 Omer Barkai1,2
Omer Barkai1,2 Robert H. Goldstein
Robert H. Goldstein Ben Katz
Ben Katz Alexander M. Binshtok
Alexander M. Binshtok- 1Department of Medical Neurobiology, Institute for Medical Research Israel-Canada, Hadassah School of Medicine, The Hebrew University-Hadassah School of Medicine, Jerusalem, Israel
- 2The Edmond and Lily Safra Center for Brain Sciences, The Hebrew University of Jerusalem, Jerusalem, Israel
Peripheral nociceptive neurons encode and convey injury-inducing stimuli toward the central nervous system. In normal conditions, tight control of nociceptive resting potential prevents their spontaneous activation. However, in many pathological conditions the control of membrane potential is disrupted, leading to ectopic, stimulus-unrelated firing of nociceptive neurons, which is correlated to spontaneous pain. We have investigated the role of KV7/M channels in stabilizing membrane potential and impeding spontaneous firing of nociceptive neurons. These channels generate low voltage-activating, noninactivating M-type K+ currents (M-current, IM), which control neuronal excitability. Using perforated-patch recordings from cultured, rat nociceptor-like dorsal root ganglion neurons, we show that inhibition of M-current leads to depolarization of nociceptive neurons and generation of repetitive firing. To assess to what extent the M-current, acting at the nociceptive terminals, is able to stabilize terminals' membrane potential, thus preventing their ectopic activation, in normal and pathological conditions, we built a multi-compartment computational model of a pseudo-unipolar unmyelinated nociceptive neuron with a realistic terminal tree. The modeled terminal tree was based on the in vivo structure of nociceptive peripheral terminal, which we assessed by in vivo multiphoton imaging of GFP-expressing nociceptive neuronal terminals innervating mice hind paw. By modifying the conductance of the KV7/M channels at the modeled terminal tree (terminal gKV7/M) we have found that 40% of the terminal gKV7/M conductance is sufficient to prevent spontaneous firing, while ~75% of terminal gKV7/M is sufficient to inhibit stimulus induced activation of nociceptive neurons. Moreover, we showed that terminal M-current reduces susceptibility of nociceptive neurons to a small fluctuations of membrane potentials. Furthermore, we simulated how the interaction between terminal persistent sodium current and M-current affects the excitability of the neurons. We demonstrated that terminal M-current in nociceptive neurons impeded spontaneous firing even when terminal Na(V)1.9 channels conductance was substantially increased. On the other hand, when terminal gKV7/M was decreased, nociceptive neurons fire spontaneously after slight increase in terminal Na(V)1.9 conductance. Our results emphasize the pivotal role of M-current in stabilizing membrane potential and hereby in controlling nociceptive spontaneous firing, in normal and pathological conditions.
Introduction
Primary sensory nociceptive neurons, which signals the CNS about the presence of noxious stimuli are mostly quiescent in the absence of injury-mediating stimuli (Reeh, 1986; Amir et al., 2002; Gudes et al., 2015; Emery et al., 2016). In models of inflammation or nerve injury, the stability of nociceptive membrane resting potential is dysregulated, leading to spontaneous or ectopic activity i.e., the activation of nociceptive fibers in the absence of noxious stimuli (Amir et al., 1999; Wu et al., 2001; Djouhri et al., 2006, 2012; Bernal et al., 2016). This ectopic activity is correlated to spontaneous pain (Djouhri et al., 2006; Kleggetveit et al., 2012; Serra et al., 2012). A particular class of potassium (K+) current, namely, Kv7/M-current (IM) due to its unique biophysical properties can serve as a stabilizer of the resting potential, a kind of intrinsic “voltage clamp” mechanism (Liu et al., 2010; Passmore et al., 2012), which dampens depolarizatory deviations hence preventing ectopic firing and therefore spontaneous pain. IM is generated by heteromeric Kv7.2/3 (KCNQ2/3) channels (Brown and Passmore, 2009), which are densely expressed at the sites of spike generation e.g., axon initial segment of central neurons (Pan et al., 2006) and terminals of peripheral nociceptive neurons (Passmore et al., 2012). These low voltage-activating (around −60 mV), non-inactivating channels underlie the slow activating and prolonged outward current, which opposes membrane depolarization (Brown and Passmore, 2009). Moreover, Kv7/M channel's activity is positively regulated by plasma membrane PtdIns(4,5)P levels (Suh and Hille, 2002; Telezhkin et al., 2012). Thus, receptors which activate the phosphoinositide lipid signaling cascade regulate IM (Yu, 1995; Selyanko and Brown, 1996; Cruzblanca et al., 1998; Wen and Levitan, 2002; Gamper and Shapiro, 2003; Linley et al., 2008). Altogether, these properties position IM suitable for controlling the resting potential, preventing ectopic firing in the absence of noxious stimuli, while allowing a shift to a more excitable states by receptor-mediated IM inhibition. Indeed, ever since it was discovered nearly 40 year ago (Brown and Adams, 1980) IM perturbations were strongly implicated in neuronal hyperexcitability underlying epilepsy and ALS (Yue and Yaari, 2004, 2006; Gu et al., 2005; Wainger et al., 2014), neuroinflammation (Tzour et al., 2016) and inflammatory, cancer and neuropathic pain (Linley et al., 2008; Liu et al., 2010; Roza et al., 2011; Zheng et al., 2013, 2015). In this context, we asked if IM in nociceptive neurons, is sufficient to maintain resting membrane potential and hence prevent spontaneous activity. In central neurons, application of the selective IM blocker, XE991 (Wang et al., 1998), or the activation of metabotropic glutamate receptors were shown to induce spontaneous firing (Shah et al., 2008; Lombardo and Harrington, 2016; Tzour et al., 2016). In peripheral nociceptive neurons, inhibition of IM by XE991 or linopridine, another IM blocker (Aiken et al., 1995) increased membrane excitability and induced membrane depolarization, but failed to induce spontaneous firing (Passmore et al., 2003; Linley et al., 2008; Liu et al., 2010). On the other hand, injection of XE991 in vivo to the hind paw led to prominent nocifencive behavior (Linley et al., 2012) and inhibition of IM in cutaneous sensory endings in ex vivo skin-nerve preparation induced ectopic activity in Aδ but not in C-fibers (Passmore et al., 2012).
Here we show that inhibition of IM by focal puff-application of low concentration of XE991 (either 3 or 10 μM) induces membrane depolarization followed by high frequency action potential firing in acutely dissociate rat nociceptor-like dorsal root ganglion (DRG) neurons. Using a multi-compartment computational model of a nociceptive neuron we demonstrate that IM, acting at nociceptive terminals is sufficient to prevent spontaneous activity of nociceptive neurons. Furthermore, IM provide a “safety zone,” such that substantial changes in persistent sodium current-mediated depolarizing conductances are required to induce spontaneous firing. Decrease in terminal IM induces spontaneous activation of nociceptive neurons after a small increase in persistent sodium current-mediated conductances, emphasizing the pivotal role of IM in controlling nociceptive excitability.
Materials and Methods
Ethical Approval
All animal procedures were approved by the Ethics Committee of the Hebrew University (Ethic number MD-15-14274-1).
Rat Lumbar DRG Cell Culture Preparation
Neurons were isolated from lumbar dorsal ganglions of 6–9 week old Sprague Dawley male rat as previously described (Binshtok et al., 2008; Nita et al., 2016). In short: lumbar DRGs (L1-L6) were removed and placed into DMEM with 1% penicillin-streptomycin, then digested in 5 mg ml−1 collagenase, 1 mg ml−1 Dispase II (Roche) and 0.25% trypsin, followed by addition of 0.25% trypsin inhibitor. Cells were triturated in the presence of DNase I (250 U) and centrifuged through 15% BSA. The cell pellet was re-suspended in 1 ml Neurobasal media, containing B27 supplement (Invitrogen), penicillin and streptomycin, 10 mM AraC, 2.5S NGF (100 ng ml−1, Promega) and GDNF (2 ng ml−1). Cells were plated onto poly-D-lysine (100 μg ml−1) and laminin (1 mg ml−1) coated 35 mm tissue culture dishes (Corning) at ~10 K cells per well, at 37°C with 5% carbon dioxide. Unless otherwise stated the materials were purchased from Sigma.
General Electrophysiology
Recordings were performed from small (~25 μm), capsaicin-sensitive (not shown) dissociated rat DRG neurons, up to 24 h after culturing. These neurons have been described in the literature to be nociceptive (Cardenas et al., 1995). Cell diameter was measured using Nikon Elements AI software (Nikon), from images acquired by a CCD camera (Q-Imaging). In experiments studying the role of IM in nociceptor-like DRG neurons (Figures 1, 2, 3A,B,E,F, Supplementary Figure 1), whole-cell membrane currents and voltages were recorded using a nystatin-based perforated patch technique (Horn and Marty, 1988) in voltage clamp and fast current-clamp modes, respectively, using a Multiclamp 700B amplifier (Molecular Devices), at room temperature (24 ± 2°C). All other experiments (Figures 3C,D, Supplementary Figure 2) were performed using voltage and current clamp in whole cell configuration. In all experiments, only the cells that showed less than 10% change in access resistance during the entire recording period were analyzed. Data were sampled at 50 kHz and were low-pass filtered at 20 kHz (-3 dB, 8 pole Bessel filter). Patch pipettes (3–5 MΩ) were pulled from borosilicate glass capillaries (1.5 mm/1.1 mm OD/ID, Sutter Instrument Co., Novato, CA, USA) on a P-1000 puller (Sutter Instrument Co.) and fire-polished (LWScientific). Access resistance was in the range of 4–8 MΩ. For voltage-clamp recordings, capacitive currents were minimized and series resistance was compensated by about 80%. Command voltage and current protocols were generated with a Digidata 1,440 A A/D interface (Molecular Devices). Data were digitized using pCLAMP 10.3 (Molecular Devices). Data averaging and peak detection were performed using Clampfit 10.3 software. Data were fitted using Origin 8 (OriginLab).
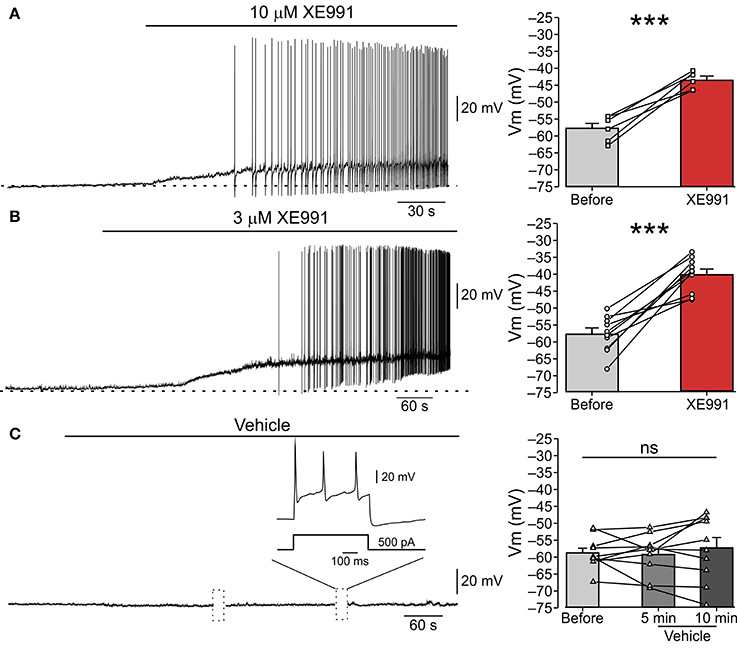
Figure 1. XE991 leads to spontaneous firing of nociceptor-like DRG neurons. (A) Left, Typical responses of nociceptor-like small (25 μm) DRG neurons to focal puff application of 10 μM XE991 (representative of 5 out of 6 experiments). Current clamp perforated-patch recordings performed from acutely dissociated DRG neurons perfused with extracellular solution. Note, membrane depolarization and onset of spike discharges shortly after application of XE991. Dashed line indicate resting potentials before drug application (−58 mV). Right, mean ± SEM (bar graphs) and individual neurons' changes in membrane potential before (gray) and after application of XE991 (red, measured before generation of first action potential). ***p < 0.001, paired Student t-test, n = 6. Note that XE991-induced depolarization in all recorded neurons. (B) Same as in (A), but 3 μM XE991 was puff-applied on the neurons. Dashed line indicate resting potential before drug application (−59 mV). Left, representative of 10/11 neurons. ***p < 0.001, paired Student t-test, n = 11. Note that 3 μM XE991 induced depolarization in all recorded neurons. (C) Control experiment. 10 min long puff application of vehicle onto nociceptor-like DRG neuron. Note, no spontaneous depolarization developed during this period, yet the cell fired normally upon injection of depolarizing current pulses (inset; representative of 9 experiments). Dotted boxes show time breaks in free-run recordings when current protocols where applied. Right, same as in (A,B), but measured 5 min and 10 min after puff application of vehicle; ns—not significant, one-way ANOVA, n = 9.
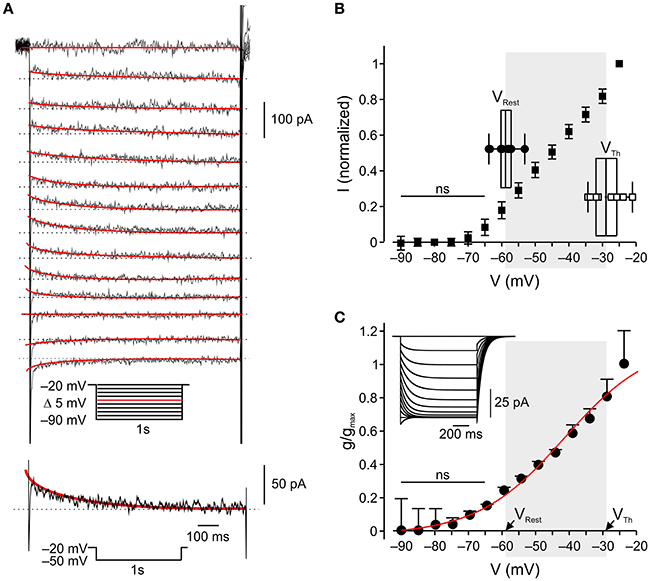
Figure 2. In nociceptor-like DRG neurons IM is outward at resting-to-threshold potential range (A). Upper, Typical voltage-clamp perforated patch recordings of IM from a nociceptor-like DRG neuron (see Methods, representative of 18/18 neurons). A family of currents was evoked by a series of 1 s, 5 mV hyperpolarizing voltage steps from a holding potential of −20 mV (voltage protocol is shown in inset). The peak current response obtained by stepping to −50 mV is shown at the bottom. The IM relaxation was fitted with a bi-exponential line (red), which was extrapolated to the beginning of the voltage step. IM amplitudes were measured as the differences between the instantaneous peak currents at command onset and the steady-state currents just before command offset (dotted line). (B) Averaged leak-subtracted peak I-V characteristics of IM recorded from nociceptor-like DRG neurons (n = 18), calculated as described in Methods. ns—not significant; one-way ANOVA comparison between the current values obtained at −90 mV (zero current level) to currents at other command voltages. Note, significant outward current at −60 mV. Insets show box charts and individual values of resting membrane potentials (VRest, n = 10) and action potential thresholds (VTh, n = 9), obtained from current clamp perforated patch recordings (see Methods). The middle line of the box charts represents the mean; the box represents 25 ~ 75% percentiles and caps delineate range within 1.5 interquartile range. The mean (in mV) is aligned to its values on the x-axis. The shadowed area indicates the range of membrane potentials between resting potential and threshold. (C) Mean ± SEM of IM activation curves (g/gmax) fitted using the Boltzmann equation (see Methods) shows the onset of activation at −60 mV (ns—not significant; one-way ANOVA comparison between the conductance values obtained at −90 mV (zero conductance level) to conductances at other command voltages) and V1/2 at −42 mV. The shadowed area indicates the range of membrane potentials between resting potential (mean, VRest) and threshold (mean, VTh) taken from (B), insets.
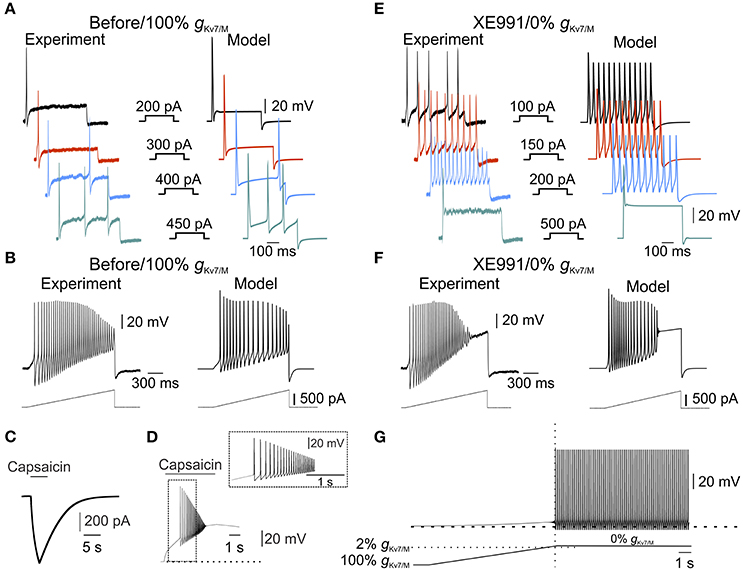
Figure 3. IM is sufficient to prevent spontaneous firing. (A) Typical voltage responses to increasing current steps (showed in the middle panel) recorded from nociceptor-like cultured DRG neuron using perforated-patch, in current clamp mode (left) or from a single-compartment model (right, see Methods). Recordings were performed on the same DRG neuron before and after application of XE991 (shown in (E), left). Traces from the model were obtained using the model with intact gKv7/M. Note the similarity between the simulated to experimental responses. (B) Same as in (A), but showing responses to 1.5 s depolarizing current ramps (250 pA/s, showed below) recorded from nociceptor-like cultured DRG neuron using perforated patch in current clamp mode (left) or from the single-compartment model (right). Recordings were performed on the same DRG neuron before and after application of XE991 (shown in (F), left). Note the similarity between the simulated and experimental responses. (C,D) Validation of the capsaicin-like stimulation on a single compartmental model. (C) Current responses evoked by capsaicin-like stimulation (5 s, 1 μM, see Methods) when applied on a single-compartment model. (D). Capsaicin-like stimulation evokes a barrage of action potentials in the simulated neuron. Dotted line indicates the membrane potentials before the stimulation (−57.85 mV). (E,F) Voltage responses to increasing current steps (E, showed in the middle panel) or current ramps (F, showed below) recorded from the same neuron shown in (A,B), 10 min after application of 3 μM XE991 (left) or from the simulated neuron with gKv7/M = 0 (right). All measurements described in panels (E,F) were performed at the native resting potential, adjusted after XE991 application or when gKv7/M = 0, by injecting appropriate repolarizing currents. Note that inhibition of IM leads to increase in neuronal excitability which was well reflected in the simulated neuron. (G) Free run recording of membrane potential in a simulated single-compartment modeled neuron during a gradual decrease in gKv7/M (the rate of decrease in gKv7/M shown below, see Methods). Dashed lines indicate resting potential before changes in gKv7/M (−57.85 mV). The vertical dotted line indicates time of first action potential. Horizontal dashed line indicates the level of gKv7/M at which the first action potential occurred. Note that in the simulated neuron, decrease in gKv7/M lead to slow depolarization followed by spontaneous firing, similar to the experimental results obtained after application of 3 and 10 μM XE991 on nociceptor-like DRG neurons (Figure 1).
Nystatin-based pipette solution (290 mOsm) for perforated patch recordings was freshly prepared in the dark every 2–3 h and contained (in mM): 140 KCl, 1.6 MgCl2, 2 EGTA, 10 HEPES, 2.5 Mg-ATP, 0.5 Na-GTP (pH = 7.4).
Nystatin (Sigma-Aldrich) was dissolved in DMSO (Sigma-Aldrich) to obtain a 50 mg ml−1 stock solution, which, after 1 min ultra-sonication, was diluted in pipette solution to obtain a working concentration of 125 μg ml−1.
The intracellular solution for measuring capsaicin-induced current contained (in mM): 145 KCl, 5 NaCl, 2.5 MgCl2, 2 MgATP, and 0.1 EGTA (pH = 7.4).
The intracellular solution for measuring capsaicin-evoked firing contained (in mM): 140 K-Aspartate, 10 NaCl, 2 MgCl2, 4 MgATP, 10 HEPES (pH = 7.4).
The extracellular solution contained (in mM): 145 NaCl, 5 KCl, 1 MgCl2, 10 HEPES, 10 D-Glucose (pH = 7.4).
Pipette potential was zeroed before seal formation and membrane potential was corrected for liquid junction potential of −4.5 mV.
Only cells that generated at least one action potential in response to current injections were then used for capsaicin application and analysis.
Current Clamp Recordings
Only neurons that demonstrated a stable resting potential and stable action potential threshold during a 5 min application of vehicle (extracellular solution) were analyzed. In some experiments we applied vehicle solution for an additional 15 min to rule out possible time-dependent changes in neuronal excitability. No changes in the neuronal excitability were observed during the period of vehicle application (n = 9).
The action potential thresholds and neuronal firing properties were assessed by analyzing the neuronal responses to a series of depolarizing steps (1–2 nA in increments of 0.05 nA each) and 1.5 s depolarizing current ramps (250 pA/s). Action potential threshold was obtained by analyzing phase plots (dV/dt) of first spike evoked by 400 pA, 500 ms step when plotted versus time or versus membrane voltage. The voltage of the threshold was measured using “first local minimum” of the function before the peak. The “first local minimum” was determined as the first minimal value of dV/dt after the peak, followed by an additional increase in the dV/dt, while analyzing the function from its positive peak to time “0.” The time for the first local minimum was defined as the time of threshold, and its voltage was then detected from the original trace. For the verification of threshold values, the thresholds were also determined from the response to the depolarizing ramps, as the potential at the onset of a clear sharp deviation from passive response with subsequent action potential generation.
Measurements of changes in action potential threshold and evoked firing, following application of XE991 were made at the original (control) resting membrane potential (Vm), maintained constant by injecting an appropriate steady current.
Voltage Clamp Recordings
To evoke IM 500 ms hyperpolarizing voltage steps incrementing by −5 mV, were applied from a holding potential of −20 mV (Halliwell and Adams, 1982; Caspi et al., 2009; Tzour et al., 2016). These steps induced slow current “relaxations” after the instantaneous inward current drops (instantaneous current), representing the slow IM deactivation. Current relaxations were fitted by bi-exponential curves (starting after the capacitance artifact) and were extrapolated back to the beginning of the hyperpolarizing command pulses. IM amplitudes were assessed as the differences between the instantaneous peak currents at command onset and the steady-state currents just before command offset.
The I-V curves were calculated according to Adams et al. (Brown and Adams, 1980) and Wang and McKinnon (1995) as follows: the intersection of I-V curves of normalized instantaneous and steady-state currents—VM, obtained from currents acquired as explained above, were measured. In our conditions VM was −75 mV (not shown). Then, the IM currents, were normalized to their maximal values and plotted vs. membrane voltages (Adams et al., 1982). The resulting I-V curve was upward shifted to align I(VM) to zero. Leak, obtained by extrapolation of the linear portion of the I-V curve between −80 to −70 mV, was then subtracted according to Passmore et al. (2003).
The conductance of IM (gKv7/M) was assessed according to Adams et al. (1982). Briefly, we averaged the currents at each command-voltage from all recorded neurons. The resulting I-V curve was upward shifted to align I(VM) to zero. Leak, obtained by extrapolation of the linear portion of the I-V curve between −80 to −70 mV, was then subtracted according to Passmore et al. (2003). The current values were then divided by the driving force and normalized to the maximal value. The data were fitted by a Boltzmann curve:
where ν1/2 is voltage at which the half-maximal activation of the channels occurs. The activation curve was best fitted with the following parameters: ν1/2 = −42mV, kl = 12, A1 = 1.13462, A2 = 1.2071.
Chemicals and Drugs
XE991 was purchased from Tocris Bioscience. Stock solutions were prepared in 10 mM and were diluted when added to the extracellular solution.
Focal Drug Application
XE991 or vehicle (extracellular solution) were focally and constantly applied onto somata of isolated DRG neurons using pressure puffs supplied by a pneumatic picopump PV820 (World Precision Instruments, Sarasota, FL, USA), connected to a fine pipette (3–5 MΩ). The pipette was placed about 25 μm away from the recorded cell.
Capsaicin (1 μM, Sigma) was puff-applied for 5 seconds via a pipette with a resistance of 2–5 MΩ which was placed 25 μm away from the recorded cell. Immediately after the puff application, capsaicin was washed out by perfusing with extracellular solution.
Computational Modeling
Simulations were performed using a small-diameter non-myelinated DRG compartmental model neuron implemented and run in NEURON simulation software (Hines and Carnevale, 1997, 2000). First, the model validation was performed by simulating a single-compartment DRG soma-like 25μm X 25 μm cylinder model (Gudes et al., 2015) with a membrane capacitance of 1μF cm−2 and membrane resistance of 10000 Ω cm−2. The simulated resting potential was −57.85 mV, which fits our experimental results (−58.15±0.9 mV). The model was adjusted to reach similarity of the simulated responses compared to experimental results. Then, the single-compartment was evolved into a multi-compartment model of pseudo-unipolar unmyelinated nociceptive neuron which includes a DRG-like soma connected to a stem axon, expanding to peripheral and central neuron axons which join at a bifurcation site (T-junction, Figure 4). Simulations were performed assuming a room temperature of 25°C, the temperature at which the experimental data were collected.
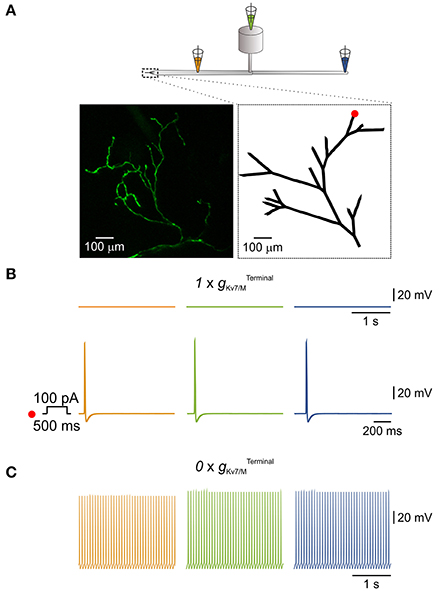
Figure 4. IM in the terminals is sufficient to prevent spontaneous activation of a simulated multi-compartment nociceptive neuron. (A) Scheme depicting the general structure of the multi-compartment model. Dotted box outlines the nociceptive terminal, enlarged in the right inset below. Orange, green, and blue pipettes indicate the sites from which the changes in membrane potentials were recorded and correspond to: peripheral axon (orange), soma (green), and central axon (blue). Insets, Left, in-vivo multiphoton imaging of nociceptive peripheral terminals and distal axons in the mouse hind paw expressing GFP (z-stack; see Methods). Right, geometry of the terminal tree based on the rendering of the image shown in left which was used in the NEURON environment to simulate nociceptive peripheral terminals and distal axons. The red dot indicates the stimulus loci. (B) Upper traces, recordings from peripheral axon (orange), soma (green), and central axon (blue) when terminal Kv7 conductance () was intact (, see Methods). Note, no spontaneous action potential firing, yet application of 100 pA depolarizing current for 500 ms at the distal terminal branch (indicated as a red dot in A, inset, right) lead to generation of a single action potential which propagated along the neuron. (C) Annulling of () lead to generation of spontaneous action potential firing at the distal axons (orange) which fully propagated along the neuron (green and blue).
Multi-Compartment Model Morphology
The morphology of the cell body was based on the single compartment model. The morphology and passive properties of the T-junction and axons were based on previous studies (Du et al., 2014; Sundt et al., 2015). The terminal morphology was based on our experimental observations (see Figure 4 and Methods below) and was constructed accordingly. The terminal tree contained a total of 27 branches with variable lengths (50–300 μm) and a diameter of 0.25 μm. The membrane resistance of the nerve ending branches was 4-fold somatic membrane resistance (Vasylyev and Waxman, 2012). To achieve morphological continuity for proper electric propagation from the terminal tree to the peripheral axon the two compartments were connected by a tapered cone-like axon with a linearly changing diameter over 100μm of length.
Active Conductances
The model includes active conductances simulating the following channels: TTX-sensitive sodium current (INattxS), TTX-s sensitive persistent sodium current (INaP), Nav1.9 TTX-r sodium channels (INav1.9) and Nav1.8 TTX-r sodium channels (INav1.8). All channels parameters of the sodium currents were adapted from Herzog et al. (Herzog et al., 2001) and Baker et al. (Baker, 2005). Three types of potassium channels included: (i) the delayed rectifier channel (IKDR) adapted from (Herzog et al., 2001); (ii) An A-type potassium channel (IKA) adapted from Miyasho et al. (Miyasho et al., 2001), whose activation and inactivation gates were shifted by 20 mV in hyperpolarized direction to closely resemble kinetics of DRG neurons (Qu and Caterina, 2016) and (iii) the Kv7/M channels which were adapted from Shah et al. (Shah et al., 2008) and tuned to our experimental results. The h-current (Ih) was also included and taken from Shah et al. (Shah et al., 2008), the slope factor was tuned according to Komagiri and Kitamura (Komagiri and Kitamura, 2003).
For simulating the excitable properties of a single-compartment neuron we used the following fixed maximal conductance parameters:
Since there is no information available regarding the distribution of channels across the nociceptive axons, for maintaining simplicity, conductances were evenly distributed in all compartments.
Kv7/M-Current Potassium Current Model
The Kv7/M current was defined as:
where gKv7/M is maximal conductance, m is the activation gate parameter, v is the membrane potential and EK is the potassium reversal potential value.
To implement our experimental results, we adjusted the voltage-dependence of steady-state activation for Kv7/M current to our findings (see Figure 2). The activation curve was fitted using the Boltzmann equation to fit the activation gate parameter:
where ν1/2 is voltage at which the half-maximal activation of channels occurs. The activation curve was best fitted with the following parameters: ν1/2 = −42mV, kl = 12, A1 = 1.13462, A2 = 1.2071. The time constants were taken from Shah et al. (Shah et al., 2008).
To simulate the currents inhibition by XE991 we decreased the value of gKv7/M as mentioned in different simulations in this study. In some experiments (Figure 3G) gKv7/M was decreased linearly with time according to:
where tinit is the time of the beginning of change in gKv7/M; tFinal is a time by which the gKv7/M = 0. The decrease of gKV7/M was modeled in a linear and fast (seconds) fashion for computational resource and simplicity reasons, it was not intended to fully replicate the time course of experimental pharmacological IM blockade.
The equilibrium potentials for Na+ (ENa) and K+ (EK) were +60mV and −85mV respectively. ERev for Ih was −20 mV. The leak reversal potential was adjusted to achieve a resting potential close to our experimental results (−58.15±0.9 mV) such that the resulted resting potential of the modeled neuron was −57.85 mV. All recordings were made after letting the simulated membrane potential reach a steady-state value.
To model terminal gKv7/M (, we used the parameters of somatic gKv7/M ( for which the values were adjusted for the area of the cable-like terminal. This value was considered the intact and was increased or decreased by multiplying it by varying factor. The relevant figures show the multiplication factor as independent variable.
Capsaicin Puff-Like Stimulus
A capsaicin-like induced current was introduced into a single simplified voltage clamp point-process with a fast exponential activation and slow exponential inactivation mimicking the experimental kinetics of puff-applied 1 μM capsaicin induced current (Nita et al., 2016):
for t ≤ tpuff
for tpuff<t
where gCap is the maximal conductance, α(x) and β(x) are the activation and inactivation functions with τα and τβ as the activation and inactivation time constants respectively. tonset and tpuff are the times at which the puff application simulation begins and the length of application respectively. ETRPV1 represents the reversal potential for TRPV1 cation channel.
The current was first tuned to fit experimental results (Supplementary Figure 2A, see also Nita et al., 2016) and then it was applied to the model neuron to verify that the induced voltage response for the experimental capsaicin-induced firing (Supplementary Figure 2A, see also Nita et al., 2016). Since our experimental recordings were obtained in DRG culture neurons which are considered to consist of only DRG soma, both adjustment and verification were performed in a soma-like model. The parameters for the capsaicin-like current were tuned to following values:
In experiments simulated voltage clamp, to avoid action potential-mediated escapes from voltage clamp, capsaicin-like currents were measured when Na(v)1.8 conductance was zeroed.
Noise Current
The noise which was injected into whole terminal tree, was based on the Ornstein-Uhlenbeck process with mean 0, and the current was taken from Olivares et al. (Olivares et al., 2015).
Where σ is square root of the steady state variance of the current amplitude; N - is a normally distributed random variable with zero mean and variance = 1 and τ is the steady-state correlation time length of the current (was fixed to 1 ms).
Terminal Excitation
When simulating an electric stimulation and recording from a single terminal, a NEURON “point process” electrode was positioned, stimulus was given and data was collected at an arbitrarily chosen single terminal branch - “Terminal (Yu, 1995)”. To avoid boundary condition problems, the stimulating electrode or capsaicin puff-like process were positioned at 15% of the branch length taken from the branch's distal ending.
In-vivo Sciatic Viral Injections
Viral injection to the sciatic nerve was performed similarly to previously described (Towne et al., 2009; Iyer et al., 2014). In short: 4-6 week old male C57/BL6 mice were anesthetized by 3% isoflurane for induction of anesthesia and kept at 1-1.5% isoflurane throughout the procedure. Mice were treated with 10 mg/kg carprofen via subcutaneous injection prior to surgery and placed on a heating pad maintained at 37 °C. After properly cleaning and disinfecting the skin of the left hind limb and lower back of mice, a 1 cm incision was made around the knee area and the sciatic nerve was exposed at the bifurcation of the Common Peroneal and Tibial nerves. A 35G beveled needle (Nanofil no. NF35BV-2, World Precision Instruments) was threaded into the Tibial branch of the nerve and 5 μl of an adeno-associated virus serotype 6 (AAV6) carrying an expression cassette for GFP under the CMV promotor (AAV6-CMV-GFP, ELSC Viral Core, The Hebrew University) was injected at a rate of 1 μl/min, using a 10 μl syringe (Hamilton Company) connected to a UltraMicroPump (UMP3) with a SYS-Micro4 Controlled WPI Syringe pump (WPI). After injection, the needle was kept inserted in the nerve for another 5 min to allow for pressure stabilization. The incision was then sutured and treated with 3% synthomycin ointment (Rekah, Israel). Mice were treated post-operatively every 12 hours for 3 days with 5 mg/kg carprofen.
In-vivo Multiphoton Z-Stack Projection of Peripheral Free Nerve Endings Innervating the Skin of The Mouse Hind Paw
Imaging was performed similarly to previously described (Yuryev and Khiroug, 2012). In short: 4 weeks post viral injection; mice were anesthetized by 3% isoflurane for induction of anesthesia and kept at 1-1.5% isoflurane throughout the experiment. Mice were placed under the microscope on a heating pad maintained at 37°C and the left hind paw was placed in between 2 glass slides with a few drips of 0.9% w/v NaCl solution between the skin of paw and glass. Hind paw and glass slides were stabilized by a Noga holding system (Noga Engineering, Ltd, Israel). Multiphoton Z-stack projection of free nerve endings in hind paw was performed using a Zeiss LSM 7 MP system mounted on a Multiphoton Axio microscope (Carl Zeiss), a Chameleon Ultra II Diod-Pumped Laser (Coherent) and Zen2010 software (Carl Zeiss). The objective used for fluorescence collection was a W Plan-Apochromat 20x/1.0 DIC CG = 0.17 M27 75 mm water immersion lens (Carl Zeiss). Laser excitation wave length was set at 900 nm and average power set to 200 mW. Emission was collected by two channel NDD PMTs, for green and red emission wave lengths. Images were acquired at a resolution of 1024 × 1024 pixels in the x, y plane (530.85x530.85 μm, 8 bit, 0.8 zoom) and z vertical steps were set at 0.5 μm (image was cropped for figure). 172 sections were acquired giving an image z projection of 86 μm. Images were further processed off-line using FIJI (ImageJ) for subtraction of red channel acquisition and linear un-mixing of the two channels to remove auto-fluorescence of surrounding skin structures.
Data Analysis
Offline analyses were performed with pCLAMP 10.3 software (Molecular Devices), Matlab and OriginPro v8. Assessment of statistical significance of differences between means was performed with Student's paired t test or repeated-measures of ANOVA, as appropriate. Data are presented as means ± SEM.
Results
Application of the IM Inhibitor, XE991, Causes Repetitive Firing of Cultured Nociceptor-Like Rat DRG Neurons
To test if IM prevents spontaneous activity of nociceptive DRG neurons we performed perforated-patch clamp recordings in current-clamp mode from acutely dissociated small (less than 25 μm in diameter), nociceptor-like DRG neurons. At resting potential (−58.15±0.9 mV, Figure 1, right “Before”), all neurons were quiescent (n = 26, Figure 1). Focal puff application of the IM blocker XE991 (10 μM; applied ~2 min after the onset of recording) caused, within 3-5 min, a substantial depolarization (12.35 ± 1.8 mV; Figure 1A, right “XE991”) in all recorded neurons followed by repetitive firing in 83% of the neurons (5/6; Figure 1A, left). This excitatory effect persisted throughout drug application (1-5 min) and in most cases was also observed during washout (not shown).
Several recent reports demonstrated that 10 μM XE991 can also affect Kv1 and Kv2/Kv9 channels (Zhong et al., 2010) and delayed rectifier (IK) and A-type (IA) potassium currents (Xia et al., 2002). We therefore performed the abovementioned experiment using focal application of a lower concentration of XE991 (3 μM) and showed similar effects on nociceptor-like DRG neuronal excitability. Puff application of 3 μM XE991 lead, within 4–6 min, to 17.58 ± 2.4 mV membrane depolarization in all neurons and repetitive firing in 91% of the neurons (10/11; Figure 1B). To reassure that the observed spontaneous activity was due to XE911 mediated IM inhibition and not due to mechanical interference caused by pressure during puff application, we recorded changes in nociceptor-like DRG neurons activity following puff application of vehicle (see Methods). Puff applied vehicle exerted no neuronal effects during sustained recordings (up to 15 min; n = 9; Figure 1C). In the latter condition the neurons displayed normal evoked firing behavior (n = 9; Figure 1C).
IM Is Outward at Resting Potential of Nociceptor-Like DRG Neurons
The abovementioned results, showing that selective inhibition of IM leads to spontaneous activity in nociceptor-like DRG neurons, indicate that IM provides sufficient outward current to prevent suprathreshold neuronal depolarization in the resting-to-threshold membrane potential range. To characterize the activation properties of the Kv7 channels underlying IM in this membrane potential range, we first measured the action potential threshold of nociceptor-like DRG neurons using perforated-patch clamp recordings in the current clamp mode. We measured spike threshold by analyzing the phase plot (dV/dt) of single action potential evoked by 500 ms steps. We further verified the action potential threshold using 500 ms current ramps (see Methods). In our experimental conditions the threshold for action potential in nociceptor-like neurons was −31.1±2.2 mV (n = 14), which was similar to the one measured using ramps (−29.4±2.2, mV n = 9, see inset for Figure 2B).
Next we characterized the voltage dependence of IM activation. We used a well-established voltage-clamp protocol in the perforated-patch whole cell configuration to isolate IM and to generate I-V and conductance slope curves (Brown and Adams, 1980; Passmore et al., 2003; Wang and McKinnon, 1995; see Methods; Figure 2A, inset). In our conditions IM was activated at −60 mV (n = 18, Figures 2B,C) similarly to what was previously shown (Passmore et al., 2003). Considering the reversal potential of IM (Figure 2B), these data indicate that IM is active and hyperpolarizatory at resting-to-threshold potentials in nociceptor-like DRG neurons.
Finally, we verified that IM in nociceptor-like DRG neurons was attenuated by 3 μM XE991 (Supplementary Figure 1).
IM Is Sufficient to Prevent Spontaneous Firing of Nociceptor-Like DRG Neurons
Our data show that XE991 abolishes IM and leads to spontaneous firing. To link between these two effects of XE991 and to show that IM is sufficient for averting nociceptive ectopic activity, we built a single-compartment computational model of a nociceptor-like DRG neuron using NEURON simulating environment (Hines and Carnevale, 1997). We used our experimental data to determine the kinetic properties of the IM-mediated conductance and tuned the I-V relationship according to the steady-state I-V curve shown in Figure 2B. We adapted the IM current model from Shah et al. (Shah et al., 2008) and adjusted its parameters to fit our findings. The properties of the resulting simulated current resembled the amplitude, kinetics and voltage dependence of IM recorded from nociceptor-like DRG neurons (Figure 2C, inset). To simulate electrical properties of DRG neurons, we based our model on previously described models of nociceptor-like DRG neurons (Gudes et al., 2015; Herzog et al., 2001; Baker, 2005; see Methods). Under these conditions the resting potential of the simulated membrane was −57.85 mV and the threshold was −30.27 mV, which were similar to the experimental values (shown in Figure 2B, insets). Importantly, the simulated responses to current steps (Figure 3A) and depolarizing ramps (Figure 3B) replicated well the experimental findings. We then validated the response of the modeled soma to “natural” stimuli. To that end, we simulated a current induced by application of capsaicin, an activator of the noxious heat-sensitive TRPV1 channel, expressed by nociceptive neurons. We and others have previously demonstrated that application of capsaicin produces depolarization of nociceptive neurons, followed by a stereotypic pattern of action potential firing (Nita et al., 2016; Blair and Bean, 2003; see also Supplementary Figure 2, here). To mimic the capsaicin-induced current, we first introduced an inward current with the activation and inactivation kinetics resembling TRPV1-induced inward current evoked by short (5s) puff application of capsaicin (1 μM) into our single-compartment model (see Methods). The stimulation of the soma with a modeled 5 s application of capsaicin (capsaicin-like stimulation) resulted in an inward current and action potential firing, which resembled the experimental data (Figures 3C,D, compare to Supplementary Figure 2A,B respectively, see also Nita et al., 2016).
Next, we explored whether our model would reflect the hyperexcitable changes following inhibition of IM. It is widely accepted that IM inhibition apart from membrane depolarization (Figure 1) leads to decrease in spike threshold and increase in firing rate (Yue and Yaari, 2004; Gu et al., 2005; Linley et al., 2008; Liu et al., 2010; Tzour et al., 2016). Similarly, in our experimental conditions inhibition of IM decreased spike threshold (−34.94±4.9 mV, n = 8) and increased action potential firing evoked either by depolarizing steps (compare Figure 3A, left to Figure 3E, left) or ramps (compare Figure 3B, left to Figure 3F, left). To examine if our model would reflect the hyperexcitability induced by IM inhibition we omitted the IM-like conductance (gKv7/M) from the model without altering any of the other parameters. The negative steady state current was applied to hyperpolarize the membrane to its initial value. In these conditions, the action potential threshold was decreased to −32.82 mV. Moreover, application of depolarizing steps or ramps accurately reflected the changes we observed in our experimental conditions when 3 μM XE991 was puff applied onto the nociceptor-like DRG neurons (Figures 3E,F). Thus, our model accurately predicts excitable properties of nociceptor-like DRG neurons, under the limitations and assumptions made (see Methods).
Finally, using this model, we show that gradual attenuation of gKv7/M (see Methods) lead to membrane depolarization followed by spontaneous action potential firing (Figure 3G). The action potential firing was induced only when at least 98% of somatic gKv7/M ( was blocked (Figure 3G, bottom). These simulated results suggest that IM is sufficient to prevent spontaneous activity of nociceptor-like DRG neurons.
IM Controls the Ectopic Activity of the Neuron, in Simulated Nociceptive Terminals
Nociceptive DRG somata robustly express Kv7 channels which underlie IM (Passmore et al., 2003). In addition, Kv7 channels are present at nociceptive peripheral terminals innervating the target organs (Passmore et al., 2012). Thus IM, as an integral part of the terminal conductance may contribute to the terminal excitability state. Indeed, subcutaneous application of XE991 into the hind paw induced increased grooming and flinching, suggesting that inhibition of IM at the terminals and distal axons leads to acute pain (Linley et al., 2008). It is therefore plausible that Kv7 channels, which are expressed at the nociceptive terminals prevent spontaneous activation of terminal and hence the ectopic activity of nociceptive neurons. To examine whether IM is sufficient to prevent spontaneous activation of nociceptive terminals we built a realistic multi-compartment model of nociceptive peripheral terminal and peripheral and central axons connected to cylindrical soma via a T-junction and stem axon (Figure 4A). To be able to closely replicate the geometry of the nociceptive terminal branches, we first assessed the structure of the nociceptive terminal tree in-vivo using multiphoton imaging of nociceptive terminal branches innervating the skin. To that end, we expressed a fluorescent marker in the sensory neurons innervating the mouse hind paw by infecting mice sciatic nerve unmyelinated C-fibers (Iyer et al., 2014) with an AAV6 carrying the expression cassette for GFP. The intact axons and terminals innervating the mice hind paw were visualized in-vivo and reconstructed in 3D, using multiphoton microscopy, two weeks after sciatic injection, as exemplified in Figure 4A, inset right. The 2D render of this 3D reconstruction was then used as a base for building a geometrical modeled structure of the terminal tree (Figure 4A, inset left).
We based some of the electrical parameter values of the model (see Methods) on data available from the literature (Pristera et al., 2012; Vasylyev and Waxman, 2012; Feng et al., 2015; Sundt et al., 2015), while some were obtained by data extrapolation. The model incorporates a repertoire of voltage-gated Na+, K+ conductances homogeneously distributed along the axons and the terminals and tuned to resemble experimental values (see Methods). To model terminal gKv7/M (, we used the parameters of somatic gKv7/M ( adjusted for the area of the cable-like terminal (see Methods). When the was intact (, see Methods) the simulated neuron was quiescent (Figure 4B, upper traces). In the latter condition, stimulation of one of the terminal branches (Figure 4A, inset right, red dot) by a 500 ms square pulse of 100 pA current, elicited activation of the distal axon which propagated along the soma and reached the central terminal (Figure 4B, lower traces). When was zeroed ( in the whole terminal tree (shown in Figure 4A, inset, right) a spontaneous firing with a rate of 13 Hz (the maximal firing rate in the model) was observed, which started at the peripheral axon and propagated throughout the neuron (Figure 4C). These data suggest that IM acting at the nociceptive terminals is sufficient to prevent spontaneous activation and therefore ectopic activity of nociceptive neurons.
We next estimated, using our multi-compartment model, what is the value of which is necessary to prevent spontaneous activation of nociceptive neurons. To that end, we gradually reduced the and examined at which the simulated neuron starts to fire spontaneously (Figure 5). We found that even after inhibition of 63% of the remaining is sufficient to prevent spontaneous firing (Figure 5A). Lowering beyond 37% elicited spontaneous firing with a frequency which progressively increases with decrease of (Figures 5A,B).
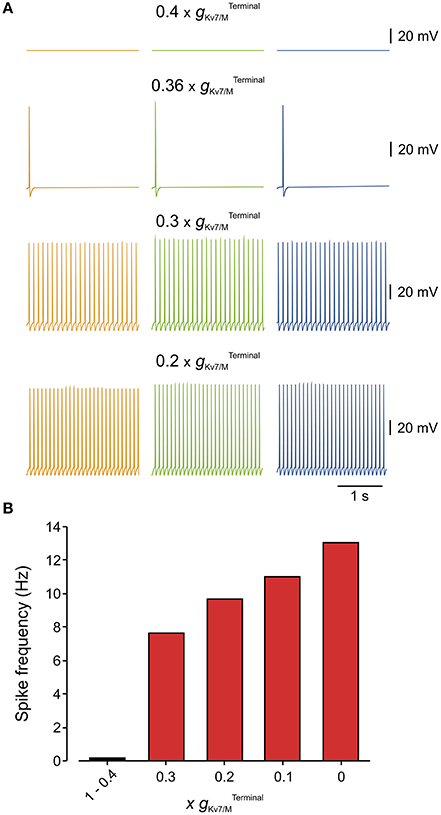
Figure 5. Only 40% of terminal gKv7/M is sufficient to prevent spontaneous firing of the modeled multi-compartment nociceptive neurons. (A) Simulated recordings of changes in membrane potentials obtained from a multi-compartment model at peripheral axon (orange), soma (green), and central axon (blue, see Figure 4). Monitoring of spontaneous firing was performed when different levels of were added to the model. Note that when 60% of was blocked () the neuron was still silent. Inhibition of 64% of () lead to spontaneous firing of 2 action potentials. Note also that the frequency of spontaneous firing increased with the decrease in (B) Bar graph plotting the spike count at different levels of inhibition (shown as a factor by which was multiplied) measured from the central axon of the multi-compartment model (blue electrode in Figure 4). At (no inhibition) till (60% inhibition) no spontaneous firing was observed, thus these values were combined into one bar.
Decrease in Terminal Kv7 Conductance Leads to Increased Responsiveness of the Simulated Nociceptive Neuron
Kv7/M channels are modulated by various signaling molecules. These channels require bound PIP2 for opening, and GqPCRs such as the muscarinic acetylcholine receptor causes IM inhibition by PLC-induced PIP2 depletion (Suh and Hille, 2002; Zhang et al., 2003; Liu et al., 2010). Moreover, both IP3-induced increases in intracellular Ca2+ concentration (Jones et al., 1995) and PKC activation (Marrion, 1994; Lee et al., 2010) have been implicated in IM inhibition. These signaling molecules and downstream signaling cascades are activated during inflammation. It is therefore plausible that during inflammation even partial inhibition of could increase the sensitivity of nociceptive neurons to incoming stimuli, thus leading to inflammatory hyperalgesia (Liu et al., 2010). We examined this hypothesis using our multi-compartment model in which we “inhibited” half of the Interestingly, although in these conditions, simulated neurons remained silent (Figure 6A, upper traces), stimulation of the terminal branch (as shown in Figure 6A, right, red dot) by a 500 ms square pulse of 100 pA current produced robust activation of the distal axon which propagated along the soma and reached the central terminal (compare Figure 6A, lower traces with Figure 4B, lower traces).
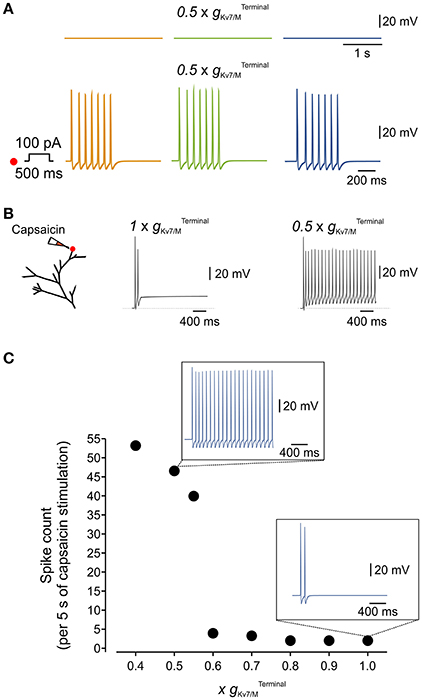
Figure 6. Terminal IM controls the responsiveness of the simulated nociceptive neuron to incoming stimuli. (A) Blockade of 50% of terminal Kv7/M channels did not lead to spontaneous activation of the simulated neuron (upper traces) but increased its responsiveness to a 100 pA, 500 ms current pulse applied to its distal terminal branch (lower traces, compare to Figure 4B). The location of stimulation is the same as in Figure 4. (B) Responses to application of capsaicin-like current. Left, Scheme of the modeled terminal showing the location of capsaicin application. Middle, recording from the simulated terminal following application of capsaicin-like stimulus when is intact. Right, the same stimuli induced a substantially stronger activation of the terminal when 50% of is inhibited. (C) Graph plotting changes in number of capsaicin-like stimulus' induced spikes (measured at the central axon during 5 sec application of capsaicin-like stimulus) with changes in (shown as a factor by which was multiplied). Insets, Simulated recordings of changes in membrane potentials obtained from a multi-compartment model at the central axon (blue electrode in Figure 4) following the same capsaicin-like stimulus (shown in (B) when intact (lower) and reduced by half (upper). Note that decrease in elicited substantially stronger capsaicin-induced firing.
Next, we examined if partial inhibition of IM would enhance the responsiveness of the simulated neuron to “natural” stimuli. We introduced the modeled TRPV1/capsaicin current (see Methods) into the terminals of the multi-compartment model. We applied a capsaicin-like stimulation onto the same terminal branch of the simulated neuron, previously used for electrical stimulation (Figure 6B, left, red dot), while varying the . In “normal” conditions, i.e. with an intact , application of the capsaicin-like stimulus induced relatively weak activation of the terminal (Figure 6B, middle) which was transmitted toward the central axon (Figure 6C, lower inset). We next examined whether inhibition of to levels which are not sufficient to induce spontaneous firing, would be sufficient to affect capsaicin-mediated responses of the multi-compartment model neuron. Inhibition of up to 30% of did not change the response of the terminal to capsaicin (not shown) and capsaicin-induced firing (Figure 6C). Further decrease of lead to a gradual increase in response to capsaicin-like stimulus (Figure 6C). Inhibition of more than 40% of induced robust, step-like increase in activation of the terminal (exemplified for in Figure 6B, right), translated into about 11 Hz firing which was fully propagated toward the central terminal of the simulated neuron (Figure 6C, exemplified for in Figure 6C, upper inset).
Terminal Kv7 Conductance Stabilizes the Membrane Potential of Modeled Nociceptive Terminals
Nociceptive membrane potential undergoes subthreshold fluctuating perturbations which are increased after nerve injury (Amir et al., 1999, 2002; Liu et al., 2000). To examine the contribution of IM in impeding spontaneous firing during membrane fluctuations we have stimulated the terminal tree of the multi-compartment model by injecting an Onstein-Uhlenbeck-based current noise (see Methods), in which the current amplitude varies normally in each time-step of 0.25 ms (Olivares et al., 2015), while changing . We varied the standard deviation of the current amplitude's normal distribution (σ) thus changing the probability of reaching a higher injected currents and causing higher membrane fluctuation amplitude (Figures 7A–D, upper traces). In normal conditions, i.e. when was intact, the modeled neuron remained quiescent despite fast membrane fluctuations at the single terminal of up to 15 mV (up to σ = 0.007; exemplified for σ = 0.004 in Figure 7A; exemplified for σ = 0.006 in Supplementary Figure 3; Figure 7F, black). When was reduced to half (Figure 7B), which, as we showed above, was not sufficient to induce spontaneous firing of the neuron (Figures 5, 6) neuronal membrane fluctuations of even 6 mV (σ = 0.004), were sufficient to elicit repetitive firing at the terminal tree which propagated toward the central terminal (Figures 7B,E, red).
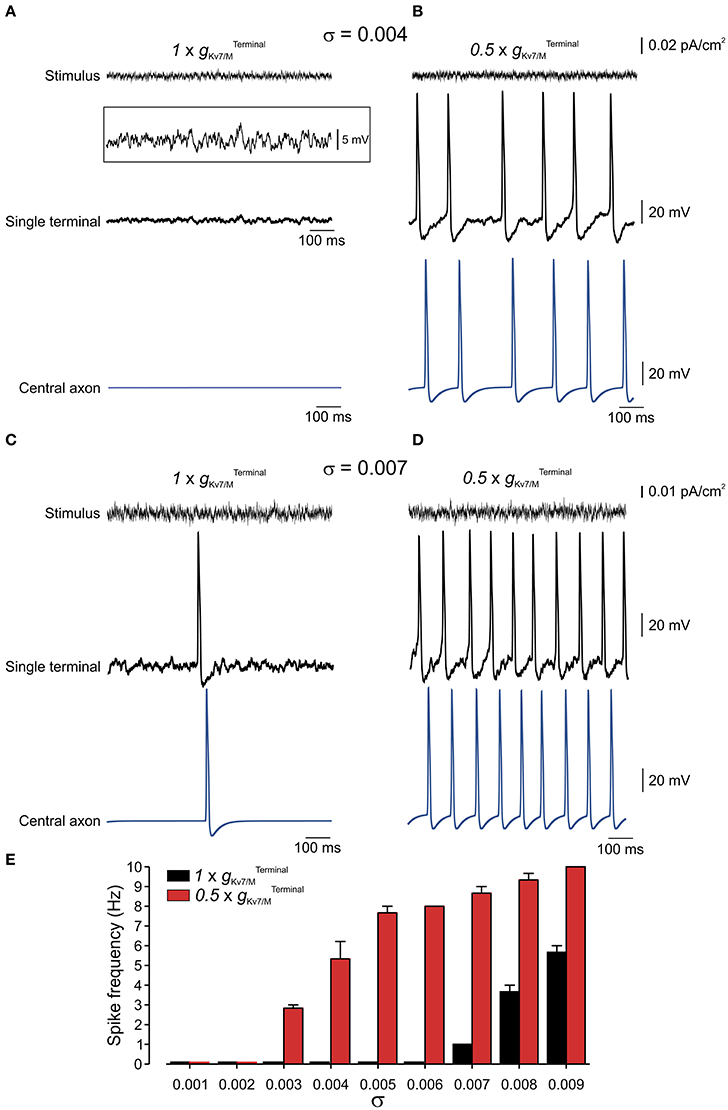
Figure 7. Terminal gKv7/M opposes membrane potential fluctuations. (A) Upper, Example of current noise trace (σ = 0.004) injected into the whole terminal tree of the multi-compartment model of the nociceptive neuron with intact Middle, simulated recordings of changes in membrane voltage in single terminal following injection of the current noise (shown above), expanded in y-axis in inset. Lower, simulated recordings of changes in membrane voltage in central axon (blue electrode in Figure 4) following injection of the current noise (shown above) to the terminal tree. Note that at this noise level no activity is elicited at either terminal or soma. (B) Same as (A) but is reduced by half . (C,D) Same as (A,B) but with σ = 0.007. (E) Graph plotting changes in spike frequency with σ of current noise when intact (black) and reduced by half (red). Each bar represents 3 trials with a random noise input (see Methods). Note, increase in spike frequency as decreases.
In suprathreshold fluctuations, in which σ was increased to evoke low frequency firing (exemplified for σ = 0.007 in Figure 7C) reduction of to half led to a substantial increase in firing frequency of nociceptive neurons (Figures 7D,E). These data predict that IM reduces susceptibility of nociceptive neurons to membrane fluctuations. When is decreased the nociceptive neuron is activated by small perturbations of 6 mV, while when is intact, the neurons will still be quiescent even upon 15 mV fluctuations.
Terminal Kv7 Conductance Affects Nociceptive Firing in Pathological Conditions
Subthreshold excitability of nociceptive neurons is controlled, among other factors, by depolarizing conductances, contributed largely by sodium channel isoform 1.9 (Na(V)1.9) which underlie persistent TTX-resistant sodium current (Herzog et al., 2001; Dib-Hajj et al., 2002; Baker, 2005). To characterize the interaction between and terminal gNa(v)1.9, we simulated, using our multi-compartmental model, the effect of changing these conductances on the excitability of a modeled nociceptive terminal and whole neuron. When was zeroed neurons remained silent if gNa(v)1.9 was inhibited (Figure 8). In our conditions, at least 65% of terminal gNa(v)1.9 were required for spontaneous firing when was zeroed. These data suggest that at subthreshold potentials IM counteracts TTX-resistant persistent sodium current, thus precluding spontaneous firing.
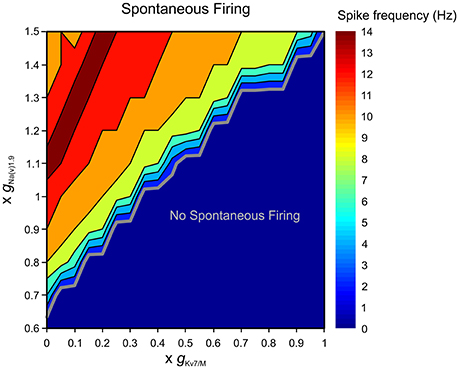
Figure 8. Terminal gKv7/M opposes gNa(v)1.9 in preventing spontaneous firing. Graphic representation of the relationship between the terminal gKv7 and gNa(v)1.9 in eliciting spontaneous firing of multi-compartmental model of nociceptive neurons. The recordings performed at the proximal axon of the modeled neuron (blue electrode in Figure 4). The thick gray line outlines the range of gKv7 and gNa(v)1.9 beneath which no spontaneous activity occurs (blue area). The frequency of the spontaneous activity is color coded (shown on the right). Note that when is intact, spontaneous firing can be elicited only when gNa(v)1.9 is increased above 140%. Note also that when is annulled, at least 65% of gNa(v)1.9 is required to induce spontaneous firing.
The enhancement of gNa(v)1.9 under pathological conditions has been shown to lead to activation of nociceptive neurons (Baker, 2005; Amaya et al., 2006; Binshtok et al., 2008; Maingret et al., 2008; Gudes et al., 2015). We examined the ability of to prevent spontaneous activity when gNa(v)1.9 was enhanced. We show that when is intact the simulated neuron remains silent even when gNa(v)1.9 is increased up to 145% (Figure 8). IM was also able to prevent stimulus-induced hyper-responsiveness when gNa(v)1.9 was increased up to 125% (Supplementary Figure 4). On the other hand, when was decreased, even a slight increase in gNa(v)1.9 resulted in spontaneous firing, such that when was reduced by half, about 10% increase in gNa(v)1.9 produced activation of modeled nociceptive neuron (Figure 8). As stated before, in these conditions, stimulus-evoked hyper-responsiveness was present even when gNa(v)1.9 was intact (see Figure 6A, Supplementary Figure 4).
Altogether these data suggest that when terminal IM is intact no spontaneous activity will occur even during substantial perturbation in depolarizing conductances in subthreshold range. It also implies that in pathological conditions spontaneous activation of nociceptive neurons could be more easily achieved by the cooperative effect of decrease in gKv7 and increase in gNa(v)1.9.
Discussion
Here we show that in peripheral nociceptive neurons IM plays a predominant role in stabilizing membrane potential at subthreshold potentials, acting to prevent ectopic activity of nociceptive neurons, in addition to its part in regulating nociceptive responsiveness to stimuli. We showed that inhibition of IM, in addition to increasing the susceptibility of nociceptive neurons to applied stimuli, also leads to spontaneous firing. Using a realistic multi-compartment computational model of peripheral nociceptive neurons we show that not all Kv7/M channels conductance at the nociceptive terminal tree is required to prevent spontaneous firing, but only ~40% of it is sufficient for this matter. We demonstrated that the “spare” Kv7/M conductance may serve to provide a “safety zone” against depolarizing membrane perturbations and to some extent to negate increasing subthreshold depolarizing conductances.
Activation of IM underlies a slow noninactivating K+ current known to be generated by KV7 channel subunits. In nociceptive neurons KV7.2/3 (KCNQ2/3) are predominately responsible for providing the majority of IM (Mucha et al., 2010; Du and Gamper, 2013). We and others have demonstrated that in nociceptive DRG neurons, these channels are activated at near resting potential such that at subthreshold potentials they produce a prominent outward current (Figure 2; see also Brown and Adams, 1980; Passmore et al., 2003), helping to keep the resting potential at hyperpolarized range. In addition to IM, the subthreshold excitability of DRG neurons is controlled by other, albeit weaker outward conductances (K2P and 4-AP sensitive voltage gated potassium channels, Du et al., 2014) which are counteracted by inward conductances: the persistent sodium current mediated largely by the Na(v)1.9 subtype of voltage gated sodium channels (Herzog et al., 2001) and hyperpolarization-activated cyclic nucleotide-gated Ih mediated mainly by HCN2 (Emery et al., 2011). Enhancement of Na(V)1.9-mediated current and activation of Ih has been shown to produce firing of DRG neurons (Baker et al., 2003; Baker, 2005; Emery et al., 2011), hence positioning active IM as a major factor to restrain membrane depolarization (Du et al., 2014) and prevent neuronal activation in lack of depolarizing stimuli. We show here, using pharmacological means, that IM-mediated conductance alone is sufficient to silence the nociceptive neurons. This conclusion is also supported by studies showing that application of the Kv7/M channels enhancers retigabine (Main et al., 2000) or NH6 (Peretz et al., 2007) lead to membrane hyperpolarization and reduces excitability of nociceptive DRG neurons (Passmore et al., 2003; Rivera-Arconada and Lopez-Garcia, 2006; Peretz et al., 2007). Moreover, retigabine reduces neuropathy-induced nocifencive behavior (Blackburn-Munro and Jensen, 2003) and prevents spontaneous firing of nociceptive fibers in models of neuropathic pain (Roza and Lopez-Garcia, 2008; Bernal et al., 2016).
Several recent reports demonstrated that inhibition of IM lead to increased responsiveness of nociceptor-like DRG neurons toward depolarizing stimuli (Passmore et al., 2003; Mucha et al., 2010; Shi et al., 2013; Du et al., 2014). IM has been shown to act as a brake on repetitive firing of nociceptive neurons such that its inhibition increases the firing rate of nociceptive neurons upon application of suprathreshold currents (Passmore et al., 2003). Moreover, inhibition of IM decreases action potential threshold together with inducing mild depolarization (Passmore et al., 2003; Linley et al., 2008). These effects together, if sufficient, could potentially lead to generation of spontaneous firing. Interestingly, although XE991-mediated spontaneous firing was reported in CA1 neurons and in spinal cord motor neurons (Shah et al., 2008; Lombardo and Harrington, 2016; Tzour et al., 2016), no XE991-mediated spontaneous firing was shown for nociceptive DRG neurons. It was suggested that XE991-induced depolarization increases the inactive fraction of sodium channels, probably via a slow inactivation mechanism (Blair and Bean, 2003; Binshtok et al., 2008; Gudes et al., 2015), which precludes action potential firing (Linley et al., 2008). If the latter is true, the differences in XE991 application could explain this discrepancy. In our experiments we used focal puff application of XE991. It could very well be that rapid access of relatively homogeneous concentrations of XE991 lead to depolarization with faster kinetics than the kinetics of sodium channels slow inactivation, rendering enough available sodium channels for action potential generation.
Kv7.2 channels are expressed both by nociceptive DRG somata and by their nerve endings (Passmore et al., 2003, 2012). XE991 injected subcutaneously at the rat's hind paw, induced nocifencive behavior (Linley et al., 2008), implying that inhibition of IM is sufficient to activate nociceptive terminals. To casually link between inhibition of IM at the soma and terminals and the firing of nociceptive neurons we have built computational models of nociceptive somata and of a whole nociceptive neuron. While building this in-silico nociceptive neuron we used data from the experimental results, when available, but for some of the parameters some assumptions had to be made. For example, since there are no available experimental data describing the various ion channel conductances at the terminal and along the axon, we assumed an equal and homogeneous distribution of the conductances between the soma, terminal and axon. We show here that lowering of terminal gKv7/M, i.e. decrease in terminal Kv7/M channels density, lead to substantial increase in nociceptive excitability. If, on the other hand, the density of the Kv7/M channels at the terminals is higher than that of the soma it will widen the “safety zone” provided by Kv7/M channels. However, other factors such as spatial distribution of Kv7/M channels and their co-localization with other channels (Battefeld et al., 2014) may affect the impact of Kv7/M channels on nociceptive excitability. In our model, for simplicity, we used a uniform distribution of all ion conductances including that of the Kv7/M channels along the terminal and axon. A recent study on cortical neurons shows that in their axon initial segment and nodes of Ranvier, Kv7/M channels are co-localized with the sodium channels (Battefeld et al., 2014). In these nodal domains, Kv7/M channels, by stabilizing the membrane resting potential, increase the availability of sodium channels, thus enhancing neuronal excitability. In the somato-dendritic domain of the cortical neurons, where Kv7/M channels are present in lower density and no co-localization with sodium channels was observed, activation of Kv7/M channels dampen neuronal excitability (Battefeld et al., 2014). It has been shown that application of retigabine onto nociceptive terminals in vivo inhibits evoked neuronal activity, whereas application of XE991 enhances it (Passmore et al., 2012), suggesting that Kv7/M channels in the nociceptive terminal tree play a restricting role on nociceptive excitability. However, it is plausible that further along the peripheral axon, where sodium channels are arranged in higher density lipid rafts (Pristera et al., 2012), or in central terminals of the nociceptive axon, Kv7/M channels are not equally distributed but co-localized with the sodium channels. In this case they may affect nociceptive excitability differently form their effect on the terminal tree or the cell body. Therefore, although our models replicate well the experimental results (Figure 3), we make no claim that it models all nociceptive neurons in all states. The purpose of our models was to show the plausibility of our interpretation of sufficiency of gKv7/M to prevent ectopic activation of nociceptive neurons. Indeed, our single and multi-compartment model predicts the effect of inhibition of somatic or terminal Kv7/M conductance by showing spontaneous firing of the simulated neuron when Kv7/M conductance has been inhibited at the soma or terminal tree (Figures 3, 4). Interestingly, our simulated data suggest that in order to remove the IM-mediated clamping from the soma of DRG-like neurons almost all (98%) Kv7/M conductance has to be inhibited while in the terminals, spontaneous firing could be induced by inhibiting 63% of Kv7/M conductance. Recent results using an immunofluorescent approach have demonstrated that in neuropathic pain models the expression level of Kv7.2 in soma of injured neurons decreased by 65% (Cisneros et al., 2015). According to our model such a decrease may not be enough to produce spontaneous firing at DRG somata, but may lead to increase of their responsiveness to applied stimuli. However, if a similar decrease in Kv7 expression occurs homogenously along the neuron including the terminal tree, 65% decrease in Kv7/M conductance could be sufficient to induce ectopic activation, which may underlie nerve-injury mediated spontaneous pain.
In yet another case of pathological pain - inflammation - proinflammatory mediators act on a variety of subthreshold conductances to increase nociceptive excitability thus leading to inflammatory pain (Baker, 2005; Amaya et al., 2006; Binshtok et al., 2008; Maingret et al., 2008; Emery et al., 2011; Gudes et al., 2015). We have shown here that IM, when intact, provides a “safety zone” which prevents spontaneous firing even when some of the subthreshold depolarizing conductances are substantially increased. However, Kv7/M channels are by themselves common targets for proinflammatory mediators (Linley et al., 2008; Liu et al., 2010; Tzour et al., 2016). The proinflammatory agent, bradykinin was shown to decrease Kv7/M conductance, albeit by less than 50% (Liu et al., 2010). Our results show that such decrease may not be sufficient to elicit spontaneous firing. However, in the inflammatory conditions bradykinin acting together with other ingredients of the inflammatory soup, also increase persistent sodium current (Maingret et al., 2008). This complex effect of inflammatory mediators on the one hand narrowing the IM-provided “safety zone” and on other hand increasing persistent sodium current could lead to ectopic activation of nociceptive neurons and to inflammatory pain.
Collectively, our data position IM as a main factor acting to prevent spontaneous firing. It could however be that this is only true for specific subtypes of nociceptive neurons. It has been recently demonstrated that application of XE991 onto ex-vivo skin-nerve preparation lead to spontaneous activity in mechanical and heat sensitive Aδ fibers but not C-fibers (Passmore et al., 2012). This suggests that IM is sufficient to silence nociceptive Aδ fibers similarly to what we have shown here. It also implies that at sensory endings of C-fibers other conductances also contribute to stable membrane potential at the subthreshold levels (see Du et al., 2014).
In conclusion, our findings in combination with those of previous studies (see Delmas and Brown, 2005) suggest that Kv7/M channels at nociceptive neurons, are critical for controlling resting membrane potential hence impeding ectopic activity and pathological pain. Our data show that in normal conditions, IM provide a “safety zone” sufficient to oppose substantial depolarizatory deviations of membrane potentials thus preventing the membrane to reach action potential threshold. Finally, our data demonstrated that spontaneous firing of nociceptive neurons in pathological conditions strongly depends on a decrease in gKv7/M, emphasizing the importance of pharmacological enhancement of gKv7/M in treatment of pathological pain.
Author Contributions
OB: Conceived the study, designed and conducted the experiments, built the model, analyzed the data and wrote the manuscript; RG and YC: Conducted the experiments, analyzed the data and wrote the manuscript; BK and SL: Analyzed the data and wrote the manuscript; AB: Conceived the study, designed the experiments, analyzed the data, supervised the project and wrote the manuscript.
Funding
Support is gratefully acknowledged from the Deutsch-Israelische Projectkooperation program of the Deutsche Forschungsgemeinschaft (DIP) grant agreement BI 1665/1-1ZI1172/12-1 (OB, RG, YK, BK, SL, AB); European Research Council under the European Union's Seventh Framework Programme (FP7/2007-2013) / ERC grant agreement n° 260914 (OB, RG, BK, SL, AB) and the Hoffman Leadership program (RHG).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling Editor declared a shared affiliation, though no other collaboration, with the authors and states that the process nevertheless met the standards of a fair and objective review.
Acknowledgments
We would like to thank Dr. Maya Groysman, the manager of ELSC viral core facility for designing and providing viruses used in this study.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fnmol.2017.00181/full#supplementary-material
Supplementary Figure 1. IM is blocked by 3 μM XE991 in nociceptor-like DRG neurons. (A) Voltage-clamp perforated patch recordings from a nociceptor-like DRG neuron using the protocol to isolate IM (see Methods and legend of Figure 2) before and 10 min after application of 3 μM XE991. Each subpanel illustrates a family of currents evoked by a series of 1 s, 5 mV hyperpolarizing voltage steps from a holding potential of −20 mV (voltage protocol is shown in inset). The dashed line indicates zero current level. Note, a decrease in the holding current. The peak current responses obtained by stepping to −50 mV are shown at the bottom of each subpanel. The red line is a bi-exponential fit of IM. The dotted line indicates the levels of steady state currents before command offset. Note XE991 inhibition of IM (representative of 4/4 experiments). (B) Subtracted trace of peak current response obtained before (shown in (A), bottom left) from those after application of XE991 (shown in (A), bottom right). Lines as in (A). (C) Bar graph comparing peak IM amplitudes measured at −50 mV (showed in (B), inset) in the two conditions as in (A); ***p < 0.001, Student t-test; n = 4.
Supplementary Figure 2. Capsaicin-induced current and voltage responses in nociceptor-like DRG neurons. (A) Representative trace of a capsaicin-induced current (1 μM) recorded evoked by 5 s puff application (representative of 10 neurons). (B) Representative trace of the changes in membrane potential in nociceptor-like neuron shows substantial depolarization followed by action potential discharge (inset: expanded time scale) induced by puff application of capsaicin (5 s, 1 μM). The dotted line indicates the membranes potential before the stimulation (−58 mV, representative of 10 neurons).
Supplementary Figure 3. Terminal gKv7/M opposes membrane potential fluctuations. (A) Upper, Example of a current noise trace (σ = 0.006) injected into the whole terminal tree of the multi-compartment model of nociceptive neuron with intact . Middle, simulated recordings of changes in membrane voltage in single terminal following injection of the current noise (showed above), expanded in y-axis in inset. Lower, simulated recordings of changes in membrane voltage in the central axon (blue electrode in Figure 4) following injection of the current noise (showed above) to the terminal tree. Note that at this noise level no activity is elicited at either terminal or soma. (B) Same as (A) but is reduced by half. Note that in this conditions same noise level elicited spontaneous firing.
Supplementary Figure 4. The limits of terminal gKv7/M in preventing stimulus induced hyper-responsiveness of modeled nociceptive neuron. Graphic representation of the relationship between the terminal gKv7 and gNa(v)1.9 in eliciting increased responsiveness to 500 ms, 100 pA square pulses. In normal conditions, when = and gNa(v)1.9 = 1 x gNa(v)1.9, this stimuli evokes 1 spike (blue area, see also Figure 4B). The recordings performed at the proximal axon of the modeled neuron (blue electrode in Figure 4). The number of spikes during 500 ms step color coded (shown on the right). Note that when , the number of spikes is increased when gNa(v)1.9 is increased above 120%.
References
Adams, P. R., Brown, D. A., and Constanti, A. (1982). M-currents and other potassium currents in bullfrog sympathetic neurones. J. Physiol. 330, 537–572. doi: 10.1113/jphysiol.1982.sp014357
Aiken, S. P., Lampe, B. J., Murphy, P. A., and Brown, B. S. (1995). Reduction of spike frequency adaptation and blockade of M-current in rat CA1 pyramidal neurones by linopirdine (DuP 996), a neurotransmitter release enhancer. Br. J. Pharmacol. 115, 1163–1168. doi: 10.1111/j.1476-5381.1995.tb15019.x
Amaya, F., Wang, H., Costigan, M., Allchorne, A. J., Hatcher, J. P., Egerton, J., et al. (2006). The voltage-gated sodium channel Na(v)1.9 is an effector of peripheral inflammatory pain hypersensitivity. J. Neurosci. 26, 12852–12860. doi: 10.1523/JNEUROSCI.4015-06.2006
Amir, R., Michaelis, M., and Devor, M. (2002). Burst discharge in primary sensory neurons: triggered by subthreshold oscillations, maintained by depolarizing afterpotentials. J. Neurosci. 22, 1187–1198. doi: 10.1046/j.1529-8027.2002.02026_10.x
Amir, R., Michaelis, M., and Devor, M. (1999). Membrane potential oscillations in dorsal root ganglion neurons: role in normal electrogenesis and neuropathic pain. J. Neurosci. 19, 8589–8596.
Baker, M. D., Chandra, S. Y., Ding, Y., Waxman, S. G., and Wood, J. N. (2003). GTP-induced tetrodotoxin-resistant Na+ current regulates excitability in mouse and rat small diameter sensory neurones. J. Physiol. 548(Pt 2), 373–382. doi: 10.1113/jphysiol.2003.039131
Baker, M. D. (2005). Protein kinase C mediates up-regulation of tetrodotoxin-resistant, persistent Na+ current in rat and mouse sensory neurones. J. Physiol. 567(Pt 3), 851–867. doi: 10.1113/jphysiol.2005.089771
Battefeld, A., Tran, B. T., Gavrilis, J., Cooper, E. C., and Kole, M. H. (2014). Heteromeric Kv7.2/7.3 channels differentially regulate action potential initiation and conduction in neocortical myelinated axons. J. Neurosci. 34, 3719–3732. doi: 10.1523/JNEUROSCI.4206-13.2014
Bernal, L., Lopez-Garcia, J. A., and Roza, C. (2016). Spontaneous activity in C-fibres after partial damage to the saphenous nerve in mice: effects of retigabine. Eur. J. Pain 20, 1335–1345. doi: 10.1002/ejp.858
Binshtok, A. M., Wang, H., Zimmermann, K., Amaya, F., Vardeh, D., Shi, L., et al. (2008). Nociceptors are interleukin-1beta sensors. J. Neurosci. 28, 14062–14073. doi: 10.1523/JNEUROSCI.3795-08.2008
Blackburn-Munro, G., and Jensen, B. S. (2003). The anticonvulsant retigabine attenuates nociceptive behaviours in rat models of persistent and neuropathic pain. Eur. J. Pharmacol. 460, 109–116. doi: 10.1016/S0014-2999(02)02924-2
Blair, N. T., and Bean, B. P. (2003). Role of tetrodotoxin-resistant Na+ current slow inactivation in adaptation of action potential firing in small-diameter dorsal root ganglion neurons. J. Neurosci. 23, 10338–10350
Brown, D. A., and Passmore, G. M. (2009). Neural KCNQ (Kv7) channels. Br. J. Pharmacol. 156, 1185–1195. doi: 10.1111/j.1476-5381.2009.00111.x
Brown, D. A., and Adams, P. R. (1980). Muscarinic suppression of a novel voltage-sensitive K+ current in a vertebrate neurone. Nature 283, 673–676. doi: 10.1038/283673a0
Cardenas, C. G., Del Mar, L. P., and Scroggs, R. S. (1995). Variation in serotonergic inhibition of calcium channel currents in four types of rat sensory neurons differentiated by membrane properties. J. Neurophysiol. 74, 1870–1879.
Caspi, A., Benninger, F., and Yaari, Y. (2009). KV7/M channels mediate osmotic modulation of intrinsic neuronal excitability. J. Neurosci. 29, 11098–11111. doi: 10.1523/JNEUROSCI.0942-09.2009
Cisneros, E., Roza, C., Jackson, N., and Lopez-Garcia, J. A. (2015). A new regulatory mechanism for Kv7.2 protein during neuropathy: enhanced transport from the soma to axonal terminals of injured sensory neurons. Front. Cell. Neurosci. 9:470. doi: 10.3389/fncel.2015.00470
Cruzblanca, H., Koh, D. S., and Hille, B. (1998). Bradykinin inhibits M current via phospholipase C and Ca2+ release from IP3-sensitive Ca2+ stores in rat sympathetic neurons. Proc. Natl. Acad. Sci. U.S.A. 95, 7151–7156. doi: 10.1073/pnas.95.12.7151
Delmas, P., and Brown, D. A. (2005). Pathways modulating neural KCNQ/M (Kv7) potassium channels. Nat. Rev. Neurosci. 6, 850–862. doi: 10.1038/nrn1785
Dib-Hajj, S., Black, J. A., Cummins, T. R., and Waxman, S. G. (2002). NaN/Nav1.9: a sodium channel with unique properties. Trends Neurosci. 25, 253–259. doi: 10.1016/S0166-2236(02)02150-1
Djouhri, L., Koutsikou, S., Fang, X., McMullan, S., and Lawson, S. N. (2006). Spontaneous pain, both neuropathic and inflammatory, is related to frequency of spontaneous firing in intact C-fiber nociceptors. J. Neurosci. 26, 1281–1292. doi: 10.1523/JNEUROSCI.3388-05.2006
Djouhri, L., Fang, X., Koutsikou, S., and Lawson, S. N. (2012). Partial nerve injury induces electrophysiological changes in conducting (uninjured) nociceptive and nonnociceptive DRG neurons: possible relationships to aspects of peripheral neuropathic pain and paresthesias. Pain 153, 1824–1836. doi: 10.1016/j.pain.2012.04.019
Du, X., Hao, H., Gigout, S., Huang, D., Yang, Y., Li, L., et al. (2014). Control of somatic membrane potential in nociceptive neurons and its implications for peripheral nociceptive transmission. Pain 155, 2306–2322. doi: 10.1016/j.pain.2014.08.025
Du, X., and Gamper, N. (2013). Potassium channels in peripheral pain pathways: expression, function and therapeutic potential. Curr. Neuropharmacol. 11, 621–640. doi: 10.2174/1570159X113119990042
Emery, E. C., Luiz, A. P., Sikandar, S., Magnúsdóttir, R., Dong, X., and Wood, J. N. (2016). In vivo characterization of distinct modality-specific subsets of somatosensory neurons using GCaMP. Sci. Adv. 2:e1600990. doi: 10.1126/sciadv.1600990
Emery, E. C., Young, G. T., Berrocoso, E. M., Chen, L., and McNaughton, P. A. (2011). HCN2 ion channels play a central role in inflammatory and neuropathic pain. Science 333, 1462–1466. doi: 10.1126/science.1206243
Feng, B., Zhu, Y., La, J. H., Wills, Z. P., and Gebhart, G. F. (2015). Experimental and computational evidence for an essential role of NaV1.6 in spike initiation at stretch-sensitive colorectal afferent endings. J. Neurophysiol. 113, 2618–2634. doi: 10.1152/jn.00717.2014
Gamper, N., and Shapiro, M. S. (2003). Calmodulin mediates Ca2+-dependent modulation of M-type K+ channels. J. Gen. Physiol. 122, 17–31. doi: 10.1085/jgp.200208783
Gu, N., Vervaeke, K., Hu, H., and Storm, J. F. (2005). Kv7/KCNQ/M and HCN/h, but not KCa2/SK channels, contribute to the somatic medium after-hyperpolarization and excitability control in CA1 hippocampal pyramidal cells. J Physiol. 566(Pt 3), 689–715. doi: 10.1113/jphysiol.2005.086835
Gudes, S., Barkai, O., Caspi, Y., Katz, B., Lev, S., and Binshtok, A. M. (2015). The role of slow and persistent TTX-resistant sodium currents in acute tumor necrosis factor- α -mediated increase in nociceptors excitability. J. Neurophysiol. 113, 601–619. doi: 10.1152/jn.00652.2014
Halliwell, J. V., and Adams, P. R. (1982). Voltage-clamp analysis of muscarinic excitation in hippocampal neurons. Brain Res. 250, 71–92. doi: 10.1016/0006-8993(82)90954-4
Herzog, R. I., Cummins, T. R., and Waxman, S. G. (2001). Persistent TTX-resistant Na+ current affects resting potential and response to depolarization in simulated spinal sensory neurons. J. Neurophysiol. 86, 1351–1364
Hines, M. L., and Carnevale, N. T. (2000). Expanding NEURON's repertoire of mechanisms with NMODL. Neural Comput. 12, 995–1007. doi: 10.1162/089976600300015475
Hines, M. L., and Carnevale, N. T. (1997). The NEURON simulation environment. Neural Comput. 9, 1179–1209. doi: 10.1162/neco.1997.9.6.1179
Horn, R., and Marty, A. (1988). Muscarinic activation of ionic currents measured by a new whole-cell recording method. J. Gen. Physiol. 92, 145–159. doi: 10.1085/jgp.92.2.145
Iyer, S. M., Montgomery, K. L., Towne, C., Lee, S. Y., Ramakrishnan, C., Deisseroth, K., et al. (2014). Virally mediated optogenetic excitation and inhibition of pain in freely moving nontransgenic mice. Nat. Biotechnol. 32, 274–278. doi: 10.1038/nbt.2834
Jones, S., Brown, D. A., Milligan, G., Willer, E., Buckley, N. J., and Caulfield, M. P. (1995). Bradykinin excites rat sympathetic neurons by inhibition of M current through a mechanism involving B2 receptors and G alpha q/11. Neuron 14, 399–405. doi: 10.1016/0896-6273(95)90295-3
Kleggetveit, I. P., Namer, B., Schmidt, R., Helas, T., Ruckel, M., Orstavik, K., et al. (2012). High spontaneous activity of C-nociceptors in painful polyneuropathy. Pain 153, 2040–2047. doi: 10.1016/j.pain.2012.05.017
Komagiri, Y., and Kitamura, N. (2003). Effect of intracellular dialysis of ATP on the hyperpolarization-activated cation current in rat dorsal root ganglion neurons. J. Neurophysiol. 90, 2115–2122. doi: 10.1152/jn.00442.2003
Lee, S. Y., Choi, H. K., Kim, S. T., Chung, S., Park, M. K., Cho, J. H., et al. (2010). Cholesterol inhibits M-type K+ channels via protein kinase C-dependent phosphorylation in sympathetic neurons. J. Biol. Chem. 285, 10939–10950. doi: 10.1074/jbc.M109.048868
Linley, J. E., Rose, K., Patil, M., Robertson, B., Akopian, A. N., and Gamper, N. (2008). Inhibition of M current in sensory neurons by exogenous proteases: a signaling pathway mediating inflammatory nociception. J. Neurosci. 28, 11240–11249. doi: 10.1523/JNEUROSCI.2297-08.2008
Linley, J. E., Pettinger, L., Huang, D., and Gamper, N. (2012). M channel enhancers and physiological M channel block. J. Physiol. 590, 793–807. doi: 10.1113/jphysiol.2011.223404
Liu, B., Linley, J. E., Du, X., Zhang, X., Ooi, L., Zhang, H., et al. (2010). The acute nociceptive signals induced by bradykinin in rat sensory neurons are mediated by inhibition of M-type K+ channels and activation of Ca2+-activated Cl- channels. J. Clin. Invest. 120, 1240–1252. doi: 10.1172/JCI41084
Liu, C. N., Michaelis, M., Amir, R., and Devor, M. (2000). Spinal nerve injury enhances subthreshold membrane potential oscillations in DRG neurons: relation to neuropathic pain. J. Neurophysiol. 84, 205–215.
Lombardo, J., and Harrington, M. A. (2016). Nonreciprocal mechanisms in up- and downregulation of spinal motoneuron excitability by modulators of KCNQ/Kv7 channels. J. Neurophysiol. 116, 2114–2124. doi: 10.1152/jn.00446.2016
Main, M. J., Cryan, J. E., Dupere, J. R., Cox, B., Clare, J. J., and Burbidge, S. A. (2000). Modulation of KCNQ2/3 potassium channels by the novel anticonvulsant retigabine. Mol. Pharmacol. 58, 253–262. doi: 10.1124/mol.58.2.253
Maingret, F., Coste, B., Padilla, F., Clerc, N., Crest, M., Korogod, S. M., et al. (2008). Inflammatory mediators increase Nav1.9 current and excitability in nociceptors through a coincident detection mechanism. J. Gen. Physiol. 131, 211–225. doi: 10.1085/jgp.200709935
Marrion, N. V. (1994). M-current suppression by agonist and phorbol ester in bullfrog sympathetic neurons. Pflugers Arch. 426, 296–303. doi: 10.1007/BF00374785
Miyasho, T., Takagi, H., Suzuki, H., Watanabe, S., Inoue, M., Kudo, Y., et al. (2001). Low-threshold potassium channels and a low-threshold calcium channel regulate Ca2+ spike firing in the dendrites of cerebellar Purkinje neurons: a modeling study. Brain Res. 891, 106–115. doi: 10.1016/S0006-8993(00)03206-6
Mucha, M., Ooi, L., Linley, J. E., Mordaka, P., Dalle, C., Robertson, B., et al. (2010). Transcriptional control of KCNQ channel genes and the regulation of neuronal excitability. J. Neurosci. 30, 13235–13245. doi: 10.1523/JNEUROSCI.1981-10.2010
Nita, I. I., Caspi, Y., Gudes, S., Fishman, D., Lev, S., Hersfinkel, M., et al. (2016). Privileged crosstalk between TRPV1 channels and mitochondrial calcium shuttling machinery controls nociception. Biochim. Biophys. Acta 1863, 2868–2880. doi: 10.1016/j.bbamcr.2016.09.009
Olivares, E., Salgado, S., Maidana, J. P., Herrera, G., Campos, M., Madrid, R., et al. (2015). TRPM8-dependent dynamic response in a mathematical model of cold thermoreceptor. PLoS ONE 10:e0139314. doi: 10.1371/journal.pone.0139314
Pan, Z., Kao, T., Horvath, Z., Lemos, J., Sul, J. Y., Cranstoun, S. D., et al. (2006). A common ankyrin-G-based mechanism retains KCNQ and NaV channels at electrically active domains of the axon. J. Neurosci. 26, 2599–2613. doi: 10.1523/JNEUROSCI.4314-05.2006
Passmore, G. M., Reilly, J. M., Thakur, M., Keasberry, V. N., Marsh, S. J., Dickenson, A. H., et al. (2012). Functional significance of M-type potassium channels in nociceptive cutaneous sensory endings. Front. Mol. Neurosci. 5:63. doi: 10.3389/fnmol.2012.00063
Passmore, G. M., Selyanko, A. A., Mistry, M., Al-Qatari, M., Marsh, S. J., Matthews, E. A., et al. (2003). KCNQ/M currents in sensory neurons: significance for pain therapy. J. Neurosci. 23, 7227–7236.
Peretz, A., Sheinin, A., Yue, C., Degani-Katzav, N., Gibor, G., Nachman, R., et al. (2007). Pre- and postsynaptic activation of M-channels by a novel opener dampens neuronal firing and transmitter release. J. Neurophysiol. 97, 283–295. doi: 10.1152/jn.00634.2006
Pristera, A., Baker, M. D., and Okuse, K. (2012). Association between tetrodotoxin resistant channels and lipid rafts regulates sensory neuron excitability. PLoS ONE 7:e40079. doi: 10.1371/journal.pone.0040079
Qu, L., and Caterina, M. J. (2016). Enhanced excitability and suppression of A-type K+ currents in joint sensory neurons in a murine model of antigen-induced arthritis. Sci. Rep. 6:28899. doi: 10.1038/srep28899
Reeh, P. W. (1986). Sensory receptors in mammalian skin in an in vitro preparation. Neurosci. Lett. 66, 141–146. doi: 10.1016/0304-3940(86)90180-1
Rivera-Arconada, I., and Lopez-Garcia, J. A. (2006). Retigabine-induced population primary afferent hyperpolarisation in vitro. Neuropharmacology 51, 756–763. doi: 10.1016/j.neuropharm.2006.05.015
Roza, C., Castillejo, S., and Lopez-Garcia, J. A. (2011). Accumulation of Kv7.2 channels in putative ectopic transduction zones of mice nerve-end neuromas. Mol. Pain 7:58. doi: 10.1186/1744-8069-7-58
Roza, C., and Lopez-Garcia, J. A. (2008). Retigabine, the specific KCNQ channel opener, blocks ectopic discharges in axotomized sensory fibres. Pain 138, 537–545. doi: 10.1016/j.pain.2008.01.031
Selyanko, A. A., and Brown, D. A. (1996). Intracellular calcium directly inhibits potassium M channels in excised membrane patches from rat sympathetic neurons. Neuron 16, 151–162. doi: 10.1016/S0896-6273(00)80032-X
Serra, J., Bostock, H., Sola, R., Aleu, J., Garcia, E., Cokic, B., et al. (2012). Microneurographic identification of spontaneous activity in C-nociceptors in neuropathic pain states in humans and rats. Pain 153, 42–55. doi: 10.1016/j.pain.2011.08.015
Shah, M. M., Migliore, M., Valencia, I., Cooper, E. C., and Brown, D. A. (2008). Functional significance of axonal Kv7 channels in hippocampal pyramidal neurons. Proc. Natl. Acad. Sci. U.S.A. 105, 7869–7874. doi: 10.1073/pnas.0802805105
Shi, L., Bian, X., Qu, Z., Ma, Z., Zhou, Y., Wang, K., et al. (2013). Peptide hormone ghrelin enhances neuronal excitability by inhibition of Kv7/KCNQ channels. Nat. Commun. 4:1435. doi: 10.1038/ncomms2439
Suh, B. C., and Hille, B. (2002). Recovery from muscarinic modulation of M current channels requires phosphatidylinositol 4,5-bisphosphate synthesis. Neuron 35, 507–520. doi: 10.1016/S0896-6273(02)00790-0
Sundt, D., Gamper, N., and Jaffe, D. B. (2015). Spike propagation through the dorsal root ganglia in an unmyelinated sensory neuron: a modeling study. J. Neurophysiol. 114, 3140–3153. doi: 10.1152/jn.00226.2015
Telezhkin, V., Brown, D. A., and Gibb, A. J. (2012). Distinct subunit contributions to the activation of M-type potassium channels by PI(4,5)P2. J. Gen. Physiol. 140, 41–53. doi: 10.1085/jgp.201210796
Towne, C., Pertin, M., Beggah, A. T., Aebischer, P., and Decosterd, I. (2009). Recombinant adeno-associated virus serotype 6 (rAAV2/6)-mediated gene transfer to nociceptive neurons through different routes of delivery. Mol. Pain 5:52. doi: 10.1186/1744-8069-5-52
Tzour, A., Leibovich, H., Barkai, O., Biala, Y., Lev, S., Yaari, Y., et al. (2016). KV 7/M channels as targets for lipopolysaccharide-induced inflammatory neuronal hyperexcitability. J. Physiol. 595, 713–738. doi: 10.1113/JP272547
Vasylyev, D. V., and Waxman, S. G. (2012). Membrane properties and electrogenesis in the distal axons of small dorsal root ganglion neurons in vitro. J. Neurophysiol. 108, 729–740. doi: 10.1152/jn.00091.2012
Wainger, B. J., Kiskinis, E., Mellin, C., Wiskow, O., Han, S. S., Sandoe, J., et al. (2014). Intrinsic membrane hyperexcitability of amyotrophic lateral sclerosis patient-derived motor neurons. Cell Rep. 7, 1–11. doi: 10.1016/j.celrep.2014.03.019
Wang, H. S., Pan, Z., Shi, W., Brown, B. S., Wymore, R. S., Cohen, I. S., et al. (1998). KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M-channel. Science 282, 1890–5395. doi: 10.1126/science.282.5395.1890
Wang, H. S., and McKinnon, D. (1995). Potassium currents in rat prevertebral and paravertebral sympathetic neurones: control of firing properties. J. Physiol. 485 (Pt 2), 319–335. doi: 10.1113/jphysiol.1995.sp020732
Wen, H., and Levitan, I. B. (2002). Calmodulin is an auxiliary subunit of KCNQ2/3 potassium channels. J. Neurosci. 22, 7991–8001.
Wu, G., Ringkamp, M., Hartke, T. V., Murinson, B. B., Campbell, J. N., Griffin, J. W., et al. (2001). Early onset of spontaneous activity in uninjured C-fiber nociceptors after injury to neighboring nerve fibers. J. Neurosci. 21, Rc140.
Xia, S., Lampe, P. A., Deshmukh, M., Yang, A., Brown, B. S., Rothman, S. M., et al. (2002). Multiple channel interactions explain the protection of sympathetic neurons from apoptosis induced by nerve growth factor deprivation. J. Neurosci. 22, 114–122.
Yu, S. P. (1995). Roles of arachidonic acid, lipoxygenases and phosphatases in calcium-dependent modulation of M-current in bullfrog sympathetic neurons. J. Physiol. 487(Pt 3), 797–811. doi: 10.1113/jphysiol.1995.sp020919
Yue, C., and Yaari, Y. (2004). KCNQ/M channels control spike afterdepolarization and burst generation in hippocampal neurons. J. Neurosci. 24, 4614–4624. doi: 10.1523/JNEUROSCI.0765-04.2004
Yue, C., and Yaari, Y. (2006). Axo-somatic and apical dendritic Kv7/M channels differentially regulate the intrinsic excitability of adult rat CA1 pyramidal cells. J. Neurophysiol. 95, 3480–3495. doi: 10.1152/jn.01333.2005
Yuryev, M., and Khiroug, L. (2012). Dynamic longitudinal investigation of individual nerve endings in the skin of anesthetized mice using in vivo two-photon microscopy. J. Biomed. Opt. 17:046007. doi: 10.1117/1.JBO.17.4.046007
Zhang, H., Craciun, L. C., Mirshahi, T., Rohacs, T., Lopes, C. M., Jin, T., et al. (2003). PIP activates KCNQ channels, and its hydrolysis underlies receptor-mediated inhibition of M currents. Neuron 37, 963–975. doi: 10.1016/S0896-6273(03)00125-9
Zheng, Q., Fang, D., Liu, M., Cai, J., Wan, Y., Han, J. S., et al. (2013). Suppression of KCNQ/M (Kv7) potassium channels in dorsal root ganglion neurons contributes to the development of bone cancer pain in a rat model. Pain 154, 434–448. doi: 10.1016/j.pain.2012.12.005
Zheng, Y., Xu, H., Zhan, L., Zhou, X., Chen, X., and Gao, Z. (2015). Activation of peripheral KCNQ channels relieves gout pain. Pain 156, 1025–1035. doi: 10.1097/j.pain.0000000000000122
Keywords: M-current, spontaneous firing, ectopic activity, nociceptors, nociceptive terminals
Citation: Barkai O, Goldstein RH, Caspi Y, Katz B, Lev S and Binshtok AM (2017) The Role of Kv7/M Potassium Channels in Controlling Ectopic Firing in Nociceptors. Front. Mol. Neurosci. 10:181. doi: 10.3389/fnmol.2017.00181
Received: 24 April 2017; Accepted: 24 May 2017;
Published: 13 June 2017.
Edited by:
Hermona Soreq, Hebrew University of Jerusalem, IsraelReviewed by:
Ilya A. Fleidervish, Ben-Gurion University of the Negev, IsraelBernard Attali, Tel Aviv University, Israel
Copyright © 2017 Barkai, Goldstein, Caspi, Katz, Lev and Binshtok. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alexander M. Binshtok, YWxleGFuZGVyYkBla21kLmh1amkuYWMuaWw=