 Destiny-Love Manecka
Destiny-Love Manecka Benoît Vanderperre
Benoît Vanderperre Edward A. Fon
Edward A. Fon Thomas M. Durcan
Thomas M. Durcan- Neurodegenerative Diseases Group and iPSC-CRISPR Core Facility, Montreal Neurological Institute, McGill University, Montreal, QC, Canada
Synucleinopathies are a family of neurodegenerative disorders that comprises Parkinson’s disease, dementia with Lewy bodies, and multiple system atrophy. Each of these disorders is characterized by devastating motor, cognitive, and autonomic consequences. Current treatments for synucleinopathies are not curative and are limited to improvement of quality of life for affected individuals. Although the underlying causes of these diseases are unknown, a shared pathological hallmark is the presence of proteinaceous inclusions containing the α-synuclein (α-syn) protein in brain tissue. In the past few years, it has been proposed that these inclusions arise from the self-templated, prion-like spreading of misfolded and aggregated forms of α-syn throughout the brain, leading to neuronal dysfunction and death. In this review, we describe how impaired protein homeostasis is a prominent factor in the α-syn aggregation cascade, with alterations in protein quality control (PQC) pathways observed in the brains of patients. We discuss how PQC modulates α-syn accumulation, misfolding and aggregation primarily through chaperoning activity, proteasomal degradation, and lysosome-mediated degradation. Finally, we provide an overview of experimental data indicating that targeting PQC pathways is a promising avenue to explore in the design of novel neuroprotective approaches that could impede the spreading of α-syn pathology and thus provide a curative treatment for synucleinopathies.
Introduction
Maintaining protein homeostasis is essential for normal cellular function and viability. This is overseen by PQC mechanisms, through the control of protein synthesis, localization, folding/refolding, degradation and formation of protein inclusions. At the post-translational level, PQC is orchestrated by several mechanisms including chaperones that maintain correct protein conformation or help refold misfolded proteins; and the UPS and ALP, which degrade proteins that are irreversibly misfolded, damaged, or are no longer required by the cell. In this review, we focus on these aspects of PQC; with other PQC pathways reviewed elsewhere (Wolff et al., 2014; Dubnikov et al., 2017). In eukaryotes, protein chaperones are essential for ensuring the correct folding of nascent proteins and refolding of misfolded proteins. Hsps or heat shock chaperones (Hscs) are a prominent group of chaperones and they can be found in the ER, mitochondria, cytoplasm or extracellular space (Hartl et al., 2011; Wyatt et al., 2013). Protein degradation through the UPS is regulated by the sequential activity of E1, E2, and E3 enzymes that conjugate primarily K48-linked ubiquitin (Ub) chains onto lysine residues in proteins destined for elimination through the 26S proteasome (Passmore and Barford, 2004). The ALP acts mainly through macroautophagy and CMA. In macroautophagy, cytoplasmic content (including soluble and aggregated proteins) is engulfed by a double-membrane to form an autophagosome that fuses with the lysosome forming an autolysosome, degrading the autophagosomal content (Bento et al., 2016). In CMA, Hsc70 specifically binds to and targets proteins containing KFERQ-like motifs to the lysosomal receptor Lamp2A for client import through the lysosomal membrane, and subsequent degradation by lysosomal hydrolases (Cuervo and Wong, 2014). In addition, chaperones and ubiquitination systems promote the spatial sequestration of misfolded proteins into inclusions (aggresome/Q-bodies) and mediate the lysosomal degradation of toxic aggregates through the aggresome–autophagy and multivesicular body pathways (Johnston et al., 1998; Sahu et al., 2011; Escusa-Toret et al., 2013; Sontag et al., 2017). These pathways are regulated by K63-linked Ub chains and are critical for the degradation of aggregated proteins including α-synuclein (α-syn) (Tanaka et al., 2004; Filimonenko et al., 2007; Tofaris et al., 2011). Inefficient PQC is implicated in protein toxicity, gain- or loss-of-function in many pathologies, including several neurodegenerative diseases known as synucleinopathies. Synucleinopathies, which include PD, LBD and MSA, are characterized by the pathologic accumulation and aggregation of α-syn (McCann et al., 2014). As some mutations altering PQC machinery are associated with familial forms of synucleinopathies and α-syn pathologic aggregates impair PQC, targeting the PQC machinery has become a promising therapeutic strategy for opposing the toxic effects of misfolded α-syn aggregates (Figure 1).
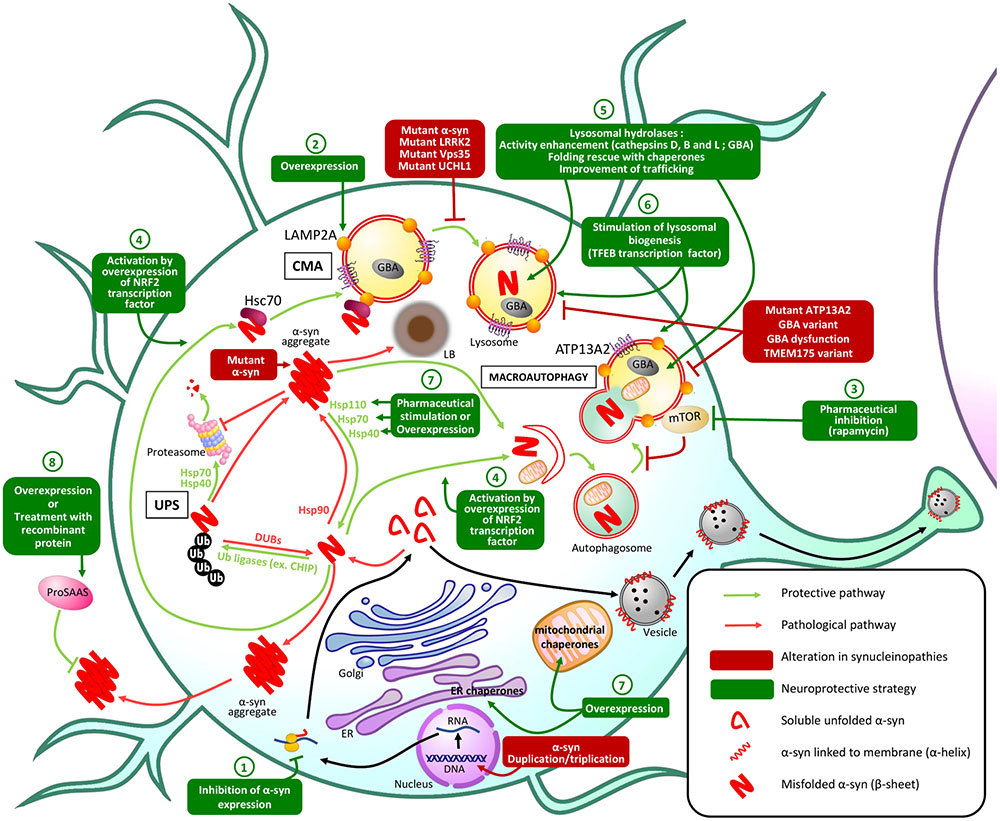
FIGURE 1. Principal PQC mechanisms involved in α-syn homeostasis and potential therapeutic approaches. In physiologic conditions, misfolded α-syn protein is degraded by PQC machinery: the UPS is responsible for ubiquitination of α-syn leading to proteasomal degradation of α-syn, while macroautophagy and CMA both lead to lysosomal degradation of misfolded α-syn. In synucleinopathies, alterations in these protective mechanisms result in the accumulation of misfolded α-syn in aggregates and Lewy bodies (LB) that lead to neuronal dysfunction and death. Genetic or pharmaceutical approaches to restore altered PQC pathways or stimulate alternative PQC pathways: (1) Inhibition of α-syn expression could prevent its pathological accumulation. (2) Overexpression of Lamp2A lysosomal receptor could increase the CMA of misfolded α-syn. (3) Pharmaceutical inhibition of mTOR, an autophagy receptor, stimulates macroautophagy preventing α-syn accumulation and aggregation. (4) Overexpression of the transcription factor NRF2 activates both macroautophagy and CMA and stimulates the lysosomal degradation of misfolded α-syn. (5) Improving lysosomal hydrolases activity or (6) stimulating lysosomal biogenesis could also enhance α-syn lysosomal degradation. α-syn aggregation can also be prevented by stimulation or overexpression of (7) endogenous or (8) secretory chaperones.
Misfolding and Spreading of α-syn in Synucleinopathies
α-syn, a 140-amino acid protein encoded by the SNCA gene, is abundant in the human brain (1% of all cytosolic proteins) (Stefanis, 2012) and is implicated in various cellular processes including vesicular trafficking, dopamine release and reuptake (Abeliovich et al., 2000; Senior et al., 2008; DeWitt and Rhoades, 2013; Burré et al., 2014). α-syn is structured into three domains: the N-terminal amphipathic domain, which allows membrane-binding; the central hydrophobic non-amyloid-β component (NAC) domain, essential for α-syn aggregation; and the acidic, negatively charged C-terminal domain, that is critical for chaperone-like activity during thermal and chemical stress (Souza et al., 2000; Ahn et al., 2006; Beyer, 2006). Under physiological conditions, α-syn is soluble and intrinsically disordered, or can adopt an N-terminal α-helix conformation with high affinity for biological membranes (Bartels et al., 2011). In synucleinopathies, α-syn follows sequential aggregation/fibrillization, starting from soluble monomers, dimers, and misfolded oligomers that aggregate into insoluble protofibrils and fibrils with an anti-parallel β-sheet structure (Dettmer et al., 2015; Rodriguez et al., 2015; Roeters et al., 2017).
Aggregation of α-syn leads to the formation of proteinaceous inclusions termed Lewy bodies (LB) and Lewy neurites (Wakabayashi et al., 1998). In PD, α-syn pathology has been shown to spread from brainstem to neocortex following a specific pattern (Braak et al., 2003). Recent evidence suggests that this is due to the prion-like, cell-to-cell propagation of α-syn aggregates (Masuda-Suzukake et al., 2013; Goedert et al., 2016). This concept was established in cells containing α-syn fibrils that secrete α-syn seeds taken up by surrounding healthy cells. In these recipient cells, exogenous protofibrils seed the aggregation of endogenous soluble α-syn monomers, causing α-syn to adopt an insoluble β-sheet conformation. This results in the formation of new α-syn seeds, which spread into neighboring cells (Volpicelli-Daley et al., 2011; Luk et al., 2012a,b; Mougenot et al., 2012).
Targeting PQC Defects as Potential Neuroprotective Strategies Against α-syn Pathology
Chaperones
The first indication that chaperones confers neuroprotection in α-syn–induced pathogenesis was Hsp70 overexpression protecting against α-syn toxicity in Drosophila (Auluck et al., 2002). Accordingly, modulating chaperone function through chemical or genetic approaches holds great therapeutic promise for synucleinopathies. Chaperones ensure the correct folding of nascent and mature protein chains (Ebrahimi-Fakhari et al., 2011; Sharma and Priya, 2017). They also prevent seeding of new aggregates and fibrillization by occluding surfaces that may serve as platforms to induce misfolding of native proteins (Hartl et al., 2011). To some extent, Hsp110, Hsp70 and Hsp40 chaperones can disassemble α-syn fibrillary aggregates in vitro (Duennwald et al., 2012; Gao et al., 2015). Enhancing disaggregase activity genetically or with pharmacological modulators could counteract α-syn aggregation (Gao et al., 2015; Jackrel and Shorter, 2015; Shorter, 2016; Sharma and Priya, 2017). Whether disaggregation occurs in vivo remains to be established, and since this process might generate soluble, potentially toxic forms of misfolded α-syn, simultaneous enhancement of α-syn degradation is likely necessary for beneficial effects.
Targeting Hsp70/Hsp90 signaling is of prime interest, not only in synucleinopathies, but also in other adult-onset proteinopathies (Pratt et al., 2015). These chaperones have opposing effects: Hsp90 stabilizes its clients, whereas Hsp70 directs them for proteasomal degradation upon Hsp90 dissociation. In yeast, cellular, or animal models of PD, inhibiting Hsp90 activity (Auluck and Bonini, 2002; Auluck et al., 2005; Putcha et al., 2010) or stimulating Hsp70 activity (Auluck et al., 2002; McLean et al., 2002; Klucken et al., 2004; Zhou et al., 2004, 2011; Shin et al., 2005; Yu et al., 2005; Batelli et al., 2008; Outeiro et al., 2008) and that of its collaborator Hsp40 (McLean et al., 2002; Fan et al., 2006) reduces α-syn oligomerization, inclusions formation, and toxicity, and diminishes α-syn levels (Table 1). Although induction of chaperone expression in various cellular locations during proteotoxic stress and its associated stress response is observed in the brains of patients affected by synucleinopathies, α-syn aggregates still accumulate, indicating that the chaperone machinery is overwhelmed. This is supported by findings that many chaperones (including Hsp70, Hsp90 and Hsp40) or mediators of the heat-shock response (HDAC6) are found in LBs (Table 1), possibly reflecting a cellular attempt to sequester soluble, harmful misfolded species of α-syn (Escusa-Toret et al., 2013). Other chaperones can also mitigate α-syn aggregation and toxicity in various models (see Table 1). Overall, it appears evident that modulation of chaperone function is an innovative therapeutic approach against α-syn toxicity. In a clinical context, where widespread α-syn aggregation has already occurred, a global increase in chaperoning activity such as stimulation of the heat-shock response (Du et al., 2014) might have a greater impact than manipulation of individual chaperones. The use of pharmacological chaperones (e.g., flavonoids or polyphenols, Caruana et al., 2011; Ren et al., 2016; Gautam et al., 2017) to prevent or revert α-syn aggregation may also complement therapeutic modulation of endogenous chaperones.
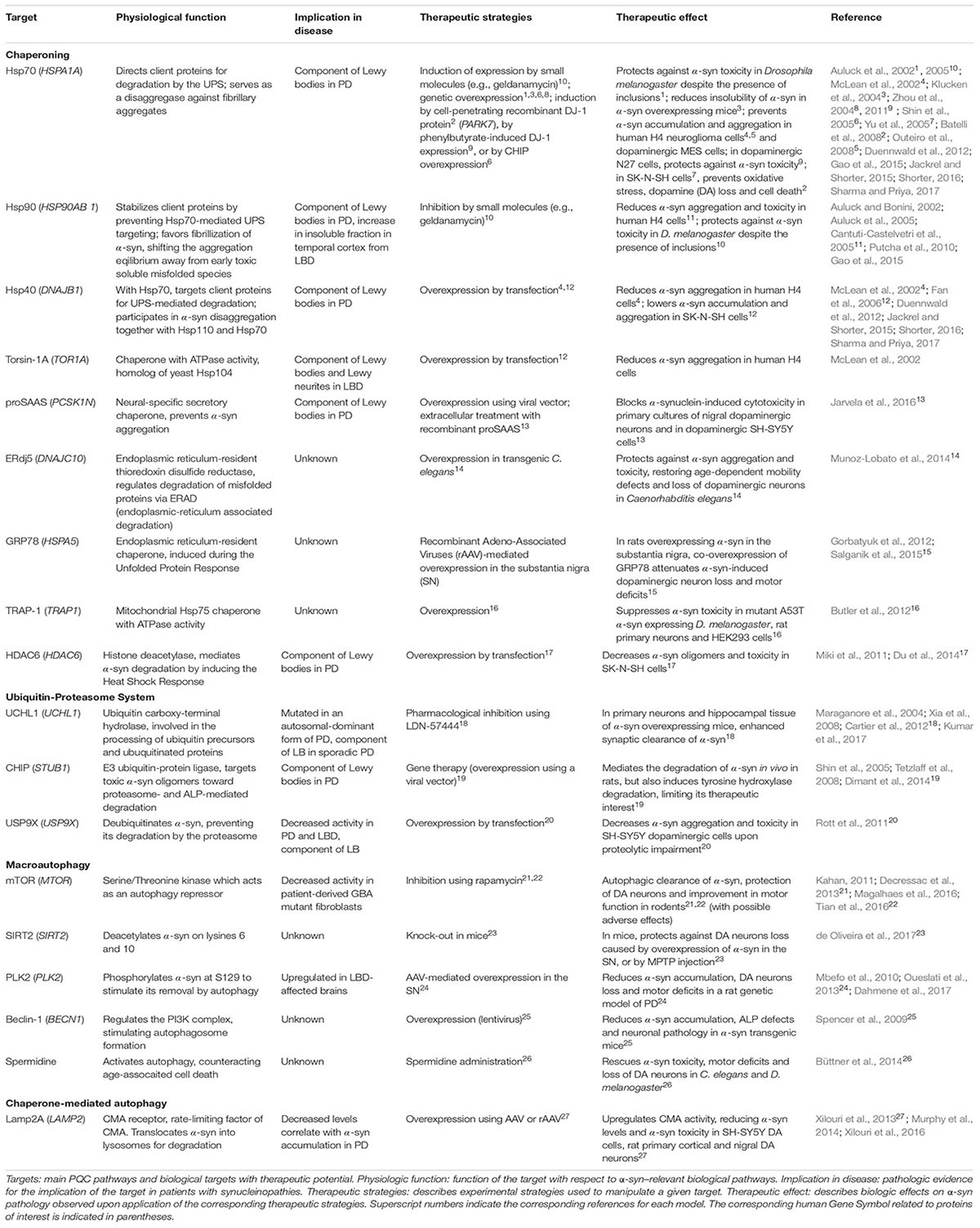
TABLE 1. Therapeutic avenues for targeting PQC pathways in the treatment of synucleinopathies.
Ubiquitin-Proteasome System
Dysfunction in the UPS contributes to α-syn pathology with proteasomal subunits and ubiquitinated α-syn observed in LBs (Ii et al., 1997; Hasegawa et al., 2002). In sporadic PD, 20S and 26S proteasome activity are reduced, and α-syn aggregates can inhibit proteasome function (Bence et al., 2001; Tanaka et al., 2001). Many dysfunctions in Ub ligases (specifically E3s) have been linked to α-syn quality control. Indeed, various E3s have been identified in LBs, including CHIP (Table 1), E6AP and SIAH (Schlossmacher et al., 2002; Liani et al., 2004; Shin et al., 2005; Mulherkar et al., 2009). CHIP, a co-chaperone with E3 Ub ligase activity, regulates α-syn proteasomal degradation, in collaboration with Hsc70, Hsp70, and Hsp90. Like CHIP, E6AP triggers α-syn degradation while SIAH monoubiquitinates α-syn to promote its aggregation (Rott et al., 2008; Mulherkar et al., 2009). Mutations in the gene encoding Parkin E3 Ub ligase are responsible for inherited PD (Olzmann et al., 2007; Lonskaya et al., 2013). Although these loss-of-function mutations are associated with PD, they do not lead to LB pathology (Shimura et al., 2000). It has been suggested that Parkin K63-linked polyubiquitination favors LB formation (Olanow et al., 2004; Kramer and Schulz-Schaeffer, 2007; Olzmann and Chin, 2008), and deficient Parkin activity would favor accumulation of earlier, potentially toxic aggregation intermediates. However, this awaits further confirmation, and to date, the role of Parkin Ub-ligase activity in mitochondrial quality control (Roberts et al., 2016) appears more relevant to PD pathogenesis than in LB formation.
Since proteasomes can degrade α-syn (Bennett et al., 1999), and regulation of α-syn ubiquitination has been implicated in PD (Liani et al., 2004; Rott et al., 2008, 2011), enhancing UPS activity could stimulate α-syn degradation and reduce aggregation-linked pathology (Opattova et al., 2015). Non-aggregated α-syn could be specifically targeted to the proteasome, thereby preventing aggregated α-syn from further inhibiting proteasome catalytic activity (Stefanis et al., 2001; Snyder et al., 2003; Chen et al., 2006). Selective enhancement of α-syn targeting to proteasomes is a more desirable approach to broader enhancement of UPS activity, which may lead to serious adverse effects. This could be achieved by increasing the activity of the specific machinery that controls the ubiquitination of α-syn, such as the druggable deubiquitinase USP9X (Rott et al., 2011, Table 1), although only inhibitors have been reported so far (Peterson et al., 2015). Deubiquitination of α-syn might redirect the α-syn burden toward the ALP, which is generally recognized as a more efficient α-syn degradation pathway than the UPS (Vogiatzi et al., 2008). It should be noted that α-syn ubiquitination can serve as a signal for lysosome-dependent degradation (Tofaris et al., 2011; Braun, 2015; Alexopoulou et al., 2016), illustrating a complex cross-talk between post-translational modifications of α-syn and cellular degradation machineries (Choi et al., 2012; Haj-Yahya et al., 2013; Shahpasandzadeh et al., 2014; Tenreiro et al., 2014; de Oliveira et al., 2017). It remains unclear which α-syn degradation pathway is favored, therefore further study is needed before a viable therapeutic strategy can be designed to enhance UPS-mediated α-syn degradation.
Autophagy-Lysosome Pathway
The ALP is thought to be the most efficient pathway for degradation of α-syn (Vogiatzi et al., 2008), with dysfunction causing accumulation and aggregation of α-syn. Defects in the ALP have been linked with an increasing number of genetic variants identified as causative or associated with PD risk (Gan-Or et al., 2015), including Vps35, a component of the retromer that mediates retrograde transport from endosomes to Golgi, the lysosomal ATPase pump ATP13A2, and LRRK2 (Ramirez et al., 2006; Usenovic et al., 2012; Orenstein et al., 2013; Kong et al., 2014; Tsunemi and Krainc, 2014; Tang et al., 2015; Follett et al., 2016). Polymorphisms in genes encoding lysosomal enzymes, acid sphingomyelinase (SMPD1 gene), and β-glucocerebrosidase (GBA, GBA1 gene), are also risk factors for synucleinopathies (Neumann et al., 2009; Dagan et al., 2015; Gan-Or et al., 2015). A reduction in GBA expression and activity is observed in the substantia nigra and cerebellum of patients with sporadic PD (Gegg et al., 2012), and the inhibition of GBA or its transporter Limp2 is sufficient to stimulate α-syn aggregation through autophagic inhibition (Rothaug et al., 2014; Du et al., 2015). Polymorphisms in the lysosomal K+ channel encoding gene TMEM175 are risk factors for PD (Nalls et al., 2014). TMEM175 deficiency causess ALP dysfunction and increased α-syn aggregation (Jinn et al., 2017). Additional ALP-related genes were just recently linked to PD (Chang et al., 2017), converging into a unifying theory for PD pathogenesis, where the ALP is challenged by defects in synaptic exocytosis, endocytosis, and endosomal trafficking, resulting in neuron dysfunction and death (Trinh and Farrer, 2013).
Macroautophagy is responsible for degrading most of the aggregated, proteasome-resistant, α-syn, and enhancing this process represents a promising therapeutic strategy (Figure 1 and Table 1). The mTOR inhibitor rapamycin activates macroautophagy, prevents α-syn accumulation and aggregation, and ameliorates motor symptoms, but adverse effects have been reported (Kahan, 2011; Li et al., 2014; Tian et al., 2016). More recently, it was shown that acetylation of α-syn increases macroautophagy-mediated degradation of α-syn aggregates, with knock-out of the α-syn deacetylase SIRT2 protects against α-syn–induced dopaminergic cell loss in vivo (de Oliveira et al., 2017, see Table 1). Independent from UPS-targeting, modulation by the deubiquitinase USP8 and the Ub-ligase Nedd4 of α-syn modification by K63-linked Ub appears to control its autophagic degradation (Braun, 2015; Alexopoulou et al., 2016). A better understanding of the specific effects of various post-translational modifications will be necessary to appropriately modulate α-syn clearance by macroautophagy.
CMA specifically degrades physiologic α-syn (which contains a KFERQ-like motif, VKKDQ), whereas pathologic α-syn inhibits CMA, thus enhancing aggregation of itself and other LB components (Martinez-Vicente et al., 2008; Vogiatzi et al., 2008; Xilouri et al., 2009). Accordingly, overexpression of certain PD-associated microRNAs is suspected to be responsible for pathologic CMA downregulation through decreased Hsc70 and Lamp2A expression. This correlates with α-syn accumulation in brains of patients with PD (Alvarez-Erviti et al., 2013; Murphy et al., 2015). CMA-mediated degradation of α-syn and LRRK2 is also impaired by mutants of these proteins that cause inherited PD (A53T and A30P α-syn mutants; G2019S and R1441C LRRK2 mutants). These mutants are recognized by Hsc70 and targeted to the lysosomal membrane, but fail to be translocated into the lysosome due to an aberrantly high affinity for Lamp2A. This impairs CMA-mediated degradation of these proteins and CMA activity, contributing to PD pathology (Cuervo et al., 2004; Orenstein et al., 2013). Deficiencies in CMA (caused by LRRK2 or VPS35 PD-associated mutations for example, Orenstein et al., 2013; Tang et al., 2015; Ho et al., 2016) cause accumulation of α-syn, favoring the emergence of aberrant α-syn species that hinder the function of the Lamp2A receptor. Lamp2A overexpression efficiently prevents α-syn burden in cellular and animal models of PD and counteracts motor deficits (see Table 1). Whether this strategy can reverse α-syn pathology in a clinical context, in a safe and effective way, still needs to be determined, especially since CMA cannot mediate the degradation of aggregated species. The role of CMA in PD pathogenesis has been reviewed recently (Sala et al., 2016), and will not be discussed further here. Notably, strategies aiming at activating both macroautophagy and CMA are also being explored (Gan et al., 2012; Lastres-Becker et al., 2016), such as overexpression of the transcription factor NRF2, which protects against α-syn pathology and increases its turnover through unknown ALP-dependent mechanisms (Skibinski et al., 2017).
A prerequisite for efficient α-syn degradation by ALP is the adequate function and hydrolytic capacity of lysosomes. Improper activity of lysosomal hydrolases, due to mutations, sorting defects, or altered lysosomal homeostasis, has emerged as a critical step in the development of PD (Trinh and Farrer, 2013; Gan-Or et al., 2015) and other synucleinopathies, particularly those associated with lysosomal storage disorders (Gaucher disease, Niemann-Pick disease; Cullen et al., 2011; Osellame and Duchen, 2013; Osellame et al., 2013). Stimulating lysosome biogenesis via activation of ALP transcriptional regulator TFEB improves autophagic α-syn clearance (Decressac et al., 2013; Kilpatrick et al., 2015), and reduces α-syn toxicity in rats (Decressac et al., 2013). Enhancing lysosomal hydrolase activity also improves α-syn degradation by the ALP, as shown for the α-syn-cleaving proteases cathepsins D, B and L (Miura et al., 2014; Lehri-Boufala et al., 2015; McGlinchey and Lee, 2015) or GBA (Yang et al., 2017). Finally, rescue of lysosomal hydrolase misfolding by small molecule chaperones (Sanchez-Martinez et al., 2016; Yang et al., 2017) or enhancement of endo-lysosomal trafficking of ALP components (Chung et al., 2013; Miura et al., 2014; Tang et al., 2015) are likely to mitigate α-syn pathology.
Other PQC Mechanisms
Other PQC mechanisms exist that are less commonly referred to in the context of α-syn pathology. α-syn synthesis could be reduced in the first place to prevent its accumulation. Several microRNAs target α-syn mRNA to reduce its expression in cell culture and in vivo (Junn et al., 2009; Doxakis, 2010; Singh and Sen, 2017). The therapeutic potential of this mechanism remains to be evaluated, especially regarding potential adverse effects of a lack of functional α-syn on the dopamine system (Abeliovich et al., 2000). Finally, unconventional secretion of misfolded proteins (misfolding-associated protein secretion, MAPS) was recently suggested to protect individual cells from misfolded proteins by delivering them to the extracellular space (Lee et al., 2016). In the context of a multicellular organism, however, this secretion might be harmful by contributing to the prion-like spreading of misfolded proteins including α-syn.
Concluding Remarks
Extensive genetic and experimental evidence indicate that PQC deficiencies influence the development of synucleinopathies. Despite the potential of several experimental strategies targeting PQC to attenuate α-syn pathology, translation into therapy is still pending. Whether these approaches will be clinically effective, where synuclein pathology is pre-existant, remains unknown. No successful clinical trial has been reported for synucleinopathies, but targeting PQC bears great promise as such strategies have proven effective to treat diseases such as cystic fibrosis or cancer (Teicher and Tomaszewski, 2015; Hegde et al., 2017). Further functional characterization of genes associated with synucleinopathies, will provide important insights regarding the molecular mechanisms that can be targeted to enhance PQC function, boost α-syn degradation, and prevent its aggregation. For patients with familial synucleinopathies, the upcoming era of personalized medicine, including the use of patient-derived induced pluripotent-stem cells and genome-editing, might allow correction of patient-specific mutations or PQC impairments. However, in sporadic cases, where genetic contributions are unknown (the majority of PD cases), simultaneous enhancement of several components of PQC machinery will likely be necessary to stop the progression, or even reverse the course, of these devastating neurodegenerative diseases.
Author Contributions
D-LM, BV, and TD: conception and organization of content of the mini-review. D-LM: design and generation of Figure 1, writing of introduction, and sections on α-syn pathology and defective PQC in synucleinopathies. BV: design and generation of Table 1, writing of section on therapeutic strategies, the abstract and concluding remarks, and assembly of manuscript. EF: overall revision. TD: in-depth editing of manuscript, and overall revision.
Funding
TD is funded by a research grant through the Michael J. Fox Foundation (award number 12241). BV is funded by a postdoctoral fellowship from the Canadian Institutes of Health Research (CIHR) Fellowship program (MFE 152571). This research was undertaken thanks in part to funding from the Canada First Research Excellence Fund, awarded to McGill University for the Healthy Brains for Healthy Lives initiative.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Drs. Trisha Rao and Lenore Beitel for their help in reading and copy-editing this manuscript.
Abbreviation
α-syn, α-synuclein; ALP, autophagy-lysosome pathway; CHIP, carboxyl terminus of Hsp70-interacting protein; CMA, chaperone-mediated autophagy; E6AP, E6 associated protein; GBA, β-glucocerebrosidase; Hsc, heat shock cognate; Hsp, heat shock protein; LB, Lewy bodies; LBD, Lewy body dementia; MPTP, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine; MSA, multiple system atrophy; NAC, non-amyloid-β component; PD, Parkinson’s disease; PQC, protein quality control; SIAH, seven in absentia homolog; Ub, ubiquitin; UPS, ubiquitin-proteasome system.
References
Abeliovich, A., Schmitz, Y., Fariñas, I., Choi-Lundberg, D., Ho, W. H., Castillo, P. E., et al. (2000). Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron 25, 239–252. doi: 10.1016/S0896-6273(00)80886-7
Ahn, M., Kim, S., Kang, M., Ryu, Y., and Doohun Kim, T. (2006). Chaperone-like activities of α-synuclein: α-Synuclein assists enzyme activities of esterases. Biochem. Biophys. Res. Commun. 346, 1142–1149. doi: 10.1016/j.bbrc.2006.05.213
Alexopoulou, Z., Lang, J., Perrett, R. M., Elschami, M., Hurry, M. E. D., Kim, H. T., et al. (2016). Deubiquitinase Usp8 regulates α-synuclein clearance and modifies its toxicity in Lewy body disease. Proc. Natl. Acad. Sci. U.S.A. 113, E4688–E4697. doi: 10.1073/pnas.1523597113
Alvarez-Erviti, L., Seow, Y., Schapira, A. H. V., Rodriguez-Oroz, M. C., Obeso, J. A., and Cooper, J. M. (2013). Influence of microRNA deregulation on chaperone-mediated autophagy and alpha-synuclein pathology in Parkinson’s disease. Cell Death Dis. 4:e545. doi: 10.1038/cddis.2013.73
Auluck, P. K., and Bonini, N. M. (2002). Pharmacological prevention of Parkinson disease in Drosophila. Nat. Med. 8, 1185–1186. doi: 10.1038/nm1102-1185
Auluck, P. K., Chan, H. Y. E., Trojanowski, J. Q., Lee, V. M. Y., and Bonini, N. M. (2002). Chaperone suppression of alpha-synuclein toxicity in a Drosophila model for Parkinson’s disease. Science 295, 865–868. doi: 10.1126/science.1067389
Auluck, P. K., Meulener, M. C., and Bonini, N. M. (2005). Mechanisms of suppression of α-synuclein neurotoxicity by geldanamycin in Drosophila. J. Biol. Chem. 280, 2873–2878. doi: 10.1074/jbc.M412106200
Bartels, T., Choi, J. G., and Selkoe, D. J. (2011). alpha-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 477, 107–110. doi: 10.1038/nature10324
Batelli, S., Albani, D., Rametta, R., Polito, L., Prato, F., Pesaresi, M., et al. (2008). DJ-1 modulates alpha-synuclein aggregation state in a cellular model of oxidative stress: relevance for Parkinson’s disease and involvement of HSP70. PLOS ONE 3:e1884. doi: 10.1371/journal.pone.0001884
Bence, N. F., Sampat, R. M., and Kopito, R. R. (2001). Impairment of the ubiquitin-proteasome system by protein aggregation. Science 292, 1552–1555. doi: 10.1126/science.292.5521.1552
Bennett, M. C., Bishop, J. F., Leng, Y., Chock, P. B., Chase, T. N., and Mouradian, M. M. (1999). Degradation of alpha-synuclein by proteasome. J. Biol. Chem. 274, 33855–33858. doi: 10.1074/jbc.274.48.33855
Bento, C. F., Renna, M., Ghislat, G., Puri, C., Ashkenazi, A., Vicinanza, M., et al. (2016). Mammalian autophagy: how does it work? Annu. Rev. Biochem. 85, 685–713. doi: 10.1146/annurev-biochem-060815-014556
Beyer, K. (2006). α-Synuclein structure, posttranslational modification and alternative splicing as aggregation enhancers. Acta Neuropathol. 112, 237–251. doi: 10.1007/s00401-006-0104-6
Braak, H., Del Tredici, K., Rüb, U., de Vos, R. A., Steur, E. N. J., and Braak, E. (2003). Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 24, 197–211. doi: 10.1016/S0197-4580(02)00065-9
Braun, R. J. (2015). Ubiquitin-dependent proteolysis in yeast cells expressing neurotoxic proteins. Front. Mol. Neurosci. 8:8. doi: 10.3389/fnmol.2015.00008
Burré, J., Sharma, M., and Südhof, T. C. (2014). α-Synuclein assembles into higher-order multimers upon membrane binding to promote SNARE complex formation. Proc. Natl. Acad. Sci. U.S.A. 111, E4274–E4283. doi: 10.1073/pnas.1416598111
Butler, E. K., Voigt, A., Lutz, A. K., Toegel, J. P., Gerhardt, E., Karsten, P., et al. (2012). The mitochondrial chaperone protein TRAP1 mitigates alpha-Synuclein toxicity. PLOS Genet. 8:e1002488. doi: 10.1371/journal.pgen.1002488
Büttner, S., Broeskamp, F., Sommer, C., Markaki, M., Habernig, L., Alavian-Ghavanini, A., et al. (2014). Spermidine protects against α-synuclein neurotoxicity. Cell Cycle 13, 3903–3908. doi: 10.4161/15384101.2014.973309
Cantuti-Castelvetri, I., Klucken, J., Ingelsson, M., Ramasamy, K., McLean, P. J., Frosch, M. P., et al. (2005). Alpha-synuclein and chaperones in dementia with Lewy bodies. J. Neuropathol. Exp. Neurol. 64, 1058–1066. doi: 10.1097/01.jnen.0000190063.90440.69
Cartier, A. E., Ubhi, K., Spencer, B., Vazquez-Roque, R. A., Kosberg, K. A., Fourgeaud, L., et al. (2012). Differential effects of UCHL1 modulation on alpha-synuclein in PD-like models of alpha-synucleinopathy. PLOS ONE 7:e34713. doi: 10.1371/journal.pone.0034713
Caruana, M., Hogen, T., Levin, J., Hillmer, A., Giese, A., and Vassallo, N. (2011). Inhibition and disaggregation of alpha-synuclein oligomers by natural polyphenolic compounds. FEBS Lett. 585, 1113–1120. doi: 10.1016/j.febslet.2011.03.046
Chang, D., Nalls, M. A., Hallgrímsdóttir, I. B., Hunkapiller, J., van der Brug, M., Cai, F., et al. (2017). A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat. Genet. doi: 10.1038/ng.3955 [Epub ahead of print].
Chen, L., Thiruchelvam, M. J., Madura, K., and Richfield, E. K. (2006). Proteasome dysfunction in aged human alpha-synuclein transgenic mice. Neurobiol. Dis. 23, 120–126. doi: 10.1016/j.nbd.2006.02.004
Choi, H. S., Liew, H., Jang, A., Kim, Y.-M., Lashuel, H., and Suh, Y.-H. (2012). Phosphorylation of alpha-synuclein is crucial in compensating for proteasomal dysfunction. Biochem. Biophys. Res. Commun. 424, 597–603. doi: 10.1016/j.bbrc.2012.06.159
Chung, C. Y., Khurana, V., Auluck, P. K., Tardiff, D. F., Mazzulli, J. R., Soldner, F., et al. (2013). Identification and rescue of α-synuclein toxicity in Parkinson patient-derived neurons. Science 342, 983–987. doi: 10.1126/science.1245296
Cuervo, A. M., Stefanis, L., Fredenburg, R., Lansbury, P. T., and Sulzer, D. (2004). Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 305, 1292–1295. doi: 10.1126/science.1101738
Cuervo, A. M., and Wong, E. (2014). Chaperone-mediated autophagy: roles in disease and aging. Cell Res. 24, 92–104. doi: 10.1038/cr.2013.153
Cullen, V., Sardi, S. P., Ng, J., Xu, Y.-H., Sun, Y., Tomlinson, J. J., et al. (2011). Acid beta-glucosidase mutants linked to Gaucher disease, Parkinson disease, and Lewy body dementia alter alpha-synuclein processing. Ann. Neurol. 69, 940–953. doi: 10.1002/ana.22400
Dagan, E., Schlesinger, I., Ayoub, M., Mory, A., Nassar, M., Kurolap, A., et al. (2015). The contribution of Niemann-Pick SMPD1 mutations to Parkinson disease in Ashkenazi Jews. Parkinsonism Relat. Disord. 21, 1067–1071. doi: 10.1016/j.parkreldis.2015.06.016
Dahmene, M., Bérard, M., and Oueslati, A. (2017). Dissecting the molecular pathway involved in PLK2 kinase-mediated α-Synuclein-selective autophagic degradation. J. Biol. Chem. 292, 3919–3928. doi: 10.1074/jbc.M116.759373
de Oliveira, R. M., Vicente Miranda, H., Francelle, L., Pinho, R., Szego, E. M., Martinho, R., et al. (2017). The mechanism of sirtuin 2-mediated exacerbation of alpha-synuclein toxicity in models of Parkinson disease. PLOS Biol. 15:e2000374. doi: 10.1371/journal.pbio.2000374
Decressac, M., Mattsson, B., Weikop, P., Lundblad, M., Jakobsson, J., and Björklund, A. (2013). TFEB-mediated autophagy rescues midbrain dopamine neurons from α-synuclein toxicity. Proc. Natl. Acad. Sci. U.S.A. 110, E1817–E1826. doi: 10.1073/pnas.1305623110
Dettmer, U., Newman, A. J., Soldner, F., Luth, E. S., Kim, N. C., von Saucken, V. E., et al. (2015). Parkinson-causing α-synuclein missense mutations shift native tetramers to monomers as a mechanism for disease initiation. Nat. Commun. 6:7314. doi: 10.1038/ncomms8314
DeWitt, D. C., and Rhoades, E. (2013). alpha-Synuclein can inhibit SNARE-mediated vesicle fusion through direct interactions with lipid bilayers. Biochemistry 52, 2385–2387. doi: 10.1021/bi4002369
Dimant, H., Zhu, L., Kibuuka, L. N., Fan, Z., Hyman, B. T., and McLean, P. J. (2014). Direct visualization of CHIP-mediated degradation of alpha-synuclein in vivo: implications for PD therapeutics. PLOS ONE 9:e92098. doi: 10.1371/journal.pone.0092098
Doxakis, E. (2010). Post-transcriptional regulation of alpha-synuclein expression by mir-7 and mir-153. J. Biol. Chem. 285, 12726–12734. doi: 10.1074/jbc.M109.086827
Du, T.-T., Wang, L., Duan, C.-L., Lu, L.-L., Zhang, J.-L., Gao, G., et al. (2015). GBA deficiency promotes SNCA/α-synuclein accumulation through autophagic inhibition by inactivated PPP2A. Autophagy 11, 1803–1820. doi: 10.1080/15548627.2015.1086055
Du, Y., Wang, F., Zou, J., Le, W., Dong, Q., Wang, Z., et al. (2014). Histone deacetylase 6 regulates cytotoxic alpha-synuclein accumulation through induction of the heat shock response. Neurobiol. Aging 35, 2316–2328. doi: 10.1016/j.neurobiolaging.2014.04.029
Dubnikov, T., Ben-Gedalya, T., and Cohen, E. (2017). Protein Quality Control in Health and Disease. Cold Spring Harb. Perspect. Biol. 9:a023523. doi: 10.1101/cshperspect.a023523
Duennwald, M. L., Echeverria, A., and Shorter, J. (2012). Small heat shock proteins potentiate amyloid dissolution by protein disaggregases from yeast and humans. PLOS Biol. 10:e1001346. doi: 10.1371/journal.pbio.1001346
Ebrahimi-Fakhari, D., Wahlster, L., and McLean, P. J. (2011). Molecular chaperones in Parkinson’s disease–present and future. J. Park. Dis. 1, 299–320.
Escusa-Toret, S., Vonk, W. I., and Frydman, J. (2013). Spatial sequestration of misfolded proteins by a dynamic chaperone pathway enhances cellular fitness during stress. Nat. Cell Biol. 15, 1231–1243. doi: 10.1038/ncb2838
Fan, G.-H., Zhou, H.-Y., Yang, H., and Chen, S.-D. (2006). Heat shock proteins reduce alpha-synuclein aggregation induced by MPP+ in SK-N-SH cells. FEBS Lett. 580, 3091–3098. doi: 10.1016/j.febslet.2006.04.057
Filimonenko, M., Stuffers, S., Raiborg, C., Yamamoto, A., Malerød, L., Fisher, E. M. C., et al. (2007). Functional multivesicular bodies are required for autophagic clearance of protein aggregates associated with neurodegenerative disease. J. Cell Biol. 179, 485–500. doi: 10.1083/jcb.200702115
Follett, J., Bugarcic, A., Yang, Z., Ariotti, N., Norwood, S. J., Collins, B. M., et al. (2016). Parkinson disease-linked Vps35 R524W mutation impairs the endosomal association of retromer and induces α-synuclein aggregation. J. Biol. Chem. 291, 18283–18298. doi: 10.1074/jbc.M115.703157
Gan, L., Vargas, M. R., Johnson, D. A., and Johnson, J. A. (2012). Astrocyte-specific overexpression of Nrf2 delays motor pathology and synuclein aggregation throughout the CNS in the alpha-synuclein mutant (A53T) mouse model. J. Neurosci. Off. J. Soc. Neurosci. 32, 17775–17787. doi: 10.1523/JNEUROSCI.3049-12.2012
Gan-Or, Z., Dion, P. A., and Rouleau, G. A. (2015). Genetic perspective on the role of the autophagy-lysosome pathway in Parkinson disease. Autophagy 11, 1443–1457. doi: 10.1080/15548627.2015.1067364
Gao, X., Carroni, M., Nussbaum-Krammer, C., Mogk, A., Nillegoda, N. B., Szlachcic, A., et al. (2015). Human Hsp70 disaggregase reverses parkinson’s-linked alpha-synuclein amyloid fibrils. Mol. Cell 59, 781–793. doi: 10.1016/j.molcel.2015.07.012
Gautam, S., Karmakar, S., Batra, R., Sharma, P., Pradhan, P., Singh, J., et al. (2017). Polyphenols in combination with beta-cyclodextrin can inhibit and disaggregate alpha-synuclein amyloids under cell mimicking conditions: a promising therapeutic alternative. Biochim. Biophys. Acta 1865, 589–603. doi: 10.1016/j.bbapap.2017.02.014
Gegg, M. E., Burke, D., Heales, S. J. R., Cooper, J. M., Hardy, J., Wood, N. W., et al. (2012). Glucocerebrosidase deficiency in substantia nigra of Parkinson disease brains. Ann. Neurol. 72, 455–463. doi: 10.1002/ana.23614
Goedert, M., Masuda-Suzukake, M., and Falcon, B. (2016). Like prions: the propagation of aggregated tau and alpha-synuclein in neurodegeneration. Brain 140(Pt 2), 266–278. doi: 10.1093/brain/aww230
Gorbatyuk, M. S., Shabashvili, A., Chen, W., Meyers, C., Sullivan, L. F., Salganik, M., et al. (2012). Glucose regulated protein 78 diminishes alpha-synuclein neurotoxicity in a rat model of Parkinson disease. Mol. Ther. J. Am. Soc. Gene Ther. 20, 1327–1337. doi: 10.1038/mt.2012.28
Haj-Yahya, M., Fauvet, B., Herman-Bachinsky, Y., Hejjaoui, M., Bavikar, S. N., Karthikeyan, S. V., et al. (2013). Synthetic polyubiquitinated alpha-Synuclein reveals important insights into the roles of the ubiquitin chain in regulating its pathophysiology. Proc. Natl. Acad. Sci. U.S.A. 110, 17726–17731. doi: 10.1073/pnas.1315654110
Hartl, F. U., Bracher, A., and Hayer-Hartl, M. (2011). Molecular chaperones in protein folding and proteostasis. Nature 475, 324–332. doi: 10.1038/nature10317
Hasegawa, M., Fujiwara, H., Nonaka, T., Wakabayashi, K., Takahashi, H., Lee, V. M.-Y., et al. (2002). Phosphorylated α-synuclein is ubiquitinated in α-synucleinopathy lesions. J. Biol. Chem. 277, 49071–49076. doi: 10.1074/jbc.M208046200
Hegde, R. N., Subramanian, A., Pothukuchi, P., Parashuraman, S., and Luini, A. (2017). Rare ER protein misfolding-mistrafficking disorders: therapeutic developments. Tissue Cell 49, 175–185. doi: 10.1016/j.tice.2017.02.001
Ho, S. L., Ho, P. W.-L., Pang, S., Liu, H., Lam, C. S.-C., Leung, M. C.-T., et al. (2016). Activation of chaperone-mediated autophagy prevents accumulation of oligomeric alpha-synuclein in LRRK2 R1441G knockin mice of Parkinson’s disease. Neurology 86:5.382.
Ii, K., Ito, H., Tanaka, K., and Hirano, A. (1997). Immunocytochemical co-localization of the proteasome in ubiquitinated structures in neurodegenerative diseases and the elderly. J. Neuropathol. Exp. Neurol. 56, 125–131. doi: 10.1097/00005072-199702000-00002
Jackrel, M. E., and Shorter, J. (2015). Engineering enhanced protein disaggregases for neurodegenerative disease. Prion 9, 90–109. doi: 10.1080/19336896.2015.1020277
Jarvela, T. S., Lam, H. A., Helwig, M., Lorenzen, N., Otzen, D. E., McLean, P. J., et al. (2016). The neural chaperone proSAAS blocks alpha-synuclein fibrillation and neurotoxicity. Proc. Natl. Acad. Sci. U.S.A. 113, E4708–E4715. doi: 10.1073/pnas.1601091113
Jinn, S., Drolet, R. E., Cramer, P. E., Wong, A. H.-K., Toolan, D. M., Gretzula, C. A., et al. (2017). TMEM175 deficiency impairs lysosomal and mitochondrial function and increases alpha-synuclein aggregation. Proc. Natl. Acad. Sci. U.S.A. 114, 2389–2394. doi: 10.1073/pnas.1616332114
Johnston, J. A., Ward, C. L., and Kopito, R. R. (1998). Aggresomes: a cellular response to misfolded proteins. J. Cell Biol. 143, 1883–1898. doi: 10.1083/jcb.143.7.1883
Junn, E., Lee, K.-W., Jeong, B. S., Chan, T. W., Im, J.-Y., and Mouradian, M. M. (2009). Repression of alpha-synuclein expression and toxicity by microRNA-7. Proc. Natl. Acad. Sci. U.S.A. 106, 13052–13057. doi: 10.1073/pnas.0906277106
Kahan, B. (2011). Toxicity spectrum of inhibitors of mammalian target of rapamycin in organ transplantation: etiology, pathogenesis and treatment. Expert Opin. Drug Saf. 10, 727–749. doi: 10.1517/14740338.2011.579898
Kilpatrick, K., Zeng, Y., Hancock, T., and Segatori, L. (2015). Genetic and chemical activation of TFEB mediates clearance of aggregated α-synuclein. PLOS ONE 10:e0120819. doi: 10.1371/journal.pone.0120819
Klucken, J., Shin, Y., Masliah, E., Hyman, B. T., and McLean, P. J. (2004). Hsp70 reduces alpha-synuclein aggregation and toxicity. J. Biol. Chem. 279, 25497–25502. doi: 10.1074/jbc.M400255200
Kong, S. M., Chan, B. K., Park, J.-S., Hill, K. J., Aitken, J. B., Cottle, L., et al. (2014). Parkinson’s disease-linked human PARK9/ATP13A2 maintains zinc homeostasis and promotes α-Synuclein externalization via exosomes. Hum. Mol. Genet. 23, 2816–2833. doi: 10.1093/hmg/ddu099
Kramer, M. L., and Schulz-Schaeffer, W. J. (2007). Presynaptic alpha-synuclein aggregates, not Lewy bodies, cause neurodegeneration in dementia with Lewy bodies. J. Neurosci. Off. J. Soc. Neurosci. 27, 1405–1410. doi: 10.1523/JNEUROSCI.4564-06.2007
Kumar, R., Jangir, D. K., Verma, G., Shekhar, S., Hanpude, P., Kumar, S., et al. (2017). S-nitrosylation of UCHL1 induces its structural instability and promotes α-synuclein aggregation. Sci. Rep. 7:44558. doi: 10.1038/srep44558
Lastres-Becker, I., Garcia-Yague, A. J., Scannevin, R. H., Casarejos, M. J., Kugler, S., Rabano, A., et al. (2016). Repurposing the NRF2 activator dimethyl fumarate as therapy against synucleinopathy in Parkinson’s disease. Antioxid. Redox Signal. 25, 61–77. doi: 10.1089/ars.2015.6549
Lee, J.-G., Takahama, S., Zhang, G., Tomarev, S. I., and Ye, Y. (2016). Unconventional secretion of misfolded proteins promotes adaptation to proteasome dysfunction in mammalian cells. Nat. Cell Biol. 18, 765–776. doi: 10.1038/ncb3372
Lehri-Boufala, S., Ouidja, M.-O., Barbier-Chassefiere, V., Henault, E., Raisman-Vozari, R., Garrigue-Antar, L., et al. (2015). New roles of glycosaminoglycans in alpha-synuclein aggregation in a cellular model of Parkinson disease. PLOS ONE 10:e0116641. doi: 10.1371/journal.pone.0116641
Li, J., Kim, S. G., and Blenis, J. (2014). Rapamycin: one drug, many effects. Cell Metab. 19, 373–379. doi: 10.1016/j.cmet.2014.01.001
Liani, E., Eyal, A., Avraham, E., Shemer, R., Szargel, R., Berg, D., et al. (2004). Ubiquitylation of synphilin-1 and α-synuclein by SIAH and its presence in cellular inclusions and Lewy bodies imply a role in Parkinson’s disease. Proc. Natl. Acad. Sci. U.S.A. 101, 5500–5505. doi: 10.1073/pnas.0401081101
Lonskaya, I., Desforges, N. M., Hebron, M. L., and Moussa, C. E.-H. (2013). Ubiquitination increases parkin activity to promote autophagic α-synuclein clearance. PLOS ONE 8:e83914. doi: 10.1371/journal.pone.0083914
Luk, K. C., Kehm, V., Carroll, J., Zhang, B., O’Brien, P., Trojanowski, J. Q., et al. (2012a). Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 338, 949–953. doi: 10.1126/science.1227157
Luk, K. C., Kehm, V. M., Zhang, B., O’Brien, P., Trojanowski, J. Q., and Lee, V. M. Y. (2012b). Intracerebral inoculation of pathological α-synuclein initiates a rapidly progressive neurodegenerative α-synucleinopathy in mice. J. Exp. Med. 209, 975–986. doi: 10.1084/jem.20112457
Magalhaes, J., Gegg, M. E., Migdalska-Richards, A., Doherty, M. K., Whitfield, P. D., and Schapira, A. H. V. (2016). Autophagic lysosome reformation dysfunction in glucocerebrosidase deficient cells: relevance to Parkinson disease. Hum. Mol. Genet. 25, 3432–3445. doi: 10.1093/hmg/ddw185
Maraganore, D. M., Lesnick, T. G., Elbaz, A., Chartier-Harlin, M. C., Gasser, T., Kruger, R., et al. (2004). UCHL1 is a Parkinson’s disease susceptibility gene. Ann. Neurol. 55, 512–521. doi: 10.1002/ana.20017
Martinez-Vicente, M., Talloczy, Z., Kaushik, S., Massey, A. C., Mazzulli, J., Mosharov, E. V., et al. (2008). Dopamine-modified α-synuclein blocks chaperone-mediated autophagy. J. Clin. Invest. 118, 777–788. doi: 10.1172/JCI32806
Masuda-Suzukake, M., Nonaka, T., Hosokawa, M., Oikawa, T., Arai, T., Akiyama, H., et al. (2013). Prion-like spreading of pathological -synuclein in brain. Brain 136, 1128–1138. doi: 10.1093/brain/awt037
Mbefo, M. K., Paleologou, K. E., Boucharaba, A., Oueslati, A., Schell, H., Fournier, M., et al. (2010). Phosphorylation of synucleins by members of the Polo-like kinase family. J. Biol. Chem. 285, 2807–2822. doi: 10.1074/jbc.M109.081950
McCann, H., Stevens, C. H., Cartwright, H., and Halliday, G. M. (2014). α-Synucleinopathy phenotypes. Parkinsonism Relat. Disord. 20(Suppl. 1), S62–S67. doi: 10.1016/S1353-8020(13)70017-8
McGlinchey, R. P., and Lee, J. C. (2015). Cysteine cathepsins are essential in lysosomal degradation of α-synuclein. Proc. Natl. Acad. Sci. U.S.A. 112, 9322–9327. doi: 10.1073/pnas.1500937112
McLean, P. J., Kawamata, H., Shariff, S., Hewett, J., Sharma, N., Ueda, K., et al. (2002). TorsinA and heat shock proteins act as molecular chaperones: suppression of alpha-synuclein aggregation. J. Neurochem. 83, 846–854. doi: 10.1046/j.1471-4159.2002.01190.x
Miki, Y., Mori, F., Tanji, K., Kakita, A., Takahashi, H., and Wakabayashi, K. (2011). Accumulation of histone deacetylase 6, an aggresome-related protein, is specific to Lewy bodies and glial cytoplasmic inclusions. Neuropathology 31, 561–568. doi: 10.1111/j.1440-1789.2011.01200.x
Miura, E., Hasegawa, T., Konno, M., Suzuki, M., Sugeno, N., Fujikake, N., et al. (2014). VPS35 dysfunction impairs lysosomal degradation of α-synuclein and exacerbates neurotoxicity in a Drosophila model of Parkinson’s disease. Neurobiol. Dis. 71, 1–13. doi: 10.1016/j.nbd.2014.07.014
Mougenot, A.-L., Nicot, S., Bencsik, A., Morignat, E., Verchère, J., Lakhdar, L., et al. (2012). Prion-like acceleration of a synucleinopathy in a transgenic mouse model. Neurobiol. Aging 33, 2225–2228. doi: 10.1016/j.neurobiolaging.2011.06.022
Mulherkar, S. A., Sharma, J., and Jana, N. R. (2009). The ubiquitin ligase E6-AP promotes degradation of α-synuclein. J. Neurochem. 110, 1955–1964. doi: 10.1111/j.1471-4159.2009.06293.x
Munoz-Lobato, F., Rodriguez-Palero, M. J., Naranjo-Galindo, F. J., Shephard, F., Gaffney, C. J., Szewczyk, N. J., et al. (2014). Protective role of DNJ-27/ERdj5 in Caenorhabditis elegans models of human neurodegenerative diseases. Antioxid. Redox Signal. 20, 217–235. doi: 10.1089/ars.2012.5051
Murphy, K. E., Gysbers, A. M., Abbott, S. K., Spiro, A. S., Furuta, A., Cooper, A., et al. (2015). Lysosomal-associated membrane protein 2 isoforms are differentially affected in early Parkinson’s disease. Mov. Disord. Off. J. Mov. Disord. Soc. 30, 1639–1647. doi: 10.1002/mds.26141
Murphy, K. E., Gysbers, A. M., Abbott, S. K., Tayebi, N., Kim, W. S., Sidransky, E., et al. (2014). Reduced glucocerebrosidase is associated with increased α-synuclein in sporadic Parkinson’s disease. Brain 137(Pt 3), 834–848. doi: 10.1093/brain/awt367
Nalls, M. A., Pankratz, N., Lill, C. M., Do, C. B., Hernandez, D. G., Saad, M., et al. (2014). Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat. Genet. 46, 989–993. doi: 10.1038/ng.3043
Neumann, J., Bras, J., Deas, E., O’Sullivan, S. S., Parkkinen, L., Lachmann, R. H., et al. (2009). Glucocerebrosidase mutations in clinical and pathologically proven Parkinson’s disease. Brain 132, 1783–1794. doi: 10.1093/brain/awp044
Olanow, C. W., Perl, D. P., DeMartino, G. N., and McNaught, K. S. P. (2004). Lewy-body formation is an aggresome-related process: a hypothesis. Lancet Neurol. 3, 496–503. doi: 10.1016/S1474-4422(04)00827-0
Olzmann, J. A., and Chin, L.-S. (2008). Parkin-mediated K63-linked polyubiquitination: a signal for targeting misfolded proteins to the aggresome-autophagy pathway. Autophagy 4, 85–87. doi: 10.4161/auto.5172
Olzmann, J. A., Li, L., Chudaev, M. V., Chen, J., Perez, F. A., Palmiter, R. D., et al. (2007). Parkin-mediated K63-linked polyubiquitination targets misfolded DJ-1 to aggresomes via binding to HDAC6. J. Cell Biol. 178, 1025–1038. doi: 10.1083/jcb.200611128
Opattova, A., Cente, M., Novak, M., and Filipcik, P. (2015). The ubiquitin proteasome system as a potential therapeutic target for treatment of neurodegenerative diseases. Gen. Physiol. Biophys. 34, 337–352. doi: 10.4149/gpb_2015024
Orenstein, S. J., Kuo, S.-H., Tasset, I., Arias, E., Koga, H., Fernandez-Carasa, I., et al. (2013). Interplay of LRRK2 with chaperone-mediated autophagy. Nat. Neurosci. 16, 394–406. doi: 10.1038/nn.3350
Osellame, L. D., and Duchen, M. R. (2013). Defective quality control mechanisms and accumulation of damaged mitochondria link Gaucher and Parkinson diseases. Autophagy 9, 1633–1635. doi: 10.4161/auto.25878
Osellame, L. D., Rahim, A. A., Hargreaves, I. P., Gegg, M. E., Richard-Londt, A., Brandner, S., et al. (2013). Mitochondria and quality control defects in a mouse model of gaucher disease—links to Parkinson’s disease. Cell Metab. 17, 941–953. doi: 10.1016/j.cmet.2013.04.014
Oueslati, A., Schneider, B. L., Aebischer, P., and Lashuel, H. A. (2013). Polo-like kinase 2 regulates selective autophagic α-synuclein clearance and suppresses its toxicity in vivo. Proc. Natl. Acad. Sci. U.S.A. 110, E3945–E3954. doi: 10.1073/pnas.1309991110
Outeiro, T. F., Putcha, P., Tetzlaff, J. E., Spoelgen, R., Koker, M., Carvalho, F., et al. (2008). Formation of toxic oligomeric alpha-synuclein species in living cells. PLOS ONE 3:e1867. doi: 10.1371/journal.pone.0001867
Passmore, L. A., and Barford, D. (2004). Getting into position: the catalytic mechanisms of protein ubiquitylation. Biochem. J. 379, 513–525. doi: 10.1042/BJ20040198
Peterson, L. F., Sun, H., Liu, Y., Potu, H., Kandarpa, M., Ermann, M., et al. (2015). Targeting deubiquitinase activity with a novel small-molecule inhibitor as therapy for B-cell malignancies. Blood 125, 3588–3597. doi: 10.1182/blood-2014-10-605584
Pratt, W. B., Gestwicki, J. E., Osawa, Y., and Lieberman, A. P. (2015). Targeting Hsp90/Hsp70-based protein quality control for treatment of adult onset neurodegenerative diseases. Annu. Rev. Pharmacol. Toxicol. 55, 353–371. doi: 10.1146/annurev-pharmtox-010814-124332
Putcha, P., Danzer, K. M., Kranich, L. R., Scott, A., Silinski, M., Mabbett, S., et al. (2010). Brain-permeable small-molecule inhibitors of Hsp90 prevent alpha-synuclein oligomer formation and rescue alpha-synuclein-induced toxicity. J. Pharmacol. Exp. Ther. 332, 849–857. doi: 10.1124/jpet.109.158436
Ramirez, A., Heimbach, A., Grundemann, J., Stiller, B., Hampshire, D., Cid, L. P., et al. (2006). Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat. Genet. 38, 1184–1191. doi: 10.1038/ng1884
Ren, R., Shi, C., Cao, J., Sun, Y., Zhao, X., Guo, Y., et al. (2016). Neuroprotective effects of a standardized flavonoid extract of safflower against neurotoxin-induced cellular and animal models of Parkinson’s disease. Sci. Rep. 6:22135. doi: 10.1038/srep22135
Roberts, R. F., Tang, M. Y., Fon, E. A., and Durcan, T. M. (2016). Defending the mitochondria: the pathways of mitophagy and mitochondrial-derived vesicles. Int. J. Biochem. Cell Biol. 79, 427–436. doi: 10.1016/j.biocel.2016.07.020
Rodriguez, J. A., Ivanova, M. I., Sawaya, M. R., Cascio, D., Reyes, F. E., Shi, D., et al. (2015). Structure of the toxic core of alpha-synuclein from invisible crystals. Nature 525, 486–490. doi: 10.1038/nature15368
Roeters, S. J., Iyer, A., Pletikapiæ, G., Kogan, V., Subramaniam, V., and Woutersen, S. (2017). Evidence for intramolecular antiparallel beta-sheet structure in alpha-synuclein fibrils from a combination of two-dimensional infrared spectroscopy and atomic force microscopy. Sci. Rep. 7:41051. doi: 10.1038/srep41051
Rothaug, M., Zunke, F., Mazzulli, J. R., Schweizer, M., Altmeppen, H., Lüllmann-Rauch, R., et al. (2014). LIMP-2 expression is critical for β-glucocerebrosidase activity and α-synuclein clearance. Proc. Natl. Acad. Sci. U.S.A. 111, 15573–15578. doi: 10.1073/pnas.1405700111
Rott, R., Szargel, R., Haskin, J., Bandopadhyay, R., Lees, A. J., Shani, V., et al. (2011). alpha-Synuclein fate is determined by USP9X-regulated monoubiquitination. Proc. Natl. Acad. Sci. U.S.A. 108, 18666–18671. doi: 10.1073/pnas.1105725108
Rott, R., Szargel, R., Haskin, J., Shani, V., Shainskaya, A., Manov, I., et al. (2008). Monoubiquitylation of α-synuclein by seven in absentia homolog (SIAH) promotes its aggregation in dopaminergic cells. J. Biol. Chem. 283, 3316–3328. doi: 10.1074/jbc.M704809200
Sahu, R., Kaushik, S., Clement, C. C., Cannizzo, E. S., Scharf, B., Follenzi, A., et al. (2011). Microautophagy of cytosolic proteins by late endosomes. Dev. Cell 20, 131–139. doi: 10.1016/j.devcel.2010.12.003
Sala, G., Marinig, D., Arosio, A., and Ferrarese, C. (2016). Role of chaperone-mediated autophagy dysfunctions in the pathogenesis of Parkinson’s Disease. Front. Mol. Neurosci. 9:157. doi: 10.3389/fnmol.2016.00157
Salganik, M., Sergeyev, V. G., Shinde, V., Meyers, C. A., Gorbatyuk, M. S., Lin, J. H., et al. (2015). The loss of glucose-regulated protein 78 (GRP78) during normal aging or from siRNA knockdown augments human alpha-synuclein (alpha-syn) toxicity to rat nigral neurons. Neurobiol. Aging 36, 2213–2223. doi: 10.1016/j.neurobiolaging.2015.02.018
Sanchez-Martinez, A., Beavan, M., Gegg, M. E., Chau, K.-Y., Whitworth, A. J., and Schapira, A. H. V. (2016). Parkinson disease-linked GBA mutation effects reversed by molecular chaperones in human cell and fly models. Sci. Rep. 6:31380. doi: 10.1038/srep31380
Schlossmacher, M. G., Frosch, M. P., Gai, W. P., Medina, M., Sharma, N., Forno, L., et al. (2002). Parkin localizes to the lewy bodies of Parkinson disease and dementia with lewy bodies. Am. J. Pathol. 160, 1655–1667. doi: 10.1016/S0002-9440(10)61113-3
Senior, S. L., Ninkina, N., Deacon, R., Bannerman, D., Buchman, V. L., Cragg, S. J., et al. (2008). Increased striatal dopamine release and hyperdopaminergic-like behaviour in mice lacking both alpha-synuclein and gamma-synuclein. Eur. J. Neurosci. 27, 947–957. doi: 10.1111/j.1460-9568.2008.06055.x
Shahpasandzadeh, H., Popova, B., Kleinknecht, A., Fraser, P. E., Outeiro, T. F., and Braus, G. H. (2014). Interplay between sumoylation and phosphorylation for protection against α-synuclein inclusions. J. Biol. Chem. 289, 31224–31240. doi: 10.1074/jbc.M114.559237
Sharma, S. K., and Priya, S. (2017). Expanding role of molecular chaperones in regulating alpha-synuclein misfolding; implications in Parkinson’s disease. Cell. Mol. Life Sci. 74, 617–629. doi: 10.1007/s00018-016-2340-9
Shimura, H., Hattori, N., Kubo, S., Mizuno, Y., Asakawa, S., Minoshima, S., et al. (2000). Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat. Genet. 25, 302–305. doi: 10.1038/77060
Shin, Y., Klucken, J., Patterson, C., Hyman, B. T., and McLean, P. J. (2005). The co-chaperone carboxyl terminus of Hsp70-interacting protein (CHIP) mediates alpha-synuclein degradation decisions between proteasomal and lysosomal pathways. J. Biol. Chem. 280, 23727–23734. doi: 10.1074/jbc.M503326200
Shorter, J. (2016). Engineering therapeutic protein disaggregases. Mol. Biol. Cell 27, 1556–1560. doi: 10.1091/mbc.E15-10-0693
Singh, A., and Sen, D. (2017). MicroRNAs in Parkinson’s disease. Exp. Brain Res. 235, 2359–2374. doi: 10.1007/s00221-017-4989-1
Skibinski, G., Hwang, V., Ando, D. M., Daub, A., Lee, A. K., Ravisankar, A., et al. (2017). Nrf2 mitigates LRRK2- and alpha-synuclein-induced neurodegeneration by modulating proteostasis. Proc. Natl. Acad. Sci. U.S.A. 114, 1165–1170. doi: 10.1073/pnas.1522872114
Snyder, H., Mensah, K., Theisler, C., Lee, J., Matouschek, A., and Wolozin, B. (2003). Aggregated and monomeric alpha-synuclein bind to the S6’ proteasomal protein and inhibit proteasomal function. J. Biol. Chem. 278, 11753–11759. doi: 10.1074/jbc.M208641200
Sontag, E. M., Samant, R. S., and Frydman, J. (2017). Mechanisms and functions of spatial protein quality control. Annu. Rev. Biochem. 86, 97–122. doi: 10.1146/annurev-biochem-060815-014616
Souza, J. M., Giasson, B. I., Lee, V. M. Y., and Ischiropoulos, H. (2000). Chaperone-like activity of synucleins. FEBS Lett. 474, 116–119. doi: 10.1016/S0014-5793(00)01563-5
Spencer, B., Potkar, R., Trejo, M., Rockenstein, E., Patrick, C., Gindi, R., et al. (2009). Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in α-synuclein models of Parkinson’s and Lewy body diseases. J. Neurosci. 29, 13578–13588. doi: 10.1523/JNEUROSCI.4390-09.2009
Stefanis, L. (2012). α-synuclein in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2:a009399. doi: 10.1101/cshperspect.a009399
Stefanis, L., Larsen, K. E., Rideout, H. J., Sulzer, D., and Greene, L. A. (2001). Expression of A53T mutant but not wild-type alpha-synuclein in PC12 cells induces alterations of the ubiquitin-dependent degradation system, loss of dopamine release, and autophagic cell death. J. Neurosci. Off. J. Soc. Neurosci. 21, 9549–9560.
Tanaka, M., Kim, Y. M., Lee, G., Junn, E., Iwatsubo, T., and Mouradian, M. M. (2004). Aggresomes formed by α-synuclein and synphilin-1 are cytoprotective. J. Biol. Chem. 279, 4625–4631. doi: 10.1074/jbc.M310994200
Tanaka, Y., Engelender, S., Igarashi, S., Rao, R. K., Wanner, T., Tanzi, R. E., et al. (2001). Inducible expression of mutant α-synuclein decreases proteasome activity and increases sensitivity to mitochondria-dependent apoptosis. Hum. Mol. Genet. 10, 919–926. doi: 10.1093/hmg/10.9.919
Tang, F.-L., Erion, J. R., Tian, Y., Liu, W., Yin, D.-M., Ye, J., et al. (2015). VPS35 in dopamine neurons is required for endosome-to-golgi retrieval of Lamp2a, a receptor of chaperone-mediated autophagy that is critical for alpha-synuclein degradation and prevention of pathogenesis of Parkinson’s disease. J. Neurosci. Off. J. Soc. Neurosci. 35, 10613–10628. doi: 10.1523/JNEUROSCI.0042-15.2015
Teicher, B. A., and Tomaszewski, J. E. (2015). Proteasome inhibitors. Biochem. Pharmacol. 96, 1–9. doi: 10.1016/j.bcp.2015.04.008
Tenreiro, S., Reimao-Pinto, M. M., Antas, P., Rino, J., Wawrzycka, D., Macedo, D., et al. (2014). Phosphorylation modulates clearance of alpha-synuclein inclusions in a yeast model of Parkinson’s disease. PLOS Genet. 10:e1004302. doi: 10.1371/journal.pgen.1004302
Tetzlaff, J. E., Putcha, P., Outeiro, T. F., Ivanov, A., Berezovska, O., Hyman, B. T., et al. (2008). CHIP targets toxic alpha-Synuclein oligomers for degradation. J. Biol. Chem. 283, 17962–17968. doi: 10.1074/jbc.M802283200
Tian, T., Sun, Y., Wu, H., Pei, J., Zhang, J., Zhang, Y., et al. (2016). Acupuncture promotes mTOR-independent autophagic clearance of aggregation-prone proteins in mouse brain. Sci. Rep. 6:19714. doi: 10.1038/srep19714
Tofaris, G. K., Kim, H. T., Hourez, R., Jung, J.-W., Kim, K. P., and Goldberg, A. L. (2011). Ubiquitin ligase Nedd4 promotes α-synuclein degradation by the endosomal–lysosomal pathway. Proc. Natl. Acad. Sci. U.S.A. 108, 17004–17009. doi: 10.1073/pnas.1109356108
Trinh, J., and Farrer, M. (2013). Advances in the genetics of Parkinson disease. Nat. Rev. Neurol. 9, 445–454. doi: 10.1038/nrneurol.2013.132
Tsunemi, T., and Krainc, D. (2014). Zn2+ dyshomeostasis caused by loss of ATP13A2/PARK9 leads to lysosomal dysfunction and alpha-synuclein accumulation. Hum. Mol. Genet. 23, 2791–2801. doi: 10.1093/hmg/ddt572
Usenovic, M., Tresse, E., Mazzulli, J. R., Taylor, J. P., and Krainc, D. (2012). Deficiency of ATP13A2 leads to lysosomal dysfunction, α-synuclein accumulation, and neurotoxicity. J. Neurosci. 32, 4240–4246. doi: 10.1523/JNEUROSCI.5575-11.2012
Vogiatzi, T., Xilouri, M., Vekrellis, K., and Stefanis, L. (2008). Wild type alpha-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J. Biol. Chem. 283, 23542–23556. doi: 10.1074/jbc.M801992200
Volpicelli-Daley, L. A., Luk, K. C., Patel, T. P., Tanik, S. A., Riddle, D. M., Stieber, A., et al. (2011). Exogenous alpha-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 72, 57–71. doi: 10.1016/j.neuron.2011.08.033
Wakabayashi, K., Hayashi, S., Kakita, A., Yamada, M., Toyoshima, Y., Yoshimoto, M., et al. (1998). Accumulation of alpha-synuclein/NACP is a cytopathological feature common to Lewy body disease and multiple system atrophy. Acta Neuropathol. 96, 445–452. doi: 10.1007/s004010050918
Wolff, S., Weissman, J. S., and Dillin, A. (2014). Differential scales of protein quality control. Cell 157, 52–64. doi: 10.1016/j.cell.2014.03.007
Wyatt, A. R., Yerbury, J. J., Ecroyd, H., and Wilson, M. R. (2013). Extracellular chaperones and proteostasis. Annu. Rev. Biochem. 82, 295–322. doi: 10.1146/annurev-biochem-072711-163904
Xia, Q., Liao, L., Cheng, D., Duong, D. M., Gearing, M., Lah, J. J., et al. (2008). Proteomic identification of novel proteins associated with Lewy bodies. Front. Biosci. J. Virtual Libr. 13:3850–3856. doi: 10.2741/2973
Xilouri, M., Brekk, O. R., Kirik, D., and Stefanis, L. (2013). LAMP2A as a therapeutic target in Parkinson disease. Autophagy 9, 2166–2168. doi: 10.4161/auto.26451
Xilouri, M., Brekk, O. R., Polissidis, A., Chrysanthou-Piterou, M., Kloukina, I., and Stefanis, L. (2016). Impairment of chaperone-mediated autophagy induces dopaminergic neurodegeneration in rats. Autophagy 12, 2230–2247. doi: 10.1080/15548627.2016.1214777
Xilouri, M., Vogiatzi, T., Vekrellis, K., Park, D., and Stefanis, L. (2009). Abberant α-synuclein confers toxicity to neurons in part through inhibition of chaperone-mediated autophagy. PLOS ONE 4:e5515. doi: 10.1371/journal.pone.0005515
Yang, S.-Y., Beavan, M., Chau, K.-Y., Taanman, J.-W., and Schapira, A. H. V. (2017). A human neural crest stem cell-derived dopaminergic neuronal model recapitulates biochemical abnormalities in GBA1 mutation carriers. Stem Cell Rep. 8, 728–742. doi: 10.1016/j.stemcr.2017.01.011
Yu, F., Xu, H., Zhuo, M., Sun, L., Dong, A., and Liu, X. (2005). Impairment of redox state and dopamine level induced by alpha-synuclein aggregation and the prevention effect of hsp70. Biochem. Biophys. Res. Commun. 331, 278–284. doi: 10.1016/j.bbrc.2005.03.148
Zhou, W., Bercury, K., Cummiskey, J., Luong, N., Lebin, J., and Freed, C. R. (2011). Phenylbutyrate up-regulates the DJ-1 protein and protects neurons in cell culture and in animal models of Parkinson disease. J. Biol. Chem. 286, 14941–14951. doi: 10.1074/jbc.M110.211029
Keywords: α-synuclein, protein quality control, Lewy body, Parkinson’s disease, chaperone, autophagy, lysosome
Citation: Manecka D-L, Vanderperre B, Fon EA and Durcan TM (2017) The Neuroprotective Role of Protein Quality Control in Halting the Development of Alpha-Synuclein Pathology. Front. Mol. Neurosci. 10:311. doi: 10.3389/fnmol.2017.00311
Received: 19 May 2017; Accepted: 14 September 2017;
Published: 27 September 2017.
Edited by:
Paola Rusmini, Università degli Studi di Milano, ItalyReviewed by:
Ralf J. Braun, University of Bayreuth, GermanyMaciej Maurycy Lalowski, University of Helsinki, Finland
Copyright © 2017 Manecka, Vanderperre, Fon and Durcan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thomas M. Durcan, dGhvbWFzLmR1cmNhbkBtY2dpbGwuY2E=
†These authors have contributed equally to this work.