 Beatrice Oehler1
Beatrice Oehler1 Milad Mohammadi1
Milad Mohammadi1 Cristina Perpina Viciano2Dagmar Hackel1
Cristina Perpina Viciano2Dagmar Hackel1 Carsten Hoffmann2,3Alexander Brack1
Carsten Hoffmann2,3Alexander Brack1 Heike L. Rittner1*
Heike L. Rittner1*- 1Department of Anesthesiology and Critical Care, University Hospital of Wuerzburg, Wuerzburg, Germany
- 2Bio-Imaging-Center/Rudolf-Virchow-Center, Institute of Pharmacology, University of Wuerzburg, Wuerzburg, Germany
- 3Institute for Molecular Cell Biology, CMB—Center for Molecular Biomedicine, University Hospital Jena, Jena, Germany
Antinociceptive pathways are activated in the periphery in inflammatory pain, for instance resolvins and opioid peptides. Resolvins are biosynthesized from omega-3 polyunsaturated fatty acids such as eicosapentaenoic acid and docosahexaenoic acid. Resolvin D1 (RvD1) and resolvin E1 (RvE1) initiate the resolution of inflammation and control of hypersensitivity via induction of anti-inflammatory signaling cascades. RvD1 binds to lipoxin A4/annexin-A1 receptor/formyl-peptide receptor 2 (ALX/FPR2), RvE1 to chemerin receptor 23 (ChemR23). Antinociception of RvD1 is mediated by interaction with transient receptor potential channels ankyrin 1 (TRPA1). Endogenous opioid peptides are synthesized and released from leukocytes in the tissue and bind to opioid receptors on nociceptor terminals. Here, we further explored peripheral mechanisms of RvD1 and chemerin (Chem), the ligand of ChemR23, in complete Freund’s adjuvant (CFA)-induced hindpaw inflammation in male Wistar rats. RvD1 and Chem ameliorated CFA-induced hypersensitivity in early and late inflammatory phases. This was prevented by peripheral blockade of the μ-opioid peptide receptor (MOR) using low dose local naloxone or by local injection of anti-β-endorphin and anti-met-enkephalin (anti-ENK) antibodies. Naloxone also hindered antinociception by the TRPA1 inhibitor HC-030031. RvD1 did not stimulate the release of β-endorphin from macrophages and neutrophils, nor did RvD1 itself activate G-proteins coupled MOR or initiate β-arrestin recruitment to the membrane. TRPA1 blockade by HC-030031 in inflammation in vivo as well as inhibition of the TRPA1-mediated calcium influx in dorsal root ganglia neurons in vitro was hampered by naloxone. Peripheral application of naloxone alone in vivo already lowered mechanical nociceptive thresholds. Therefore, either a perturbation of the balance of endogenous pro- and antinociceptive mechanisms in early and late inflammation, or an interaction of TRPA1 and opioid receptors weaken the antinociceptive potency of RvD1 and TRPA1 blockers.
Introduction
Resolvins are lipid mediators that rise during resolution of acute inflammation from poly-unsaturated fatty acids and docosahexanoic acid (Serhan, 2014). Resolvins such as resolvin D1 (RvD1) and resolvin E1 (RvE1) facilitate the resolution of inflammation (Ji et al., 2011) in, for example, sepsis, asthma, atherosclerosis, and osteoarthritis (Merched et al., 2008; Xu and Ji, 2011; Chiang et al., 2015; Provoost et al., 2016; Huang et al., 2017). All known resolvin receptors are G protein-coupled receptors (GPCR). They interfere with immune cell functions to promote resolution. RvD1 binds to the lipoxin A4/annexin-A1 receptor/formyl-peptide receptor 2 (ALX/FPR2) as well as to the G-protein-coupled receptor 32 (GPR32; Krishnamoorthy et al., 2010). RvE1 and chemerin (Chem), an endogenous peptide agonist, bind to the Chem receptor 23 (ChemR23; Arita et al., 2005). Intracellular pro-inflammatory signaling cascades are downregulated by resolvin receptor activation. Chem, a 14 kDa protein secreted in an inactive form as pro-Chem and activated through cleavage of the C-terminus by serine proteases as activated in inflammation and coagulation (Xu et al., 2010).
Transient receptor potential channels of the ankyrin 1 (TRPA1) and vanilloid subtype (TRPV1) are chemosensors in nociceptors (Gangadharan and Kuner, 2013). TRPV1 is activated by heat (>42°C), acidosis, endogenous lipidergic pro-inflammatory messengers such as leukotrienes and natural pungent compounds such as capsaicin (Tominaga et al., 1998). TRPA1 responds to exogenous irritants like allyl isothiocyanate (AITC; Bautista et al., 2006) and endogenous lipids like oxidized phospholipids found in complete Freund’s adjuvant (CFA)-induced hindpaw inflammation.
Resolvins were the first endogenous TRPA1 and TRPV1 channel inhibitors described (Park et al., 2011b; Serhan, 2014; Lim et al., 2015). Apart from their inflammation resolving properties, resolvins have antinociceptive properties: intraplantar and intrathecal application of RvD1 and RvE1 reduce thermal hypersensitivity and local inflammation induced by carrageenan, CFA, or formalin without changing basal nociceptive thresholds (Xu et al., 2010). Intrathecal administration of RvD1 before surgery increases nociceptive thresholds. When administered on the first postoperative day, animals showed the same pain as before surgery (Huang et al., 2011). Beside of TRPA1 and TRPV1, antinociception by RvD1 is mediated by inhibition of TRPV3 and TRPV4, also expressed in peripheral nerve endings in the skin and muscles (Bang et al., 2010; Xu et al., 2010). Specifically, spinal opioid receptors are not involved in RvE1-evoked antinociception (Xu et al., 2010; Park et al., 2011a).
In the early phase of inflammation, neutrophils are the predominant leukocyte population, during the late phase, macrophages are more prevalent (Brack et al., 2004). Endogenous opioids like met-enkephalin and β-endorphin are produced and released from neutrophils and monocytes/macrophages (Rittner et al., 2001, 2006, 2009; Sauer et al., 2014; Wang et al., 2014). They bind to the G-protein-coupled μ-opioid receptor (MOR) expressed in the peripheral nociceptive system (Mambretti et al., 2016; Corder et al., 2017; Spahn et al., 2017). MOR activates several signaling cascades including Gi/o-protein-dependent and -independent signaling pathways such as activation of adenylate cyclase, phosphorylation by protein kinases, activation of specific potassium calcium channels, and β-arrestin recruitment (Mambretti et al., 2016). Naloxone, a competitive antagonist of MOR, as well as antibodies against endogenous opioid peptides like anti-met-enkephalin (anti-ENK) or anti-β-endorphin (anti-END)—applied locally—prevent e.g., formyl-peptide-induced antinociception (Rittner et al., 2009; Wang et al., 2014).
In our study here, we further investigated peripheral antinociceptive properties of RvD1 and Chem, an agonist to the same receptor as RvE1, in rats in the early and late phase of local inflammation and deciphered its local mechanism. We hypothesized that RvD1 and Chem interact with opioid receptor activation on peripheral nerve terminals and/or TRPA1 activation.
Materials and Methods
Reagents and Chemicals
The following reagents were used: AITC, HC-030031, naloxone, DAMGO, IgG, fMLP and ionomycin (Sigma-Aldrich, Taufkirchen, Germany), RvD1 (Cayman, Ann Arbor, MI, USA), Chem (R&D Systems Inc., Minneapolis, MN, USA), anti-END and anti-ENK (Bachem, Weil am Rhein, Germany), anti-PMN serum (Accurate Chemical and Scientific Corporation, Westbury, NY, USA), and CFA (Calbiochem, San Diego, USA/BD Bioscience, San Jose, CA, USA). Dimethyl sulfoxide or aqueous physiological solutions served as solvents.
Chemicals and reagents for cell culture experiments included gentamycin, penicillin, streptomycin, nerve growth factor (NGF), IST liquid media supplement (100×), HEPES (4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid, N-(2-Hydroxyethyl)piperazine-N′-(2-ethanesulfonic acid)), poly-L-lysine, DMEM/F12, fetal bovine serum (Life Technologies GmbH, Darmstadt, Germany), and Effectene transfection reagent (Qiagen, Hilden, Germany).
In Vivo Studies
Animals
This study was carried out in accordance with the recommendations of International Association for the Study of Pain (IASP). The protocol was approved by the animal care committee of the provincial government of Wuerzburg (55.2-2531.01-5/13). Male Wistar rats weighing 180–220 g were injected intraplantarly under brief isoflurane anesthesia as described below. Animals were randomly assigned to treatments by numbers within a cage.
Measurement of Nociceptive Thresholds
Mechanical thresholds were determined using the paw pressure algesiometer (modified Randall-Selitto test; Ugo Basile, Comerio, Italy; Hackel et al., 2012). The pressure required to elicit paw withdrawal (paw pressure threshold, PPT) was determined by a blinded investigator. Averages from three measurements per treatment were calculated. Baseline measurements were obtained before and 2 h or 96 h after intraplantar injection of 150 μl CFA. After indicated time points, RvD1, Chem, or HC-030031 dissolved in 150 μl 0.9% saline were applied. PPT were determined 15, 45 and 180 min thereafter. In selected experiments, 0.56 ng naloxone (NLX), anti-END (2 μg) or anti-ENK (1.25 μg; Rittner et al., 2006) dissolved in 0.9% saline were injected intraplantarly before or together with TRP channel antagonists, RvD1 or Chem. Anti-PMN (80 μl) was injected i.p. 15–18 h before CFA to deplete neutrophils (Rittner et al., 2006). Doses were chosen based on pilot experiments and on the literature (Xu et al., 2010; Liu et al., 2016).
In Vitro Studies
Primary Culture of Dorsal Root Ganglia Neurons (DRG)
Preparation of Dorsal Root Ganglia (DRG) from adult wildtype mice were carried out as described (Schulze et al., 2013). DRG neurons were grown at a density of 7 × 103 cells per glass cover slip coated with poly-L-lysine (20 μg/ml) and cultured at 37°C, 5% CO2 atmosphere for 1 day. Measurements were performed the following day. Medium contained 100 ng/ml NGF (Sigma-Aldrich, Taufkirchen, Germany).
Calcium Imaging
For ratiometric single cell calcium analysis, DRGs were labeled with Fura-2/AM in imaging solution (in mM): 134 NaCl, 6 KCl, 1 MgCl2, 1 CaCl2, 10 HEPES, 5.5 glucose, pH 7.4 adjusted with NaOH (Oehler et al., 2012, 2017). All measurements were performed at room temperature using a Nikon TE2000-E microscope. Fura-2/AM was excited with a Lambda DG4/17 wavelength switch (Sutter Instruments, Novato, CA, USA). Time-lapse image series were acquired with a cooled EMCCD Andor iXon camera (Andor Technology Ltd., Belfast, UK) controlled by NIS Elements Software (Nikon, Düsseldorf, Germany). Objective: CFI S-Fluor 10×/0.5 (Nikon). Image series were analyzed with ImageJ 1.46r, time series analyzer V2.0 plugin (Rasband, W.S., ImageJ, U.S. National Institutes of Health, Bethesda, MD, USA). AITC was used as TRPA1 agonist and β-endorphin as MOR agonist. The mean of basal fluorescence intensity was determined for each measurement. Number of reacting cells (%) was calculated by 1.5-fold increase of mean basal fluorescent intensity after stimulation. The area under curve (AUC) was taken from the mean of five individual experiments. Intervals correspond to the stimulation period of AITC.
β-Endorphin Release
Rat neutrophils were isolated by lavage of the peritoneal cavity with 2 mM EDTA/phosphate buffered saline (PBS) 4 h after i.p. injection of 20 ml 1% oyster glycogen (Rittner et al., 2006), macrophages 4 days after i.p. injection of 3% thioglycollate (Hackel et al., 2013). Neutrophils or macrophages (107 cells in Hanks balanced salt solution (HBSS)) were treated with cytochalasin B (5 μg/ml) at 37°C for 5 min for preactivation (Rittner et al., 2009). Subsequently, RvD1 or ionomycin (10 μM) and fMLP (1 μM) dissolved in HBSS containing bestatin, aprotinin, and thiorphan, was added. To terminate stimulation, cells were cooled on ice. β-endorphin was measured in the supernatant (β-endorphin (rat) EIA Kit, Phoenix Pharmaceuticals, Burlingame, CA, USA)). Absorbance was measured at 450 nm (Magellan V5.01 software, Tecan, Crailsheim, Germany).
Kinetic Measurements of Gαi Activation
HEK-293 cells were cultured in DMEM, high glucose, 10% fetal calf serum (Biochrom, Berlin, Germany), 100 Uml−1 penicillin G, and 100 μgml−1 streptomycin sulfate. For kinetic measurements of Gαi1, Gαi2 and Gαi3 activation, HEK-293 cells were seeded on 40 mm poly-D-lysin-coated plates 1 day before transfection. Transient transfection with 0.63 μg of untagged MOR and 0.63 μg of the corresponding subtype of G protein sensor, either Gαi1, Gαi2 or Gαi3 (pGβ1-2A-yellow fluorescent protein (YFP)-Gγ2-IRES-Gαi-mTq2 cDNA; van Unen et al., 2016) per dish and Effectene transfection reagent (Qiagen, Hilden, Germany) was performed according to manufacturer’s instructions. FRET-measurements were accomplished the next day. During the experiment, cells were superfused with measuring buffer (140 mM NaCl, 5.4 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 10 mM HEPES, pH 7.3) supplemented with the ligand RvD1 or DAMGO at the indicated concentration at indicated time points, using a microfluidic pipette (Ainla et al., 2012; Fluicell, Gothenburg, Sweden).
Förster resonance energy transfer (FRET) measurements were performed as previously described (Hoffmann et al., 2005) with slight modifications. In brief, FRET measurements were performed on an inverted microscope (Zeiss Axiovert 200, Zeiss, Jena, Germany) equipped with an oil immersion 63× objective lens and a dual-emission photometric system (Till Photonics, Gräfelfing, Germany). The transfected cells were excited with light from a polychrome IV (Till Photonics) at a frequency of 10 Hz with 20 ms illumination out of a total time of 100 ms. Emission of cyan fluorescent protein (CFP, 480 ± 20 nm) and YFP (535 ± 15 nm), and the FRET ratio (FYFP/FCFP) were monitored simultaneously (beam splitter DCLP 505 nm) upon excitation at 436 ± 10 nm (beam splitter DCLP 460 nm). Fluorescence signals were detected by photodiodes, digitalized using an analog-digital converter (Digidata 1440A, Axon Instruments, Sunnyvale, CA, USA) and stored with Clampex 9.0 software (Science Products, Hofheim, Germany). Processing of the data was done in Origin 2017 (Additive, Friedrichsdorf, Germany) and GraphPad Prism (La Jolla, CA, USA).
β-Arrestin-2 Recruitment Assays
HEK-293 cells were seeded on 24 mm glass coverslips coated with poly-D-lysine for 20 min, placed in 6-well plates. Twenty-four hours later, cells were transiently transfected with three plasmids using Effectene: 2.6 μg of human MOP-CFP, 0.5 μg of β-arrestin-2-YFP, and 1.0 μg of GRK2 per dish (Hoffmann et al., 2008; Mambretti et al., 2016). All constructs were in pcDNA3. The day after, the medium was changed and cells analyzed 48 h after transfection.
The experiment was performed in a Leica TCS SP2 scanning microscope (Leica, Wetzlar, Germany) as previously described (Hoffmann et al., 2005). Images were taken with a HCX PL APO CS 63× 1.32 oil objective. In brief, CFP was excited with using 458 nm, and emission was detected from 465–500 nm while YFP was excited with the 514 nm line of the argon laser, and emission was detected from 525–600 nm. The following settings were kept constant over all the measurements: 512 × 512-pixel format, line average 2, frame average 2400 Hz. Pictures (time series) were taken at 30 s intervals for 20 or 30 min. MOP-CFP was useful to specifically target cells which express the receptor in the membrane.
β-arrestin translocation to the membrane was quantified using Leica Confocal SP2 Software. Regions of interest, ROIs, were selected in the cytosol of the cells and quantified over time. In order to correct for photobleaching, control regions were selected (whole cells) and quantified too. Upon correction, the fluorescence intensity values were normalized to the value right before the recruitment takes place and plotted against time.
Statistical Analysis
In vivo data are presented as mean ± SEM. Either two-way ANOVA with repeated measurements (behavior experiments), one-way ANOVA (β-endorphin release) or student’s t-tests (calcium imaging) were performed with SigmaPlot12 or Origin 2017.
Results
Local Antinociception of RvD1 and Chem in Early and Late CFA-Induced Hind Paw Inflammation
Intrathecal applications of RvD1 and Chem reduce nociceptive behavior after formalin injection and thermal hypersensitivity in short-term (2–4 h) carrageenan-induced hindpaw inflammation (Xu et al., 2010). Here, we asked whether these lipid mediators also act peripherally on sustained mechanical inflammatory hypersensitivity in a prolonged inflammatory model (CFA-induced hindpaw inflammation). Mechanical nociceptive thresholds decreased 2 h and 96 h after intraplantar CFA injection when RvD1 and Chem were applied intraplantarly at 2 h (Figures 1A,C) and 96 h (Figures 1B,D) after CFA application, respectively. PPTs were determined 15, 45 and 180 min after intraplantar antagonist injection. Both, RvD1 and Chem significantly reduced CFA-induced mechanical hypersensitivity. The highest concentration of Chem (100 ng) and 20 ng RvD1 were most effective in early as well as in late CFA-induced inflammation. Antinociception lasted for up 45 min and returned to baseline hypersensitivity after 3 h. Higher doses of RvD1 did not results in more effective or longer antinociception of RvD1. No change was seen in the contralateral paw (data not shown).
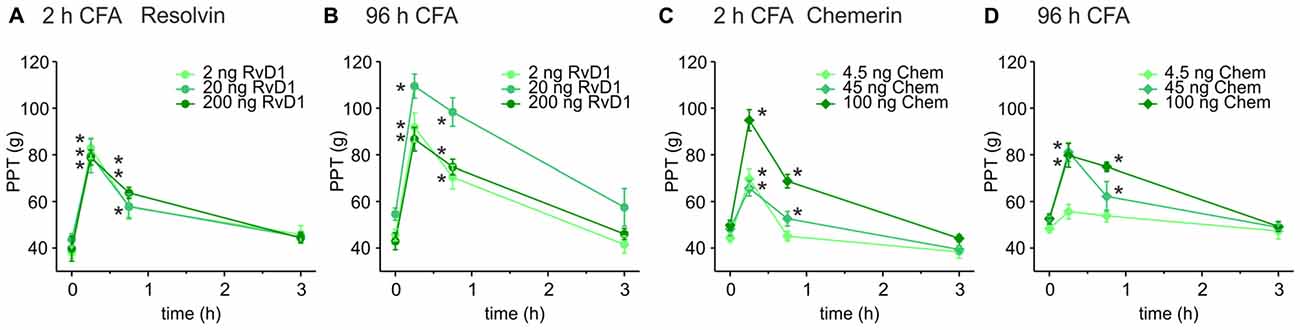
Figure 1. Antinociception by resolvin D1 (RvD1) and chemerin (Chem) in early and late states of complete freund’s adjuvant (CFA)-induced hind paw inflammation in rats. Antinociceptive effects of RvD1 (A,B) and Chem (C,D) were measured in the early (2 h; A,C) and the late phase (96 h; B,D) of CFA-evoked hind paw inflammation in male Wistar rats. Mechanical hypersensitivity (paw pressure thresholds (PPTs); in g) was evaluated at indicated time points after intraplantar injection of RvD1 or Chem. Baseline values were obtained before injection of RvD1 or Chem. Dose-dependency was evaluated by injection of three different concentrations of RvD1 and Chem (see legends; n = 4–6 per group, mean ± SEM, two-way ANOVA RM post hoc Holm-Sidak, *p < 0.05 vs. baseline pre-injection values, 0 h).
Opioid Receptor and Ligand Participation in RvD1- or Chem-Generated Antinociception
Next, we evaluated whether opioid receptors shape antinociception by RvD1 or Chem. Intraplantar injection of RvD1 or Chem together with naloxone either completely abolished or significantly reduced antinociception in early (Figures 2A,B) and late CFA-induced hind paw inflammation (Figures 2F,G). Since RvD1 is a TRPA1 antagonist, we replicated the experiment with the TRPA1 antagonist HC-030031 (Figure 2C). Similar to RvD1, intraplantar naloxone prevented the HC-030031-induced rise in mechanical nociceptive thresholds.
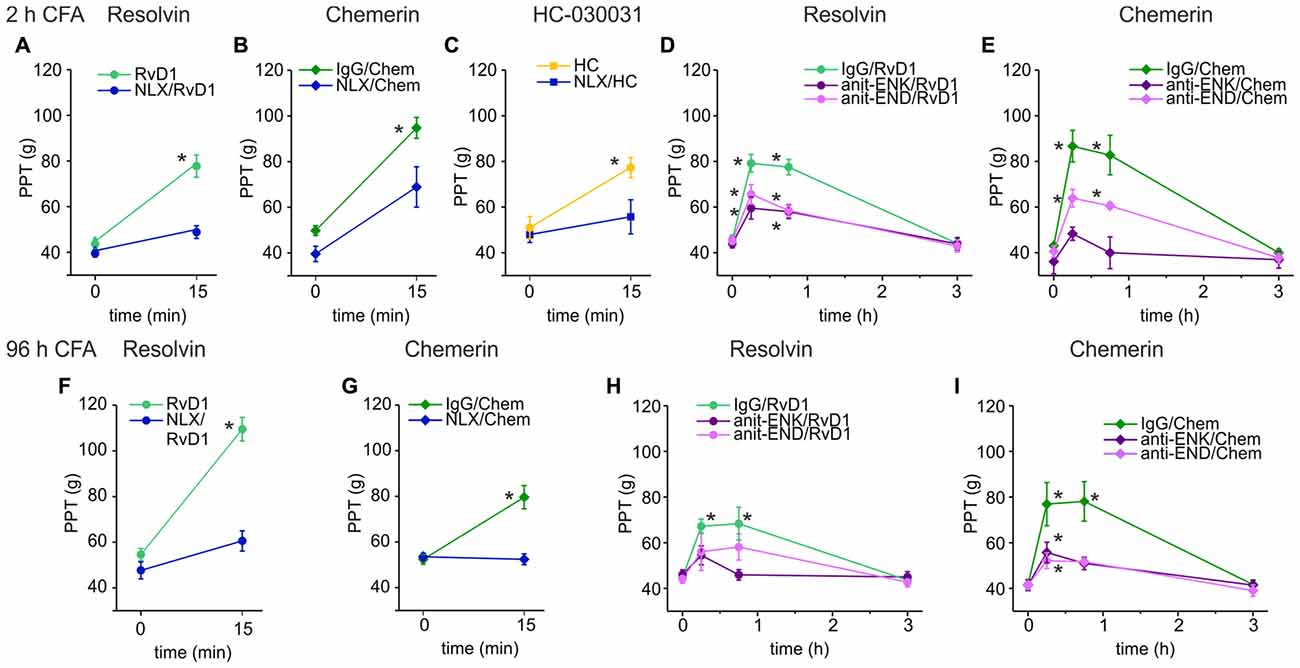
Figure 2. Reversal of RvD1-mediated antinociception by peripheral blockade of the μ-opioid peptide receptor (MOR) or of endogenous opioid peptides in rats. Antinociception by RvD1 (A,D,F,H), Chem (B,E,G,I), and HC-030031 (HC, C) in CFA-mediated hind paw inflammation on mechanical hypersensitivity was reduced by local co-application of naloxone (A–C,F,G) or local co-injection of antibodies against the endogenous opioid peptides β-endorphin (END) and met-enkephalin (ENK; anti-END/anti-ENK; D,E,H,I) at indicated time points in the early (A–E) and the late phase (F–I) of CFA-induced inflammation. Baseline inflammatory hypersensitivity was measured before injection of RvD1, Chem, or HC at CFA-time points indicated above the graphs (n = 6 per group, mean ± SEM; two-way ANOVA RM post hoc Holm-Sidak, *p < 0.05 compared to baseline pre-injection values, 0 h).
Peripheral opioid receptors are activated by release of endogenous opioid peptides secreted from leukocytes (Rittner et al., 2009). To define the contribution of endogenous ligands of MOR, antibodies against the opioid peptides anti-END and anti-ENK (Figures 2D,E,H,I), were injected locally together with RvD1 or Chem. IgG isotype antibodies served as controls to exclude unspecific effects. Both antibodies, anti-END and anti-ENK, significantly reduced RvD1- and Chem-mediated antinociception. In summary, peripheral opioid receptor activation by endogenous opioid peptides interacts with antinociceptive properties of RvD1, Chem and the TRPA1 antagonist HC-030031.
Role of Leukocytes for RvD1-Mediated Peripheral Antinociception
To further investigate this pathway, we assessed the involvement of immune cells and their release of opioid peptides. In early CFA-induced (2 h) hind paw inflammation, systemic injection of an anti-polymorphonuclear neutrophil (anti-PMN) serum depleted neutrophils from the circulation and from the inflamed paw (Rittner et al., 2006, 2009). This treatment reduced antinociception evoked by RvD1 by 60% (Figure 3A). However, in vitro β-endorphin liberation from immune cells like rat peritoneal neutrophils or macrophages was not significantly increased after RvD1 stimulation independent of the dose (Figures 3B,C). As a positive control, stimulation with the chemotactic peptide, fMLP, or ionomycin lead to 2–4 times higher concentrations of released β-endorphin.
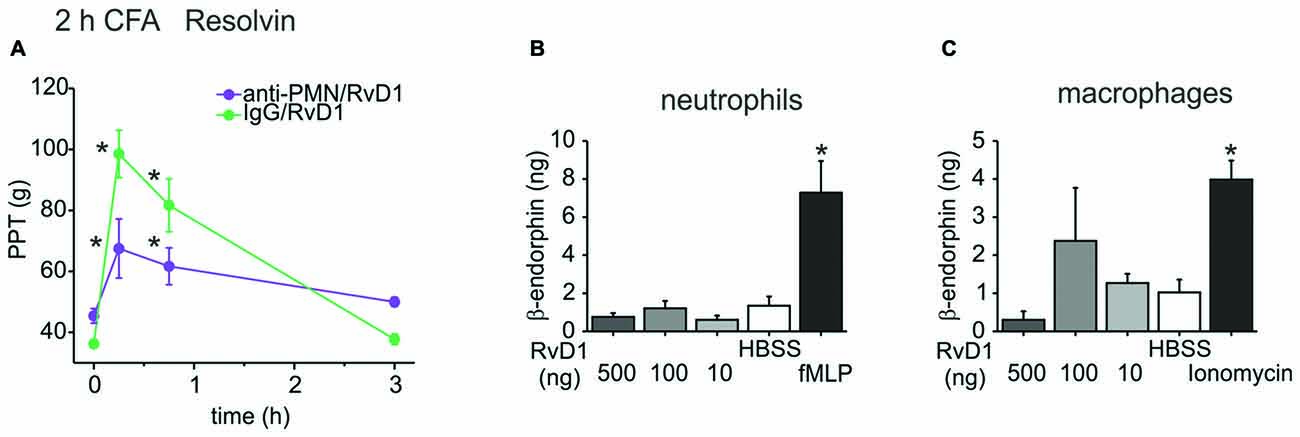
Figure 3. Leukocyte dependency of RvD1-mediated antinociception in early states of CFA-induced hind paw inflammation. Antinociception by RvD1 (A) was prevented in part in leukocyte-depleted Wistar rats. Leukocyte depletion was achieved by systemic anti-PMN (i.p.) treatment as described before (Rittner et al., 2006), IgG isotype antibody served as control. Mechanical hypersensitivity was measured at indicated time points before and after local RVD1 injection. (n = 4–6 per group, mean ± SEM; two-way ANOVA RM post hoc Holm-Sidak, *p < 0.05 compared to baseline pre-injection values, 0 h). (B,C) Release of β-endorphin from peritoneal neutrophils (B) and macrophages (C) of Wistar rats was not stimulated by different concentrations of RvD1. β-endorphin release (ng/ml per 1 × 107 cells) was measured with a commercially available ELISA after stimulation of isolated cells by RvD1 at indicated concentrations. Hank’s balanced salt solution (HBSS) served as negative control, fMLP (B; 1 μM) or ionomycin (C; 10 μM) as positive control (n = 2–8 per group, mean ± SEM, one-way ANOVA post hoc Holm-Sidak, *p < 0.05 compared to HBSS solvent control).
No Direct Activation of MOR by RvD1
Because of the evidence for the involvement of opioids in RvD1- and Chem-mediated antinociception and the preference of resolvins to interact with GPCR, we used an in vitro model to assess MOR agonist-properties. HEK-293 cells were co-transfected with untagged MOR and a G protein FRET sensor subtype: Gαi1, Gαi2, or Gαi3, respectively (Figure 4A). The synthetic opioid peptide DAMGO, which has a high affinity to MOR, served as positive control. While DAMGO reduced the FRET ratio in all experiments, RvD1 did not alter the signal. Neither did RvD1 reduced the FRET ratio nor did it prevent DAMGO-induced lowering of FRET ratios in a Gαi subunits. Application of buffer solution washed out the compounds and restored basal values.
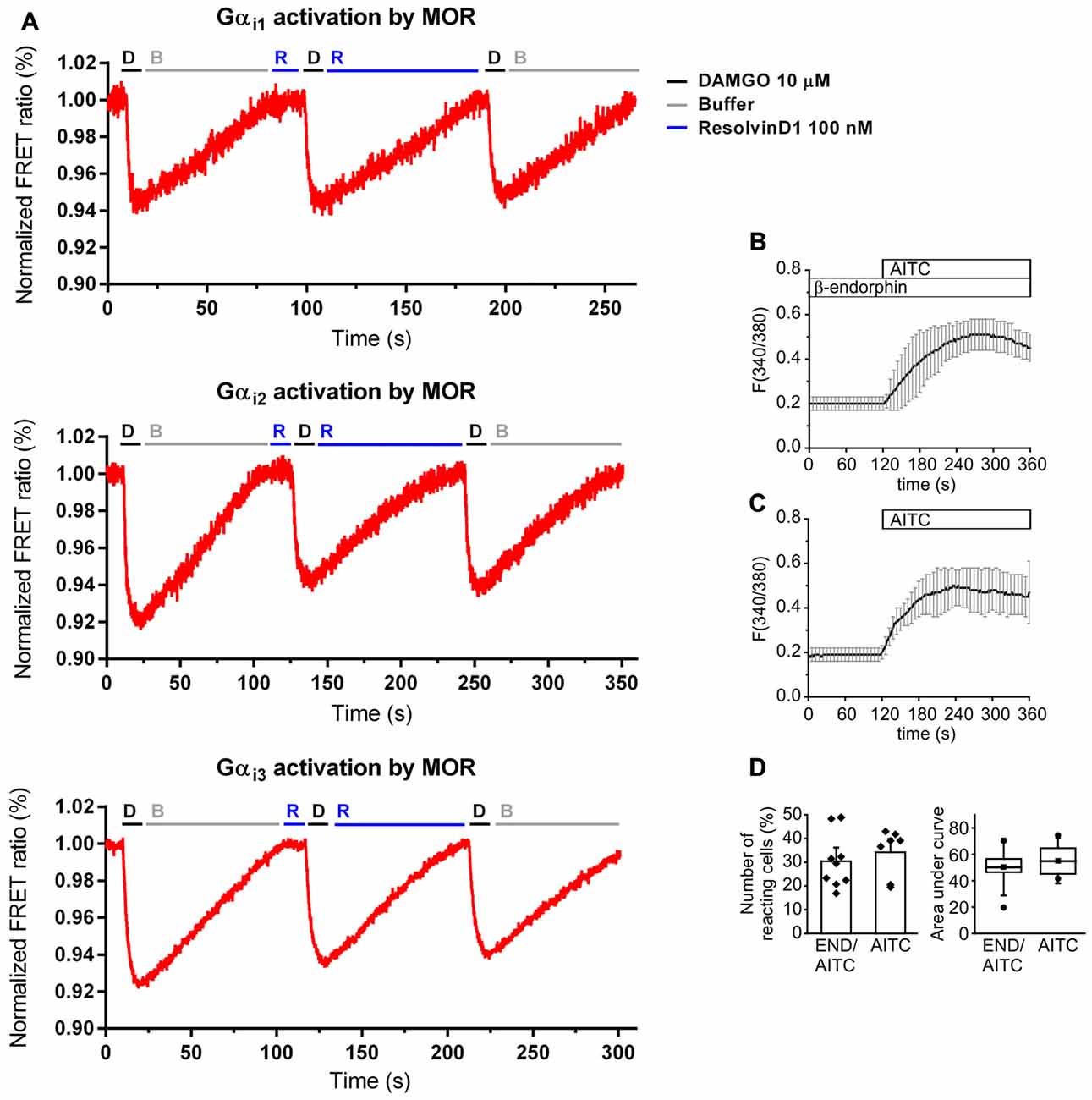
Figure 4. No activation of MOR by RvD1 in HEK-293 and lack of alteration of transient receptor potential channels ankyrin 1 (TRPA1) activity by β-endophin in Dorsal Root Ganglia (DRG) neurons. (A) Activation or blockade of MOR by RvD1 (100 nM) was assessed by a Förster resonance energy transfer (FRET) assay in HEK-293 cells co-transfected with MOR and Gα subunits as indicated. Representative measurements are shown. Fluorescence intensity is given as FRET ratio normalized to baseline. DAMGO (10 μM) served as positive control. (B) Fura-2/AM-based single cell calcium imaging (ratio F(340/380)) in dissected, cultured DRGs of adult mice. Preincubation of the DRGs in β-endorphin (100 nM, 5 min) did not further increase intracellular calcium upon TRPA1 stimulation with allyl isothiocyanate (AITC ; 10 μM). Traces represent mean ± SD of 7–9 independent measurements per group. (C,D) Statistical analysis of the number of reacting cells (C) and the area under curve (D) demonstrate no significant differences between the treatment groups. (AITC: n = 7; pretreatment with β-endorphin: n = 9; mean ± SEM, student’s t-test, p < 0.05).
Unaltered TRPA1-Induced Calcium Influx by β-endorphin
Next, we tested whether endogenous β-endorphin modulate TRPA1 activity in DRGs. Cultured adult DRG neurons from of C57BL/6 mice were loaded with Fura/2-AM, a ratiometric calcium dye. Neurons were incubated in β-endorphin before stimulation with allyl isothiocyanate (AITC), a specific TRPA1 agonist (Figure 4B). β-endorphin neither increased nor decreased the number of TRPA1-positive neurons (Figure 4C) or the area under curve (Figure 4D).
RvD1 does Not Recruit β-Arrestin
To assess whether β-arrestin is crucial for TRP channel blockade by resolvins, we transiently transfected HEK-293 cells with β-arrestin-2-YFP, GRK2 and MOR-CFP (Figure 5). FRET measurements were performed. While DAMGO induced a translocation of β-arrestin to the membrane (Figures 5A,D), RvD1 failed (Figures 5B,D). Furthermore, RvD1 did not block the DAMGO-induced β-arrestin recruitment (Figures 5C,E).
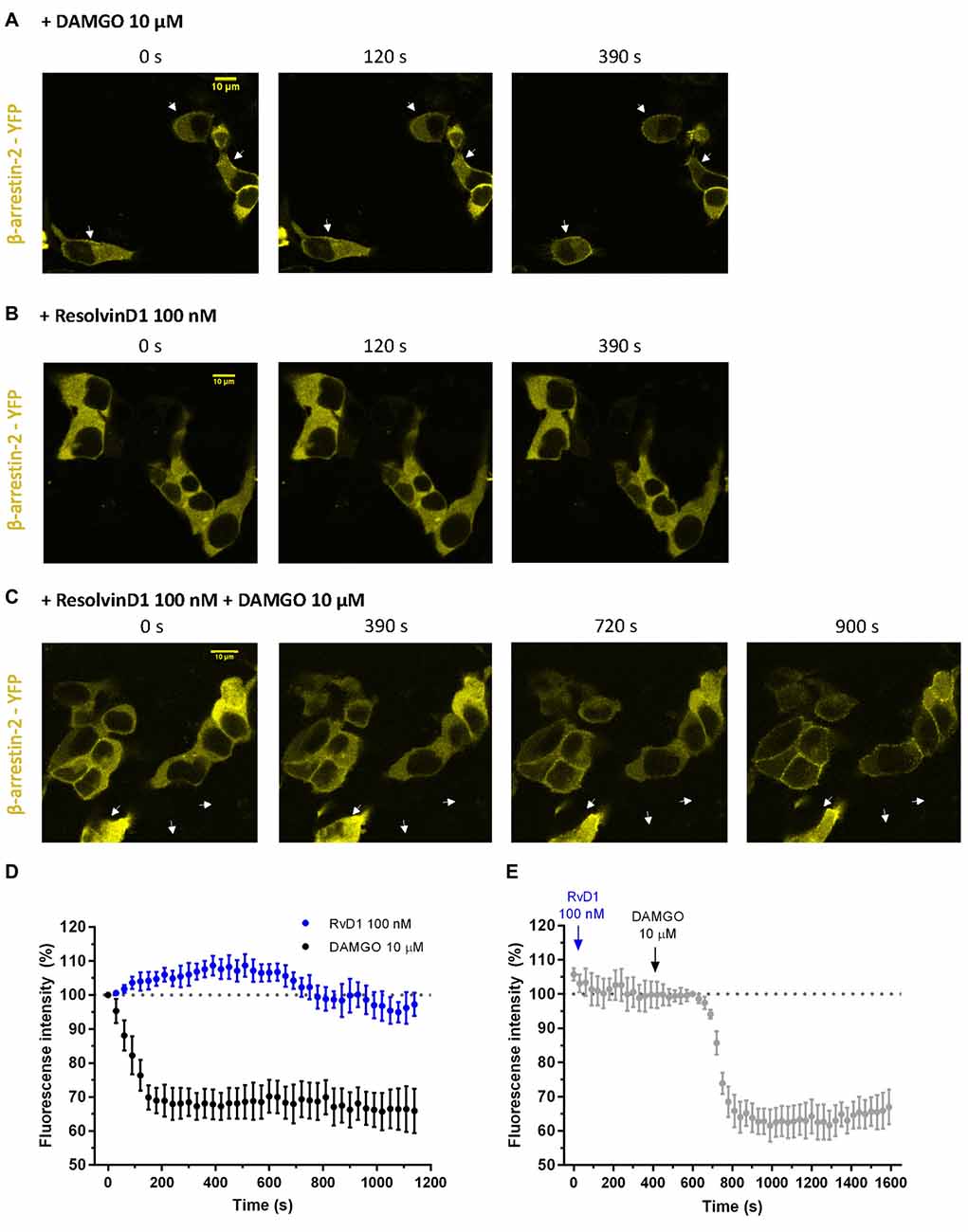
Figure 5. β-arrestin-2 recruitment mediated by MOP in response to DAMGO or/and ResolvinD1. Representative images from HEK-293 cells transfected with MOR-CFP, GRK2, and β-arrestin-2-yellow fluorescent protein (YFP), are shown at different time points (0, 120, 390, 720 or 900 s) upon application of the indicated concentration of ligand: 10 μM DAMGO (A), 100 nM RvD1 (B) or 100 nM RvD1 followed by addition of 10 μM DAMGO after 450 s (C). Arrows indicate β-arrestin-2 recruitment. (D,E) Images were quantified and corrected for photobleaching of the sample (DAMGO = 7 cells; RvD1 = 8 cells; RvD1 + DAMGO = 10 cells).
Deficit of Endogenous Peripheral MOR Antinociception Impairs TRPA1 Antinociceptive Properties
Endogenous opioid peptides are tonically secreted and mediate thermal antinociception in early inflammation (Rittner et al., 2009). Here, we proposed that the balance of endogenous pro- and anti-hyperalgesic mediators shapes mechanical hypersensitivity. Hence, application of antagonists affects mechanical nociceptive thresholds. In early CFA-evoked inflammation, intraplantar injection of naloxone (blue lines, 1st arrow) further lowered mechanical PPTs (Figures 6A,C). No further decrease of the nociceptive thresholds by naloxone was observed in late inflammation (Figures 6B,D). Subsequently, rats receive an intraplantar co-injection of the TRPA1 antagonist RvD1 (Figures 6A,B) or HC-030031 (Figures 6C,D) and naloxone (2nd arrow). However, the TRPA1 inhibitors failed to ameliorate hypersensitivity in presence of naloxone. Mechanical nociceptive thresholds in the naloxone group were significantly lower than in the control groups where rats only received the TRPA1 inhibitor RvD1 or HC-030031.
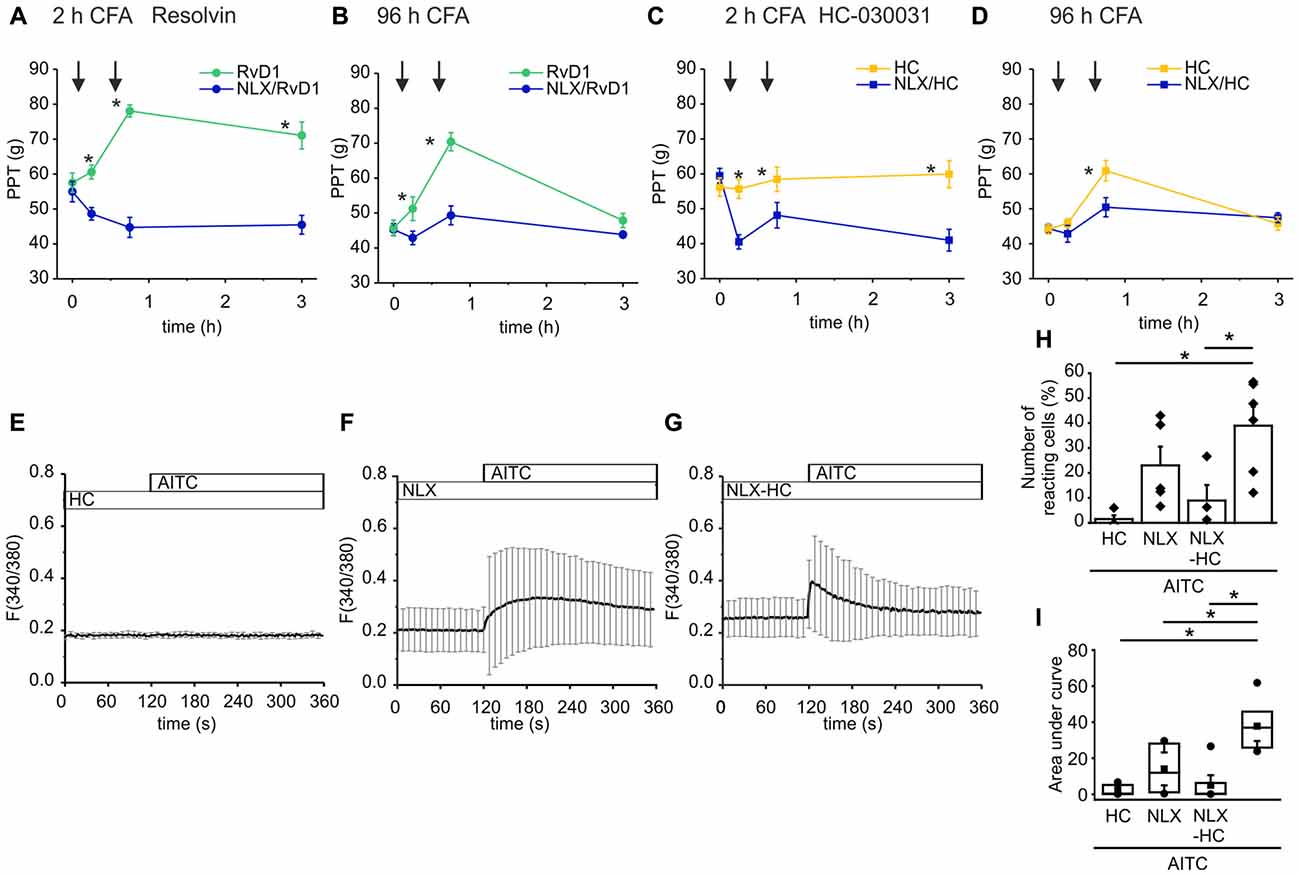
Figure 6. Naloxone reverses antagonism of RvD1 and HC-030031 in vivo and impairs TRPA1 blockade in vitro. (A–D) Measurements of PPTs (g) in the early (A,C) and the late phase (B,D) of CFA-induced inflammation. First naloxone was injected (1st arrow, blue lines) or saline (yellow/green lines). Subsequently, naloxone plus RvD1 (A,B) or HC-030031 (C,D) were injected in the corresponding naloxone group (blue lines; 2nd arrow) while only the TRPA1 antagonists RvD1 (A,B; green) or HC-030031 (C,D; yellow) were applied in the control groups. Mechanical nociceptive thresholds were obtained at indicated time points. (n = 5 per group, mean ± SEM; two-way ANOVA RM post hoc Holm-Sidak, *p < 0.05 naloxone + RvD1/HC-030031 vs. solvent + RvD1/HC-030031). (E–G) Representative measurements of calcium transients in DRG neurons pre-incubated for 5 min in HC-030031 (10 μM), naloxone (10 μM) or HC-030031 plus naloxone before stimulation with AITC (10 μM) at indicated time points. Mean ± S.D. (H,I) Statistical analysis of the number of reacting cells (%; H) area under curve (taken from 160 s to 340 s; I). AITC: n = 6; pretreatment with HC-030031 n = 4; pretreatment with naloxone: n = 5; pretreatment with HC-030031 plus naloxone: n = 7; mean ± SEM, One-way ANOVA post hoc Holm-Sidak, p < 0.05.
To mimic the reversal of TRPA1 blockade by naloxone in vitro, we measured calcium transients in DRG neurons upon stimulation with AITC. Pre-incubation of the neuronal cells in HC-030031 prevented the TRPA1-mediated intracellular calcium increase evoked by AITC (Figure 6E). Pre-incubation of the cells with naloxone diminished the AITC-elicited calcium transients (Figures 6G,F). The activation pattern turned out to be more in-homogenous. While in some experiments, the number of activated cells was reduced compare to stimulation with AITC (Figure 6H) the area under curve was lowered significantly (Figure 6I). On the other hand, naloxone impaired the blockade of TRPA1 by HC-030031. The AITC-induced calcium increase was restored when cells we pre-treated with HC-030031 and naloxone at the same time (Figure 6D). Since only some neuronal cells respond to the AITC stimulus, the area under curve did not increase significantly.
Discussion
In this study, local RvD1 and Chem raise mechanical nociceptive thresholds in the early neutrophil dominated phase as well as in the late macrophage phase of CFA-induced inflammation. Local blockade of MOR or opioid peptides as well as neutrophil depletion prevent antinociception by RvD1 and Chem. Both, RvD1 and Chem do not stimulate neutrophils or macrophages to secret opioid peptides nor does RvD1 directly activate MOR in vitro. Therefore, we hypothesize that either an altered balance of pro- and antinociceptive mediators or intracellular pathways by interaction of GPCR and TRPA1 are responsible. In vitro, β-endorphin does not sensitize or desensitize TRPA1 in DRG neurons, but naloxone hampers TRPA1 blockade by HC-030031. In vivo, local MOR blockade further decreases mechanical nociceptive thresholds shifting the thresholds towards a further enhanced hypersensitivity. Subsequently, antinociception by RvD1 and HC-030031, the most specific TRPA1 blocker, was diminished. These findings support the hypothesis of either a dysbalance of endogenous pro- and antinociceptive mechanisms or an interaction of TRPA1 and opioid receptors.
Antinociception by Resolvins
Up to now, most of the studies analyzed central antinociceptive effects of resolvins. For example, intrathecal application of RvD1 and RvE1 increases thermal nociceptive thresholds after CFA (Xu et al., 2010). Spinal RvD1 elicits potent antinociceptive effect in lumbar disc herniation (Liu et al., 2016), prevents long-term hypersensitivity after thoracotomy (Wang and Strichartz, 2017) as well as chronic pancreatitis-induced visceral pain (Quan-Xin et al., 2012) and 17(R)-HDoHE reverses pain behavior in two models of osteoarthritis pain (Huang et al., 2017). Less is known about peripheral effects and about mechanical hypersensitivity: intraplantar RvD1 and RvE1 reduced carrageen-induced thermal hypersensitivity and pain behavior evoked by capsaicin or AITC, respectively (Park et al., 2011b). Here, we describe peripheral antinociception by RvD1 and Chem in early and late CFA-induced inflammation on mechanical nociceptive thresholds.
Four different antinociceptive mechanisms by resolvins have been implicated: first, in the resolution phase of inflammation in the inflamed tissue, resolvins downregulate the recruitment and function of immune cells (Serhan, 2014) and thereby reduce pain. GPCR activation diminishes the secretion of pro-inflammatory cytokines and chemokines (Ji et al., 2011; Serhan, 2014; Lim et al., 2015). Low concentrations of RvD1 specifically limit the infiltration of neutrophils, polarize monocytes and increase phagocytosis of apoptotic cells by macrophages via GPR32 (Arita et al., 2007; Cash et al., 2008; Merched et al., 2008; Krishnamoorthy et al., 2010, 2012; Norling et al., 2012; Schmid et al., 2016). The function of Chem is more diverse: Chem binding to Chem23R is a potent macrophage chemoattractant protein (Hart and Greaves, 2010). On the other hand, Chem inhibits pain behavior in the second phase of the formalin test when injected intrathecally (Xu et al., 2010) or CFA-induced hypersensitivity as shown here. Rapid relief of hypersensitivity observed in vivo appeared to be mediated by receptor activation/inhibition independent of intercellular actions such as recruitment or activation of innate immune cells.
Second, intrathecal application of resolvins reduces spinal inflammation by inhibition of TNF-α and IL-1β, stimulation of IL-10 and TGF-β1, and attenuation of NFκB/p65 and p-ERK expression in a dose-dependent manner (Liu et al., 2016).
Third, activation of resolvin receptors inhibits TRP activity. Several groups suggest that resolvin-mediated blockade of TRP channels acts via a G-protein regulated mechanism (Choi and Hwang, 2016). Pathways include a phosphorylation cascade inhibiting the adenylate cyclase or ERK (Park et al., 2011b; Serhan, 2014; Lim et al., 2015) or β-arrestin coupled to resolvin receptors (Krishnamoorthy et al., 2010). Indeed, β-arrestin recruitment could lead to increased TRP channel sensitization or desensitization (Por et al., 2013; Rowan et al., 2014). Coupling of resolvin receptors with TRP channels in nociceptor terminals would be needed to be supported by further experimental evidence. Since ChemR23 and TRPV1 are co-expressed in small diameter DRGs, it is likely that RvE1-mediated inhibition of TRPV1 (Xu et al., 2010) arises upon ChemR23 stimulation (Jo et al., 2016). Taken together, the exact mechanism of resolvin-mediated TRP channel blockade needs further exploration.
Fourth, interactions with opioid receptors have been explored. MOR did not mediate the central antinociceptive action of RvE1, as the MOR antagonist naloxone did not block (Xu et al., 2010). In fact, RvD1 is not a MOR agonist in vitro because is neither activates Gi nor recruits β-arrestin directly in MOR-expressing HEK-293. So, MOR is not the GPCR for the ligand RvD1.
In our studies, we not only observed an interaction of resolvins with MOR but also TRPA1 with MOR. In recent years, protein-protein connections have been analyzed in depth supporting the complexity of the pain pathway (Rouwette et al., 2015). Protein-protein interactions include receptors, kinases or adapter proteins. For example, A-kinase anchor protein 79/150 (AKAP79/150) binds to TRPV1, Tmem100, a membrane adaptor protein, potentiates TRPA1 activity (Weng et al., 2015) and annexin A2 impairs TRPA1 availability in the plasma membrane (Avenali et al., 2014). Therefore, it is possible that MOR and TRPA1 interact. In addition, N-methyl-D-aspartate (NMDA) receptor are a possible link between opioid receptors and TRPV1 (Mao, 1999; Lee et al., 2012).
Naloxone as well as naloxone-methiodide are considered to be non-specific opioid receptor antagonists, because they can reverse antinociceptive effects of MOR, DOR and κ-opioid receptors (KOR; Labuz et al., 2007). Since the in vivo system is very complex, conclusions drawn from in vitro experiments have to be evaluated carefully. Mimicking the in vivo situation in in vitro setups is limited as recently shown by matching the data of in vivo and in vitro calcium imaging in the DRG (Emery et al., 2016). The comparison of naloxone effects in vivo and in vitro is speculative. In vivo, it could be suggested that inhibitory effects of endogenous opioids released from immune cells in inflammation are blocked by naloxone. In our study here, endogenous opioids seem to interact in the periphery. Naloxone prevented antinociception by RvD1, Chem or HC-030031 in CFA-induced inflammation. Thus, the endogenous pro- and anti-nociceptive balance is disturbed. In addition, we observed the reduction of inhibitory effects of TRPA1 inhibitors in the presence of naloxone in DRG neurons in vitro. Viewing from the pharmacological site, relatively high doses of naloxone in vitro (e.g., 10 μM) could also possibly directly block TRPA1.
Naloxone per se reverses opioid-mediated analgesia. Whether the application of naloxone aggravates pain or prevents inhibitory effects of other nociceptor blockers in the periphery, as shown here, has not been studied so far. Indeed, interactions of TRPA1 and opioid receptors have been explored before in a model of visceral pain (Pereira et al., 2013). Hypersensitivity of the abdomen after ifosfamide-induced hemorrhagic cystitis was lessened by systemic application of the TRPA1 antagonist HC-030031. These authors observed only a small and insignificant reversal of HC-030031-antinociception by naloxone. However, they used a much higher dose applied systemically (2 mg/kg i.p. vs. 0.56 ng intraplantar), which presumably blocks central opioid receptors, and a different model of pain.
Opioid Peptide Release from Immune Cells
Opioid peptides like β-endorphin, met-enkephalin and dynorphin are secreted from neutrophils or monocytes/macrophages upon stimulation (Plein and Rittner, 2017) by Chemokines (Rittner et al., 2006), bacterial products (Rittner et al., 2009; Sauer et al., 2014) or exogenous opioids itself (Celik et al., 2016; Labuz et al., 2016). Opioid peptide release is constantly present in inflamed tissue, because intraplantar injection of anti-opioid peptide antibodies or naloxone further lower thermal nociceptive thresholds (Rittner et al., 2009). Similarly, anti-PMN serum depleting neutrophils (Rittner et al., 2006, 2009) or cytotoxic drugs like cyclophosphamide depleting immune cells further worsen mechanical hypersensitivity in rats with CFA-induced hindpaw inflammation (Sauer et al., 2014). We have extensively used peritoneal neutrophils and macrophages for in vitro experiments before and found matching results between in vivo and in vitro (Rittner et al., 2006, 2009; Sauer et al., 2014). To our knowledge immune cells isolated from the inflamed paw tissue have not been used for release of opioid peptides from immune cells so far. Schreiter et al. (2012) used membranes from infiltrating immune cells to analyze aminopeptidase N and neutral enkephalinase activity. Celik et al. (2016) employed immune cells isolated from the nerve to study release of opioid peptides in vitro. Nevertheless, based on our results, we argue that even cells from the tissue would not release opioid peptides in response to resolvin. Furthermore, we now provide evidence that local naloxone also reduces mechanical nociceptive thresholds measured by Randall-Selitto test especially in early stages of CFA-induced hindpaw inflammation. We did not observe this at later time points possibly due to more pronounced hypersensitivity and detection thresholds of the Randall-Selitto test.
Next, we proposed that RvD1 stimulates the release of opioid peptides. RvD1 augments synthesis and release of anti-inflammatory cytokines while it decreases the production of proinflammatory cytokines. RvE1 rises intracellular Ca2+ levels (Arita et al., 2007). We have previously shown that the Gαi/o-Gβγ protein-PLC-IP3 receptors-intracellular Ca2+ pathways regulate the opioid peptide release within the cell (Rittner et al., 2006, 2009; Sauer et al., 2014; Celik et al., 2016). Therefore, we asked whether RvD1 or Chem could stimulate release of opioid peptides via intracellular calcium increase. However, in vitro RvD1 was not able to stimulate the release of β-endorphin in peritoneal macrophages or neutrophils. One explanation could be that RvD1 rather decreases arachidonic acid or cAMP mediated intracellular Ca2+ increase (Fredman et al., 2014). Alternatively, RvD1 is not strong enough in vitro, since cells were preactivated by recruitment into the peritoneal cavity and with cytochalasin. Taken together, RvD1-induced opioid peptide release seems not to be a prominent mechanism in vivo. Instead, we provide evidence that in vivo blockade of MOR shifts the balance of pro-nociceptive and anti-nociceptive pathways towards enhanced hypersensitivity. Therefore, although antinociceptive TRPA1 pathways are blocked by RvD1 or specific blockers, this only reverses the intense hypersensitivity but it is not potent enough to normalize mechanical nociceptive thresholds.
Future Clinical Implications
Resolvins are very promising candidates to treat inflammatory pain. Interestingly, these endogenous lipid derivatives can contribute to the resolution of two different pathologic processes, pain and inflammation via differential cellular targets and biological mechanisms. No adverse effects have been reported so far in in vivo studies. Although conceivably, premature termination of inflammation or pain could be harmful. Unfortunately, a proof of concept clarifying the relevant molecular mechanisms for pain inhibition has not been provided yet (Choi and Hwang, 2016). It remains to be deciphered how resolvins bind to the GPCR and how they transduce their signals. Further, the interaction with TRPV1 and TRPA1 as well as its interaction with MOR is so far not clear and needs further studies.
In the recent years, longer acting resolvins have been developed as promising drugs. Furthermore, diets rich in fish and fish oil as well as omega-3 polyunsaturated fatty acids might not only be protective in inflammatory conditions but also ameliorate pain. These aspects offer new perspectives towards the development of novel targeted interventions for the treatment of inflammatory, postoperative or neuropathic pain.
Author Contributions
BO conducted calcium imaging experiments and analyzed all data. DH and MM performed the behavioral studies. CPV and CH tested the contribution of MOR. AB provided conceptual expertise and contributed to manuscript preparation. BO and HLR interpreted the results and wrote the manuscript.
Funding
This study was supported by funds from the University of Wuerzburg as well as the Interdisciplinary Center for Clinical Research (IZKF) of the University Hospital of Wuerzburg. CPV was supported by the Marie Curie Initial Training Networks (ITN) “ONCORNET” grant agreement number 641833.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Michael Sendtner and Robert Blum, Institute for Clinical Neurobiology, University Hospital of Würzburg provided the calcium imaging setup and the cell culture facilities. We thank Ying Wang who measured the β-endorphin release.
Abbreviations
ALX/FRP2, lipoxin A (4)/Annexin-A1 receptor formyl-peptide receptor 2; CFA, complete Freund’s adjuvant; CFP, cyan fluorescent protein; Chem, chemerin; GPCR, G-protein-coupled receptor; GPR32, G-protein-coupled receptor 32; HBSS, Hank’s balanced salt solution; MOR, μ-opioid peptide receptor; PBS, phosphate buffered saline; PPT, paw pressure threshold; RvD1, resolvin D1; RvD2, resolvin D2; RvE1, resolvin E1; TRP, transient receptor potential; TRPA1, TRP, ankyrin 1; TRPV1, TRP, vanilloid 1; YFP, yellow fluorescent protein.
References
Ainla, A., Jeffries, G. D., Brune, R., Orwar, O., and Jesorka, A. (2012). A multifunctional pipette. Lab Chip 12, 1255–1261. doi: 10.1039/c2lc20906c
Arita, M., Bianchini, F., Aliberti, J., Sher, A., Chiang, N., Hong, S., et al. (2005). Stereochemical assignment, antiinflammatory properties and receptor for the omega-3 lipid mediator resolvin E1. J. Exp. Med. 201, 713–722. doi: 10.1084/jem.20042031
Arita, M., Ohira, T., Sun, Y. P., Elangovan, S., Chiang, N., and Serhan, C. N. (2007). Resolvin E1 selectively interacts with leukotriene B4 receptor BLT1 and ChemR23 to regulate inflammation. J. Immunol. 178, 3912–3917. doi: 10.4049/jimmunol.178.6.3912
Avenali, L., Narayanan, P., Rouwette, T., Cervellini, I., Sereda, M., Gomez-Varela, D., et al. (2014). Annexin A2 regulates TRPA1-dependent nociception. J. Neurosci. 34, 14506–14516. doi: 10.1523/JNEUROSCI.1801-14.2014
Bang, S., Yoo, S., Yang, T. J., Cho, H., Kim, Y. G., and Hwang, S. W. (2010). Resolvin D1 attenuates activation of sensory transient receptor potential channels leading to multiple anti-nociception. Br. J. Pharmacol. 161, 707–720. doi: 10.1111/j.1476-5381.2010.00909.x
Bautista, D. M., Jordt, S. E., Nikai, T., Tsuruda, P. R., Read, A. J., Poblete, J., et al. (2006). TRPA1 mediates the inflammatory actions of environmental irritants and proalgesic agents. Cell 124, 1269–1282. doi: 10.1016/j.cell.2006.02.023
Brack, A., Rittner, H. L., Machelska, H., Leder, K., Mousa, S. A., Schäfer, M., et al. (2004). Control of inflammatory pain by chemokine-mediated recruitment of opioid-containing polymorphonuclear cells. Pain 112, 229–238. doi: 10.1016/j.pain.2004.08.029
Cash, J. L., Hart, R., Russ, A., Dixon, J. P., Colledge, W. H., Doran, J., et al. (2008). Synthetic chemerin-derived peptides suppress inflammation through ChemR23. J. Exp. Med. 205, 767–775. doi: 10.1084/jem.20071601
Celik, M. Ö., Labuz, D., Henning, K., Busch-Dienstfertig, M., Gaveriaux-Ruff, C., Kieffer, B. L., et al. (2016). Leukocyte opioid receptors mediate analgesia via Ca2+-regulated release of opioid peptides. Brain Behav. Immun. 57, 227–242. doi: 10.1016/j.bbi.2016.04.018
Chiang, N., Dalli, J., Colas, R. A., and Serhan, C. N. (2015). Identification of resolvin D2 receptor mediating resolution of infections and organ protection. J. Exp. Med. 212, 1203–1217. doi: 10.1084/jem.20150225
Choi, G., and Hwang, S. W. (2016). Modulation of the activities of neuronal ion channels by fatty acid-derived pro-resolvents. Front. Physiol. 7:523. doi: 10.3389/fphys.2016.00523
Corder, G., Tawfik, V. L., Wang, D., Sypek, E. I., Low, S. A., Dickinson, J. R., et al. (2017). Loss of mu opioid receptor signaling in nociceptors, but not microglia, abrogates morphine tolerance without disrupting analgesia. Nat. Med. 23, 164–173. doi: 10.1038/nm.4262
Emery, E. C., Luiz, A. P., Sikandar, S., Magnúsdóttir, R., Dong, X., and Wood, J. N. (2016). In vivo characterization of distinct modality-specific subsets of somatosensory neurons using GCaMP. Sci. Adv. 2:e1600990. doi: 10.1126/sciadv.1600990
Fredman, G., Ozcan, L., Spolitu, S., Hellmann, J., Spite, M., Backs, J., et al. (2014). Resolvin D1 limits 5-lipoxygenase nuclear localization and leukotriene B4 synthesis by inhibiting a calcium-activated kinase pathway. Proc. Natl. Acad. Sci. U S A 111, 14530–14535. doi: 10.1073/pnas.1410851111
Gangadharan, V., and Kuner, R. (2013). Pain hypersensitivity mechanisms at a glance. Dis. Model. Mech. 6, 889–895. doi: 10.1242/dmm.011502
Hackel, D., Krug, S. M., Sauer, R. S., Mousa, S. A., Bócker, A., Pflücke, D., et al. (2012). Transient opening of the perineurial barrier for analgesic drug delivery. Proc. Natl. Acad. Sci. U S A 109, E2018–E2027. doi: 10.1073/pnas.1120800109
Hackel, D., Pflücke, D., Neumann, A., Viebahn, J., Mousa, S., Wischmeyer, E., et al. (2013). The connection of monocytes and reactive oxygen species in pain. PLoS One 8:e63564. doi: 10.1371/journal.pone.0063564
Hart, R., and Greaves, D. R. (2010). Chemerin contributes to inflammation by promoting macrophage adhesion to VCAM-1 and fibronectin through clustering of VLA-4 and VLA-5. J. Immunol. 185, 3728–3739. doi: 10.4049/jimmunol.0902154
Hoffmann, C., Gaietta, G., Bünemann, M., Adams, S. R., Oberdorff-Maass, S., Behr, B., et al. (2005). A FlAsH-based FRET approach to determine G protein-coupled receptor activation in living cells. Nat. Methods 2, 171–176. doi: 10.1038/nmeth742
Hoffmann, C., Ziegler, N., Reiner, S., Krasel, C., and Lohse, M. J. (2008). Agonist-selective, receptor-specific interaction of human P2Y receptors with β-arrestin-1 and -2. J. Biol. Chem. 283, 30933–30941. doi: 10.1074/jbc.M801472200
Huang, J., Burston, J. J., Li, L., Ashraf, S., Mapp, P. I., Bennett, A. J., et al. (2017). Targeting the D series resolvin receptor system for the treatment of osteoarthritis pain. Arthritis Rheumatol. 69, 996–1008. doi: 10.1002/art.40001
Huang, L., Wang, C. F., Serhan, C. N., and Strichartz, G. (2011). Enduring prevention and transient reduction of postoperative pain by intrathecal resolvin D1. Pain 152, 557–565. doi: 10.1016/j.pain.2010.11.021
Ji, R. R., Xu, Z. Z., Strichartz, G., and Serhan, C. N. (2011). Emerging roles of resolvins in the resolution of inflammation and pain. Trends Neurosci. 34, 599–609. doi: 10.1016/j.tins.2011.08.005
Jo, Y. Y., Lee, J. Y., and Park, C. K. (2016). Resolvin E1 inhibits substance P-induced potentiation of TRPV1 in primary sensory neurons. Mediators Inflamm. 2016:5259321. doi: 10.1155/2016/5259321
Krishnamoorthy, S., Recchiuti, A., Chiang, N., Fredman, G., and Serhan, C. N. (2012). Resolvin D1 receptor stereoselectivity and regulation of inflammation and proresolving microRNAs. Am. J. Pathol. 180, 2018–2027. doi: 10.1016/j.ajpath.2012.01.028
Krishnamoorthy, S., Recchiuti, A., Chiang, N., Yacoubian, S., Lee, C. H., Yang, R., et al. (2010). Resolvin D1 binds human phagocytes with evidence for proresolving receptors. Proc. Natl. Acad. Sci. U S A 107, 1660–1665. doi: 10.1073/pnas.0907342107
Labuz, D., Mousa, S. A., Schafer, M., Stein, C., and Machelska, H. (2007). Relative contribution of peripheral versus central opioid receptors to antinociception. Brain Res. 1160, 30–38. doi: 10.1016/j.brainres.2007.05.049
Labuz, D., Spahn, V., Celik, M. Ö., and Machelska, H. (2016). Opioids and TRPV1 in the peripheral control of neuropathic pain—Defining a target site in the injured nerve. Neuropharmacology 101, 330–340. doi: 10.1016/j.neuropharm.2015.10.003
Lee, J., Chung, M. K., and Ro, J. Y. (2012). Activation of NMDA receptors leads to phosphorylation of TRPV1 S800 by protein kinase C and A-Kinase anchoring protein 150 in rat trigeminal ganglia. Biochem. Biophys. Res. Commun. 424, 358–363. doi: 10.1016/j.bbrc.2012.07.008
Lim, J. Y., Park, C. K., and Hwang, S. W. (2015). Biological roles of resolvins and related substances in the resolution of pain. Biomed Res. Int. 2015:830930. doi: 10.1155/2015/830930
Liu, Z. H., Miao, G. S., Wang, J. N., Yang, C. X., Fu, Z. J., and Sun, T. (2016). Resolvin D1 inhibits mechanical hypersensitivity in sciatica by modulating the expression of nuclear factor-kappaB, phospho-extracellular signal-regulated kinase and pro- and antiinflammatory cytokines in the spinal cord and dorsal root ganglion. Anesthesiology 124, 934–944. doi: 10.1097/ALN.0000000000001010
Mambretti, E. M., Kistner, K., Mayer, S., Massotte, D., Kieffer, B. L., Hoffmann, C., et al. (2016). Functional and structural characterization of axonal opioid receptors as targets for analgesia. Mol. Pain 12:1744806916628734. doi: 10.1177/1744806916628734
Mao, J. (1999). NMDA and opioid receptors: their interactions in antinociception, tolerance and neuroplasticity. Brain Res. Rev. 30, 289–304. doi: 10.1016/s0165-0173(99)00020-x
Merched, A. J., Ko, K., Gotlinger, K. H., Serhan, C. N., and Chan, L. (2008). Atherosclerosis: evidence for impairment of resolution of vascular inflammation governed by specific lipid mediators. FASEB J. 22, 3595–3606. doi: 10.1096/fj.08-112201
Norling, L. V., Dalli, J., Flower, R. J., Serhan, C. N., and Perretti, M. (2012). Resolvin D1 limits polymorphonuclear leukocyte recruitment to inflammatory loci: receptor-dependent actions. Arterioscler. Thromb. Vasc. Biol. 32, 1970–1978. doi: 10.1161/ATVBAHA.112.249508
Oehler, B., Kistner, K., Martin, C., Schiller, J., Mayer, R., Mohammadi, M., et al. (2017). Inflammatory pain control by blocking oxidized phospholipid-mediated TRP channel activation. Sci. Rep. 7:5447. doi: 10.1038/s41598-017-05348-3
Oehler, B., Scholze, A., Schaefer, M., and Hill, K. (2012). TRPA1 is functionally expressed in melanoma cells but is not critical for impaired proliferation caused by allyl isothiocyanate or cinnamaldehyde. Naunyn Schmiedebergs Arch. Pharmacol. 385, 555–563. doi: 10.1007/s00210-012-0747-x
Park, C. K., Lü, N., Xu, Z. Z., Liu, T., Serhan, C. N., and Ji, R. R. (2011a). Resolving TRPV1- and TNF-α-mediated spinal cord synaptic plasticity and inflammatory pain with neuroprotectin D1. J. Neurosci. 31, 15072–15085. doi: 10.1523/JNEUROSCI.2443-11.2011
Park, C. K., Xu, Z. Z., Liu, T., Lü, N., Serhan, C. N., and Ji, R. R. (2011b). Resolvin D2 is a potent endogenous inhibitor for transient receptor potential subtype V1/A1, inflammatory pain, and spinal cord synaptic plasticity in mice: distinct roles of resolvin D1, D2, and E1. J. Neurosci. 31, 18433–18438. doi: 10.1523/JNEUROSCI.4192-11.2011
Pereira, L. M., Lima-Júnior, R. C., Bem, A. X., Teixeira, C. G., Grassi, L. S., Medeiros, R. P., et al. (2013). Blockade of TRPA1 with HC-030031 attenuates visceral nociception by a mechanism independent of inflammatory resident cells, nitric oxide and the opioid system. Eur. J. Pain 17, 223–233. doi: 10.1002/j.1532-2149.2012.00177.x
Plein, L. M., and Rittner, H. L. (2017). Opioids and the immune system—friend or foe. Br. J. Pharmacol. doi: 10.1111/bph.13750 [Epub ahead of print].
Por, E. D., Gomez, R., Akopian, A. N., and Jeske, N. A. (2013). Phosphorylation regulates TRPV1 association with β-arrestin-2. Biochem. J. 451, 101–109. doi: 10.1042/BJ20121637
Provoost, S., De Grove, K. C., Fraser, G. L., Lannoy, V. J., Tournoy, K. G., Brusselle, G. G., et al. (2016). Pro- and anti-inflammatory role of ChemR23 signaling in pollutant-induced inflammatory lung responses. J. Immunol. 196, 1882–1890. doi: 10.4049/jimmunol.1501113
Quan-Xin, F., Fan, F., Xiang-Ying, F., Shu-Jun, L., Shi-Qi, W., Zhao-Xu, L., et al. (2012). Resolvin D1 reverses chronic pancreatitis-induced mechanical allodynia, phosphorylation of NMDA receptors and cytokines expression in the thoracic spinal dorsal horn. BMC Gastroenterol. 12:148. doi: 10.1186/1471-230x-12-148
Rittner, H. L., Brack, A., Machelska, H., Mousa, S. A., Bauer, M., Schäfer, M., et al. (2001). Opioid peptide expressing leukocytes—Identification, recruitment and simultaneously increasing inhibiton of inflammatory pain. Anesthesiology 95, 500–508. doi: 10.1097/00000542-200108000-00036
Rittner, H. L., Hackel, D., Voigt, P., Mousa, S., Stolz, A., Labuz, D., et al. (2009). Mycobacteria attenuate nociceptive responses by formyl peptide receptor triggered opioid peptide release from neutrophils. PLoS Pathog. 5:e1000362. doi: 10.1371/journal.ppat.1000362
Rittner, H. L., Labuz, D., Schaefer, M., Mousa, S. A., Schulz, S., Schäfer, M., et al. (2006). Pain control by CXCR2 ligands through Ca2+-regulated release of opioid peptides from polymorphonuclear cells. FASEB J. 20, 2627–2629. doi: 10.1096/fj.06-6077fje
Rouwette, T., Avenali, L., Sondermann, J., Narayanan, P., Gomez-Varela, D., and Schmidt, M. (2015). Modulation of nociceptive ion channels and receptors via protein-protein interactions: implications for pain relief. Channels (Austin) 9, 175–185. doi: 10.1080/19336950.2015.1051270
Rowan, M. P., Bierbower, S. M., Eskander, M. A., Szteyn, K., Por, E. D., Gomez, R., et al. (2014). Activation of mu opioid receptors sensitizes transient receptor potential vanilloid type 1 (TRPV1) via β-arrestin-2-mediated cross-talk. PLoS One 9:e93688. doi: 10.1371/journal.pone.0093688
Sauer, R. S., Hackel, D., Morschel, L., Sahlbach, H., Wang, Y., Mousa, S. A., et al. (2014). Toll like receptor (TLR)-4 as a regulator of peripheral endogenous opioid-mediated analgesia in inflammation. Mol. Pain 10:10. doi: 10.1186/1744-8069-10-10
Schmid, M., Gemperle, C., Rimann, N., and Hersberger, M. (2016). Resolvin D1 polarizes primary human macrophages toward a proresolution phenotype through GPR32. J. Immunol. 196, 3429–3437. doi: 10.4049/jimmunol.1501701
Schreiter, A., Gore, C., Labuz, D., Fournie-Zaluski, M. C., Roques, B. P., Stein, C., et al. (2012). Pain inhibition by blocking leukocytic and neuronal opioid peptidases in peripheral inflamed tissue. FASEB J. 26, 5161–5171. doi: 10.1096/fj.12-208678
Schulze, A., Oehler, B., Urban, N., Schaefer, M., and Hill, K. (2013). Apomorphine is a bimodal modulator of TRPA1 channels. Mol. Pharmacol. 83, 542–551. doi: 10.1124/mol.112.081976
Serhan, C. N. (2014). Pro-resolving lipid mediators are leads for resolution physiology. Nature 510, 92–101. doi: 10.1038/nature13479
Spahn, V., Del Vecchio, G., Labuz, D., Rodriguez-Gaztelumendi, A., Massaly, N., Temp, J., et al. (2017). A nontoxic pain killer designed by modeling of pathological receptor conformations. Science 355, 966–969. doi: 10.1126/science.aai8636
Tominaga, M., Caterina, M. J., Malmberg, A. B., Rosen, T. A., Gilbert, H., Skinner, K., et al. (1998). The cloned capsaicin receptor integrates multiple pain-producing stimuli. Neuron 21, 531–543. doi: 10.1016/s0896-6273(00)80564-4
van Unen, J., Stumpf, A. D., Schmid, B., Reinhard, N. R., Hordijk, P. L., Hoffmann, C., et al. (2016). A new generation of FRET sensors for robust measurement of gαi1, Gαi2 and Gαi3 activation kinetics in single Cells. PLoS One 11:e0146789. doi: 10.1371/journal.pone.0146789
Wang, Y., Gehringer, R., Mousa, S. A., Hackel, D., Brack, A., and Rittner, H. L. (2014). CXCL10 controls inflammatory pain via opioid peptide-containing macrophages in electroacupuncture. PLoS One 9:e94696. doi: 10.1371/journal.pone.0094696
Wang, J. C., and Strichartz, G. R. (2017). Prevention of chronic post-thoracotomy pain in rats by intrathecal resolvin D1 and D2: effectiveness of perioperative and delayed drug delivery. J. Pain 18, 535–545. doi: 10.1016/j.jpain.2016.12.012
Weng, H. J., Patel, K. N., Jeske, N. A., Bierbower, S. M., Zou, W., Tiwari, V., et al. (2015). Tmem100 is a regulator of TRPA1-TRPV1 complex and contributes to persistent pain. Neuron 85, 833–846. doi: 10.1016/j.neuron.2014.12.065
Xu, Z. Z., and Ji, R. R. (2011). Resolvins are potent analgesics for arthritic pain. Br. J. Pharmacol. 164, 274–277. doi: 10.1111/j.1476-5381.2011.01348.x
Keywords: resolvin, opioid receptors, opioid peptides, transient receptor potential channels, inflammation, animals, pain behavior
Citation: Oehler B, Mohammadi M, Perpina Viciano C, Hackel D, Hoffmann C, Brack A and Rittner HL (2017) Peripheral Interaction of Resolvin D1 and E1 with Opioid Receptor Antagonists for Antinociception in Inflammatory Pain in Rats. Front. Mol. Neurosci. 10:242. doi: 10.3389/fnmol.2017.00242
Received: 28 April 2017; Accepted: 18 July 2017;
Published: 03 August 2017.
Edited by:
Hermona Soreq, Hebrew University of Jerusalem, IsraelReviewed by:
Sangsu Bang, Duke University, United StatesNader Ghasemlou, Queen’s University, Canada
Copyright © 2017 Oehler, Mohammadi, Perpina Viciano, Hackel, Hoffmann, Brack and Rittner. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Heike L. Rittner, cml0dG5lcl9oQHVrdy5kZQ==