 Yonglu Tian1,2†
Yonglu Tian1,2† Chaojuan Yang1†
Chaojuan Yang1† Shujiang Shang1†Yijun Cai3†Xiaofei Deng4†Jian Zhang1
Shujiang Shang1†Yijun Cai3†Xiaofei Deng4†Jian Zhang1 Feng Shao5Desheng Zhu1Yunbo Liu6Guiquan Chen7
Feng Shao5Desheng Zhu1Yunbo Liu6Guiquan Chen7 Jing Liang4*
Jing Liang4* Qiang Sun3*
Qiang Sun3* Zilong Qiu3*
Zilong Qiu3* Chen Zhang1,8*
Chen Zhang1,8*- 1State Key Laboratory of Membrane Biology, School of Life Sciences, Peking University-IDG/McGovern Institute for Brain Research, Peking University, Beijing, China
- 2Peking-Tsinghua Center for Life Sciences, Academy for Advanced Interdisciplinary Studies, Peking University, Beijing, China
- 3CAS Key Laboratory of Primate Neurobiology, Institute of Neuroscience, Chinese Academy of Sciences, Shanghai, China
- 4Key Laboratory of Mental Health, Institute of Psychology, Chinese Academy of Sciences, Beijing, China
- 5Department of Psychology, Peking University, Beijing, China
- 6Institute of Laboratory Animal Science, Peking Union Medical College/Chinese Academy of Medical Sciences, Beijing, China
- 7MOE Key Laboratory of Model Animal for Disease Study, Model Animal Research Center, Nanjing University, Nanjing, China
- 8Key Laboratory for Neuroscience, Ministry of Education/National Health and Family Planning Commission, Peking University, Beijing, China
Fragile X syndrome (FXS) is a neurodevelopmental disorder caused by mutations in the FMR1 gene that inactivate expression of the gene product, the fragile X mental retardation 1 protein (FMRP). In this study, we used clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas9) technology to generate Fmr1 knockout (KO) rats by disruption of the fourth exon of the Fmr1 gene. Western blotting analysis confirmed that the FMRP was absent from the brains of the Fmr1 KO rats (Fmr1exon4-KO). Electrophysiological analysis revealed that the theta-burst stimulation (TBS)–induced long-term potentiation (LTP) and the low-frequency stimulus (LFS)–induced long-term depression (LTD) were decreased in the hippocampal Schaffer collateral pathway of the Fmr1exon4-KO rats. Short-term plasticity, measured as the paired-pulse ratio, remained normal in the KO rats. The synaptic strength mediated by the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR) was also impaired. Consistent with previous reports, the Fmr1exon4-KO rats demonstrated an enhanced 3,5-dihydroxyphenylglycine (DHPG)–induced LTD in the present study, and this enhancement is insensitive to protein translation. In addition, the Fmr1exon4-KO rats showed deficits in the probe trial in the Morris water maze test. These results demonstrate that deletion of the Fmr1 gene in rats specifically impairs long-term synaptic plasticity and hippocampus-dependent learning in a manner resembling the key symptoms of FXS. Furthermore, the Fmr1exon4-KO rats displayed impaired social interaction and macroorchidism, the results consistent with those observed in patients with FXS. Thus, Fmr1exon4-KO rats constitute a novel rat model of FXS that complements existing mouse models.
Introduction
Fragile X syndrome (FXS) is the most common heritable cause of mental retardation and intellectual disability in humans (Pieretti et al., 1991). The prevalence of FXS is about 1 in 4,000 men and 1 in 6,000–8,000 women (de Vries et al., 1997). Approximately 85% of male and 25% of female patients with FXS show significant intellectual and developmental disability (Lozano et al., 2016). Fragile X mental retardation protein (FMRP) is enriched in the brain and testes (Devys et al., 1993; Bakker et al., 2000), in accordance with the mental retardation and macroorchidism exhibited by most patients with FXS (Hagerman, 1987; Martin and Arici, 2008; Saldarriaga et al., 2014).
The most studied FXS animal model is the Fmr1 knockout (KO) mouse, which is generated by disrupting either exon 5 (The Dutch-Belgian Fragile X Consortium, 1994) or exon 1 and the promoter region (Mientjes et al., 2006) of the Fmr1 gene. Both Fmr1 KO mouse lines lack FMRP in the brain and show diverse behavioral phenotypes and synaptic physiology deficits, some of which recapitulate the clinical symptoms of patients with FXS [reviewed in (Kazdoba et al., 2014)]. Fmr1 KO mice with transgenic expression of the human FMR1 gene have demonstrated reduced anxiety and increased exploratory behavior in addition to the correction of some KO behavior phenotypes (Peier et al., 2000; Spencer et al., 2008). Mouse models with expansion of the CGG trinucleotide repeat in the Fmr1 gene have also been developed to mimic the genetic changes observed in humans with FXS (Bontekoe et al., 2001). However, mouse FXS models have yielded mixed results or failed to reproduce several core FXS clinical phenotypes, including global cognitive dysfunction. For example, Fmr1 KO mice showed normal behavior in the probe trial in the Morris water maze test, with the exception of a subtle change during the reversal trial when the platform was changed to the opposite position (The Dutch-Belgian Fragile X Consortium, 1994; Kooy et al., 1996). In the radial arm maze test, Fmr1 KO mice exhibited a normal working memory in comparison with that of the wild-type (WT) mice (Yan et al., 2004). Long-term potentiation (LTP) is a major type of long-lasting synaptic plasticity and is associated with learning and memory. Protein synthesis-dependent late-phase LTP in the hippocampus of Fmr1 KO mice is still controversial (Hu et al., 2008; Shang et al., 2009; Koga et al., 2015).
Rats are genetically more similar than mice to humans. The usage of rats in scientific research began in the middle of the 19th century (Baker et al., 1979). Rats are widely used in studies of neurological disorders, such as epilepsy, depression, Parkinson’s disease, stroke, and vascular brain disorders (Kerkerian-Le Goff et al., 2009; Melani et al., 2010; Bailey et al., 2011; Nabika et al., 2012; Tayebati et al., 2012; Liao et al., 2013; Russo et al., 2013). Inactivation of the Fmr1 gene in rats via zinc finger nuclease (ZFN) technology targeting of the junction region between intron 7 and exon 8 was recently reported (Hamilton et al., 2014) in a three-chamber test in which 21 amino acids were deleted from the FMRP. This line of KO rats (Fmr1exon8-KO) displayed social dysfunction, an autism-related phenotype. Further studies of Fmr1exon8-KO rats revealed abnormal neuronal morphology in the superior olivary complex and impaired sound processing (Engineer et al., 2014; Ruby et al., 2015), as well as increased metabotropic glutamate receptor (mGluR)-dependent hippocampal long-term depression (Till et al., 2015). Juvenile Fmr1exon8-KO rats showed dysfunction in regulating the circuit state in the visual cortex (Berzhanskaya et al., 2016, 2017).
Fragile X mental retardation 1 protein is highly expressed in neurons, and dysregulation of FMRP causes impairment of synaptic strength and neural circuit development. In the present study, a KO rat model was generated by specifically targeting exon 4 using clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas9) technology, ensuring that regions downstream of exon 4, including the full RNA binding sequence, were not translated. We examine the physiology in hippocampal CA1 pyramidal neurons of the Fmr1exon4-KO rat. Loss of FMRP can lead to deficits in basal synaptic transmission and long-term synaptic plasticity, including theta burst stimulation (TBS)–induced LTP, a low-frequency stimulus (LFS)–induced long-term depression (LTD), and a 3,5-dihydroxyphenylglycine (DHPG)–induced LTD in the Fmr1exon4-KO rat. The knockout (KO) Fmr1 gene in rats also contributes to abnormal cognitive behaviors.
Materials and Methods
Animals
Fmr1 KO rats were produced by the CRISPR/Cas9 method and maintained in the laboratory animal center of Peking University. This line was created via the outbred Sprague-Dawley background. The KO rat lines were maintained with heterozygous female and WT male breeding pairs. The genotypes of the animals were identified. KO and WT rats aged 8–12 weeks were used in the study. The rats were kept in a temperature- and relative-humidity-controlled environment (22 ± 2°C, 40–70%) with a 12-h light/dark cycle and free access to food and water. All animal studies were conducted in accordance with the Guide for the Care and Use of Laboratory Animals (8th edition) and approved by the Institutional Animal Care and Use Committee of Peking University. All tests were performed using WT and KO littermates derived from breeding heterozygous female rats with Sprague-Dawley WT male rats. All behavioral tests were conducted in a temperature-controlled (24 ± 2°C) test room between 14:00 and 18:00. After each test, the apparatus and the test area were cleaned with 75% ethanol to remove olfactory cues. In all the behavioral assays, the light intensity was 15–20 lx, and the sound intensity was less than 60 dB. All the behavior tests and electrophysiological measurements were performed in a blinded manner.
DNA Analysis and Genotyping
DNA was obtained from rat-toe tissue samples by incubation with 500 μg/mL proteinase K (Amresco, Solon, OH, United States) in 400 μL lysis buffer [10 mM Tris–HCl, 5 mM EDTA, 0.2% sodium dodecyl sulfate (SDS), 200 mM NaCl] for 6–8 h at 55°C. After incubation, 400 μL isopropyl alcohol was added to precipitate DNA. The suspensions were centrifuged at 13,000 rpm for 10 min, after which the supernatant was removed. Next, 1 mL of 70% ethanol was added to the sample, which was centrifuged at 13,000 rpm for 10 min, after which all ethanol was removed, and the tube was dried. The DNA was dissolved in 100 μL 5 mM Tris buffer (pH 8.0) for 30 min at 55°C. PCR genotyping was performed using 2× Taq PCR Mix (Aidlab, Beijing, China), Fmr1 forward primer (5′-CCG TGA GTT CTC AAG TTG TTT CCA-3′), and Fmr1 reverse primer (5′-GGG ATT AAG AGC ATG CAT CAC CAT-3′). Polymerase chain reaction (PCR) was performed with the following protocol on a MyCycler Thermal CyclerTM (Bio-Rad, Hercules, CA, United States): 95°C for 4 min, 95°C for 30 s, 60°C for 30 s (lowered 0.5°C per cycle), 72°C for 30 min (30 cycles); 95°C for 30 s, 45 °C for 30 s, 72°C for 30 min (30 cycles); 72°C for 7 min, and a final hold at 4°C. PCR products were run on 1% agarose gel. The amplicon was approximately 500 bp. The amplicon was sequenced to determine the genotypes of the rats.
Western Blot Analysis
The brains of the WT and Fmr1 KO rats were homogenized in phosphate-buffered saline (PBS) containing 0.1 mM ethylene glycol-bis (β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA), 1 mM phenylmethylsulfonyl fluoride (PMSF), 1 μg/mL pepstatin, 1 μg/mL leupeptin, 2 μg/mL protinin, and 1% Triton X-100. Proteins in the homogenate were extracted for 2 h at 4°C, after which insoluble material was removed with centrifugation (1 h at 100,000 × g). Protein concentrations were determined using a BCA Protein Assay Kit (Thermo Scientific, Carlsbad, CA, United States). Each protein sample (100 μg) was boiled in sodium dodecyl sulfate (SDS)-loading buffer, subjected to electrophoresis on a 10% SDS-polyacrylamide gel, and electroblotted onto a nitrocellulose membrane as described previously (Wei et al., 2017). FMRP was detected using a rabbit antibody (Cell Signaling Technology, Danvers, MA, United States, #4317, 1:1,000) as the primary antibody. GAPDH was detected using a rabbit antibody (Abmart, Shanghai, China, P30008, 1:1,000) as the primary antibody. IRDye 800CW-labeled anti-rabbit IgG was used as the secondary antibody and was detected with an Odyssey Infrared Imager System (LI-COR, Lincoln, NE, United States).
RNA Isolation and Quantitative RT-PCR
Total RNA was isolated from the hippocampus and cortex samples that were collected from three WT rats and three Fmr1 KO rats at approximately 8–12 weeks of age. The samples were homogenized in a glass-Teflon® homogenizer according to the protocol supplied with TRIzol® Reagent (Life Technologies, Carlsbad, CA, United States). The concentration of RNA was measured with spectrophotometry. The reaction volume consisted of 2 μg of total RNA, 5× buffer (Takara, Kusatsu, Japan), Rt enzyme mix (Takara), oligo (dT) (Takara), Random6 primer (Takara), and RNase-free H2O (to a final volume of 20 μL). The amplification program was as follows: 37°C for 15 min, 85°C for 5 s, and a final hold at 4°C. Quantitative PCR was carried out in an MX 3000PTM (Agilent Stratagene, Palo Alto, CA, United States) real-time PCR system with 2× SYBR Green qPCR Mix (Aidlab, PC3302) using designed primers. Three primer pairs were designed for the Fmr1 amplicon: a pair crossing exons 1, 2, and 3 (forward primer: 5′-GGC TCC AAT GGC GCT TTC TA-3′; reverse primer: 5′-TAA CCT ACA GGT GGT GGG-3′); a pair crossing exons 4 and 5 (forward primer: 5′-TAA CCT ACA GGT GGT GGG-3′); reverse primer: 5′-TGT GAC AAT TTC ATT GTA TG-3′); and a pair crossing exons 7 and 8 (forward primer: 5′-GAA ATG AAG AAG CCA GTA A-3′; reverse primer: 5′-AAT CAA TAG CAG TGA CCC-3′). GAPDH was used as an internal control (forward primer: 5′-CCT GGA GAA ACC TGC CAA GTA T-3′; reverse primer: 5′-CCC TCA GAT GCC TGC TTC A-3′) (Mientjes et al., 2006). Relative expression levels were calculated using the 2-Δ ΔCT method.
Three-Chamber Sociability Test
The experiment was executed as described previously (Chung et al., 2015; Xu et al., 2015; Lo et al., 2016). WT (n = 8) and KO (n = 11) male rats aged 8–12 weeks were tested in a three-chamber apparatus (40 cm × 34 cm × 24 cm) with each side chamber connected to the middle chamber by a corridor (10 cm × 10 cm × 15 cm). Before the test day, the animals were allowed to habituate the environment for 60 min. At the beginning of the test, each rat was placed into the middle chamber and allowed to move freely through all three chambers for 5 min. For the sociability tested, a novel rat (stranger1) locked in a small cage was placed in one of the side chambers, and an empty cage of the same size and design was placed in the other side chamber. The test animal was monitored and allowed to explore both chambers for 10 min, and the total time spent in each chamber was measured. The intruder was randomly assigned to one of the side chambers to avoid a side bias. In the social novelty tested, a new unfamiliar rat (stranger2) was enclosed in the cage that had been empty during the sociability test. All model rats were male and were the same age as the testing rat but had no previous contact with each other. Data were analyzed with one-way analysis of variance (ANOVA), and a two-sided Student’s t-test was used to perform the preference index analysis.
Assessment of Motor Activity Using a Force-Plate Actometer
A force-plate actometer (Bioanalytical Systems, West Lafayette, IN, United States) was used as an open field to evaluate hyperactivity and motor function. The actometer consisted of a Plexiglas® enclosure (33 cm high), a 44 cm × 44 cm plate, four force transducers, and a recording and analysis system. The area was defined as the center point to 11.64 cm, and the outer area was defined as the zone from 11.64 to 44.00 cm. The animals were placed in a force-plate actometer chamber (44 cm × 44 cm) in a dark and sound-attenuating cabinet for 60 min. Data were collected and stored during time units of 40.96 frames, with a sampling frequency of 100 points/s. The distance traveled, the tremor index, focused stereotypy, bouts of low mobility (BLM; 10 s within a 20 mm radius), and time spent in the center field were recorded. The temperature of the test room was controlled (24 ± 2°C). Before the test, rats were allowed to adapt to the environment for 1 h.
Morris Water Maze Assay
Tests were conducted in a circular black tank 150 cm in diameter containing 22 cm of water (24 ± 2°C). A circular platform (8 cm in diameter) was placed 2 cm beneath the water level. The swim paths of the rats were tracked, digitized, and stored for later behavioral analysis using Ethovision (Noldus, Wageningen, Netherlands). The water maze was divided into four quadrants (I, II, III, and IV). The rats were given four trials per day (30 min inter-trial intervals, ITIs) for four consecutive days during the spatial learning phase. During the learning phase, each animal was randomly placed in a different quadrant, with the exception of the quadrant where the platform was placed in each trial. The maximum trial length was 60 s. When a rat did not find the platform within 60 s, the latency time was calculated as 60 s. After the rats were taken out of the pool, they were dried with towels and returned to their cages. The platform was removed during the probe test. During the reverse training phase, the platform was placed in the third quadrant, which was opposite that used during the learning phase.
Elevated Plus Maze
The elevated plus maze (EPM) was used to assess anxiety-like behavior. The black-painted maze consisted of four arms (50 cm length × 10 cm width). Two opposite open arms without walls and two opposite closed arms with 35 cm high walls formed a “+” shape. The maze was elevated 76 cm above the floor by four metal legs under each arm. Each rat was placed at the junction of the open and closed arms, facing an open arm. The rat was allowed to freely explore the entire maze for 5 min. The time spent in the open arms and closed arms were recorded using the Xeye Aba V3.2 tracking system.
Slice Physiology
Hippocampal slices (400 μm) were produced from 8-week-old male WT and Fmr1 KO rats as previously (Wei et al., 2016). Animals were anesthetized using pentobarbital (10 mg/mL, 0.1 mL/10 g) and euthanized via decapitation. The brain was quickly removed to an ice-cold dissection solution with a pH of 7.3–7.4. The solution contained 213 mM sucrose, 10 mM glucose, 3 mM KCl, 1 mM NaH2PO4, 0.5 mM CaCl2, 5 mM MgCl2, and 26 mM NaHCO3. Transverse slices were cut in ice-cold dissection solution on a vibrating blade microtome (Leica VT-1200s, Wetzlar, Germany). Slices were maintained for 1 h at room temperature in artificial cerebrospinal fluid (ACSF) containing the following: 10 mM glucose, 125 mM NaCl, 5 mM KCl, 2 mM NaH2PO4, 2.6 mM CaCl2, 1.3 mM MgCl2, and 26 mM NaHCO3 (pH 7.3–7.4). The ACSF and dissection solution were gassed with 95% O2 and 5% CO2. For the recordings, slices were individually transferred to the recording chamber and mounted on the stage of an upright microscope (Olympus BX51WI, Tokyo, Japan). The bathing solution was kept at room temperature and constantly exchanged through a gravity-driven perfusion system with a flow rate of approximately 2 mL/min during the experiment. Stimuli was delivered to the slice via a concentric bipolar electrode (CBBEB75, FHC, Bowdoin, ME, United States). Microelectrodes filled with ACSF (4–7 MΩ) were used to record field excitatory postsynaptic potentials (fEPSPs) from the stratum radiatum of the CA1 region. An EPC10 Patch Clamp Amplifier (HEKA, Lambrecht, Germany) was used to record fEPSPs, the values of which were calculated by measuring the onset (a 30–70% rising phase) slope of the fEPSP. TBSs were used to induce LTP as described previously (Zhang C. et al., 2009; Zhang et al., 2010). Each TBS was composed of five episodes of stimulation delivered at 0.1 Hz, whereas each episode contained 10 stimuli trains of five pulses (100 Hz) delivered at 5 Hz. The average response was expressed as a percentage of the pre-TBS response. A LTD was induced with low-frequency stimulation (1 Hz, 900 pulses) or DHPG (100 μM, 10 min, Tocris, Bristol, United Kingdom). Anisomycin (20 μM, MedChemExpress, Monmouth Junction, NJ, United States) was added to the ACSF 1 h before recording and throughout the recordings. The synaptic ratio was calculated as the percentage of the second fEPSP slope vs. the first slope in individual slices.
Histology
Cresyl violet (Nissl) staining was used to evaluate the cytoarchitecture in the hippocampal regions. The rats were deeply anesthetized with tribromoethanol (240 mg/kg, Sigma–Aldrich, St. Louis, MO, United States) and transcardially with 4% paraformaldehyde (PFA) (w/v) in PBS. The brains were removed and dipped into fresh 4% PFA for an additional 48 to 72 h to be post-fixed at room temperature. Then the samples were embedded in paraffin and sectioned. Four-micron-thick sections were used for staining. The tissue slides were dried for 30 min at 55°C and then rewarmed at room temperature. The sections were washed at the time for distilled water and stained with a 0.5% Cresyl violet solution for 10 min. Then, the sections were washed again with distilled water, dehydrated in a graded ethanol series (95%, 1 min; 95%, 30 s; 100%, 1 min; and 100%, 1 min), and subsequently soaked three times in xylene, 5 min per time. Using the mounting medium, the sections were covered with a coverslip. Finally, an Axio Scan.Z1 (Zeiss, Oberkochen, Germany) digital slide scanner with an X20 objective was used for image acquisition.
Results
Generation of Fmr1 KO Rats with CRISPR/Cas9-Mediated Genome Editing
In the present study, the CRISPR/Cas9 system was used to introduce deletions or mutations in exon 4 of the Fmr1 gene in rats (Figure 1A). Sanger sequencing showed that one of the offspring lines carried a deletion of five amino acids and a G-A mutation in the Fmr1 gene (Figure 1B). This genetic modification resulted in a frame-shift starting from the second Agenet-like 2 domain in FMRP (Figure 1C). RT-PCR analyses of the expression of the Fmr1 transcript in the hippocampus and the cortex were conducted using three pairs of primers: one pair upstream of exon 4 of the Fmr1 gene in rats and two pairs downstream of the gene. Similar to the expression of the Fmr1 transcript in KO mice (Mientjes et al., 2006), expression of the Fmr1 transcript in Fmr1exon4-KO rats was approximately 18.58–33.78% of that of the WT rats (Figures 1D,E). The primers targeting the sequence upstream of exon 4 yielded statistically significant increases in the transcript expression levels in the hippocampus that were slightly higher than those of the two primer pairs targeting regions downstream of exon 4 (Figure 1E). Western blotting, using specific anti-FMRP antibodies, confirmed the presence of FMRP in the cortex and hippocampus lysate of the WT [XFmr1(+)Y] rats but not in that of the KO rats [XFmr1(-)Y] (Figure 1F and Supplementary Figure S1, cortex: WT, 0.21 ± 0.02, KO, 0.06 ± 0.01, n = 3, p < 0.01; hippocampus: WT, 0.29 ± 0.03, KO, 0.03 ± 0.02, n = 3, p < 0.01). When XFmr1(+)Y rats were crossed with XFmr1(+)XFmr1(-) rats, 50.24% of the male offspring were KO rats (109 of 211 male animals from eight breeding pairs), which is consistent with the expected Mendelian ratio. The male KO rats showed normal development curves (Figure 1G). The brains of the KO rats exhibited normal histology of hippocampal and neuron densities (Figures 1H,I, CA1: WT, 2533 ± 163.3, KO, 2578 ± 164.8, n = 3, p > 0.05; CA3: WT, 1711 ± 145.7, KO, 1644 ± 104.2, n = 3, p > 0.05; DG: WT, 5289 ± 185.9, KO, 5467 ± 312.7, n = 3, p > 0.05), suggesting that FMRP deletion did not cause prenatal lethality or pervasive developmental deficits.
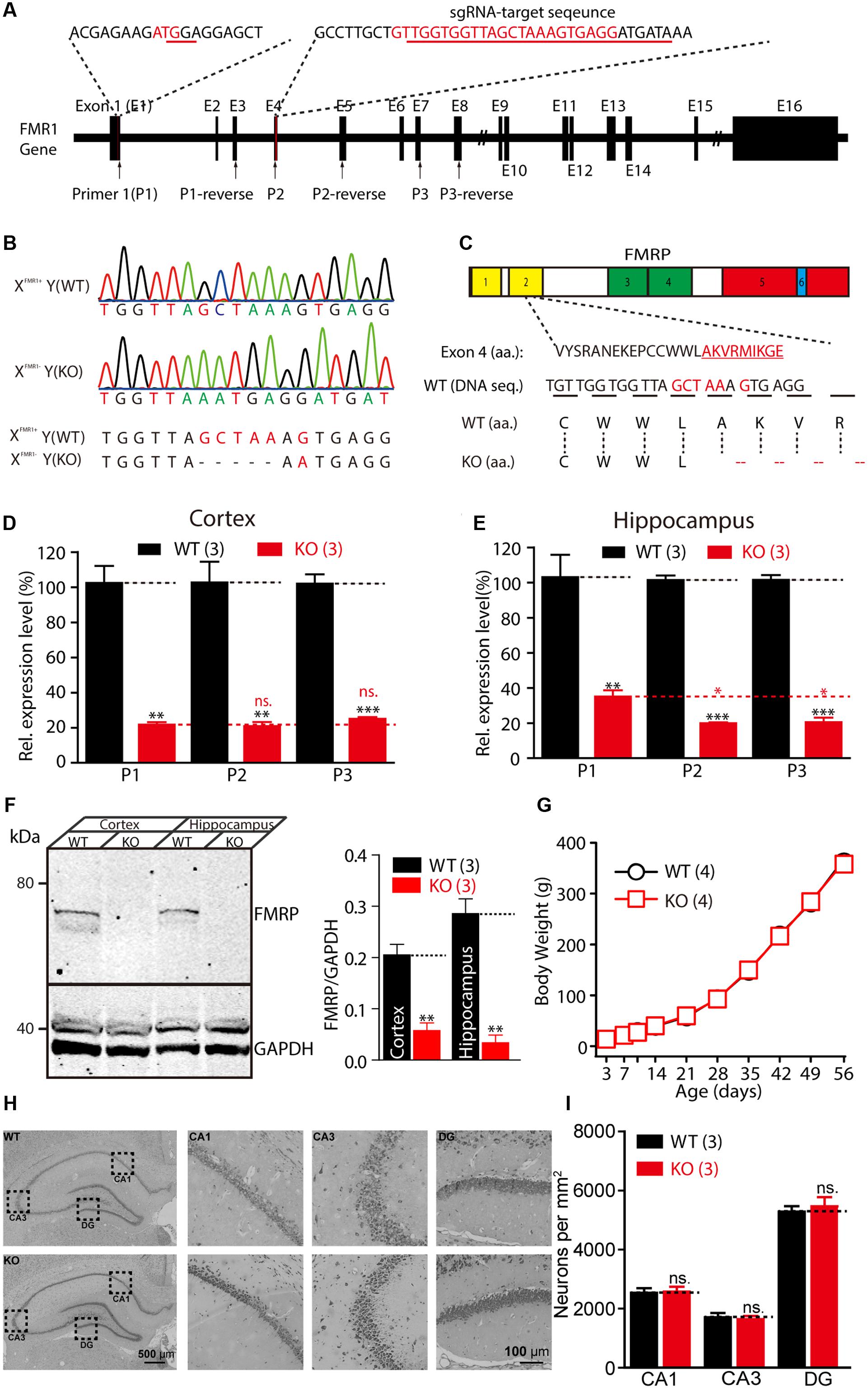
FIGURE 1. Generation of Fmr1 knockout (KO) rats using the CRISPR/Cas9 method. (A) Targeting of the Fmr1 gene. (B) Genotypes of the Fmr1 KO rats were determined with sequencing of polymerase chain reaction (PCR) products amplified from tail DNA. (C) Localization of the deletion in FMRP. Domains in FMRP: Agenet-like 1 (no. 1, yellow), Agenet-like 2 (no. 2, yellow), KH 1 (no. 3, green), and KH 2 (no. 4, green), interaction with RANBP9 (no. 5, red) and RNA-binding RGG box (no. 6, blue). (D,E) Relative expression levels of Fmr1 transcripts in the cerebral cortex (D) and hippocampus (E) were measured. Student’s t-test was used to compare the expression levels of the two groups. (F) Fmr1 KO brain lacking expression of the fragile X mental retardation 1 protein (FMRP). Brain homogenates (100 μg) were subjected to western blot analysis (left). Normalized expression levels of FMRP in rat brain homogenate (right). (G) Fmr1 KO rats showed a normal developmental curve. (H) Hippocampal regions stained with Nissl staining and the cellular layer in CA1, CA3, and DG region of the hippocampus of the WT and KO rats. (I) Neuron densities were calculated in the CA1, CA3, and DG region of hippocampus. All data are presented as mean ± standard error of the mean (SEM). (∗∗∗p < 0.001, ∗∗p < 0.01, ∗p < 0.05; ns., not significant, two-sided Student’s t-test was used).
Impaired Basal Synaptic Transmission and Synaptic Plasticity in Fmr1 KO Rats
Patients with FXS exhibit severe mental retardation that is caused by synaptic dysfunction. We first examined synaptic transmission and plasticity in the hippocampal Schaffer collateral pathway in acute slice preparation. Extracellular recordings were performed to monitor the fEPSP elicited by the stimulation of Schaffer collateral/CA1 glutamatergic fibers. The slope of the input-output curve of the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR)–mediated fEPSP was statistically significantly decreased in the Fmr1 KO rats compared with that of the WT littermate controls (Figure 2A, slopeWT = 0.015 ± 0.001, slopeKO = 0.009 ± 0.001, p < 0.001), demonstrating impairment of the basal synaptic transmission at the CA3–CA1 excitatory synapses. Next, we measured the synaptic facilitation induced by two identical stimuli separated by various intervals, which is an indicator of short-term plasticity. As shown in Figure 2B, paired-pulse facilitation was normal in the Fmr1 KO rats when compared with that of the control rats. We next examined the effect of FMRP inactivation in CA3 neurons on TBS-induced LTP in the Schaffer collateral pathway. The amplitude of TBS-induced LTP (slope averaged 50–60 min post-TBS stimulation) was markedly impaired in the Fmr1 KO rats (Figure 2C, WT: 277.8 ± 30.3%; KO: 183.4 ± 18.5%, t = 2.770, df = 18, p < 0.01), demonstrating a critical role of FMRP in regulating LTP at CA3–CA1 synapses. Therefore, we next asked whether the loss of FMRP in rats alters the maintenance of long-term depression. The LFS-induced LTD was statistically significantly reduced in Fmr1exon4-KO rats (Figure 2D, WT: 73.5 ± 7.2%; KO: 116.2 ± 13.7%, t = 2.752, df = 18, p < 0.05). The magnitude of the mGluRs-dependent LTD elicited by directly activating group I mGluRs with the agonist DHPG was statistically significantly greater in slices from the Fmr1 KO rats compared with their control littermates (Figure 2E, WT: 71.7 ± 5.7%; KO: 42.7 ± 8.1%, t = 2.948, df = 14, p < 0.05). Moreover, treating the slice with anisomycin (20 μM), a protein synthesis inhibitor, prevented the maintenance of the mGluR-dependent LTD of the WT rats (98.38 ± 7.602%) but not that of the KO rats (49.49 ± 7.954%, Figure 2F). This result suggested that a DHPG-induced LTD does not require protein synthesis in Fmr1exon4-KO rats. Thus, FMRP differentially regulates the LTP and LTD in the hippocampal CA3-CA1 synapses in an induction-specific manner.
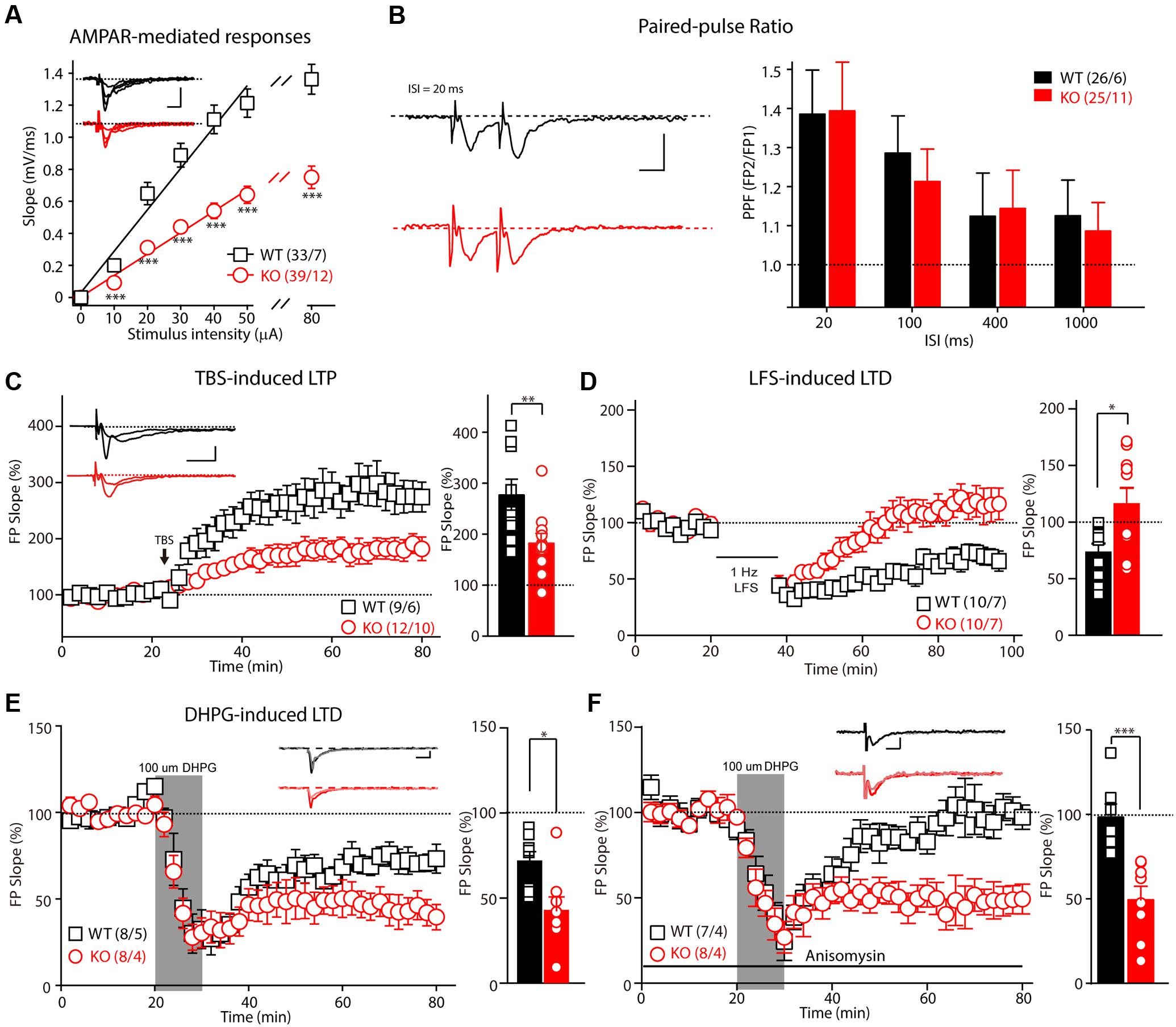
FIGURE 2. Impaired basic synaptic transmission and long-term plasticity in hippocampal CA3–CA1 synapses in Fmr1 KO rats. (A) The α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor(AMPAR)–mediated field excitatory postsynaptic potential (fEPSP) and the input-output curve were reduced in 8-week-old Fmr1 knockout (KO) rats. The initial slopes of the evoked fEPSP were plotted as a function of the stimulus intensity. (B) Left: Representative fEPSP traces from the control and Fmr1 KO rats evoked by two consecutive stimuli with a 20-ms ISI. Right: Paired-pulse facilitation was normal in the 8-week-old KO rats. (C) The theta-burst stimulation (TBS)–induced long-term potentiation (LTP) was impaired in the 8-week-old KO rats. The inset (left panel) shows representative traces before and after LTP induction. The mean fEPSP slopes averaged 50–60 min after LTP induction in the wild-type (WT) and KO rats (Student’s t-test; right). (D) The low-frequency stimulus (LFS)–induced long-term depression (LTD) was impaired in the 8-week-old KO rats. (E) Fmr1 KO rats showed an enhanced 3,5-dihydroxyphenylglycine (DHPG)–induced LTD (left: represented traces of WT and KO rats). (F) The protein synthesis inhibitor anisomycin blocks the DHPG-induced LTD of the WT rats but has no effect on the Fmr1 KO rats. All data are presented as mean ± standard error of the mean (SEM). The scale bars represent 10 ms, 1 mV (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001; ns., not significant. The number of hippocampal slices (left) and rats (right) used in each experiment is indicated in parentheses).
Fmr1 KO Rats Exhibit Altered Learning in the Morris Water Maze Test
Altered Fmr1 gene function is the major cause of mental retardation in patients with FXS. To test whether deletion of the Fmr1 gene affects the spatial learning ability of KO rats, their performance was evaluated in a Morris water maze. The training time comprised 1-min periods over 4 days (Figure 3A). The escape latency and the travel distance of the WT and KO rats were statistically significantly reduced on days 1–4 (the training period) compared with these measurements on the test days [Figures 3B–D; escape latency: F(3,48) = 64.655, p < 0.001; distance traveled: F(3,48) = 64.941, p < 0.001; swimming speed: F(3,48) = 45.650, p < 0.001]. Furthermore, in the KO rats, the escape latency was lengthened in comparison with that of the WT animals [Figure 3B, genotype F(1,16) = 3.435, p = 0.082], the distance traveled before finding the hidden platform was longer [Figure 3C, genotype F(1,16) = 6.083, p = 0.025], and swimming speed was statistically significantly increased [Figure 3D, genotype F(1,16) = 7.186, p = 0.016]. After 4 days of training, the rats were tested in the same maze but without the platform (the probe test phase of memory; Figure 3E). As shown in Figure 3F, the KO rats crossed the target area statistically significantly less often than did the WT controls (WT: 9.43 ± 0.48, n = 7; KO: 6.55 ± 0.39, n = 11, p < 0.001); this difference was not due to impaired motor function, as the swimming speeds of the rats of each genotype did not differ statistically significantly (Figure 3G, WT: 16.38 ± 0.39, n = 7; KO: 16.55 ± 0.52, n = 11, p > 0.05). The time in each quadrant of the KO and WT littermate controls was not statistically significantly different (Figures 3H,I: WT: 38.17 ± 2.42, n = 7, KO: 35.99 ± 2.40, n = 11, p > 0.05; II: WT: 24.31 ± 3.60, n = 7, KO: 21.63 ± 2.64, n = 11, p > 0.05; III: WT: 19.25 ± 2.07, n = 7, KO: 20.46 ± 2.12, n = 11, p > 0.05; IV: WT: 18.26 ± 2.38, n = 7; KO: 21.92 ± 2.83, n = 11, p > 0.05). The WT and KO rats spent more time in the target quadrant. After the probe test phase, the rats were subjected to reverse learning, and the hidden platform was switched to the opposite quadrant to test behavioral flexibility (Figure 3I). The WT rats consistently found the new hidden platform more quickly than did the KO rats, but there was no statistically significant difference between the two groups [Figures 3J–L; escape latency: reversal training day, F(3,48) = 24.566, p < 0.001; genotype, F(1,16) = 2.605, p = 0.126; distance traveled: reversal training day, F(3,48) = 27.113, p < 0.001; genotype, F(1,16) = 3.160, p = 0.094; swimming speed: reversal training day, F(3,48) = 60.115, p < 0.001; genotype, F(1,16) = 1.089, p = 0.312]. These results show that deletion of FMRP in rats impaired their ability to obtain spatial learning and memory and to maintain a normal ability to retain memories.
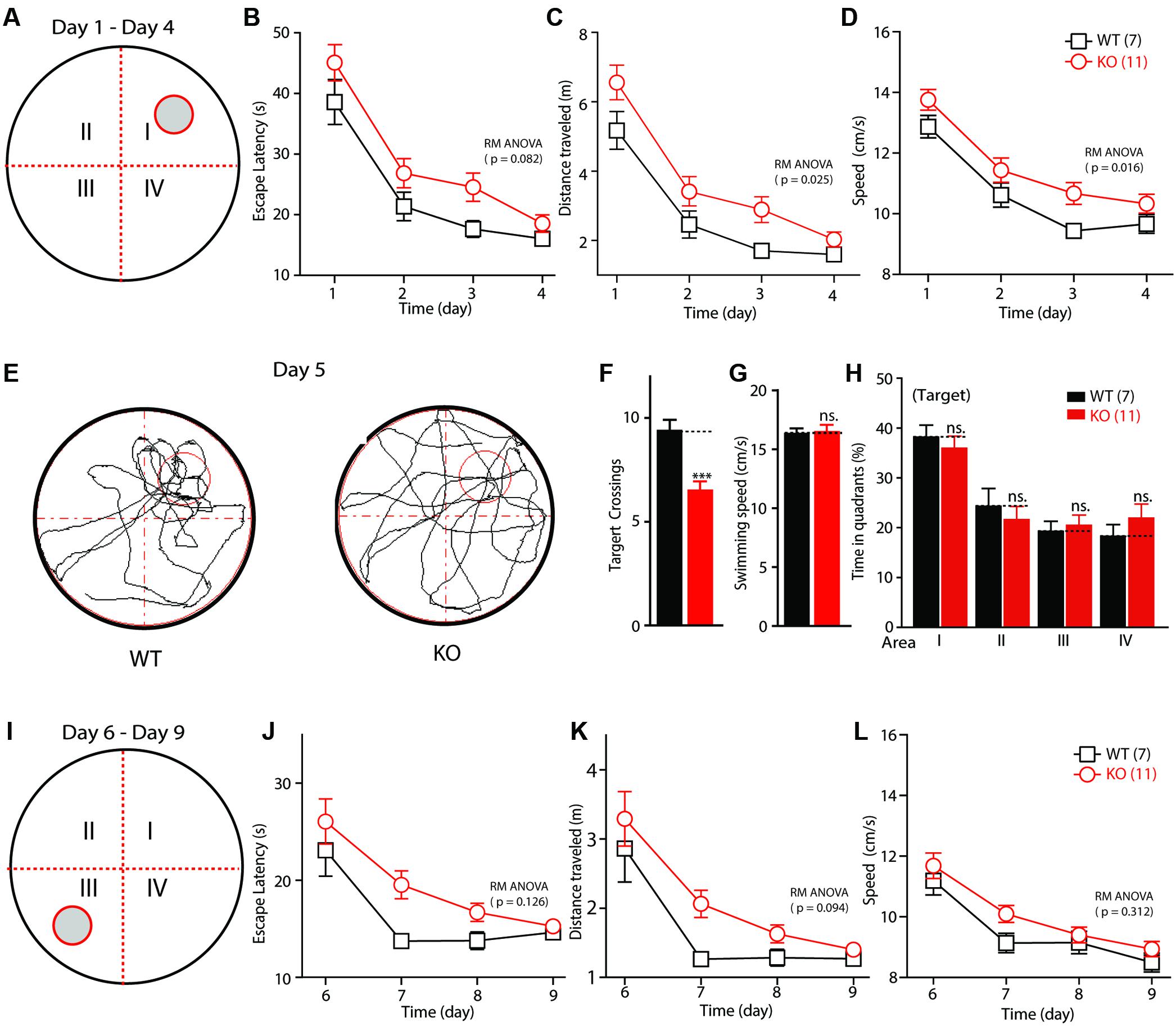
FIGURE 3. Impaired spatial learning and memory in Fmr1 KO rats in the Morris water maze. (A) Schematic diagram of the apparatus. (B) Plot of escape latency. (C) Plot of travel distance. (D) Plot of swimming speed required for the rats to find the visible platform during the learning phase. (E) Representative movement traces. The empty red circles (left) indicate the location of the former platform, which was removed in the probe test. (F) Summary of the number of crosses of the former platform area. (G) Swimming speed in the probe test. (H) Time in each quadrant during the probe test (p < 0.001 target quadrant vs. Q2, Q3, or Q4). (I) Schematic diagram of the apparatus. The visible platform was placed in the third quadrant, which was opposite to that used during the learning phase (the first quadrant). (J) Plot of escape latency. (K) Plot of travel distance. (L) Plot of swimming speed required for the rats to find the visible platform during the reverse learning phase. All data are presented as mean ± SEM. Repeated-measures analysis of variance (RM ANOVA) was used to evaluate the difference between the two curves in (A–D) and (J–L). Student’s t-test was used to compare the two groups (F,G). Post hoc analysis was used to analyze the statistically significant ANOVA results (H) (∗∗∗p < 0.001; ns., not significant).
Fmr1 KO Rats Display Impaired Social Interaction
Social dysfunction has been observed in patients with FXS and the animal models of FXS. To determine whether Fmr1 KO rats display social deficits, we monitored the behavior of the rats in the three-chamber apparatus (Nadler et al., 2004), in which the social approach of a rat toward a stranger rat trapped in a wire cage can be measured. We first tested the WT and KO rats in three empty chambers; the two genotypes showed no difference (Figures 4A,B, WT: left: 98.86 ± 7.4, center: 93.9 ± 4.5, right: 107.2 ± 4.8, p > 0.05; KO: left: 93.7 ± 6.5, center: 96.2 ± 6.1, right: 110.1 ± 9.5, p > 0.05). In the sociability test, a novel object was placed in one side chamber, and a novel, same-sex rat (stranger1) was placed in the other side of the chamber. The WT and KO rats showed normal performance as measured by the amount of time spent in each chamber (Figure 4C, WT: stranger1: 346.3 ± 46.5 s, object: 177.9 ± 36.2 s, p < 0.05; KO: stranger1: 391.8 ± 41.7 s, object: 147.4 ± 35.5 s, p < 0.001), the preference index derived from these parameters (Figure 4D, WT: 28.06 ± 13.7, KO: 40.72 ± 12.8, p > 0.05), and the frequency of subject entry into each side chamber from the center chamber (Figure 4E, WT: stranger1: 6.4 ± 1.5, object: 5.8 ± 1.5, p > 0.05; KO: stranger1: 5.5 ± 0.9, object: 4.6 ± 0.9 s, p > 0.05). However, in the social novelty test, when the inanimate object was replaced with another stranger rat (stranger2), stranger1 was observed to be a familiar stimulus. The Fmr1 KO rats spent less time with stranger2 compared to the amount of time spent with stranger1 (Figure 4F, WT: stranger1: 168.7.3 ± 34.0 s, stranger2: 356.0 ± 44.0 s, p < 0.05; KO: stranger1: 336.3 ± 53.2 s, stranger2: 183.5 ± 41.1 s, p < 0.05). The preference index derived from these parameters was also different (Figure 4G, WT: 31.23 ± 12.9, KO: -25.46 ± 15.4, p < 0.05), but the frequency of subject entry into each side chamber from the center chamber of the KO rats was similar to that obtained from the WT rats (Figure 4H, WT: stranger1: 4.4 ± 1.3, stranger2: 4.1 ± 1.0, p > 0.05; KO: stranger1: 4.3 ± 0.9, stranger2: 4.1 ± 1.0, p > 0.05). These results suggest that Fmr1 KO rats are impaired in terms of social novelty recognition but display normal sociability or social anxiety.
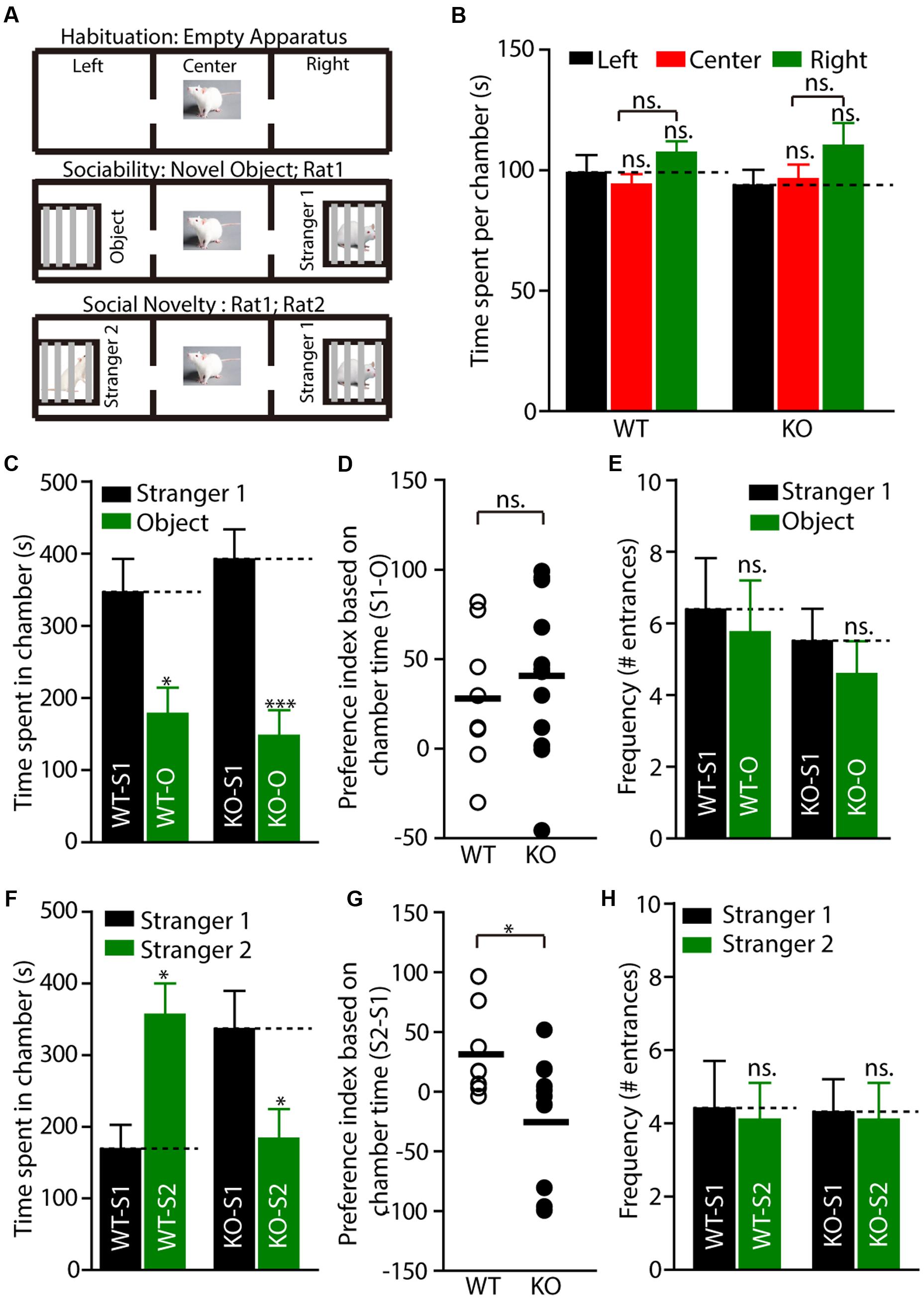
FIGURE 4. Fmr1 KO rats display impaired social interaction in the three-chamber test. (A) Diagram of the testing apparatus with two outer side chambers, each housing a novel object or stranger rat behind perforated Plexiglas, and the center chamber where the subject was started. Two doorways allowed the subject to move freely between all chambers. (B) A WT or KO subject was allowed to explore the apparatus. The mean total duration each subject spent per chamber (including time at partitions within side chambers) is shown, WT: n = 8; KO: n = 12. (C–E) Quantification of the results in (A) (middle), as shown by the amount of time spent in chamber (C) with a novel rat (stranger1, S1) vs. an inanimate object (O), or the preference index derived from the numerical difference between the time spent in chamber (D) with S1 and O divided by total time spent × 100. Frequency of subject entry into each side chamber from the center chamber is shown (E). (F–H) Quantification of the results in (A) (bottom). S2 (stranger2) and S1 (stranger1). All data are presented mean ± SEM. [∗p < 0.05, ∗∗∗p < 0.001; ns., not significant; one-way analysis of variance (ANOVA) and two-sided Student’s t-test were used].
Fmr1 KO Rats Demonstrate Normal Locomotor Activity and Normal Anxiety Levels
To test whether motor dysfunction might contribute to the learning deficits observed in the Morris water maze test, the locomotor activity of Fmr1 KO rats was measured on a force-plate actometer (Fowler et al., 2001). The travel distances and the number of BLM of the KO and WT littermate controls were not statistically significantly different (Figures 5A,B, distance: WT: 133.25 ± 8.93 m, n = 7; KO: 136.64 ± 15.25 m, n = 11, p > 0.05; BLM: WT: 197.25 ± 20.45, n = 7; KO: 199.33 ± 19.9, n = 11, p > 0.05). Furthermore, the KO rats spent essentially the same percentage of time in the center area (23.2 cm × 23.2 cm; Figure 5C left, WT: 18.82 ± 2.83, n = 7; KO: 17.39 ± 6.26, n = 11, p > 0.05) and made a similar number of leaps over the center (Figure 5C right, WT: 830.43 ± 141.42, n = 7; KO: 601.64 ± 136.52, n = 11, p > 0.05). The tremor index of the KO and WT rats (calculated from the power spectra data using Fourier analysis) was also not statistically significantly different (Figure 5D, tremor index 1: WT: 0.18 ± 0.03, n = 7; KO: 0.17 ± 0.03, n = 11, p > 0.05; tremor index 2: WT: -0.41 ± 0.08, n = 7; KO: -0.30 ± 0.10, n = 11, p > 0.05). The frequency of stereotypical behavior declined over time after the WT and KO rats were placed on the force plate, but there were no genotypic differences [Figure 5E, genotype, F(1,160) = 0.1110, p = 0.7395; time block, F(9,160) = 1.310, p = 0.2354]. These results demonstrate that deletion of FMRP in rats had no detectable effects on motor function.
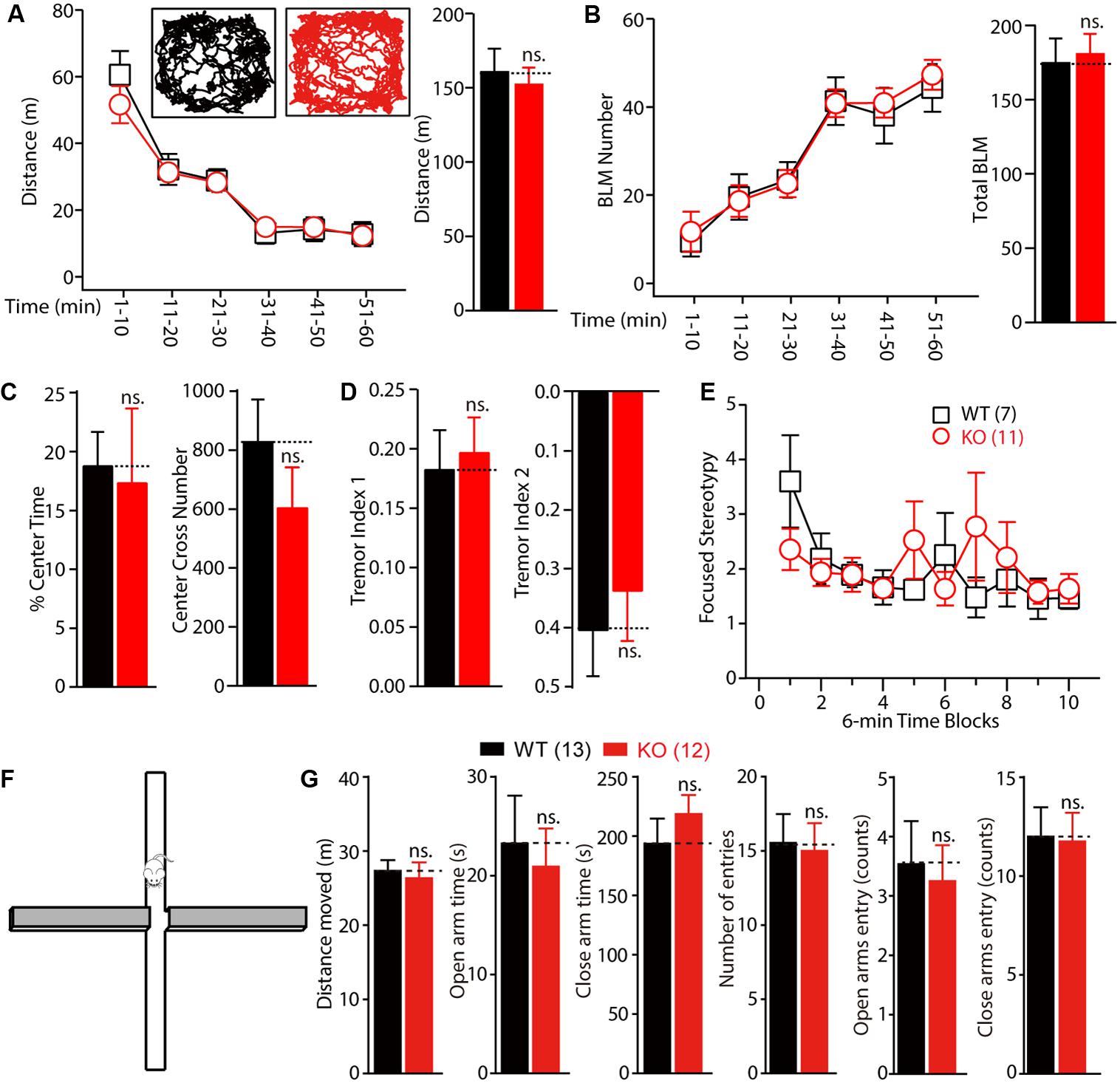
FIGURE 5. Normal locomotor activity and anxiety of Fmr1 KO rats in force plate actometer and elevated plus maze test. (A,B) Fmr1 KO rats showed normal exploratory behavior (A) and bouts of low mobility (BLM) (B) on the force plate. Rats were allowed to move freely on the force plate actometer for 60 min. Distance traveled and BLM were quantified every 10 min (left) and for the total 60 min period (right). The inset shows the representative movement trajectories of the WT and KO rats. (C) The percentage of time spent in the center field of the force plate and the number of crosses of the center field were measured. (D) KO rats showed normal whole-body tremor, which was quantified from the force variation data. (E) The focused stereotypy scores of the KO rats were similar to those of their WT littermate controls. The focused stereotypy scores were calculated from the intense rhythmic movement data of the rats on the force plate. (F) Diagram of the elevated plus maze test apparatus. (G) The distance the rats moved, in open and close arm time; there was no statistically significant difference in the open and close arm entries between the WT and KO rats. All data are presented as mean ± standard error of the mean (SEM). (ns., not significant, two-way ANOVA and two-sided Student’s t-test were used).
To explore whether the KO rats showed anxiety and hyperactivity, we performed the EPM test. The time spent in open arms and close arms were not different between genotypes (Figures 5F,G, open arms: WT vs. KO, 23.25 ± 4.85 s vs. 20.93 ± 3.83 s; close arms: WT vs. KO, 193.50 ± 21.09 s vs. 218.50 ± 16.09 s, WT: n = 13, KO: n = 12). These results indicated that the Fmr1 KO rats showed normal anxiety level with the WT rats.
Fmr1 KO Rats Show Macroorchidism
Macroorchidism, one of the hallmark symptoms experienced by patients with FXS, is also observed in several FXS animal models (Hamilton et al., 2014). Therefore, we examined the weight of the testes from 5-week-old to 5-month-old rats. As mentioned above, there were no differences in the average body weights of the WT and KO rats (Figure 6A left: WT 152.92 ± 6.10 g, n = 5; KO 147.26 ± 3.41 g, n = 5, p > 0.05; Figure 6B left: WT 511.26 ± 9.79 g, n = 9; KO 519.39 ± 8.89 g, n = 10, p > 0.05). No differences were observed in the testes of KO rats at the age of 5 weeks, in either net weight (Figure 6A middle: total: WT 1.4 ± 0.04 g, KO 1.4 ± 0.05 g, p > 0.05; left: WT 0.72 ± 0.04 g, KO 0.72 ± 0.04 g, p > 0.05; right: WT 0.68 ± 0.02 g, KO 0.70 ± 0.03 g, p > 0.05) or organ relative weight (Figure 6A right: total: WT 0.92 ± 0.02%, KO 0.97 ± 0.04%, p > 0.05; left: WT 0.47 ± 0.01%, KO 0.49 ± 0.02%, p > 0.05; right: WT 0.45 ± 0.02%, KO 0.48 ± 0.03%, p > 0.05). However, the net weight of the 5-month-old Fmr1 KO rats’ testes was statistically significantly heavier than that of the WT rats (Figure 6B middle: total: WT 3.77 ± 0.12 g, KO 4.37 ± 0.11 g, p < 0.01; left: WT 1.85 ± 0.07 g, KO 2.17 ± 0.06 g, p < 0.01; right: WT 1.92 ± 0.06 g, KO 2.20 ± 0.06 g, p < 0.01), and the organ relative weight was also heavier than that of the WT rats (Figure 6B right: total: WT 0.74 ± 0.02%, KO 0.84 ± 0.02%, p < 0.01; left: WT 0.36 ± 0.01%, KO 0.42 ± 0.01%, p < 0.01; right: WT 0.38 ± 0.01%, KO 0.42 ± 0.01%, p < 0.01). These results demonstrate that Fmr1 KO rats display macroorchidism at the age of 5 months.
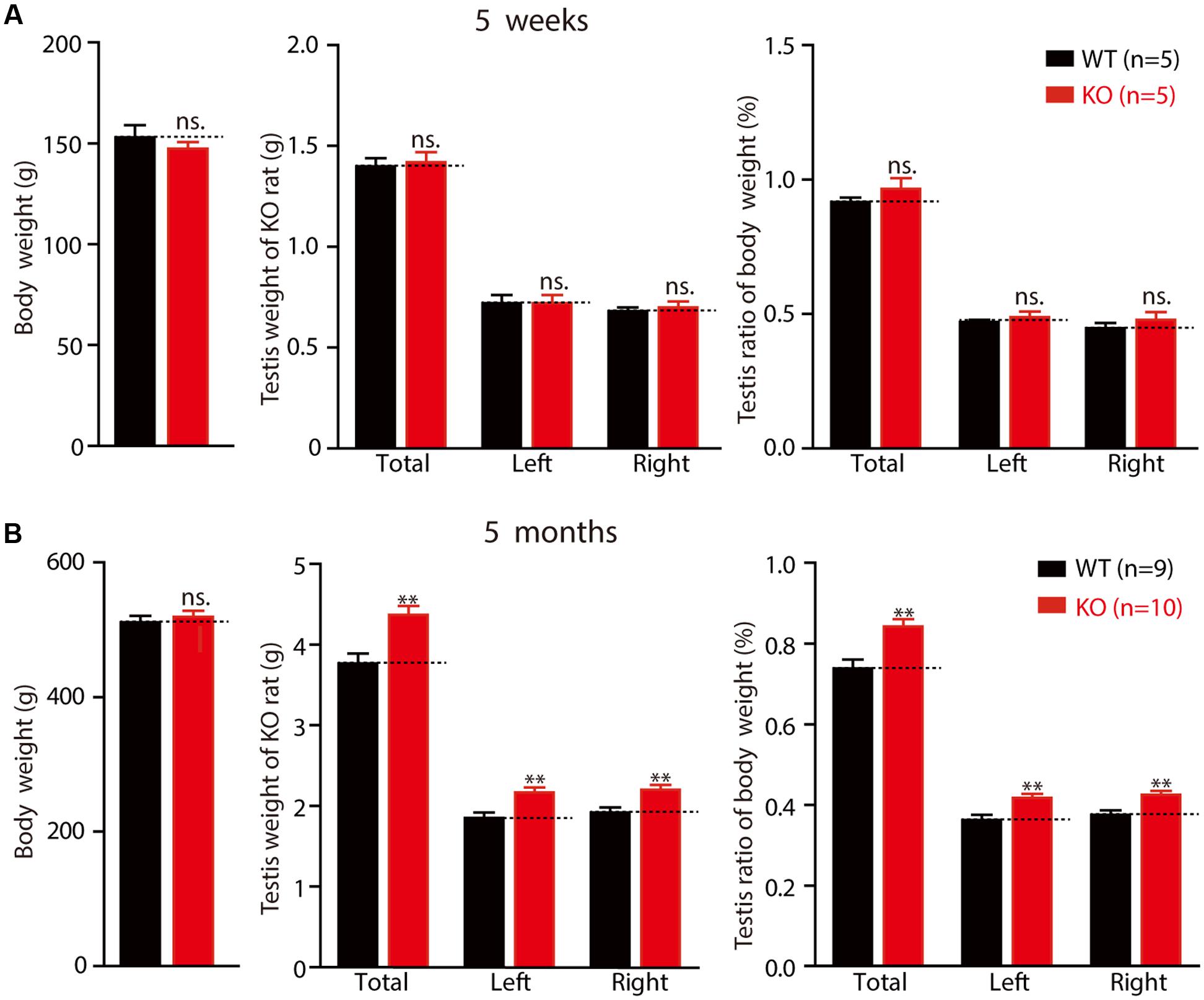
FIGURE 6. Fmr1 KO rats exhibit macroorchidism. (A) Fmr1 KO rats’ body weight, testis weight and testis ratio of body in 5-week-old Fmr1 KO rats. (B) Fmr1 KO rats’ body weight, testis weight and testis ratio of body in 5-month-old Fmr1 KO rats. All data are presented as mean ± SEM (∗∗p < 0.01, two-sided Student’s t-test was used).
Discussion
In this study, we identified hippocampal physiology, hippocampal-dependent, and social behavior of Fmr1 KO rats, which were generated by creating a five amino acid deletion in exon 4 of the Fmr1 gene of a rat using the CRISPR/CAS9 method. FMRP consists of several protein domains: nuclear localization signal (NLS), two hnRNP-K-homology (KH) domains (KH1 and KH2), nuclear export signal (NES), and arginine-glycine-glycine (RGG box). FMRP regulates protein translation by binding to approximately 4% of the mRNA in the mammalian brain through the two KH domains and the RGG box (Ashley et al., 1993; Siomi et al., 1993). In the present study, the genomic modification caused a frame-shift, but not an in-frame deletion in exon 4, ensuring that the remaining transcript, if any, would not generate a truncated FMRP with functional domains (Figures 1A–C). Western blotting analysis confirmed the absence of FMRP in the brains of KO rats (Figure 1F). The Fmr1exon4-KO rats exhibited grossly normal development (Figure 1G) and normal locomotor activity, as reflected by normal activity in the open field test and the force plate tests and a normal anxiety level in the EPM test (Figure 5). These observations demonstrate that inactivation of the Fmr1 gene in rats does not cause global motor dysfunction.
The normal function of the hippocampus, including LTP at the hippocampal Schaffer collateral pathway, is essential for learning during the Morris water maze test (Nosten-Bertrand et al., 1996; Lu et al., 1997; Moser et al., 1998; Jia et al., 2001). In Fmr1 KO mice, LTP induced by either TBS or high-frequency stimulation remained unaltered at the hippocampal Schaffer collateral pathway (Godfraind et al., 1996; Zhang J. et al., 2009; Yun and Trommer, 2011; Bostrom et al., 2015), while the LTD was consistently enhanced in the Fmr1 KO mouse and rat models (Godfraind et al., 1996; Huber et al., 2002; Li et al., 2002; Till et al., 2015). The dependence of the hippocampal LTP on FMRP had not been investigated in rats before this study. In the present study, electrophysiological analysis revealed that the TBS-induced LTP was severely reduced in hippocampal CA3–CA1 synapses, providing a plausible explanation for the learning deficits observed in the Fmr1 KO rats (Figure 2D). Furthermore, basal synaptic transmission, as measured by the slope of the input–output curve, was statistically significantly reduced in the Fmr1 KO rats, whereas short-term plasticity, a presynaptic phenomenon, was unchanged. These data imply that postsynaptic plasticity might be affected more severely than presynaptic plasticity at Fmr1 KO synapses. The Fmr1exon4-KO rats exhibited an enhanced DHPG-induced LTD (Figure 2E), and this enhancement is independent of protein synthesis (Figure 2F), similar to previous studies of KO mice (Nosyreva and Huber, 2006; Till et al., 2015). Consistent with previous studies with another line of Fmr1 KO rats, which was generated by SAGE Lab using ZFN technology to target intron 7 and exon 8 of Fmr1 (Hamilton et al., 2014), both lines exhibited an enhanced DHPG-induced LTD, and social dysfunction in the three-chamber test (Till et al., 2015). Interestingly, Fmr1exon4-KO rats exhibited some distinct disease-related symptoms, including a reduced TBS-induced LTP (Figure 2C) and an LFS-induced LTD (Figure 2D) and learning disability in Morris water maze test (Figure 3). FMRP regulates the translation of the protein that is necessary for the induction and expression of synaptic plasticity and can impact synaptic plasticity through FMRP’s control of protein translation (Sidorov et al., 2013). Loss of FMRP may to some extent impact the interaction of the protein with AMPAR trafficking and then result in a reduced LTP and LTD through a postsynaptic mechanism.
In the Morris water maze test, an apparatus used to measure hippocampus-dependent spatial learning and memory (Schenk and Morris, 1985; Terry, 2009), Fmr1exon4-KO rats demonstrated slower learning and statistically significantly poorer performance during the probe test phase and the reversal leaning phase. These results are consistent with the fact that the majority of patients with FXS are diagnosed with a learning disability (Skinner et al., 2005; Hall et al., 2008). The probe trial difference in the Fmr1exon4-KO rats is intriguing and has not been observed in most of the Fmr1 KO mouse lines. Studies using Fmr1 KO mice have consistently revealed normal trial performance, whereas mixed results have been reported with respect to the memory acquisition and reversal learning processes (The Dutch-Belgian Fragile X Consortium, 1994; Kooy et al., 1996; D’Hooge et al., 1997; Paradee et al., 1999; Baker et al., 2010; Uutela et al., 2012). Results obtained using Fmr1 KO mice do not correspond well with clinical observations of the symptoms of patients with FXS. Another explanation for differences in mouse and rat models of FXS might be differences in behavior-training paradigms, which seem to contribute to the performance difference in the probe trial. The Fmr1exon8-KO rats showed normal performance in the Morris water maze test when trained with an enhanced training paradigm (Till et al., 2015), suggesting that Fmr1 KO rats may maintain spatial learning ability to some extent but have difficulty with complex spatial learning tasks. In the present study, we increased the training difficulty by hiding the platform underneath the water throughout the experiments, instead of using a visible platform as in the previous report (Till et al., 2015). Moreover, two FMR1 paralogs, FXR1P and FXR2P, share a high domain homology with FMRP in mammals (Kaufmann et al., 2002). Functional compensation by Fxr1p and Fxr2p in the KO rats may also allow them to perform relatively well in easy tasks. Cooperation of FMRP and FXR2P in regulating synaptic plasticity has been observed in comparisons of Fmr1 knockout, Fxr2p knockout, and Fmr1/Fxr2p double-knockout mice (Zhang J. et al., 2009). Therefore, it might be worthwhile to examine the cognitive ability and synaptic plasticity of Fmr1- and Fxrps-compound mutant rats. Thus, based on behavior and electrophysiological phenotypes, Fmr1exon4-KO rats constitute an ideal model with which to further explore the mechanisms underlying cognitive impairment in patients with FXS, which are directly related to the pathogenesis of FXS.
Patients with FXS exhibit abnormalities in social, communication, and stereotypic behaviors. In this study, Fmr1exon4-KO rats displayed normal social recognition but abnormal social novelty behavior in the three-chamber test. In the sociability test, the wild-type and Fmr1exon4-KO rats preferred to explore the first novel rat (stranger1) over an object relatively, and there was a lack of genotype effect. However, the Fmr1exon4-KO rats spent less time with the novel rat (stranger 2) in the social novelty test compared to the control rats, which is consistent with previous reports (McNaughton et al., 2008; Liu and Smith, 2009; Mines et al., 2010; Heitzer et al., 2013). These results are analogous to the abnormalities in individuals with FXS who display social withdrawal and anxiety (Demark et al., 2003; Cohen et al., 2005; Hatton et al., 2006). These results indicate that basal synaptic transmission in the Schaffer collateral pathway of Fmr1exon4-KO rats is deficit. The loss of long-term plasticity may constitute an essential mechanism in the Morris water maze test.
Author Contributions
YT, CY, SS, YC, and XD contributed equally to this work. YT, CY, SS, YC, and XD carried out the experiments. JZ, FS, DZ, YL, GC, JL, QS, ZQ, and CZ contributed to the planning of the work. ZQ and CZ wrote the paper.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grants from the National Key Research and Development Program of China (2017YFA0105201, 2012YQ03026004 and 2014CB942804), the National Science Foundation of China (31670842), Beijing Municipal Science and Technology Commission (Z161100002616021, Z161100000216154), and the Seeding Grant for Medicine and Life Sciences of Peking University (Grant 2014-MB-11).
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fnmol.2017.00269/full#supplementary-material
FIGURE S1 | Full image of western blot.
References
Ashley, C. T., Sutcliffe, J. S., Kunst, C. B., Leiner, H. A., Eichler, E. E., Nelson, D. L., et al. (1993). Human and murine FMR-1: alternative splicing and translational initiation downstream of the CGG-repeat. Nat. Genet. 4, 244–251. doi: 10.1038/ng0793-244
Bailey, E. L., Smith, C., Sudlow, C. L., and Wardlaw, J. M. (2011). Is the spontaneously hypertensive stroke prone rat a pertinent model of sub cortical ischemic stroke? A systematic review. Int. J. Stroke 6, 434–444. doi: 10.1111/j.1747-4949.2011.00659.x
Baker, H. J., Lindsey, J. R., and Weisbroth, S. H. (1979). The Laboratory Rat. New York, NY: Academic Press.
Baker, K. B., Wray, S. P., Ritter, R., Mason, S., Lanthorn, T. H., and Savelieva, K. V. (2010). Male and female Fmr1 knockout mice on C57 albino background exhibit spatial learning and memory impairments. Genes Brain Behav. 9, 562–574. doi: 10.1111/j.1601-183X.2010.00585.x
Bakker, C. E., de Diego Otero, Y., Bontekoe, C., Raghoe, P., Luteijn, T., Hoogeveen, A. T., et al. (2000). Immunocytochemical and biochemical characterization of FMRP, FXR1P, and FXR2P in the mouse. Exp. Cell Res. 258, 162–170. doi: 10.1006/excr.2000.4932
Berzhanskaya, J., Phillips, M. A., Gorin, A., Lai, C., Shen, J., and Colonnese, M. T. (2017). Disrupted Cortical State Regulation in a Rat Model of Fragile X Syndrome. Cereb. Cortex 27, 1386–1400. doi: 10.1093/cercor/bhv331
Berzhanskaya, J., Phillips, M. A., Shen, J., and Colonnese, M. T. (2016). Sensory hypo-excitability in a rat model of fetal development in Fragile X Syndrome. Sci. Rep. 6:30769. doi: 10.1038/srep30769
Bontekoe, C. J., Bakker, C. E., Nieuwenhuizen, I. M., van der Linde, H., Lans, H., de Lange, D., et al. (2001). Instability of a (CGG)98 repeat in the Fmr1 promoter. Hum. Mol. Genet. 10, 1693–1699. doi: 10.1093/hmg/10.16.1693
Bostrom, C. A., Majaess, N. M., Morch, K., White, E., Eadie, B. D., and Christie, B. R. (2015). Rescue of NMDAR-dependent synaptic plasticity in Fmr1 knock-out mice. Cereb. Cortex 25, 271–279. doi: 10.1093/cercor/bht237
Chung, W., Choi, S. Y., Lee, E., Park, H., Kang, J., Choi, Y., et al. (2015). Social deficits in IRSp53 mutant mice improved by NMDAR and mGluR5 suppression. Nat. Neurosci. 18, 435–443. doi: 10.1038/nn.3927
Cohen, D., Pichard, N., Tordjman, S., Baumann, C., Burglen, L., Excoffier, E., et al. (2005). Specific genetic disorders and autism: clinical contribution towards their identification. J. Autism Dev. Disord. 35, 103–116. doi: 10.1007/s10803-004-1038-2
de Vries, B. B., van den Ouweland, A. M., Mohkamsing, S., Duivenvoorden, H. J., Mol, E., Gelsema, K., et al. (1997). Screening and diagnosis for the fragile X syndrome among the mentally retarded: an epidemiological and psychological survey. Collaborative Fragile X Study Group. Am. J. Hum. Genet. 61, 660–667. doi: 10.1086/515496
Demark, J. L., Feldman, M. A., and Holden, J. J. (2003). Behavioral relationship between autism and fragile x syndrome. Am. J. Ment. Retard. 108, 314–326. doi: 10.1352/0895-8017(2003)108<314:BRBAAF>2.0.CO;2
Devys, D., Lutz, Y., Rouyer, N., Bellocq, J. P., and Mandel, J. L. (1993). The FMR-1 protein is cytoplasmic, most abundant in neurons and appears normal in carriers of a fragile X premutation. Nat. Genet. 4, 335–340. doi: 10.1038/ng0893-335
D’Hooge, R., Nagels, G., Franck, F., Bakker, C. E., Reyniers, E., Storm, K., et al. (1997). Mildly impaired water maze performance in male Fmr1 knockout mice. Neuroscience 76, 367–376. doi: 10.1016/S0306-4522(96)00224-2
Engineer, C. T., Centanni, T. M., Im, K. W., Rahebi, K. C., Buell, E. P., and Kilgard, M. P. (2014). Degraded speech sound processing in a rat model of fragile X syndrome. Brain Res. 1564, 72–84. doi: 10.1016/j.brainres.2014.03.049
Fowler, S. C., Birkestrand, B. R., Chen, R., Moss, S. J., Vorontsova, E., Wang, G., et al. (2001). A force-plate actometer for quantitating rodent behaviors: illustrative data on locomotion, rotation, spatial patterning, stereotypies, and tremor. J. Neurosci. Methods 107, 107–124. doi: 10.1016/S0165-0270(01)00359-4
Godfraind, J. M., Reyniers, E., De Boulle, K., D’Hooge, R., De Deyn, P. P., Bakker, C. E., et al. (1996). Long-term potentiation in the hippocampus of fragile X knockout mice. Am. J. Med. Genet. 64, 246–251. doi: 10.1002/(SICI)1096-8628(19960809)64:2<246::AID-AJMG2>3.0.CO;2-S
Hagerman, R. J. (1987). Fragile X syndrome. Curr. Probl. Pediatr. 17, 621–674. doi: 10.1016/0045-9380(87)90011-9
Hall, S. S., Burns, D. D., Lightbody, A. A., and Reiss, A. L. (2008). Longitudinal changes in intellectual development in children with Fragile X syndrome. J. Abnorm. Child Psychol. 36, 927–939. doi: 10.1007/s10802-008-9223-y
Hamilton, S. M., Green, J. R., Veeraragavan, S., Yuva, L., McCoy, A., Wu, Y., et al. (2014). Fmr1 and Nlgn3 knockout rats: novel tools for investigating autism spectrum disorders. Behav. Neurosci. 128, 103–109. doi: 10.1037/a0035988
Hatton, D. D., Sideris, J., Skinner, M., Mankowski, J., Bailey, D. B. Jr., Roberts, J., et al. (2006). Autistic behavior in children with fragile X syndrome: prevalence, stability, and the impact of FMRP. Am. J. Med. Genet. A 140A, 1804–1813. doi: 10.1002/ajmg.a.31286
Heitzer, A. M., Roth, A. K., Nawrocki, L., Wrenn, C. C., and Valdovinos, M. G. (2013). Brief report: altered social behavior in isolation-reared Fmr1 knockout mice. J. Autism Dev. Disord. 43, 1452–1458. doi: 10.1007/s10803-012-1670-1
Hu, H., Qin, Y., Bochorishvili, G., Zhu, Y., van Aelst, L., and Zhu, J. J. (2008). Ras signaling mechanisms underlying impaired GluR1-dependent plasticity associated with fragile X syndrome. J. Neurosci. 28, 7847–7862. doi: 10.1523/JNEUROSCI.1496-08.2008
Huber, K. M., Gallagher, S. M., Warren, S. T., and Bear, M. F. (2002). Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc. Natl. Acad. Sci. U.S.A. 99, 7746–7750. doi: 10.1073/pnas.122205699
Jia, Z., Lu, Y. M., Agopyan, N., and Roder, J. (2001). Gene targeting reveals a role for the glutamate receptors mGluR5 and GluR2 in learning and memory. Physiol. Behav. 73, 793–802. doi: 10.1016/S0031-9384(01)00516-9
Kaufmann, W. E., Cohen, S., Sun, H. T., and Ho, G. (2002). Molecular phenotype of Fragile X syndrome: FMRP, FXRPs, and protein targets. Microsc. Res. Tech. 57, 135–144. doi: 10.1002/jemt.10066
Kazdoba, T. M., Leach, P. T., Silverman, J. L., and Crawley, J. N. (2014). Modeling fragile X syndrome in the Fmr1 knockout mouse. Intractable Rare Dis. Res. 3, 118–133. doi: 10.5582/irdr.2014.01024
Kerkerian-Le Goff, L., Bacci, J. J., Jouve, L., Melon, C., and Salin, P. (2009). Impact of surgery targeting the caudal intralaminar thalamic nuclei on the pathophysiological functioning of basal ganglia in a rat model of Parkinson’s disease. Brain Res. Bull. 78, 80–84. doi: 10.1016/j.brainresbull.2008.08.010
Koga, K., Liu, M. G., Qiu, S., Song, Q., O’Den, G., Chen, T., et al. (2015). Impaired presynaptic long-term potentiation in the anterior cingulate cortex of Fmr1 knock-out mice. J. Neurosci. 35, 2033–2043. doi: 10.1523/JNEUROSCI.2644-14.2015
Kooy, R. F., D’Hooge, R., Reyniers, E., Bakker, C. E., Nagels, G., De Boulle, K., et al. (1996). Transgenic mouse model for the fragile X syndrome. Am. J. Med. Genet. 64, 241–245. doi: 10.1002/(SICI)1096-8628(19960809)64:2<241::AID-AJMG1>3.0.CO;2-X
Li, J., Pelletier, M. R., Perez Velazquez, J. L., and Carlen, P. L. (2002). Reduced cortical synaptic plasticity and GluR1 expression associated with fragile X mental retardation protein deficiency. Mol. Cell. Neurosci. 19, 138–151. doi: 10.1006/mcne.2001.1085
Liao, S. J., Huang, R. X., Su, Z. P., Zeng, J. S., Mo, J. W., Pei, Z., et al. (2013). Stroke-prone renovascular hypertensive rat as an animal model for stroke studies: from artery to brain. J. Neurol. Sci. 334, 1–5. doi: 10.1016/j.jns.2013.07.2517
Liu, Z. H., and Smith, C. B. (2009). Dissociation of social and nonsocial anxiety in a mouse model of fragile X syndrome. Neurosci. Lett. 454, 62–66. doi: 10.1016/j.neulet.2009.02.066
Lo, S. C., Scearce-Levie, K., and Sheng, M. (2016). Characterization of social behaviors in caspase-3 deficient mice. Sci. Rep. 6:18335. doi: 10.1038/srep18335
Lozano, R., Azarang, A., Wilaisakditipakorn, T., and Hagerman, R. J. (2016). Fragile X syndrome: a review of clinical management. Intractable Rare Dis. Res. 5, 145–157. doi: 10.5582/irdr.2016.01048
Lu, Y. M., Jia, Z., Janus, C., Henderson, J. T., Gerlai, R., Wojtowicz, J. M., et al. (1997). Mice lacking metabotropic glutamate receptor 5 show impaired learning and reduced CA1 long-term potentiation (LTP) but normal CA3 LTP. J. Neurosci. 17, 5196–5205.
Martin, J. R., and Arici, A. (2008). Fragile X and reproduction. Curr. Opin. Obstet. Gynecol. 20, 216–220. doi: 10.1097/GCO.0b013e3282fe7254
McNaughton, C. H., Moon, J., Strawderman, M. S., Maclean, K. N., Evans, J., and Strupp, B. J. (2008). Evidence for social anxiety and impaired social cognition in a mouse model of fragile X syndrome. Behav. Neurosci. 122, 293–300. doi: 10.1037/0735-7044.122.2.293
Melani, A., Cipriani, S., Corti, F., and Pedata, F. (2010). Effect of intravenous administration of dipyridamole in a rat model of chronic cerebral ischemia. Ann. N. Y. Acad. Sci. 1207, 89–96. doi: 10.1111/j.1749-6632.2010.05732.x
Mientjes, E. J., Nieuwenhuizen, I., Kirkpatrick, L., Zu, T., Hoogeveen-Westerveld, M., Severijnen, L., et al. (2006). The generation of a conditional Fmr1 knock out mouse model to study Fmrp function in vivo. Neurobiol. Dis. 21, 549–555. doi: 10.1016/j.nbd.2005.08.019
Mines, M. A., Yuskaitis, C. J., King, M. K., Beurel, E., and Jope, R. S. (2010). GSK3 influences social preference and anxiety-related behaviors during social interaction in a mouse model of fragile X syndrome and autism. PLoS ONE 5:e9706. doi: 10.1371/journal.pone.0009706
Moser, E. I., Krobert, K. A., Moser, M. B., and Morris, R. G. (1998). Impaired spatial learning after saturation of long-term potentiation. Science 281, 2038–2042. doi: 10.1126/science.281.5385.2038
Nabika, T., Ohara, H., Kato, N., and Isomura, M. (2012). The stroke-prone spontaneously hypertensive rat: still a useful model for post-GWAS genetic studies? Hypertens. Res. 35, 477–484. doi: 10.1038/hr.2012.30
Nadler, J. J., Moy, S. S., Dold, G., Trang, D., Simmons, N., Perez, A., et al. (2004). Automated apparatus for quantitation of social approach behaviors in mice. Genes Brain Behav. 3, 303–314. doi: 10.1111/j.1601-183X.2004.00071.x
Nosten-Bertrand, M., Errington, M. L., Murphy, K. P., Tokugawa, Y., Barboni, E., Kozlova, E., et al. (1996). Normal spatial learning despite regional inhibition of LTP in mice lacking Thy-1. Nature 379, 826–829. doi: 10.1038/379826a0
Nosyreva, E. D., and Huber, K. M. (2006). Metabotropic receptor-dependent long-term depression persists in the absence of protein synthesis in the mouse model of fragile X syndrome. J. Neurophysiol. 95, 3291–3295. doi: 10.1152/jn.01316.2005
Paradee, W., Melikian, H. E., Rasmussen, D. L., Kenneson, A., Conn, P. J., and Warren, S. T. (1999). Fragile X mouse: strain effects of knockout phenotype and evidence suggesting deficient amygdala function. Neuroscience 94, 185–192. doi: 10.1016/S0306-4522(99)00285-7
Peier, A. M., McIlwain, K. L., Kenneson, A., Warren, S. T., Paylor, R., and Nelson, D. L. (2000). (Over)correction of FMR1 deficiency with YAC transgenics: behavioral and physical features. Hum. Mol. Genet. 9, 1145–1159. doi: 10.1093/hmg/9.8.1145
Pieretti, M., Zhang, F. P., Fu, Y. H., Warren, S. T., Oostra, B. A., Caskey, C. T., et al. (1991). Absence of expression of the FMR-1 gene in fragile X syndrome. Cell 66, 817–822. doi: 10.1016/0092-8674(91)90125-I
Ruby, K., Falvey, K., and Kulesza, R. J. (2015). Abnormal neuronal morphology and neurochemistry in the auditory brainstem of Fmr1 knockout rats. Neuroscience 303, 285–298. doi: 10.1016/j.neuroscience.2015.06.061
Russo, E., Citraro, R., Davoli, A., Gallelli, L., Di Paola, E. D., and De Sarro, G. (2013). Ameliorating effects of aripiprazole on cognitive functions and depressive-like behavior in a genetic rat model of absence epilepsy and mild-depression comorbidity. Neuropharmacology 64, 371–379. doi: 10.1016/j.neuropharm.2012.06.039
Saldarriaga, W., Tassone, F., Gonzalez-Teshima, L. Y., Forero-Forero, J. V., Ayala-Zapata, S., and Hagerman, R. (2014). Fragile X syndrome. Colomb. Med. 45, 190–198.
Schenk, F., and Morris, R. G. (1985). Dissociation between components of spatial memory in rats after recovery from the effects of retrohippocampal lesions. Exp. Brain Res. 58, 11–28. doi: 10.1007/BF00238949
Shang, Y., Wang, H., Mercaldo, V., Li, X., Chen, T., and Zhuo, M. (2009). Fragile X mental retardation protein is required for chemically-induced long-term potentiation of the hippocampus in adult mice. J. Neurochem. 111, 635–646. doi: 10.1111/j.1471-4159.2009.06314.x
Sidorov, M. S., Auerbach, B. D., and Bear, M. F. (2013). Fragile X mental retardation protein and synaptic plasticity. Mol. Brain 6:15. doi: 10.1186/1756-6606-6-15
Siomi, H., Siomi, M. C., Nussbaum, R. L., and Dreyfuss, G. (1993). The protein product of the fragile X gene, FMR1, has characteristics of an RNA-binding protein. Cell 74, 291–298. doi: 10.1016/0092-8674(93)90420-U
Skinner, M., Hooper, S., Hatton, D. D., Roberts, J., Mirrett, P., Schaaf, J., et al. (2005). Mapping nonverbal IQ in young boys with fragile X syndrome. Am. J. Med. Genet. A 132A, 25–32. doi: 10.1002/ajmg.a.30353
Spencer, C. M., Graham, D. F., Yuva-Paylor, L. A., Nelson, D. L., and Paylor, R. (2008). Social behavior in Fmr1 knockout mice carrying a human FMR1 transgene. Behav. Neurosci. 122, 710–715. doi: 10.1037/0735-7044.122.3.710
Tayebati, S. K., Tomassoni, D., and Amenta, F. (2012). Spontaneously hypertensive rat as a model of vascular brain disorder: microanatomy, neurochemistry and behavior. J. Neurol. Sci. 322, 241–249. doi: 10.1016/j.jns.2012.05.047
Terry, A. V. Jr. (2009). “Spatial navigation (water maze) tasks,” in Methods of Behavior Analysis in Neuroscience, 2nd Edn, ed. J. J. Buccafusco (Boca Raton, FL: CRC Press).
The Dutch-Belgian Fragile X Consortium. (1994). Fmr1 knockout mice: a model to study fragile X mental retardation. The Dutch-Belgian Fragile X Consortium. Cell 78, 23–33.
Till, S. M., Asiminas, A., Jackson, A. D., Katsanevaki, D., Barnes, S. A., Osterweil, E. K., et al. (2015). Conserved hippocampal cellular pathophysiology but distinct behavioural deficits in a new rat model of FXS. Hum. Mol. Genet. 24, 5977–5984. doi: 10.1093/hmg/ddv299
Uutela, M., Lindholm, J., Louhivuori, V., Wei, H., Louhivuori, L. M., Pertovaara, A., et al. (2012). Reduction of BDNF expression in Fmr1 knockout mice worsens cognitive deficits but improves hyperactivity and sensorimotor deficits. Genes Brain Behav. 11, 513–523. doi: 10.1111/j.1601-183X.2012.00784.x
Wei, M., Jia, M., Zhang, J., Yu, L., Zhao, Y., Chen, Y., et al. (2017). The inhibitory effect of alpha/beta-hydrolase domain-containing 6 (ABHD6) on the surface targeting of GluA2- and GluA3-containing AMPA receptors. Front. Mol. Neurosci. 10:55. doi: 10.3389/fnmol.2017.00055
Wei, M., Zhang, J., Jia, M., Yang, C., Pan, Y., Li, S., et al. (2016). alpha/beta-Hydrolase domain-containing 6 (ABHD6) negatively regulates the surface delivery and synaptic function of AMPA receptors. Proc. Natl. Acad. Sci. U.S.A. 113, E2695–E2704. doi: 10.1073/pnas.1524589113
Xu, X. J., Zhang, H. F., Shou, X. J., Li, J., Jing, W. L., Zhou, Y., et al. (2015). Prenatal hyperandrogenic environment induced autistic-like behavior in rat offspring. Physiol. Behav. 138, 13–20. doi: 10.1016/j.physbeh.2014.09.014
Yan, Q. J., Asafo-Adjei, P. K., Arnold, H. M., Brown, R. E., and Bauchwitz, R. P. (2004). A phenotypic and molecular characterization of the fmr1-tm1Cgr fragile X mouse. Genes Brain Behav. 3, 337–359. doi: 10.1111/j.1601-183X.2004.00087.x
Yun, S. H., and Trommer, B. L. (2011). Fragile X mice: reduced long-term potentiation and N-Methyl-D-aspartate receptor-mediated neurotransmission in dentate gyrus. J. Neurosci. Res. 89, 176–182. doi: 10.1002/jnr.22546
Zhang, C., Wu, B., Beglopoulos, V., Wines-Samuelson, M., Zhang, D., Dragatsis, I., et al. (2009). Presenilins are essential for regulating neurotransmitter release. Nature 460, 632–636. doi: 10.1038/nature08177
Zhang, D., Zhang, C., Ho, A., Kirkwood, A., Sudhof, T. C., and Shen, J. (2010). Inactivation of presenilins causes pre-synaptic impairment prior to post-synaptic dysfunction. J. Neurochem. 115, 1215–1221. doi: 10.1111/j.1471-4159.2010.07011.x
Keywords: FXS, hippocampus, long-term plasticity, spatial learning, intellectual disability
Citation: Tian Y, Yang C, Shang S, Cai Y, Deng X, Zhang J, Shao F, Zhu D, Liu Y, Chen G, Liang J, Sun Q, Qiu Z and Zhang C (2017) Loss of FMRP Impaired Hippocampal Long-Term Plasticity and Spatial Learning in Rats. Front. Mol. Neurosci. 10:269. doi: 10.3389/fnmol.2017.00269
Received: 10 June 2017; Accepted: 09 August 2017;
Published: 28 August 2017.
Edited by:
Eunjoon Kim, Institiue for Basic Science (IBS) and Korea Advanced Institute of Science and Technology (KAIST), KoreaReviewed by:
Kihoon Han, Korea University College of Medicine, South KoreaHyunsoo Shawn Je, Duke-NUS Medical School, Singapore
Copyright © 2017 Tian, Yang, Shang, Cai, Deng, Zhang, Shao, Zhu, Liu, Chen, Liang, Sun, Qiu and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jing Liang, bGlhbmdqQHBzeWNoLmFjLmNu Qiang Sun, cXN1bkBpb24uYWMuY24= Zilong Qiu, enFpdUBpb24uYWMuY24= Chen Zhang, Y2guemhhbmdAcGt1LmVkdS5jbg==
†These authors have contributed equally to this work.