 Paola Rusmini1†
Paola Rusmini1† Riccardo Cristofani1†
Riccardo Cristofani1† Mariarita Galbiati1
Mariarita Galbiati1 Maria E. Cicardi1Marco Meroni1Veronica Ferrari1Giulia Vezzoli1Barbara Tedesco1Elio Messi1Margherita Piccolella1
Maria E. Cicardi1Marco Meroni1Veronica Ferrari1Giulia Vezzoli1Barbara Tedesco1Elio Messi1Margherita Piccolella1 Serena Carra2
Serena Carra2 Valeria Crippa1,3†
Valeria Crippa1,3† Angelo Poletti1,4*†
Angelo Poletti1,4*†- 1Dipartimento di Scienze Farmacologiche e Biomolecolari (DiSFeB), Centro di Eccellenza sulle Malattie Neurodegenerative, Università degli Studi di Milano, Milano, Italy
- 2Dipartimento di Scienze Biomediche, Metaboliche e Neuroscienze, Università di Modena e Reggio Emilia, Modena, Italy
- 3C. Mondino National Neurological Institute, Pavia, Italy
- 4Centro Interuniversitario sulle Malattie Neurodegenerative, Università degli Studi di Firenze, Roma Tor Vergata, Milano, Italy
Amyotrophic lateral sclerosis (ALS) and spinal and bulbar muscular atrophy (SBMA) are two motoneuron diseases (MNDs) characterized by aberrant protein behavior in affected cells. In familial ALS (fALS) and in SBMA specific gene mutations lead to the production of neurotoxic proteins or peptides prone to misfold, which then accumulate in form of aggregates. Notably, some of these proteins accumulate into aggregates also in sporadic ALS (sALS) even if not mutated. To prevent proteotoxic stresses detrimental to cells, misfolded and/or aggregated proteins must be rapidly removed by the protein quality control (PQC) system. The small heat shock protein B8 (HSPB8) is a chaperone induced by harmful events, like proteasome inhibition. HSPB8 is expressed both in motoneuron and muscle cells, which are both targets of misfolded protein toxicity in MNDs. In ALS mice models, in presence of the mutant proteins, HSPB8 is upregulated both in spinal cord and muscle. HSPB8 interacts with the HSP70 co-chaperone BAG3 and enhances the degradation of misfolded proteins linked to sALS, or causative of fALS and of SBMA. HSPB8 acts by facilitating autophagy, thereby preventing misfolded protein accumulation in affected cells. BAG3 and BAG1 compete for HSP70-bound clients and target them for disposal to the autophagy or proteasome, respectively. Enhancing the selective targeting of misfolded proteins by HSPB8-BAG3-HSP70 to autophagy may also decrease their delivery to the proteasome by the BAG1-HSP70 complex, thereby limiting possible proteasome overwhelming. Thus, approaches aimed at potentiating HSPB8-BAG3 may contribute to the maintenance of proteostasis and may delay MNDs progression.
Introduction
Motoneuron diseases (MNDs) are neurodegenerative diseases (NDs) in which cortical and/or spinal motoneurons are affected. They appear in sporadic or familial forms; little is known on alterations inducing sporadic MNDs, while specific gene mutations are responsible for altered RNA or protein functions in familial MNDs pathogenesis. Mutations may affect RNA/protein synthesis or activity (loss-of-function) or induce neurotoxicity (gain-of-functions). Amyotrophic lateral sclerosis (ALS) and spinal and bulbar muscular atrophy (SBMA) are MNDs mainly associated with gain-of-functions in proteins which become resistant to folding or conformationally unstable, leading to unfolding/misfolding. Misfolded proteins are prone to aggregate and neurotoxic impairing several cellular functions causing cell death. To prevent misfolded proteins toxicity, cells activate a protein quality control (PQC) system, which surveys protein folding and clears damaged substrates. The PQC system is crucial to counteract the neurotoxic events triggered by misfolded proteins and thus should be considered as a potential target for therapeutic intervention to ameliorate MNDs course.
The Protein Quality Control System
The PQC system is composed of molecular chaperones and degradative pathways.
Chaperones, like the heat shock proteins (HSPs), are often constitutively expressed, but also over-induced by different cell stresses (Morimoto, 2006), and include more than 150 members (grouped on the basis of their size (small HSPs, HSP40s, HSP60s, HSP70s, HSP90s and HSP100), structure and function; Kampinga and Craig, 2010). Several chaperones require co-chaperones, which act as nucleotide exchange factors (NEFs), like the BCL2-associated athanogene (BAG) family of proteins (Takayama and Reed, 2001). Several chaperones and co-chaperones are mutated in NDs or other diseases characterized by neuronal loss (Smith et al., 2015), suggesting that they are protective against neurodegeneration. Chaperones directly assist the proper folding of nascent proteins or refold denatured existing proteins. When folding fails, chaperones route unfolded, partially folded or misfolded proteins to degradation. During cell stress (i.e., presence of misfolded proteins), some HSPBs, like heat shock protein B8 (HSPB8), limit the levels of aberrant proteins escaping degradation, which can accumulate in cells (Crippa et al., 2010b; Minoia et al., 2014; Cristofani et al., 2017a; Figure 1).
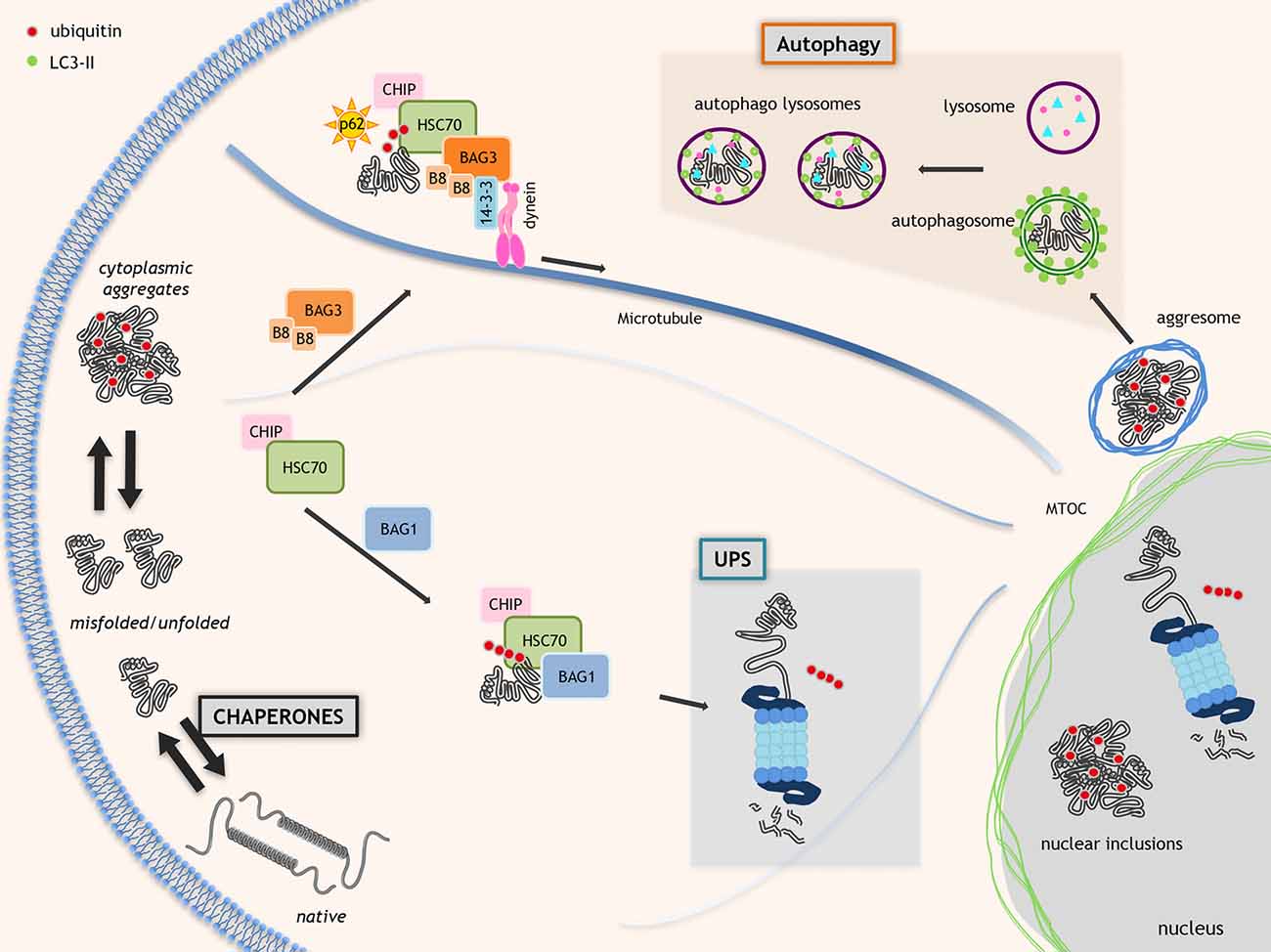
Figure 1. Proteostasis in neurodegenerative disorders. Molecular chaperones assist proteins to acquire the proper folding. When folding fails, the chaperones allow the ubiquitination and route misfolded proteins to degradative systems. This process is mediated by the HSC70-CHIP complex that interacts with nucleotide exchange factor (NEF)/BCL2-associated athanogenes (BAGs; HSC70 co-chaperones). BAG1 routes misfolded proteins to ubiquitin proteasome system (UPS). Alternatively, BAG3 and specific chaperone heat shock protein B8 (HSPB8; B8 in the figure) promote the degradation of HSC70 substrates via autophagy. The HSC70-CHIP interaction with BAG1 inhibits the HSP70 chaperone activity and allows misfolded proteins polyubiquitination by HSC70-binding co-factor CHIP resulting in misfolded proteins degradation via UPS. BAG3 interacts with dynein and 14-3-3 protein moving misfolded proteins to microtubule organization center (MTOC) where aggresomes are assembled. Polyubiquitinated proteins linked to HSC70-BAG3 are recognized by SQSTM1/p62 and its interaction with LC3 allows misfolded proteins insertion into autophagosomes.
The degradative pathways include the ubiquitin-proteasome system (UPS) and the autophagy (also the unfolded protein response (UPR), which relies on a specific endoplasmic reticulum associated degradation (ERAD) is part of the PQC but extensively reviewed recently (Volpi et al., 2017)). UPS and autophagy are in a finely orchestrated equilibrium, controlled by specific chaperones and co-chaperones (Gamerdinger et al., 2011a; Lilienbaum, 2013; Minoia et al., 2014; Behl, 2016; Cristofani et al., 2017a). UPS has low capacity, but high selectivity for monomeric misfolded proteins. An example of chaperone complex targeting misfolded proteins to UPS is formed by BAG1, HSP70 and the E3-ubiquitin ligase CHIP/STUB1 (Figure 1). Autophagy has high capacity, but low selectivity for substrates degrading oligo-/hetero-meric species, aggregates and damaged organelles (Klionsky et al., 2016). An example of chaperone complex targeting misfolded proteins to autophagy is the chaperone-assisted selective autophagy (CASA) complex, composed of HSPB8, BAG3, HSP70 and CHIP/STUB1. CASA complex interacts with the autophagy receptor SQSTM1/p62 which binds both ubiquitinated proteins and lipidated LC3 (LC3-II) targeting proteins to autophagosomes for degradation (Klionsky et al., 2016; Figure 1).
An imbalance of these two degradative pathways plays deleterious effects in several NDs (Kakkar et al., 2014; Ciechanover and Kwon, 2015; Nikoletopoulou et al., 2015; Senft and Ronai, 2015; Xilouri and Stefanis, 2015).
ALS and SBMA as Models to Study Misfolded Proteins in MNDs
ALS is a MND involving brain motor cortex, brainstem and anterior horn spinal cord motoneurons. Neurons of the fronto-temporal brain regions could be involved (Robberecht and Philips, 2013), causing mixed motor and cognitive phenotype (ALS with fronto-temporal dementia or FTD). Other cells, like glial cells (astrocytes (Trotti et al., 1999; Boillée et al., 2006; Nagai et al., 2007), oligodendrocytes (Philips et al., 2013), Schwann cells (Lobsiger et al., 2009; Turner et al., 2010), microglia (Philips and Robberecht, 2011)), and muscle cells (Musarò, 2010; Onesto et al., 2011; Galbiati et al., 2014) modulate disease progression, affecting motoneuron functions and survival capability.
ALS are mainly sporadic (sALS), with only 15% familial (fALS) forms, clinically indistinguishable from sALS. fALS are linked to specific gene mutations (e.g., superoxide dismutase-1 (SOD1), TAR DNA-binding protein 43 (TDP-43), fused-in-sarcoma/translocated-in-liposarcoma (FUS/TLS), Sequestosome-1 (SQSTM1/p62), Optineurin (OPTN-1), Ubiquilin (UBQLN-2), Valosin Containing Protein (VCP), TANK Binding Kinase 1 (TBK1) and several others (see Taylor et al., 2016 for review). Many of these gene products are autophagy-related proteins, key players of the PQC system (Ju et al., 2009; Tresse et al., 2010; Seguin et al., 2014; Taylor et al., 2016), or mislocalize and aggregate exerting proteotoxicity (Robberecht and Philips, 2013; Taylor et al., 2016). Some of these proteins are prone to aggregate in sALS even in their wild-type form (e.g., TDP-43, FUS, SQSTM1/p62, OPTN-1, UBQLN-2, etc.), suggesting that the mutation exacerbates their natural propensity to misfold (Neumann et al., 2006; Daoud et al., 2009; Bosco and Landers, 2010) and that common toxic mechanisms are involved in fALS and sALS. About 50% of fALS are linked to GGGGCC hexanucleotide (or G4C2) repeat expansions in the C9ORF72 gene (Al-Sarraj et al., 2011; DeJesus-Hernandez et al., 2011; Renton et al., 2011); this repeat undergoes a non-canonical repeat-associated non-ATG (RAN) translation, which generates five different dipeptides (DPRs), highly aggregation-prone (Ash et al., 2013; Lashley et al., 2013; Mori et al., 2013).
SBMA differs from ALS by a slower progression rate and involves lower motoneurons, dorsal root ganglia (DRG) sensory neurons, muscle cells and different androgen-target cells in reproductive tissues. Glial cells or microglia are not involved (La Spada et al., 1991; Fischbeck, 1997; Sorarù et al., 2008; Boyer et al., 2013; Malena et al., 2013; Cortes et al., 2014; Lieberman et al., 2014). SBMA is linked to a CAG repeat expansion in the androgen receptor (AR) gene, which codes an elongated polyglutamine tract (polyQ) in the AR protein (ARpolyQ; La Spada et al., 1991). ARpolyQ tends to misfold acquiring neurotoxic properties (Poletti, 2004), but only after binding to its ligand testosterone (Stenoien et al., 1999; Simeoni et al., 2000; Katsuno et al., 2002, 2003). Testosterone triggers conformational changes required for AR activation possibly impaired by the polyQ.
Several studies prove that misfolded proteins accumulation in fALS, sALS or SBMA alters the degradative pathways. The UPS could be overwhelmed by an excess of misfolded proteins or clogged by the polyQ (Ciechanover and Kwon, 2015; Rusmini et al., 2016); the autophagic flux could be blocked by misfolded protein aggregates (Rusmini et al., 2013). However, the molecular steps altered by misfolded proteins in these pathways are poorly understood. Several chaperones enhance misfolded protein degradation either/both by facilitating their proteasomal degradation or/and by limiting autophagic flux alterations (Balchin et al., 2016; Rusmini et al., 2016; van Noort et al., 2017; Charmpilas et al., 2017).
The HSPB8 Functions and Its Role in ALS and SBMA
HSPB8 is a chaperone widely distributed in several (if not all) human tissues, even if at different expression levels. In addition, HSPB8 upregulation may protect in ALS and SBMA (Carra et al., 2005, 2013; Crippa et al., 2010b; Rusmini et al., 2013). Notably, HSPB8 mutations cause Charcot-Marie-Tooth type 2L disease, hereditary distal motor neuropathy type II (dHMN-II) or distal myopathy (Fontaine et al., 2006; Irobi et al., 2010; Ghaoui et al., 2016), diseases involving motoneurons and/or muscle cells. These mutations impair the chaperone HSPB8 activity (Kwok et al., 2011), suggesting it has a crucial role in preserving motoneuron function and viability. Here, we review experimental findings in support of this hypothesis.
Interestingly, within the spinal cord, HSPB8 is specifically found in motoneurons, and its expression declines with age (Crippa et al., 2010b) suggesting that motoneurons might become more vulnerable to misfolded protein toxicity during aging. In cultured motoneurons, HSPB8 expression is greatly induced by proteasome impairment (Figure 2; Crippa et al., 2010a,b), a condition generally occurring in MNDs. In addition, HSPB8 mRNA expression is higher in autopsy specimens of ALS patient spinal cord than in age-matched controls (Anagnostou et al., 2010). We found a robust increase of HSPB8 protein levels in anterior horn spinal cord motoneurons surviving at end-stages of disease in transgenic (tg) ALS SOD1-G93A mice compared to wild-type mice (Crippa et al., 2010b), and this upregulation correlates with the presence of diffuse and non-aggregated mutant SOD (Crippa et al., 2010b).
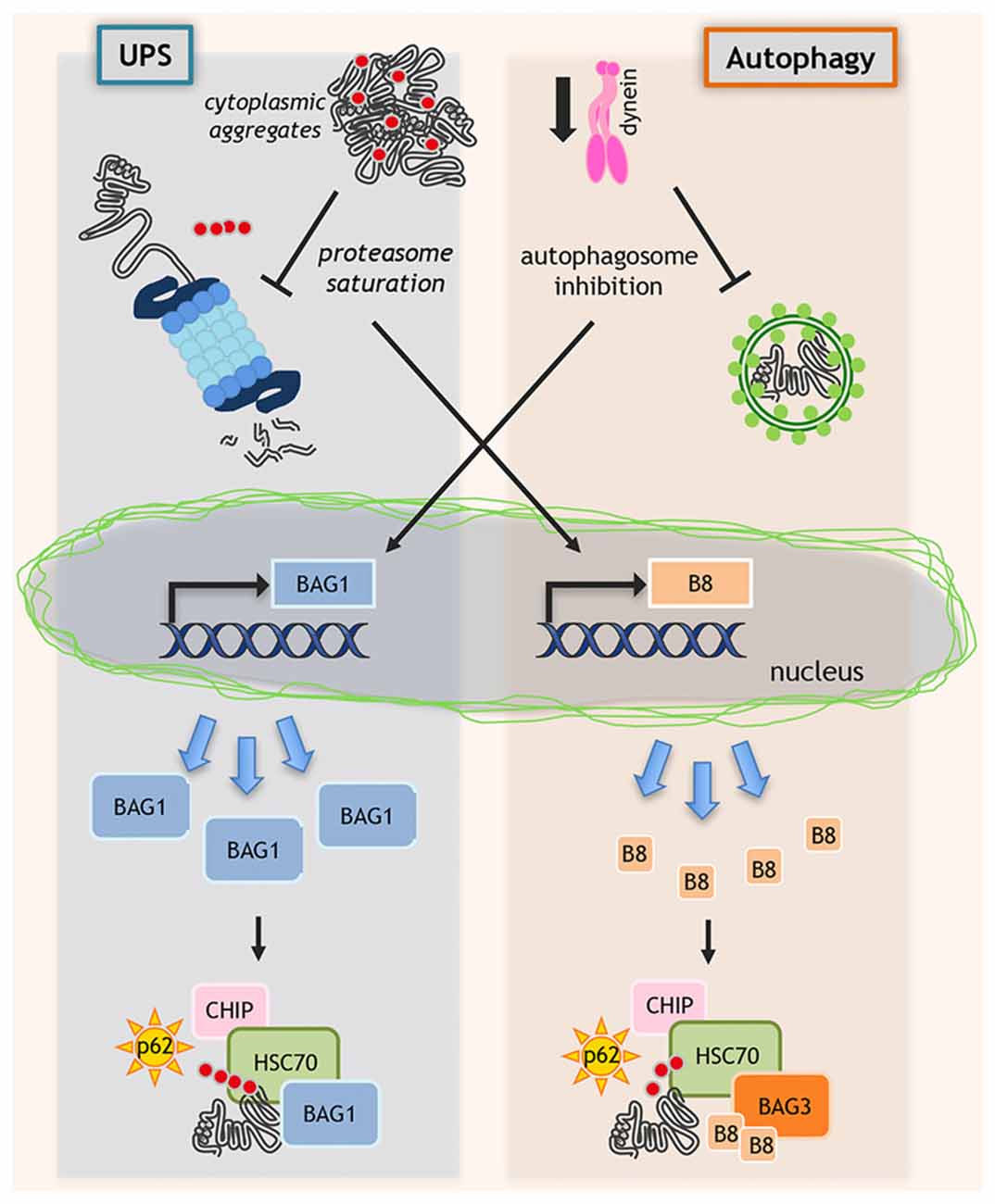
Figure 2. Regulation of protein quality control (PQC) system. When misfolded proteins cannot be efficiently removed by degradative pathways, misfolded proteins may accumulate and block ubiquitin proteasome system (UPS) and autophagy. In this context, proteasome saturation by misfolded proteins increases the transcription of HSPB8 (B8 in the figure) that, together with its partners BAG3 and HSC70, routes misfolded proteins to autophagy. In parallel, when dynein-mediated transport is blocked and autophagosomes formation is inhibited, still unknown factors activate the de novo transcription of BAG1, which binds to HSP70/CHIP and routes misfolded proteins to UPS.
HSPB8 is also highly expressed in muscle. Indeed, the genome-wide tissue analysis of RNA and protein expression (available at the Human Protein Atlas portal1) reports that in human skeletal muscle HSPB8 mRNA is expressed at high level, while the protein is expressed at medium levels if compared to other human tissues (Uhlén et al., 2015). In mice, HSPB8 expression in skeletal muscle dramatically increases during disease progression in ALS (Carra et al., 2013; Crippa et al., 2013a,b), and SBMA (Rusmini et al., 2015) mice. Since in these two MNDs, both motoneurons and myoblasts are target of misfolded protein toxicity, the increased HSPB8 expression may contribute to enhance the aberrant proteins clearance from muscle to improve cell survival. This hypothesis is supported by a recent report showing that the protein ICP10PK, the herpes simplex virus type 2 (HSV-2) homolog of HSPB8, delays disease onset and slows down progression rate of tg ALS SOD1-G93A mice (Aurelian et al., 2012). These affects are associated to reduced damages at neuromuscular junctions, and to increased motoneuronal survival (Aurelian et al., 2012). We demonstrated that the overexpression of HSP67Bc, the fly functional ortholog of HSPB8, exerts protective effects in two Drosophila melanogaster models of ALS. HSP67Bc overexpression prevented the mislocalization of a neurotoxic mutant TDP-43 protein (Ritson et al., 2010), while HSP67Bc downregulation correlated with increased TDP-43 and polyubiquitinated proteins accumulation, worsening the eye phenotype, of mutant TDP-43 flies (Crippa et al., 2016). HSP67Bc also rescued from pupae lethality flies overexpressing the ALS-associated 35 kDa TDP-43 fragment (TDP-35; Crippa et al., 2016). Thus, HSPB8 upregulation protects against misfolded protein-mediated toxicity in ALS models.
At cellular levels, the HSPB8 protective effects associate to its capability to facilitate misfolded proteins autophagic degradation. Indeed, HSPB8 could remove the blockage of autophagic flux found in several NDs (Rusmini et al., 2013, 2016; Giorgetti et al., 2015; Crippa et al., 2016). The HSPB8 pro-degradative activity was demonstrated with several different neuropathogenic proteins, like polyQ proteins (ARpolyQ, huntingtin-polyQ, ataxin-3-polyQ), beta-amyloid, alpha-synuclein, ALS proteins mutant SOD1 and TDP-43 fragments (Chávez Zobel et al., 2003; Wilhelmus et al., 2006; Carra et al., 2008a,b; Crippa et al., 2010b, 2016; Bruinsma et al., 2011; Seidel et al., 2012; Rusmini et al., 2013), and on five different RAN translated DPRs from the C9Orf72 gene linked to ALS and FTD (Cristofani et al., 2017b). In most cases, HSPB8 down-regulation resulted in increased accumulation of these mutant proteins and DPRs, supporting its role in the misfoded protein clearance (Crippa et al., 2010b, 2016; Rusmini et al., 2013; Cristofani et al., 2017b).
Moreover, increasing genetic and experimental evidences suggest that the RNA-protein inclusions accumulating in ALS and similar NDs may arise from the conversion of dynamic ribonucleoprotein complexes, stress granules (SGs), into amyloid-like aggregates. In particular, misfolded proteins accumulating in SGs promote their conversion into aggregates. Intriguingly, the PQC system response named “granulostasis”, surveys SG composition and maintains their dynamic behavior. One of the key players of granulostasis is the HSPB8-BAG3-HSP70 complex (Ganassi et al., 2016; Carra et al., 2017; Mateju et al., 2017).
The Molecular Mediators of The HSPB8 Pro-Autophagic Activity in Motoneurons
In line with the proposed pro-autophagic activity of HSPB8, autophagy, but not proteasome inhibition blocks, the HSPB8 pro-degradative activity (Crippa et al., 2010b, 2016; Rusmini et al., 2013). Mechanistically, HSPB8 facilitates the autophagic clearance of misfolded protein by associating with BAG3 (in a 2:1 ratio) and HSP70 (Carra et al., 2008b). The HSPB8-BAG3-HSP70 complex allows the cargoes delivery (i.e. misfolded proteins) to autophagy for degradation (Figure 1). BAG3 is essential in the complex, and its loss leads to a fast HSPB8 degradation (Carra et al., 2008b). In physiological conditions, the HSPB8-BAG3-HSP70 complex is essential at muscle level for Z-disk maintenance, where it is induced in response of acute physical exercise and of repeated mechanical stimulation (Ulbricht et al., 2015). These conditions generate a large excess of damaged proteins (e.g., actin) by post-translational modifications (carbonylation, nitrosylation), and the HSPB8-BAG3-HSP70 complex recognizes these damaged proteins and, by interacting via HSP70, with the E3-ubiquitin ligase CHIP/STUB1, forms the CASA complex. Here, CHIP/STUB1 ubiquitinates the target substrate allowing its SQSTM1/p62 recognition and insertion into the autophagosomes for degradation (Arndt et al., 2010). In skeletal muscle, the CASA complex directly interacts with DNAJB6 (of the DNAJ/Hsp40 family; Sarparanta et al., 2012), an HSP70 co-chaperone that suppresses aggregation of several misfolded proteins involved in NDs (Hageman et al., 2010). Indeed, DNAJB6 mutations cause Limb-girdle muscular dystrophies (LGMDs) in which a less-effective anti-aggregation activity of DNAJB6 has been found. In LGMDs patient muscle biopsies, DNAJB6 and CASA complex proteins aggregation and accumulation are observed, suggesting that the pathogenesis is also mediated by CASA dysfunctions (Sandell et al., 2016).
We demonstrated an important role of the CASA complex in motoneurons under pathological conditions, since in ALS cell models, this complex clears misfolded mutant SOD1 accumulating into motoneurons (Crippa et al., 2010a,b). While HSPB8 appears to be the limiting factor of this complex, BAG3 mediates its formation acting as a scaffold which interacts by its N-terminus with HSPB8 and by its C-terminus with HSP70 (which binds CHIP/STUB1). BAG3 contains PXXP motif, adjacent to the BAG domain, which binds dynein (Merabova et al., 2015), and two binding sites for the 14-3-3 protein, which stabilizes the BAG3 and dynein interaction (Mccollum et al., 2010; Behl, 2011, 2016; Gamerdinger et al., 2011a,b; Xu et al., 2013; Jia et al., 2014; Merabova et al., 2015). Dynein allows the efficient transport of the entire BAG3-multi-heteromeric complex at the site of autophagosomes assembly (Arndt et al., 2010; Crippa et al., 2010b; Merabova et al., 2015; Figure 1).
Interestingly, when dynein mediated transport is genetically (siRNAs) or pharmacologically (EHNA) blocked, the HSP70-CHIP cannot bind HSPB8-BAG3 complex to dispose misfolded proteins (ARpolyQ, mutant SOD1 and truncated TDP-43) via autophagy (Cristofani et al., 2017a). Here, still unknown factors activate the de novo transcription of another NEF/BAG, the BAG1, which binds HSP70/CHIP re-routing misfolded proteins to UPS (Behl, 2011, 2016; Gamerdinger et al., 2011a; Cristofani et al., 2017a). Indeed, BAG1 exogenous overexpression facilitates proteasomal removal of ARpolyQ. When dynein transport is inhibited, the pro-degradative BAG1 activity is blocked by proteasome inhibitors, but not by autophagy blockers (Cristofani et al., 2017a; Figure 2).
The BAG3/BAG1 ratio is the key factor that determines the fate of misfolded protein degradation via UPS or autophagy. HSPB8, being a chaperone “holder”, would bind to client proteins keeping them in a competent state for further processing by the BAG3-HSP70 complex. Increases in HSPB8 levels restore a deficient autophagic flux and ensure proper targeting of misfolded proteins to autophagy for clearance (Figure 2).
HSPB8-BAG3 Induction as Possible Therapeutic Approach for MNDs
Since HSPB8 overexpression is sufficient to restore autophagy, HSPB8 acts as a limiting factor for misfolded proteins autophagic degradation. Thus, small molecules acting as HSPB8 inducers may be of therapeutic interest in MNDs (Figure 2). Indeed, estrogens are physiological HSPB8 inducers, and selective estrogen receptor modulators (SERM) may differentially control its expression (Sun et al., 2007; Piccolella et al., 2017). To find FDA-approved drugs and natural compounds inducers of HSPB8 expression, we performed a high throughput screening (HTS) based on the human HSPB8 promoter controlling luciferase expression. We identified colchicine and doxorubicin as potent HSPB8 inducers and autophagy facilitators of removal of insoluble TDP-43 species (Crippa et al., 2016). Colchicine and derivatives might represent useful compounds to be tested in ALS models. Besides colchicine, which may have side effects, we found another HSPB8 inducer, the autophagic stimulator trehalose (Rusmini et al., 2013), already tested with positive results in several mice models of NDs (Tanaka et al., 2004; Davies et al., 2006; Rodríguez-Navarro et al., 2010; Perucho et al., 2012; Schaeffer and Goedert, 2012; Castillo et al., 2013; Du et al., 2013; Sarkar et al., 2014; Zhang et al., 2014; He et al., 2016).
Recently, it was shown that trehalose was also able to upregulate BAG3 expression (Lei et al., 2015). Whether trehalose protective effect is partly mediated by HSPB8 and BAG3 induction, in mice has still to be tested and represents an attractive hypothesis for future studies. Other compounds able to induce BAG3 expression has not been tested in MNDs models. Several evidences demonstrate that besides its role in autophagy, both HSPB8 and BAG3 modulate intracellular pathways involved in apoptosis or development altered in several tumors, and pharmacological BAG3 upregulation is obtained with proteasome inhibitors, TNF-related apoptosis-inducing ligand, fludarabine, cytosine arabinoside and etoposide, compounds used in chemotherapy, indicating BAG3 as a mediator of therapy resistance (Romano et al., 2003; Chiappetta et al., 2007; Rapino et al., 2014).
Thus, from preclinical studies, the pharmacological induction of key molecules like HSPB8 or BAG3 represents a novel and attractive target to treat MNDs. However, given the ability of these proteins to modulate major biological processes, the side-effects of their induction on other intracellular pathways remain to be elucidated.
Conclusions
Data collected in recent years clearly demonstrated that an increased PQC system activity protects against proteotoxic stresses induced by MNDs associated with the accumulation of misfolded proteins. Amongst the several PQC system components, the chaperone HSPB8 has attracted great attention, since it exerts a potent pro-degradative activity on misfolded proteins facilitating their removal via autophagy and preventing their intracellular accumulation. HSPB8 acts in a molecular complex involved in the fine tuning of the equilibrium between the UPS and autophagy. By this mechanism, HSPB8 also prevents the delivery of excessive amounts of misfolded proteins to UPS by routing them to autophagy. Based on these findings, the pharmacological induction of HSPB8 in MND affected cells should represent a promising therapeutic approach to counteract disease onset and progression.
Author Contributions
PR, RC, MG, SC, VC and AP designed and wrote the manuscript and critically discussed all sections of the minireview article. In addition RC, prepared the figures. MEC, MM, VF, GV, BT, EM, and MP critically revised the important intellectual content of the manuscript and the figures. All authors have provided final approval of the version to be published.
Funding
The following grants are gratefully acknowledged: Fondazione Telethon, Italy (grant no. GGP14039 to AP); Fondazione Cariplo, Italy (grant no. 2014-0686 to AP and SC); Fondazione AriSLA, Italy (grant no. ALS_HSPB8 to AP and SC; ALS_Granulopathy to AP and SC); Association Française contre les Myopathies, France (AFM Telethon grant no. 16406 to AP); Università degli Studi di Milano e piano di sviluppo UNIMI—linea B (to PR); Italian Ministry of Health (MinSal; grant no. GR-2011-02347198 to VC and SC); Fondazione Regionale per la Ricerca Biomedica (FRRB; TRANS_ALS, Rif. grant no. 2015-0023 to AP), Regione Lombardia, Italy (to AP); MIUR Rita Levi Montalcini (to SC); Italian Ministry of University and Research (MIUR), PRIN—Progetti di ricerca di interesse nazionale (grant no. 2015LFPNMN to AP and SC); European Molecular Biology Organization (EMBO), short term fellowship (grant no. 537-2015 to RC). This is an EU Joint Programme—Neurodegenerative Disease Research (JPND) project. The project is supported through the following funding organizations under the aegis of JPND—www.jpnd.eu. This project has received funding from the European Union’s Horizon 2020 research and innovation programme under grant agreement No 643417 (Grant ID: 01ED1601A, CureALS to AP and SC).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
References
Al-Sarraj, S., King, A., Troakes, C., Smith, B., Maekawa, S., Bodi, I., et al. (2011). p62 positive, TDP-43 negative, neuronal cytoplasmic and intranuclear inclusions in the cerebellum and hippocampus define the pathology of C9orf72-linked FTLD and MND/ALS. Acta Neuropathol. 122, 691–702. doi: 10.1007/s00401-011-0911-2
Anagnostou, G., Akbar, M. T., Paul, P., Angelinetta, C., Steiner, T. J., and de Belleroche, J. (2010). Vesicle associated membrane protein B (VAPB) is decreased in ALS spinal cord. Neurobiol. Aging 31, 969–985. doi: 10.1016/j.neurobiolaging.2008.07.005
Arndt, V., Dick, N., Tawo, R., Dreiseidler, M., Wenzel, D., Hesse, M., et al. (2010). Chaperone-assisted selective autophagy is essential for muscle maintenance. Curr. Biol. 20, 143–148. doi: 10.1016/j.cub.2009.11.022
Ash, P. E., Bieniek, K. F., Gendron, T. F., Caulfield, T., Lin, W. L., Dejesus-Hernandez, M., et al. (2013). Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 77, 639–646. doi: 10.1016/j.neuron.2013.02.004
Aurelian, L., Laing, J. M., and Lee, K. S. (2012). H11/HspB8 and its herpes simplex virus type 2 homologue ICP10PK share functions that regulate cell life/death decisions and human disease. Autoimmune. Dis. 2012:395329. doi: 10.1155/2012/395329
Balchin, D., Hayer-Hartl, M., and Hartl, F. U. (2016). In vivo aspects of protein folding and quality control. Science 353:aac4354. doi: 10.1126/science.aac4354
Behl, C. (2011). BAG3 and friends: co-chaperones in selective autophagy during aging and disease. Autophagy 7, 795–798. doi: 10.4161/auto.7.7.15844
Behl, C. (2016). Breaking BAG: the co-chaperone BAG3 in health and disease. Trends Pharmacol. Sci. 37, 672–688. doi: 10.1016/j.tips.2016.04.007
Boillée, S., Vande Velde, C., and Cleveland, D. W. (2006). ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron 52, 39–59. doi: 10.1016/j.neuron.2006.09.018
Bosco, D. A., and Landers, J. E. (2010). Genetic determinants of amyotrophic lateral sclerosis as therapeutic targets. CNS Neurol. Disord. Drug Targets 9, 779–790. doi: 10.2174/187152710793237494
Boyer, J. G., Murray, L. M., Scott, K., De Repentigny, Y., Renaud, J. M., and Kothary, R. (2013). Early onset muscle weakness and disruption of muscle proteins in mouse models of spinal muscular atrophy. Skelet. Muscle 3:24. doi: 10.1186/2044-5040-3-24
Bruinsma, I. B., Bruggink, K. A., Kinast, K., Versleijen, A. A., Segers-Nolten, I. M., Subramaniam, V., et al. (2011). Inhibition of α-synuclein aggregation by small heat shock proteins. Proteins 79, 2956–2967. doi: 10.1002/prot.23152
Carra, S., Alberti, S., Arrigo, P. A., Benesch, J. L., Benjamin, I. J., Boelens, W., et al. (2017). The growing world of small heat shock proteins: from structure to functions. Cell Stress Chaperones 22, 601–611. doi: 10.1007/s12192-017-0787-8
Carra, S., Rusmini, P., Crippa, V., Giorgetti, E., Boncoraglio, A., Cristofani, R., et al. (2013). Different anti-aggregation and pro-degradative functions of the members of the mammalian sHSP family in neurological disorders. Philos. Trans. R Soc. Lond. B Biol. Sci. 368:20110409. doi: 10.1098/rstb.2011.0409
Carra, S., Seguin, S. J., Lambert, H., and Landry, J. (2008a). HspB8 chaperone activity toward poly(Q)-containing proteins depends on its association with Bag3, a stimulator of macroautophagy. J. Biol. Chem. 283, 1437–1444. doi: 10.1074/jbc.M706304200
Carra, S., Seguin, S. J., and Landry, J. (2008b). HspB8 and Bag3: a new chaperone complex targeting misfolded proteins to macroautophagy. Autophagy 4, 237–239. doi: 10.4161/auto.5407
Carra, S., Sivilotti, M., Chávez Zobel, A. T., Lambert, H., and Landry, J. (2005). HspB8, a small heat shock protein mutated in human neuromuscular disorders, has in vivo chaperone activity in cultured cells. Hum. Mol. Genet. 14, 1659–1669. doi: 10.1093/hmg/ddi174
Castillo, K., Nassif, M., Valenzuela, V., Rojas, F., Matus, S., Mercado, G., et al. (2013). Trehalose delays the progression of amyotrophic lateral sclerosis by enhancing autophagy in motoneurons. Autophagy 9, 1308–1320. doi: 10.4161/auto.25188
Charmpilas, N., Kyriakakis, E., and Tavernarakis, N. (2017). Small heat shock proteins in ageing and age-related diseases. Cell Stress Chaperones 22, 481–492. doi: 10.1007/s12192-016-0761-x
Chávez Zobel, A. T., Loranger, A., Marceau, N., Thériault, J. R., Lambert, H., and Landry, J. (2003). Distinct chaperone mechanisms can delay the formation of aggresomes by the myopathy-causing R120G αB-crystallin mutant. Hum. Mol. Genet. 12, 1609–1620. doi: 10.1093/hmg/ddg173
Chiappetta, G., Ammirante, M., Basile, A., Rosati, A., Festa, M., Monaco, M., et al. (2007). The antiapoptotic protein BAG3 is expressed in thyroid carcinomas and modulates apoptosis mediated by tumor necrosis factor-related apoptosis-inducing ligand. J. Clin. Endocrinol. Metab. 92, 1159–1163. doi: 10.1210/jc.2006-1712
Ciechanover, A., and Kwon, Y. T. (2015). Degradation of misfolded proteins in neurodegenerative diseases: therapeutic targets and strategies. Exp. Mol. Med. 47:e147. doi: 10.1038/emm.2014.117
Cortes, C. J., Ling, S. C., Guo, L. T., Hung, G., Tsunemi, T., Ly, L., et al. (2014). Muscle expression of mutant androgen receptor accounts for systemic and motor neuron disease phenotypes in spinal and bulbar muscular atrophy. Neuron 82, 295–307. doi: 10.1016/j.neuron.2014.03.001
Crippa, V., Boncoraglio, A., Galbiati, M., Aggarwal, T., Rusmini, P., Giorgetti, E., et al. (2013a). Differential autophagy power in the spinal cord and muscle of transgenic ALS mice. Front. Cell. Neurosci. 7:234. doi: 10.3389/fncel.2013.00234
Crippa, V., Galbiati, M., Boncoraglio, A., Rusmini, P., Onesto, E., Giorgetti, E., et al. (2013b). Motoneuronal and muscle-selective removal of ALS-related misfolded proteins. Biochem. Soc. Trans. 41, 1598–1604. doi: 10.1042/BST20130118
Crippa, V., Carra, S., Rusmini, P., Sau, D., Bolzoni, E., Bendotti, C., et al. (2010a). A role of small heat shock protein B8 (HspB8) in the autophagic removal of misfolded proteins responsible for neurodegenerative diseases. Autophagy 6, 958–960. doi: 10.4161/auto.6.7.13042
Crippa, V., Sau, D., Rusmini, P., Boncoraglio, A., Onesto, E., Bolzoni, E., et al. (2010b). The small heat shock protein B8 (HspB8) promotes autophagic removal of misfolded proteins involved in amyotrophic lateral sclerosis (ALS). Hum. Mol. Genet. 19, 3440–3456. doi: 10.1093/hmg/ddq257
Crippa, V., Cicardi, M. E., Ramesh, N., Seguin, S. J., Ganassi, M., Bigi, I., et al. (2016). The chaperone HSPB8 reduces the accumulation of truncated TDP-43 species in cells and protects against TDP-43-mediated toxicity. Hum. Mol. Genet. 25, 3908–3924. doi: 10.1093/hmg/ddw232
Cristofani, R., Crippa, V., Rusmini, P., Cicardi, M. E., Meroni, M., Licata, N. V., et al. (2017a). Inhibition of retrograde transport modulates misfolded protein accumulation and clearance in motoneuron diseases. Autophagy doi: 10.1080/15548627.2017.1308985 [Epub ahead of print].
Cristofani, R., Crippa, V., Vezzoli, G., Rusmini, P., Galbiati, M., Cicardi, M. E., et al. (2017b). The small heat shock protein B8 (HSPB8) efficiently removes aggregating species of dipeptides produced in C9ORF72-related neurodegenerative diseases. Cell Stress Chaperones doi: 10.1007/s12192-017-0806-9 [Epub ahead of print].
Daoud, H., Valdmanis, P. N., Kabashi, E., Dion, P., Dupré, N., Camu, W., et al. (2009). Contribution of TARDBP mutations to sporadic amyotrophic lateral sclerosis. J. Med. Genet. 46, 112–114. doi: 10.1136/jmg.2008.062463
Davies, J. E., Sarkar, S., and Rubinsztein, D. C. (2006). Trehalose reduces aggregate formation and delays pathology in a transgenic mouse model of oculopharyngeal muscular dystrophy. Hum. Mol. Genet. 15, 23–31. doi: 10.1093/hmg/ddi422
DeJesus-Hernandez, M., Mackenzie, I. R., Boeve, B. F., Boxer, A. L., Baker, M., Rutherford, N. J., et al. (2011). Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256. doi: 10.1016/j.neuron.2011.09.011
Du, J., Liang, Y., Xu, F., Sun, B., and Wang, Z. (2013). Trehalose rescues Alzheimer’s disease phenotypes in APP/PS1 transgenic mice. J. Pharm. Pharmacol. 65, 1753–1756. doi: 10.1111/jphp.12108
Fontaine, J. M., Sun, X., Hoppe, A. D., Simon, S., Vicart, P., Welsh, M. J., et al. (2006). Abnormal small heat shock protein interactions involving neuropathy-associated HSP22 (HSPB8) mutants. FASEB J. 20, 2168–2170. doi: 10.1096/fj.06-5911fje
Galbiati, M., Crippa, V., Rusmini, P., Cristofani, R., Cicardi, M. E., Giorgetti, E., et al. (2014). ALS-related misfolded protein management in motor neurons and muscle cells. Neurochem. Int. 79, 70–78. doi: 10.1016/j.neuint.2014.10.007
Gamerdinger, M., Carra, S., and Behl, C. (2011a). Emerging roles of molecular chaperones and co-chaperones in selective autophagy: focus on BAG proteins. J. Mol. Med. 89, 1175–1182. doi: 10.1007/s00109-011-0795-6
Gamerdinger, M., Kaya, A. M., Wolfrum, U., Clement, A. M., and Behl, C. (2011b). BAG3 mediates chaperone-based aggresome-targeting and selective autophagy of misfolded proteins. EMBO Rep. 12, 149–156. doi: 10.1038/embor.2010.203
Ganassi, M., Mateju, D., Bigi, I., Mediani, L., Poser, I., Lee, H. O., et al. (2016). A surveillance function of the HSPB8-BAG3-HSP70 chaperone complex ensures stress granule integrity and dynamism. Mol. Cell 63, 796–810. doi: 10.1016/j.molcel.2016.07.021
Ghaoui, R., Palmio, J., Brewer, J., Lek, M., Needham, M., Evila, A., et al. (2016). Mutations in HSPB8 causing a new phenotype of distal myopathy and motor neuropathy. Neurology 86, 391-398 doi: 10.1212/WNL.0000000000002556
Giorgetti, E., Rusmini, P., Crippa, V., Cristofani, R., Boncoraglio, A., Cicardi, M. E., et al. (2015). Synergic prodegradative activity of bicalutamide and trehalose on the mutant androgen receptor responsible for spinal and bulbar muscular atrophy. Hum. Mol. Genet. 24, 64–75. doi: 10.1093/hmg/ddu419
Hageman, J., Rujano, M. A., van Waarde, M. A., Kakkar, V., Dirks, R. P., Govorukhina, N., et al. (2010). A DNAJB chaperone subfamily with HDAC-dependent activities suppresses toxic protein aggregation. Mol. Cell 37, 355–369. doi: 10.1016/j.molcel.2010.01.001
He, Q., Koprich, J. B., Wang, Y., Yu, W. B., Xiao, B. G., Brotchie, J. M., et al. (2016). Treatment with trehalose prevents behavioral and neurochemical deficits produced in an AAV α-synuclein rat model of Parkinson’s disease. Mol. Neurobiol. 53, 2258–2268. doi: 10.1007/s12035-015-9173-7
Irobi, J., Almeida-Souza, L., Asselbergh, B., De Winter, V., Goethals, S., Dierick, I., et al. (2010). Mutant HSPB8 causes motor neuron-specific neurite degeneration. Hum. Mol. Genet. 19, 3254–3265. doi: 10.1093/hmg/ddq234
Jia, B., Wu, Y., and Zhou, Y. (2014). 14-3-3 and aggresome formation: implications in neurodegenerative diseases. Prion 8, 173–177. doi: 10.4161/pri.28123
Ju, J. S., Fuentealba, R. A., Miller, S. E., Jackson, E., Piwnica-Worms, D., Baloh, R. H., et al. (2009). Valosin-containing protein (VCP) is required for autophagy and is disrupted in VCP disease. J. Cell Biol. 187, 875–888. doi: 10.1083/jcb.200908115
Kakkar, V., Meister-Broekema, M., Minoia, M., Carra, S., and Kampinga, H. H. (2014). Barcoding heat shock proteins to human diseases: looking beyond the heat shock response. Dis. Model. Mech. 7, 421–434. doi: 10.1242/dmm.014563
Kampinga, H. H., and Craig, E. A. (2010). The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat. Rev. Mol. Cell Biol. 11, 579–592. doi: 10.1038/nrm2941
Katsuno, M., Adachi, H., Doyu, M., Minamiyama, M., Sang, C., Kobayashi, Y., et al. (2003). Leuprorelin rescues polyglutamine-dependent phenotypes in a transgenic mouse model of spinal and bulbar muscular atrophy. Nat. Med. 9, 768–773. doi: 10.1038/nm878
Katsuno, M., Adachi, H., Kume, A., Li, M., Nakagomi, Y., Niwa, H., et al. (2002). Testosterone reduction prevents phenotypic expression in a transgenic mouse model of spinal and bulbar muscular atrophy. Neuron 35, 843–854. doi: 10.1016/S0896-6273(02)00834-6
Klionsky, D. J., Abdelmohsen, K., Abe, A., Abedin, M. J., Abeliovich, H., Acevedo Arozena, A., et al. (2016). Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 12, 1–222. doi: 10.1080/15548627.2015.1100356
Kwok, A. S., Phadwal, K., Turner, B. J., Oliver, P. L., Raw, A., Simon, A. K., et al. (2011). HspB8 mutation causing hereditary distal motor neuropathy impairs lysosomal delivery of autophagosomes. J. Neurochem. 119, 1155–1161. doi: 10.1111/j.1471-4159.2011.07521.x
La Spada, A. R., Wilson, E. M., Lubahn, D. B., Harding, A. E., and Fischbeck, K. H. (1991). Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature 352, 77–79. doi: 10.1038/352077a0
Lashley, T., Hardy, J., and Isaacs, A. M. (2013). RANTing about C9orf72. Neuron 77, 597–598. doi: 10.1016/j.neuron.2013.02.009
Lei, Z., Brizzee, C., and Johnson, G. V. (2015). BAG3 facilitates the clearance of endogenous tau in primary neurons. Neurobiol. Aging 36, 241–248. doi: 10.1016/j.neurobiolaging.2014.08.012
Lieberman, A. P., Yu, Z., Murray, S., Peralta, R., Low, A., Guo, S., et al. (2014). Peripheral androgen receptor gene suppression rescues disease in mouse models of spinal and bulbar muscular atrophy. Cell Rep. 7, 774–784. doi: 10.1016/j.celrep.2014.02.008
Lilienbaum, A. (2013). Relationship between the proteasomal system and autophagy. Int. J. Biochem. Mol. Biol. 4, 1–26.
Lobsiger, C. S., Boillee, S., Mcalonis-Downes, M., Khan, A. M., Feltri, M. L., Yamanaka, K., et al. (2009). Schwann cells expressing dismutase active mutant SOD1 unexpectedly slow disease progression in ALS mice. Proc. Natl. Acad. Sci. U S A 106, 4465–4470. doi: 10.1073/pnas.0813339106
Malena, A., Pennuto, M., Tezze, C., Querin, G., D’ascenzo, C., Silani, V., et al. (2013). Androgen-dependent impairment of myogenesis in spinal and bulbar muscular atrophy. Acta Neuropathol. 126, 109–121. doi: 10.1007/s00401-013-1122-9
Mateju, D., Franzmann, T. M., Patel, A., Kopach, A., Boczek, E. E., Maharana, S., et al. (2017). An aberrant phase transition of stress granules triggered by misfolded protein and prevented by chaperone function. EMBO J. 36, 1669–1687. doi: 10.15252/embj.201695957
Mccollum, A. K., Casagrande, G., and Kohn, E. C. (2010). Caught in the middle: the role of Bag3 in disease. Biochem. J. 425, e1–e3. doi: 10.1042/BJ20091739
Merabova, N., Sariyer, I. K., Saribas, A. S., Knezevic, T., Gordon, J., Turco, M. C., et al. (2015). WW domain of BAG3 is required for the induction of autophagy in glioma cells. J. Cell. Physiol. 230, 831–841. doi: 10.1002/jcp.24811
Minoia, M., Boncoraglio, A., Vinet, J., Morelli, F. F., Brunsting, J. F., Poletti, A., et al. (2014). BAG3 induces the sequestration of proteasomal clients into cytoplasmic puncta: implications for a proteasome-to-autophagy switch. Autophagy 10, 1603–1621. doi: 10.4161/auto.29409
Mori, K., Weng, S. M., Arzberger, T., May, S., Rentzsch, K., Kremmer, E., et al. (2013). The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 339, 1335–1338. doi: 10.1126/science.1232927
Morimoto, R. I. (2006). Stress, aging and neurodegenerative disease. N. Engl. J. Med. 355, 2254–2255. doi: 10.1056/NEJMcibr065573
Musarò, A. (2010). State of the art and the dark side of amyotrophic lateral sclerosis. World J. Biol. Chem. 1, 62–68. doi: 10.4331/wjbc.v1.i5.62
Nagai, M., Re, D. B., Nagata, T., Chalazonitis, A., Jessell, T. M., Wichterle, H., et al. (2007). Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat. Neurosci. 10, 615–622. doi: 10.1038/nn1876
Neumann, M., Sampathu, D. M., Kwong, L. K., Truax, A. C., Micsenyi, M. C., Chou, T. T., et al. (2006). Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133. doi: 10.1126/science.1134108
Nikoletopoulou, V., Papandreou, M. E., and Tavernarakis, N. (2015). Autophagy in the physiology and pathology of the central nervous system. Cell Death Differ. 22, 398–407. doi: 10.1038/cdd.2014.204
Onesto, E., Rusmini, P., Crippa, V., Ferri, N., Zito, A., Galbiati, M., et al. (2011). Muscle cells and motoneurons differentially remove mutant SOD1 causing familial amyotrophic lateral sclerosis. J. Neurochem. 118, 266–280. doi: 10.1111/j.1471-4159.2011.07298.x
Perucho, J., Casarejos, M. J., Gomez, A., Solano, R. M., De Yébenes, J. G., and Mena, M. A. (2012). Trehalose protects from aggravation of amyloid pathology induced by isoflurane anesthesia in APP(swe) mutant mice. Curr. Alzheimer Res. 9, 334–343. doi: 10.2174/156720512800107573
Philips, T., Bento-Abreu, A., Nonneman, A., Haeck, W., Staats, K., Geelen, V., et al. (2013). Oligodendrocyte dysfunction in the pathogenesis of amyotrophic lateral sclerosis. Brain 136, 471–482. doi: 10.1093/brain/aws339
Philips, T., and Robberecht, W. (2011). Neuroinflammation in amyotrophic lateral sclerosis: role of glial activation in motor neuron disease. Lancet Neurol. 10, 253–263. doi: 10.1016/S1474-4422(11)70015-1
Piccolella, M., Crippa, V., Cristofani, R., Rusmini, P., Galbiati, M., Cicardi, M. E., et al. (2017). The small heat shock protein B8 (HSPB8) modulates proliferation and migration of breast cancer cells. Oncotarget 8, 10400–10415. doi: 10.18632/oncotarget.14422
Poletti, A. (2004). The polyglutamine tract of androgen receptor: from functions to dysfunctions in motor neurons. Front. Neuroendocrinol. 25, 1–26. doi: 10.1016/j.yfrne.2004.03.001
Rapino, F., Jung, M., and Fulda, S. (2014). BAG3 induction is required to mitigate proteotoxicity via selective autophagy following inhibition of constitutive protein degradation pathways. Oncogene 33, 1713–1724. doi: 10.1038/onc.2013.110
Renton, A. E., Majounie, E., Waite, A., Simón-Sánchez, J., Rollinson, S., Gibbs, J. R., et al. (2011). A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268. doi: 10.1016/j.neuron.2011.09.010
Ritson, G. P., Custer, S. K., Freibaum, B. D., Guinto, J. B., Geffel, D., Moore, J., et al. (2010). TDP-43 mediates degeneration in a novel drosophila model of disease caused by mutations in VCP/p97. J. Neurosci. 30, 7729–7739. doi: 10.1523/JNEUROSCI.5894-09.2010
Robberecht, W., and Philips, T. (2013). The changing scene of amyotrophic lateral sclerosis. Nat. Rev. Neurosci. 14, 248–264. doi: 10.1038/nrn3430
Rodríguez-Navarro, J. A., Rodríguez, L., Casarejos, M. J., Solano, R. M., Gómez, A., Perucho, J., et al. (2010). Trehalose ameliorates dopaminergic and tau pathology in parkin deleted/tau overexpressing mice through autophagy activation. Neurobiol. Dis. 39, 423–438. doi: 10.1016/j.nbd.2010.05.014
Romano, M. F., Festa, M., Petrella, A., Rosati, A., Pascale, M., Bisogni, R., et al. (2003). BAG3 protein regulates cell survival in childhood acute lymphoblastic leukemia cells. Cancer Biol. Ther. 2, 508–510. doi: 10.4161/cbt.2.5.524
Rusmini, P., Crippa, V., Cristofani, R., Rinaldi, C., Cicardi, M. E., Galbiati, M., et al. (2016). The role of the protein quality control system in SBMA. J. Mol. Neurosci. 58, 348–364. doi: 10.1007/s12031-015-0675-6
Rusmini, P., Crippa, V., Giorgetti, E., Boncoraglio, A., Cristofani, R., Carra, S., et al. (2013). Clearance of the mutant androgen receptor in motoneuronal models of spinal and bulbar muscular atrophy. Neurobiol. Aging 34, 2585–2603. doi: 10.1016/j.neurobiolaging.2013.05.026
Rusmini, P., Polanco, M. J., Cristofani, R., Cicardi, M. E., Meroni, M., Galbiati, M., et al. (2015). Aberrant autophagic response in the muscle of a knock-in mouse model of spinal and bulbar muscular atrophy. Sci. Rep. 5:15174. doi: 10.1038/srep15174
Sandell, S., Huovinen, S., Palmio, J., Raheem, O., Lindfors, M., Zhao, F., et al. (2016). Diagnostically important muscle pathology in DNAJB6 mutated LGMD1D. Acta Neuropathol. Commun. 4:9. doi: 10.1186/s40478-016-0276-9
Sarkar, S., Chigurupati, S., Raymick, J., Mann, D., Bowyer, J. F., Schmitt, T., et al. (2014). Neuroprotective effect of the chemical chaperone, trehalose in a chronic MPTP-induced Parkinson’s disease mouse model. Neurotoxicol 44, 250–262. doi: 10.1016/j.neuro.2014.07.006
Sarparanta, J., Jonson, P. H., Golzio, C., Sandell, S., Luque, H., Screen, M., et al. (2012). Mutations affecting the cytoplasmic functions of the co-chaperone DNAJB6 cause limb-girdle muscular dystrophy. Nat. Genet. 44, 450–455, S1–S2. doi: 10.1038/ng.1103
Schaeffer, V., and Goedert, M. (2012). Stimulation of autophagy is neuroprotective in a mouse model of human tauopathy. Autophagy 8, 1686–1687. doi: 10.4161/auto.21488
Seguin, S. J., Morelli, F. F., Vinet, J., Amore, D., De Biasi, S., Poletti, A., et al. (2014). Inhibition of autophagy, lysosome and VCP function impairs stress granule assembly. Cell Death Differ. 21, 1838–1851. doi: 10.1038/cdd.2014.103
Seidel, K., Vinet, J., Den Dunnen, W. F., Brunt, E. R., Meister, M., Boncoraglio, A., et al. (2012). The HSPB8-BAG3 chaperone complex is upregulated in astrocytes in the human brain affected by protein aggregation diseases. Neuropathol. Appl. Neurobiol. 38, 39–53. doi: 10.1111/j.1365-2990.2011.01198.x
Senft, D., and Ronai, Z. A. (2015). UPR, autophagy and mitochondria crosstalk underlies the ER stress response. Trends Biochem. Sci. 40, 141–148. doi: 10.1016/j.tibs.2015.01.002
Simeoni, S., Mancini, M. A., Stenoien, D. L., Marcelli, M., Weigel, N. L., Zanisi, M., et al. (2000). Motoneuronal cell death is not correlated with aggregate formation of androgen receptors containing an elongated polyglutamine tract. Hum. Mol. Genet. 9, 133–144. doi: 10.1093/hmg/9.1.133
Smith, H. L., Li, W., and Cheetham, M. E. (2015). Molecular chaperones and neuronal proteostasis. Semin. Cell Dev. Biol. 40, 142–152. doi: 10.1016/j.semcdb.2015.03.003
Sorarù, G., D’ascenzo, C., Polo, A., Palmieri, A., Baggio, L., Vergani, L., et al. (2008). Spinal and bulbar muscular atrophy: skeletal muscle pathology in male patients and heterozygous females. J. Neurol. Sci. 264, 100–105. doi: 10.1016/j.jns.2007.08.012
Stenoien, D. L., Cummings, C. J., Adams, H. P., Mancini, M. G., Patel, K., DeMartino, G., et al. (1999). Polyglutamine-expanded androgen receptors form aggregates that sequester heat shock proteins, proteasome components and SRC-1 and are suppressed by the HDJ-2 chaperone. Hum. Mol. Genet. 8, 731–741. doi: 10.1093/hmg/8.5.731
Sun, X., Fontaine, J. M., Bartl, I., Behnam, B., Welsh, M. J., and Benndorf, R. (2007). Induction of Hsp22 (HspB8) by estrogen and the metalloestrogen cadmium in estrogen receptor-positive breast cancer cells. Cell Stress Chaperones 12, 307–319. doi: 10.1379/csc-276.1
Takayama, S., and Reed, J. C. (2001). Molecular chaperone targeting and regulation by BAG family proteins. Nat. Cell Biol. 3, E237–E241. doi: 10.1038/ncb1001-e237
Tanaka, M., Machida, Y., Niu, S., Ikeda, T., Jana, N. R., Doi, H., et al. (2004). Trehalose alleviates polyglutamine-mediated pathology in a mouse model of Huntington disease. Nat. Med. 10, 148–154. doi: 10.1038/nm985
Taylor, J. P., Brown, R. H., and Cleveland, D. W. (2016). Decoding ALS: from genes to mechanism. Nature 539, 197–206. doi: 10.1038/nature20413
Tresse, E., Salomons, F. A., Vesa, J., Bott, L. C., Kimonis, V., Yao, T. P., et al. (2010). VCP/p97 is essential for maturation of ubiquitin-containing autophagosomes and this function is impaired by mutations that cause IBMPFD. Autophagy 6, 217–227. doi: 10.4161/auto.6.2.11014
Trotti, D., Rolfs, A., Danbolt, N. C., Brown, R. H. Jr., and Hediger, M. A. (1999). SOD1 mutants linked to amyotrophic lateral sclerosis selectively inactivate a glial glutamate transporter. Nat. Neurosci. 2:848. doi: 10.1038/12227
Turner, B. J., Ackerley, S., Davies, K. E., and Talbot, K. (2010). Dismutase-competent SOD1 mutant accumulation in myelinating Schwann cells is not detrimental to normal or transgenic ALS model mice. Hum. Mol. Genet. 19, 815–824. doi: 10.1093/hmg/ddp550
Uhlén, M., Fagerberg, L., Hallström, B. M., Lindskog, C., Oksvold, P., Mardinoglu, A., et al. (2015). Proteomics. tissue-based map of the human proteome. Science 347:1260419. doi: 10.1126/science.1260419
Ulbricht, A., Gehlert, S., Leciejewski, B., Schiffer, T., Bloch, W., and Höhfeld, J. (2015). Induction and adaptation of chaperone-assisted selective autophagy CASA in response to resistance exercise in human skeletal muscle. Autophagy 11, 538–546. doi: 10.1080/15548627.2015.1017186
van Noort, J. M., Bugiani, M., and Amor, S. (2017). Heat shock proteins: old and novel roles in neurodegenerative diseases in the central nervous system. CNS Neurol. Disord. Drug Targets 16, 244–256. doi: 10.2174/1871527315666161031125317
Volpi, V. G., Touvier, T., and D’antonio, M. (2017). Endoplasmic reticulum protein quality control failure in myelin disorders. Front. Mol. Neurosci. 9:162. doi: 10.3389/fnmol.2016.00162
Wilhelmus, M. M., Boelens, W. C., Otte-Höller, I., Kamps, B., Kusters, B., Maat-Schieman, M. L., et al. (2006). Small heat shock protein HspB8: its distribution in Alzheimer’s disease brains and its inhibition of amyloid-beta protein aggregation and cerebrovascular amyloid-beta toxicity. Acta Neuropathol. 111, 139–149. doi: 10.1007/s00401-005-0030-z
Xilouri, M., and Stefanis, L. (2015). Chaperone mediated autophagy to the rescue: a new-fangled target for the treatment of neurodegenerative diseases. Mol. Cell. Neurosci. 66, 29–36. doi: 10.1016/j.mcn.2015.01.003
Xu, Z., Graham, K., Foote, M., Liang, F., Rizkallah, R., Hurt, M., et al. (2013). 14-3-3 protein targets misfolded chaperone-associated proteins to aggresomes. J. Cell Sci. 126, 4173–4186. doi: 10.1242/jcs.126102
Keywords: motoneuron diseases, amyotrophic lateral sclerosis, spinal and bulbar muscular atrophy, proteasome, autophagy, chaperones, misfolded proteins, HSPB8
Citation: Rusmini P, Cristofani R, Galbiati M, Cicardi ME, Meroni M, Ferrari V, Vezzoli G, Tedesco B, Messi E, Piccolella M, Carra S, Crippa V and Poletti A (2017) The Role of the Heat Shock Protein B8 (HSPB8) in Motoneuron Diseases. Front. Mol. Neurosci. 10:176. doi: 10.3389/fnmol.2017.00176
Received: 04 April 2017; Accepted: 18 May 2017;
Published: 21 June 2017.
Edited by:
Eva Zerovnik, Jožef Stefan Institute, SloveniaReviewed by:
Anat Ben-Zvi, Ben-Gurion University of the Negev, IsraelAndrew P. Lieberman, University of Michigan, United States
Andre Patrick Arrigo, Claude Bernard University Lyon 1, France
Copyright © 2017 Rusmini, Cristofani, Galbiati, Cicardi, Meroni, Ferrari, Vezzoli, Tedesco, Messi, Piccolella, Carra, Crippa and Poletti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Angelo Poletti, YW5nZWxvLnBvbGV0dGlAdW5pbWkuaXQ=
† These authors have contributed equally to this work.