 Charlotte Veyrat-Durebex1,2
Charlotte Veyrat-Durebex1,2 Pascal Reynier1,2
Pascal Reynier1,2 Vincent Procaccio1,2Rudolf Hergesheimer3Philippe Corcia3,4Christian R. Andres3,5
Vincent Procaccio1,2Rudolf Hergesheimer3Philippe Corcia3,4Christian R. Andres3,5 Hélène Blasco2,3,5*
Hélène Blasco2,3,5*- 1Département de Biochimie et Génétique, Centre Hospitalier Universitaire, Angers, France
- 2INSERM 1083, CNRS, Equipe Mitolab, Institut MITOVASC, UMR 6015, Université d’Angers, Angers, France
- 3INSERM U930, Université François Rabelais de Tours, Tours, France
- 4Service de Neurologie, Centre Hospitalier Universitaire de Tours, Tours, France
- 5Laboratoire de Biochimie et Biologie Moléculaire, Centre Hospitalier Universitaire de Tours, Tours, France
A ketogenic diet (KD) is a normocaloric diet composed by high fat (80–90%), low carbohydrate, and low protein consumption that induces fasting-like effects. KD increases ketone body (KBs) production and its concentration in the blood, providing the brain an alternative energy supply that enhances oxidative mitochondrial metabolism. In addition to its profound impact on neuro-metabolism and bioenergetics, the neuroprotective effect of specific polyunsaturated fatty acids and KBs involves pleiotropic mechanisms, such as the modulation of neuronal membrane excitability, inflammation, or reactive oxygen species production. KD is a therapy that has been used for almost a century to treat medically intractable epilepsy and has been increasingly explored in a number of neurological diseases. Motor function has also been shown to be improved by KD and/or medium-chain triglyceride diets in rodent models of Alzheimer’s disease, Parkinson’s disease, amyotrophic lateral sclerosis, and spinal cord injury. These studies have proposed that KD may induce a modification in synaptic morphology and function, involving ionic channels, glutamatergic transmission, or synaptic vesicular cycling machinery. However, little is understood about the molecular mechanisms underlying the impact of KD on motor function and the perspectives of its use to acquire the neuromuscular effects. The aim of this review is to explore the conditions through which KD might improve motor function. First, we will describe the main consequences of KD exposure in tissues involved in motor function. Second, we will report and discuss the relevance of KD in pre-clinical and clinical trials in the major diseases presenting motor dysfunction.
Introduction
The KD, tested for the first time in 1921 for intractable childhood epilepsy, is based on a normocaloric, high fat, adequate-protein, and low-carbohydrate diet resulting in the production of KBs (Keene, 2006). Different types of KDs have been described. The classic ketogenic therapy is based on a diet providing 90% of calories from long-chain fatty acids, a restricted protein portion (1 g/kg/day), and minimal carbohydrates. Traditionally, the diet is comprised of four parts fat, mainly LCTs, for one part carbohydrates and proteins. The ratio can be modified to 3:1, 2:1, or 1:1, respectively, similar to the modified Atkins diet (Kossoff et al., 2003). The MCTs diet is also proposed with 60% of calories from octanoate and decanoate that are more ketogenic than LCTs (Huttenlocher, 1976). The last alternative to a ketogenic therapy is the low glycemic index diet characterized by higher amounts of carbohydrates with low glycemic index (Coppola et al., 2011).
Despite the underlying, unclear mechanisms, KD is considered to be a “neuroketotherapeutic” (Koppel and Swerdlow, 2017). The efficacy of KD in drug-resistant epilepsy in children and adult patients has been proven for almost a century (Stafstrom and Rho, 2012) with more than 50% reduction in seizures for intractable childhood epilepsy (Lefevre and Aronson, 2000). KD has progressively gained interest for the treatment of other diseases as a stand-alone metabolic therapy or as part of a general, therapeutic strategy (Paoli et al., 2014). Various mechanisms have been advocated to explain the anti-convulsive and neuroprotective effects of KD, such as a decrease in glucose metabolism due to the increase in lipid oxidation, a reduction in ROS production, an increase in ATP, and modulations of neuronal membrane excitability, inflammation, oxidative stress, and mitochondrial function (Gasior et al., 2006; Elia et al., 2017).
Thus, KD is expected to be highly relevant in diseases characterized by any of these mechanisms. For example, motor dysfunction, involving the nervous system, muscles and tendons, observed in neuromuscular diseases or as a component of various pathological conditions, may benefit from such treatment. As non-pharmacological management is rarely considered and little data has been published on dietary therapies, we have focused our review on the potential benefit of such KD therapies on motor function. Firstly, we will describe the neuroprotective effects of KD, especially in tissues involved in motor function. Secondly, we will present and discuss pre-clinical and clinical trials of KD for diseases presenting a motor dysfunction. Finally, we will present some perspectives of other new therapeutics, based on metabolic factors targeting energy metabolism.
Protective Effects of KD on the Neuromuscular System
The effects of KD administration on the neuromuscular system come through different mechanisms. For one, KD can directly induce metabolic shifts due to the high blood levels of KBs and to the restriction of carbohydrate intake (Danial et al., 2013). KD can also modify nutrient-integrating pathways, such as the mTOR pathway, involved in autophagy and mitophagy-related mitochondrial renewal. Finally, KD might have potential, indirect roles, such as effects on neurotransmission, oxidative stress, and inflammatory mechanisms. Figure 1 sums up the cellular mechanisms induced by KD.
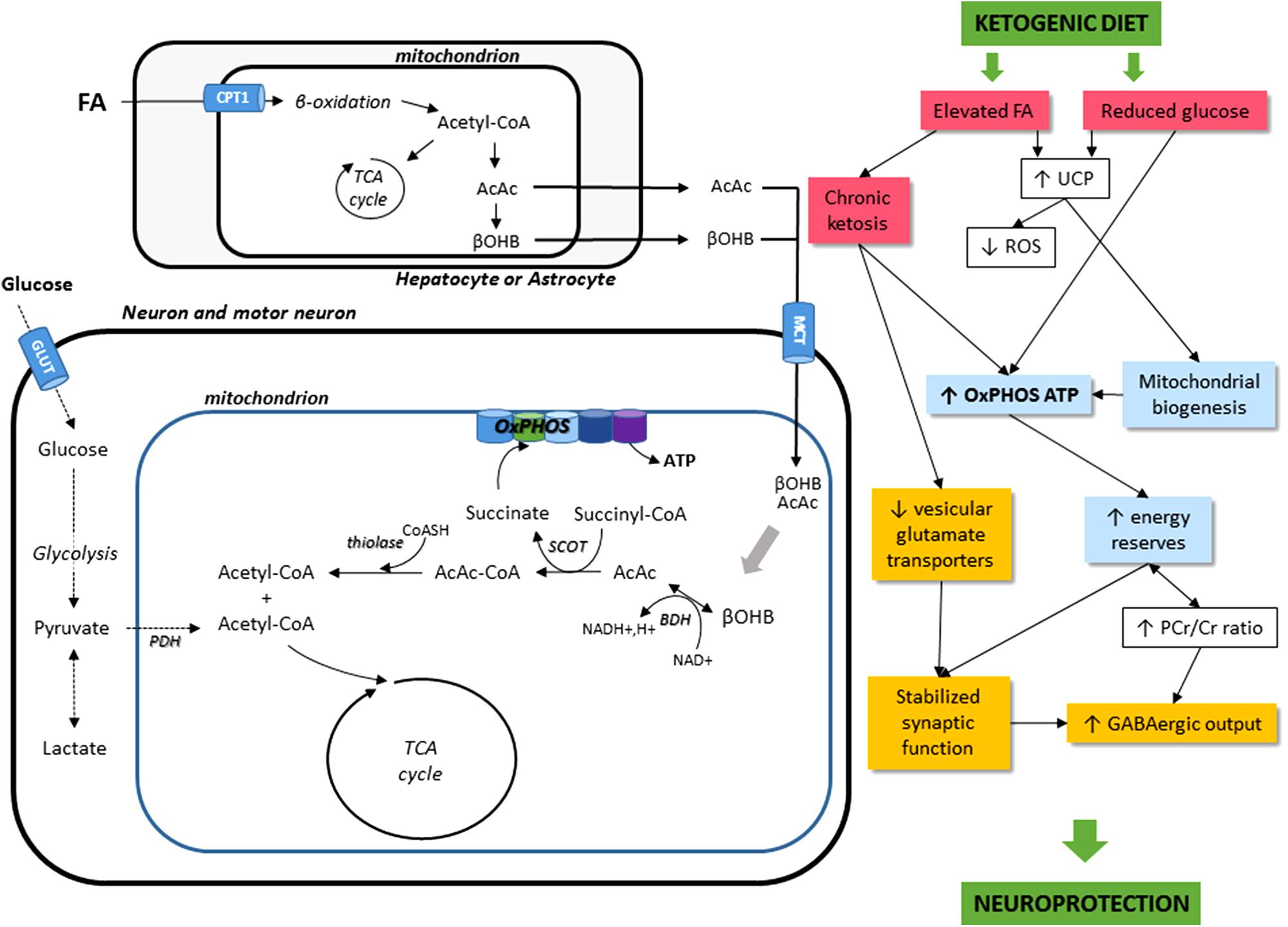
FIGURE 1. Schematic illustration of the main cellular mechanisms in the KD on the neuromuscular system. The main energetic substrate of the nervous system is glucose, but alternative substrates exist, for instance pyruvate (Gonzalez et al., 2005), lactate (Dienel, 2012), and βOHB (Kunnecke et al., 1993; Achanta and Rae, 2017). Most KBs (β-hydroxybutyrate and acetoacetate AcAc) used by the brain are supplied by the liver, but they can also be synthesized in astrocytes, which are the only cell type in the brain capable of oxidizing fatty acids (FA). KBs enter cells via the monocarboxylate transporter (MCT). Then, acetyl-CoA can enter the TCA cycle to produce ATP. KD leads to a decrease in glycolytic ATP generation and an increase in ATP generation in mitochondria. FA, fatty acids; CPT1, carnitine palmitoyltransferase 1; AcAc, acetoacetate; βOHB, β-hydroxybutyrate; OxPHOS, oxidative phosphorylation; TCA cycle, tricaboxylic acid cycle; ATP, adenosine triphosphate; BDH, beta-hydroxybutyrate dehydrogenase; SCOT, succinyl-CoA:3-ketoacid coenzyme A transferase; PDH, pyruvate dehydrogenase; GLUT, glucose transporters; UCP, uncoupling proteins; ROS, reactive oxygen species; PCr/Cr, phosphocreatine/creatine; GABA, gamma-aminobutyric acid.
Metabolism Switch
Ketogenic diet has a high impact in tissues with a high-energy requirement and with challenges from modifications in metabolic substrates, such as the neuromuscular system. The brain represents 2% of one’s body weight but consumes about 20% of the body’s energy stores (Belanger et al., 2011) in order to fuel the processes of neurotransmitter production/recycling, vesicular trafficking, maintenance of ion gradients for the propagation of action potentials, and memory, to name a few. Similarly, in the resting state, 20% of the energy expenditure is devoted to muscle (Gallagher et al., 1998), and it can largely increase with muscle contractions, for example for transforming organelles, enzymatic activities, intracellular signaling, and transcriptional responses (Coffey et al., 2007).
Ketogenic diet promotes KBs (acetoacetate and β-hydroxybutyrate) production in the liver from acetyl-CoA formed during mitochondrial β-oxidation of fatty acids. Some of the acetyl-CoA enters the TCA cycle, and the excess is used to form acetoacetate, which could be converted to βOHB by βOHB dehydrogenase (BDH) enzyme or spontaneously converted to acetone (Newman and Verdin, 2014). KBs are transported to other tissues (brain, muscle, and heart) through the blood and used as fuel, especially in the brain (Kunnecke et al., 1993; Rae et al., 2012; Achanta and Rae, 2017). It has also been reported that astrocytes can produce KBs from fatty acids (Auestad et al., 1991) and leucine (Bixel and Hamprecht, 1995). Astrocytes present the same preference for fatty acids (rather than glucose) as metabolic fuel and have enzymatic machinery similar to that of cultured hepatocytes (Guzman and Blazquez, 2001).
β-Hydroxybutyrate and acetoacetate enter cells via the MCT and provide an alternative substrate for brain. Several studies seem to highlight that KBs are a preferred carbon source under certain conditions (LaManna et al., 2009; Lund et al., 2011; Zhang Y. et al., 2013; Chowdhury et al., 2014; Valente-Silva et al., 2015), but these finding remain controversial (McKenna, 2012; Achanta et al., 2017). Some studies evoked that KBs provide more efficient energy source than glucose, even for the brain. They are metabolized faster than glucose and bypass the glycolytic pathway by directly entering the TCA cycle, whereas glucose has to first undergo glycolysis (Veech et al., 2001; LaManna et al., 2009; Elamin et al., 2017). Consequently, KBs lead to a decrease in glycolytic ATP generation and an increase in ATP generation by mitochondrial oxidation (Kim et al., 2010; Steriade et al., 2014; Hyatt et al., 2016). Moreover, Elamin et al. (2017) showed that whereas both glucose and KBs produce two molecules of acetyl-CoA, glucose reduces four molecules of NAD+ and KBs reduce either one (for conversion of BOHB in acetoacetate), or none (for conversion of acetoacetate in acetyl-CoA) during acetyl-CoA synthesis. KD is associated with a coordinated upregulation of hippocampal genes encoding metabolic and mitochondrial enzymes (Bough et al., 2006). KD also leads to fatty acid-mediated activation of peroxisome proliferator-activated receptor α (PPARα) that inhibits glycolysis and fatty acid synthesis, promotes the transcription of ketogenic enzymes, promoting mitochondrial and peroxisomal fatty acid oxidation (Cullingford, 2004). Moreover, the cellular energy reserve is increased as a result from a higher phosphocreatine/creatine ratio in the hippocampal tissue (Bough et al., 2006).
Taken altogether, neurons possess a better resistance and adaptive ability to metabolic stress and challenges, both having to do with a more energy-efficient fuel source and a larger mitochondrial load based on a stimulation of mitochondrial biogenesis (Bough et al., 2006; Srivastava et al., 2012, 2013).
Antioxidant Effects
The accumulation of certain metabolites and hypoxia produced during muscular contractions, along with the high energetic requirement of the brain, might increase ROS production through the mitochondrial electron transport chain (Morales-Alamo and Calbet, 2014). Despite possible, positive effects of ROS production on mitochondrial adaptations (Ji et al., 2016), the deleterious effect of oxidative stress must be controlled, primarily in the brain (Islam, 2017). Veech (2014) reported a decrease in free radical production through the reduction of coenzyme Q, induced by KBs. Importantly, a decrease in oxidative stress and an increase in mitochondrial glutathione and glutathione peroxidase activity were observed during KD, which may protect tissues from injury (Ziegler et al., 2003; Bough et al., 2006; Stafford et al., 2010; Stafstrom and Rho, 2012; Greco et al., 2016). As ROS generation partially reflects mitochondrial function, the decrease in ROS production may be a result of an effect on NADH oxidation or on calcium overload (Kim et al., 2007; Maalouf et al., 2007). Some authors have described that, in KD, there is an elevated production of mitochondrial uncoupling proteins, thereby decreasing ROS levels probably via fatty acids found elevated in treated patients (Fraser et al., 2003; Sullivan et al., 2004; Maalouf et al., 2007). Carbohydrate restriction also induces stress response proteins, leading to a lowering of ROS and the maintenance of mitochondrial function (Lee et al., 1999). Tieu et al. (2003) observed a beneficial effect of KD in restoring mitochondrial Complex I function following inhibition by pharmacological agents. These results point out a central role of KD in oxidative stress management associated with the mitochondria.
Synaptic Transmission
Numerous reports have suggested that the anticonvulsive mechanisms behind ketosis are based on a metabolic shift between the neurotransmitters GABA and glutamate, resulting in an increased inhibition and/or decreased excitation (Bough and Rho, 2007; Yudkoff et al., 2007). This could spark attention toward neuromuscular transmission (Diana et al., 2017). Indeed, KD can induce an increase in glutamate decarboxylase expression in the striatum of rats (Cheng et al., 2004) and an alteration of astrocytic GABA degradation via a modification of GABA transaminase activity (Suzuki et al., 2009). Moreover, GABA content is increased by KBs in rat brain synaptosomes (Erecinska et al., 1996), rat hippocampus (Calderon et al., 2017), and in the brain of patients (Wang et al., 2003). Some authors have reported the inhibition of glutamatergic, synaptic transmission (Danial et al., 2013; Lutas and Yellen, 2013), which implies a blockade of vesicular glutamate transporters (Juge et al., 2010). This interference with glutamate-mediated toxicity, implied in neuronal injury, could explain the interest for KD in neurological diseases.
Signaling Pathways
Ketogenic diet could modulate crucial mechanisms in cellular homeostasis. For example, mTOR and AMPK pathways involved in cell proliferation, energetic metabolism, or protein biosynthesis could be implicated. KD induces the binding of insulin and free IGF-1 to their specific tyrosine kinase receptors and activates the phosphatidylinositol-3 kinase (PI3K)-Akt-mammalian target of rapamycin complex 1 (mTORC1). However, this effect is counteracted by the decrease in the intracellular ATP/AMP ratio and the activation of liver kinase B1 (LKB1)-AMP-activated protein kinase (AMPK) signaling, inhibiting mTORC1 (Newman and Verdin, 2014). Such effects may be essential for motor dysfunction in ALS, for example, as deregulation in mTOR and AMPK signaling pathways has been described in this disease (Saxena et al., 2013; Perera and Turner, 2016).
Anti-inflammatory Effects
Fasting and KD have been associated with effects on inflammatory mechanisms (Stamp et al., 2005; Dupuis et al., 2015). Some authors have observed an elevated expression of cytokine interferon-γ in the hippocampus of rats, thus protecting cells from excitotoxicity (Lee et al., 2006). Moreover, fatty acids activate PPARα which, in turn, inhibits the pro-inflammatory NF-κB and cytokines, such as IL-6 and TNFα (Cullingford, 2004; Nandivada et al., 2016). βOHB inhibits the NLRP3 inflammasome, which controls the activation of caspase-1 and the release of the pro-inflammatory cytokines, IL-1β and IL-18 (Youm et al., 2015; Goldberg et al., 2017).
Use of KD in Motor Dysfunction
The beneficial effect of KD has been hypothesized in various diseases, such as epilepsy, metabolic defects, cancers, autism, depression, migraines, narcolepsy, Parkinson’s disease, and Alzheimer’s disease (Vanitallie et al., 2005; Baranano and Hartman, 2008; Newport et al., 2015). Also, a therapeutic effect has been proposed for diseases with substantial motor dysfunction, as reported in Table 1 summing the main pre-clinical and clinical evaluations of KD in these diseases.
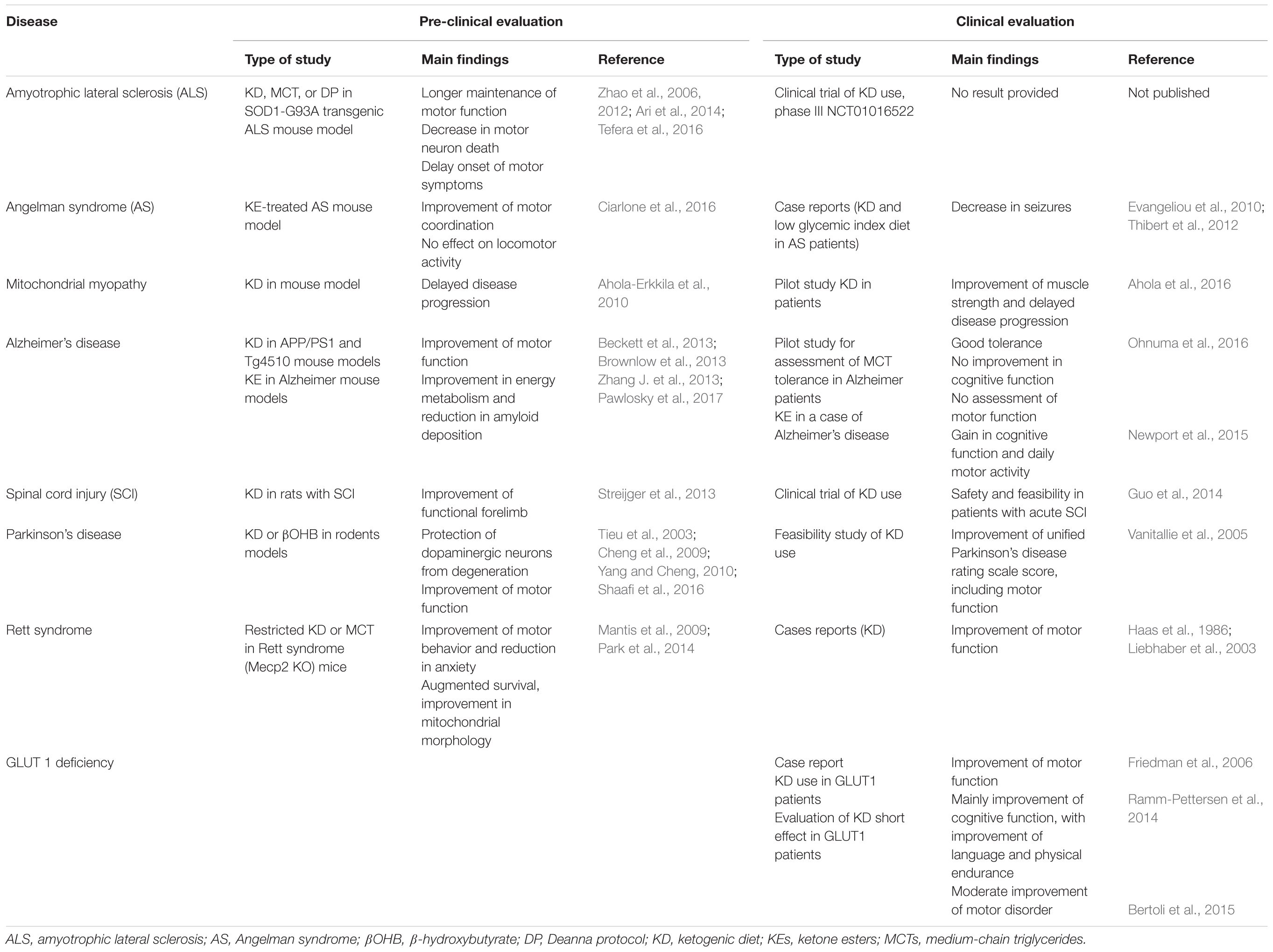
TABLE 1. Main preclinical and clinical evaluations of KD and treatments derived from the KD in diseases with motor dysfunction.
Amyotrophic Lateral Sclerosis (ALS)
Rational of the Use of KD in ALS
Amyotrophic lateral sclerosis is a fatal, neurodegenerative condition characterized by motor neuron degeneration that leads to progressive motor weakness and death between 2 and 5 years from onset (Korner et al., 2013). Many molecular mechanisms have been involved in ALS pathophysiology, but the starting point of the disease remains unclear. Drug development has yielded few successes, and the prognosis has not changed dramatically since the first reports, nearly 150 years ago.
Regarding studies on ALS mouse models, Siva (2006) proposed KD as a promising strategy to slow down the progression of ALS, especially through the increase in mitochondrial function. The main mechanisms related to mitochondrial malfunction and ALS have been recently reviewed (Carri et al., 2017).
The heterogeneous effects of KD on the neuromuscular system could support the interest in KD treatment in a heterogeneous disease, such as ALS. In fact, many hypotheses have been raised on ALS pathophysiology. Among the known gene mutations associated with ALS, TAR DNA-binding protein (TARDPB) gene is related with neuronal density of mitochondria and cristae formation (Xu et al., 2010; Stribl et al., 2014), SOD1 is associated with oxidative stress, and CHCHD10 is involved in oxidative phosphorylation and the maintenance of mitochondrial cristae morphology (Bannwarth et al., 2014). Other authors have described a decreased activity in mitochondrial Complex I in skeletal muscle and the spinal cord of ALS patients (Vielhaber et al., 2000). Moreover, several reports have highlighted metabolic alterations in this disease, enabling a possible role of therapeutics targeting energetic metabolism (Dupuis et al., 2008; Blasco et al., 2016). Various studies have directly observed shifts in TCA cycle intermediates, such as decreased C4-intermediate levels in ALS mice (Niessen et al., 2007), or decreased oxidative phosphorylation and ATP production in cellular models, transgenic mice and ALS patients (Jung et al., 2002; Mattiazzi et al., 2002; Menzies et al., 2002; Wiedemann et al., 2002; Browne et al., 2006). GABA imbalance and glutamate neurotransmission are also involved in ALS pathophysiology and may be relevant to modulate (Blasco et al., 2014; Diana et al., 2017). Likewise, the roles of neuro-inflammation and oxidative stress have often been indicated in in vitro and in vivo experiments, justifying a therapeutic approach also focused on these mechanisms (Liu and Wang, 2017; Liu et al., 2017). All these mechanisms involved in ALS pathophysiology could justify the use of KD as therapy in this disease.
Evaluation of KD in ALS
Zhao et al. (2006) reported in a study that a higher motor neuron survival and an improved motor function resulted from KD administration through the gain in mitochondrial energy production in SOD1-G93A transgenic ALS mice. They noticed that KD-fed mice maintained motor function longer than SOD1-mutant mice fed a SD and had more intact motor neurons in the lumbar spinal cord. They also demonstrated that βOHB prevented rotenone-mediated inhibition of mitochondrial Complex I (Zhao et al., 2006). Zhao et al. (2012) confirmed the promising therapeutic approach by way of improvement in mitochondrial function and motor neuron count in an ALS mouse model following administration of caprylic acid, a MCT precursor of KB.
The link between lipid concentrations and survival remains an enigma in ALS. Nevertheless, a recent study has revealed that higher caloric intake improves survival and that low cholesterol may end up being deleterious in ALS patients (Blasco et al., 2017). A phase III clinical trial evaluating the safety and the tolerance of KD in ALS patients fed through a gastrostomy tube has been conducted in the United States (clinicaltrials.gov NCT01016522), but results have not yet been published. The primary outcome measure is to evaluate the prevention of malnutrition in ALS patients but not to identify the benefit in the disease’s prognosis.
Angelman Syndrome (AS)
Rational of the Use of KD in AS
Angelman syndrome is a devastating, neurological disorder with no treatment. AS patients suffer from motor dysfunction, intellectual disability, frequent smiling and laughter, lack of speech, and severe seizures. This syndrome is due to an alteration in the E3 ubiquitin ligase (Margolis et al., 2015). The main pathophysiological pathways concern an impairment in synaptic plasticity, deregulated dopaminergic and GABAergic neurons, abnormal mTOR signaling in the cerebellum, abnormal cell contact signaling and excitation/inhibition imbalance (Bi et al., 2016). It has been shown, as well, that the stimulation of mitochondrial biogenesis by KD improves the hippocampal deficits in AS mice (Bough et al., 2006; Su et al., 2011).
Evaluation of KD in AS
Ciarlone et al. (2016) studied the effect of KEs, precursors of KBs, in AS mouse models. They investigated behavioral and metabolic outcomes, as well as cognitive and motor functions in AS and control mice either receiving KEs or not.
The major findings highlighted that KEs supplementation improves motor coordination but does not affect general locomotor activity in AS mice. The average latency for falling on the accelerating rotarod was significantly increased in AS mice treated with KE compared to untreated AS mice, but the performances did not reach those of the control mice. KE-treated AS mice showed a significant increase in the latency on the wire hang test compared to AS mice, but the latency remained inferior to WT mice for which KE had no effect. The severity of the hindlimb clasping score was significantly decreased in KE-treated AS mice compared to untreated AS mice.
Both the KD and the low glycemic index diets have been administered to patients and have illustrated promising results in seizures of AS (Evangeliou et al., 2010; Thibert et al., 2012), suggesting that the benefit rather stems from carbohydrate restriction and not from increasing blood KB levels (Gano et al., 2014).
Mitochondrial Myopathy
Mitochondrial disorders are clinically and genetically heterogeneous diseases with a neuromuscular component caused by mutations either in mitochondrial DNA or in nuclear genes encoding mitochondrial proteins. Interestingly, KD has showed a relevant effect on a mouse model for late-onset mitochondrial myopathy characterized by generalized muscle weakness (Ahola-Erkkila et al., 2010). KD decreased the amount of Cytochrome c Oxidase-negative muscle fibers and protected mitochondrial ultrastructure in the muscle. A recent study suggested that the use of KD in mitochondrial myopathy enhanced muscle strength and delayed the progression of the disease but might induce muscle damage in a subpopulation (Ahola et al., 2016).
Other Diseases with a Neuromuscular Component
Various studies have supported a regeneration in motor performance with KD in rodent models of Alzheimer’s disease, spinal cord injury, Parkinson’s disease, and Rett syndrome. Various mouse models of Alzheimer’s disease (i.e., mice carrying mutations in amyloid precursor peptide, APP, and/or presenilin, PS, as models of amyloid deposition, and Tg4510 mouse model as a model of tau deposition) have presented an improved latency for falling on Rotarod apparatus under KD without a reduction in β-amyloid or tau accumulation (Beckett et al., 2013; Brownlow et al., 2013). KD improved forelimb motor function in rodents after spinal cord injury with maintenance of the functional benefits when returning to a SD after 12 weeks of KD (Streijger et al., 2013). A number of studies have suggested that KD and βOHB administration protect dopaminergic neurons from degeneration (Kashiwaya et al., 2000) and improve motor function in rodent models of Parkinson’s disease (Tieu et al., 2003; Cheng et al., 2009; Yang and Cheng, 2010; Shaafi et al., 2016). Mantis et al. (2009) illustrated a recovery in motor behavior in Rett syndrome mice, principally by caloric restriction in the diet.
The use of KD in these diseases presenting a neuromuscular component was also studied in human patients. A pilot study of the use of KD in Parkinson’s patients showed a global enhancement in the Unified Parkinson’s Disease Rating Scale scores in all five of its patients, including motor function (Vanitallie et al., 2005). Furthermore, case reports showed improvement in motor functions in patients with Rett syndrome (Haas et al., 1986; Liebhaber et al., 2003) and patients with GLUT1 deficiency (Friedman et al., 2006). Two other studies have used KD in patients with GLUT1 deficiency, mainly to assess short-term effects of its use, including the amelioration of cognitive function. Yet, both of them also highlighted a moderate improvement of motor disorders (Ramm-Pettersen et al., 2014; Bertoli et al., 2015). It should be noted that ketogenic treatments have been studied in patients with Alzheimer’s disease, but mainly for the assessment on the improvement in cognitive functions (Reger et al., 2004; Henderson et al., 2009).
What Importance Should be Given to KD to Improve Motor Dysfunction?
With respect to the neuroprotective effect of KD involving the different mechanisms previously cited and largely involved in pathophysiological mechanisms of various neurological and neuromuscular diseases, the theoretical benefit of KD is not doubtful. As KD and fasting share similar, potentially beneficial effects, we suspect that fasting could be considered as a therapy. However, this question was raised in a trial evaluating KD versus an intermittent fasting regimen in mice undergoing acute seizure tests (Hartman et al., 2010). Although both strategies were associated with anticonvulsive properties, the mechanisms were different, underlining the need for further studies to fully understand the different strategies.
Contrary to other indications such as epilepsy or cognitive dysfunction, little clinical data is available to assess beneficial effects of KD on motor function. Importantly, KD also has short and long-term, adverse effects regardless of the disease. The classic KD has showed more problems of tolerability than the various modified diets. Major short-term side effects are gastro-intestinal disturbances (which could lead to poor compliance), acidosis, and hypoglycemia (Dhamija et al., 2013; Branco et al., 2016). Long-term side effects include hypercholesterolemia, nephrolithiasis, and premature heart disease (Dhamija et al., 2013). Some authors showed that 15 months of KD in children with intractable epilepsy induced a declining linear growth status with constant weight and resting energy expenditure (Groleau et al., 2014). Clinicians and dietitians currently attempt to prevent KD side effects using gradual initiation of the KD (Bergqvist et al., 2005), alternative diets, such as the modified Atkins diet and the low glycemic index diet, or administering supplements, such as selenium, citrate, calcium, and vitamin D (Bergqvist et al., 2003, 2007; McNally et al., 2009).
The balance between efficacy and toxicity of KD provides substantial promise for such treatment for motor impairment, but it is probably insufficient when administered alone. However, the lack of double-blind, randomized control studies accurately measuring the effects on motor function prevents one from obtaining clear conclusions about this treatment. As some findings about the neuroprotective effect of KD are contradictory, we have to consider the standardization of human and animal protocols. A greater understanding and a better clinical evaluation of KD effects would cause the KD treatment to merit more attention.
Perspectives of Metabolism-Based Therapeutics in the Management of Neuromuscular Diseases
Medium-Chain Triglycerides (MCTs)
Medium-chain triglycerides have been studied as an alternative to KD. In the context of ALS, Zhao et al. (2012) reported the recovery of motor function after restoration of energy metabolism in an ALS mouse model treated with MCT (caprylic triglyceride). Although the results were highly promising in enhancing motor performances, there was no effect on animal survival. Nonetheless, this study shows potential for clinical trials. Furthermore, a recent study revealed that triheptanoin, another MCT, protected lumbar motor neurons and delayed onset of motor symptoms in an ALS mouse model (Tefera et al., 2016). Triheptanoin supplementation was also tested in Rett syndrome mice. The results revealed an augmented survival in triheptanoin-fed mice, a gain in social interactions and motor function, an improvement in mitochondrial morphology in skeletal muscle (Park et al., 2014). A diet with MCT was also tested in patients with Alzheimer’s disease, primarily for the evaluation of cognitive function (Ohnuma et al., 2016).
Deanna Protocol
The Deanna protocol (DP) is a metabolic therapy that provides alternative, energetic fuels. The DP is essentially comprised of arginine alpha-ketoglutarate (AAKG) and other molecules, such as ubiquinol, MCTs, and gamma-aminobutyric acid. The beneficial role of ubiquinol is based on its role in the electron transport chain in mitochondria and ATP production.
Ari et al. (2014) reported that DP would increase motor function and survival in the SOD1-G93A ALS mouse model. ALS mice were fed a SD, KD, or either diets containing ingredients of the DP. SD+DP-fed mice showed a better neurological score than the controls, and KD-fed mice exhibited better motor performance in all motor function tests compared to the controls. The Rotarod test revealed better motor performance in KD mice and the grip test also highlighted better performance in KD and SD+DP mice compared to controls. The Paw Grip Endurance test showed higher motor performance in KD or SD+DP mice than in controls. Significantly, ALS mice fed SD+DP or KD+DP had a remarkable, extended survival time. Authors have suggested that AAKG enhances blood flow, increases muscle protein synthesis, and ameliorates muscle strength though the metabolic role of α-ketoglutarate. MCTs could have a similar function though their conversion to KBs and acetyl-CoA. The production of energetic intermediates bypassing transport and metabolism of glucose, probably deregulated in ALS, may increase motor function in this disease.
Ketone Esters (KEs)
One therapeutic goal is to replace the KD, and its strict requirements for observance, with dietary supplements that could generate sustained ketosis. KEs are considered to be substitutes for KD and are suitable for oral treatment, compared to KBs. Ingestion of KEs can directly increase blood levels of KBs without the delay observed in KD or fasting (Hashim and VanItallie, 2014). The main KEs used are 1,3-butanediol monoester of βOHB and glyceryl-tri-3-hydroxybutyrate (3GHB) (Brunengraber, 1997). Oral and intravenous administering of KEs demonstrated that they were safe and well-tolerated in animals and humans (Clarke et al., 2012a,b), which makes them an interesting complementary treatment.
Ketone ester supplementation has been shown to relieve symptoms in AS mouse models (Ciarlone et al., 2016). Positive effects of KEs have been observed in Alzheimer’s mouse models with an improvement in energy metabolism and a reduction in amyloid-β deposition (Zhang J. et al., 2013; Pawlosky et al., 2017). A study in one patient with Alzheimer’s disease treated with KEs exhibited a gain in cognitive function and physical activity (Newport et al., 2015).
Conclusion
The perspective of the use of KD in a variety of diseases has been growing these past recent years. This adjuvant therapy has shown interesting potential in neurological and neuromuscular diseases. The obtained experimental results point out the neuroprotective role of the increase of KBs levels and the reduction of blood glucose, in association with the involvement of various signaling pathways (e.g., IGF-1/AKT/mTOR, AMPK) and effects on inflammation and oxidative status. The utility of KD and its variations for the treatment of neuromuscular diseases suggest a central mechanism in restoring energy metabolism, especially in disease with an impairment of glucose metabolism. Although various studies have suggested a positive effect of KD in a number of neuromuscular diseases, several obstacles remain to be tackled before these findings can be applied widely in the clinic, and further research is necessary to elucidate the mechanisms that mediate the neuroprotective effects. Indeed, several points remain unexplored, as for example the direct effects of KBs on gene expression. Many questions remain unanswered: is there a neuroprotective effect of KD in all conditions, pathological or physiological? If the KD can be beneficial for the brain, can it be deleterious for other organs? How long an exposure to the diet is necessary to confer long-term benefit? Does a diet monitoring improved efficacy? It should also be noted that many studies on KD were performed on animal models, highlighted that clinical trials of its use in affected patients are essential.
The effects of KD involve numerous mechanisms highlighting specific pathways that can represent interesting therapeutic approaches. Despite the well-documented advantage of KD in the treatment of several diseases, the adverse effects should also be acknowledged. More and more studies search for alternative treatments or diets to KD, presenting similar effects with fewer adverse side effects and fewer daily constraints for patients. However, these diets might represent an exceptional option as a co-adjuvant therapy.
Author Contributions
CV-D and HB wrote the manuscript and designed the review. RH helped write new paragraphs of the manuscript to perform responses to reviewers. CV-D and RH edited the manuscript following reviewers comments. CV-D, PR, VP, RH, PC, CA, and HB have been involved in drafting the manuscript or revising it critically for important intellectual content. All authors contributed to the conception of this review article. All authors read and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer YN and handling Editor declared their shared affiliation.
Abbreviations
ALS, amyotrophic lateral sclerosis; AS, Angelman syndrome; ATP, adenosine triphosphate; βOHB, β-hydroxybutyrate; DP, Deanna protocol; GABA, gamma amino-butyric acid; KBs, ketone bodies; KD, ketogenic diet; KEs, ketone esters; LCTs, long-chain triglycerides; MCTs, medium-chain triglycerides; MCT, monocarboxylate transporter; NADH, nicotinamide adenine dinucleotide; ROS, reactive oxygen species; SD, standard diet; TCA, tricarboxylic acid.
References
Achanta, L. B., and Rae, C. D. (2017). Beta-hydroxybutyrate in the brain: one molecule, multiple mechanisms. Neurochem. Res. 42, 35–49. doi: 10.1007/s11064-016-2099-2
Achanta, L. B., Rowlands, B. D., Thomas, D. S., Housley, G. D., and Rae, C. D. (2017). Beta-hydroxybutyrate boosts mitochondrial and neuronal metabolism but is not preferred over glucose under activated conditions. Neurochem. Res. 42, 1710–1723. doi: 10.1007/s11064-017-2228-6
Ahola, S., Auranen, M., Isohanni, P., Niemisalo, S., Urho, N., Buzkova, J., et al. (2016). Modified Atkins diet induces subacute selective ragged-red-fiber lysis in mitochondrial myopathy patients. EMBO Mol. Med. 8, 1234–1247. doi: 10.15252/emmm.201606592
Ahola-Erkkila, S., Carroll, C. J., Peltola-Mjosund, K., Tulkki, V., Mattila, I., Seppanen-Laakso, T., et al. (2010). Ketogenic diet slows down mitochondrial myopathy progression in mice. Hum. Mol. Genet. 19, 1974–1984. doi: 10.1093/hmg/ddq076
Ari, C., Poff, A. M., Held, H. E., Landon, C. S., Goldhagen, C. R., Mavromates, N., et al. (2014). Metabolic therapy with Deanna Protocol supplementation delays disease progression and extends survival in amyotrophic lateral sclerosis (ALS) mouse model. PLOS ONE 9:e103526. doi: 10.1371/journal.pone.0103526
Auestad, N., Korsak, R. A., Morrow, J. W., and Edmond, J. (1991). Fatty acid oxidation and ketogenesis by astrocytes in primary culture. J. Neurochem. 56, 1376–1386. doi: 10.1111/j.1471-4159.1991.tb11435.x
Bannwarth, S., Ait-El-Mkadem, S., Chaussenot, A., Genin, E. C., Lacas-Gervais, S., Fragaki, K., et al. (2014). A mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvement. Brain 137(Pt 8), 2329–2345. doi: 10.1093/brain/awu138
Baranano, K. W., and Hartman, A. L. (2008). The ketogenic diet: uses in epilepsy and other neurologic illnesses. Curr. Treat Options Neurol. 10, 410–419. doi: 10.1007/s11940-008-0043-8
Beckett, T. L., Studzinski, C. M., Keller, J. N., Paul Murphy, M., and Niedowicz, D. M. (2013). A ketogenic diet improves motor performance but does not affect beta-amyloid levels in a mouse model of Alzheimer’s disease. Brain Res. 1505, 61–67. doi: 10.1016/j.brainres.2013.01.046
Belanger, M., Allaman, I., and Magistretti, P. J. (2011). Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metab. 14, 724–738. doi: 10.1016/j.cmet.2011.08.016
Bergqvist, A. G., Chee, C. M., Lutchka, L., Rychik, J., and Stallings, V. A. (2003). Selenium deficiency associated with cardiomyopathy: a complication of the ketogenic diet. Epilepsia 44, 618–620. doi: 10.1046/j.1528-1157.2003.26102.x
Bergqvist, A. G., Schall, J. I., Gallagher, P. R., Cnaan, A., and Stallings, V. A. (2005). Fasting versus gradual initiation of the ketogenic diet: a prospective, randomized clinical trial of efficacy. Epilepsia 46, 1810–1819. doi: 10.1111/j.1528-1167.2005.00282.x
Bergqvist, A. G., Schall, J. I., and Stallings, V. A. (2007). Vitamin D status in children with intractable epilepsy, and impact of the ketogenic diet. Epilepsia 48, 66–71. doi: 10.1111/j.1528-1167.2006.00803.x
Bertoli, S., Neri, I. G., Trentani, C., Ferraris, C., De Amicis, R., Battezzati, A., et al. (2015). Short-term effects of ketogenic diet on anthropometric parameters, body fat distribution, and inflammatory cytokine production in GLUT1 deficiency syndrome. Nutrition 31, 981–987. doi: 10.1016/j.nut.2015.02.017
Bi, X., Sun, J., Ji, A. X., and Baudry, M. (2016). Potential therapeutic approaches for Angelman syndrome. Expert Opin. Ther. Targets 20, 601–613. doi: 10.1517/14728222.2016.1115837
Bixel, M. G., and Hamprecht, B. (1995). Generation of ketone bodies from leucine by cultured astroglial cells. J. Neurochem. 65, 2450–2461. doi: 10.1046/j.1471-4159.1995.65062450.x
Blasco, H., Mavel, S., Corcia, P., and Gordon, P. H. (2014). The glutamate hypothesis in ALS: pathophysiology and drug development. Curr. Med. Chem. 21, 3551–3575. doi: 10.2174/0929867321666140916120118
Blasco, H., Patin, F., Andres, C. R., Corcia, P., and Gordon, P. H. (2016). Amyotrophic Lateral Sclerosis, 2016: existing therapies and the ongoing search for neuroprotection. Expert Opin. Pharmacother. 17, 1669–1682. doi: 10.1080/14656566.2016.1202919
Blasco, H., Patin, F., Molinier, S., Vourc’h, P., Le Tilly, O., Bakkouche, S., et al. (2017). A decrease in blood cholesterol after gastrostomy could impact survival in ALS. Eur. J. Clin. Nutr. 71, 1133–1135. doi: 10.1038/ejcn.2017.54
Bough, K. J., and Rho, J. M. (2007). Anticonvulsant mechanisms of the ketogenic diet. Epilepsia 48, 43–58. doi: 10.1111/j.1528-1167.2007.00915.x
Bough, K. J., Wetherington, J., Hassel, B., Pare, J. F., Gawryluk, J. W., Greene, J. G., et al. (2006). Mitochondrial biogenesis in the anticonvulsant mechanism of the ketogenic diet. Ann. Neurol. 60, 223–235. doi: 10.1002/ana.20899
Branco, A. F., Ferreira, A., Simoes, R. F., Magalhaes-Novais, S., Zehowski, C., Cope, E., et al. (2016). Ketogenic diets: from cancer to mitochondrial diseases and beyond. Eur. J. Clin. Invest. 46, 285–298. doi: 10.1111/eci.12591
Browne, S. E., Yang, L., DiMauro, J. P., Fuller, S. W., Licata, S. C., and Beal, M. F. (2006). Bioenergetic abnormalities in discrete cerebral motor pathways presage spinal cord pathology in the G93A SOD1 mouse model of ALS. Neurobiol. Dis. 22, 599–610. doi: 10.1016/j.nbd.2006.01.001
Brownlow, M. L., Benner, L., D’Agostino, D., Gordon, M. N., and Morgan, D. (2013). Ketogenic diet improves motor performance but not cognition in two mouse models of Alzheimer’s pathology. PLOS ONE 8:e75713. doi: 10.1371/journal.pone.0075713
Brunengraber, H. (1997). Potential of ketone body esters for parenteral and oral nutrition. Nutrition 13, 233–235. doi: 10.1016/S0899-9007(96)00409-1
Calderon, N., Betancourt, L., Hernandez, L., and Rada, P. (2017). A ketogenic diet modifies glutamate, gamma-aminobutyric acid and agmatine levels in the hippocampus of rats: a microdialysis study. Neurosci. Lett. 642, 158–162. doi: 10.1016/j.neulet.2017.02.014
Carri, M. T., D’Ambrosi, N., and Cozzolino, M. (2017). Pathways to mitochondrial dysfunction in ALS pathogenesis. Biochem. Biophys. Res. Commun. 483, 1187–1193. doi: 10.1016/j.bbrc.2016.07.055
Cheng, B., Yang, X., An, L., Gao, B., Liu, X., and Liu, S. (2009). Ketogenic diet protects dopaminergic neurons against 6-OHDA neurotoxicity via up-regulating glutathione in a rat model of Parkinson’s disease. Brain Res. 1286, 25–31. doi: 10.1016/j.brainres.2009.06.060
Cheng, C. M., Hicks, K., Wang, J., Eagles, D. A., and Bondy, C. A. (2004). Caloric restriction augments brain glutamic acid decarboxylase-65 and -67 expression. J. Neurosci. Res. 77, 270–276. doi: 10.1002/jnr.20144
Chowdhury, G. M., Jiang, L., Rothman, D. L., and Behar, K. L. (2014). The contribution of ketone bodies to basal and activity-dependent neuronal oxidation in vivo. J. Cereb. Blood Flow Metab. 34, 1233–1242. doi: 10.1038/jcbfm.2014.77
Ciarlone, S. L., Grieco, J. C., D’Agostino, D. P., and Weeber, E. J. (2016). Ketone ester supplementation attenuates seizure activity, and improves behavior and hippocampal synaptic plasticity in an Angelman syndrome mouse model. Neurobiol. Dis. 96, 38–46. doi: 10.1016/j.nbd.2016.08.002
Clarke, K., Tchabanenko, K., Pawlosky, R., Carter, E., Knight, N. S., Murray, A. J., et al. (2012a). Oral 28-day and developmental toxicity studies of (R)-3-hydroxybutyl (R)-3-hydroxybutyrate. Regul. Toxicol. Pharmacol. 63, 196–208. doi: 10.1016/j.yrtph.2012.04.001
Clarke, K., Tchabanenko, K., Pawlosky, R., Carter, E., Todd King, M., Musa-Veloso, K., et al. (2012b). Kinetics, safety and tolerability of (R)-3-hydroxybutyl (R)-3-hydroxybutyrate in healthy adult subjects. Regul. Toxicol. Pharmacol. 63, 401–408. doi: 10.1016/j.yrtph.2012.04.008
Coffey, V. G., Reeder, D. W., Lancaster, G. I., Yeo, W. K., Febbraio, M. A., Yaspelkis, B. B., et al. (2007). Effect of high-frequency resistance exercise on adaptive responses in skeletal muscle. Med. Sci. Sports Exerc. 39, 2135–2144. doi: 10.1249/mss.0b013e31815729b6
Coppola, G., D’Aniello, A., Messana, T., Di Pasquale, F., della Corte, R., Pascotto, A., et al. (2011). Low glycemic index diet in children and young adults with refractory epilepsy: first Italian experience. Seizure 20, 526–528. doi: 10.1016/j.seizure.2011.03.008
Cullingford, T. E. (2004). The ketogenic diet; fatty acids, fatty acid-activated receptors and neurological disorders. Prostaglandins Leukot. Essent. Fatty Acids 70, 253–264. doi: 10.1016/j.plefa.2003.09.008
Danial, N. N., Hartman, A. L., Stafstrom, C. E., and Thio, L. L. (2013). How does the ketogenic diet work? Four potential mechanisms. J. Child Neurol. 28, 1027–1033. doi: 10.1177/0883073813487598
Dhamija, R., Eckert, S., and Wirrell, E. (2013). Ketogenic diet. Can. J. Neurol. Sci. 40, 158–167. doi: 10.1017/S0317167100013676
Diana, A., Pillai, R., Bongioanni, P., O’Keeffe, A. G., Miller, R. G., and Moore, D. H. (2017). Gamma aminobutyric acid (GABA) modulators for amyotrophic lateral sclerosis/motor neuron disease. Cochrane Database. Syst. Rev. 1:Cd006049. doi: 10.1002/14651858.CD006049.pub2
Dienel, G. A. (2012). Brain lactate metabolism: the discoveries and the controversies. J. Cereb. Blood Flow Metab. 32, 1107–1138. doi: 10.1038/jcbfm.2011.175
Dupuis, L., Corcia, P., Fergani, A., Gonzalez De Aguilar, J. L., Bonnefont-Rousselot, D., Bittar, R., et al. (2008). Dyslipidemia is a protective factor in amyotrophic lateral sclerosis. Neurology 70, 1004–1009. doi: 10.1212/01.wnl.0000285080.70324.27
Dupuis, N., Curatolo, N., Benoist, J. F., and Auvin, S. (2015). Ketogenic diet exhibits anti-inflammatory properties. Epilepsia 56, e95–e98. doi: 10.1111/epi.13038
Elamin, M., Ruskin, D. N., Masino, S. A., and Sacchetti, P. (2017). Ketone-based Metabolic Therapy: Is Increased NAD(+) a Primary Mechanism? Front. Mol. Neurosci. 10:377. doi: 10.3389/fnmol.2017.00377
Elia, M., Klepper, J., Leiendecker, B., and Hartmann, H. (2017). Ketogenic diets in the treatment of epilepsy. Curr. Pharm. Des. doi: 10.2174/1381612823666170809101517 [Epub ahead of print].
Erecinska, M., Nelson, D., Daikhin, Y., and Yudkoff, M. (1996). Regulation of GABA level in rat brain synaptosomes: fluxes through enzymes of the GABA shunt and effects of glutamate, calcium, and ketone bodies. J. Neurochem. 67, 2325–2334. doi: 10.1046/j.1471-4159.1996.67062325.x
Evangeliou, A., Doulioglou, V., Haidopoulou, K., Aptouramani, M., Spilioti, M., and Varlamis, G. (2010). Ketogenic diet in a patient with Angelman syndrome. Pediatr. Int. 52, 831–834. doi: 10.1111/j.1442-200X.2010.03118.x
Fraser, D. D., Whiting, S., Andrew, R. D., Macdonald, E. A., Musa-Veloso, K., and Cunnane, S. C. (2003). Elevated polyunsaturated fatty acids in blood serum obtained from children on the ketogenic diet. Neurology 60, 1026–1029. doi: 10.1212/01.WNL.0000049974.74242.C6
Friedman, J. R., Thiele, E. A., Wang, D., Levine, K. B., Cloherty, E. K., Pfeifer, H. H., et al. (2006). Atypical GLUT1 deficiency with prominent movement disorder responsive to ketogenic diet. Mov. Disord. 21, 241–245. doi: 10.1002/mds.20660
Gallagher, D., Belmonte, D., Deurenberg, P., Wang, Z., Krasnow, N., Pi-Sunyer, F. X., et al. (1998). Organ-tissue mass measurement allows modeling of REE and metabolically active tissue mass. Am. J. Physiol. 275(2 Pt 1), E249–E258. doi: 10.1152/ajpendo.1998.275.2.E249
Gano, L. B., Patel, M., and Rho, J. M. (2014). Ketogenic diets, mitochondria, and neurological diseases. J. Lipid Res. 55, 2211–2228. doi: 10.1194/jlr.R048975
Gasior, M., Rogawski, M. A., and Hartman, A. L. (2006). Neuroprotective and disease-modifying effects of the ketogenic diet. Behav. Pharmacol. 17, 431–439. doi: 10.1097/00008877-200609000-00009
Goldberg, E. L., Asher, J. L., Molony, R. D., Shaw, A. C., Zeiss, C. J., Wang, C., et al. (2017). Beta-hydroxybutyrate deactivates neutrophil NLRP3 inflammasome to relieve gout flares. Cell Rep. 18, 2077–2087. doi: 10.1016/j.celrep.2017.02.004
Gonzalez, S. V., Nguyen, N. H., Rise, F., and Hassel, B. (2005). Brain metabolism of exogenous pyruvate. J. Neurochem. 95, 284–293. doi: 10.1111/j.1471-4159.2005.03365.x
Greco, T., Glenn, T. C., Hovda, D. A., and Prins, M. L. (2016). Ketogenic diet decreases oxidative stress and improves mitochondrial respiratory complex activity. J. Cereb. Blood Flow Metab. 36, 1603–1613. doi: 10.1177/0271678x15610584
Groleau, V., Schall, J. I., Stallings, V. A., and Bergqvist, C. A. (2014). Long-term impact of the ketogenic diet on growth and resting energy expenditure in children with intractable epilepsy. Dev. Med. Child Neurol. 56, 898–904. doi: 10.1111/dmcn.12462
Guo, C., Zhou, J., Wu, X., Jiang, H., Lu, K., Chen, J., et al. (2014). A clinical trial of ketogenic diet in patients with acute spinal cord injury: safety and feasibility. Nan Fang Yi Ke Da Xue Xue Bao 34, 571–575.
Guzman, M., and Blazquez, C. (2001). Is there an astrocyte-neuron ketone body shuttle? Trends Endocrinol. Metab. 12, 169–173.
Haas, R. H., Rice, M. A., Trauner, D. A., and Merritt, T. A. (1986). Therapeutic effects of a ketogenic diet in Rett syndrome. Am. J. Med. Genet. Suppl. 1, 225–246. doi: 10.1002/ajmg.1320250525
Hartman, A. L., Zheng, X., Bergbower, E., Kennedy, M., and Hardwick, J. M. (2010). Seizure tests distinguish intermittent fasting from the ketogenic diet. Epilepsia 51, 1395–1402. doi: 10.1111/j.1528-1167.2010.02577.x
Hashim, S. A., and VanItallie, T. B. (2014). Ketone body therapy: from the ketogenic diet to the oral administration of ketone ester. J. Lipid Res. 55, 1818–1826. doi: 10.1194/jlr.R046599
Henderson, S. T., Vogel, J. L., Barr, L. J., Garvin, F., Jones, J. J., and Costantini, L. C. (2009). Study of the ketogenic agent AC-1202 in mild to moderate Alzheimer’s disease: a randomized, double-blind, placebo-controlled, multicenter trial. Nutr. Metab. 6:31. doi: 10.1186/1743-7075-6-31
Huttenlocher, P. R. (1976). Ketonemia and seizures: metabolic and anticonvulsant effects of two ketogenic diets in childhood epilepsy. Pediatr. Res. 10, 536–540. doi: 10.1203/00006450-197605000-00006
Hyatt, H. W., Kephart, W. C., Holland, A. M., Mumford, P., Mobley, C. B., Lowery, R. P., et al. (2016). A ketogenic diet in rodents elicits improved mitochondrial adaptations in response to resistance exercise training compared to an isocaloric western diet. Front. Physiol. 7:533. doi: 10.3389/fphys.2016.00533
Islam, M. T. (2017). Oxidative stress and mitochondrial dysfunction-linked neurodegenerative disorders. Neurol. Res. 39, 73–82. doi: 10.1080/01616412.2016.1251711
Ji, L. L., Kang, C., and Zhang, Y. (2016). Exercise-induced hormesis and skeletal muscle health. Free Radic. Biol. Med. 98, 113–122. doi: 10.1016/j.freeradbiomed.2016.02.025
Juge, N., Gray, J. A., Omote, H., Miyaji, T., Inoue, T., Hara, C., et al. (2010). Metabolic control of vesicular glutamate transport and release. Neuron 68, 99–112. doi: 10.1016/j.neuron.2010.09.002
Jung, C., Higgins, C. M., and Xu, Z. (2002). A quantitative histochemical assay for activities of mitochondrial electron transport chain complexes in mouse spinal cord sections. J. Neurosci. Methods 114, 165–172. doi: 10.1016/S0165-0270(01)00524-6
Kashiwaya, Y., Takeshima, T., Mori, N., Nakashima, K., Clarke, K., and Veech, R. L. (2000). D-beta-hydroxybutyrate protects neurons in models of Alzheimer’s and Parkinson’s disease. Proc. Natl. Acad. Sci. U.S.A. 97, 5440–5444. doi: 10.1073/pnas.97.10.5440
Keene, D. L. (2006). A systematic review of the use of the ketogenic diet in childhood epilepsy. Pediatr. Neurol. 35, 1–5. doi: 10.1016/j.pediatrneurol.2006.01.005
Kim, D. Y., Davis, L. M., Sullivan, P. G., Maalouf, M., Simeone, T. A., van Brederode, J., et al. (2007). Ketone bodies are protective against oxidative stress in neocortical neurons. J. Neurochem. 101, 1316–1326. doi: 10.1111/j.1471-4159.2007.04483.x
Kim, D. Y., Vallejo, J., and Rho, J. M. (2010). Ketones prevent synaptic dysfunction induced by mitochondrial respiratory complex inhibitors. J. Neurochem. 114, 130–141. doi: 10.1111/j.1471-4159.2010.06728.x
Koppel, S. J., and Swerdlow, R. H. (2017). Neuroketotherapeutics: A modern review of a century-old therapy. Neurochem. Int. doi: 10.1016/j.neuint.2017.05.019 [Epub ahead of print].
Korner, S., Kollewe, K., Ilsemann, J., Muller-Heine, A., Dengler, R., Krampfl, K., et al. (2013). Prevalence and prognostic impact of comorbidities in amyotrophic lateral sclerosis. Eur. J. Neurol. 20, 647–654. doi: 10.1111/ene.12015
Kossoff, E. H., Krauss, G. L., McGrogan, J. R., and Freeman, J. M. (2003). Efficacy of the Atkins diet as therapy for intractable epilepsy. Neurology 61, 1789–1791. doi: 10.1212/01.WNL.0000098889.35155.72
Kunnecke, B., Cerdan, S., and Seelig, J. (1993). Cerebral metabolism of [1,2-13C2]glucose and [U-13C4]3-hydroxybutyrate in rat brain as detected by 13C NMR spectroscopy. NMR Biomed. 6, 264–277. doi: 10.1002/nbm.1940060406
LaManna, J. C., Salem, N., Puchowicz, M., Erokwu, B., Koppaka, S., Flask, C., et al. (2009). Ketones suppress brain glucose consumption. Adv. Exp. Med. Biol. 645, 301–306. doi: 10.1007/978-0-387-85998-9_45
Lee, J., Bruce-Keller, A. J., Kruman, Y., Chan, S. L., and Mattson, M. P. (1999). 2-Deoxy-D-glucose protects hippocampal neurons against excitotoxic and oxidative injury: evidence for the involvement of stress proteins. J. Neurosci. Res. 57, 48–61. doi: 10.1002/(SICI)1097-4547(19990701)57:1<48::AID-JNR6>3.0.CO;2-L
Lee, J., Kim, S. J., Son, T. G., Chan, S. L., and Mattson, M. P. (2006). Interferon-gamma is up-regulated in the hippocampus in response to intermittent fasting and protects hippocampal neurons against excitotoxicity. J. Neurosci. Res. 83, 1552–1557. doi: 10.1002/jnr.20831
Lefevre, F., and Aronson, N. (2000). Ketogenic diet for the treatment of refractory epilepsy in children: a systematic review of efficacy. Pediatrics 105:E46. doi: 10.1542/peds.105.4.e46
Liebhaber, G. M., Riemann, E., and Baumeister, F. A. (2003). Ketogenic diet in Rett syndrome. J. Child Neurol. 18, 74–75. doi: 10.1177/08830738030180011801
Liu, J., and Wang, F. (2017). Role of neuroinflammation in amyotrophic lateral sclerosis: cellular mechanisms and therapeutic implications. Front. Immunol. 8:1005. doi: 10.3389/fimmu.2017.01005
Liu, Z., Zhou, T., Ziegler, A. C., Dimitrion, P., and Zuo, L. (2017). Oxidative stress in neurodegenerative diseases: from molecular mechanisms to clinical applications. Oxid Med. Cell Longev. 2017:2525967. doi: 10.1155/2017/2525967
Lund, T. M., Obel, L. F., Risa, O., and Sonnewald, U. (2011). Beta-hydroxybutyrate is the preferred substrate for GABA and glutamate synthesis while glucose is indispensable during depolarization in cultured GABAergic neurons. Neurochem. Int. 59, 309–318. doi: 10.1016/j.neuint.2011.06.002
Lutas, A., and Yellen, G. (2013). The ketogenic diet: metabolic influences on brain excitability and epilepsy. Trends Neurosci. 36, 32–40. doi: 10.1016/j.tins.2012.11.005
Maalouf, M., Sullivan, P. G., Davis, L., Kim, D. Y., and Rho, J. M. (2007). Ketones inhibit mitochondrial production of reactive oxygen species production following glutamate excitotoxicity by increasing NADH oxidation. Neuroscience 145, 256–264. doi: 10.1016/j.neuroscience.2006.11.065
Mantis, J. G., Fritz, C. L., Marsh, J., Heinrichs, S. C., and Seyfried, T. N. (2009). Improvement in motor and exploratory behavior in Rett syndrome mice with restricted ketogenic and standard diets. Epilepsy Behav. 15, 133–141. doi: 10.1016/j.yebeh.2009.02.038
Margolis, S. S., Sell, G. L., Zbinden, M. A., and Bird, L. M. (2015). Angelman syndrome. Neurotherapeutics 12, 641–650. doi: 10.1007/s13311-015-0361-y
Mattiazzi, M., D’Aurelio, M., Gajewski, C. D., Martushova, K., Kiaei, M., Beal, M. F., et al. (2002). Mutated human SOD1 causes dysfunction of oxidative phosphorylation in mitochondria of transgenic mice. J. Biol. Chem. 277, 29626–29633. doi: 10.1074/jbc.M203065200
McKenna, M. C. (2012). Substrate competition studies demonstrate oxidative metabolism of glucose, glutamate, glutamine, lactate and 3-hydroxybutyrate in cortical astrocytes from rat brain. Neurochem. Res. 37, 2613–2626. doi: 10.1007/s11064-012-0901-3
McNally, M. A., Pyzik, P. L., Rubenstein, J. E., Hamdy, R. F., and Kossoff, E. H. (2009). Empiric use of potassium citrate reduces kidney-stone incidence with the ketogenic diet. Pediatrics 124, e300–e304. doi: 10.1542/peds.2009-0217
Menzies, F. M., Cookson, M. R., Taylor, R. W., Turnbull, D. M., Chrzanowska-Lightowlers, Z. M., Dong, L., et al. (2002). Mitochondrial dysfunction in a cell culture model of familial amyotrophic lateral sclerosis. Brain 125(Pt 7), 1522–1533. doi: 10.1093/brain/awf167
Morales-Alamo, D., and Calbet, J. A. (2014). Free radicals and sprint exercise in humans. Free Radic. Res. 48, 30–42. doi: 10.3109/10715762.2013.825043
Nandivada, P., Fell, G. L., Pan, A. H., Nose, V., Ling, P. R., Bistrian, B. R., et al. (2016). Eucaloric ketogenic diet reduces hypoglycemia and inflammation in mice with endotoxemia. Lipids 51, 703–714. doi: 10.1007/s11745-016-4156-7
Newman, J. C., and Verdin, E. (2014). Ketone bodies as signaling metabolites. Trends Endocrinol. Metab. 25, 42–52. doi: 10.1016/j.tem.2013.09.002
Newport, M. T., VanItallie, T. B., Kashiwaya, Y., King, M. T., and Veech, R. L. (2015). A new way to produce hyperketonemia: use of ketone ester in a case of Alzheimer’s disease. Alzheimers Dement. 11, 99–103. doi: 10.1016/j.jalz.2014.01.006
Niessen, H. G., Debska-Vielhaber, G., Sander, K., Angenstein, F., Ludolph, A. C., Hilfert, L., et al. (2007). Metabolic progression markers of neurodegeneration in the transgenic G93A-SOD1 mouse model of amyotrophic lateral sclerosis. Eur. J. Neurosci. 25, 1669–1677. doi: 10.1111/j.1460-9568.2007.05415.x
Ohnuma, T., Toda, A., Kimoto, A., Takebayashi, Y., Higashiyama, R., Tagata, Y., et al. (2016). Benefits of use, and tolerance of, medium-chain triglyceride medical food in the management of Japanese patients with Alzheimer’s disease: a prospective, open-label pilot study. Clin. Interv. Aging 11, 29–36. doi: 10.2147/cia.s95362
Paoli, A., Bianco, A., Damiani, E., and Bosco, G. (2014). Ketogenic diet in neuromuscular and neurodegenerative diseases. Biomed. Res. Int. 2014:474296. doi: 10.1155/2014/474296
Park, M. J., Aja, S., Li, Q., Degano, A. L., Penati, J., Zhuo, J., et al. (2014). Anaplerotic triheptanoin diet enhances mitochondrial substrate use to remodel the metabolome and improve lifespan, motor function, and sociability in MeCP2-null mice. PLOS ONE 9:e109527. doi: 10.1371/journal.pone.0109527
Pawlosky, R. J., Kemper, M. F., Kashiwaya, Y., King, M. T., Mattson, M. P., and Veech, R. L. (2017). Effects of a dietary ketone ester on hippocampal glycolytic and tricarboxylic acid cycle intermediates and amino acids in a 3xTgAD mouse model of Alzheimer’s disease. J. Neurochem. 141, 195–207. doi: 10.1111/jnc.13958
Perera, N. D., and Turner, B. J. (2016). AMPK signalling and defective energy metabolism in amyotrophic lateral sclerosis. Neurochem. Res. 41, 544–553. doi: 10.1007/s11064-015-1665-3
Rae, C., Fekete, A. D., Kashem, M. A., Nasrallah, F. A., and Broer, S. (2012). Metabolism, compartmentation, transport and production of acetate in the cortical brain tissue slice. Neurochem. Res. 37, 2541–2553. doi: 10.1007/s11064-012-0847-5
Ramm-Pettersen, A., Stabell, K. E., Nakken, K. O., and Selmer, K. K. (2014). Does ketogenic diet improve cognitive function in patients with GLUT1-DS? A 6- to 17-month follow-up study. Epilepsy Behav. 39, 111–115. doi: 10.1016/j.yebeh.2014.08.015
Reger, M. A., Henderson, S. T., Hale, C., Cholerton, B., Baker, L. D., Watson, G. S., et al. (2004). Effects of beta-hydroxybutyrate on cognition in memory-impaired adults. Neurobiol. Aging 25, 311–314. doi: 10.1016/s0197-4580(03)00087-3
Saxena, S., Roselli, F., Singh, K., Leptien, K., Julien, J. P., Gros-Louis, F., et al. (2013). Neuroprotection through excitability and mTOR required in ALS motoneurons to delay disease and extend survival. Neuron 80, 80–96. doi: 10.1016/j.neuron.2013.07.027
Shaafi, S., Najmi, S., Aliasgharpour, H., Mahmoudi, J., Sadigh-Etemad, S., Farhoudi, M., et al. (2016). The efficacy of the ketogenic diet on motor functions in Parkinson’s disease: a rat model. Iran J. Neurol. 15, 63–69.
Siva, N. (2006). Can ketogenic diet slow progression of ALS? Lancet Neurol. 5:476. doi: 10.1016/S1474-4422(06)70462-8
Srivastava, S., Baxa, U., Niu, G., Chen, X., and Veech, R. L. (2013). A ketogenic diet increases brown adipose tissue mitochondrial proteins and UCP1 levels in mice. IUBMB Life 65, 58–66. doi: 10.1002/iub.1102
Srivastava, S., Kashiwaya, Y., King, M. T., Baxa, U., Tam, J., Niu, G., et al. (2012). Mitochondrial biogenesis and increased uncoupling protein 1 in brown adipose tissue of mice fed a ketone ester diet. FASEB J. 26, 2351–2362. doi: 10.1096/fj.11-200410
Stafford, P., Abdelwahab, M. G., Kim, D. Y., Preul, M. C., Rho, J. M., and Scheck, A. C. (2010). The ketogenic diet reverses gene expression patterns and reduces reactive oxygen species levels when used as an adjuvant therapy for glioma. Nutr. Metab. 7:74. doi: 10.1186/1743-7075-7-74
Stafstrom, C. E., and Rho, J. M. (2012). The ketogenic diet as a treatment paradigm for diverse neurological disorders. Front. Pharmacol. 3:59. doi: 10.3389/fphar.2012.00059
Stamp, L. K., James, M. J., and Cleland, L. G. (2005). Diet and rheumatoid arthritis: a review of the literature. Semin. Arthritis Rheum. 35, 77–94. doi: 10.1016/j.semarthrit.2005.05.001
Steriade, C., Andrade, D. M., Faghfoury, H., Tarnopolsky, M. A., and Tai, P. (2014). Mitochondrial encephalopathy with lactic acidosis and stroke-like episodes (MELAS) may respond to adjunctive ketogenic diet. Pediatr. Neurol. 50, 498–502. doi: 10.1016/j.pediatrneurol.2014.01.009
Streijger, F., Plunet, W. T., Lee, J. H., Liu, J., Lam, C. K., Park, S., et al. (2013). Ketogenic diet improves forelimb motor function after spinal cord injury in rodents. PLOS ONE 8:e78765. doi: 10.1371/journal.pone.0078765
Stribl, C., Samara, A., Trumbach, D., Peis, R., Neumann, M., Fuchs, H., et al. (2014). Mitochondrial dysfunction and decrease in body weight of a transgenic knock-in mouse model for TDP-43. J. Biol. Chem. 289, 10769–10784. doi: 10.1074/jbc.M113.515940
Su, H., Fan, W., Coskun, P. E., Vesa, J., Gold, J. A., Jiang, Y. H., et al. (2011). Mitochondrial dysfunction in CA1 hippocampal neurons of the UBE3A deficient mouse model for Angelman syndrome. Neurosci. Lett. 487, 129–133. doi: 10.1016/j.neulet.2009.06.079
Sullivan, P. G., Rippy, N. A., Dorenbos, K., Concepcion, R. C., Agarwal, A. K., and Rho, J. M. (2004). The ketogenic diet increases mitochondrial uncoupling protein levels and activity. Ann. Neurol. 55, 576–580. doi: 10.1002/ana.20062
Suzuki, Y., Takahashi, H., Fukuda, M., Hino, H., Kobayashi, K., Tanaka, J., et al. (2009). Beta-hydroxybutyrate alters GABA-transaminase activity in cultured astrocytes. Brain Res. 1268, 17–23. doi: 10.1016/j.brainres.2009.02.074
Tefera, T. W., Wong, Y., Barkl-Luke, M. E., Ngo, S. T., Thomas, N. K., McDonald, T. S., et al. (2016). Triheptanoin protects motor neurons and delays the onset of motor symptoms in a mouse model of amyotrophic lateral sclerosis. PLOS ONE 11:e0161816. doi: 10.1371/journal.pone.0161816
Thibert, R. L., Pfeifer, H. H., Larson, A. M., Raby, A. R., Reynolds, A. A., Morgan, A. K., et al. (2012). Low glycemic index treatment for seizures in Angelman syndrome. Epilepsia 53, 1498–1502. doi: 10.1111/j.1528-1167.2012.03537.x
Tieu, K., Perier, C., Caspersen, C., Teismann, P., Wu, D. C., Yan, S. D., et al. (2003). D-beta-hydroxybutyrate rescues mitochondrial respiration and mitigates features of Parkinson disease. J. Clin. Invest. 112, 892–901. doi: 10.1172/jci18797
Valente-Silva, P., Lemos, C., Kofalvi, A., Cunha, R. A., and Jones, J. G. (2015). Ketone bodies effectively compete with glucose for neuronal acetyl-CoA generation in rat hippocampal slices. NMR Biomed. 28, 1111–1116. doi: 10.1002/nbm.3355
Vanitallie, T. B., Nonas, C., Di Rocco, A., Boyar, K., Hyams, K., and Heymsfield, S. B. (2005). Treatment of Parkinson disease with diet-induced hyperketonemia: a feasibility study. Neurology 64, 728–730. doi: 10.1212/01.wnl.0000152046.11390.45
Veech, R. L. (2014). Ketone ester effects on metabolism and transcription. J. Lipid Res. 55, 2004–2006. doi: 10.1194/jlr.R046292
Veech, R. L., Chance, B., Kashiwaya, Y., Lardy, H. A., and Cahill, GF Jr (2001). Ketone bodies, potential therapeutic uses. IUBMB Life 51, 241–247. doi: 10.1080/152165401753311780
Vielhaber, S., Kunz, D., Winkler, K., Wiedemann, F. R., Kirches, E., Feistner, H., et al. (2000). Mitochondrial DNA abnormalities in skeletal muscle of patients with sporadic amyotrophic lateral sclerosis. Brain 123(Pt 7), 1339–1348. doi: 10.1093/brain/123.7.1339
Wang, Z. J., Bergqvist, C., Hunter, J. V., Jin, D., Wang, D. J., Wehrli, S., et al. (2003). In vivo measurement of brain metabolites using two-dimensional double-quantum MR spectroscopy–exploration of GABA levels in a ketogenic diet. Magn. Reson. Med. 49, 615–619. doi: 10.1002/mrm.10429
Wiedemann, F. R., Manfredi, G., Mawrin, C., Beal, M. F., and Schon, E. A. (2002). Mitochondrial DNA and respiratory chain function in spinal cords of ALS patients. J. Neurochem. 80, 616–625. doi: 10.1046/j.0022-3042.2001.00731.x
Xu, Y. F., Gendron, T. F., Zhang, Y. J., Lin, W. L., D’Alton, S., Sheng, H., et al. (2010). Wild-type human TDP-43 expression causes TDP-43 phosphorylation, mitochondrial aggregation, motor deficits, and early mortality in transgenic mice. J. Neurosci. 30, 10851–10859. doi: 10.1523/jneurosci.1630-10.2010
Yang, X., and Cheng, B. (2010). Neuroprotective and anti-inflammatory activities of ketogenic diet on MPTP-induced neurotoxicity. J. Mol. Neurosci. 42, 145–153. doi: 10.1007/s12031-010-9336-y
Youm, Y. H., Nguyen, K. Y., Grant, R. W., Goldberg, E. L., Bodogai, M., Kim, D., et al. (2015). The ketone metabolite beta-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat. Med. 21, 263–269. doi: 10.1038/nm.3804
Yudkoff, M., Daikhin, Y., Melo, T. M., Nissim, I., and Sonnewald, U. (2007). The ketogenic diet and brain metabolism of amino acids: relationship to the anticonvulsant effect. Annu. Rev. Nutr. 27, 415–430. doi: 10.1146/annurev.nutr.27.061406.093722
Zhang, J., Cao, Q., Li, S., Lu, X., Zhao, Y., Guan, J. S., et al. (2013). 3-Hydroxybutyrate methyl ester as a potential drug against Alzheimer’s disease via mitochondria protection mechanism. Biomaterials 34, 7552–7562. doi: 10.1016/j.biomaterials.2013.06.043
Zhang, Y., Kuang, Y., Xu, K., Harris, D., Lee, Z., LaManna, J., et al. (2013). Ketosis proportionately spares glucose utilization in brain. J. Cereb. Blood Flow Metab. 33, 1307–1311. doi: 10.1038/jcbfm.2013.87
Zhao, W., Varghese, M., Vempati, P., Dzhun, A., Cheng, A., Wang, J., et al. (2012). Caprylic triglyceride as a novel therapeutic approach to effectively improve the performance and attenuate the symptoms due to the motor neuron loss in ALS disease. PLOS ONE 7:e49191. doi: 10.1371/journal.pone.0049191
Zhao, Z., Lange, D. J., Voustianiouk, A., MacGrogan, D., Ho, L., Suh, J., et al. (2006). A ketogenic diet as a potential novel therapeutic intervention in amyotrophic lateral sclerosis. BMC Neurosci. 7:29. doi: 10.1186/1471-2202-7-29
Keywords: ketogenic diet, motor function, motor neuron, β-hydroxybutyrate, ketone bodies, neuromuscular diseases
Citation: Veyrat-Durebex C, Reynier P, Procaccio V, Hergesheimer R, Corcia P, Andres CR and Blasco H (2018) How Can a Ketogenic Diet Improve Motor Function? Front. Mol. Neurosci. 11:15. doi: 10.3389/fnmol.2018.00015
Received: 29 September 2017; Accepted: 10 January 2018;
Published: 26 January 2018.
Edited by:
Karsten Hiller, Technische Universitat Braunschweig, GermanyReviewed by:
David Ruskin, Trinity College, United StatesYannic Nonnenmacher, Technische Universitat Braunschweig, Germany
Copyright © 2018 Veyrat-Durebex, Reynier, Procaccio, Hergesheimer, Corcia, Andres and Blasco. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hélène Blasco, aGVsZW5lLmJsYXNjb0B1bml2LXRvdXJzLmZy