 Alexei A. Surguchev
Alexei A. Surguchev Andrei Surguchov
Andrei Surguchov- 1Department of Surgery, Section of Otolaryngology, Yale School of Medicine, Yale University, New Haven, CT, United States
- 2Department of Neurology, University of Kansas Medical Center, Kansas City, KS, United States
Synuclein family consists of three members, α, β, and γ-synuclein. Due to their involvement in human diseases, they have been thoroughly investigated for the last 30 years. Since the first synuclein identification and description, members of this family are found in all vertebrates. Sequencing of their genes indicates high evolutionary conservation suggesting important function(s) of these proteins. They are small naturally unfolded proteins prone to aggregate, easily change their conformation, and bind to the membranes. The genes for α, β, and γ-synuclein have different chromosomal localization and a well preserved general organization composed of five coding exons of similar size. Three genes encoding synucleins are present in the majority of vertebrates, however, a variable number of synuclein genes are described in fishes of different species. An important question concerns their normal function in cells and tissues. α-Synuclein is implicated in the regulation of synaptic activity through regulation of synaptic vesicle release, while the physiological functions of two other members of the family is understood less clearly. Here we discuss recent results describing their role in the regulation of gene expression.
Introduction
Since the discovery of the first synuclein by Maroteaux et al. (1988), the members of the synuclein family attract growing attention primarily as proteins implicated in neurodegenerative (α-synuclein) and neoplastic (γ-synuclein) diseases. The majority from almost 9,000 publications describe how these naturally unfolded proteins aggregate and propagate between neurons (α-synuclein), or cause misregulation of intracellular signaling pathways in oncological transformation (γ-synuclein). β-Synuclein, the third member of the family, received much less attention. It is less prone to form insoluble aggregates and presumably plays a protective role against α-synucleinopathies (Hashimoto et al., 2001). Since the discovery of synucleins, researchers investigate mostly α-synuclein for its association with synucleinopathies, i.e., PD, PD dementia, DLB and multiple system atrophy.
During the late 1990s the popular theory to explain mechanisms causing α-synuclein aggregation in cellular milieu was a concept of macromolecular crowding (Minton, 1993, 1998; Uversky et al., 2001). According to this concept, total cytoplasmic concentration of proteins and other macromolecules can either reach the high level of 400 g/l causing macromolecular crowding (Uversky et al., 2001, 2002; Shtilerman et al., 2002) or be subjected to excluded volume effect (Ellis, 2001) in the neuronal cytoplasm. This induces α-synuclein and other naturally unfolded protein aggregation, which may lead to the formation of toxic aggregates, fibrils, and protofibrils.
While synuclein’s role in pathology is thoroughly and systematically investigated, their normal physiological functions still remain debated despite almost 30 years of research. Some progress in unveiling α-synuclein physiological role is achieved due to the discovery of its role in synapsis. Several research teams have demonstrated that α-synuclein is implicated in the modulation of synaptic activity through modulation of vesicle release (Chandra et al., 2005; Burré et al., 2010; Bendor et al., 2013; Zaltieri et al., 2015). Numerous studies demonstrate α-synuclein’s participation in vesicle trafficking, chaperoning of SNARE complex assembly (Bonini and Giasson, 2005; Chandra et al., 2005; Burré et al., 2010; Diao et al., 2013), regulation of synaptic vesicles endocytosis, presynaptic terminal topography (Vargas et al., 2014, 2017), and modulation of dopamine release (Abeliovich et al., 2000). However, the progress in understanding the physiology of β and γ-synucleins is much more limited. β-Synuclein inhibits α-synuclein aggregation caused by several types of lipids as well as α-synuclein nucleation (Brown et al., 2016). γ-Synuclein possesses chaperon activity, is involved in the regulation of monoamine homeostasis and cytoskeleton structure rearrangement. Moreover, it may have similar functions as α–synuclein in the process of synaptic vesicle turnover, neurotransmitter release (Ninkina et al., 2012; Kokhan et al., 2013), and regulation of signaling pathways (Surguchov et al., 1999, 2001, 2017; Surgucheva et al., 2006, 2008; Surguchov, 2008, 2015).
Despite the information presented above, many reviews consider the normal functions of α-synuclein to be unclear and vague, creating an impression that it does not have its own function in living cells, and settles for a supporting role in several pathways. In addition to synaptic functions, α-synuclein participation in several cellular processes has been described, e.g., suppression of apoptosis, regulation of glucose level, modulation of calmodulin level, maintenance of PUFA level, antioxidation, neuronal differentiation, and regulation of dopamine biosynthesis (Emamzadeh, 2016). However, it appears that in all these processes, synucleins are not the major players, but just “ramblers in a crowd” with limited authority and jurisdictions. Still the emerging evidence suggests, that synucleins may have a very important non-canonical function unrelated to their role in synapsis. The nuclear localization of α-synuclein and several new findings point to its essential role in the nucleus related to the regulation of gene expression.
Synucleins As Modulators of Specific Gene Expression
Gradually new results accumulate, and point out that our knowledge about synuclein’s functions is incomplete, their “social network” is larger than it is currently assumed, and they play a more essential dynamic role in the regulation of intracellular processes. The current understanding of their role in cellular processes is slowly shifting from “ramblers in the crowd” to “cops regulating traffic.” This regulatory role is performed through their interactions with key players governing gene expression, i.e., nucleic acids, transcription and translation factors, and histones. Moreover, synucleins may play a role in epigenetic regulation of cellular processes. Epigenetics applies to external modifications to DNA that alter expression of specific genes without changes in the DNA sequence. It can involve several mechanisms, e.g., DNA methylation, histone modifications, and RNA-associated silencing.
While the researchers still keep working out the details of synuclein’s functions in synapsis and a role in vesicles formation, another page of their story emerges, attracting increasing attention. Several new methods demonstrate that synucleins are “more social” that it has been assumed earlier, and find novel partners in synuclein’s “social network” not only among proteins, but also nucleic acids. Below we present several examples of synuclein’s regulatory role in gene expression.
The first hint suggesting that synucleins regulate gene expression came from the analysis of the effect of their upregulation in cell cultures (Seo et al., 2002; Baptista et al., 2003). Although the effect of elevated synucleins level on expression of specific genes was significant, the exact molecular mechanisms was often unknown. In several studies a direct effect of synucleins on transcription in the nucleus is demonstrated, other publications suggest hypothetic mechanisms explaining this effect. For example, α-synuclein downregulates c-Jun N-terminal kinase protecting cells against oxidative stress, upregulates caveolin-1 expression, and downregulates ERK, affecting the pathogenesis of PD (Hashimoto et al., 2003). In another publication α-synuclein’s effect in regulating apoptosis is reported. It reduced Bcl-xL expression and increased BAX expression (Seo et al., 2002).
In several studies a substitution of one amino acid in α-synuclein dramatically changed its regulatory properties suggesting that this effect was mediated by conformational alterations. For example, α-synuclein altered expression of several families of genes including genes responsible for apoptosis, stress response, transcription regulation, and membrane proteins (Baptista et al., 2003). Significant changes in expression levels are also found for genes responsible for the regulation of dopamine synthesis. Reduced expression of the orphan nuclear receptor Nurr1 suggests that the synergetic regulation of dopamine synthesis occurs through this transcription factor. These alterations in mRNAs levels identified by microarray experiments occurred on transcriptional level and were validated by quantitative RT-PCR. Interestingly, the expression levels of four genes were regulated in opposite directions in cells overexpressing wild-type protein or α-synuclein with A53T substitution (Baptista et al., 2003).
Overexpression of wild-type or mutant α-synuclein interferes with DBH transcription regulation by CRE element in catecholaminergic neurons (Kim et al., 2014). In the nucleus α-synuclein interacts with the DBH promoter including the CRE element, which interferes with forskolin-induced transcription factor CREB binding to the CRE region. Thus, α-synuclein attenuates CRE-mediated transcription of DBH. This interaction is physiologically important, because CREB plays an essential role in survival of neurons by controlling the transcription of several genes implicated in cell protection, such as BDNF (Chalovich et al., 2006). Remarkably, mutant α-synuclein demonstrates higher tendency to nuclear translocation and interaction with the DBH promoter than the wild type.
Another example confirming that the effect of α-synuclein on transcription of specific gene may differ as a result of a single amino acid substitution is recently described (Segura-Ulate et al., 2017). Oligodendroglial cells OLN-93 stably expressing either a human wild type or a mutant A53E α-synuclein (multiple system atrophy associated mutant) reduced BDNF mRNA to practically unmeasurable levels. At the same time, another MSA-associated α-synuclein mutant, caused only a small reduction in BDNF mRNA. Therefore, point mutations in α-synuclein may change not only its physico-chemical properties and propensity to aggregation, but also alter its regulatory function (Segura-Ulate et al., 2017). Moreover, in dopaminergic neurons α-synuclein also reduces the expression of PKCδ to inhibit apoptosis, by decreasing enzymatic activity of p300 HAT.
This effect results in neuroprotection in a α-synuclein expressing dopaminergic cell model as a result of exposure to the Parkinsonian neurotoxicant MPP+. This mechanism involves modulation of both NFκB and p300 signaling pathways in transgenic mice and in neuronal culture (Jin et al., 2011). The effect of α-synuclein on histone acetylation is further described in Chapter 6.
α-Synuclein is colocalized with transcription factor Elk-1 and interacts with MAP kinase pathway (Iwata et al., 2001a,b). These findings point to the role of α-synuclein in the modulation of transcription and signaling pathways.
Another member of the synuclein family, γ-synuclein also interacts with Elk-1 (Surguchov et al., 1999). Furthermore, it plays a role as a modulator of matrix metalloproteinases (MMP-9 and MMP-2) expression upregulating both MMP-9 protein level and activity (3.2 to 7.1-fold, respectively). This upregulation takes place on transcriptional level via the activation of the AP-1 cis-elements in MMP-9 promoter (Surgucheva et al., 2003). Further studies demonstrated direct binding of γ-synuclein to transcription factors JunB, MECP2, CREB1, PPAR-gamma, TCEA1, and ATF3 (Surgucheva and Surguchov, 2008). Recent study using yeast two-hybrid screening, identified another transcriptional regulator – polyC binding protein 1 (PCBP1) as a γ-synuclein interacting protein (Hunkele et al., 2016).
Synuclein – DNA Interactions
α-Synuclein Binding to DNA
The existence of α-synuclein in the nucleus first found by Maroteaux et al. (1988) and later confirmed by many other studies, encouraged researchers to examine synuclein–DNA interaction and to investigate its possible functions. Several research groups demonstrated that α-synuclein could bind directly to a single copy DNA; this binding is especially efficient if DNA is active in the process of transcription and is not bound to histones (Hegde and Jagannatha Rao, 2003; Hegde et al., 2010; Vasudevaraju et al., 2012). This binding occurs preferentially to GC-box-like sequences (Vasudevaraju et al., 2012; Ma et al., 2014) and may alter properties of both protein and DNA. In particular, α-synuclein induces DNA damage by changing its stability, conformation, and by causing DNA nicking (Padmaraju et al., 2011). In turn, DNA can itself modulate α-synuclein folding (Hegde and Rao, 2007). After binding with double- or single-stranded DNA α-synuclein acquires a highly structured conformation. Circular dichroism studies show that the α-helical content of α-synuclein increases from 5 to 64% upon binding to DNA, whereas the random coil decreases from 95 to 33% (Hegde and Rao, 2007).
Interaction of α-synuclein with linear or supercoiled double-stranded DNA (dsDNA) protects DNA from digestion by restriction endonucleases. Complexes between α-synuclein and DNA as well as assembly of wild-type α-synuclein into fibrils in the presence of linear DNA are revealed by electron microscopy (Cherny et al., 2004).
The presence of synucleins in the nucleus and their binding to DNA provides a possibility that such interaction affects transcription regulation and may change neuronal function (Ma et al., 2014; Surguchov, 2014). In several studies such functional consequences of synuclein – DNA interaction have been demonstrated. For example, in neuroglioma, α-synuclein binds to DNA, and regulates the transcription of genes controlling ubiquitination and other biochemical processes linked to PD (Martins et al., 2011).
DNA Binding Is a Common Feature of Several Amyloidogenic Proteins
α-Synuclein is not a unique amyloidogenic protein possessing DNA-binding ability. Amyloid-beta (Aβ) peptides and prion proteins also have high DNA binding capacity, suggesting that DNA binding may be a common property of amyloidogenic proteins (Hegde et al., 2010). Binding of α-synuclein and other amyloidogenic proteins to DNA may affect normal DNA functions and cause genetic stress altering the normal pattern of gene expression (Jiménez, 2010). Results supporting this unifying hypothesis were obtained from the investigation which identified DNA aptamers that specifically bind to α-synuclein (Tsukakoshi et al., 2010, 2012). In this study eight aptamers specific for α-synuclein monomers, oligomers, and fibrils were characterized by a competitive screening method. Their nucleotide sequences are not conserved, but all possess guanine-rich sequences which form scaffolds-like G-quadruplex structures. Importantly, some of the aptamers recognizing α-synuclein, are also specific for Aβ oligomers. Dissociation constant for such aptamers binding is in nano- to picomolar range. The structure of these aptamers may give a clue for the search of new medications and biomarkers specific for neurodegenerative diseases. Notably, the aptamer could recognize not only the primary structure of α-synuclein, but also its conformation (Tsukakoshi et al., 2012).
Several studies demonstrate that α-synuclein is able to bind directly to promoter region of specific genes and affect their transcription. For example, α-synuclein binding to a promoter of the mitochondrial transcriptional co-activator PGC-1α, which reduces its expression in response to oxidative stress is described (Siddiqui et al., 2012).
Another member of the synuclein family, γ-synuclein also modulates genes expression by binding to the promoter region of specific genes. For instance, upregulation of matrix metalloproteinases-9 (MMP-9) expression and activity is mediated by γ-synuclein binding to AP-1 sites at the promoter region of the MMP-9 gene (Surgucheva et al., 2003). Under stress conditions a translocation of γ-synuclein to the nucleus decreases outgrowth of neurites more efficiently than α-synuclein overexpression. Thus, γ-synuclein may alter its intracellular localization in response to stress and make appropriate alterations in the gene expression pattern (Surgucheva et al., 2003, 2006).
Binding of Synuclein to RNA
α-Synuclein binds its own mRNA and prevents initiation of translation (Zanzoni et al., 2013). Interaction of proteins with cognate transcript is a known regulatory mechanism modulating gene expression at the translational level. Usually such mRNA contains a riboswitch – a regulatory segment that interacts with a small molecule, affecting the translation of the proteins encoded by the mRNA (Tucker and Breaker, 2005). However, no riboswitches have been described for synucleins yet. Such autoregulatory control is important for assurance of optimal protein expression levels, and abolition of this normal feedback may lead to various negative consequences (Tai et al., 2004; Hasnat et al., 2007). Autogenous interactions alter gene expression at the translational level and when protein production is elevated, binding to mRNA has a significant inhibitory effect on translation efficiency.
To calculate the propensity of proteins to bind RNA, catRAPID approach was put forward which allows to predict the incidence of autogenous associations in the human proteome (Zanzoni et al., 2013). This method demonstrates that α-synuclein easily binds with cognate mRNA, inhibiting its translation, preventing overexpression and thus supporting the optimal level of protein expression. Since aggregation is intrinsically concentration dependent, it is likely that autogenous interactions play a crucial role in controlling protein homeostasis.
Several recently developed methods are useful to investigate the role of RNA-protein interactions in the pathogenesis of human diseases. CatRAPID is a theoretical framework, which predicts the binding ability of protein and RNA molecules based on physico-chemical properties of nucleotide and amino acid chains. This includes hydrogen bonding, secondary structure, and van der Waals’ forces to predict protein–RNA associations with a high confidence (Bellucci et al., 2011; Cirillo et al., 2013). This approach presents a novel tool in the development of RNA aptamers, a useful therapeutic instrument for the diagnostics and management of neurodegenerative diseases (Lee et al., 2006).
Garcia-Esparcia et al. (2015) described significant alterations in the machinery of protein synthesis at the specific regions of the brains of PD patients which are region- and stage-dependent. These alterations include 18S and 28S rRNA, expression of several mRNAs encoding ribosomal proteins, and altered level of translation factors eIF3 and eEF2. These alterations occur in substantia nigra and in the cerebral cortex and may be linked to a significant elevation of α-synuclein oligomers. However, there is no direct evidence to date proving this association.
Thus, there is growing understanding that interaction of α-synuclein with RNA and other protein-RNA interactions are involved in PD and other neurodegenerative diseases (Anthony and Gallo, 2010; Cirillo et al., 2013).
Data From Yeast Help to Expand α-Synuclein’s Human Interactome Demonstrating Its Critical Role in the Regulation of Translation
New methodologies are required to reveal how synucleins orchestrate the expression of other genes, to identify new binding partners, and to examine mechanisms of synuclein’s toxicity. Classical methods, such as knockdown of a single member or all three members of the synuclein family, give only limited information about synuclein’s function, confirming their role in synapse structure and physiology. Synuclein’s deficiency causes age-dependent neuronal dysfunction, impaired survival and extensive alterations in synaptic dopamine neurotransmission in the nigrostriatal system (Greten-Harrison et al., 2010; Anwar et al., 2011). The results of these experiments also confirmed the existence of overlapping functions in synuclein family members (Anwar et al., 2011). These findings were important for understanding of synuclein pathology in PD and aging, but synuclein biology and their normal physiological functions remained poorly understood.
An important clue for better understanding of a protein’s physiological function is the elucidation of its physical interactions. To recognize the normal biological functions and role of α-synuclein in neuropathology, several transgenic and viral overexpression models were developed in various organisms, including roundworms (nematode), fruit flies, rodents, and non-human primates. Amazingly, significant progress in the identification of new synuclein binding partners was made in budding yeast Saccharomyces cerevisiae, the organism without CNS or brain. Moreover, it does not even have α-synuclein ortholog. Yeast Saccharomyces cerevisiae have repeatedly been shown to be suitable for studies of high eukaryotic cell biology, for example, programmed cell death, mitochondria biology (Ter-Avanesyan et al., 1982; Grandin and Charbonneau, 1999; Knott et al., 2008), vesicular trafficking (Littleton and Bellen, 1995), secretory pathway (Malhotra and Emr, 2002), as a model to study neuroprotection at the cellular level (Scheper and Hoozemans, 2009). The method used in the laboratory of Susan Lindquist (Khurana and Lindquist, 2010) and other labs (Braun et al., 2010; Tenreiro et al., 2017) also helped to get better understanding of the mechanisms of α-synuclein toxicity. The ‘humanized’ yeast models for synucleinopathies recapitulate the fundamental properties of the pathology on molecular and cellular level, identified in human diseases. This provides the rationale for engaging the powerful analytical instrument for various high throughput screening approaches in yeast. In yeast, genome-wide analyses can be performed, which enables the un-biased identification of genes and pathways critically modulating (either executing or preventing) the toxicity of α-synuclein (Lan et al., 2011; Tardiff et al., 2014; Khurana et al., 2017). In addition, comprehensive transcriptomic data may be collected from yeast cells expressing various levels of α-synuclein. Usually genetic screens identify response regulators, whereas transcriptomic profiling assays reveal components of metabolic processes. The ResponseNet computational method combines data from the genetic screens and gene expression analyses with the comprehensive knowledge on protein (and gene) interaction information in public yeast data bases (Yeger-Lotem et al., 2009). This generated a novel level of information on the functional α-synuclein gene/protein interaction network. The ResponseNet method allowed to reveal cellular pathways that responded to the toxicity of α-synuclein mapping both previously identified, and unrecognized pathways responding to α-synuclein toxicity. In the response to α-synuclein toxicity four de novo predictions identified by ResponseNet were validated, i.e., the nitrosative stress, the TOR pathway, the disruption of the sterol biosynthesis pathway and the mode-of-action of the genetic suppressor Gip2 (Yeger-Lotem et al., 2009).
Recently a new important method, TransposeNet, was developed allowing to combine genome-wide genetic screens in yeast with the comprehensive protein and gene interaction information available in yeast databases, using an improved computational method (Khurana et al., 2017). The method allows identification of protein–protein and protein–DNA interactions using a yeast interactome data. With this approach a functional protein/gene interaction network for α-synuclein has been described. An important step in this method is a transposition of α-synuclein interaction network into a human α-synuclein interaction network. This transposition is based on a significantly improved identification of human homologs of yeast genes, and on the information of the protein and gene interaction networks known from both the human system and from yeast. A very important result brought by the TransposeNet is that the mRNA translation subnetwork is relevant for α-synuclein toxicity in patient-derived neurons.
The pivotal role of mRNA translation for α-synuclein toxicity was further substantiated by a biochemical method enriching our knowledge about α-synuclein interactome (Chung et al., 2017). Here, the authors used APEX (from ascorbate peroxidase), a method based on the extremely short-lived radicals which covalently react and label amino acids in their immediate proximity (Martell et al., 2012). This method identifies even transient protein–protein interactions in living neurons by mass spectrometry. The method relies on fusing α-synuclein with APEX, which oxidizes phenol derivatives to phenol radicals (Figure 1).
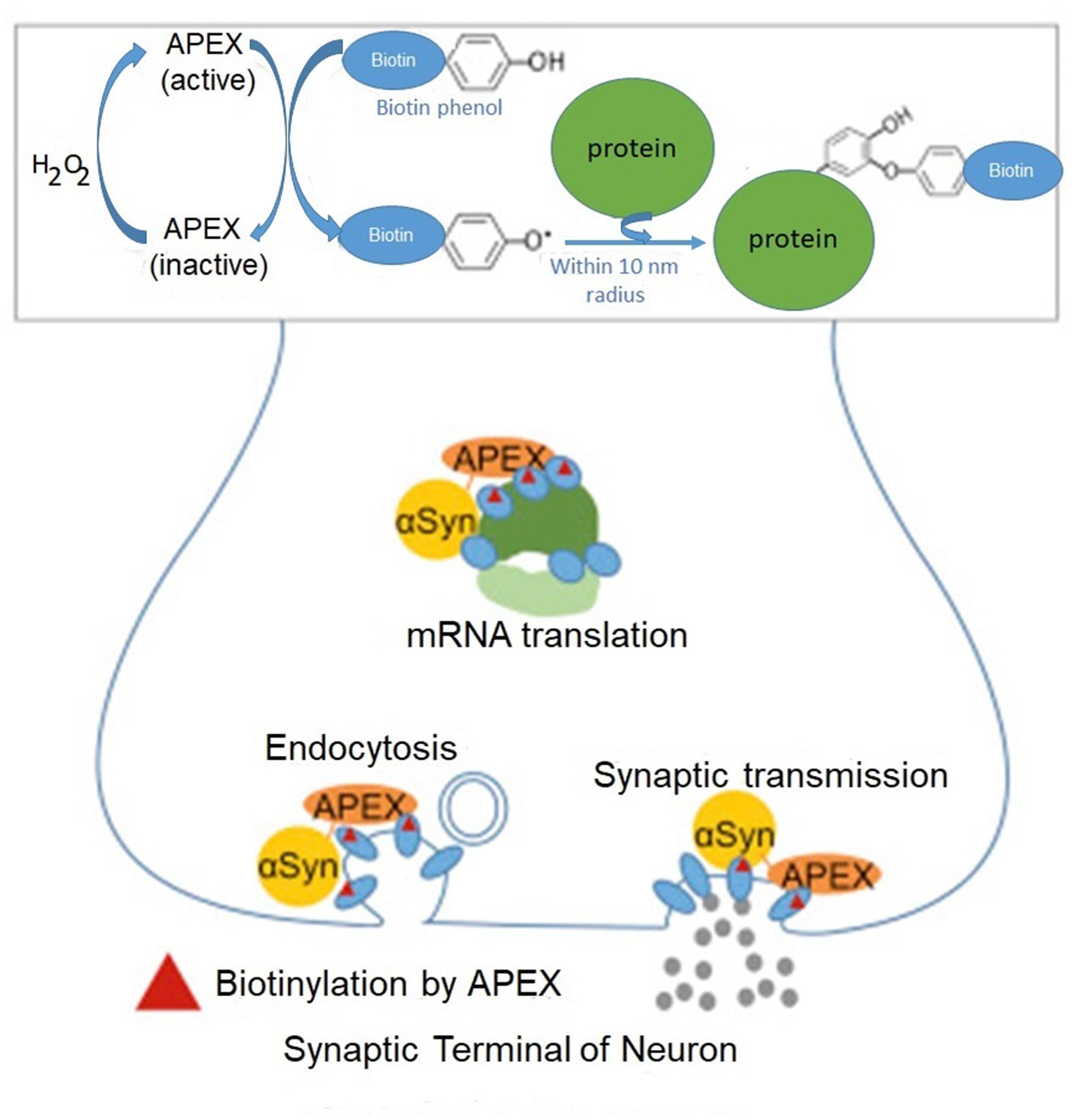
FIGURE 1. Identification of α-synuclein-interacting proteins by peroxidase labeling technique coupled with mass spectrometry. A genetically engineered peroxidase APEX tagged to the C-terminal of wild-type α-synuclein defines the protein network in the vicinity (10 nm) of α-synuclein in living neurons. In addition to anticipated α-synuclein-interacting proteins involved in synaptic transmission and endocytosis, this method reveals several mRNA binding proteins implicated in mRNA translation. Adapted from Chung et al. (2017) with permission from the copyright holder.
Rat primary cortical neurons were transduced with α-synuclein fused to APEX2, a catalytically superior version of APEX. As a result, α-synuclein interacting proteins were identified by mass spectrometry. In initial screening the authors detected 225 proteins in living neurons which interacted with α-synuclein. Many of these interacting proteins were present in complexes with α-synuclein. This study in addition to anticipated interacting proteins involved in synaptic transmission and endocytosis, identifies several proteins implicated in mRNA metabolism (RNA binding, processing and translation factors). Among them are ten proteins with various functions in translation: EIF3C, EIF3D, CARS, EEF1B2, DARS, EIF3L, EEF2, RPS10, EEF1D and MARS (Figure 2).
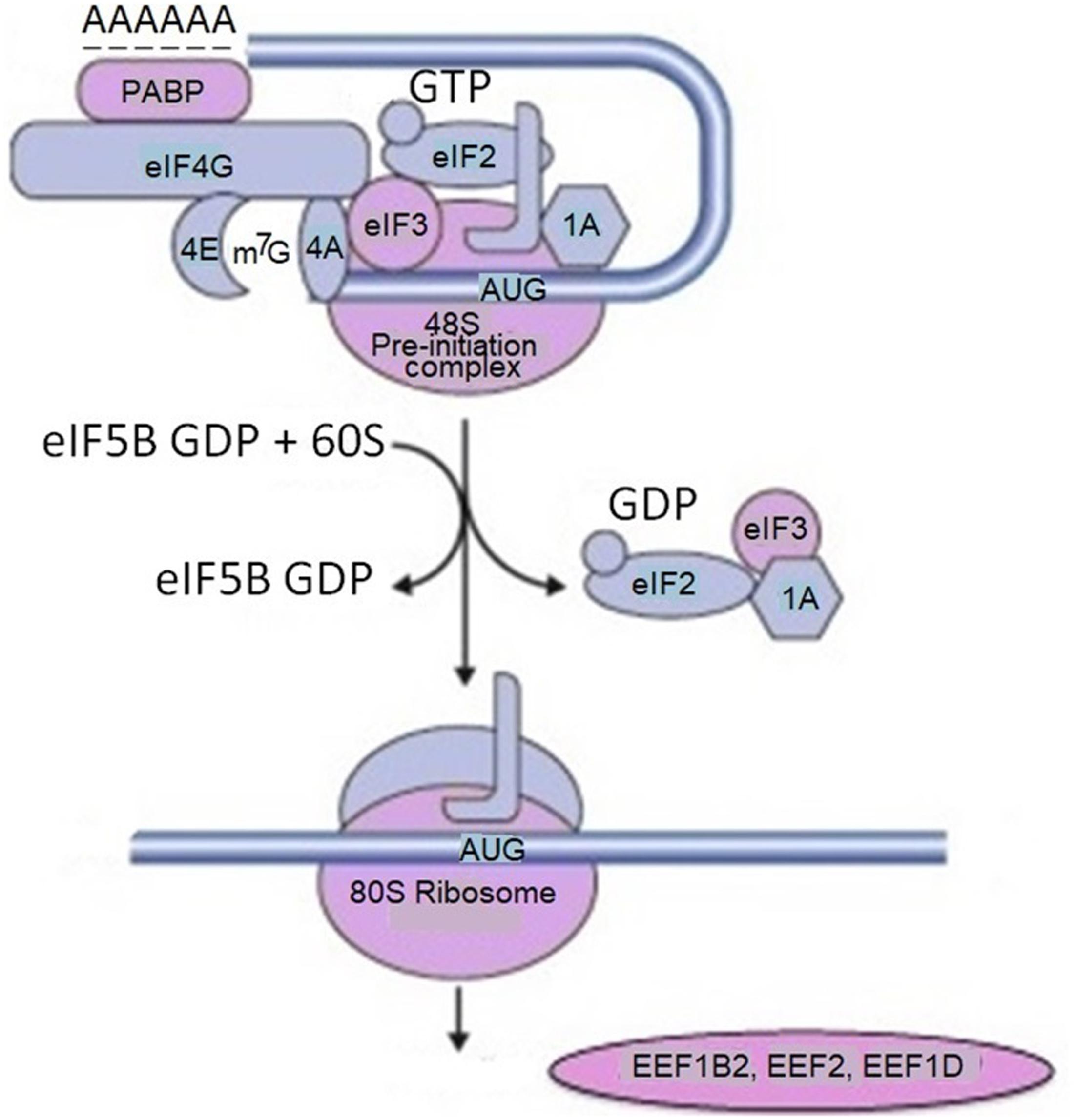
FIGURE 2. Proteins involved in mRNA translation labeled by α-synuclein-APEX. mRNA translation-related proteins are tested for physical interaction with α-synuclein using the membrane-2-hybrid (MYTH) technique. For example, multiple subunits of translation initiation factor EIF3 are labeled with α-synuclein-APEX and stain positive for MYTH. From the list of mRNA translation hits in the APEX neuron study, five were available for testing by MYTH. Four of those, namely EIF3C, EIF3L, RPS12, and PABPC1 were confirmed. These data imply that mRNA translation machinery elements and binding proteins are in close proximal interactions with α-synuclein. Adapted from Chung et al. (2017) with permission from the copyright holder.
Polyadenylate-binding protein 1 (PABPC1) – an mRNA binding protein which facilitates mRNA transport out of the nucleus, degradation, translation, and stability is also identified as α-synuclein interacting protein. The authors assume that α-synuclein physically associates with translation factors and sequesters them. This could occur in the synapse, where α-synuclein is localized, and the local mRNA translation plays a key role in the synaptic plasticity. This approach links many Parkinsonism and neurodegenerative disease risk factors to α-synuclein toxicity through specific molecular pathways. The most important of them are mRNA metabolism, translation, and vesicle trafficking. In this network, α-synuclein is associated with genetic modifiers related to mRNA translation, including initiation factors. In particular, it binds to translation initiation factor 4 gamma 1 (EIF4G1) and the poly(A)-binding protein (PAB or PABPC1). The latter one binds to the 3′ poly(A) tail of mRNA and is involved in poly(A) shortening and translation initiation. In this network α-synuclein is also associated with several ribosomal components. Overexpression of these genetic modifiers in the mRNA translation and mRNA processing pathways suppressed α-synuclein toxicity in bioscreen, while genetic experiments in various disease models revealed that their effects were specific. As a result, α-synuclein screens and network analysis identified a robust effect of α-synuclein toxicity on bulk mRNA translation in cellular models of synucleinopathy, which was not attributable to an ER stress response (Khurana et al., 2017). The link of α-synuclein with EIF4G1 presumably is involved in pathological alterations in PD and other neurodegenerative disease, since this translation initiation factor is linked to both PD (Chartier-Harlin et al., 2011) and Lewy body dementia (Fujioka et al., 2013). EIF4G1 functions as a scaffold in the eIF4F initiation complex, recruiting other components of translation machinery, i.e., ribosomes and tRNAs to the 5′ cap of mRNA (Sonenberg and Dever, 2003).
α-Synuclein Regulates Cellular Processes by Epigenetic Mechanisms
Interaction of α-Synuclein with Histones
The first evidence of α-synuclein interaction with histones was published almost 15 years ago (Goers et al., 2003). The initial data describing this interaction were obtained from the studies of neurotoxicity. After injections of the herbicide paraquat to mice α-synuclein was colocalized with histones in the nuclei of nigral neurons (Goers et al., 2003; Cherny et al., 2004). α-Synuclein formed a tight complex with histones with a molecular mass of 48,700 Da, with a stoichiometry of 2:1 (α-synuclein/histone), and a dissociation constant of about 1 μM. Hypothetically, histones enriched with arginine and lysine residues act as scaffolds bringing together molecules of α-synuclein with high content of acidic amino acids at C-terminal domain. The authors assume that translocation into the nucleus and binding of histones is one of the mechanisms underlying α-synuclein toxicity (Goers et al., 2003).
Physiological Consequences of α-Synuclein-Histone Interaction
Further studies demonstrate that interaction of α-synuclein with histones has an important physiological function. Binding of α-synuclein to histones decreases the histone H3 acetylation and reduces acetylation in HAT assays (Kontopoulos et al., 2006). Two α-synuclein mutations, A30P and A53T, that cause familial PD, display increased probability of localization in the nucleus. These data point to an importance of further research on histone deacetylase inhibitors as a potential target for the treatment of this disease.
Several lines of evidence show that interaction of α-synuclein with histones may also alter transcription of specific genes, representing epigenetic mechanism of gene expression regulation. α-Synuclein overexpression enhances mono- and dimethylation of histone H3K9, resulting in an increase in methylated form of this histone at the SNAP25 promoter, presumably disturbing SNARE complex assembly and fusion of synaptic vesicles (Sugeno et al., 2016). Histone modification is an epigenetic mechanism which has a unique role in the cell, modulating the transactivation or repression of particular genes. Such mechanism may contribute to synaptic dysfunction occurring in PD (Sugeno et al., 2016).
Another group examined the role of histones in α-synuclein aggregation in cells experiencing apoptosis and in neuronal cells with nuclear membrane defects causing leakiness. The authors found that histones H1 and H3 released into the cytoplasm during apoptosis interact with α-synuclein to form cytoplasmic aggregates and play a role as a proaggregant factor. Importantly, their results demonstrate that histone-induced α-synuclein aggregates are transmissible to neurons both in vitro and in vivo. Histone-induced α-synuclein aggregates could spread to neurons and seed α-synuclein aggregation. Histone H1 less tightly associated with DNA is more important in the formation of pathological forms of α-synuclein. Histone-induced aggregates contain α-synuclein oligomers of variable size, including protofibrils and mature fibrils. The authors hypothesize that endogenous histones might facilitate internalization of α-synuclein aggregates and favor cell-to-cell propagation similar to the widely used transfection reagent lipofectamine (Jiang et al., 2017).
Importantly, binding of α-synuclein to histones not only affects histone modifications (Figure 3), but also it accelerates α-synuclein fibrillation.
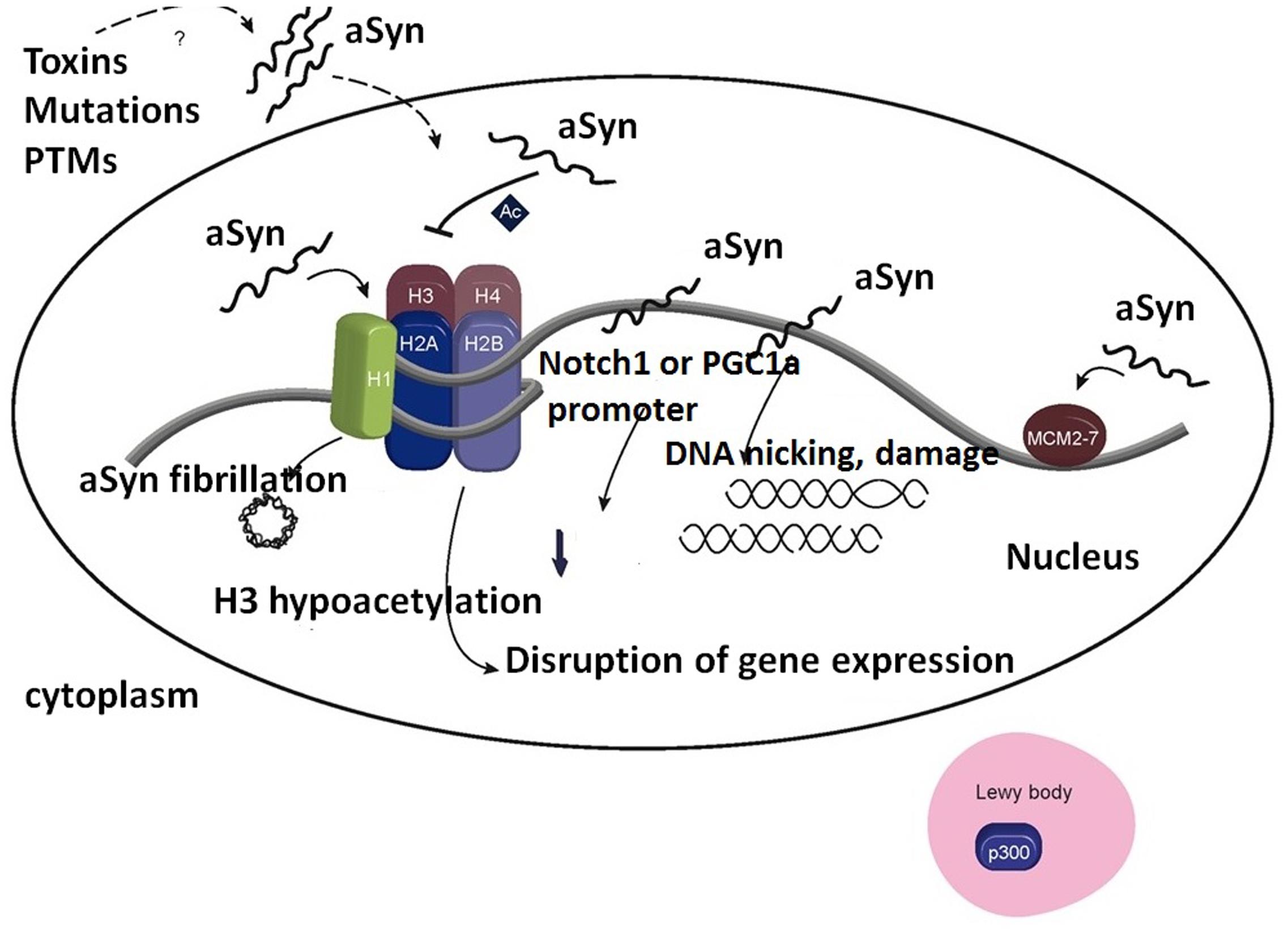
FIGURE 3. Association of α-synuclein with histones in the cell nucleus induces formation of α-synuclein fibrils. Importantly, inhibition of H3 histone acetylation by α-synuclein affects the expression level of genes responsible for cell survival. Furthermore, association of α-synuclein with p300 decreases HAT activity. Binding of α-synuclein to DNA affects the expression of several genes, e.g., NOTCH1 and PGC-1A, and may lead to DNA damage. It allows the preservation of associated MCM2-7 complexes on the chromatin. Ac, acetylation; PTMs, post-translational modifications. Adapted from Pavlou et al. (2017) with permission from the copyright holder.
Synuclein binding with histone H1 or the other core histones, causes the enhancement of α-synuclein fibrillation. α-Synuclein is able to reduce H3 acetylation, affecting the expression of several genes responsible for cell survival (Pavlou et al., 2017).
Effect of α-Synuclein on DNA Methylation
Another mechanism of α-synuclein participation in epigenetic regulation is realized via DNA methylation. α-Synuclein associates with Dnmt1 causing mislocalization and retention of Dnmt1 in the cytoplasm of neuronal cells (Desplats et al., 2011). As a result methylation in the regulatory regions of specific genes in PD and DLB brains is significantly reduced. The effect is observed both in post-mortem human brain samples and in brains of animal models of PD/DLB. Importantly, overexpression of another member of the synuclein family, β-synuclein does not cause cytoplasmic retention and Dnmt1 is not immunoprecipitated by anti-β-synuclein antibody, confirming the specificity of association between α-synuclein with Dnmt1. However, this effect could be reversed by lentivirus-mediated overexpression of Dnmt1. The finding of the inverse relation between α-synuclein oligomerization and Dnmt1 content in the nucleus suggests that the sequestration of Dnmt1 is increased by α-synuclein aggregation (Desplats et al., 2011). The loss of Dnmt1 protein from the cell nuclei is described in brains of patients with PD, DLB, and brains from transgenic mice that overexpress α-synuclein. In turn, these changes lead to the alterations in methylation pattern at the promoters of α-synuclein and several other genes, associated with neurodegenerative diseases (Desplats et al., 2011). A reduction in Dnmt1 causing mislocalization has been described in PD patient’s brains. Another team (Jowaed et al., 2010) found that methylation in intron 1 decreases α-synuclein expression, whereas inhibition of DNA methylation upregulates expression. Furthermore, methylation of intron 1 in α-synuclein gene is reduced in DNA from sporadic PD patients’ brain, including substantia nigra, putamen, and cortex. These results confirm epigenetic regulation of α-synuclein expression in PD (Jowaed et al., 2010). This data suggest another possible mechanism of α-synuclein interference with pathogenic processes of PD.
Epigenetic Mechanisms Regulating α-Synuclein Expression May Affect Its Regulatory Functions
As discussed in chapter 2, α-synuclein role in gene expression regulation depends on its intracellular concentration which in turn is regulated by epigenetic mechanisms controlling its levels. These mechanisms include methylation of α-synuclein promoter, post-translational modifications of histones, and epigenetic mechanisms based on non-coding RNAs (Pavlou et al., 2017).
Epigenetic mechanisms that control α-synuclein expression level may affect both its aggregation state and regulatory properties. Importantly, methylation level of CpG sites in α-synuclein gene in leukocytes correlates with the level in brain cells and therefore this analysis may be used as informative biomarker for prognosis of neurodegenerative diseases (Tan et al., 2014; Schmitt et al., 2015).
α-Synuclein and Chromatin Remodeling
The molecular interactions described above in Chapter 6, including binding of α-synuclein with histones, effect on DNA methylation, and epigenetic mechanisms could all induce chromatin remodeling.
Association of α-synuclein with histones may disturb nucleosome structure by reducing the availability of free histones for binding with DNA, or by affecting histone PTMs and leading to alterations in the pattern of gene transcription. In post-mortem PD brains the association of α-synuclein and chromatin is higher than in control samples. Furthermore, α-synuclein binds to the promoter of the master mitochondrial transcription activator, PGC1 alpha (PGC-1α), which is downregulated in PD brains (Siddiqui et al., 2012; Pavlou et al., 2017). The role of α-synuclein in chromatin remodeling is discussed in a recent review (Pavlou et al., 2017).
Conclusion
Here we discuss recent findings showing that in addition to their role in synapses, synucleins have non-canonical functions, and are involved in the regulation of essential cellular processes. In particular, the results presented here are consistent with a significant, although not completely understood role of synucleins in the regulation of gene expression. These mechanisms include both direct interaction of synucleins with DNA, transcription and translation factors, and less direct intervention, for example, via their effect on histone acetylation. Thus, synucleins may play a passive role of “ramblers in the crowd,” but in response to stress or changing environmental conditions they become involved in the modulation of specific protein expression via mechanisms described in Chapters 2–4 and 6. Further research is required to unveil the details of these intimate mechanisms.
Author Contributions
AAS wrote, and edited the manuscript; made substantial contributions to the conception of the work; made a final approval; and agreed to be accountable for all aspects of the work. AS wrote a part of the manuscript; made analysis and interpretation of data; made a final approval of the version to be published; and agreed to be accountable for all aspects of the work.
Funding
Some of the work by AS was conducted at the Kansas City VA Medical Center, Kansas City, MO, United States, with support from VA Merit Review grants 1I01BX000361 and the Glaucoma Foundation grant QB42308.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgment
We acknowledge VA Central Office for Merit Review grants and the Glaucoma Foundation for support.
Abbreviations
Aβ, amyloid beta; BDNF, brain-derived neurotrophic factor; DBH, dopamine-β-hydroxylase; DLB, dementia with Lewy Bodies; DNMT, DNA methyltransferase; eEF2, translation elongation factor 2; eIF3, translation initiation factor 3; HAT, histone acetyltransferase; MMP, matrix metalloproteinase; PD, Parkinson’s disease; PKCδ, protein kinase Cδ.
References
Abeliovich, A., Schmitz, Y., Fariñas, I., Choi-Lundberg, D., Ho, W. H., Castillo, P. E., et al. (2000). Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron 25, 239–252. doi: 10.1016/S0896-6273(00)80886-7
Anthony, K., and Gallo, J.-M. (2010). Aberrant RNA processing events in neurological disorders. Brain Res. 1338, 67–77. doi: 10.1016/j.brainres.2010.03.008
Anwar, S., Peters, O., Millership, S., Ninkina, N., Doig, N., Connor-Robson, N., et al. (2011). Functional alterations to the nigrostriatal system in mice lacking all three members of the synuclein family. J. Neurosci. 31, 7264–7274. doi: 10.1523/JNEUROSCI.6194-10.2011
Baptista, M. J., O’Farrell, C., Daya, S., Ahmad, R., Miller, D. W., Hardy, J., et al. (2003). Co-ordinate transcriptional regulation of dopamine synthesis genes by alpha-synuclein in human neuroblastoma cell lines. J. Neurochem. 85, 957–968. doi: 10.1046/j.1471-4159.2003.01742.x
Bellucci, A., Navarria, L., Falarti, E., Zaltieri, M., Bono, F., Collo, G., et al. (2011). Redistribution of DAT/α-synuclein complexes visualized by “in situ” proximity ligation assay in transgenic mice modelling early Parkinson’s disease. PLoS ONE 6:e27959. doi: 10.1371/journal.pone.0027959
Bendor, J. T., Logan, T. P., and Edwards, R. H. (2013). The function of α-synuclein. Neuron 79, 1044–1066. doi: 10.1016/j.neuron.2013.09.004
Bonini, N. M., and Giasson, B. I. (2005). Snaring the function of alpha-synuclein. Cell 123, 359–361. doi: 10.1016/j.cell.2005.10.017
Braun, R. J., Büttner, S., Ring, J., Kroemer, G., and Madeo, F. (2010). Nervous yeast: modeling neurotoxic cell death. Trends Biochem. Sci. 35, 135–144. doi: 10.1016/j.tibs.2009.10.005
Brown, J. W., Buell, A. K., Michaels, T. C., Meisl, G., Carozza, J., Flagmeier, P., et al. (2016). β-Synuclein suppresses both the initiation and amplification steps of α-synuclein aggregation via competitive binding to surfaces. Sci. Rep. 6:36010. doi: 10.1038/srep36010
Burré, J., Sharma, M., Tsetsenis, T., Buchman, V., Etherton, M. R., and Südhof, T. C. (2010). Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 329, 1663–1667. doi: 10.1126/science.1195227
Chalovich, E. M., Zhu, J. H., Caltagarone, J., Bowser, R., and Chu, C. T. (2006). Functional repression of cAMP response element in 6-hydroxydopamine-treated neuronal cells. J. Biol. Chem. 281, 17870–17881. doi: 10.1074/jbc.M602632200
Chandra, S., Gallardo, G., Fernández-Chacón, R., Schlüter, O. M., and Südhof, T. C. (2005). Alpha-synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell 123, 383–396. doi: 10.1016/j.cell.2005.09.028
Chartier-Harlin, M. C., Dachsel, J. C., Vilariño-Güell, C., Lincoln, S. J., Leprêtre, F., Hulihan, M. M., et al. (2011). Translation initiator EIF4G1 mutations in familial Parkinson disease. Am. J. Hum. Genet. 89, 398–406. doi: 10.1016/j.ajhg.2011.08.009
Cherny, D., Hoyer, W., Subramaniam, V., and Jovin, T. M. (2004). Double-stranded DNA stimulates the fibrillation of alpha-synuclein in vitro and is associated with the mature fibrils: an electron microscopy study. J. Mol. Biol. 344, 929–938. doi: 10.1016/j.jmb.2004.09.096
Chung, C. Y., Khurana, V., Yi, S., Sahni, N., Loh, K. H., Auluck, P. K., et al. (2017). In situ peroxidase labeling and mass-spectrometry connects alpha-synuclein directly to endocytic trafficking and mRNA metabolism in neurons. Cell Syst. 4, 242. e4–250. e4. doi: 10.1016/j.cels.2017.01.002
Cirillo, D., Agostini, F., Klus, P., Marchese, D., Rodriguez, S., Bolognesi, B., et al. (2013). Neurodegenerative diseases: quantitative predictions of protein-RNA interactions. RNA 19, 129–140. doi: 10.1261/rna.034777.112
Desplats, P., Spencer, B., Coffee, E., Patel, P., Michael, S., Patrick, C., et al. (2011). Alpha-synuclein sequesters Dnmt1 from the nucleus: a novel mechanism for epigenetic alterations in Lewy body diseases. J. Biol. Chem. 286, 9031–9037. doi: 10.1074/jbc.C110.212589
Diao, J., Burré, J., Vivona, S., Cipriano, D. J., Sharma, M., Kyoung, M., et al. (2013). Native α-synuclein induces clustering of synaptic-vesicle mimics via binding to phospholipids and synaptobrevin-2/VAMP2. eLife 2:e00592. doi: 10.7554/eLife.00592
Ellis, R. J. (2001). Macromolecular crowding: obvious but underappreciated. Trends Biochem. Sci. 26, 597–604. doi: 10.1016/S0968-0004(01)01938-7
Emamzadeh, F. N. (2016). Alpha-synuclein structure, functions, and interactions. J. Res. Med. Sci. 21:29. doi: 10.4103/1735-1995.181989
Fujioka, S., Sundal, C., Strongosky, A. J., Castanedes, M. C., Rademakers, R., Ross, O. A., et al. (2013). Sequence variants in eukaryotic translation initiation factor 4-gamma (eIF4G1) are associated with Lewy body dementia. Acta Neuropathol. 125, 425–438. doi: 10.1007/s00401-012-1059-4
Garcia-Esparcia, P., Hernández-Ortega, K., Koneti, A., Gil, L., Delgado-Morales, R., Castaño, E., et al. (2015). Altered machinery of protein synthesis is region- and stage-dependent and is associated with α-synuclein oligomers in Parkinson’s disease. Acta Neuropathol. Commun. 3:76. doi: 10.1186/s40478-015-0257-4
Goers, J., Manning-Bog, A. B., McCormack, A. L., Millett, I. S., Doniach, S., Di Monte, D. A., et al. (2003). Nuclear localization of α-synuclein and its interaction with histones. Biochemistry 42, 8465–8471. doi: 10.1021/bi0341152
Grandin, N., and Charbonneau, M. (1999). Dbf2 is implicated in a Cbt1-dependent pathway following a shift from glucose to galactose or non-fermentable carbon sources in Saccharomyces cerevisiae. Mol. Gen. Genet. 261, 402–407.
Greten-Harrison, B., Polydoro, M., Morimoto-Tomita, M., Diao, L., Williams, A. M., Nie, E. H., et al. (2010). αβγ-Synuclein triple knockout mice reveal age-dependent neuronal dysfunction. Proc. Natl. Acad. Sci. U.S.A. 107, 19573–19578. doi: 10.1073/pnas.1005005107
Hashimoto, M., Rockenstein, E., Mante, M., Mallory, M., and Masliah, E. (2001). Beta-Synuclein inhibits alpha-synuclein aggregation: a possible role as an anti-parkinsonian factor. Neuron 32, 213–223. doi: 10.1016/S0896-6273(01)00462-7
Hashimoto, M., Takenouchi, T., Rockenstein, E., and Masliah, E. (2003). Alpha-synuclein up-regulates expression of caveolin-1 and down-regulates extracellular signal-regulated kinase activity in B103 neuroblastoma cells: role in the pathogenesis of Parkinson’s disease. J. Neurochem. 85, 1468–1479. doi: 10.1046/j.1471-4159.2003.01791.x
Hasnat, A., Bichenkova, E., Yu, X., Arnold, J. R., Fisher, J., Fedorova, O., et al. (2007). Fluorescence spectroscopic and (19)f NMR studies of human thymidylate synthase with its cognate RNA. J. Biomol. Struct. Dyn. 25, 253–270. doi: 10.1080/07391102.2007.10507174
Hegde, M. L., and Jagannatha Rao, K. S. (2003). Challenges and complexities of alpha-synuclein toxicity: new postulates in unfolding the mystery associated with Parkinson’s disease. Arch. Biochem. Biophys. 418, 169–178. doi: 10.1016/j.abb.2003.08.015
Hegde, M. L., and Rao, K. S. (2007). DNA induces folding in alpha-synuclein: understanding the mechanism using chaperone property of osmolytes. Arch. Biochem. Biophys. 464, 57–69. doi: 10.1016/j.abb.2007.03.042
Hegde, M. L., Vasudevaraju, P., and Rao, K. J. (2010). DNA induced folding/fibrillation of alpha-synuclein: new insights in Parkinson’s disease. Front. Biosci. 15, 418–436. doi: 10.2741/3628
Hunkele, A., Sultan, H., Ikalina, F. A., Liu, A. H., Nahar-Gohad, P., and Ko, J. L. (2016). Identification of gamma-synuclein as a new PCBP1-interacting protein. Neurol. Res. 38, 1064–1078. doi: 10.1179/1743132815Y.0000000091
Iwata, A., Maruyama, M., Kanazawa, I., and Nukina, N. (2001a). alpha-Synuclein affects the MAPK pathway and accelerates cell death. J. Biol. Chem. 276, 45320–45329.
Iwata, A., Miura, S., Kanazawa, I., Sawada, M., and Nukina, N. (2001b). alpha-Synuclein forms a complex with transcription factor Elk-1. J. Neurochem. 77, 239–252.
Jiang, P., Gan, M., Yen, S. H., McLean, P. J., and Dickson, D. W. (2017). Histones facilitate α-synuclein aggregation during neuronal apoptosis. Acta Neuropathol. 133, 547–558. doi: 10.1007/s00401-016-1660-z
Jiménez, J. S. (2010). Protein-DNA interaction at the origin of neurological diseases: a hypothesis. J. Alzheimers Dis. 22, 375–391. doi: 10.3233/JAD-2010-100189
Jin, H., Kanthasamy, A., Ghosh, A., Yang, Y., Anantharam, V., and Kanthasamy, A. G. (2011). α-Synuclein negatively regulates protein kinase Cδ expression to suppress apoptosis in dopaminergic neurons by reducing p300 histone acetyltransferase activity. J. Neurosci. 31, 2035–2051. doi: 10.1523/JNEUROSCI.5634-10.2011
Jowaed, A., Schmitt, I., Kaut, O., and Wüllner, U. (2010). Methylation regulates alpha-synuclein expression and is decreased in Parkinson’s disease patients’ brains. J. Neurosci. 30, 6355–6359. doi: 10.1523/JNEUROSCI.6119-09.2010
Khurana, V., and Lindquist, S. (2010). Modelling neurodegeneration in Saccharomyces cerevisiae: Why cook with baker’s yeast? Nat. Rev. Neurosci. 11, 436–449. doi: 10.1038/nrn2809
Khurana, V., Peng, J., Chung, C. Y., Auluck, P. K., Fanning, S., Tardiff, D. F., et al. (2017). Genome-scale networks link neurodegenerative disease genes to α-synuclein through specific molecular pathways. Cell Syst. 4, 157.e14–170. e14. doi: 10.1016/j.cels.2016.12.011
Kim, S., Park, J. M., Moon, J., and Choi, H. J. (2014). Alpha-synuclein interferes with cAMP/PKA-dependent upregulation of dopamine β-hydroxylase and is associated with abnormal adaptive responses to immobilization stress. Exp. Neurol. 252, 63–74. doi: 10.1016/j.expneurol.2013.11.009
Knott, A. B., Perkins, G., Schwarzenbacher, R., and Bossy-Wetzel, E. (2008). Mitochondrial fragmentation in neurodegeneration. Nat. Rev. Neurosci. 9, 505–518. doi: 10.1038/nrn2417
Kokhan, V. S., Van’kin, G. I., Bachurin, S. O., and Shamakina, I. Y. (2013). Differential involvement of the gamma-synuclein in cognitive abilities on the model of knockout mice. BMC Neurosci. 14:53. doi: 10.1186/1471-2202-14-53
Kontopoulos, E., Parvin, J. D., and Feany, M. B. (2006). α-synuclein acts in the nucleus to inhibit histone acetylation and promote neurotoxicity. Hum. Mol. Genet. 15, 3012–3023. doi: 10.1093/hmg/ddl243
Lan, A., Smoly, I. Y., Rapaport, G., Lindquist, S., Fraenkel, E., and Yeger-Lotem, E. (2011). ResponseNet: revealing signaling and regulatory networks linking genetic and transcriptomic screening data. Nucleic Acids Res. 39, W424–W429. doi: 10.1093/nar/gkr359
Lee, J. F., Stovall, G. M., and Ellington, A. D. (2006). Aptamer therapeutics advance. Curr. Opin. Chem. Biol. 10, 282–289. doi: 10.1016/j.cbpa.2006.03.015
Littleton, J. T., and Bellen, H. J. (1995). Synaptotagmin controls and modulates synaptic-vesicle fusion in a Ca2+ dependent manner. Trends Neurosci. 18, 177–183. doi: 10.1016/0166-2236(95)93898-8
Ma, K. L., Song, L. K., Yuan, Y. H., Zhang, Y., Yang, J. L., Zhu, P., et al. (2014). α-Synuclein is prone to interaction with the GC-box-like sequence in vitro. Cell Mol. Neurobiol. 34, 603–609. doi: 10.1007/s10571-014-0046-9
Malhotra, V., and Emr, S. D. (2002). Rothman and Schekman SNAREd by lasker for trafficking. Cell 111, 1–3. doi: 10.1016/S0092-8674(02)01008-5
Maroteaux, L., Campanelli, J. T., and Scheller, R. H. (1988). Synuclein: a neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J. Neurosci. 8, 2804–2815.
Martell, J. D., Deerinck, T. J., Sancak, Y., Poulos, T. L., Mootha, V. K., Sosinsky, G. E., et al. (2012). Engineered ascorbate peroxidase as a genetically encoded reporter for electron microscopy. Nat. Biotechnol. 30, 1143–1148. doi: 10.1038/nbt.2375
Martins, M., Rosa, A., Guedes, L. C., Fonseca, B. V., Gotovac, K., Violante, S., et al. (2011). Convergence of miRNA expression profiling, α-synuclein interaction and GWAS in Parkinson’s disease. PLoS ONE 6:e25443. doi: 10.1371/journal.pone.0025443
Minton, A. P. (1993). Macromolecular crowding and molecular recognition. J. Mol. Recognit. 6, 211–214. doi: 10.1002/jmr.300060410
Minton, A. P. (1998). Molecular crowding: analysis of effects of high concentrations of inert cosolutes on biochemical equilibria and rates in terms of volume exclusion. Methods Enzymol. 295, 127–149. doi: 10.1016/S0076-6879(98)95038-8
Ninkina, N., Peters, O. M., Connor-Robson, N., Lytkina, O., Sharfeddin, E., and Buchman, V. L. (2012). Contrasting effects of α-synuclein and γ-synuclein on the phenotype of cysteine string protein α (CSPα) null mutant mice suggest distinct function of these proteins in neuronal synapses. J. Biol. Chem. 287, 44471–44477. doi: 10.1074/jbc.M112.422402
Padmaraju, V., Bhaskar, J. J., Prasada Rao, U. J., Salimath, P. V., and Rao, K. S. (2011). Role of advanced glycation on aggregation and DNA binding properties of α-synuclein. J. Alzheimers Dis. 24(Suppl. 2), 211–221. doi: 10.3233/JAD-2011-101965
Pavlou, M. A., Pinho, R., Paiva, I., and Outeiro, T. F. (2017). The yin and yang of α-synuclein-associated epigenetics in Parkinson’s disease. Brain 140, 878–886. doi: 10.1093/brain/aww227
Scheper, W., and Hoozemans, J. J. (2009). Endoplasmic reticulum protein quality control in neurodegenerative disease: the good, the bad and the therapy. Curr. Med. Chem. 16, 615–626. doi: 10.2174/092986709787458506
Schmitt, I., Kaut, O., Khazneh, H., deBoni, L., Ahmad, A., Berg, D., et al. (2015). L-dopa increases α-synuclein DNA methylation in Parkinson’s disease patients in vivo and in vitro. Mov. Disord. 30, 1794–1801. doi: 10.1002/mds.26319
Segura-Ulate, I., Yang, B., Vargas-Medrano, J., and Perez, R. G. (2017). FTY720 (Fingolimod) reverses α-synuclein-induced downregulation of brain-derived neurotrophic factor mRNA in OLN-93 oligodendroglial cells. Neuropharmacology 117, 149–157. doi: 10.1016/j.neuropharm.2017.01.028
Seo, J. H., Rah, J. C., Choi, S. H., Shin, J. K., Min, K., Kim, H. S., et al. (2002). Alpha-synuclein regulates neuronal survival via Bcl-2 family expression and PI3/Akt kinase pathway. FASEB J. 16, 1826–1828.
Shtilerman, M. D., Ding, T. T., and Lansbury, P. T. Jr. (2002). Molecular crowding accelerates fibrillization of alpha-synuclein: could an increase in the cytoplasmic protein concentration induce Parkinson’s disease? Biochemistry 41, 3855–3860.
Siddiqui, A., Chinta, S. J., Mallajosyula, J. K., Rajagopolan, S., Hanson, I., Rane, A., et al. (2012). Selective binding of nuclear α-synuclein to the PGC1alpha promoter under conditions of oxidative stress may contribute to losses in mitochondrial function: implications for Parkinson’s disease. Free Radic. Biol. Med. 53, 993–1003. doi: 10.1016/j.freeradbiomed.2012.05.024
Sonenberg, N., and Dever, T. E. (2003). Eukaryotic translation initiation factors and regulators. Curr. Opin. Struct. Biol. 13, 56–63. doi: 10.1016/S0959-440X(03)00009-5
Sugeno, N., Jäckel, S., Voigt, A., Wassouf, Z., Schulze-Hentrich, J., and Kahle, P. J. (2016). α-Synuclein enhances histone H3 lysine-9 dimethylation and H3K9me2-dependent transcriptional responses. Sci. Rep. 6:36328. doi: 10.1038/srep36328
Surgucheva, I., McMahon, B., and Surguchov, A. (2006). gamma-Synuclein has a dynamic intracellular localization. Cell Motil. Cytoskeleton 63, 447–458. doi: 10.1002/cm.20135
Surgucheva, I., Shestopalov, V. I., and Surguchov, A. (2008). Effect of γ-synuclein silencing on apoptotic pathways in retinal ganglion cells. J. Biol. Chem. 283, 36377–36385. doi: 10.1074/jbc.M806660200
Surgucheva, I., and Surguchov, A. (2008). gamma-Synuclein: cell-type specific promoter activity and binding to transcription factors. J. Mol. Neurosci. 35, 267–271. doi: 10.1007/s12031-008-9074-6
Surgucheva, I. G., Sivak, J. M., Fini, M. E., Palazzo, R. E., and Surguchov, A. P. (2003). Effect of gamma-synuclein overexpression on matrix metalloproteinases in retinoblastoma Y79 cells. Arch. Biochem. Biophys. 410, 167–176. doi: 10.1016/S0003-9861(02)00664-1
Surguchov, A. (2008). Molecular and cellular biology of synucleins. Int. Rev. Cell Mol. Biol. 270, 225–317. doi: 10.1016/S1937-6448(08)01406-8
Surguchov, A. (2015). Intracellular dynamics of synucleins: here, there and everywhere. Int. Rev. Cell Mol. Biol. 320, 103–169. doi: 10.1016/bs.ircmb.2015.07.007
Surguchov, A., Palazzo, R. E., and Surgucheva, I. (2001). Gamma synucleins: subcellular localization in neuronal cells and effect on signal transduction. Cell Motil. Cytoskeleton 2001, 218–228. doi: 10.1002/cm.1035
Surguchov, A., Surgucheva, I., Sharma, M., and Singh, V. (2017). New putative epigenetic mechanism of epilepsy. Front. Neurol. 8:3. doi: 10.3389/fneur.2017.00003
Surguchov, A., Surgucheva, I., Solessio, E., and Baehr, W. (1999). Synoretin–a new protein belonging to the synuclein family. Mol. Cell. Neurosci. 13, 95–103. doi: 10.1006/mcne.1999.0735
Tai, N., Schmitz, J. C., Liu, J., Lin, X., Bailly, M., Chen, T. M., et al. (2004). Translational autoregulation of thymidylate synthase and dihydrofolate reductase. Front. Biosci. 9, 2521–2526. doi: 10.2741/1413
Tan, Y. Y., Wu, L., Zhao, Z. B., Wang, Y., Xiao, Q., Liu, J., et al. (2014). Methylation of α-synuclein and leucine-rich repeat kinase 2 in leukocyte DNA of Parkinson’s disease patients. Parkinsonism Relat. Disord. 20, 308–313. doi: 10.1016/j.parkreldis.2013.12.002
Tardiff, D. F., Khurana, V., Chung, C. Y., and Lindquist, S. (2014). From yeast to patient neurons and back again: powerful new discovery platform. Mov. Disord. 10, 1231–1240. doi: 10.1002/mds.25989
Tenreiro, S., Franssens, V., Winderickx, J., and Outeiro, T. F. (2017). Yeast models of Parkinson’s disease-associated molecular pathologies. Curr. Opin. Genet. Dev. 44, 74–83. doi: 10.1016/j.gde.2017.01.013
Ter-Avanesyan, M. D., Zimmermann, J., Inge-Vechtomov, S. G., Sudarikov, A. B., Smirnov, V. N., and Surguchov, A. P. (1982). Ribosomal recessive suppressors cause a respiratory deficiency in yeast Saccharomyces cerevisiae. Mol. Gen. Genet. 185, 319–323. doi: 10.1007/BF00330805
Tsukakoshi, K., Abe, K., Sode, K., and Ikebukuro, K. (2012). Selection of DNA aptamers that recognize α-synuclein oligomers using a competitive screening method. Anal. Chem. 84, 5542–5547. doi: 10.1021/ac300330g
Tsukakoshi, K., Harada, R., Sode, K., and Ikebukuro, K. (2010). Screening of DNA aptamer which binds to alpha-synuclein. Biotechnol. Lett. 32, 643–648. doi: 10.1007/s10529-010-0200-5
Tucker, B. J., and Breaker, R. R. (2005). Riboswitches as versatile gene control elements. Curr. Opin. Struct. Biol. 15, 342–348. doi: 10.1016/j.sbi.2005.05.003
Uversky, V. N., Li, J., and Fink, A. L. (2001). Metal-triggered structural transformations, aggregation, and fibrillation of human alpha-synuclein. J. Biol. Chem. 276, 44284–44296. doi: 10.1074/jbc.M105343200
Uversky, V. N., M Cooper, E., Bower, K. S., Li, J., and Fink, A. L. (2002). Accelerated alpha-synuclein fibrillation in crowded milieu. FEBS Lett. 515, 99–103. doi: 10.1016/S0014-5793(02)02446-8
Vargas, K. J., Makani, S., Davis, T., Westphal, T. H., Castillo, P. E., and Chandra, S. S. (2014). Synucleins regulate the kinetics of synaptic vesicle endocytosis. J. Neurosci. 34, 9364–9376. doi: 10.1523/JNEUROSCI.4787-13.2014
Vargas, K. J., Schrod, N., Davis, T., Fernandez-Busnadiego, R., Taguchi, Y. V., Laugks, U., et al. (2017). Synucleins have multiple effects on presynaptic architecture. Cell Rep. 18, 161–173. doi: 10.1016/j.celrep.2016.12.023
Vasudevaraju, P., Guerrero, E., Hegde, M. L., Collen, T. B., Britton, G. B., and Rao, K. S. (2012). New evidence on α-synuclein and Tau binding to conformation and sequence specific GC∗ rich DNA: relevance to neurological disorders. J. Pharm. Bioallied Sci. 4, 112–117. doi: 10.4103/0975-7406.94811
Yeger-Lotem, E., Riva, L., Su, L. J., Gitler, A. D., Cashikar, A. G., King, O. D., et al. (2009). Bridging high-throughput genetic and transcriptional data reveals cellular responses to alpha-synuclein toxicity. Nat. Genet. 41, 16–23. doi: 10.1038/ng.337
Zaltieri, M., Grigoletto, J., Longhena, F., Navarria, L., Favero, G., Castrezzati, S., et al. (2015). α-Synuclein and synapsin III cooperatively regulate synaptic function in dopamine neurons. J. Cell Sci. 128, 2231–2243. doi: 10.1242/jcs.157867
Keywords: gene expression, synucleins, epigenetic regulation, transcription factors, translation factors, DNA methylation, histone modifications, neurodegeneration
Citation: Surguchev AA and Surguchov A (2017) Synucleins and Gene Expression: Ramblers in a Crowd or Cops Regulating Traffic? Front. Mol. Neurosci. 10:224. doi: 10.3389/fnmol.2017.00224
Received: 20 May 2017; Accepted: 29 June 2017;
Published: 13 July 2017.
Edited by:
Ivette Martinez Sandoval, Michigan State University, United StatesReviewed by:
Ralf J. Braun, University of Bayreuth, GermanySubhrangshu Guhathakurta, University of Central Florida, United States
Copyright © 2017 Surguchev and Surguchov. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andrei Surguchov, YXN1cmd1Y2hvdkBrdW1jLmVkdQ==