 R. Michelle Saré
R. Michelle Saré Lee Harkless
Lee Harkless Carolyn B. Smith
Carolyn B. Smith- Section on Neuroadaptation and Protein Metabolism, Department of Health and Human Services, National Institute of Mental Health (NIMH), National Institutes of Health (NIH), Bethesda, MD, United States
In patients with fragile X syndrome (FXS), sleep problems are commonly observed but are not well characterized. In animal models of FXS (dfmr1 and Fmr1 knockout (KO)/Fxr2 heterozygote) circadian rhythmicity is affected, but sleep per se has not been examined. We used a home-cage monitoring system to assess total sleep time in both light and dark phases in Fmr1 KO mice at different developmental stages. Fmr1 KOs at P21 do not differ from controls, but genotype × phase interactions in both adult (P70 and P180) groups are statistically significant indicating that sleep in Fmr1 KOs is reduced selectively in the light phase compared to controls. Our results show the emergence of abnormal sleep in Fmr1 KOs during the later stages of brain maturation. Treatment of adult Fmr1 KO mice with a GABAB agonist, R-baclofen, did not restore sleep duration in the light phase. In adult (P70) Fmr1 KO/Fxr2 heterozygote animals, total sleep time was further reduced, once again in the light phase. Our data highlight the importance of the fragile X genes (Fmr1 and Fxr2) in sleep physiology and confirm the utility of these mouse models in enhancing our understanding of sleep disorders in FXS.
Introduction
Fragile X syndrome (FXS) is an X-linked disorder caused by a CGG repeat expansion in the 5′UTR of FMR1 resulting in gene silencing. FXS is a major inherited cause of intellectual disability and is also associated with autistic-like behaviors. Sleep abnormalities are a common symptom reported in patients with FXS (Picchioni et al., 2014) and are correlated with the severity of the behavioral phenotypes of the disorder (Kronk et al., 2010). The exact nature of sleep abnormalities is not well understood. The studies reporting sleep characteristics in FXS patients are confounded by the large age range of subjects studied, the variety of methods by which sleep is assessed, and medications used in the patients studied (Musumeci et al., 1995; Gould et al., 2000; Miano et al., 2008; Kronk et al., 2010).
Animal models of FXS provide a system in which many of the confounds of clinical studies can be avoided. Moreover, preclinical studies in animal models are critical to the investigation of efficacy of pharmacological interventions. To date, studies in animal models have focused more on circadian rhythm abnormalities than sleep deficiencies. In Drosophila models of FXS (dfmr1), an absence of circadian rhythm has been demonstrated (Dockendorff et al., 2002; Inoue et al., 2002; Sekine et al., 2008). In addition, two studies of the dmfr1 model have shown that sleep is also dysregulated (Bushey et al., 2009; van Alphen et al., 2013). In mice, Fmr1 deletion alone (Fmr1 knockout, KO) results in a slightly shorter period length measured in free running mice housed in constant darkness (Zhang et al., 2008). In contrast to Drosophila, mammals also express Fmr1 paralogs, Fxr1 and Fxr2; both paralogs can interact with Fmr1 (Zhang et al., 1995). Mice with mutations in both Fmr1 and Fxr2 have exaggerated behavioral phenotypes (Spencer et al., 2006) and a loss of circadian rhythm (Zhang et al., 2008). Sleep in these mice, however, has not been characterized.
Here, we report results of our studies of sleep in Fmr1 KO mice. We studied mice at three ages, P21, P70 and P180, to determine the developmental course of sleep deficiencies. We also examined sleep in Fmr1 KO/Fxr2 Heterozygous (Het) animals at P70 to determine if the phenotype was made worse by the addition of an Fxr2 mutation. We assessed sleep by means of a non-invasive home-cage monitoring-based system (Pack et al., 2007). Our results suggest that sleep disturbances increase over the lifecycle of Fmr1 KO mice. At weaning (P21), total sleep time was not affected. At P70, Fmr1 KO mice had reduced sleep in the light phase compared to controls. This phenotype persisted at P180 and was not rescued by treatment with a GABAB agonist, R-baclofen. Additionally, P70 Fmr1 KO/Fxr2 Het animals had a further decrease in sleep in the light phase compared to Fmr1 single mutants. These findings highlight the utility of Fmr1 KO mice to understand sleep in FXS.
Materials and Methods
Animals
All mice were group housed in a standard housing environment with up to five mice per cage (except during sleep analysis) in a climate-controlled central facility with a 12:12 h (6:00 AM–6:00 PM) light:dark environment. Food and water were available to mice ad libitum. All procedures were carried out in accordance with the National Institutes of Health Guidelines on the Care and Use of Animals and approved by the National Institute of Mental Health Animal Care and Use Committee.
Fmr1 KO Breeding
These studies were conducted on male Fmr1 hemizygous KO animals (Fmr1 KO) and control littermates (on a C57BL/6J background), generated in house through Het female and WT male breeding pairs. Genotyping of mouse tail DNA by PCR amplification was previously described (Qin et al., 2002). In separate groups of animals, studies were initiated at 20–22 days of age (P21), 60–80 days of age (P70), or 170–190 days of age (P180).
Fmr1 KO/Fxr2 Het Breeding
These studies were conducted on male Fmr1 hemizygous KO animals, Fxr2+/+ (Fmr1 KO/Fxr2 WT) and Fmr1 hemizygous KO animals, Fxr2+/− (Fmr1 KO/Fxr2 Het) on a C57BL/6J background. Studies were conducted at 60–80 days of age. These mice were generated from female Fmr1−/−, Fxr2+/− and male Fmr1 hemizygous, Fxr2+/− breeder pairs kindly provided by David Nelson (Baylor College of Medicine). The following primers were used to genotype Fxr2: (1) 5′-GTG ACA GTT TCC TGC TTT ACA GTC C; (2) 5′-TCT GCC TGC TTC CTG AGT GTT G; and (3) 5′-CGC CTT CTA TCG CCT TCT TGA C. Cycling conditions were as follows: 94°C for 5 min, (94°C for 60 s 54°C for 30 s and 72°C for 45 s) × 30 cycles, and 72°C for 7 min.
Home-Cage Assessment of Sleep
Sleep was assessed by home-cage activity monitoring. Mice were singly housed in a clean standard cage surrounded by a rectangular arena of oppositely positioned infrared emitters and sensors (Comprehensive Laboratory Animal Monitoring System (CLAMS); Columbus Instruments, Columbus, OH, USA). Photobeams were spaced 0.5 inches apart on the x and y planes to assess activity on a high-resolution grid. The CLAMS software discriminated between fine movements (multiple breaks of the same beam) and locomotor activity (breaking two adjacent beams). For the analysis, the sums of fine and locomotor activities were used. The CLAMS software detected beam breaks in 10 s epochs. A mouse was considered inactive if there was no xy movement over the 10 s epoch, and 40 s of such inactivity was recorded as sleep. Validation of these measures as indicators of sleep in C57BL/6J mice was reported previously (Pack et al., 2007). The total amount of time asleep was separated into light phase (time asleep between 6:00 AM and 6:00 PM) and dark phase (time asleep between 6:00 PM and 6:00 AM) and then recorded as a percentage of the 12 h total time per phase. Sleep was analyzed for each 24 h period. For Fmr1 KO mice, sleep was analyzed for six consecutive days. For Fmr1 KO/Fxr2 Het mice, sleep was analyzed for three consecutive days.
R-Baclofen Treatment
In a separate group of animals, we used 18 Fmr1 KO animals at 6 months of age to assess sleep duration prior to and during R-baclofen treatment. Mice were given saline injections, i.p., at 6:00 AM for 9 days. Sleep was assessed during the last 4 days of saline injections. R-baclofen was obtained from Seaside Therapeutics (Cambridge, MA, USA), dissolved in saline, and administered at 1.5 mg/kg i.p. at 6:00 AM for 2 days following the 9 days of saline injections (Days 10–11). The average sleep durations in the light and dark phases during saline injections were compared (Days 7–9) with sleep durations during R-baclofen injections (Days 10–11).
Statistical Analysis
Data were analyzed by means of a mixed model repeated measures (RM) ANOVA. The between subjects’ variable was genotype. The within subjects’ variables were day and phase (light, dark). A criterion of p ≤ 0.05 was considered to be statistically significant. These results are indicated with an “*”. We compared sleep duration during saline injections with sleep duration during R-baclofen injections by means of a paired t-test.
Results
Habituation to Home-Cage Monitoring
Recording commenced as soon as animals were housed in monitoring cages and continued for 6 days. We assessed the effect of day (habituation effect) on sleep in control and Fmr1 KO mice at P21, P70 and P180. At P21, neither the day × phase nor the genotype × phase interaction was statistically significant. Furthermore, neither the main effect of day nor genotype was statistically significant. The main effect of phase was statistically significant. As expected in nocturnal animals, percent sleep time was longer in the light phase. Sleep was stable across the 6-day recording period and did not differ by genotype (Figure 1A). At P70, the day × phase interaction was statistically significant (p < 0.001). Post hoc t-tests revealed that sleep duration on Day 1 differed from Days 2–6 in the light phase only. The genotype × phase interaction was also statistically significant (p = 0.002; Figure 1B), indicating that Fmr1 KO mice had a shorter sleep duration in the light phase. At P180, the day × phase interaction was statistically significant (p < 0.001). Post hoc t-tests revealed that Day 1 was different from all other days only in the light phase. The genotype × phase interaction was also statistically significant (p = 0.004; Figure 1C), indicating that the Fmr1 KO mice had a shorter sleep duration in the light phase. These results show that habituation to the housing condition occurred during the first 24 h period, particularly in the adult animals, and that habituation was similar in both control and Fmr1 KO mice.
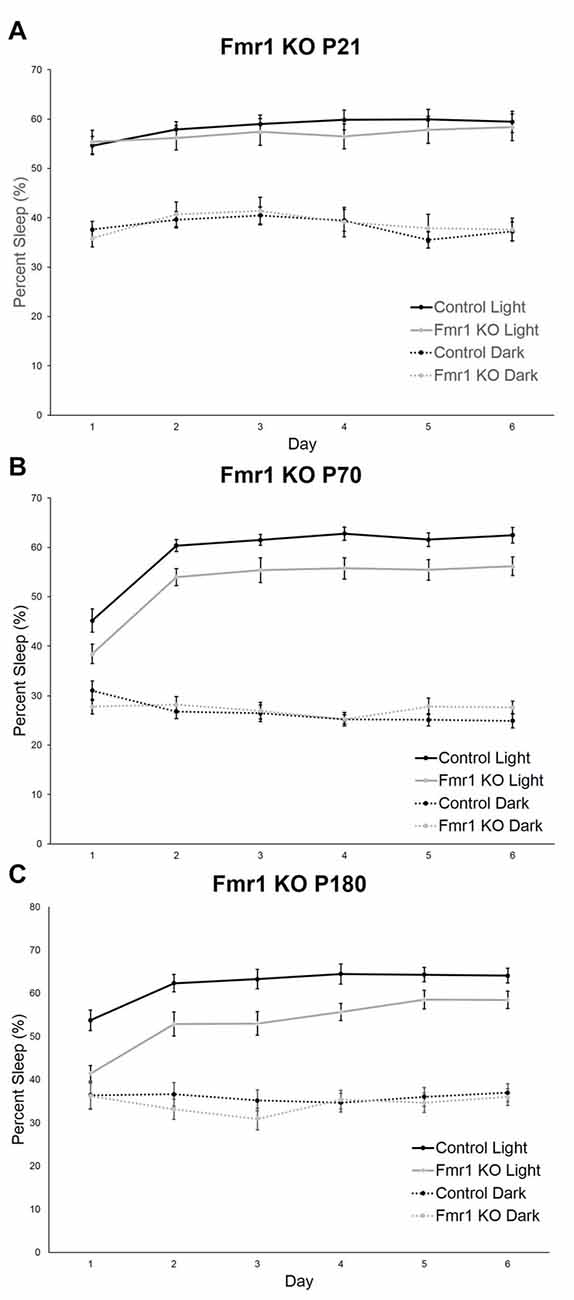
Figure 1. Habituation effect in control and Fmr1 knockout (KO) mice across the 6-day testing period in the light and dark phases. Points are the means ± standard error of the mean (SEM). (A) At P21, there were no differences in genotype or in day in either phase. (B) At P70, the day × phase interaction was statistically significant (p < 0.001). Post hoc t-tests indicate that Day 1 differed from all other days in the light phase only. This habituation was not affected by genotype. (C) At P180, the day × phase interaction was statistically significant (p < 0.001). Post hoc t-tests indicate that Day 1 differed from all other days in the light phase only. This was not affected by genotype.
Developmental Course of Sleep Deficiencies in Fmr1 KO Mice
The genotype × phase interaction in P70 and P180 animals indicates that there are differences between the genotypes in sleep time that depend on phase. To eliminate the effect of habituation, we confirmed these effects by analyzing the genotype × phase effects on average sleep time over Days 2–6.
We assessed sleep in juvenile control (n = 19) and Fmr1 KO (n = 23) mice at P21. At this age, neither the main effect of genotype nor the genotype × phase interaction were statistically significant (Table 1). Mean percent times asleep were similar for both genotypes in both phases (Figure 2A).
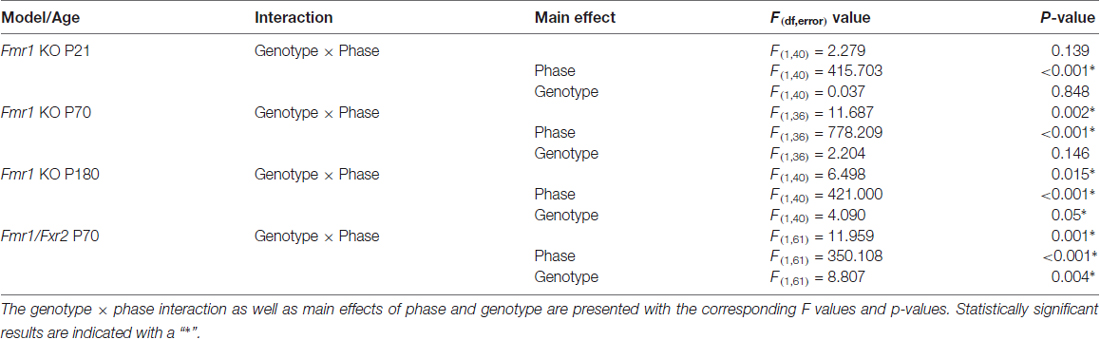
Table 1. Post hoc ANOVA results of average sleep times across Days 2–6 for the models presented.
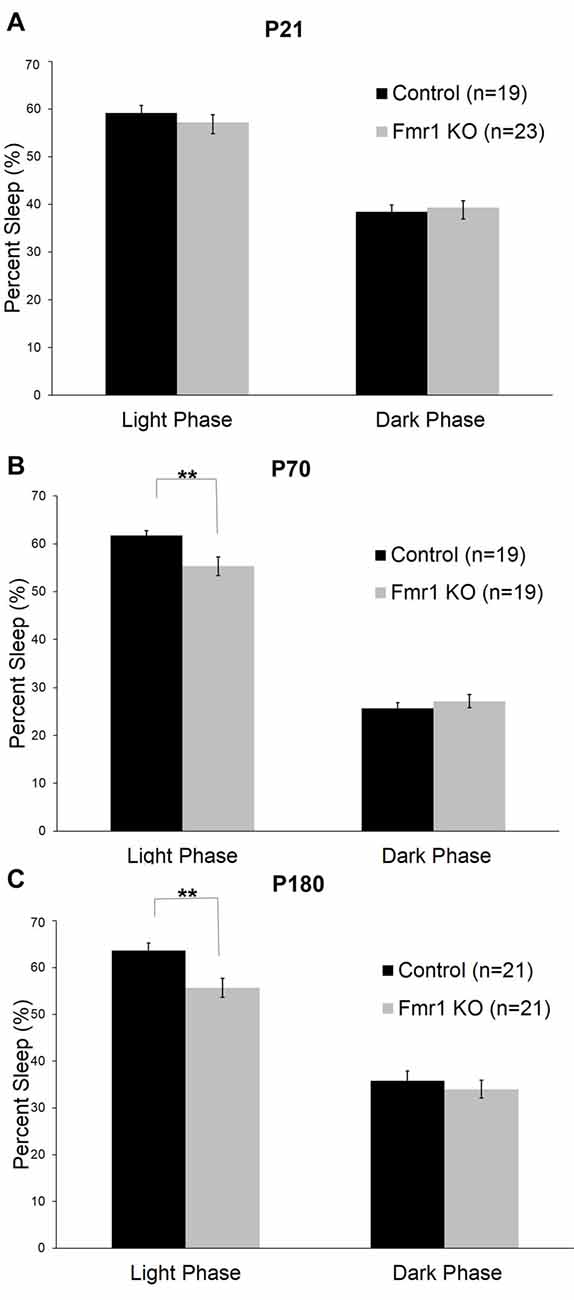
Figure 2. Sleep duration in control and Fmr1 KO mice in light and dark phases. Bars are the means ± SEM of sleep averaged across Days 2–6 of the number of animals indicated in parentheses. For each variable, full results of repeated measures (RM) ANOVA are reported in Table 1. **Denotes p < 0.01. (A) At P21, there were no differences between the genotypes in sleep duration in light and dark phases. (B) At P70, the genotype × phase interaction was statistically significant (p < 0.001). Post hoc t-tests indicate that Fmr1 KO (n = 19) animals had less sleep than controls (n = 19) in the light phase (p = 0.005). (C) At P180, the genotype × phase interaction was statistically significant (p = 0.015). Post hoc t-tests indicate that Fmr1 KO animals (n = 21) than controls (n = 21) in the light phase (p = 0.004).
At P70 (young adult; Figure 2B), the phase × genotype interaction was statistically significant (p < 0.001; Table 1). Post hoc t-tests indicate that Fmr1 KO mice (n = 19) had significantly (p = 0.007) reduced sleep only in the light phase compared to controls (n = 19). Mean differences were 6.39%.
At P180 (adult), the phase × genotype interaction was statistically significant (p = 0.015; Table 1; Figure 2C). Post hoc t-tests indicate that Fmr1 KO mice (n = 21) slept less than controls (n = 21) in the light phase only (p = 0.004). Mean differences were 8.0%.
We asked if the sleep deficits in Fmr1 KO mice at P180 could be reversed by treatment with R-baclofen, a GABAB agonist. R-baclofen treatment reverses other behavioral and physiological phenotypes in adult Fmr1 KO mice (Henderson et al., 2012; Qin et al., 2015a). We administered R-baclofen by daily i.p. injections (1.5 mg/kg). We used a within subjects’ design, i.e., sleep behavior was monitored in mice during daily i.p. injections of normal saline for 3 days followed by 2 days of daily R-baclofen i.p. injections. Prior to sleep monitoring, mice were acclimated to daily i.p. injections of saline for 6 days and 1 day of acclimation to the home-cage monitoring system. The sleep deficit in the light phase was not reversed by treatment with R-baclofen (57.7% sleep in the light phase during saline injections compared to 58.7% sleep during R-baclofen injections; p = 0.67, paired t-test). Moreover, R-baclofen did not affect sleep duration in the dark phase.
Sleep Deficiencies in Fmr1/Fxr2 Mice: Effects of Additional Fxr Deletion
We asked if the Fxr2 paralog was involved in sleep regulation in Fmr1 KO mice. The absence of Fxr2 in Fmr1 KO mice exacerbates circadian rhythm abnormalities (Zhang et al., 2008). To see if this role of Fxr2 in circadian rhythm extends to sleep, we studied Fmr1 KO mice with (Fmr1 KO/Fxr2 WT) or haploinsufficient (Fmr1 KO/Fxr2 Het) for Fxr2 at P70. We found that the phase × genotype interaction was statistically significant (p = 0.001; Table 1). In the light phase, Fmr1 KO/Fxr2 Het animals slept significantly less than Fmr1 KO/Fxr2 WT animals (p < 0.001; mean difference of 10%), but in the dark phase percent times were similar for both genotypes (Figure 3).
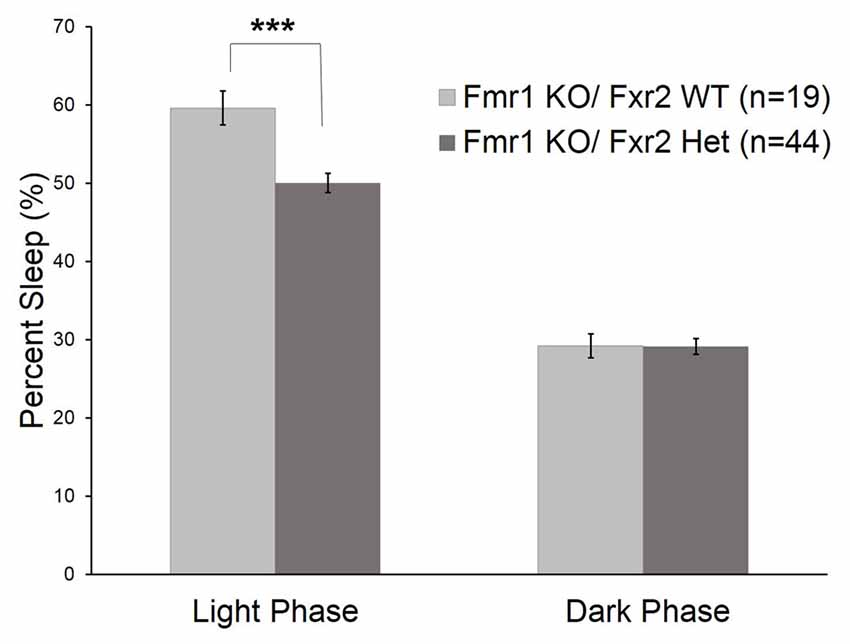
Figure 3. Sleep duration in Fmr1 KO/Fxr2 WT (n = 19) and Fmr1 KO/Fxr2 Heterozygous (Het; n = 44) mice at P70. Bars are the means ± SEM of sleep averaged across Days 2–3 on the number of animals indicated in parentheses. Full results of RM ANOVA are reported in Table 1. The genotype × phase interaction was statistically significant (p = 0.001), indicating that total sleep time in Fmr1 KO/Fxr2 Het animals was reduced compared with Fmr1 KO/Fxr2 WT animals in the light phase (p < 0.001). ***p < 0.001.
Discussion
The results of our study show that Fmr1 expression plays a role in the regulation of sleep physiology, and that its influence becomes apparent in adulthood. Additionally, Fxr2, an Fmr1 paralog, appears to have a further influence on sleep physiology in mice. Our data highlight sleep physiology as an important phenotype in FXS that needs further characterization in patients. Moreover, abundant data support the importance of sleep in behavior and brain function (Picchioni et al., 2014; Kreutzmann et al., 2015; Saré et al., 2016a). Accordingly, correction of sleep abnormalities in FXS patients offers a promising therapeutic strategy. The effects of such therapies on sleep and ultimately on behavioral outcomes can be tested in FXS mouse models.
There are several strengths to our current studies, as well as a few limitations. First, we conducted a cross-sectional study to investigate sleep across development. We had good statistical power for our analysis. We controlled for several of the variables problematic in human clinical studies. Our animals were well matched for age; we used the same measure of sleep across all studies; all animals had not had any previous exposure to drugs. However, because of the nature of our study, we only have information about total sleep time, and we cannot measure sleep stages or sleep bout duration, which could inform us whether sleep fragmentation was occurring. For these questions, an electroencephalogram (EEG) study would be informative.
Hyperactivity is one of the common phenotypes detected in the Fmr1 KO mouse model in both the active (Saré et al., 2016b) and inactive phases (Liu et al., 2011). Although hyperactivity and reduced sleep might be mediated by a similar mechanism, it is important to note that we are not detecting hyperactivity, per se. First, hyperactivity is traditionally assessed in a novel open-field environment larger than a typical mouse home-cage, whereas we are assessing sleep in the home-cage. Both tests use beam breaks to detect movement of the animal, but the criteria are different. A continuous measure of the number of beam breaks is used to measure activity in the open field. In the home-cage monitoring system, an animal is considered awake if it breaks a single beam in a 40 s epoch or if it breaks numerous beams in a 40 s epoch. Hyperactivity and decreased sleep duration may go hand in hand, but it is also possible that a hyperactive animal has the same number or even fewer awake epochs than a more sedentary animal.
Our finding of decreased sleep time in the light phase in Fmr1 KO mice contrasts with results in the dfmr1 model, in which sleep duration was increased (Inoue et al., 2002; Bushey et al., 2009). These phenotypic differences could reflect the absence of both Fmr1 paralogs, Fxr1 and Fxr2, in flies. In our study, both Fmr1 KO and Fmr1 KO /Fxr2 Het mice had decreased sleep time in the light phase. The effects of loss of Fxr1 in mice could not be tested due to the poor viability of Fxr1 KO mice (Mientjes et al., 2004). Reduced sleep duration only in the light phase (the animal’s inactive phase) suggests that the sleep effect is modulated by circadian rhythms, and that sleep deficiency and circadian rhythm disruption are linked in Fmr1 KO animals. These results align with the circadian rhythm disruption previously reported in Fmr1 KO animals (Zhang et al., 2008).
Although we did not detect sleep deficiencies in Fmr1 KO mice at P21 by activity monitoring, there is electrophysiological and calcium imaging evidence that cortical neuronal firing and synchrony during sleep are abnormally high in Fmr1 KO mice at P14–P16 suggesting that Up/Down states are not normal (Gonçalves et al., 2013). How this may progress into circadian alterations and reduced sleep in the light phase is unknown. However, circadian rhythm in P21 animals is not as defined as in adult animals (Hagenauer et al., 2009), which may mask potential differences between genotypes. Additionally, at P21, the mouse brain is still developing, and is thought to be somewhat equivalent to a human of around 3 years old. By P70, the mouse brain is mature and equivalent to about a 20 year old human (Semple et al., 2013). It is during this period from P21 to P70 that Fmr1 KO mice develop a statistically significant genotype × phase interaction, suggestive of a circadian rhythm disruption, implying that this abnormality unfolds during brain maturation. It is interesting to note that most behavioral abnormalities reported in Fmr1 KO mice have been assessed at 2 months of age and later (Yan et al., 2004; Spencer et al., 2005; Zhao et al., 2005; Moon et al., 2006; Liu and Smith, 2009; Liu et al., 2011; Ding et al., 2014; Qin et al., 2015a,b), suggesting that sleep/circadian rhythm problems may develop before other behavioral impairments. The timing of the development of sleep abnormalities in the mouse may inform the timing of screening for sleep problems in FXS children. It also may help to determine the best window for treatment.
Although the differences in total sleep time between Fmr1 KO and control mice are relatively small (6% in the light phase in P70 animals and 8% in the light phase in P180 animals), these differences may very well be biologically significant. Sleep has an important role in brain development and plasticity (Picchioni et al., 2014; Kreutzmann et al., 2015). Studies of chronic partial sleep loss have revealed that deficits are similar to those observed in acute total sleep deprivation. These deficits were in areas of cognition and neurobehavioral function (Van Dongen et al., 2003). Chronic sleep restriction in mice leads to long-lasting effects on behavior, even after restoration of normal sleep (Saré et al., 2016a). These behavioral changes could be mediated by changes in plasticity in the brain that are not recovered during subsequent sleep periods. Studies have shown that cellular processes implicated in plasticity such as myelination, cellular stress and neurogenesis are affected by sleep restriction and may not recover even after regaining sleep (Tung et al., 2005; Picchioni et al., 2014; Kreutzmann et al., 2015). The consequences of chronically reduced sleep in the light phase in FXS may be an important contributor to the brain and behavioral manifestations of the disorder.
Given the potential impact of sleep deficits in the unfolding of fragile X phenotypes, it may be important to determine the mechanisms by which sleep is dysregulated in the Fmr1 KO mice. There are two processes controlling the drive to sleep. One is by means of the circadian clock and the other is a homeostatic drive (Borbély and Achermann, 1999). In their study of circadian rhythm, Zhang et al. examined the expression of clock genes involved in circadian rhythm in both Fmr1 KO and Fmr1/Fxr2 double Het animals. They found that both models showed rhythmicity in the clock genes in the superchiasmatic nucleus (SCN). However, Fmr1 KO/Fxr2 Het animals did show increased expression of Cry1 at the beginning of the active phase (Zhang et al., 2008). Given that both Fmr1 KO and Fmr1 KO/Fxr2 Het animals show decreased sleep, this mechanism is unlikely to account for the change in sleep duration. Downstream of the SCN, in the liver which is noted as the peripheral clock, regulation of Bmal1, mPer1, mPer2 and Npas2 in the Fmr1/Fxr2 Het animals was altered relative to controls (Zhang et al., 2008). Again, as these changes did not occur in Fmr1 KO mice, they cannot fully explain the reduced sleep phenotype. Both Fmr1 KO and Fmr1/Fxr2 Het animals did have increased expression of Cry1 in the liver at the beginning of the active phase (Zhang et al., 2008), so it is possible that Cry1 regulation may contribute to the reduced sleep in both Fmr1 KO and Fmr1 KO/Fxr2 Het animals.
The other process controlling sleep is the homeostatic drive. Understanding the molecular mechanisms that manage the homeostatic regulation of sleep in Fmr1 KO mice is much more difficult because the process is less understood. One mechanism of sleep initiation, particularly nonREM sleep, is activation of GABA receptors (Lancel, 1999). It has been shown that Fmr1 KO mice have downregulation of both GABAA and GABAB receptors (Pacey et al., 2011; Liu et al., 2013). In our study, R-baclofen, a GABAB agonist, did not improve sleep in adult Fmr1 KO mice. We controlled for effects of i.p. injections and acclimation to the monitoring system on sleep duration. We only tested mice at 6 months of age, and it is possible that the treatment might be effective in younger mice. Based on our results, we think it unlikely that GABAB receptors are involved in the sleep deficits observed in Fmr1 KO and Fmr1 KO/Fxr2 Het animals. Future work will address the role of GABAA receptors in mediating the sleep deficits in Fmr1 KO and Fmr1 KO/Fxr2 Het animals.
Our findings highlight the importance of Fmr1 and Fxr2 in the regulation of sleep in adult mice. With loss of Fmr1 (either alone or in combination with Fxr2), adult animals have reduced total sleep time in the light phase. Our data in conjunction with clinical reports (Musumeci et al., 1995; Gould et al., 2000) suggest that patients without FMR1 expression are likely to have chronically reduced night-time sleep. Our findings in Fmr1 KO mice suggest that sleep problems (such as reduced sleep) should be more thoroughly examined in FXS patients and considered as targets for therapeutic intervention. Additionally, these studies show that Fmr1 KO mice and Fmr1 KO/Fxr2 Het mice may be useful for further examining the consequences and potential treatments for the sleep problems in FXS.
Author Contributions
RMS designed the study, performed experiments, analyzed data, interpreted the data and drafted the manuscript; LH performed experiments; ML performed experiments and drafted the manuscript; AT performed experiments; CAS performed experiments; CBS designed the study, interpreted the data, and drafted the manuscript. All authors read and approved the final version of the manuscript.
Funding
This research was supported by the Intramural Research Program of the National Institute of Mental Health (NIMH) ZIA MH000889-36 DIRP. RMS was also supported by an Autism Speaks Postdoctoral Fellowship 8679 and a FRAXA Postdoctoral Fellowship.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Zengyan Xia for the genotyping of the mice. We would also like to thank David Nelson for providing the breeding pairs for the Fmr1 KO/Fxr2 Het mice. Finally, we also thank the NIH Fellows Editorial Board for editorial assistance.
Abbreviations
CLAMS, comprehensive laboratory animal monitoring system; FXS, Fragile X syndrome; SEM, standard error of the mean.
References
Borbély, A. A., and Achermann, P. (1999). Sleep homeostasis and models of sleep regulation. J. Biol. Rhythms. 14, 557–568.
Bushey, D., Tononi, G., and Cirelli, C. (2009). The Drosophila fragile X mental retardation gene regulates sleep need. J. Neurosci. 29, 1948–1961. doi: 10.1523/JNEUROSCI.4830-08.2009
Ding, Q., Sethna, F., and Wang, H. (2014). Behavioral analysis of male and female Fmr1 knockout mice on C57BL/6 background. Behav. Brain Res. 271, 72–78. doi: 10.1016/j.bbr.2014.05.046
Dockendorff, T. C., Su, H. S., McBride, S. M., Yang, Z., Choi, C. H., Siwicki, K. K., et al. (2002). Drosophila lacking dfmr1 activity show defects in circadian output and fail to maintain courtship interest. Neuron 34, 973–984. doi: 10.1016/s0896-6273(02)00724-9
Gonçalves, J. T., Anstey, J. E., Golshani, P., and Portera-Cailliau, C. (2013). Circuit level defects in the developing neocortex of Fragile X mice. Nat. Neurosci. 16, 903–909. doi: 10.1038/nn.3415
Gould, E. L., Loesch, D. Z., Martin, M. J., Hagerman, R. J., Armstrong, S. M., and Huggins, R. M. (2000). Melatonin profiles and sleep characteristics in boys with fragile X syndrome: a preliminary study. Am. J. Med. Genet. 95, 307–315. doi: 10.1002/1096-8628(20001211)95:4<307::aid-ajmg3>3.3.co;2-v
Hagenauer, M. H., Perryman, J. I., Lee, T. M., and Carskadon, M. A. (2009). Adolescent changes in the homeostatic and circadian regulation of sleep. Dev. Neurosci. 31, 276–284. doi: 10.1159/000216538
Henderson, C., Wijetunge, L., Kinoshita, M. N., Shumway, M., Hammond, R. S., Postma, F. R., et al. (2012). Reversal of disease-related pathologies in the fragile X mouse model by selective activation of GABAB receptors with arbaclofen. Sci. Transl. Med. 4:152ra128. doi: 10.1126/scitranslmed.3004218
Inoue, S., Shimoda, M., Nishinokubi, I., Siomi, M. C., Okamura, M., Nakamura, A., et al. (2002). A role for the Drosophila fragile X-related gene in circadian output. Curr. Biol. 12, 1331–1335. doi: 10.1016/s0960-9822(02)01036-9
Kreutzmann, J. C., Havekes, R., Abel, T., and Meerlo, P. (2015). Sleep deprivation and hippocampal vulnerability: changes in neuronal plasticity, neurogenesis and cognitive function. Neuroscience 309, 173–190. doi: 10.1016/j.neuroscience.2015.04.053
Kronk, R., Bishop, E. E., Raspa, M., Bickel, J. O., Mandel, D. A., and Bailey, D. B. Jr. (2010). Prevalence, nature, and correlates of sleep problems among children with fragile X syndrome based on a large scale parent survey. Sleep 33, 679–687. doi: 10.1093/sleep/33.5.679
Lancel, M. (1999). Role of GABAA receptors in the regulation of sleep: initial sleep responses to peripherally administered modulators and agonists. Sleep 22, 33–42. doi: 10.1093/sleep/22.1.33
Liu, Z. H., Chuang, D. M., and Smith, C. B. (2011). Lithium ameliorates phenotypic deficits in a mouse model of fragile X syndrome. Int. J. Neuropsychopharmacol. 14, 618–630. doi: 10.1017/s1461145710000520
Liu, B., Li, L., Chen, J., Wang, Z., Li, Z., and Wan, Q. (2013). Regulation of GABAA receptors by fragile X mental retardation protein. Int. J. Physiol. Pathophysiol. Pharmacol. 5, 169–176.
Liu, Z. H., and Smith, C. B. (2009). Dissociation of social and nonsocial anxiety in a mouse model of fragile X syndrome. Neurosci. Lett. 454, 62–66. doi: 10.1016/j.neulet.2009.02.066
Miano, S., Bruni, O., Elia, M., Scifo, L., Smerieri, A., Trovato, A., et al. (2008). Sleep phenotypes of intellectual disability: a polysomnographic evaluation in subjects with Down syndrome and Fragile-X syndrome. Clin. Neurophysiol. 119, 1242–1247. doi: 10.1016/j.clinph.2008.03.004
Mientjes, E. J., Willemsen, R., Kirkpatrick, L. L., Nieuwenhuizen, I. M., Hoogeveen-Westerveld, M., Verweij, M., et al. (2004). Fxr1 knockout mice show a striated muscle phenotype: implications for Fxr1p function in vivo. Hum. Mol. Genet. 13, 1291–1302. doi: 10.1093/hmg/ddh150
Moon, J., Beaudin, A. E., Verosky, S., Driscoll, L. L., Weiskopf, M., Levitsky, D. A., et al. (2006). Attentional dysfunction, impulsivity, and resistance to change in a mouse model of fragile X syndrome. Behav. Neurosci. 120, 1367–1379. doi: 10.1037/0735-7044.120.6.1367
Musumeci, S. A., Ferri, R., Elia, M., Dalgracco, S., Scuderi, C., Stefanini, M. C., et al. (1995). Sleep neurophyisology in fragile X syndrome patients. Dev. Brain Dysf. 8, 218–222.
Pacey, L. K., Tharmalingam, S., and Hampson, D. R. (2011). Subchronic administration and combination metabotropic glutamate and GABAB receptor drug therapy in fragile X syndrome. J. Pharmacol. Exp. Ther. 338, 897–905. doi: 10.1124/jpet.111.183327
Pack, A. I., Galante, R. J., Maislin, G., Cater, J., Metaxas, D., Lu, S., et al. (2007). Novel method for high-throughput phenotyping of sleep in mice. Physiol. Genomics 28, 232–238. doi: 10.1152/physiolgenomics.00139.2006
Picchioni, D., Reith, R. M., Nadel, J. L., and Smith, C. B. (2014). Sleep, plasticity and the pathophysiology of neurodevelopmental disorders: the potential roles of protein synthesis and other cellular processes. Brain Sci. 4, 150–201. doi: 10.3390/brainsci4010150
Qin, M., Huang, T., Kader, M., Krych, L., Xia, Z., Burlin, T., et al. (2015a). R-baclofen reverses a social behavior deficit and elevated protein synthesis in a mouse model of fragile X syndrome. Int. J. Neuropsychopharmacol. 18:pyv034. doi: 10.1093/ijnp/pyv034
Qin, M., Zeidler, Z., Moulton, K., Krych, L., Xia, Z., and Smith, C. B. (2015b). Endocannabinoid-mediated improvement on a test of aversive memory in a mouse model of fragile X syndrome. Behav. Brain Res. 291, 164–171. doi: 10.1016/j.bbr.2015.05.003
Qin, M., Kang, J., and Smith, C. B. (2002). Increased rates of cerebral glucose metabolism in a mouse model of fragile X mental retardation. Proc. Natl. Acad. Sci. U S A 99, 15758–15763. doi: 10.1073/pnas.242377399
Saré, R. M., Levine, M., Hildreth, C., Picchioni, D., and Smith, C. B. (2016a). Chronic sleep restriction during development can lead to long-lasting behavioral effects. Physiol. Behav. 155, 208–217. doi: 10.1016/j.physbeh.2015.12.019
Saré, R. M., Levine, M., and Smith, C. B. (2016b). Behavioral phenotype of Fmr1 knock-out mice during active phase in an altered light/dark cycle. eNeuro 3:ENEURO.0035-16.2016. doi: 10.1523/ENEURO.0035-16.2016
Sekine, T., Yamaguchi, T., Hamano, K., Siomi, H., Saez, L., Ishida, N., et al. (2008). Circadian phenotypes of Drosophila fragile X mutants in alternative genetic backgrounds. Zoolog. Sci. 25, 561–571. doi: 10.2108/zsj.25.561
Semple, B. D., Blomgren, K., Gimlin, K., Ferriero, D. M., and Noble-Haeusslein, L. J. (2013). Brain development in rodents and humans: identifying benchmarks of maturation and vulnerability to injury across species. Prog. Neurobiol. 106–107, 1–16. doi: 10.1016/j.pneurobio.2013.04.001
Spencer, C. M., Alekseyenko, O., Serysheva, E., Yuva-Paylor, L. A., and Paylor, R. (2005). Altered anxiety-related and social behaviors in the Fmr1 knockout mouse model of fragile X syndrome. Genes Brain Behav. 4, 420–430. doi: 10.1111/j.1601-183x.2005.00123.x
Spencer, C. M., Serysheva, E., Yuva-Paylor, L. A., Oostra, B. A., Nelson, D. L., and Paylor, R. (2006). Exaggerated behavioral phenotypes in Fmr1/Fxr2 double knockout mice reveal a functional genetic interaction between Fragile X-related proteins. Hum. Mol. Genet. 15, 1984–1994. doi: 10.1093/hmg/ddl121
Tung, A., Takase, L., Fornal, C., and Jacobs, B. (2005). Effects of sleep deprivation and recovery sleep upon cell proliferation in adult rat dentate gyrus. Neuroscience 134, 721–723. doi: 10.1016/j.neuroscience.2005.06.008
van Alphen, B., Yap, M. H., Kirszenblat, L., Kottler, B., and van Swinderen, B. (2013). A dynamic deep sleep stage in Drosophila. J. Neurosci. 33, 6917–6927. doi: 10.1523/JNEUROSCI.0061-13.2013
Van Dongen, H. P., Maislin, G., Mullington, J. M., and Dinges, D. F. (2003). The cumulative cost of additional wakefulness: dose-response effects on neurobehavioral functions and sleep physiology from chronic sleep restriction and total sleep deprivation. Sleep 26, 117–126. doi: 10.1093/sleep/26.2.117
Yan, Q. J., Asafo-Adjei, P. K., Arnold, H. M., Brown, R. E., and Bauchwitz, R. P. (2004). A phenotypic and molecular characterization of the fmr1-tm1Cgr fragile X mouse. Genes Brain Behav. 3, 337–359. doi: 10.1111/j.1601-183x.2004.00087.x
Zhang, J., Fang, Z., Jud, C., Vansteensel, M. J., Kaasik, K., Lee, C. C., et al. (2008). Fragile X-related proteins regulate mammalian circadian behavioral rhythms. Am. J. Hum. Genet. 83, 43–52. doi: 10.1016/j.ajhg.2008.06.003
Zhang, Y., O’Connor, J. P., Siomi, M. C., Srinivasan, S., Dutra, A., Nussbaum, R. L., et al. (1995). The fragile X mental retardation syndrome protein interacts with novel homologs FXR1 and FXR2. EMBO J. 14, 5358–5366.
Keywords: sleep disruption, home-cage monitoring system, Fragile X, Fmr1, Fxr2
Citation: Saré RM, Harkless L, Levine M, Torossian A, Sheeler CA and Smith CB (2017) Deficient Sleep in Mouse Models of Fragile X Syndrome. Front. Mol. Neurosci. 10:280. doi: 10.3389/fnmol.2017.00280
Received: 23 May 2017; Accepted: 18 August 2017;
Published: 01 September 2017.
Edited by:
Regina Dahlhaus, University of Erlangen-Nuremberg, GermanyReviewed by:
Norio K. Ishida, National Institute of Advanced Industrial Science and Technology, JapanHansen Wang, University of Toronto, Canada
Copyright © 2017 Saré, Harkless, Levine, Torossian, Sheeler and Smith. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carolyn B. Smith, YmVlYmVAbWFpbC5uaWguZ292