester numerous metastable proteins with essential cellular functions. Cell 144, 67–78. doi: 10.1016/j.cell.2010.11.050
O’Rourke, E. J., Kuballa, P., Xavier, R., and Ruvkun, G. (2013). ω-6 polyunsaturated fatty acids extend life span through the activation of autophagy. Genes Dev. 27, 429–440. doi: 10.1101/gad.205294.112
O’Rourke, E. J., Soukas, A. A., Carr, C. E., and Ruvkun, G. (2009). C. elegans major fats are stored in vesicles distinct from lysosome-related organelles. Cell Metab. 10, 430–435. doi: 10.1016/j.cmet.2009.10.002
Orr, H. T., and Zoghbi, H. Y. (2007). Trinucleotide repeat disorders. Annu. Rev. Neurosci. 30, 575–621. doi: 10.1146/annurev.neuro.29.051605.113042
Ratnappan, R., Amrit, F. R., Chen, S. W., Gill, H., Holden, K., Ward, J., et al. (2014). Germline signals deploy NHR-49 to modulate fatty-acid β-oxidation and desaturation in somatic tissues of C. elegans. PLoS Genet. 10:e1004829. doi: 10.1371/journal.pgen.1004829
Seah, N. E., de Magalhaes Filho, C. D., Petrashen, A. P., Henderson, H. R., Laguer, J., Gonzalez, J., et al. (2016). Autophagy-mediated longevity is modulated by lipoprotein biogenesis. Autophagy 12, 261–272. doi: 10.1080/15548627.2015.1127464
Shai, N., Shemesh, N., and Ben-Zvi, A. (2014). Remodeling of proteostasis upon transition to adulthood is linked to reproduction onset. Curr. Genomics 15, 122–129. doi: 10.2174/1389202915666140221005023
Shemesh, N., Shai, N., and Ben-Zvi, A. (2013). Germline stem cell arrest inhibits the collapse of somatic proteostasis early in Caenorhabditis elegans adulthood. Aging Cell 12, 814–822. doi: 10.1111/acel.12110
Sherman, M. Y., and Muchowski, P. J. (2003). Making yeast tremble: yeast models as tools to study neurodegenerative disorders. Neuromolecular Med. 4, 133–146. doi: 10.1385/nmm:4:1-2:133
Steinbaugh, M. J., Narasimhan, S. D., Robida-Stubbs, S., Moronetti Mazzeo, L. E., Dreyfuss, J. M., Hourihan, J. M., et al. (2015). Lipid-mediated regulation of SKN-1/Nrf in response to germ cell absence. Elife 4:e07836. doi: 10.7554/eLife.07836
Suhr, S. T., Senut, M. C., Whitelegge, J. P., Faull, K. F., Cuizon, D. B., and Gage, F. H. (2001). Identities of sequestered proteins in aggregates from cells with induced polyglutamine expression. J. Cell Biol. 153, 283–294. doi: 10.1083/jcb.153.2.283
Taylor, R. C., and Dillin, A. (2011). Aging as an event of proteostasis collapse. Cold Spring Harb. Perspect. Biol. 3:a004440. doi: 10.1101/cshperspect.a004440
Taylor, R. C., and Dillin, A. (2013). XBP-1 is a cell-nonautonomous regulator of stress resistance and longevity. Cell 153, 1435–1447. doi: 10.1016/j.cell.2013.05.042
Tian, Y., Garcia, G., Bian, Q., Steffen, K. K., Joe, L., Wolff, S., et al. (2016). Mitochondrial stress induces chromatin reorganization to promote longevity and UPRmt. Cell 165, 1197–1208. doi: 10.1016/j.cell.2016.04.011
Tyedmers, J., Mogk, A., and Bukau, B. (2010). Cellular strategies for controlling protein aggregation. Nat. Rev. Mol. Cell Biol. 11, 777–788. doi: 10.1038/nrm2993
van Oosten-Hawle, P., Porter, R. S., and Morimoto, R. I. (2013). Regulation of organismal proteostasis by transcellular chaperone signaling. Cell 153, 1366–1378. doi: 10.1016/j.cell.2013.05.015
Venkatraman, P., Wetzel, R., Tanaka, M., Nukina, N., and Goldberg, A. L. (2004). Eukaryotic proteasomes cannot digest polyglutamine sequences and release them during degradation of polyglutamine-containing proteins. Mol. Cell 14, 95–104. doi: 10.1016/s1097-2765(04)00151-0
Vilchez, D., Morantte, I., Liu, Z., Douglas, P. M., Merkwirth, C., Rodrigues, A. P., et al. (2012). RPN-6 determines C. elegans longevity under proteotoxic stress conditions. Nature 489, 263–268. doi: 10.1038/nature11315
Vilchez, D., Saez, I., and Dillin, A. (2014). The role of protein clearance mechanisms in organismal ageing and age-related diseases. Nat. Commun. 5:5659. doi: 10.1038/ncomms6659
Voisine, C., and Hart, A. C. (2004). Caenorhabditis elegans as a model system for triplet repeat diseases. Methods Mol. Biol. 277, 141–160. doi: 10.1385/1-59259-804-8:141
Walther, D. M., Kasturi, P., Zheng, M., Pinkert, S., Vecchi, G., Ciryam, P., et al. (2015). Widespread proteome remodeling and aggregation in aging C. elegans. Cell 161, 919–932. doi: 10.1016/j.cell.2015.03.032
Wang, D., Hou, L., Nakamura, S., Su, M., Li, F., Chen, W., et al. (2017). LIN-28 balances longevity and germline stem cell number in Caenorhabditis elegans through let-7/AKT/DAF-16 axis. Aging Cell 16, 113–124. doi: 10.1111/acel.12539
Wang, M. C., O’Rourke, E. J., and Ruvkun, G. (2008). Fat metabolism links germline stem cells and longevity in C. elegans. Science 322, 957–960. doi: 10.1126/science.1162011
Watts, J. L., and Browse, J. (2006). Dietary manipulation implicates lipid signaling in the regulation of germ cell maintenance in C. elegans. Dev. Biol. 292, 381–392. doi: 10.1016/j.ydbio.2006.01.013
Williams, G. C. (1957). Pleiotropy, natural selection, and the evolution of senescence. Evolution 11, 398–411. doi: 10.2307/2406060
Yerbury, J. J., Ooi, L., Dillin, A., Saunders, D. N., Hatters, D. M., Beart, P. M., et al. (2016). Walking the tightrope: proteostasis and neurodegenerative disease. J. Neurochem. 137, 489–505. doi: 10.1111/jnc.13575
Yu, A., Shibata, Y., Shah, B., Calamini, B., Lo, D. C., and Morimoto, R. I. (2014). Protein aggregation can inhibit clathrin-mediated endocytosis by chaperone competition. Proc. Natl. Acad. Sci. U S A 111, E1481–E1490. doi: 10.1073/pnas.1321811111
Keywords: aging, arachidonic acid (AA), Caenorhabditis elegans, lipl-4, neurodegenerative diseases, proteostasis, polyglutamine (polyQ) diseases, reproduction
Citation: Shemesh N, Shai N, Meshnik L, Katalan R and Ben-Zvi A (2017) Uncoupling the Trade-Off between Somatic Proteostasis and Reproduction in Caenorhabditis elegans Models of Polyglutamine Diseases. Front. Mol. Neurosci. 10:101. doi: 10.3389/fnmol.2017.00101
Received: 05 February 2017; Accepted: 24 March 2017;
Published: 20 April 2017.
Edited by:
Serena Carra, University of Modena and Reggio Emilia, ItalyReviewed by:
Martin Lothar Duennwald, University of Western Ontario, CanadaPatricija Van Oosten-Hawle, University of Leeds, UK
Copyright © 2017 Shemesh, Shai, Meshnik, Katalan and Ben-Zvi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anat Ben-Zvi, YW5hdGJ6QGJndS5hYy5pbA==