 Regina Dahlhaus
Regina Dahlhaus- Institute for Biochemistry, Emil-Fischer Centre, University of Erlangen-Nürnberg, Erlangen, Germany
The Fragile X Syndrome (FXS) is one of the most common forms of inherited intellectual disability in all human societies. Caused by the transcriptional silencing of a single gene, the fragile x mental retardation gene FMR1, FXS is characterized by a variety of symptoms, which range from mental disabilities to autism and epilepsy. More than 20 years ago, a first animal model was described, the Fmr1 knock-out mouse. Several other models have been developed since then, including conditional knock-out mice, knock-out rats, a zebrafish and a drosophila model. Using these model systems, various targets for potential pharmaceutical treatments have been identified and many treatments have been shown to be efficient in preclinical studies. However, all attempts to turn these findings into a therapy for patients have failed thus far. In this review, I will discuss underlying difficulties and address potential alternatives for our future research.
Introduction
The tremendous advance that has taken place in life sciences during the last decades has opened a variety of options and opportunities for research as well as for human societies, in particular in the field of genetics. One of these advances was the invention of the Crispr-Cas system (Crispr; reviewed in Donohoue et al., 2017; Huang et al., 2017; Petersen, 2017). The technique allows for a fast and relatively precise gene editing in a variety of different organisms ranging from plants and insects to vertebrates and primates including human cell lines and embryos. Being relatively efficient and easy to use, the system promises much progress not only for our understanding of complex biological systems, but also for the treatment of genetic disorders.
Of foremost interest in this context are monogenetic diseases with limited treatment options for patients, such as the Fragile X Syndrome (FXS), a nonetheless strikingly complex autism spectrum >disorder (ASD). Combined with the advance of genetic screening methods for persons at risk (reviewed in Rajan-Babu and Chong, 2016) and assisted reproduction services, the Crispr technique opens not only new horizons, but also raises many ethical concerns, although there is no common agreement on ethical standards among mankind and not all people are sharing the concerns.
For our future research, it will therefore be important to critically evaluate what we are able to achieve, what we have achieved, and, on a society based level, what we do want to achieve. This article will review and discuss important results as well as ideas from the FXS field, and in particular address underlying difficulties arising from the current mouse models.
The Fragile X Syndrome — of Men
Phenotype
Affecting approximately 1 in 7000 males and 11,000 females (meta-analysis: Hunter et al., 2014), FXS represents one of the most frequent forms of monogenetically determined mental retardation in all human populations and ethnic groups (reviewed in Tzschach and Ropers, 2007). In the vast majority of cases, the disease is caused by the transcriptional silencing of a single gene on the X chromosome, the Fragile X Mental Retardation gene FMR1. In consequence, expression of the encoded protein FMRP is lost (reviewed in Saldarriaga et al., 2014; Usdin and Kumari, 2015).
FXS patients display a variety of intellectual deficits ranging from mild learning impairments to severe cognitive disabilities, but also autistic behaviors such as aggression, social anxiety and stereotypic acting characterize the disease (reviewed in Saldarriaga et al., 2014; Gross et al., 2015b; Figure 1). Men are in general more severely affected than women, achieving only average IQs1 of 40–50, whereas women mainly present with mild to moderate cognitive impairments and an average IQ of about 80, though their abilities may range from severe deficits to superior performances (Freund et al., 1993; de Vries et al., 1996; Lewis et al., 2006; Chaste et al., 2012, reviewed in Huddleston et al., 2014). However, even men can be high functioning (Basuta et al., 2015).
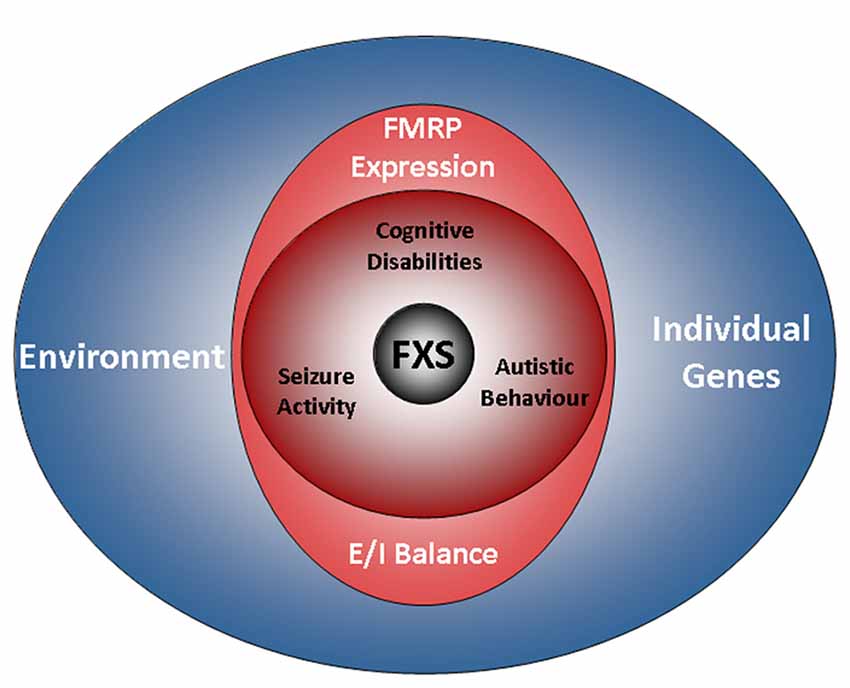
Figure 1. The FXS world. The drawing illustrates factors influencing the disease. A characteristic of FXS is the presence of a broad range of deficits with a high degree of individual variation. The phenotype of the disorder includes cognitive disabilities as well as autistic behaviors and epilepsy. Aside from a loss of FMRP expression, the genetic background and environmental factors are emerging as determinants of the disease. However, while residual FMRP expression is known to correlate with the cognitive performance of FXS patients, the impact of individual genes and the relevance of the environment are less well understood (also see the section “Phenotype”). Recent findings indicate that autistic behaviors and epilepsy are influenced by the E/I balance, but not by residual FMRP expression. FXS, Fragile X Syndrome; FMRP, Fragile X Mental Retardation Protein; E/I balance, balance of excitation and inhibition in neuronal networks.
Shyness, poor eye contact and attention difficulties are particularly characteristic to young women with FXS (FMR1−/–), whereas increased aggression and the use of rote phrases are more typical to men (Murphy and Abbeduto, 2007; Hartley et al., 2011). In their adult life, women are mostly affected by deficits in interpersonal skills, while weak functional skills primarily concern men, though reduced social interaction skills are also prominent in men (Hartley et al., 2011). As a result, only 9% of the men affected by FXS achieve a high or very high level of independence in adult life, whereas 44% of the FXS women reach such a level. Contrary to most other X-linked diseases, approximately 35% of the women carrying a single mutated allele only (FMR1–/+) also demonstrate cognitive disabilities (Hagerman et al., 1992).
The impairments observed in FXS patients are not uniform though, but rather specific to certain capabilities: several studies demonstrated that FXS patients perform particularly weak in tasks requiring abstract item reasoning, attention, the solution of new problems and goal-directed actions, as well as in tasks relying on short-term memory and visual-motor coordination. By contrast, FXS patients usually demonstrate normal skills in vocabulary knowledge, although they show a cluttered and less complex speech (Hanson et al., 1986; Dykens et al., 1987; Maes et al., 1994; Loesch et al., 2004; Lewis et al., 2006; Roberts et al., 2007; Van der Molen et al., 2010).
Recent research has linked some of the variability observed in the cognitive phenotype of FXS patients to residual FMRP expression and mosaic expression patterns (Kaufmann et al., 1999; Dyer-Friedman et al., 2002; Loesch et al., 2003, 2004; Pretto et al., 2014; Basuta et al., 2015). For instance, the cognitive abilities of FXS patients were shown to strongly correlate with FMRP expression levels, even when full-scale IQ scores are used. Although this holds true for most of the cases, exceptions exist: Govaerts and colleagues reported a case, in which residual FMRP expression could not explain the good cognitive performance observed (Govaerts et al., 2007), thus implying a role for individual genetic factors and/or environmental effects (cp.2 Figure 1).
Further studies indeed support the significance of environmental factors in FXS (Dyer-Friedman et al., 2002; Kuo et al., 2002; Glaser et al., 2003): in particular maternal warmth and responsivity were demonstrated to ameliorate maladaptive as well as autistic behaviors, whereas maternal depression and criticism were indicated to increase FXS symptoms in children (Greenberg et al., 2012; Robinson et al., 2016; Smith et al., 2016). Contrary to cognitive deficits, autistic behaviors display no correlation with residual FMRP expression (Glaser et al., 2003; Pretto et al., 2014).
Particularly in children, seizures are frequent as well, affecting about 45% of the adolescent patients between 1 year and 14 years of age (Cowley et al., 2016; Figure 1). After the age of 20, seizure activity decreases, resulting in an overall prevalence of about 24% (Sabaratnam et al., 2001), although there is considerable variation among studies. Interestingly, some data suggest that the attention deficits observed in FXS patients are related to seizure activity (Cowley et al., 2016).
Just like autistic behaviors, seizure activity was found not to correlate with residual FMRP expression (Pretto et al., 2014). Although an imbalance of excitatory and inhibitory neuronal activity has been associated with seizure activity and autistic behaviors in several ASDs as well as in corresponding animal models (reviewed in Frye et al., 2016; Uzunova et al., 2016; Lee et al., 2017), data on FXS patients are rare. Using EEG3 electrodes, increases in event related potentials were found in the auditory cortex of FXS children, suggesting enhanced excitability (Castrén et al., 2003). EEG-studies of oscillatory dynamics in males with FXS identified impaired theta oscillations indicative of an imbalance in excitatory and inhibitory neuronal circuit activity (Van der Molen and Van der Molen, 2013; Van der Molen et al., 2014) and implied a lack of coordination in information processing. Notably, two studies also found decreased activations, one in prefrontal regions and one in the fusiform gyrus (Dalton et al., 2008; Holsen et al., 2008). These findings suggest that FXS patients experience brain-region and most likely circuit-specific imbalances in neuronal excitation.
Genotype
Microsatellites — Sources of Complexity
The molecular mechanisms leading to the silencing of the FMR1 gene during embryonic development are complex and result from expansions in the length of a microsatellite located in the 5′UTR4 of FMR1 (Fu et al., 1991; Pieretti et al., 1991; Verkerk et al., 1991; Eiges et al., 2007; Bar-Nur et al., 2012). In healthy individuals, the sequence consists of CGG/CCG tandem tracts and includes approximately 6–44 repeats, whereas FXS patients show more than 200 repeats. FMR1 alleles containing 45–54 repeats are classified as intermediate, and 55–200 repeats as pre-mutation alleles (Figure 2). Contrary to regular tandem tracts, pre-mutation alleles are meiotically as well as mitotically unstable and may turn into full-mutation alleles within one generation, if transmitted by a female (Fu et al., 1991; Heitz et al., 1992; Yu et al., 1992).
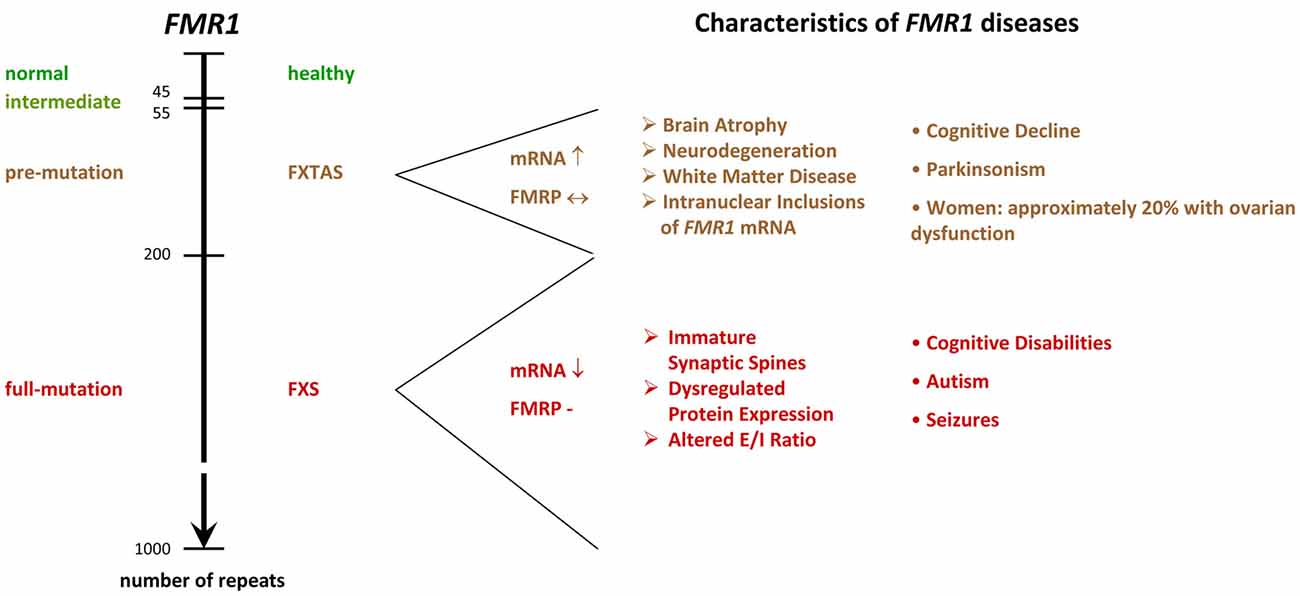
Figure 2. FMR1−/y diseases. The scheme shows the relation between microsatellite length (number of repeats) and phenotype. While healthy individuals show 6–44 tandem tracts in the 5′ UTR of their FMR1 gene, FXS patients display more than 200 repeats. Alleles containing 45–54 repeats are classified as intermediate, and 55–200 repeats as pre-mutation alleles. Premutation alleles give rise to a neurodegenerative disorder called FXTAS, which presents with parkinsonism and brain atrophy. FXTAS typically manifests in individuals over the age of 50. FMR1: Fragile X Mental Retardation gene. FMRP: Fragile X Mental Retardation Protein. FXS: Fragile X Syndrome. FXTAS: Fragile X-associated Tremor/Ataxia Syndrome. E/I balance: balance of excitation and inhibition in neuronal networks, UTR: untranslated region. ↓: decreased levels in the diseased condition. ↔ similar levels in normal and diseased conditions. ↑ increased levels in diseased conditions.
Although microsatellites are often associated with diseases (reviewed in Nelson et al., 2013; Zhang and Ashizawa, 2017), they turned out to play crucial roles in many species, but in particular in humans. Due to their high variability, tandem tracts are thought to serve as a substrate for evolution (reviewed in Kashi and King, 2006; Hannan, 2012; Plohl et al., 2012). Most microsatellites are nonetheless only maintained by chance and total microsatellite numbers are rather species or clade specific than related to vertebrate evolution (Buschiazzo and Gemmell, 2010; Adams et al., 2016). Yet, tract length polymorphism turned out to be a major source for the emergence of variability and complexity in species: recent research revealed that tandem tracts located in regulatory regions contribute to the genesis of complexity (Liu H. et al., 2012; Namdar-Aligoodarzi et al., 2015). By triggering the formation of secondary structures in DNA as well as RNA molecules in a length dependent manner, these repeats redress the transcription and translation efficiency and, in doing so, control the expression level of their proteins (reviewed in Kashi and King, 2006; Pezer et al., 2012; Sawaya et al., 2012; also see e.g., Zumwalt et al., 2007; Halder et al., 2009; Vinces et al., 2009; Quilez et al., 2016). In consequence, the protein stoichiometry is altered, leading to modifications in a variety of protein interactions, while the functionality of the protein itself is preserved. It is therefore no surprise that tandem tracts are preferentially located in the proximity of transcription starts (Vinces et al., 2009; Sawaya et al., 2013; Liang K. C. et al., 2015), that human promoter regions are particularly rich in microsatellites (Sawaya et al., 2013) and that the polymorphism and complexity of tandem tracts significantly increase in primates and humans (Zhang et al., 2004; Mohammadparast et al., 2014; Sabino et al., 2014; Bilgin Sonay et al., 2015; Namdar-Aligoodarzi et al., 2015; Ohadi et al., 2015; Rezazadeh et al., 2015).
Moreover, tandem tract polymorphism turned out to be astonishingly abundant in genes involved in the development of the nervous system (Riley and Krieger, 2009). Indeed, some studies even associated repeat variations with individual differences in behavioral traits—not only in humans (Gerra et al., 2005; Larsen et al., 2010; Simmons and Roney, 2011; Berry et al., 2013; Durdiaková et al., 2013; Valomon et al., 2014; Votinov et al., 2015), but also in birds (Stuber et al., 2016) and other mammals (Hammock and Young, 2005; Lucarelli et al., 2017). In addition, Bagshaw and colleagues showed that polymorphic microsatellites of genes involved in human personality traits and social behavior are able to integrate interactions with the environment, in this case maternal smoking, which caused anti-social acts in carriers of certain TBR1 alleles (Bagshaw et al., 2017). Their finding is in line with an earlier study showing that a CGG repeat variant of the glutathion peroxidase 1 gene is protective for autism (Ming et al., 2010). Indeed, it has been found that prenatal oxidative stress, such as caused by environmental toxicants, is involved in the establishment of autism, in particular when occurring in certain sensitive stages of embryogenesis (reviewed in Chauhan and Chauhan, 2006; Landrigan, 2010; Wells et al., 2016; Heyer and Meredith, 2017).
These findings illustrate that microsatellite polymorphism is an important component of individuality, complexity, neuronal development and gene-environment interactions. Since FMRP itself is developmentally regulated (Hinds et al., 1993) and was found to function in neuronal migration, differentiation and dendritic spine maturation (Hinton et al., 1991; Irwin et al., 2001; Saffary and Xie, 2011; Telias et al., 2013; La Fata et al., 2014; Khalfallah et al., 2017), it seems therefore possible that the microsatellite of FMR1 might have currently unrecognized functions in the individual peculiarities characteristic to FXS.
To Silence or Not to Silence Expanded FMR1?
Transcript toxicity in humans
Mirroring the results on microsatellites and protein expression, the different FMR1 alleles indeed give rise to different expression patterns: pre-mutation carriers are characterized by enhanced mRNA, but normal or slightly reduced protein levels, since the elongated transcripts are inefficiently translated, but heavily transcribed (Tassone et al., 2000a,b; Kenneson et al., 2001; Primerano et al., 2002; Ludwig et al., 2014), whereas full-mutations cause FMRP deficiency due to DNA hypermethylation, Histone modification and subsequent heterochromatin formation (Pieretti et al., 1991; Sutcliffe et al., 1992; Hornstra et al., 1993; Coffee et al., 1999, 2002; Kumari and Usdin, 2010). Some residual mRNA is nonetheless still present in many men with FXS, but the mRNA is not translated (Tassone et al., 2001), probably due to secondary structure formation in the tandem tract.
It is noteworthy that pre-mutation carriers often develop a neurodegenerative disorder called the Fragile X-associated Tremor/Ataxia Syndrome (FXTAS, Figure 2), which presents with neurodegeneration, parkinsonism and brain atrophy, and which is associated with primary ovarian insufficiency in females (reviewed in Botta-Orfila et al., 2016; Hagerman and Hagerman, 2016). FXTAS is believed to arise from a toxicity of elongated mRNA transcripts and/or of a cryptic FMR1 protein derived from CGG repeat triggered non-ATG translation (Handa et al., 2005; Hashem et al., 2009; Chen et al., 2010; Todd et al., 2013). Since the transcript levels are markedly reduced in FXS patients, these findings suggest that the silencing of full-mutation alleles in FXS serves to prevent from toxic effects, however, clear evidence for a toxicity of the full-mutation mRNA or the cryptic protein is missing. Indeed, the identification of several healthy and non-mosaic individuals carrying unmethylated, normally expressing full-mutation alleles (Smeets et al., 1995; Pietrobono et al., 2005; Tabolacci et al., 2008) argues against the idea of mutation-triggered toxicity in humans. Nonetheless, two cases were identified, where expression of a full-mutation gene caused severe FXTAS (Loesch et al., 2012; Santa Maria et al., 2014), thus supporting the idea that a fully mutated FMR1 transcript can have toxic effects and that elongated transcripts are causative for FXTAS, but not for FXS, although some mRNA is present in many FXS patients (Tassone et al., 2001).
These apparently conflicting cases illustrate that individual genes and/or environmental effects may overcome the typical mechanisms and phenotypes observed in FXS. The relevance of the latter is further emphasized by the fact that alcohol abuse seemed to be involved in the case of severe FXTAS reported by Loesch and colleagues (Loesch et al., 2012). Recent studies revealed that alcohol is in fact exaggerating behavioral problems such as aggression and impulsivity in FXS patients (Salcedo-Arellano et al., 2016) and accelerating neurological deterioration in FXTAS (Muzar et al., 2014). It might be for these negative effects that, in contrast to cases of high-functioning autism, where patients used drinking to cope with social anxiety (Lalanne et al., 2015), alcoholism is at least in FXS patients rare (cp. Salcedo-Arellano et al., 2016).
For our future research, it will therefore be important to address questions such as: Why may fully mutated FMR1 transcripts have toxic effects in one case, but not in the other? How does the environment influence the underlying mechanisms? Why may it be better to have a fully mutated gene silenced, and thus FXS, than unsilenced, and eventually FXTAS — from an evolutionary point of view? A deeper understanding of the relevance of genetic individuality is required before attempting a reactivation of FMR1 in patients (reviewed in Tabolacci et al., 2016b) — in particular, since full-mutation mRNA is not necessarily translated (Tassone et al., 2001; Dolskiy et al., 2017) and since transcribed alleles are able to cause FXTAS under circumstances currently unknown (Loesch et al., 2012; Santa Maria et al., 2014).
A question of secondary structures?
An alternative explanation for the silencing of full-mutation alleles is DNA stability. During germ cell generation as well as during early phases of embryogenesis and prior to the silencing of FMR1, CGG repeats may expand or contract through mechanisms under debate (reviewed in Mor-Shaked and Eiges, 2016; Gerhardt, 2017). Despite some uncertainty about the exact molecular events that cause repeat instability, it is believed that the formation of secondary DNA structures during recombination, DNA replication and DNA repair leads to the addition or deletion of repeats. Since no repeat instability has been observed in FMR1 postnatally (Reyniers et al., 1993, 1999; Wöhrle et al., 1993), it is thought that the instability is related to events of the embryogenesis.
Little is known about the factors that could contribute to the instability of the CGG repeats in FMR1. It has been noticed that the number of repeats, the content of interspersed AGG and the haplotype are able to influence the stability of FMR1 (Oberlé et al., 1991; Eichler et al., 1994; Gunter et al., 1998; Hirst and White, 1998; Taylor et al., 1999; Larsen et al., 2000; Dombrowski et al., 2002; Nolin et al., 2003, 2013, 2015; Yrigollen et al., 2012, 2013, 2014; Avitzour et al., 2014; Weiss et al., 2014). AGG interruptions, for instance, have been indicated to support the stability of FMR1 by reducing secondary structure formation (Weisman-Shomer et al., 2000; Jarem et al., 2010) and promoting appropriate DNA conformations (Jarem et al., 2010) as well as adequate DNA packing (Mulvihill et al., 2005; Volle and Delaney, 2013). Furthermore, the number of repeats was shown to directly correlate with the instability of the tandem tracts (Oberlé et al., 1991; Eichler et al., 1994; Taylor et al., 1999; Avitzour et al., 2014): it is assumed that the size of G-rich tracts directly correlates with the formation of secondary structures and polymerase slippage during replication (Mornet et al., 1996; Freudenreich et al., 1997; Weitzmann et al., 1997; Hirst and White, 1998). In line with this idea, the total length of the CGG repeat allele turned out to be the best predictor for the risk of transmission (Yrigollen et al., 2014).
Aside from internal genetic properties, trans-acting factors have also been postulated to affect the stability of FMR1 during mitosis and meiosis (Mornet et al., 1996; Nolin et al., 1996, 1999), but although it seems plausible that other genes might impact on the stability of FMR1, studies are rare. In an attempt to identify such trans-acting factors, Xu et al. (2013) analyzed two microarray sets containing data on the transcript expression in FXS patients and controls, and found a significant down-regulation of DNA damage/repair pathway transcripts, thus implying that impaired DNA repair pathways may support FMR1 instability in FXS patients.
Remarkably, environmental factors have also been found to influence the instability of FMR1: maternal age was recently related to increased instability (Yrigollen et al., 2014) and oxidative stress was demonstrated to interfere with the stability of FMR1 (Adihe Lokanga et al., 2014). The latter finding is in line with other studies showing that different kinds of stress can induce instability in microsatellites (Chatterjee et al., 2015; Wu et al., 2017) and that oxidized DNA can trigger repeat expansion or contraction (Lai et al., 2013; Cilli et al., 2016). Given that chronic alcoholism causes oxidative stress (reviewed in Wu et al., 2006; Hernández et al., 2016), the data imply that alcohol abuse could contribute to the consolidation of FMR1 diseases. Interestingly, a study conducted by Kogan and colleagues found alcoholism to be significantly more common in families of pre-mutation carriers than in control families (Kogan et al., 2008). Although this association does not tell whether the disease (FXTAS) is causing the alcoholism, for example by overcharging family members who care for their affected relatives, or whether the alcoholism is causing the disease (or both), the facts that maternal alcoholism seems to increase the risk for FXS (mentioned in Hagerman et al., 2010) and that foetuses are particularly vulnerable to alcohol (reviewed in Henderson et al., 1999; Dennery, 2007) support a role for alcohol abuse in the emergence of FXS. Much more research is needed to establish the relation of toxins, such as derived from smoking or drinking, oxidative stress, microsatellite instability and the consolidation of FMR1 diseases.
Repeat numbers exceeding 200 tandems trigger the epigenetic silencing of FMR1 by initiating an abnormal 5′-C-phosphate-G-3′ methylation and repressive Histone modifications in the promoter region (e.g., Coffee et al., 1999, 2002; Chandler et al., 2003; Kumari and Usdin, 2010; Brasa et al., 2016). As a consequence, CpG5 islands flanking the repeats as well as the repeats itself, which also function as a CpG island, become hypermethylated and render the gene inactive (Hansen et al., 1992). Studies in human FXS cell lines aiming to reactivate FMR1 by either changing repressive Histone modifications (Kumari and Usdin, 2016; Dolskiy et al., 2017) or decreasing CpG methylation (Chiurazzi et al., 1998; Coffee et al., 2002; Tabolacci et al., 2005, 2016a), suggest that DNA methylation is the primary cause for gene inactivity, while repressive Histone methylations have a supportive function.
The molecular mechanisms by which CGG expansions trigger the epigenetic silencing of FMR1 are currently not well understood though (for a review, please see Usdin and Kumari, 2015). It is thought that transient unpairing of the DNA during replication, transcription or repair provides an opportunity for the repeat region of FMR1 to form secondary structures such as hairpins or G-quadruplexes (Fry and Loeb, 1994; Kettani et al., 1995; Mitas et al., 1995; Usdin and Woodford, 1995; Patel et al., 2000; Loomis et al., 2014). Similar structures occur in FMR1 transcripts (Handa et al., 2003; Napierala et al., 2005; Zumwalt et al., 2007; Malgowska et al., 2014). Studies showed that these secondary structures hinder replication, transcription and translation (Fry and Loeb, 1994; Nadel et al., 1995; Usdin and Woodford, 1995; Subramanian et al., 1996) and that structure-disrupting proteins are able to alleviate the situation (Fukuda et al., 2005; Khateb et al., 2007), thus suggesting that the formation of secondary structures is troubling the cells.
Not all structures are alike though: R-loops, a DNA-RNA hybrid formed during transcription, were recently indicated to prevent gene silencing by protecting DNA from de novo methylation (Ginno et al., 2012). Moreover, R-loops were shown to cause chromosome decondensation and transcription activation (Powell et al., 2013). Since FMR1 has been observed to give rise to R-loops (Groh et al., 2014; Loomis et al., 2014), this data could nicely explain the enhanced expression of pre-mutation alleles observed in FXTAS, but experimental evidence is missing. In fact, Groh and colleagues found that the formation of R-loops on FMR1 impedes gene expression (Groh et al., 2014), thus suggesting that R-loop formation is involved in the silencing of FMR1. Their data are in line with another study demonstrating that promoter-bound FMR1 transcripts containing tandem tracts induce the silencing of FMR1 (Colak et al., 2014).
The relevance of the number of repeats required to build R-loops and the role of the respective loop size are not yet clear though, since R-loops were found to form on normal, pre-mutation and full-mutation alleles (Colak et al., 2014; Groh et al., 2014; Loomis et al., 2014). It seems therefore possible that the role of R-loops in transcription regulation depends on the loop size: Loomis and colleagues provided evidence that the expansion of FMR1 repeats causes an enhanced formation of aggrandized loops, which tend to form higher-order structures. These structures distinguish FMR1 from other CpG islands containing promoters (Ginno et al., 2012, 2013) and could finally trigger repeat instability and hypermethylation (Loomis et al., 2014). Hence, R-loops formed on FMR1 could enhance gene expression until they exceed a specific size and form secondary structures. Previous research on the methylation of different loci indeed indicated that it is the higher-order structures that trigger DNA methylation (Smrzka et al., 1995; Paoloni-Giacobino et al., 2007; Gentry and Meyer, 2013).
These findings imply that CpG methylation could serve to limit the formation of secondary structures. Nuclear magnetic resonance analyses revealed CpG methylations to decrease the dynamics of the DNA backbone (Geahigan et al., 2000), while molecular dynamic investigations illustrated DNA methylation to increase the rigidity of the DNA by steric hinderance and hydrophobicity (Derreumaux et al., 2001). Using density functional theory and nuclear magnetic resonance measurements, Taqi and colleagues further demonstrated that cytosine methylation impairs the conformational flexibility of short ssDNA6 molecules and their ability to form secondary structures (Taqi et al., 2012). Although these data support the idea that hypermethylation of FMR1 could serve to prevent the formation of secondary structures, more evidence is required.
Further studies showed that the hypermethylation of FMR1 indeed correlates with enhanced tract stability during mitosis (Gläser et al., 1999; Wöhrle et al., 2001; Nichol Edamura et al., 2005). Remarkably, Zhou and colleagues observed that cells carrying fully mutated and hypermethylated alleles outcompete those carrying alleles with less repeats and no methylation when co-cultured, resulting in a loss of these cells (Zhou et al., 2016). Since no differences were seen in the viability of both cell lines, toxic effects are unlikely to account for the disappearing of cells with unsilenced alleles. The reasons for this effect remain to be investigated though.
The data imply that hypermethylation of FMR1 should occur when cells start to divide a lot. Studies using fetal tissues showed that hypermethylation is established between the 10th and 12th week of gestation, but FMR1 may remain partly active for some time (Devys et al., 1992; Sutcliffe et al., 1992; Suzumori et al., 1993; Iida et al., 1994; Willemsen et al., 2002; reviewed in Mor-Shaked and Eiges, 2018). Models of cell division in human embryos do not support high rates of mitosis during this time though, they rather show a decline in division rates (Luecke et al., 1999). Looking at the brain in specific, the situation is yet different: By the 9th week of gestation, neuronal tube formation is completed and shortly after, at the 12th week of gestation, neurogenesis as well as neuronal migration will reach their first peak (reviewed in Linderkamp et al., 2009). Nerve cells will then be proliferating at rates of about 15 million per hour (reviewed in Ackerman, 1992), thus implying that the silencing of FMR1 could be related to cell line-specific changes serving in neurogenesis. Such a mechanism could be important to maintain the correct timing and pace during neuronal development, which is essential to establish the complex connections that characterize the brain.
Indeed, when Khalfallah and colleagues induced differentiation in a murine embryonic stem cell line lacking FMRP (shFmr1 ES), they found an accelerated generation of both, progenitor and neuronal cells during the first steps of neurogenesis (Khalfallah et al., 2017; reviewed in Bardoni et al., 2017; Westmark, 2017). Their experiments further revealed that this phenotype is due to enhanced expression of a target of FMRP, APP7, which is able to accelerate neurogenesis following cleavage into the A-beta peptide. This mechanism might also provide an alternative or additional explanation for the observation that embryonic stem cells carrying a hypermethylated FMR1 gene outcompete cells with the active gene as it was found by Zhou and colleagues (Zhou et al., 2016). It will be interesting to see how these changes affect neuronal maturation and signaling and how the findings of Khalfallah and colleagues relate to neuronal development in humans.
Notably, studies employing human embryonic stem cells (hESCs) showed that the epigenetic silencing of FMR1 may occur prior to differentiation (Avitzour et al., 2014; Zhou et al., 2016) or upon differentiation (Eiges et al., 2007): of 11 hESC lines, 7 showed some levels of stable hypermethylation prior to differentiation. It is not yet clear though, when and how the hypermethylation is established. Current models suggest that fully expanded genes first acquire abnormal methylation patterns before or during embryo implantation and that FMR1 silencing is achieved after the blastocyst stage. Microsatellites that escape the initial methylation changes are believed to remain unmethylated. Since standard reprogramming procedures serving to generate induced pluripotent stem cells (iPSCs) from skin fibroblasts are unable to remove the exaggerated methylation marks inactivating FMR1 (Urbach et al., 2010; Sheridan et al., 2011; Doers et al., 2014), hypermethylation is thought to be stable and irreversible once it is established.
This is not necessarily the case though: removing CGG repeats and the immediate 5′-flanking region from fully mutated microsatellites in the FMR1 gene of a male iPSC line by the CRISPR/Cas9 system, Park and colleagues were able to restore FMR1 gene expression as well as FMRP protein levels in a pair of clones (Park et al., 2015). The reactivation of FMR1 led to a stable expression of the gene throughout differentiation into mature neurons. These findings are further supported by the previously mentioned study of Zhou and colleagues, who showed that hypermethylation present in embryonic stem cells is dynamic (Zhou et al., 2016): alleles containing over 400 repeats may contract to smaller repeat numbers, resulting in a permanent reactivation even when more than 200 tandems are present. Moreover, de Esch and colleagues (de Esch et al., 2014) observed that the reprogramming of fibroblasts from an atypical individual carrying an active full mutation FMR1 gene with 330 repeats into iPSCs recurrently resulted in a complete inactivation of the gene. Taken together, these studies not only illustrate the dynamics of the system, but also suggest that repeat length is not the only factor influencing the silencing of FMR1 and that other factors, such as neurodevelopmental stage are involved. Indeed, the fact that the brother of the atypical individual also carried an unsilenced full-mutation implies that maternal-parental components, which were not present in fibroblasts or inactivated during the reprogramming procedure, and/or specific environmental factors caused their FMR1 genes to remain active despite the high repeat length.
Aside from tandem repeat polymorphism, mutations in the coding region of FMR1 have also been associated with the occurrence of FXS in patients (Quartier et al., 2017): a deletion of the last exon, which is giving rise to a truncated FMRP isoform, was recently identified in three brothers meeting FXS criteria (Hagerman’s scores = 15). Moreover, two splice variants were detected in two unrelated patients showing the same outcome in the test, and several missense mutations have been identified in FXS patients (Siomi et al., 1994; Handt et al., 2014; Myrick et al., 2014). Although these cases represent rare exemptions, they nonetheless demonstrate that the correct functioning of FMRP is central to FXS.
The Fragile X Syndrome — of Mice
The Genotype of Mice or the Charm of Simplicity
Mice are much different from humans, they are much smaller, live much shorter and have much less to learn. Nonetheless, mice and men are sharing almost 99% of their genes (Waterston et al., 2002) as well as most physiological functions and pathogenic mechanisms (see for example the reviews of Tecott, 2003; Elefteriou and Yang, 2011; Van der Weyden and Adams, 2013; Jacobs et al., 2013; Eilam, 2014; Hoehndorf et al., 2014; Vandamme, 2014; Lubojemska et al., 2016). Indeed, even their aging was recently shown to match human senescence surprisingly well (Dutta and Sengupta, 2016). Since mice are also easy to keep, they became the most widely used model organism in life sciences after their first documented employment almost 500 years ago (reviewed in Paigen, 2003; Goodman et al., 2015): of nearly 11.5 million animals used for scientific purposes in the European Union in 2011, 61% were mice (European Commission, 2010). Despite these massive research efforts, most attempts to translate the outcomes to humans have failed. In the FXS field for instance, more than 70 studies reporting rescues (excluding reviews) have been published on pubmed during the last 12 years, 63 clinical trials are registered on ClinicalTrials.gov, and not a single treatment is available for patients yet (2nd of October 2017; current state reviewed in Ligsay and Hagerman, 2016). Although several positive outcomes were observed during the trials, indicating at least some progress toward a better understanding of the disease and a treatment for patients, discrepancies between the data obtained in men and mice were common: benefits experienced by patients were often very subtle, limited to subgroups, outside the outcome measures or simply absent. How much wishful-thinking is involved in our mouse models?
Some facts on men and mice (for more details, please see Table 1):
• mice are 3000× smaller
• mice and humans diverted 75 million years ago (Waterston et al., 2002)
• laboratory mice are highly inbred
• their genome is approximately 14% smaller, probably due to deletions (Waterston et al., 2002)
• the DNA sequence identity is only 40% (Waterston et al., 2002)
• the average substitution rate in mice is twofold higher than in humans (Waterston et al., 2002)
• manipulations of their genome may induce mutations in their microsatellite sequences (Zuo et al., 2012; Du et al., 2013)
• mouse-specific promoter and enhancer regions are significantly enriched in repetitive sequences (Yue et al., 2014)
• only 12.6% of the murine DNA are associated with regulatory functions such as transcription factor binding, chromatin organization etc. (humans: 20%; Yue et al., 2014)
• approximately 50% of the regulatory sequences have no identifiable orthologs in human (Cheng et al., 2014; Yue et al., 2014)
• 38.5% of mouse-specific transcription enhancers do not show activity in human ES cells (Yue et al., 2014)
• mice have dozens of local gene family expansions related to reproduction, immunity and olfaction (Waterston et al., 2002)

Table 1. Differences between men and mice.
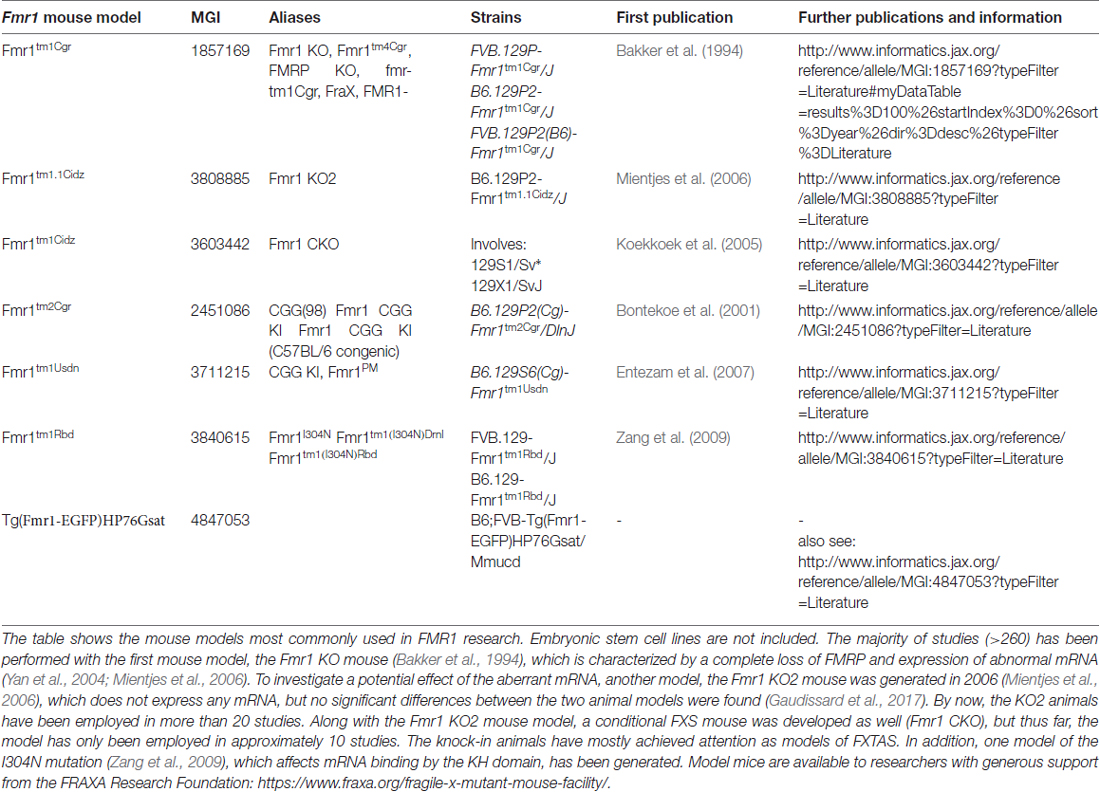
Table 2. Fragile X Syndrome and Fragile X-associated Tremor/Ataxia Syndrome model mice.
Taken together, these studies show that although men and mice share many features, they differ in a variety of aspects when more detailed data are included (Figure 3). It is now widely accepted that the characteristics of mice and men mostly arise from alterations in the mechanisms controlling gene expression, in particular from variations and polymorphisms of cis-elements (reviewed in Wittkopp and Kalay, 2011).
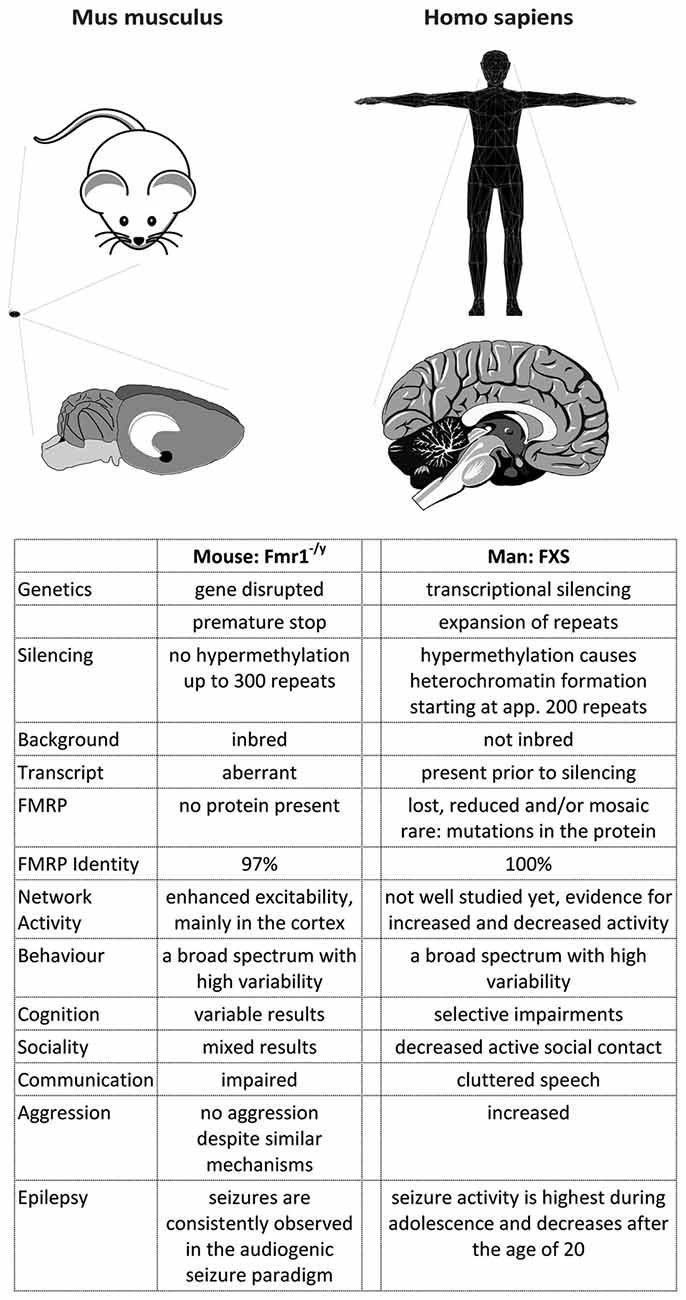
Figure 3. A comparison of FXS in mice and men. The figure summarizes some major differences between FXS model mice and patients. Most differences arise from the development of the cortex in primates, which caused a rewiring inside the cortex as well as between the cortex and the hippocampus (and potentially other brain regions). Consequently, the behavioral phenotype observed in men and mice does not match very well, although the mouse model recapitulates many biochemical aspects of the disease. In addition, the complex genetics of the disease cannot be modeled in mice, probably due to a more relaxed gene expression control in this species.
Genetics of the FXS Model Mouse
The fundamental difference between commonly used FXS model mice (Fmr1−/y mice8) and patients is that mice never have any FMRP, beginning with their very first moment of existence, whereas in patients, the gene is active at least until the 10th week of gestation (Devys et al., 1992; Sutcliffe et al., 1992; Suzumori et al., 1993; Iida et al., 1994; Willemsen et al., 2002). Moreover, patients display mosaic expression patterns (Kaufmann et al., 1999; Dyer-Friedman et al., 2002; Loesch et al., 2003, 2004; Govaerts et al., 2007; Pretto et al., 2014; Basuta et al., 2015) as well as residual full-length mRNA (Tassone et al., 2001), but mice don’t.
In order to overcome this dissatisfying situation, several CGG repeat knock-in mice were made to mimic the genotype found in humans (e.g., Bontekoe et al., 1997, 2001; Lavedan et al., 1997, 1998; Baskaran et al., 2002; Peier and Nelson, 2002; Fleming et al., 2003; Brouwer et al., 2007; Entezam et al., 2007; Alam et al., 2010). Similar to human pre-mutation carriers (Tassone et al., 2000a,b; Kenneson et al., 2001; Primerano et al., 2002; Ludwig et al., 2014), the knock-in mice display a direct correlation between the mRNA transcript level and the repeat length as well as an indirect correlation between the repeat length and the protein level, showing significant variation between individual animals (Ludwig et al., 2014).
In terms of pathological features, the knock-in mice reflect several biochemical, histological and behavioral symptoms of FXTAS patients (Willemsen et al., 2003; Van Dam et al., 2005; Entezam et al., 2007; Hunsaker et al., 2010, 2011; Wenzel et al., 2010; Cunningham et al., 2011) and pre-mutation transcripts were found toxic in mice (Handa et al., 2005; Hashem et al., 2009; Chen et al., 2010; Hukema et al., 2014). However, no explicit data on behavioral deficits or toxic effects were reported for the full-mutation situation (200–350 repeats; Entezam et al., 2007; Hunsaker et al., 2010, 2011; Ludwig et al., 2014) in specific, thus suggesting that contrary to humans, 350 repeats still represent a pre-mutation situation in mice.
Although some instability has been recognized in mice harboring pre-mutation alleles of about 100–260 repeats (Bontekoe et al., 2001; Peier and Nelson, 2002; Brouwer et al., 2007; Entezam et al., 2007), and based on these models, transacting mechanisms such as mismatch repair and transcript coupled repair were identified to support instability of the gene in mice (Zhao and Usdin, 2014; Zhao X. N. et al., 2015), instability is apparently not a major phenotype of the mouse (Bontekoe et al., 1997; Lavedan et al., 1997, 1998; Peier and Nelson, 2002; Fleming et al., 2003).
This situation first changed, when Baskaran and colleagues introduced the SV40 origin of replication along with their transcript to the gene (Baskaran et al., 2002) in order to exclude nucleosome formation. The transgene is driving the expression of FMR1 Exon 1 by the SV40 early promoter encoded in the SV40 origin, which excludes chromatin formation at the transgenic locus. Exon 1 contains 26 copies of the CGG repeat together with the translational FMR1 start codon. As a result, the transgene obtained an open chromatin structure compared to the normally nucleosome-flanked CGG repeats seen in other mouse models (Datta et al., 2011) and is therefore more prone to instability (cp. Oostra, 1998). Thereby, Baskaran and colleagues achieved expansions from 26 to 350 repeats within three generations only; however, the DNA remained unmethylated (Alam et al., 2010). Sadly, these results are in line with two more recent studies which also achieved significant expansions (120–230 repeats; Brouwer et al., 2007; Entezam et al., 2007), but no methylation and no inactivation of Fmr1. Given that the expression of the full-mutation mRNA did apparently not cause unusual severe FXTAS or even FXS phenotypes, and given that the full-mutation alleles were neither hypermethylated nor inactivated or significantly unstable, these data support the idea that in mice, 200–350 repeats still represent a pre-mutation situation.
Taken together, these studies further suggest that the mechanisms leading to the inactivation of FMR1 might be different in mice and humans (cp. the section “A Question of Secondary Structures?”). Indeed, Matsuo and colleagues found that CpG islands of men and mice have different properties: comparing 23 orthologous genes, they discovered that mice almost always have less pronounced islands, or even none at all. The authors speculate that the CpG islands in mice might have eroded during evolution as an adaption to the mouse’s small body mass and short life-span by allowing for a more relaxed control of gene activity (Matsuo et al., 1993). Their study is in line with an earlier report that also detected low numbers of CpG islands in mice (Aïssani and Bernardi, 1991), thus implying that mice may simply not have the capabilities to methylate genes the same way humans do, causing fully mutated Fmr1 to remain active even if humanized transgenes are employed (e.g., Lavedan et al., 1998). This hypothesis is supported by comparative in vivo footprinting analyses across several human and mouse CpG islands that demonstrated striking differences in the protein-DNA interactions of both species (Cuadrado et al., 2001, reviewed in Antequera, 2003). Furthermore, Fu and colleagues found that the murine and the human DNA methyltransferase DNMT1, which is responsible for the maintenance of methylation patterns by preferentially adding methyl groups to hemi-methylated CpG sites, differ in their processivity (Fu et al., 2012). In particular, they demonstrate a high level of processivity for human DNMT1 at FMR1, which is possibly not achieved by the murine enzyme as it has much longer non-association tracts in vivo.
Phenotypes of the Fmr1−/y Mouse
FMRP Functions to Regulate the mRNA Metabolism
Studying FMRP functions in humans is challenging. It is therefore no surprise, that most of the corresponding knowledge we acquired is derived from mouse models. However, recent advances in stem cell research contributed significantly to our understanding of the disease.
In healthy individuals, FMRP is widely expressed, yet most abundant in testes and brain, where it is present at particular high levels throughout the cerebral cortex, the hippocampus and the Purkinje cell layer as well as the granular layer of the cerebellum (Devys et al., 1993; Hinds et al., 1993; Bakker et al., 2000, also see http://mouse.brain-map.org/gene/show/14042 and http://www.gensat.org/GeneProgressTracker.jsp?gensatGeneID=339). FMRP has been detected in glial and neuronal cells (Wang et al., 2004; Gholizadeh et al., 2015), but its special importance in synaptic signaling has drawn most attention on its function in neurons. Here, FMRP is primarily located in the cytosol and the nucleus (Feng et al., 1997), but it has also been found along dendrites, axons and at synaptic sites (Weiler et al., 1997; Greenough et al., 2001; Antar et al., 2006; Akins et al., 2017). Indeed, FMRP travels between these locations through microtubules (Feng et al., 1997; Antar et al., 2005).
The main function of FMRP is to regulate the mRNA metabolism. Interacting with five different RNA-motifs (U-Pentameres, Kissing complex, SosLip, G-quartets, G-rich regions), the protein may associate with a diversity of mRNAs, approximately 4% of all mRNAs in the mammalian brain (Brown et al., 2001; Miyashiro et al., 2003; Darnell et al., 2011). Detailed biochemical studies revealed that thereby, FMRP is able to regulate not only mRNA transport (Dictenberg et al., 2008; Estes et al., 2008) and stability (Bagni and Greenough, 2005; De Rubeis and Bagni, 2010), but also mRNA translation (Ceman et al., 2003; Zalfa et al., 2003; Bechara et al., 2009). Furthermore, FMRP was found to influence the microRNA-pathway, thus gaining further control on the expression of its target proteins (reviewed in Kenny and Ceman, 2016).
Inside the nucleus, FMRP localizes to active transcription sites, where it binds to nascent mRNA (Eberhart et al., 1996; Kim et al., 2009) and may even take action in alternative splicing, since G-quartets present in the mRNA of FMRP itself were found to function as exonic splicing enhancers (Didiot et al., 2008). Studies of the transcription and splicing machinery actually revealed brain-region as well as cell type specific alterations in Fmr1−/y mice (Derlig et al., 2013) and showed that loss of FMRP results in aberrant transcriptional regulation (Korb et al., 2017). Furthermore, FMRP was found to bind to DNA and to function in DNA damage response (Alpatov et al., 2014) as well as in Heterochromatin organization (Tan et al., 2016). These findings suggest that FMRP may control every step of protein expression from DNA organization to translation.
FXS and Group 1 Metabotropic Glutamate Receptors
On the other hand, FMRP itself turned out to be under control of mGluR1/59 signaling cascades (Narayanan et al., 2008), hence allowing for an activity dependent regulation of the mRNA metabolism in various aspects by FMRP. Detailed studies on the function of mGluRs in FXS have lead to the advance of the mGluR-Theory (Huber et al., 2002; Bear et al., 2004; Dolen et al., 2007; Nakamoto et al., 2007): the theory states that FMRP normally acts as a repressor of mRNA translation downstream of group 1 mGluRs, which is released after mGluR activation and thereby induces the translation of proteins required for the expression of LTD. Hence, in the absence of FMRP, persistent and mGluR stimulation independent synthesis of LTD-proteins causes ongoing AMPAR10 internalization. The theory further posits that exaggerated mGluR-signaling, perhaps as a consequence of malfunctioning feedback inhibition, is causing many of the symptoms observed in FXS.
There is indeed good evidence for this theory: the pharmacological down-regulation of mGluR5 signaling has been shown to improve a variety of typical symptoms in Fmr1−/y mice, including aberrant neuronal morphology, hyperactivity, social behavior, seizure susceptibility and learning and memory (Yan et al., 2005; de Vrij et al., 2008; Levenga et al., 2011; Su et al., 2011; Michalon et al., 2012; Vinueza Veloz et al., 2012; Gantois et al., 2013; de Esch et al., 2015) and so has a genetic reduction of group 1 mGluRs (Dolen et al., 2007; de Esch et al., 2015). One group reported some contradictory findings (Thomas et al., 2011), but the promise of the results prompted clinical trials with mGluR antagonists such as AFQ056 (Novartis) and RO4917523 (Roche) to down-regulate the exaggerated mGluR signaling. Although patients treated with these substances were initially reported to experience some behavioral improvements (Jacquemont et al., 2011), all trials were discontinued during phases IIb/III since the studies did not show any significant improvements in abnormal behaviors compared to placebo (reviewed in Scharf et al., 2015). It has been speculated that cross-reactions with other drugs used to treat FXS patients, the long time of perpetuation in adult patients, irreversible changes during early brain development and difficulties in the outcome measures might have caused the failure, but the underlying reasons have remained unclear.
Remarkably, several targets of FMRP belong to the two major mGluR-signaling cascades controlling the expression of related proteins, the ERK11 - and the mTOR12 - pathway: ERK, PI3K13, PIKE14, GSK315 and mTOR are target mRNAs of FMRP (Darnell et al., 2011; Ascano et al., 2012). Both signaling pathways turned out to be exaggerated not only in the hippocampus of Fmr1−/y mice (ERK pathway: Hou et al., 2006; Michalon et al., 2012; mTOR pathway: Sharma et al., 2010; Liu Z. H. et al., 2012; Bhattacharya et al., 2016; Choi et al., 2016), but also in humans (Weng et al., 2008; Hoeffer et al., 2012; Wang X. et al., 2012; Kumari et al., 2014; Pellerin et al., 2016).
There has been some dispute about the status of ERK activation in FXS though (mice: Hu et al., 2008; Gross et al., 2010; Osterweil et al., 2010; patients: Yrigollen et al., 2016), but pathway-specific inhibitors of ERK-signaling constantly rescued characteristic deficits in FXS models (Chuang et al., 2005; Osterweil et al., 2010; Wang X. et al., 2012), thus supporting the idea that enhanced mGluR1/5 cascades are causative for some symptoms of the disease. Further studies found that inhibition of ERK-signaling with Metformin (Gantois et al., 2017) or Lovastatin (Osterweil et al., 2013) indeed ameliorates many deficits in Fmr1−/y mice. Strikingly though, mTOR inhibition with Rapamycin turned out to induce adverse effects on sleep and social behavior in both, control and Fmr1−/y mice (Saré et al., 2018), although positive effects of Rapamycin were reported for the BTBR T+Itpr3tf/J mouse model of autism (Burket et al., 2014). In fact, some data even suggest that mTOR inhibition with Rapamycin might cause neurodegeneration (Lin et al., 2013). The hopes are therefore now on clinical trials with Lovastatin (Caku et al., 2014; Pellerin et al., 2016) and Metformin (Dy et al., 2018).
Brain Region Specific Mechanisms in FXS
All the above mentioned strategies to amend FXS suffer from the same difficulty though; contrary to drugs, the outlined mechanisms only apply to the hippocampus. There is evidence that the signaling mechanisms of the cortical and hippocampal networks are differentially affected by the loss of FMRP. In cortical synaptosomes of Fmr1−/y mice for example, mTOR activity was found normal (Sawicka et al., 2016), whereas in the hippocampus, mTOR signaling is exaggerated (Sharma et al., 2010; Liu Z. H. et al., 2012; Bhattacharya et al., 2016; Choi et al., 2016). Cortical ERK was demonstrated to be erroneously deactivated following mGluR stimulation in Fmr1−/y mice (Kim et al., 2008), whereas it was illustrated to be normal (Osterweil et al., 2010) or even over-activated in hippocampal tissue (Hou et al., 2006; Michalon et al., 2012). Remarkably, the study of Sawicka and colleagues showed that ERK signaling in the neocortex of Fmr1−/y mice is impinging on ribosomal protein S6, which usually receives input from mTOR, though in this study, cortical ERK activity was found exaggerated.
The data on patients are mostly derived from fibroblasts (Kumari et al., 2014; Yrigollen et al., 2016) or thrombocytes (Weng et al., 2008; Pellerin et al., 2016), making conclusions on the characteristics of specific brain regions difficult. Using post-mortem tissue, one study detected no differences in ERK activation in the frontal lobe between FXS patients and controls (Hoeffer et al., 2012), while another one found increased levels of phosphorylated ERK in the frontal cortex of patients (Wang X. et al., 2012). Despite the unclear status of ERK activation in the cortex, these studies suggest that mGluR1/5 signaling pathways are functioning differently in the cortex and the hippocampus. In line with this notion, a recent study of mTOR activity and exercise demonstrated that the activation status of mTOR is depending on the brain region, the cell type (neuron or glia) and the type of exercise (sedentary, voluntary or forced; Lloyd et al., 2017).
Long term potentiation
Further support for the relevance of brain region specific mechanisms in FXS comes from a variety of studies on synaptic plasticity. Synaptic plasticity is mostly investigated by the induction of LTP16 or LTD17, two paradigms which are considered cellular models of learning and memory (reviewed in Kandel, 2001; Bliss et al., 2003; Neves et al., 2008). Both, LTP and LTD, ultimately depend on the modulation of synaptic signaling and have been studied intensively in Fmr1−/y mice using manifold induction protocols (please see Table 3 for more details).
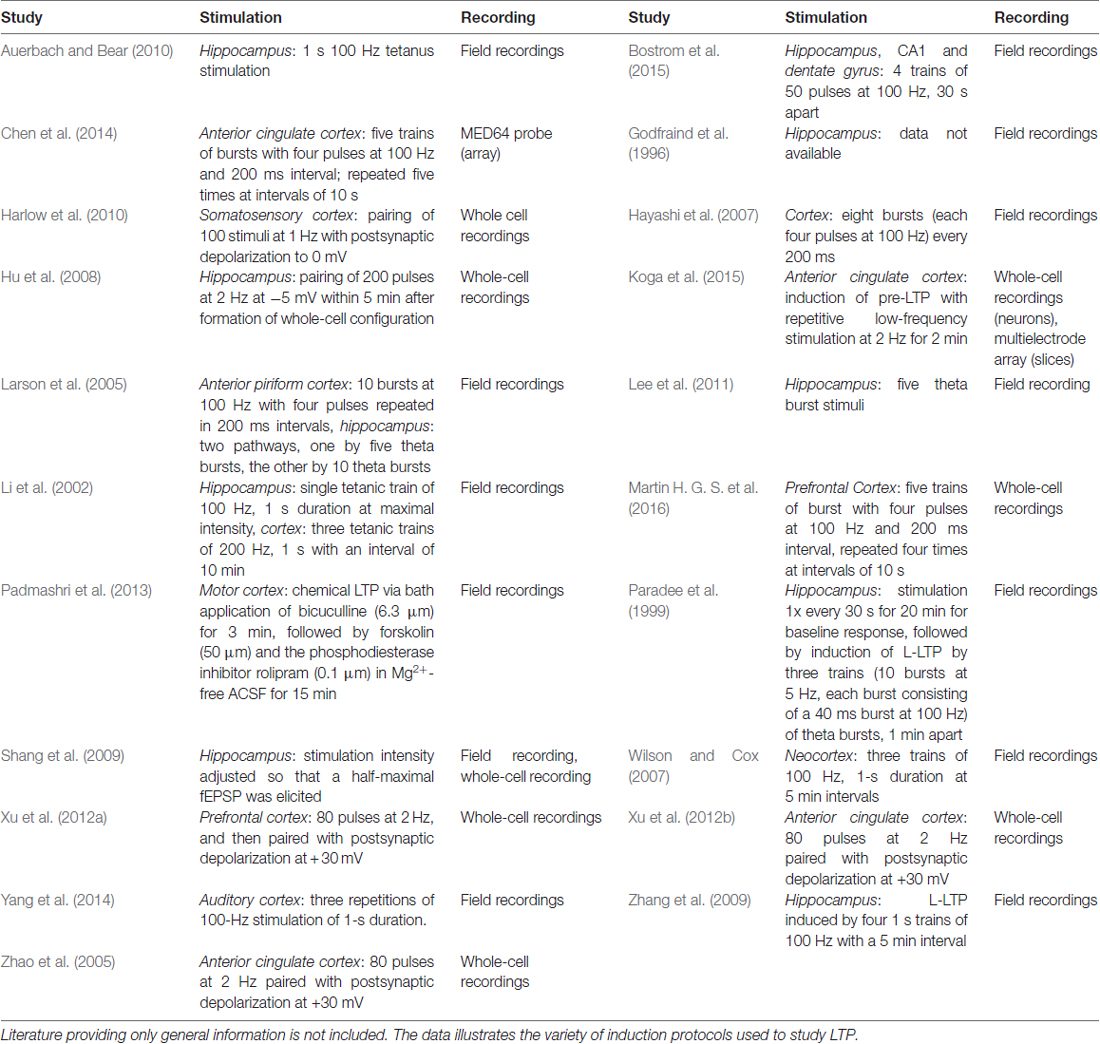
Table 3. LTP protocols used in different studies.
While some protocols have led to the discovery of disturbances in certain forms and aspects of hippocampal LTP (Hu et al., 2008; Shang et al., 2009; Lee et al., 2011), most examinations found the expression of hippocampal LTP in Fmr1−/y mice to be normal (Godfraind et al., 1996; Paradee et al., 1999; Li et al., 2002; Larson et al., 2005; Zhang et al., 2009; Auerbach and Bear, 2010). Two of these studies also investigated cortical LTP: while they observed no abnormalities in hippocampal LTP, both detected significantly impaired LTP in the cortex of Fmr1−/y mice (Li et al., 2002; Larson et al., 2005). The result is line with several studies demonstrating defective LTP in different regions of the cortex (Zhao et al., 2005; Hayashi et al., 2007; Wilson and Cox, 2007; Harlow et al., 2010; Xu et al., 2012b; Padmashri et al., 2013; Yang et al., 2014; Koga et al., 2015).
LTP includes two distinct phases, an early phase (E-LTP), which does not require protein synthesis, and a late phase (L-LTP), which depends on protein synthesis and gene expression (Frey et al., 1993; Abel et al., 1997, reviewed in Kandel, 2001, 2009). While L-LTP is unaffected in the hippocampus of Fmr1−/y mice (Paradee et al., 1999; Zhang et al., 2009), it is blocked in the cingulate cortex (Chen et al., 2014). Since pharmacological inhibition of mGluR5 or GSK3 rescued L-LTP in the cingulate cortex of Fmr1−/y mice, these results show that exaggerated mGluR signaling is involved. Indeed, two other studies also reported rescues of cortical LTP based on mGluR1/5 antagonists (Xu et al., 2012a; Martin H. G. S. et al., 2016), but the effects of mGluR5 or GSK3 inhibition on (L-)LTP in the hippocampus were never investigated although hippocampal L-LTP is known to depend on mGluR1/5 activation (Riedel and Reymann, 1996; Francesconi et al., 2004; Neyman and Manahan-Vaughan, 2008; Fan, 2013).
The differences between the hippocampus and the cortex are further emphasized by the fact that age-related deficits in LTP were discovered specifically in the cortex (Larson et al., 2005; Martin H. G. S. et al., 2016). While Fmr1−/y mice older than 6 months displayed significant defects in the expression of cortical LTP, the mice never displayed any impairment in hippocampal LTP. In fact, Bostrom et al. (2015) even demonstrated differences within the hippocampus itself: while the loss of FMRP caused impairments in NMDAR18-dependent LTP in the dentate gyrus, NMDAR-LTP was found normal in the CA119 region of the hippocampus.
Long term depression
The most prominent and best studied plasticity model in FXS is mGluR-LTD, a type of LTD that depends on the activation of mGluR1/5, protein synthesis and the internalization of AMPA receptors. However, while numerous studies demonstrated enhanced mGluR-LTD in the hippocampus of Fmr1−/y mice (Huber et al., 2002; Hou et al., 2006; Nosyreva and Huber, 2006; Volk et al., 2007; Park et al., 2008; Ronesi and Huber, 2008; Zhang et al., 2009; Auerbach and Bear, 2010; Choi et al., 2011; Bhattacharya et al., 2012; Michalon et al., 2012; Niere et al., 2012; Costa et al., 2015; Toft et al., 2016; Thomson et al., 2017), nobody studied mGluR-LTD in the cortex yet. There is evidence though that cortical mGluR-LTD is existing (reviewed in Kang and Kaang, 2016). Using a protocol to induce spike-time-dependent plasticity (STD-LTP or STD-LTD) in the neocortex of Fmr1−/y mice, Desai and colleagues found no impairment in LTD, but a significant reduction in LTP (Desai et al., 2006). Experiments with MPEP20, a mGluR5 antagonist, revealed that cortical STD-LTP is not depending on mGluR5 activation, whereas cortical STD-LTD is. Remarkably, the application of anisomycin, an inhibitor of protein synthesis, revealed that cortical STD-LTD does not require protein synthesis, thus suggesting that FMRP is not necessary for cortical STD-LTD despite the dependence on mGluR5. Sadly though, STD-LTD has not been investigated in the hippocampus yet.
It is worth noting that the biological relevance of mGluR-LTD has recently been questioned, arguing that mGluR-LTD in the absence of previous LTP is artificial (discussed in Jones, 2017). Nonetheless, since more than 15 years of mGluR-LTD research in Fmr1−/y mice did not provide any evidence for abnormal mGluR-LTD in the cortex, it seems likely that the mechanisms underlying this form of plasticity differ among brain regions.
Keeping the balance
FXS is associated with a vast misregulation of protein expression, not only in the context of mGluR signaling, but also with respect to proteins regulating other aspects of neuronal excitability such as Calmodulin or Neuroligin for instance (Liao et al., 2008; Matic et al., 2014; Kalinowska et al., 2015; Tang et al., 2015). Although it is not clear yet whether abnormal mGluR-LTD directly impacts on the balance of excitatory and inhibitory activity in neuronal networks (E/I balance), mGluR signaling is able to alter the excitability of neurons by increasing the intrinsic conductance (Bianchi et al., 2009; Tang and Alger, 2015). Furthermore, FMRP itself may bind to ion channels such as Calcium, Slack21 and BK22 channels, thereby providing an additional level of FMRP mediated control (Brown et al., 2010; Deng et al., 2013; Ferron et al., 2014).
Deviations in the balance of excitation and inhibition have been associated with seizure activity, hypersensitivity and cognitive deficits in several ASDs and animal models (reviewed in Frye et al., 2016; Uzunova et al., 2016; Lee et al., 2017; e.g., Orekhova et al., 2008; Tebartz van Elst et al., 2014; Robertson et al., 2016), including Fmr1−/y mice (e.g., D’Hulst et al., 2006; Zhong et al., 2009; Dahlhaus and El-Husseini, 2010; Aguilar-Valles et al., 2015; Deng and Klyachko, 2016). Using these animals, in particular neocortical circuits have been shown to experience enhanced excitation (Gibson et al., 2008; Goncalves et al., 2013; Patel et al., 2013; Zhang Y. et al., 2014; Westmark et al., 2016), partially due to inhibitory deficits (Selby et al., 2007) and potentially supported by inhibitory dysfunctions in the cortico-hippocampal pathway and inhibitory defects in feed-forward circuits (Wahlstrom-Helgren and Klyachko, 2015).
Indeed, not only glutamatergic signaling has been demonstrated to be altered in Fmr1−/y mice, but also gabaergic and dopaminergic mechanisms have been found malfunctioning. Studies showed that Fmr1−/y mice experience diminished GABAa/b23 receptor expression, reduced GABA release, decreased dopamine receptor expression and malfunctioning interneurons (D’Hulst et al., 2006; Selby et al., 2007; Pacey et al., 2011b; Paluszkiewicz et al., 2011; Henderson et al., 2012; Heulens et al., 2012; Patel et al., 2013; Paul et al., 2013; Berzhanskaya et al., 2016a; Kang et al., 2017). Deficiencies in inhibitory conductance are therefore characteristic to many circuits of the murine FXS brain including circuits of the striatum, amygdala, hippocampus, subiculum and the somatosensory as well as the prefrontal cortex (Centonze et al., 2008; Curia et al., 2009; Olmos-Serrano et al., 2010; Paluszkiewicz et al., 2011; Vislay et al., 2013; Martin et al., 2014; Sabanov et al., 2016). In addition, the loss of interactions between FMRP and BK channels causes uncontrolled Glutamate release, altered action potential waveforms and exaggerated excitability (Zhang Y. et al., 2014; Myrick et al., 2015; Deng and Klyachko, 2016).
Recent research indicates that the alterations in the E/I balance may include brain region and circuit specific mechanisms. For instance, while dendrites of hippocampal neurons from Fmr1−/y mice display increased HCN1-channel24 expression and reduced input resistance (Brager et al., 2012), dendrites of cortical layer 5 neurons show the opposite (Zhang Y. et al., 2014). The intrinsic membrane excitability of cortical layer 4 excitatory neurons is exaggerated (Gibson et al., 2008), while that of excitatory hippocampal neurons is normal (Deng et al., 2013; Luque et al., 2017). Hippocampal neurons demonstrate significantly longer action potential durations and higher firing frequencies in the absence of FMRP than under normal conditions (Luque et al., 2017), whereas layer 2/3 neurons in the prefontral cortex present significantly narrower and taller action potentials in Fmr1−/y mice than in their wildtype litter mates (Routh et al., 2017). Although both alterations indicate enhanced excitability, the sharpened action potentials observed the cortex of Fmr1−/y mice are in contrast with the broadened action potentials seen in the hippocampus. Since two studies even demonstrated increased inhibition early in development (Berzhanskaya et al., 2016b; Truszkowski et al., 2016), the data show that the alterations in neuronal activity depend on the specific circuit, aspect and age.
Despite the high complexity of the system, several studies were able to rescue the aberrant neuronal activity based on restorations of mGluR1/5 signaling (Meredith et al., 2011; Ronesi et al., 2012; Westmark et al., 2016; Aloisi et al., 2017), GABA signaling (Olmos-Serrano et al., 2010, 2011; Martin B. S. et al., 2016; Kang et al., 2017) or ion channel function (Zhang Y. et al., 2014; Deng and Klyachko, 2016; Aloisi et al., 2017). In doing so, phenotypes such as epileptiform activity (Ronesi et al., 2012; Zhang Y. et al., 2014; Deng and Klyachko, 2016; Westmark et al., 2016), hyperactivity (Olmos-Serrano et al., 2011; Ronesi et al., 2012) and hypersensitivity (Zhang Y. et al., 2014) were normalized. A few studies have also investigated network oscillations in FXS model animals (Gibson et al., 2008; Goncalves et al., 2013; Rotschafer and Razak, 2013; Radwan et al., 2016; Westmark et al., 2016; Berzhanskaya et al., 2017). In these experiments, cortical neurons displayed increased synchrony in their activity as well as a threefold higher firing rate during Up states (Gibson et al., 2008; Goncalves et al., 2013; Westmark et al., 2016), increased high-frequency as well as reduced low-frequency power during rest (Berzhanskaya et al., 2017) and elevated responses to auditory stimuli (Rotschafer and Razak, 2013).
The findings support the theory that neuronal hyperexcitability is a leading cause for many symptoms of FXS and fit well with the Intense World Theory of Autism (reviewed in Markram and Markram, 2010), which posits that hyperactive micro-networks cause many of the cognitive deficits characteristic to ASDs, in particular hypersensitivity, hyperattention, hyperemotionality and seizure susceptibility. Although the studies of oscillatory dynamics in FXS patients currently available mostly confirm the idea of imbalanced circuit activity (Castrén et al., 2003; Dalton et al., 2008; Holsen et al., 2008; Van der Molen and Van der Molen, 2013; Van der Molen et al., 2014; Wang et al., 2017), not all of them are in favor of the theory: two studies detected decreased activity, one in prefrontal regions and one in the fusiform gyrus (Dalton et al., 2008; Holsen et al., 2008), while a third study found intracortical inhibition in FXS patients to be normal (Oberman et al., 2010).
From Mice to Men
Literature Summary
Most rescues reported from Fmr1−/y mice are based on investigations of the hippocampus (Table 4). Some studies included data on further brain regions or transferred approaches to cortical regions, analyzing different aspects of mGluR signaling for example; however, only 4 studies were found that focussed on the cortex in terms of their strategy and their experiments. No studies could be identified that are reporting behavioral rescues based on mechanisms characteristic to the amygdala, cerebellum, striatum or any other brain region in specific, although the available data argue for different deficits in different brain regions (see for instance Chen et al., 2014; Bostrom et al., 2015; Sawicka et al., 2016; Lloyd et al., 2017) and despite the fact that rescues of neuronal activity have been reported for the amygdala (Olmos-Serrano et al., 2010, 2011; Suvrathan et al., 2010; Martin B. S. et al., 2016). These findings show that there is a significant imbalance between the current hippocampus centered investigations and the multifarious mechanisms found in the brain of wildtype as well as FXS model animals.
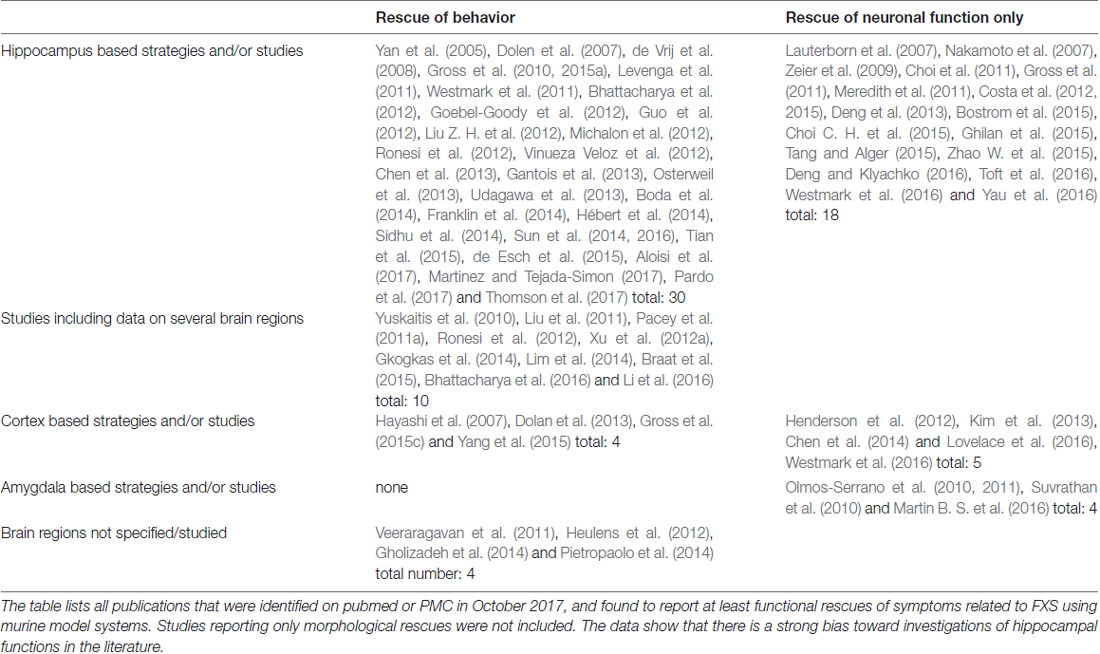
Table 4. Brain-region bias in FXS research.
The Cortex, the Hippocampus and FXS
Problems in translating findings from mice to men are a common phenomenon in brain research. A major difficulty arises from the development of the cerebral cortex in primates and in particular in humans. Recent research revealed that the cognitive performance of vertebrates is best reflected by a combination of the number of neurons, neuron density and axonal conduction velocity in the cortex (reviewed in Dicke and Roth, 2016). In this regard, mice are not well set, since their cerebral cortex exhibits only a relatively low density of neurons: if mice would have a brain the size of a human, their cerebral cortex would contain 2 billion neurons, whereas the human cerebral cortex does in fact have 16 billion (cp. Herculano-Houzel, 2009). However, neuron density is not the only disadvantage mice are confronted with:
• the neocortex constitutes 80% of the human brain (Azevedo et al., 2009), but only 40% of a mouse brain (reviewed in Herculano-Houzel, 2009)
• while mice have about 20 different cortical areas, humans have more than 200 (Kaas, 1989, 2011; Changizi and Shimojo, 2005)
• most cortical areas in mice are related to sensory and motor functions, whereas the majority of the human areas function in association (reviewed in Buckner and Krienen, 2013)
• some parts of the human neocortex are specifically enlarged compared to the rest of the neocortex:
- the prefrontal cortex and area 10 (Semendeferi et al., 2002, 2011), which is important to higher cognitive functions,
- the insula, which usually functions in sensory information processing, but has new areas in humans that are functioning in empathy and social awareness (reviewed in Keysers et al., 2010) and
- the posterior parietal cortex (reviewed in Orban et al., 2006), which is important to planning, imitation and the highly skilled use of tools.
• by contrast, the primary sensory and motor areas have maintained their relative sizes during the evolution from mice to men (Hill et al., 2010; Preuss, 2011)
• several cell types found in the human brain do not exist in mice (reviewed in Buckner and Krienen, 2013).
In line with these findings, rodents perform relatively poor in behavioral tasks when compared to monkeys possessing similar sized brains (e.g., capybara (Macdonald, 1981) and capuchin monkey (Anderson et al., 2013; De Moraes et al., 2014; Takahashi M. et al., 2015)), thus suggesting that primate brains have properties, which are significantly different from those of rodents.
This notion is emphasized by an elegant study of Han and colleagues (Han et al., 2013), who showed that human glia cells are much more competent than murine glia cells when it comes to supporting brain functions: replacing the glia of mice with human cells, they observed that the engrafted mice were able to propagate calcium signals three times faster than allografted mice, exhibited sharply enhanced LTPs and performed excellent in cognition tests. Indeed, astrocytes are able to coordinate and modulate neural signal transmission (reviewed in Allen, 2014; Allen and Eroglu, 2017), but human astrocytes differ from their murine counterparts in that human astrocytes are larger, more complex and more diverse, have much more synaptic contacts and are more efficient in calcium signaling (Andriezen, 1893; Colombo, 1996; Colombo et al., 1997, 2000; Reisin and Colombo, 2002; Oberheim et al., 2009).
Remarkably, this is particularly true for the cerebral cortex: there are at least four morphologically distinct astrocyte classes within the primate cortex, as compared to two in rodents. The two novel classes are interlaminar astrocytes and varicose projection astrocytes (Andriezen, 1893, reviewed in Vasile et al., 2017). Varicose projection astrocytes have hitherto been observed only in cortical layers 5–6 of humans and chimpanzees, but the human cells are more complex. By contrast, interlaminar astrocytes reside in upper cortical layers and extend long processes to cortical layers 3 and 4 (Colombo, 1996; Colombo et al., 1997; Reisin and Colombo, 2002; Korzhevskii et al., 2005; Oberheim et al., 2009). Although interlaminar astrocytes are present in both, monkeys and men, humans have higher numbers of interlaminar astrocytes. Despite the fact that more than 120 years have passed since their initial description, the role of these primate-specific astrocytes is still elusive.
Nonetheless, astroglia seem to have a role in FXS: though FMRP is predominantly expressed in neurons at all ages, it is also seen in oligodendrocyte precursor cells and astrocytes. While neurons display a gradual decrease in FMRP expression during development (Davidovic et al., 2011; Bonaccorso et al., 2015), astrocytes and oligodendrocyte precursor cells mainly express FMRP during early and mid-postnatal stages of brain maturation (Wang et al., 2004; Gholizadeh et al., 2015), thus suggesting a role for glia and FMRP in development. Indeed, it was shown that FMRP deficient astrocytes cause developmental delays in dendrite maturation and synaptic protein expression of hippocampal wildtype neurons (Jacobs et al., 2010, 2016), whereas normal astrocytes prevent abnormal dendritic development in hippocampal FXS neurons (Jacobs and Doering, 2010).
Investigations on the underlying mechanisms are also still in their infancy. In line with the role of FMRP in regulating protein expression, it was recently shown that the astrocyte-secreted factors Hevin25 and SPARC26, which function to control excitatory synapse development, display abnormal expression patterns in hippocampal and cortical tissues from Fmr1−/y mice (Wallingford et al., 2017): while hippocampal Hevin expression is gradually increasing from a reduced expression at P727 to elevated levels at P21, Hevin expression in the cortex displays only a transient increase at P14. SPARC, on the other hand, shows a modest decrease at P7 and P14 in the cortex, but no abnormalities in the hippocampus. Emphasizing the relevance of this data, Cheng and colleagues were able to demonstrate that astrocyte-conditioned medium is sufficient to prevent morphological deficits in hippocampal neurons cultured from FXS model mice (Cheng et al., 2016). Furthermore, Higashimori and colleagues showed that a selective knock-out of astroglial FMRP in mice modestly increases spine density and size in cortical neurons, whereas a selective re-expression is able to attenuate the abnormal spine morphology characteristic for FXS (Higashimori et al., 2016). The group also described a normalization of the FXS-typical gain in body weight, but no data on cognitive performances were reported. Studies with human astroglia would be desirable, but are currently missing.
The expansion of neocortical areas in primates caused a profound rewiring of the cortex. As a consequence, the human association cortex lacks the strict hierarchical organization of circuits seen in rodents, although certain projections follow the canonical form (e.g., parieto-prefrontal projections), but is instead characterized by multiple, large-scale distributed and highly interwoven networks termed non-canonical circuits. These circuits are highly active during cognitive performances (reviewed in Goldman-Rakic, 1988; Buckner and Krienen, 2013; Margulies, 2017).
Notably, not only the internal pathways of the cortex changed during the evolution from mice to men, also the wiring between the cortex and the hippocampus was modified: in men, the hippocampus preferentially connects to cortical association networks, whereas in mice, it preferentially associates with sensory networks (Bergmann et al., 2016). Although there are still two conserved parallel pathways between the cortex and the hippocampus in mice and men, which transfer object and context related information (reviewed in Ranganath and Ritchey, 2012), the finding of Bergmann and colleagues is of quiet some significance since cortical-hippocampal pathways are required for important brain functions including spatial working memory (studies in rodents and humans; reviewed in Sigurdsson and Duvarci, 2016), long-term memory (studies in rodents, primates and humans; reviewed in Sigurdsson and Duvarci, 2016), motivation and emotion (studies in rodents; reviewed in Sigurdsson and Duvarci, 2016), and social recognition (studies in rodents and humans; reviewed in Bicks et al., 2015). Indeed, weaknesses in working memory performance, in particular when requiring abstract item reasoning, are characteristic to FXS patients (Munir et al., 2000; Cornish et al., 2001; Kwon et al., 2001; Ornstein et al., 2008; Baker et al., 2011; Wang et al., 2013), but not to model mice (Leach et al., 2016). While Fmr1−/y mice perform as well as their wildtype littermates even when their working memory is significantly challenged (Leach et al., 2016), individuals with FXS are unable to modulate activation of the prefrontal and parietal cortex in response to an increasing working memory load (Kwon et al., 2001), implying a lack of circuit control. Indeed, a fMRI28 study showed that decreased levels of FMRP correlate with decreases in parahippocampal activation and reduced connectivity between the hippocampus and the prefrontal cortex in patients (Wang J. M. et al., 2012).
Emotional and social difficulties are frequently observed in individuals with FXS as well (Cohen et al., 1988; Mazzocco et al., 1994; Hall et al., 2009; Cordeiro et al., 2011; Kim et al., 2014), but the data on mice are inconsistent (reviewed in Kazdoba et al., 2014). Similar to the impairments observed in working memory performance, social deficits were associated with impairments in the activation of prefrontal regions in individuals with FXS (Holsen et al., 2008). Taken together, these data demonstrate that the cortical and cortical-hippocampal circuits, which characterize the human brain, are critical to FXS and cannot be appropriately modeled in mice. Even though mice may perform the same tasks as men, it is obligatory to investigate whether they also do it in the same way.
In the hippocampus, the situation is slightly different. Current research does not support major differences in the structure and function of hippocampi from mice and men (reviewed in Clark and Squire, 2013). A study comparing human and murine spatial navigation showed that both species use the same strategies (Eilam, 2014) to solve this basic and evolutionary old task. Spatial navigation in mammals is assumed to rely on hippocampal place cells, entorhinal grid cells (reviewed in Moser et al., 2008) and hippocampal theta oscillations, albeit neuronal firing in distant brain regions such as the somatosensory or prefrontal cortex is phase-locked to hippocampal theta oscillations (reviewed in Jacobs, 2013). Interestingly, the frequencies of these theta oscillations differ among species: while rats typically display oscillations at 4–8 Hz, human theta oscillations have a frequency of approximately 1–4 Hz (reviewed in Jacobs, 2013).
In line with this data, both, Fmr1−/y mice as well as FXS patients were found to show elevated error rates in the Hebb-Williams maze (MacLeod et al., 2010), a test that analyses spatial memory performance in humans and rodents under comparable conditions. In a follow-up study, the group was able to rescue the deficit in Fmr1−/y mice using MPEP, an antagonist of mGluR5 (Gandhi et al., 2014), however, contrary to humans, mice did not show increased latencies, implying differences between FXS individuals and model mice. The authors suggest that the discrepancy might arise from differences in the presentation of the maze, that is real vs. virtual, but since the human control group faced the same difficulty, this would rather argue for a different navigation strategy in FXS individuals, a strategy, that relies more on real information, such as obtained from walking, touching or smelling for instance. Indeed, difficulties in abstract thinking are characteristic for FXS patients, but not for model mice, which do as well as their wildtype litter mates in tests employing touch screens (cp. the review of Huddleston et al., 2014 and Leach et al., 2016). Since the Hebb-Williams maze is rarely used for rodents and was not employed to measure outcomes in clinical trials, conclusions are difficult to draw, in particular in the light of the role of cortical connections in human cognition and of the mixed results obtained from mice (reviewed in Kazdoba et al., 2014). However, even under conditions clearly favoring a translation between mice and men, such as in spatial navigation paradigms, the rodent model apparently reflects the situation in humans only partially.
Alternatives
Poor translatability represents indeed a major issue in most preclinical brain research. In order to overcome this obstacle, touch screen paradigms have recently been developed for rodents, which center on the Cambridge Neuropsychological Test Automated Battery for humans (reviewed in Horner et al., 2013; Hvoslef-Eide et al., 2016; Kangas and Bergman, 2017). Although these assays analyze specific cognitive abilities known to be affected in patients and achieve a high degree of standardization as well as high throughputs, Fmr1−/y mice failed to recapitulate the working memory impairments characteristic to individuals with FXS in corresponding tests even when their cognitive performance was challenged in a non-match to position task with increasing delays (Leach et al., 2016). The reasons are elusive. It is possible that the excessive pre-training as well as the correction trials required for mice affect the interpretation of results. Also, since working memory relies on the prefrontal cortex, differences in the cognitive processing can be expected, but studies are missing. More research is required to validate the neurocognitive mechanisms underlying each behavioral paradigm in our species of interest and to identify routes as well as boundaries of translation across species.
The outlined data illustrate that the current FXS mouse models fail to mirror important genetic as well as behavioral aspects of FXS, probably due to the evolutionary distance of mice and men. To address this issue, two rat models were recently developed, Fmr1 KO29 rats (Engineer et al., 2014; Till et al., 2015; Berzhanskaya et al., 2016b; Kenkel et al., 2016) and Fmr1 exon4 KO rats (Tian et al., 2017). The investigations revealed that the new model animals indeed recapitulate many features of the murine model, including enhanced basal protein synthesis, exaggerated hippocampal mGluR-LTD, elevated dendritic spine densities and cortical hyper-excitability (Till et al., 2015; Berzhanskaya et al., 2016b; Tian et al., 2017). However and much to a surprise, the rats neither reflect the behavioral phenotype of mice very well (Till et al., 2015), nor reproduce the symptoms of FXS patients any better than the murine model: although the animals demonstrated deficits in hippocampus-dependent learning (Till et al., 2015; Tian et al., 2017), they failed to display defects in spatial reference memory and reversal learning (Till et al., 2015), thereby contrasting not only the majority of data obtained from Fmr1−/y mice (D’Hooge et al., 1997; Paradee et al., 1999; Van Dam et al., 2000; Baker et al., 2010, reviewed in Kazdoba et al., 2014), but also the weak performance of FXS patients in tasks requiring the solution of new problems (Dykens et al., 1987; Maes et al., 1994; Loesch et al., 2004; Lewis et al., 2006; Van der Molen et al., 2010). Instead, these results rather imply that there is a clade-dependent component in the behavioral phenotype observed in FXS, causing the same molecular and cellular changes to give rise to different behavioral phenotypes. Given the high number of proteins affected by the loss of FMRP, numerous options exist, through which the genetic background may impact on the symptoms of the disorder. It is indeed long known that the specific phenotype of FXS model mice varies significantly among strains (reviewed in Bernardet and Crusio, 2006; Kazdoba et al., 2014). Further studies would be needed to characterize the performance of FXS model rats in cognition as well as sociality in more detail and to explain certain discrepancies between the two rat models (see Till et al., 2015; Tian et al., 2017), but it might not be worth the effort.
Considering the evolution of cognition in primates, the logical model organism for human brain function would be a closely related primate. Although virus-mediated transgenesis (Sasaki et al., 2009; Liu et al., 2016; Park et al., 2016) or ZFN30/TALEN31-driven genome editing (Sato et al., 2016) have been used to generate primate models, the invention of Crispr-mediated genome editing and its successful employment in primates (Niu et al., 2014; Chen et al., 2015; Tu et al., 2017; Zhao et al., 2017; Zuo et al., 2017) and even in (nonviable) human embryos (Liang P. et al., 2015; Tang et al., 2017) have brought this option within much closer reach. Compared to rodents, primates have several advantages when it comes to modeling cognition (for a comprehensive review, please also see “Why primate models matter”; Phillips et al., 2014), in particular with respect to ASDs:
• the cell types and circuits seen in primates are more similar to those found in humans, which is relevant to many cognitive tasks, but particularly important to spatial working memory and social recognition (see Hopkins, 2013; Frey et al., 2014; Neubert et al., 2014; Morecraft et al., 2015; Wilson et al., 2015a; and the section: “The Cortex, the Hippocampus and FXS”)
• the prolonged prenatal development of the cortex, which is characteristic to humans and primates, is not present in rodents, hampering neuro-developmental studies in rodents
• group living, co-operative behavior and cultural intelligence are much more sophisticated in primates (cp. the section: “The Cortex, the Hippocampus and FXS” and Decasien et al., 2017; Street et al., 2017)
• the basic communication features characteristic to human language are already present in primates, including the ability to utilize symbolization, basic semantic representation, categorical representation and rudimentary grammar (Moore et al., 2003, 2005, 2006; Joly et al., 2012; Morrill et al., 2012; Ghazanfar et al., 2013; Wilson et al., 2013, 2015b)
• many tests developed to analyze cognition in humans can easily be adapted for primates, e.g., eye-tracking to study abnormal gaze (Machado and Nelson, 2011; Rosati et al., 2016), a typical symptom of autism spectrum disorders, or computerized cognition tests (Spinelli et al., 2004; Harris et al., 2007; Barner et al., 2008; Diester and Nieder, 2010; Jones et al., 2010; Takemoto et al., 2011; Verrico et al., 2011; Beran et al., 2012, 2015; Brosnan et al., 2012; Evans and Beran, 2012; Basile and Hampton, 2013; Klein et al., 2013; Bramlett-Parker and Washburn, 2016; Oikonomidis et al., 2017), thereby facilitating the translation of results
Taken together, these studies show that many features characterizing human behavior and typically affected in ASDs, such as communication capabilities and social skills or abstract thinking for instance, are represented in primates. A primate model would hence facilitate investigations addressing core symptoms of the autism phenotype, which are absent or only rudimentary developed in rodents.
There is another important feature of the disease, which cannot be modeled in mice: hypermethylation. Contrary to mice, in which most CpG-islands have apparently eroded during evolution (Aïssani and Bernardi, 1991; Matsuo et al., 1993), primates show methylation patterns highly similar to those of humans. Approximately 90% of the methylation patterns are in fact conserved between men and chimpanzees (Martin et al., 2011; Molaro et al., 2011; Pai et al., 2011), in particular at CpG-islands and promoter regions (Illingworth et al., 2010; Hernando-Herraez et al., 2013, 2015; Long et al., 2013). However, when comparing those genes that are differentially methylated, human genes turned out to exhibit lower levels of promoter methylation than genes from chimpanzees (Gama-Sosa et al., 1983; Zeng et al., 2012), suggesting that gene expression control is less strict in humans. Since a disproportional high number of differentially methylated genes is associated with human diseases (Zeng et al., 2012; Fukuda et al., 2013; Hernando-Herraez et al., 2013), it seems possible that the reduced control of certain genes, which finally made us human, also makes us more susceptible to certain pathologies such as senescence and Alzheimer’s disease, disorders, which are not present in our closest relatives, the great apes (reviewed in Finch and Austad, 2015; Lowenstine et al., 2016). Notably, though some of the genes shown to be differentially methylated in humans are known for their association with ASDs, such as GABAa and GABAb receptors for instance (Zeng et al., 2012; Hernando-Herraez et al., 2013), FMR1 was not associated with human-specific methylation patterns yet.
Despite these promising premises, primate models also have some disadvantages when compared to rodent models: Working with primates, in particular with great apes, is expensive by all means, it requires a lot of training and experience, bears the risk of transmitting diseases and the longevity of great apes hampers investigations of senescence. Significant n-numbers are often hard to achieve. Hence, primate species which are small, relatively short lived and easy to keep could help to overcome these obstacles.
The evolutionary next closest alternatives to great apes are old world monkeys. Consisting of about 160 species, old world monkeys represent the largest group of primates. Among them are the macaque monkeys, which include two species, that have been used in science for a long time, the crab-eating (alias: cynomolgus; M. fascicularis) and the rhesus macaque (M. mulatta). These two species are the widest spread primates next to humans and are categorized as species with Least Concerns by the Red List. Facilitating housing and breeding, macaques live in large multi-male, multi-female groups, mature at the age of app. 3.5 years (great apes: 6–10 years) and have a gestation period of 160–180 days (great apes: 225–270 days), giving birth to one infant per parturition (reviewed in Lindburg, 1991).
Notably, crab-eating macaques have recently been used to model ASDs (Liu et al., 2016; Zhao et al., 2017). Liu and colleagues expressed human MeCP232 in the macaque brain to mimic an ASD resulting from duplications of the MECP2-containing genome segment, whereas Zhao and colleagues introduced deletions to SHANK333, which are known to cause ASD. Remarkably, both studies found striking differences compared to the corresponding mouse models: while the MECP2-overexpression model demonstrated clear autism-like behaviors in primates, but not in mice, the SHANK3-deletion model revealed significant differences in the expression pattern of Shank3, and found an impaired neurogenesis in the prefrontal cortex of macaques.
Despite these promising results, macaque models also have some disadvantages: The monkeys are carriers of Herpes B viruses and may transmit this potentially fatal disease to humans. Their long adolescence (3.5 years) hampers breeding, and their longevity (approximately 30 years in captivity) is clearly not supporting studies of senescence. Although highly developed, their social system is not in favor of ASD research as well, since macaques live in large multi-male/female groups, whereas humans live in (extended) family groups. As a result, proactive prosociality, which is believed to be based on shared infant care, is well developed in humans, but not in chimpanzees or macaques (Burkart and van Schaik, 2013).
For these reasons, marmosets have recently garnered interest as model animals for studying brain function (reviewed in Saito, 2015; Tokuno et al., 2015), for a general review on the species, please see (Schiel and Souto, 2017). Also known as zaris, these new world monkeys split from the primate lineage approximately 40 million years ago, 35 million years before humans and chimpanzees separated. Compared to great apes and macaques, marmosets have the advantage of being small, easy to handle, easy to breed and less expensive: with a life span of about 12 years in captivity, marmosets mature more quickly than great apes or macaques, starting reproduction at 18 months of age and giving birth to twins after a gestation period of 140–150 days. Contrary to macaques, marmosets do not represent a natural reservoir of Herpes B viruses.
Though lissencephalic (smooth), their brain shares many features of the human brain, and their cerebral cortex shows the neuronal architecture of all primates (Bendor and Wang, 2005; Burman et al., 2006; Elston et al., 2006; Burman and Rosa, 2009; de la Mothe et al., 2012; Chaplin et al., 2013), from which only humans differ in that neurons of their prefrontal cortex are more spiny and more complex than those of their primate relatives (Elston et al., 2006). Consequently, marmosets have the high cognitive abilities characteristic to all primates: they are not only able to perform true imitation (Bugnyar and Huber, 1997; Voelkl and Huber, 2007), transposition and generalization (Yamazaki et al., 2014) but to also solve string problems (Halsey et al., 2006; Gagne et al., 2012) and to understand physical causality (Yamazaki et al., 2011). Considering the weaknesses of FXS individuals in abstract item reasoning and in addressing new problems, the cognitive abilities of marmosets might help to develop tasks which directly translate results between the two species.
Of particular interest for the design of ASD models is the sociality of marmosets. In a convergent evolution to humans, marmosets developed a high level of group living (reviewed in Graham, 2016). At a state of sophistication not known from other mammals including most primates, marmosets demonstrate altruistic behaviors such as proactive prosociality and third-party reciprocity (Burkart et al., 2007; Burkart and van Schaik, 2013), social learning during infancy (Dell’Mour et al., 2009) as well as adulthood (Caldwell and Whiten, 2004), and behavioral adaptations to social environments (Koski and Burkart, 2015). Similar to humans, marmosets live in extended family groups, pair-bond and care for their offspring in a cooperative manner (Digby and Barreto, 1993; Sousa et al., 2005; Birnie et al., 2013; Yamamoto et al., 2014). Since parental care and in particular maternal warmth are known to influence FXS symptoms in children (Dyer-Friedman et al., 2002; Kuo et al., 2002; Glaser et al., 2003; Greenberg et al., 2012; Robinson et al., 2016; Smith et al., 2016), the latter could be useful to further investigate the role of the social environment for FXS.
Like other primates, marmosets mostly rely on vocalizations (Miller and Wang, 2006; Chen et al., 2009; Miller et al., 2010; Watson and Caldwell, 2010; Bakker et al., 2014; Kato et al., 2014; Agamaite et al., 2015) and visual cues (de Boer et al., 2013; Massen et al., 2016) for their communication, although social grooming (Lazaro-Perea et al., 2004) and scent marking (Epple, 1970; Massen et al., 2016) are present as well. Since ASDs are characterized by gaze avoidance, it is particularly interesting that marmosets are able to use a variety of facial expressions for communication (Kemp and Kaplan, 2013) and to gain information by geometrical gaze following, even from human experimenters (Burkart and Heschl, 2006). Analyzing the face scanning patterns performed by marmosets, Kotani et al. (2017) showed that these primates primarily view the eye region during contact and that this behavior can be employed to evaluate the influence of drugs.
Due to the dense vegetation of their natural habitat, vocalizations are unusual rich in marmosets, even for primates (Epple, 1968; Morrill et al., 2013; Agamaite et al., 2015). Marmoset conversations involve a cooperative vocal control and require the infants to learn when to talk and when to listen (Takahashi et al., 2013, 2016; Choi J. Y. et al., 2015; Chow et al., 2015), thereby paralleling human development. Indeed, recent research revealed that marmoset infants need to transform their babbling and crying into mature vocalizations in order to properly communicate with other group members (Margaret Elowson et al., 1998; Pistorio et al., 2006; Takahashi D. Y. et al., 2015; Ghazanfar and Zhang, 2016) and that this transformation requires social reinforcement from caregivers (Margaret Elowson et al., 1998; Takahashi et al., 2017). Given the ability of marmosets to even learn grammar (Wilson et al., 2013) and the cluttered speech observed in individuals with FXS (Hanson et al., 1986; Belser and Sudhalter, 2001; Roberts et al., 2007; Klusek et al., 2014), the high level of vocal communication in marmosets could be valuable to the development of treatments for FXS and ASD patients.
Though their sociality and their cognitive abilities make marmosets a promising model organism for FXS and ASD research, it is just this very same point, which also bears the disadvantages. The high level of communication and understanding of social interactions increases the risk that relations between researchers and experimental animals arise, which may impact on experiments (cp. Herzog, 2002), for example, if individual animals dislike or prefer being handled or cared for by a certain person and subsequent stress levels influence the outcome of an experiment. Since the n-numbers used in primate research are at the minimum, such effects might easily turn out “significant”.
Another important concern is morality. Although all mammals are able to feel pain, stress and fear in a similar way, no matter whether they are rodents, primates or of any other clade, the high cognitive capabilities and social skills of primates have raised particular ethical concerns regarding their use in biomedical research (cp. the reviews of Coors et al., 2010; Phillips et al., 2014; Zhou, 2014; Bailey and Taylor, 2016; Arnason, 2017). As a consequence, many countries have implemented policies and regulations to ensure the physiological as well as psychological well-being of all research animals (Pereira et al., 2004; Luy, 2007; Hansen et al., 2017), and the standards are particularly high for primates (see Tardif et al., 2013; Weiss and Hampshire, 2015). However, the ethical viewpoints often differ, not only within a society (cp. Buckley et al., 2011; Phillips et al., 2014; Arnason, 2017 vs. Bailey and Taylor, 2016), but also among societies (cp. Cyranoski, 2004; Zhang X. L. et al., 2014), causing the hurdles for the approval of a project to vary significantly between countries and sometimes even within countries. The same project might thus be approved within a few weeks in one country, within a few months in another, and not in years in the next.
These circumstances raise several questions:
Is it ethically justified to approve therapies developed in countries with lower animal welfare standards in countries with higher standards, considering that such approvals would torpedo all animal welfare efforts? Is it ethically justified to deny patients a therapy developed in a country, which had lower animal care standards? Is it ethically justified to have wealthy people traveling to countries offering corresponding therapies, while others cannot afford to do so?
Moreover: can cognitive abilities indeed be a measure to define welfare standards and requirements? Where is the red line? These two questions are intimately connected, since the use of cognitive capabilities as an argument for increased standards in turn causes diminished abilities to result in the opposite. Since everything goes the way of least constrains, this is the point where the seemingly high moral standards cause ethics and science to start loosing ground: is it ethically and scientifically justified to use thousands of mice in preclinical research, to enrol hundreds of patients in clinical studies, to put patients at risk, albeit the risk might be marginal, and to cause hopes and disappointments in their families, just to learn how hard it is to find out how to keep the Concorde flying from studying a biplane?
Author Contributions
RD wrote the manuscript.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
I would like to acknowledge support by the Deutsche Forschungsgemeinschaft and the Friedrich-Alexander-Universität Erlangen-Nürnberg (FAU) within the funding pogramme Open Access Publishing.
Footnotes
- ^ intelligence quotient scores
- ^ compare
- ^ Electroencephalography
- ^ untranslated region
- ^ 5′-C-phosphate-G-3′
- ^ single-stranded DNA
- ^ Amyloid-Precursor-Protein
- ^ In 2006, an inducible Fmr1−/y mouse model was generated to address aspects related to developmental or regional expression patterns, but the animals have only been used in a very few studies thus far, possibly due to limitations in the availability of appropriate Cre-lines. For more information, please refer to Table 2.
- ^ metabotropic glutamate receptors 1 and 5, = group 1 mGluRs
- ^ α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor
- ^ extracellular signal-regulated kinase
- ^ mammalian target of rapamycin
- ^ Phosphoinositid-3-Kinase
- ^ PI3K Enhancer
- ^ Glycogen synthase kinase 3
- ^ long term potentiation
- ^ long term depression
- ^ N-methyl-D-aspartate receptor
- ^ Cornu Ammonis region 1
- ^ 2-Methyl-6-(phenylethynyl) pyridine
- ^ sodium-activated potassium channel Slack
- ^ large-conductance calcium-activated potassium channels
- ^ gamma-aminobutyric acid
- ^ hyperpolarization-activated cyclic nucleotide-gated cation channel
- ^ high endothelial venule protein, also known as SPARC like protein
- ^ secreted protein, acidic and rich in cysteine
- ^ postnatal day 7
- ^ functional magnetic resonance imaging
- ^ knock-out
- ^ zinc-finger nucleases
- ^ transcription activator-like effector nucleases
- ^ methyl CpG binding protein 2
- ^ SH3 and multiple ankyrin repeat domains 3, alias: proline-rich protein 2
References
Abel, T., Nguyen, P. V., Barad, M., Deuel, T. A., Kandel, E. R., and Bourtchouladze, R. (1997). Genetic demonstration of a role for PKA in the late phase of LTP and in hippocampus-based long-term memory. Cell 88, 615–626. doi: 10.1016/s0092-8674(00)81904-2
Adams, R. H., Blackmon, H., Reyes-Velasco, J., Schield, D. R., Card, D. C., Andrew, A. L., et al. (2016). Microsatellite landscape evolutionary dynamics across 450 million years of vertebrate genome evolution. Genome 59, 295–310. doi: 10.1139/gen-2015-0124
Adihe Lokanga, R., Zhao, X.-N., Entezam, A., and Usdin, K. (2014). X inactivation plays a major role in the gender bias in somatic expansion in a mouse model of the fragile X-related disorders: implications for the mechanism of repeat expansion. Hum. Mol. Genet. 23, 4985–4994. doi: 10.1093/hmg/ddu213
Agamaite, J. A., Chang, C. J., Osmanski, M. S., and Wang, X. (2015). A quantitative acoustic analysis of the vocal repertoire of the common marmoset (Callithrix jacchus). J. Acoust. Soc. Am. 138, 2906–2928. doi: 10.1121/1.4934268
Aguilar-Valles, A., Matta-Camacho, E., Khoutorsky, A., Gkogkas, C., Nader, K., Lacaille, J. C., et al. (2015). Inhibition of group I metabotropic glutamate receptors reverses autistic-like phenotypes caused by deficiency of the translation repressor eIF4E binding protein 2. J. Neurosci. 35, 11125–11132. doi: 10.1523/JNEUROSCI.4615-14.2015
Aïssani, B., and Bernardi, G. (1991). CpG islands: features and distribution in the genomes of vertebrates. Gene 106, 173–183. doi: 10.1016/0378-1119(91)90197-j
Akins, M. R., Berk-Rauch, H. E., Kwan, K. Y., Mitchell, M. E., Shepard, K. A., Korsak, L. I., et al. (2017). Axonal ribosomes and mRNAs associate with fragile X granules in adult rodent and human brains. Hum. Mol. Genet. 26, 192–209. doi: 10.1093/hmg/ddw381
Alam, M. P., Datta, S., Majumdar, S., Mehta, A. K., Baskaran, S., Gulati, N., et al. (2010). Comparative analysis of DNA methylation in transgenic mice with unstable CGG repeats from FMR1 gene. Epigenetics 5, 241–248. doi: 10.4161/epi.5.3.11417
Allen, N. J. (2014). Astrocyte regulation of synaptic behavior. Annu. Rev. Cell Dev. Biol. 30, 439–463. doi: 10.1146/annurev-cellbio-100913-013053
Allen, N. J., and Eroglu, C. (2017). Cell biology of astrocyte-synapse interactions. Neuron 96, 697–708. doi: 10.1016/j.neuron.2017.09.056
Aloisi, E., Le Corf, K., Dupuis, J., Zhang, P., Ginger, M., Labrousse, V., et al. (2017). Altered surface mGluR5 dynamics provoke synaptic NMDAR dysfunction and cognitive defects in Fmr1 knockout mice. Nat. Commun. 8:1103. doi: 10.1038/s41467-017-01191-2
Alpatov, R., Lesch, B. J., Nakamoto-Kinoshita, M., Blanco, A., Chen, S., Stutzer, A., et al. (2014). A chromatin-dependent role of the fragile X mental retardation protein FMRP in the DNA damage response. Cell 157, 869–881. doi: 10.1016/j.cell.2014.03.040
Anderson, J. R., Takimoto, A., Kuroshima, H., and Fujita, K. (2013). Capuchin monkeys judge third-party reciprocity. Cognition 127, 140–146. doi: 10.1016/j.cognition.2012.12.007
Andriezen, W. L. (1893). The neuroglia elements in the human brain. Br. Med. J. 2, 227–230. doi: 10.1136/bmj.2.1700.227
Antar, L. N., Dictenberg, J. B., Plociniak, M., Afroz, R., and Bassell, G. J. (2005). Localization of FMRP-associated mRNA granules and requirement of microtubules for activity-dependent trafficking in hippocampal neurons. Genes Brain Behav. 4, 350–359. doi: 10.1111/j.1601-183x.2005.00128.x
Antar, L. N., Li, C., Zhang, H., Carroll, R. C., and Bassell, G. J. (2006). Local functions for FMRP in axon growth cone motility and activity-dependent regulation of filopodia and spine synapses. Mol. Cell. Neurosci. 32, 37–48. doi: 10.1016/j.mcn.2006.02.001
Antequera, F. (2003). Structure, function and evolution of CpG island promoters. Cell. Mol. Life Sci. 60, 1647–1658. doi: 10.1007/s00018-003-3088-6
Arnason, G. (2017). The ethical justification for the use of non-human primates in research: the Weatherall report revisited. J. Med. Ethics doi: 10.1136/medethics-2016-103827 [Epub ahead of print].
Ascano, M., Mukherjee, N., Bandaru, P., Miller, J. B., Nusbaum, J. D., Corcoran, D. L., et al. (2012). FMRP targets distinct mRNA sequence elements to regulate protein expression. Nature 492, 382–386. doi: 10.1038/nature11737
Auerbach, B. D., and Bear, M. F. (2010). Loss of the fragile X mental retardation protein decouples metabotropic glutamate receptor dependent priming of long-term potentiation from protein synthesis. J. Neurophysiol. 104, 1047–1051. doi: 10.1152/jn.00449.2010
Avitzour, M., Mor-Shaked, H., Yanovsky-Dagan, S., Aharoni, S., Altarescu, G., Renbaum, P., et al. (2014). FMR1 epigenetic silencing commonly occurs in undifferentiated fragile X-affected embryonic stem cells. Stem Cell Rep. 3, 699–706. doi: 10.1016/j.stemcr.2014.09.001
Azevedo, F. A., Carvalho, L. R., Grinberg, L. T., Farfel, J. M., Ferretti, R. E., Leite, R. E., et al. (2009). Equal numbers of neuronal and nonneuronal cells make the human brain an isometrically scaled-up primate brain. J. Comp. Neurol. 513, 532–541. doi: 10.1002/cne.21974
Bagni, C., and Greenough, W. T. (2005). From mRNP trafficking to spine dysmorphogenesis: the roots of fragile X syndrome. Nat. Rev. Neurosci. 6, 376–387. doi: 10.1038/nrn1667
Bagshaw, A. T., Horwood, L. J., Fergusson, D. M., Gemmell, N. J., and Kennedy, M. A. (2017). Microsatellite polymorphisms associated with human behavioural and psychological phenotypes including a gene-environment interaction. BMC Med. Genet. 18:12. doi: 10.1186/s12881-017-0374-y
Bailey, J., and Taylor, K. (2016). Non-human primates in neuroscience research: the case against its scientific necessity. Altern. Lab. Anim. 44, 43–69.
Baker, S., Hooper, S., Skinner, M., Hatton, D., Schaaf, J., Ornstein, P., et al. (2011). Working memory subsystems and task complexity in young boys with Fragile X syndrome. J. Intellect. Disabil. Res. 55, 19–29. doi: 10.1111/j.1365-2788.2010.01343.x
Baker, K. B., Wray, S. P., Ritter, R., Mason, S., Lanthorn, T. H., and Savelieva, K. V. (2010). Male and female Fmr1 knockout mice on C57 albino background exhibit spatial learning and memory impairments. Genes Brain Behav. 9, 562–574. doi: 10.1111/j.1601-183x.2010.00585.x
Bakker, C. E., De Diego Otero, Y., Bontekoe, C., Raghoe, P., Luteijn, T., Hoogeveen, A. T., et al. (2000). Immunocytochemical and biochemical characterization of FMRP, FXR1P and FXR2P in the mouse. Exp. Cell Res. 258, 162–170. doi: 10.1006/excr.2000.4932
Bakker, J., van Nijnatten, T. J., Louwerse, A. L., Baarends, G., Arndt, S. S., and Langermans, J. A. (2014). Evaluation of ultrasonic vocalizations in common marmosets (Callithrix jacchus) as a potential indicator of welfare. Lab Anim. (NY) 43, 313–320. doi: 10.1038/laban.568
Bakker, C. E., Verheij, C., Willemsen, R., van der Helm, R., Oerlemans, F., Vermey, M., et al. (1994). Fmr1 knockout mice: a model to study fragile X mental retardation. The Dutch-Belgian Fragile X Consortium. Cell 78, 23–33. doi: 10.1016/0092-8674(94)90569-x
Bar-Nur, O., Caspi, I., and Benvenisty, N. (2012). Molecular analysis of FMR1 reactivation in fragile-X induced pluripotent stem cells and their neuronal derivatives. J. Mol. Cell. Biol. 4, 180–183. doi: 10.1093/jmcb/mjs007
Barak, M. M., Lieberman, D. E., and Hublin, J. J. (2013). Of mice, rats and men: trabecular bone architecture in mammals scales to body mass with negative allometry. J. Struct. Biol. 183, 123–131. doi: 10.1016/j.jsb.2013.04.009
Bardoni, B., Capovilla, M., and Lalli, E. (2017). Modeling Fragile X syndrome in neurogenesis: an unexpected phenotype and a novel tool for future therapies. Neurogenesis 4:e1270384. doi: 10.1080/23262133.2016.1270384
Barner, D., Wood, J., Hauser, M., and Carey, S. (2008). Evidence for a non-linguistic distinction between singular and plural sets in rhesus monkeys. Cognition 107, 603–622. doi: 10.1016/j.cognition.2007.11.010
Basile, B. M., and Hampton, R. R. (2013). Dissociation of active working memory and passive recognition in rhesus monkeys. Cognition 126, 391–396. doi: 10.1016/j.cognition.2012.10.012
Baskaran, S., Datta, S., Mandal, A., Gulati, N., Totey, S., Anand, R. R., et al. (2002). Instability of CGG repeats in transgenic mice. Genomics 80, 151–157. doi: 10.1006/geno.2002.6813
Basuta, K., Schneider, A., Gane, L., Polussa, J., Woodruff, B., Pretto, D., et al. (2015). High functioning male with fragile X syndrome and fragile X-associated tremor/ataxia syndrome. Am. J. Med. Genet. A 167A, 2154–2161. doi: 10.1002/ajmg.a.37125
Bear, M. F., Huber, K. M., and Warren, S. T. (2004). The mGluR theory of fragile X mental retardation. Trends Neurosci. 27, 370–377. doi: 10.1016/j.tins.2004.04.009
Bechara, E. G., Didiot, M. C., Melko, M., Davidovic, L., Bensaid, M., Martin, P., et al. (2009). A novel function for fragile X mental retardation protein in translational activation. PLoS Biol. 7:e1000016. doi: 10.1371/journal.pbio.1000016
Belser, R. C., and Sudhalter, V. (2001). Conversational characteristics of children with fragile X syndrome: repetitive speech. Am. J. Ment. Retard. 106, 28–38. doi: 10.1352/0895-8017(2001)106<0028:ccocwf>2.0.co;2
Bendor, D., and Wang, X. (2005). The neuronal representation of pitch in primate auditory cortex. Nature 436, 1161–1165. doi: 10.1038/nature03867
Beran, M. J., Evans, T. A., Klein, E. D., and Einstein, G. O. (2012). Rhesus monkeys (Macaca mulatta) and capuchin monkeys (Cebus apella) remember future responses in a computerized task. J. Exp. Psychol. Anim. Behav. Process. 38, 233–243. doi: 10.1037/a0027796
Beran, M. J., Parrish, A. E., Futch, S. E., Evans, T. A., and Perdue, B. M. (2015). Looking ahead? Computerized maze task performance by chimpanzees (Pan troglodytes), rhesus monkeys (Macaca mulatta), capuchin monkeys (Cebus apella), and human children (Homo sapiens). J. Comp. Psychol. 129, 160–173. doi: 10.1037/a0038936
Bergmann, E., Zur, G., Bershadsky, G., and Kahn, I. (2016). The organization of mouse and human cortico-hippocampal networks estimated by intrinsic functional connectivity. Cereb. Cortex 26, 4497–4512. doi: 10.1093/cercor/bhw327
Bernardet, M., and Crusio, W. E. (2006). Fmr1 KO mice as a possible model of autistic features. ScientificWorldJournal 6, 1164–1176. doi: 10.1100/tsw.2006.220
Berry, D., Deater-Deckard, K., McCartney, K., Wang, Z., and Petrill, S. A. (2013). Gene-environment interaction between dopamine receptor D4 7-repeat polymorphism and early maternal sensitivity predicts inattention trajectories across middle childhood. Dev. Psychopathol. 25, 291–306. doi: 10.1017/s095457941200106x
Berzhanskaya, J., Phillips, M. A., Gorin, A., Lai, C., Shen, J., and Colonnese, M. T. (2016a). Disrupted cortical state regulation in a rat model of fragile X syndrome. Cereb. Cortex 5.
Berzhanskaya, J., Phillips, M. A., Shen, J., and Colonnese, M. T. (2016b). Sensory hypo-excitability in a rat model of fetal development in Fragile X Syndrome. Sci. Rep. 6:30769. doi: 10.1038/srep30769
Berzhanskaya, J., Phillips, M. A., Gorin, A., Lai, C., Shen, J., and Colonnese, M. T. (2017). Disrupted cortical state regulation in a rat model of fragile X syndrome. Cereb. Cortex 27, 1386–1400. doi: 10.1093/cercor/bhv331
Bhattacharya, A., Kaphzan, H., Alvarez-Dieppa, A. C., Murphy, J. P., Pierre, P., and Klann, E. (2012). Genetic removal of p70 S6 kinase 1 corrects molecular, synaptic and behavioral phenotypes in fragile X syndrome mice. Neuron 76, 325–337. doi: 10.1016/j.neuron.2012.07.022
Bhattacharya, A., Mamcarz, M., Mullins, C., Choudhury, A., Boyle, R. G., Smith, D. G., et al. (2016). Targeting translation control with p70 S6 kinase 1 inhibitors to reverse phenotypes in fragile X syndrome mice. Neuropsychopharmacology 41, 1991–2000. doi: 10.1038/npp.2015.369
Bianchi, R., Chuang, S. C., Zhao, W., Young, S. R., and Wong, R. K. (2009). Cellular plasticity for group I mGluR-mediated epileptogenesis. J. Neurosci. 29, 3497–3507. doi: 10.1523/JNEUROSCI.5447-08.2009
Bicks, L. K., Koike, H., Akbarian, S., and Morishita, H. (2015). Prefrontal cortex and social cognition in mouse and man. Front. Psychol. 6:1805. doi: 10.3389/fpsyg.2015.01805
Bilgin Sonay, T., Carvalho, T., Robinson, M. D., Greminger, M. P., Krützen, M., Comas, D., et al. (2015). Tandem repeat variation in human and great ape populations and its impact on gene expression divergence. Genome Res. 25, 1591–1599. doi: 10.1101/gr.190868.115
Birnie, A. K., Taylor, J. H., Cavanaugh, J., and French, J. A. (2013). Quality of maternal and paternal care predicts later stress reactivity in the cooperatively-breeding marmoset (Callithrix geoffroyi). Psychoneuroendocrinology 38, 3003–3014. doi: 10.1016/j.psyneuen.2013.08.011
Bliss, T. V., Collingridge, G. L., and Morris, R. G. (2003). Introduction. Long-term potentiation and structure of the issue. Philos. Trans. R. Soc. Lond. B Biol. Sci. 358, 607–611. doi: 10.1098/rstb.2003.1282
Boda, B., Mendez, P., Boury-Jamot, B., Magara, F., and Muller, D. (2014). Reversal of activity-mediated spine dynamics and learning impairment in a mouse model of Fragile X syndrome. Eur. J. Neurosci. 39, 1130–1137. doi: 10.1111/ejn.12488
Bonaccorso, C. M., Spatuzza, M., Di Marco, B., Gloria, A., Barrancotto, G., Cupo, A., et al. (2015). Fragile X mental retardation protein (FMRP) interacting proteins exhibit different expression patterns during development. Int. J. Dev. Neurosci. 42, 15–23. doi: 10.1016/j.ijdevneu.2015.02.004
Bontekoe, C. J., Bakker, C. E., Nieuwenhuizen, I. M., van der Linde, H., Lans, H., de Lange, D., et al. (2001). Instability of a (CGG)98 repeat in the Fmr1 promoter. Hum. Mol. Genet. 10, 1693–1699. doi: 10.1093/hmg/10.16.1693
Bontekoe, C. J., de Graaff, E., Nieuwenhuizen, I. M., Willemsen, R., and Oostra, B. A. (1997). FMR1 premutation allele (CGG)81 is stable in mice. Eur. J. Hum. Genet. 5, 293–298.
Bostrom, C. A., Majaess, N. M., Morch, K., White, E., Eadie, B. D., and Christie, B. R. (2015). Rescue of NMDAR-dependent synaptic plasticity in Fmr1 knock-out mice. Cereb. Cortex 25, 271–279. doi: 10.1093/cercor/bht237
Botta-Orfila, T., Tartaglia, G. G., and Michalon, A. (2016). Molecular pathophysiology of fragile X-associated tremor/ataxia syndrome and perspectives for drug development. Cerebellum 15, 599–610. doi: 10.1007/s12311-016-0800-2
Braat, S., D’Hulst, C., Heulens, I., De Rubeis, S., Mientjes, E., Nelson, D. L., et al. (2015). The GABAA receptor is an FMRP target with therapeutic potential in fragile X syndrome. Cell Cycle 14, 2985–2995. doi: 10.4161/15384101.2014.989114
Brager, D. H., Akhavan, A. R., and Johnston, D. (2012). Impaired dendritic expression and plasticity of h-channels in the fmr1−/y mouse model of fragile X syndrome. Cell Rep. 1, 225–233. doi: 10.1016/j.celrep.2012.02.002
Bramlett-Parker, J., and Washburn, D. A. (2016). Can rhesus monkey learn executive attention? Behav. Sci. 6:E26. doi: 10.3390/bs6020011
Brasa, S., Mueller, A., Jacquemont, S., Hahne, F., Rozenberg, I., Peters, T., et al. (2016). Reciprocal changes in DNA methylation and hydroxymethylation and a broad repressive epigenetic switch characterize FMR1 transcriptional silencing in fragile X syndrome. Clin. Epigenetics 8:15. doi: 10.1186/s13148-016-0181-x
Brosnan, S. F., Wilson, B. J., and Beran, M. J. (2012). Old World monkeys are more similar to humans than New World monkeys when playing a coordination game. Proc. Biol. Sci. 279, 1522–1530. doi: 10.1098/rspb.2011.1781
Brouwer, J. R., Mientjes, E. J., Bakker, C. E., Nieuwenhuizen, I. M., Severijnen, L. A., Van der Linde, H. C., et al. (2007). Elevated Fmr1 mRNA levels and reduced protein expression in a mouse model with an unmethylated Fragile X full mutation. Exp. Cell Res. 313, 244–253. doi: 10.1016/j.yexcr.2006.10.002
Brown, V., Jin, P., Ceman, S., Darnell, J. C., O’Donnell, W. T., Tenenbaum, S. A., et al. (2001). Microarray identification of FMRP-associated brain mRNAs and altered mRNA translational profiles in fragile X syndrome. Cell 107, 477–487. doi: 10.1016/s0092-8674(01)00568-2
Brown, M. R., Kronengold, J., Gazula, V. R., Chen, Y., Strumbos, J. G., Sigworth, F. J., et al. (2010). Fragile X mental retardation protein controls gating of the sodium-activated potassium channel Slack. Nat. Neurosci. 13, 819–821. doi: 10.1038/nn.2563
Buckley, L. A., Chapman, K., Burns-Naas, L. A., Todd, M. D., Martin, P. L., and Lansita, J. A. (2011). Considerations regarding nonhuman primate use in safety assessment of biopharmaceuticals. Int. J. Toxicol. 30, 583–590. doi: 10.1177/1091581811415875
Buckner, R. L., and Krienen, F. M. (2013). The evolution of distributed association networks in the human brain. Trends Cogn. Sci. 17, 648–665. doi: 10.1016/j.tics.2013.09.017
Bugnyar, T., and Huber, L. (1997). Push or pull: an experimental study on imitation in marmosets. Anim. Behav. 54, 817–831. doi: 10.1006/anbe.1996.0497
Burket, J. A., Benson, A. D., Tang, A. H., and Deutsch, S. I. (2014). Rapamycin improves sociability in the BTBR T+Itpr3tf/J mouse model of autism spectrum disorders. Brain Res. Bull. 100, 70–75. doi: 10.1016/j.brainresbull.2013.11.005
Burkart, J. M., Fehr, E., Efferson, C., and van Schaik, C. P. (2007). Other-regarding preferences in a non-human primate: common marmosets provision food altruistically. Proc. Natl. Acad. Sci. U S A 104, 19762–19766. doi: 10.1073/pnas.0710310104
Burkart, J., and Heschl, A. (2006). Geometrical gaze following in common marmosets (Callithrix jacchus). J. Comp. Psychol. 120, 120–130. doi: 10.1037/0735-7036.120.2.120
Burkart, J. M., and van Schaik, C. (2013). Group service in macaques (Macaca fuscata), capuchins (Cebus apella) and marmosets (Callithrix jacchus): a comparative approach to identifying proactive prosocial motivations. J. Comp. Psychol. 127, 212–225. doi: 10.1037/a0026392
Burman, K. J., Palmer, S. M., Gamberini, M., and Rosa, M. G. (2006). Cytoarchitectonic subdivisions of the dorsolateral frontal cortex of the marmoset monkey (Callithrix jacchus) and their projections to dorsal visual areas. J. Comp. Neurol. 495, 149–172. doi: 10.1002/cne.20837
Burman, K. J., and Rosa, M. G. (2009). Architectural subdivisions of medial and orbital frontal cortices in the marmoset monkey (Callithrix jacchus). J. Comp. Neurol. 514, 11–29. doi: 10.1002/cne.21976
Buschiazzo, E., and Gemmell, N. J. (2010). Conservation of human microsatellites across 450 million years of evolution. Genome Biol. Evol. 2, 153–165. doi: 10.1093/gbe/evq007
Caku, A., Pellerin, D., Bouvier, P., Riou, E., and Corbin, F. (2014). Effect of lovastatin on behavior in children and adults with fragile X syndrome: an open-label study. Am. J. Med. Genet. A 164A, 2834–2842. doi: 10.1002/ajmg.a.36750
Caldwell, C. A., and Whiten, A. (2004). Testing for social learning and imitation in common marmosets, Callithrix jacchus, using an artificial fruit. Anim. Cogn. 7, 77–85. doi: 10.1007/s10071-003-0192-9
Castrén, M., Pääkkönen, A., Tarkka, I. M., Ryynänen, M., and Partanen, J. (2003). Augmentation of auditory N1 in children with fragile X syndrome. Brain Topogr. 15, 165–171. doi: 10.1023/A:1022606200636
Ceman, S., O’Donnell, W. T., Reed, M., Patton, S., Pohl, J., and Warren, S. T. (2003). Phosphorylation influences the translation state of FMRP-associated polyribosomes. Hum. Mol. Genet. 12, 3295–3305. doi: 10.1093/hmg/ddg350
Centonze, D., Rossi, S., Mercaldo, V., Napoli, I., Ciotti, M. T., De Chiara, V., et al. (2008). Abnormal striatal GABA transmission in the mouse model for the fragile X syndrome. Biol. Psychiatry 63, 963–973. doi: 10.1016/j.biopsych.2007.09.008
Chandler, S. P., Kansagra, P., and Hirst, M. C. (2003). Fragile X (CGG)n repeats induce a transcriptional repression in cis upon a linked promoter: evidence for a chromatin mediated effect. BMC Mol. Biol. 4:3. doi: 10.1186/1471-2199-4-3
Changizi, M. A., and Shimojo, S. (2005). Parcellation and area-area connectivity as a function of neocortex size. Brain Behav. Evol. 66, 88–98. doi: 10.1159/000085942
Chaplin, T. A., Yu, H. H., Soares, J. G., Gattass, R., and Rosa, M. G. (2013). A conserved pattern of differential expansion of cortical areas in simian primates. J. Neurosci. 33, 15120–15125. doi: 10.1523/JNEUROSCI.2909-13.2013
Chaste, P., Betancur, C., Gerard-Blanluet, M., Bargiacchi, A., Kuzbari, S., Drunat, S., et al. (2012). High-functioning autism spectrum disorder and fragile X syndrome: report of two affected sisters. Mol. Autism 3:5. doi: 10.1186/2040-2392-3-5
Chatterjee, N., Lin, Y., Santillan, B. A., Yotnda, P., and Wilson, J. H. (2015). Environmental stress induces trinucleotide repeat mutagenesis in human cells. Proc. Natl. Acad. Sci. U S A 112, 3764–3769. doi: 10.1073/pnas.1421917112
Chauhan, A., and Chauhan, V. (2006). Oxidative stress in autism. Pathophysiology 13, 171–181. doi: 10.1016/j.pathophys.2006.05.007
Chen, H. C., Kaplan, G., and Rogers, L. J. (2009). Contact calls of common marmosets (Callithrix jacchus): influence of age of caller on antiphonal calling and other vocal responses. Am. J. Primatol. 71, 165–170. doi: 10.1002/ajp.20636
Chen, T., Lu, J. S., Song, Q., Liu, M. G., Koga, K., Descalzi, G., et al. (2014). Pharmacological rescue of cortical synaptic and network potentiation in a mouse model for fragile X syndrome. Neuropsychopharmacology 39, 1955–1967. doi: 10.1038/npp.2014.44
Chen, X., Sun, W., Pan, Y., Yang, Q., Cao, K., Zhang, J., et al. (2013). Lithium ameliorates open-field and elevated plus maze behaviors and brain phospho-glycogen synthase kinase 3-β expression in fragile X syndrome model mice. Neurosciences 18, 356–362.
Chen, Y., Tassone, F., Berman, R. F., Hagerman, P. J., Hagerman, R. J., Willemsen, R., et al. (2010). Murine hippocampal neurons expressing Fmr1 gene premutations show early developmental deficits and late degeneration. Hum. Mol. Genet. 19, 196–208. doi: 10.1093/hmg/ddp479
Chen, Y., Zheng, Y., Kang, Y., Yang, W., Niu, Y., Guo, X., et al. (2015). Functional disruption of the dystrophin gene in rhesus monkey using CRISPR/Cas9. Hum. Mol. Genet. 24, 3764–3774. doi: 10.1093/hmg/ddv120
Cheng, C., Lau, S. K. M., and Doering, L. C. (2016). Astrocyte-secreted thrombospondin-1 modulates synapse and spine defects in the fragile X mouse model. Mol. Brain 9:74. doi: 10.1186/s13041-016-0256-9
Cheng, Y., Ma, Z., Kim, B.-H., Wu, W., Cayting, P., Boyle, A. P., et al. (2014). Principles of regulatory information conservation between mouse and human. Nature 515, 371–375. doi: 10.1038/nature13985
Chiurazzi, P., Pomponi, M. G., Willemsen, R., Oostra, B. A., and Neri, G. (1998). in vitro reactivation of the FMR1 gene involved in fragile X syndrome. Hum. Mol. Genet. 7, 109–113. doi: 10.1093/hmg/7.1.109
Choi, C. H., Schoenfeld, B. P., Bell, A. J., Hinchey, P., Kollaros, M., Gertner, M. J., et al. (2011). Pharmacological reversal of synaptic plasticity deficits in the mouse model of fragile X syndrome by group II mGluR antagonist or lithium treatment. Brain Res. 1380, 106–119. doi: 10.1016/j.brainres.2010.11.032
Choi, C. H., Schoenfeld, B. P., Bell, A. J., Hinchey, J., Rosenfelt, C., Gertner, M. J., et al. (2016). Multiple drug treatments that increase cAMP signaling restore long-term memory and aberrant signaling in fragile X syndrome models. Front. Behav. Neurosci. 10:136. doi: 10.3389/fnbeh.2016.00136
Choi, C. H., Schoenfeld, B. P., Weisz, E. D., Bell, A. J., Chambers, D. B., Hinchey, J., et al. (2015). PDE-4 inhibition rescues aberrant synaptic plasticity in Drosophila and mouse models of fragile X syndrome. J. Neurosci. 35, 396–408. doi: 10.1523/JNEUROSCI.1356-12.2015
Choi, J. Y., Takahashi, D. Y., and Ghazanfar, A. A. (2015). Cooperative vocal control in marmoset monkeys via vocal feedback. J. Neurophysiol. 114, 274–283. doi: 10.1152/jn.00228.2015
Chow, C. P., Mitchell, J. F., and Miller, C. T. (2015). Vocal turn-taking in a non-human primate is learned during ontogeny. Proc. Biol. Sci. 282:20150069. doi: 10.1098/rspb.2015.0069
Chuang, S. C., Zhao, W., Bauchwitz, R., Yan, Q., Bianchi, R., and Wong, R. K. (2005). Prolonged epileptiform discharges induced by altered group I metabotropic glutamate receptor-mediated synaptic responses in hippocampal slices of a fragile X mouse model. J. Neurosci. 25, 8048–8055. doi: 10.1523/JNEUROSCI.1777-05.2005
Cilli, P., Ventura, I., Minoprio, A., Meccia, E., Martire, A., Wilson, S. H., et al. (2016). Oxidized dNTPs and the OGG1 and MUTYH DNA glycosylases combine to induce CAG/CTG repeat instability. Nucleic Acids Res. 44, 5190–5203. doi: 10.1093/nar/gkw170
Clark, R. E., and Squire, L. R. (2013). Similarity in form and function of the hippocampus in rodents, monkeys, and humans. Proc. Natl. Acad. Sci. U S A 110, 10365–10370. doi: 10.1073/pnas.1301225110
Coffee, B., Zhang, F., Ceman, S., Warren, S. T., and Reines, D. (2002). Histone modifications depict an aberrantly heterochromatinized FMR1 gene in fragile x syndrome. Am. J. Hum. Genet. 71, 923–932. doi: 10.1086/342931
Coffee, B., Zhang, F., Warren, S. T., and Reines, D. (1999). Acetylated histones are associated with FMR1 in normal but not fragile X-syndrome cells. Nat. Genet. 22, 98–101. doi: 10.1038/8807
Cohen, I. L., Fisch, G. S., Sudhalter, V., Wolf-Schein, E. G., Hanson, D., Hagerman, R., et al. (1988). Social gaze, social avoidance, and repetitive behavior in fragile X males: a controlled study. Am. J. Ment. Retard. 92, 436–446.
Colak, D., Zaninovic, N., Cohen, M. S., Rosenwaks, Z., Yang, W. Y., Gerhardt, J., et al. (2014). Promoter-bound trinucleotide repeat mRNA drives epigenetic silencing in fragile X syndrome. Science 343, 1002–1005. doi: 10.1126/science.1245831
Colombo, J. A. (1996). Interlaminar astroglial processes in the cerebral cortex of adult monkeys but not of adult rats. Acta Anat. 155, 57–62. doi: 10.1159/000147790
Colombo, J. A., Fuchs, E., Härtig, W., Marotte, L. R., and Puissant, V. (2000). “Rodent-like” and “primate-like” types of astroglial architecture in the adult cerebral cortex of mammals: a comparative study. Anat. Embryol. 201, 111–120. doi: 10.1007/pl00008231
Colombo, J. A., Gayol, S., Yañez, A., and Marco, P. (1997). Immunocytochemical and electron microscope observations on astroglial interlaminar processes in the primate neocortex. J. Neurosci. Res. 48, 352–357. doi: 10.1002/(sici)1097-4547(19970515)48:4<352::aid-jnr7>3.3.co;2-d
Coors, M. E., Glover, J. J., Juengst, E. T., and Sikela, J. M. (2010). The ethics of using transgenic non-human primates to study what makes us human. Nat. Rev. Genet. 11, 658–662. doi: 10.1038/nrg2864
Cordeiro, L., Ballinger, E., Hagerman, R., and Hessl, D. (2011). Clinical assessment of DSM-IV anxiety disorders in fragile X syndrome: prevalence and characterization. J. Neurodev. Disord. 3, 57–67. doi: 10.1007/s11689-010-9067-y
Cornish, K., Munir, F., and Wilding, J. (2001). A neuropsychological and behavioural profile of attention deficits in fragile X syndrome. Rev. Neurol. 33, S24–S29.
Costa, L., Sardone, L. M., Lacivita, E., Leopoldo, M., and Ciranna, L. (2015). Novel agonists for serotonin 5-HT7 receptors reverse metabotropic glutamate receptor-mediated long-term depression in the hippocampus of wild-type and Fmr1 KO mice, a model of fragile X syndrome. Front. Behav. Neurosci. 9:65. doi: 10.3389/fnbeh.2015.00065
Costa, L., Spatuzza, M., D’Antoni, S., Bonaccorso, C. M., Trovato, C., Musumeci, S. A., et al. (2012). Activation of 5-HT7 serotonin receptors reverses metabotropic glutamate receptor-mediated synaptic plasticity in wild-type and Fmr1 knockout mice, a model of Fragile X syndrome. Biol. Psychiatry 72, 924–933. doi: 10.1016/j.biopsych.2012.06.008
Cowley, B., Kirjanen, S., Partanen, J., and Castrén, M. L. (2016). Epileptic electroencephalography profile associates with attention problems in children with fragile X syndrome: review and case series. Front. Hum. Neurosci. 10:353. doi: 10.3389/fnhum.2016.00353
Cuadrado, M., Sacristan, M., and Antequera, F. (2001). Species-specific organization of CpG island promoters at mammalian homologous genes. EMBO Rep. 2, 586–592. doi: 10.1093/embo-reports/kve131
Cunningham, C. L., Martínez Cerdeño, V., Navarro Porras, E., Prakash, A. N., Angelastro, J. M., Willemsen, R., et al. (2011). Premutation CGG-repeat expansion of the Fmr1 gene impairs mouse neocortical development. Hum. Mol. Genet. 20, 64–79. doi: 10.1093/hmg/ddq432
Curia, G., Papouin, T., Seguela, P., and Avoli, M. (2009). Downregulation of tonic GABAergic inhibition in a mouse model of fragile X syndrome. Cereb. Cortex 19, 1515–1520. doi: 10.1093/cercor/bhn159
Cyranoski, D. (2004). China takes steps to secure pole position in primate research. Nature 432:3. doi: 10.1038/432003a
Dahlhaus, R., and El-Husseini, A. (2010). Altered neuroligin expression is involved in social deficits in a mouse model of the fragile X syndrome. Behav. Brain Res. 208, 96–105. doi: 10.1016/j.bbr.2009.11.019
Dalton, K. M., Holsen, L., Abbeduto, L., and Davidson, R. J. (2008). Brain function and gaze fixation during facial-emotion processing in fragile X and autism. Autism Res. 1, 231–239. doi: 10.1002/aur.32
Darnell, J. C., Van Driesche, S. J., Zhang, C., Hung, K. Y., Mele, A., Fraser, C. E., et al. (2011). FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 146, 247–261. doi: 10.1016/j.cell.2011.06.013
Datta, S., Alam, M. P., Majumdar, S. S., Mehta, A. K., Maiti, S., Wadhwa, N., et al. (2011). Nucleosomal occupancy and CGG repeat expansion: a comparative analysis of triplet repeat region from mouse and human fragile X mental retardation gene 1. Chromosome Res. 19, 445–455. doi: 10.1007/s10577-011-9206-7
Davidovic, L., Navratil, V., Bonaccorso, C. M., Catania, M. V., Bardoni, B., and Dumas, M. E. (2011). A metabolomic and systems biology perspective on the brain of the fragile X syndrome mouse model. Genome Res. 21, 2190–2202. doi: 10.1101/gr.116764.110
de Boer, R. A., Overduin-de Vries, A. M., Louwerse, A. L., and Sterck, E. H. (2013). The behavioral context of visual displays in common marmosets (Callithrix jacchus). Am. J. Primatol. 75, 1084–1095. doi: 10.1002/ajp.22167
Decasien, A. R., Williams, S. A., and Higham, J. P. (2017). Primate brain size is predicted by diet but not sociality. Nat. Ecol. Evol. 1:112. doi: 10.1038/s41559-017-0112
de Esch, C. E., Ghazvini, M., Loos, F., Schelling-Kazaryan, N., Widagdo, W., Munshi, S. T., et al. (2014). Epigenetic characterization of the FMR1 promoter in induced pluripotent stem cells from human fibroblasts carrying an unmethylated full mutation. Stem Cell Rep. 3, 548–555. doi: 10.1016/j.stemcr.2014.07.013
de Esch, C. E., van Den Berg, W. E., Buijsen, R. A., Jaafar, I. A., Nieuwenhuizen-Bakker, I. M., Gasparini, F., et al. (2015). Fragile X mice have robust mGluR5-dependent alterations of social behaviour in the Automated Tube Test. Neurobiol. Dis. 75, 31–39. doi: 10.1016/j.nbd.2014.12.021
de la Mothe, L. A., Blumell, S., Kajikawa, Y., and Hackett, T. A. (2012). Cortical connections of auditory cortex in marmoset monkeys: lateral belt and parabelt regions. Anat. Rec. Hoboken 295, 800–821. doi: 10.1002/ar.22451
De Moraes, B. L., Da Silva Souto, A., and Schiel, N. (2014). Adaptability in stone tool use by wild capuchin monkeys (Sapajus libidinosus). Am. J. Primatol. 76, 967–977. doi: 10.1002/ajp.22286
De Rubeis, S., and Bagni, C. (2010). Fragile X mental retardation protein control of neuronal mRNA metabolism: insights into mRNA stability. Mol. Cell. Neurosci. 43, 43–50. doi: 10.1016/j.mcn.2009.09.013
de Vries, B. B., Wiegers, A. M., Smits, A. P., Mohkamsing, S., Duivenvoorden, H. J., Fryns, J. P., et al. (1996). Mental status of females with an FMR1 gene full mutation. Am. J. Hum. Genet. 58, 1025–1032.
de Vrij, F. M., Levenga, J., van der Linde, H. C., Koekkoek, S. K., De Zeeuw, C. I., Nelson, D. L., et al. (2008). Rescue of behavioral phenotype and neuronal protrusion morphology in Fmr1 KO mice. Neurobiol. Dis. 31, 127–132. doi: 10.1016/j.nbd.2008.04.002
Dell’Mour, V., Range, F., and Huber, L. (2009). Social learning and mother’s behavior in manipulative tasks in infant marmosets. Am. J. Primatol. 71, 503–509. doi: 10.1002/ajp.20682
Demetrius, L. (2006). Aging in mouse and human systems: a comparative study. Ann. N Y Acad. Sci. 1067, 66–82. doi: 10.1196/annals.1354.010
Deng, P. Y., and Klyachko, V. A. (2016). Genetic upregulation of BK channel activity normalizes multiple synaptic and circuit defects in a mouse model of fragile X syndrome. J. Physiol. 594, 83–97. doi: 10.1113/JP271031
Deng, P. Y., Rotman, Z., Blundon, J. A., Cho, Y., Cui, J., Cavalli, V., et al. (2013). FMRP regulates neurotransmitter release and synaptic information transmission by modulating action potential duration via BK channels. Neuron 77, 696–711. doi: 10.1016/j.neuron.2013.03.027
Dennery, P. A. (2007). Effects of oxidative stress on embryonic development. Birth Defects Res. C Embryo Today 81, 155–162. doi: 10.1002/bdrc.20098
Derlig, K., Gießl, A., Brandstätter, J. H., Enz, R., and Dahlhaus, R. (2013). Identification and characterisation of simiate, a novel protein linked to the fragile X syndrome. PLoS One 8:e83007. doi: 10.1371/journal.pone.0083007
Derreumaux, S., Chaoui, M., Tevanian, G., and Fermandjian, S. (2001). Impact of CpG methylation on structure, dynamics and solvation of cAMP DNA responsive element. Nucleic Acids Res. 29, 2314–2326. doi: 10.1093/nar/29.11.2314
Desai, N. S., Casimiro, T. M., Gruber, S. M., and Vanderklish, P. W. (2006). Early postnatal plasticity in neocortex of Fmr1 knockout mice. J. Neurophysiol. 96, 1734–1745. doi: 10.1152/jn.00221.2006
Devys, D., Biancalana, V., Rousseau, F., Boué, J., Mandel, J. L., and Oberlé, I. (1992). Analysis of full fragile X mutations in fetal tissues and monozygotic twins indicate that abnormal methylation and somatic heterogeneity are established early in development. Am. J. Med. Genet. 43, 208–216. doi: 10.1002/ajmg.1320430134
Devys, D., Lutz, Y., Rouyer, N., Bellocq, J. P., and Mandel, J. L. (1993). The FMR-1 protein is cytoplasmic, most abundant in neurons and appears normal in carriers of a fragile X premutation. Nat. Genet. 4, 335–340. doi: 10.1038/ng0893-335
D’Hooge, R., Nagels, G., Franck, F., Bakker, C. E., Reyniers, E., Storm, K., et al. (1997). Mildly impaired water maze performance in male Fmr1 knockout mice. Neuroscience 76, 367–376. doi: 10.1016/s0306-4522(96)00224-2
D’Hulst, C., De Geest, N., Reeve, S. P., Van Dam, D., De Deyn, P. P., Hassan, B. A., et al. (2006). Decreased expression of the GABAA receptor in fragile X syndrome. Brain Res. 22, 238–245. doi: 10.1016/j.brainres.2006.08.115
Dicke, U., and Roth, G. (2016). Neuronal factors determining high intelligence. Philos. Trans. R. Soc. Lond. B Biol. Sci. 371:20150180. doi: 10.1098/rstb.2015.0180
Dictenberg, J. B., Swanger, S. A., Antar, L. N., Singer, R. H., and Bassell, G. J. (2008). A direct role for FMRP in activity-dependent dendritic mRNA transport links filopodial-spine morphogenesis to fragile X syndrome. Dev. Cell 14, 926–939. doi: 10.1016/j.devcel.2008.04.003
Didiot, M. C., Tian, Z. X., Schaeffer, C., Subramanian, M., Mandel, J. L., and Moine, H. (2008). The G-quartet containing FMRP binding site in FMR1 mRNA is a potent exonic splicing enhancer. Nucleic Acids Res. 36, 4902–4912. doi: 10.1093/nar/gkn472
Diester, I., and Nieder, A. (2010). Numerical values leave a semantic imprint on associated signs in monkeys. J. Cogn. Neurosci. 22, 174–183. doi: 10.1162/jocn.2009.21193
Digby, L. J., and Barreto, C. E. (1993). Social organization in a wild population of Callithrix jacchus. I. Group composition and dynamics. Folia Primatol. 61, 123–134. doi: 10.1159/000156739
Doers, M. E., Musser, M. T., Nichol, R., Berndt, E. R., Baker, M., Gomez, T. M., et al. (2014). iPSC-derived forebrain neurons from FXS individuals show defects in initial neurite outgrowth. Stem Cells Dev. 23, 1777–1787. doi: 10.1089/scd.2014.0030
Dolan, B. M., Duron, S. G., Campbell, D. A., Vollrath, B., Shankaranarayana Rao, B. S., Ko, H. Y., et al. (2013). Rescue of fragile X syndrome phenotypes in Fmr1 KO mice by the small-molecule PAK inhibitor FRAX486. Proc. Natl. Acad. Sci. U S A 110, 5671–5676. doi: 10.1073/pnas.1219383110
Dolen, G., Osterweil, E., Rao, B. S., Smith, G. B., Auerbach, B. D., Chattarji, S., et al. (2007). Correction of fragile X syndrome in mice. Neuron 56, 955–962. doi: 10.1016/j.neuron.2007.12.001
Dolenšek, J., Rupnik, M. S., and Stožer, A. (2015). Structural similarities and differences between the human and the mouse pancreas. Islets 7:e1024405. doi: 10.1080/19382014.2015.1024405
Dolskiy, A. A., Pustylnyak, V. O., Yarushkin, A. A., Lemskaya, N. A., and Yudkin, D. V. (2017). Inhibitors of histone deacetylases are weak activators of the FMR1 gene in fragile X syndrome cell lines. Biomed Res. Int. 2017:3582601. doi: 10.1155/2017/3582601
Dombrowski, C., Levesque, S., Morel, M. L., Rouillard, P., Morgan, K., and Rousseau, F. (2002). Premutation and intermediate-size FMR1 alleles in 10572 males from the general population: loss of an AGG interruption is a late event in the generation of fragile X syndrome alleles. Hum. Mol. Genet. 11, 371–378. doi: 10.1093/hmg/11.4.371
Donohoue, P. D., Barrangou, R., and May, A. P. (2017). Advances in industrial biotechnology using CRISPR-cas systems. Trends Biotechnol. 36, 134–146. doi: 10.1016/j.tibtech.2017.07.007
Du, X., Cui, J., Wang, C., Huo, X., Lu, J., Li, Y., et al. (2013). Detected microsatellite polymorphisms in genetically altered inbred mouse strains. Mol. Genet. Genomics 288, 309–316. doi: 10.1007/s00438-013-0751-y
Durdiaková, J., Lakatošová, S., Kubranská, A., Laznibatová, J., Ficek, A., Ostatniková, D., et al. (2013). Mental rotation in intellectually gifted boys is affected by the androgen receptor CAG repeat polymorphism. Neuropsychologia 51, 1693–1698. doi: 10.1016/j.neuropsychologia.2013.05.016
Dutta, S., and Sengupta, P. (2016). Men and mice: relating their ages. Life Sci. 152, 244–248. doi: 10.1016/j.lfs.2015.10.025
Dy, A. B. C., Tassone, F., Eldeeb, M., Salcedo-Arellano, M. J., Tartaglia, N., and Hagerman, R. (2018). Metformin as targeted treatment in fragile X syndrome. Clin. Genet. 93, 216–222. doi: 10.1007/978-3-642-21649-7_17
Dyer-Friedman, J., Glaser, B., Hessl, D., Johnston, C., Huffman, L. C., Taylor, A., et al. (2002). Genetic and environmental influences on the cognitive outcomes of children with fragile X syndrome. J. Am. Acad. Child Adolesc. Psychiatry 41, 237–244. doi: 10.1097/00004583-200203000-00002
Dykens, E. M., Hodapp, R. M., and Leckman, J. F. (1987). Strengths and weaknesses in the intellectual functioning of males with fragile X syndrome. Am. J. Ment. Defic. 92, 234–236.
Eberhart, D. E., Malter, H. E., Feng, Y., and Warren, S. T. (1996). The fragile X mental retardation protein is a ribonucleoprotein containing both nuclear localization and nuclear export signals. Hum. Mol. Genet. 5, 1083–1091. doi: 10.1093/hmg/5.8.1083
Eichler, E. E., Holden, J. J., Popovich, B. W., Reiss, A. L., Snow, K., Thibodeau, S. N., et al. (1994). Length of uninterrupted CGG repeats determines instability in the FMR1 gene. Nat. Genet. 8, 88–94. doi: 10.1038/ng0994-88
Eiges, R., Urbach, A., Malcov, M., Frumkin, T., Schwartz, T., Amit, A., et al. (2007). Developmental study of fragile X syndrome using human embryonic stem cells derived from preimplantation genetically diagnosed embryos. Cell Stem Cell 1, 568–577. doi: 10.1016/j.stem.2007.09.001
Eilam, D. (2014). Of mice and men: building blocks in cognitive mapping. Neurosci. Biobehav. Rev. 47, 393–409. doi: 10.1016/j.neubiorev.2014.09.010
Elefteriou, F., and Yang, X. (2011). Genetic mouse models for bone studies—strengths and limitations. Bone 49, 1242–1254. doi: 10.1016/j.bone.2011.08.021
Elston, G. N., Benavides-Piccione, R., Elston, A., Zietsch, B., Defelipe, J., Manger, P., et al. (2006). Specializations of the granular prefrontal cortex of primates: implications for cognitive processing. Anat. Rec. A Discov. Mol. Cell. Evol. Biol. 288, 26–35. doi: 10.1002/ar.a.20278
Engineer, C. T., Centanni, T. M., Im, K. W., Rahebi, K. C., Buell, E. P., and Kilgard, M. P. (2014). Degraded speech sound processing in a rat model of fragile X syndrome. Brain Res. 20, 72–84. doi: 10.1016/j.brainres.2014.03.049
Entezam, A., Biacsi, R., Orrison, B., Saha, T., Hoffman, G. E., Grabczyk, E., et al. (2007). Regional FMRP deficits and large repeat expansions into the full mutation range in a new Fragile X premutation mouse model. Gene 395, 125–134. doi: 10.1016/j.gene.2007.02.026
Epple, G. (1968). Comparative studies on vocalization in marmoset monkeys (Hapalidae). Folia Primatol. 8, 1–40. doi: 10.1159/000155129
Epple, G. (1970). Quantitative studies on scent marking in the marmoset (Callithrix jacchus). Folia Primatol. 13, 48–62. doi: 10.1159/000155308
Estes, P. S., O’Shea, M., Clasen, S., and Zarnescu, D. C. (2008). Fragile X protein controls the efficacy of mRNA transport in Drosophila neurons. Mol. Cell. Neurosci. 39, 170–179. doi: 10.1016/j.mcn.2008.06.012
European Commission. (2010). Seventh Report on the Statistics on the Number of Animals Used for Experimental and Other Scientific Purposes in the Member States of the European Union SEC(2010) 1107/final 2. Brussels: European Commission. Available online at: http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=SWD:2013:0497:FIN:EN:PDF
Evans, T. A., and Beran, M. J. (2012). Monkeys exhibit prospective memory in a computerized task. Cognition 125, 131–140. doi: 10.1016/j.cognition.2012.07.012
Fan, W. (2013). Group I metabotropic glutamate receptors modulate late phase long-term potentiation in hippocampal CA1 pyramidal neurons: comparison of apical and basal dendrites. Neurosci. Lett. 553, 132–137. doi: 10.1016/j.neulet.2013.08.030
Feng, Y., Gutekunst, C. A., Eberhart, D. E., Yi, H., Warren, S. T., and Hersch, S. M. (1997). Fragile X mental retardation protein: nucleocytoplasmic shuttling and association with somatodendritic ribosomes. J. Neurosci. 17, 1539–1547.
Ferron, L., Nieto-Rostro, M., Cassidy, J. S., and Dolphin, A. C. (2014). Fragile X mental retardation protein controls synaptic vesicle exocytosis by modulating N-type calcium channel density. Nat. Commun. 5:3628. doi: 10.1038/ncomms4628
Finch, C. E., and Austad, S. N. (2015). Commentary: is Alzheimer’s disease uniquely human? Neurobiol. Aging 36, 553–555. doi: 10.1016/j.neurobiolaging.2014.10.025
Finkel, T., and Holbrook, N. J. (2000). Oxidants, oxidative stress and the biology of ageing. Nature 408, 239–247. doi: 10.1038/35041687
Fleming, K., Riser, D. K., Kumari, D., and Usdin, K. (2003). Instability of the fragile X syndrome repeat in mice: the effect of age, diet and mutations in genes that affect DNA replication, recombination and repair proficiency. Cytogenet Genome Res. 100, 140–146. doi: 10.1159/000072848
Francesconi, W., Cammalleri, M., and Sanna, P. (2004). The metabotropic glutamate receptor 5 is necessary for late-phase long-term potentiation in the hippocampal CA1 region. Brain Res. 1022, 12–18. doi: 10.1016/j.brainres.2004.06.060
Franklin, A. V., King, M. K., Palomo, V., Martinez, A., McMahon, L. L., and Jope, R. S. (2014). Glycogen synthase kinase-3 inhibitors reverse deficits in long-term potentiation and cognition in fragile X mice. Biol. Psychiatry 75, 198–206. doi: 10.1016/j.biopsych.2013.08.003
Freudenreich, C. H., Stavenhagen, J. B., and Zakian, V. A. (1997). Stability of a CTG/CAG trinucleotide repeat in yeast is dependent on its orientation in the genome. Mol. Cell. Biol. 17, 2090–2098. doi: 10.1128/mcb.17.4.2090
Freund, L. S., Reiss, A. L., and Abrams, M. T. (1993). Psychiatric disorders associated with fragile X in the young female. Pediatrics 91, 321–329.
Frey, U., Huang, Y. Y., and Kandel, E. R. (1993). Effects of cAMP simulate a late stage of LTP in hippocampal CA1 neurons. Science 260, 1661–1664. doi: 10.1126/science.8389057
Frey, S., Mackey, S., and Petrides, M. (2014). Cortico-cortical connections of areas 44 and 45B in the macaque monkey. Brain Lang. 131, 36–55. doi: 10.1016/j.bandl.2013.05.005
Fry, M., and Loeb, L. A. (1994). The fragile X syndrome d(CGG)n nucleotide repeats form a stable tetrahelical structure. Proc. Natl. Acad. Sci. U S A 91, 4950–4954. doi: 10.1073/pnas.91.11.4950
Frye, R. E., Casanova, M. F., Fatemi, S. H., Folsom, T. D., Reutiman, T. J., Brown, G. L., et al. (2016). Neuropathological mechanisms of seizures in autism spectrum disorder. Front. Neurosci. 10:192. doi: 10.3389/fnins.2016.00192
Fu, A. Q., Genereux, D. P., Stoger, R., Burden, A. F., Laird, C. D., and Stephens, M. (2012). Statistical inference of in vivo properties of human DNA methyltransferases from double-stranded methylation patterns. PLoS One 7:19. doi: 10.1371/journal.pone.0032225
Fu, Y. H., Kuhl, D. P., Pizzuti, A., Pieretti, M., Sutcliffe, J. S., Richards, S., et al. (1991). Variation of the CGG repeat at the fragile X site results in genetic instability: resolution of the Sherman paradox. Cell 67, 1047–1058. doi: 10.1016/0092-8674(91)90283-5
Fukuda, H., Katahira, M., Tanaka, E., Enokizono, Y., Tsuchiya, N., Higuchi, K., et al. (2005). Unfolding of higher DNA structures formed by the d(CGG) triplet repeat by UP1 protein. Genes Cells 10, 953–962. doi: 10.1111/j.1365-2443.2005.00896.x
Fukuda, K., Ichiyanagi, K., Yamada, Y., Go, Y., Udono, T., Wada, S., et al. (2013). Regional DNA methylation differences between humans and chimpanzees are associated with genetic changes, transcriptional divergence and disease genes. J. Hum. Genet. 58, 446–454. doi: 10.1038/jhg.2013.55
Gagne, M., Levesque, K., Nutile, L., and Locurto, C. (2012). Performance on patterned string problems by common marmosets (Callithrix jacchus). Anim. Cogn. 15, 1021–1030. doi: 10.1007/s10071-012-0511-0
Gama-Sosa, M. A., Midgett, R. M., Slagel, V. A., Githens, S., Kuo, K. C., Gehrke, C. W., et al. (1983). Tissue-specific differences in DNA methylation in various mammals. Biochim. Biophys. Acta 740, 212–219. doi: 10.1016/0167-4781(83)90079-9
Gandhi, R. M., Kogan, C. S., and Messier, C. (2014). 2-Methyl-6-(phenylethynyl) pyridine (MPEP) reverses maze learning and PSD-95 deficits in Fmr1 knock-out mice. Front. Cell. Neurosci. 8:70. doi: 10.3389/fncel.2014.00070
Gantois, I., Khoutorsky, A., Popic, J., Aguilar-Valles, A., Freemantle, E., Cao, R., et al. (2017). Metformin ameliorates core deficits in a mouse model of fragile X syndrome. Nat. Med. 23, 674–677. doi: 10.1038/nm.4335
Gantois, I., Pop, A. S., de Esch, C. E., Buijsen, R. A., Pooters, T., Gomez-Mancilla, B., et al. (2013). Chronic administration of AFQ056/Mavoglurant restores social behaviour in Fmr1 knockout mice. Behav. Brain Res. 239, 72–79. doi: 10.1016/j.bbr.2012.10.059
Gaudissard, J., Ginger, M., Premoli, M., Memo, M., Frick, A., and Pietropaolo, S. (2017). Behavioral abnormalities in the Fmr1-KO2 mouse model of fragile X syndrome: the relevance of early life phases. Autism Res. 10, 1584–1596. doi: 10.1002/aur.1814
Geahigan, K. B., Meints, G. A., Hatcher, M. E., Orban, J., and Drobny, G. P. (2000). The dynamic impact of CpG methylation in DNA. Biochemistry 39, 4939–4946. doi: 10.1021/bi9917636
Gentry, M., and Meyer, P. (2013). An 11bp region with stem formation potential is essential for de novo DNA methylation of the RPS element. PLoS One 8:e63652. doi: 10.1371/journal.pone.0063652
Gerhardt, J. (2017). Epigenetic modifications in human fragile X pluripotent stem cells; Implications in fragile X syndrome modeling. Brain Res. 1656, 55–62. doi: 10.1016/j.brainres.2015.10.004
Gerra, G., Garofano, L., Pellegrini, C., Bosari, S., Zaimovic, A., Moi, G., et al. (2005). Allelic association of a dopamine transporter gene polymorphism with antisocial behaviour in heroin-dependent patients. Addict. Biol. 10, 275–281. doi: 10.1080/13556210500223769
Gharib, S. A., Nguyen, E., Altemeier, W. A., Shaffer, S. A., Doneanu, C. E., Goodlett, D. R., et al. (2010). Of mice and men: comparative proteomics of bronchoalveolar fluid. Eur. Respir. J. 35, 1388–1395. doi: 10.1183/09031936.00089409
Ghazanfar, A. A., Morrill, R. J., and Kayser, C. (2013). Monkeys are perceptually tuned to facial expressions that exhibit a theta-like speech rhythm. Proc. Natl. Acad. Sci. U S A 110, 1959–1963. doi: 10.1073/pnas.1214956110
Ghazanfar, A. A., and Zhang, Y. S. (2016). The autonomic nervous system is the engine for vocal development through social feedback. Curr. Opin. Neurobiol. 40, 155–160. doi: 10.1016/j.conb.2016.07.016
Ghilan, M., Hryciw, B. N., Brocardo, P. S., Bostrom, C. A., Gil-Mohapel, J., and Christie, B. R. (2015). Enhanced corticosteroid signaling alters synaptic plasticity in the dentate gyrus in mice lacking the fragile X mental retardation protein. Neurobiol. Dis. 77, 26–34. doi: 10.1016/j.nbd.2015.01.008
Gholizadeh, S., Arsenault, J., Xuan, I. C., Pacey, L. K., and Hampson, D. R. (2014). Reduced phenotypic severity following adeno-associated virus-mediated Fmr1 gene delivery in fragile X mice. Neuropsychopharmacology 39, 3100–3111. doi: 10.1038/npp.2014.167
Gholizadeh, S., Halder, S. K., and Hampson, D. R. (2015). Expression of fragile X mental retardation protein in neurons and glia of the developing and adult mouse brain. Brain Res. 30, 22–30. doi: 10.1016/j.brainres.2014.11.023
Gibson, J. R., Bartley, A. F., Hays, S. A., and Huber, K. M. (2008). Imbalance of neocortical excitation and inhibition and altered UP states reflect network hyperexcitability in the mouse model of fragile X syndrome. J. Neurophysiol. 100, 2615–2626. doi: 10.1152/jn.90752.2008
Ginno, P. A., Lim, Y. W., Lott, P. L., Korf, I., and Chedin, F. (2013). GC skew at the 5′ and 3′ ends of human genes links R-loop formation to epigenetic regulation and transcription termination. Genome Res. 23, 1590–1600. doi: 10.1101/gr.158436.113
Ginno, P. A., Lott, P. L., Christensen, H. C., Korf, I., and Chedin, F. (2012). R-loop formation is a distinctive characteristic of unmethylated human CpG island promoters. Mol. Cell 45, 814–825. doi: 10.1016/j.molcel.2012.01.017
Gkogkas, C. G., Khoutorsky, A., Cao, R., Jafarnejad, S. M., Prager-Khoutorsky, M., Giannakas, N., et al. (2014). Pharmacogenetic inhibition of eIF4E-dependent Mmp9 mRNA translation reverses fragile X syndrome-like phenotypes. Cell Rep. 9, 1742–1755. doi: 10.1016/j.celrep.2014.10.064
Glaser, B., Hessl, D., Dyer-Friedman, J., Johnston, C., Wisbeck, J., Taylor, A., et al. (2003). Biological and environmental contributions to adaptive behavior in fragile X syndrome. Am. J. Med. Genet. A 15, 21–29. doi: 10.1002/ajmg.a.10549
Gläser, D., Wöhrle, D., Salat, U., Vogel, W., and Steinbach, P. (1999). Mitotic behavior of expanded CGG repeats studied on cultured cells: further evidence for methylation-mediated triplet repeat stability in fragile X syndrome. Am. J. Med. Genet. 84, 226–228. doi: 10.1002/(sici)1096-8628(19990528)84:3<226::aid-ajmg12>3.3.co;2-w
Godfraind, J. M., Reyniers, E., De Boulle, K., D’Hooge, R., De Deyn, P. P., Bakker, C. E., et al. (1996). Long-term potentiation in the hippocampus of fragile X knockout mice. Am. J. Med. Genet. 64, 246–251. doi: 10.1002/(sici)1096-8628(19960809)64:2<246::aid-ajmg2>3.0.co;2-s
Goebel-Goody, S. M., Wilson-Wallis, E. D., Royston, S., Tagliatela, S. M., Naegele, J. R., and Lombroso, P. J. (2012). Genetic manipulation of STEP reverses behavioral abnormalities in a fragile X syndrome mouse model. Genes Brain Behav. 11, 586–600. doi: 10.1111/j.1601-183x.2012.00781.x
Goldman-Rakic, P. S. (1988). Topography of cognition: parallel distributed networks in primate association cortex. Annu. Rev. Neurosci. 11, 137–156. doi: 10.1146/annurev.neuro.11.1.137
Goncalves, J. T., Anstey, J. E., Golshani, P., and Portera-Cailliau, C. (2013). Circuit level defects in the developing neocortex of Fragile X mice. Nat. Neurosci. 16, 903–909. doi: 10.1038/nn.3415
Goodman, J., Chandna, A., and Roe, K. (2015). Trends in animal use at US research facilities. J. Med. Ethics 41, 567–569. doi: 10.1136/medethics-2014-102404
Govaerts, L. C., Smit, A. E., Saris, J. J., Vanderwerf, F., Willemsen, R., Bakker, C. E., et al. (2007). Exceptional good cognitive and phenotypic profile in a male carrying a mosaic mutation in the FMR1 gene. Clin. Genet. 72, 138–144. doi: 10.1111/j.1399-0004.2007.00829.x
Graham, D. M. (2016). Exploring social cognition with marmosets. Lab Anim. (NY) 45:249. doi: 10.1038/laban.1049
Greenberg, J., Seltzer, M., Baker, J., Smith, L., Warren, S. F., Brady, N., et al. (2012). Family environment and behavior problems in children, adolescents, and adults with fragile X syndrome. Am. J. Intellect. Dev. Disabil. 117, 331–346. doi: 10.1352/1944-7558-117.4.331
Greenough, W. T., Klintsova, A. Y., Irwin, S. A., Galvez, R., Bates, K. E., and Weiler, I. J. (2001). Synaptic regulation of protein synthesis and the fragile X protein. Proc. Natl. Acad. Sci. U S A 98, 7101–7106. doi: 10.1073/pnas.141145998
Groh, M., Lufino, M. M., Wade-Martins, R., and Gromak, N. (2014). R-loops associated with triplet repeat expansions promote gene silencing in Friedreich ataxia and fragile X syndrome. PLoS Genet. 10:e1004318. doi: 10.1371/journal.pgen.1004318
Gross, C., Chang, C. W., Kelly, S. M., Bhattacharya, A., McBride, S. M., Danielson, S. W., et al. (2015a). Increased expression of the PI3K enhancer PIKE mediates deficits in synaptic plasticity and behavior in fragile X syndrome. Cell Rep. 11, 727–736. doi: 10.1016/j.celrep.2015.03.060
Gross, C., Hoffmann, A., Bassell, G. J., and Berry-Kravis, E. M. (2015b). Therapeutic strategies in fragile X syndrome: from bench to bedside and back. Neurotherapeutics 12, 584–608. doi: 10.1007/s13311-015-0355-9
Gross, C., Raj, N., Molinaro, G., Allen, A. G., Whyte, A. J., Gibson, J. R., et al. (2015c). Selective role of the catalytic PI3K subunit p110β in impaired higher order cognition in fragile X syndrome. Cell Rep. 11, 681–688. doi: 10.1016/j.celrep.2015.03.065
Gross, C., Nakamoto, M., Yao, X., Chan, C. B., Yim, S. Y., Ye, K., et al. (2010). Excess phosphoinositide 3-kinase subunit synthesis and activity as a novel therapeutic target in fragile X syndrome. J. Neurosci. 30, 10624–10638. doi: 10.1523/JNEUROSCI.0402-10.2010
Gross, C., Yao, X., Pong, D. L., Jeromin, A., and Bassell, G. J. (2011). Fragile X mental retardation protein regulates protein expression and mRNA translation of the potassium channel Kv4.2. J. Neurosci. 31, 5693–5698. doi: 10.1523/JNEUROSCI.6661-10.2011
Gunter, C., Paradee, W., Crawford, D. C., Meadows, K. A., Newman, J., Kunst, C. B., et al. (1998). Re-examination of factors associated with expansion of CGG repeats using a single nucleotide polymorphism in FMR1. Hum. Mol. Genet. 7, 1935–1946. doi: 10.1093/hmg/7.12.1935
Guo, W., Murthy, A. C., Zhang, L., Johnson, E. B., Schaller, E. G., Allan, A. M., et al. (2012). Inhibition of GSK3β improves hippocampus-dependent learning and rescues neurogenesis in a mouse model of fragile X syndrome. Hum. Mol. Genet. 21, 681–691. doi: 10.1093/hmg/ddr501
Hagerman, R. J., and Hagerman, P. (2016). Fragile X-associated tremor/ataxia syndrome—features, mechanisms and management. Nat. Rev. Neurol. 12, 403–412. doi: 10.1038/nrneurol.2016.82
Hagerman, R., Hoem, G., and Hagerman, P. (2010). Fragile X and autism: intertwined at the molecular level leading to targeted treatments. Mol. Autism 1:12. doi: 10.1186/2040-2392-1-12
Hagerman, R. J., Jackson, C., Amiri, K., Silverman, A. C., O’Connor, R., and Sobesky, W. (1992). Girls with fragile X syndrome: physical and neurocognitive status and outcome. Pediatrics 89, 395–400.
Halder, K., Wieland, M., and Hartig, J. S. (2009). Predictable suppression of gene expression by 5′-UTR-based RNA quadruplexes. Nucleic Acids Res. 37, 6811–6817. doi: 10.1093/nar/gkp696
Hall, S. S., Lightbody, A. A., Huffman, L. C., Lazzeroni, L. C., and Reiss, A. L. (2009). Physiological correlates of social avoidance behavior in children and adolescents with fragile X syndrome. J. Am. Acad. Child Adolesc. Psychiatry 48, 320–329. doi: 10.1097/CHI.0b013e318195bd15
Halsey, L. G., Bezerra, B. M., and Souto, A. S. (2006). Can wild common marmosets (Callithrix jacchus) solve the parallel strings task? Anim. Cogn. 9, 229–233. doi: 10.1007/s10071-006-0016-9
Hammock, E. A., and Young, L. J. (2005). Microsatellite instability generates diversity in brain and sociobehavioral traits. Science 308, 1630–1634. doi: 10.1126/science.1111427
Han, X., Chen, M., Wang, F., Windrem, M., Wang, S., Shanz, S., et al. (2013). Forebrain engraftment by human glial progenitor cells enhances synaptic plasticity and learning in adult mice. Cell Stem Cell 12, 342–353. doi: 10.1016/j.stem.2012.12.015
Handa, V., Goldwater, D., Stiles, D., Cam, M., Poy, G., Kumari, D., et al. (2005). Long CGG-repeat tracts are toxic to human cells: implications for carriers of Fragile X premutation alleles. FEBS Lett. 579, 2702–2708. doi: 10.1016/j.febslet.2005.04.004
Handa, V., Saha, T., and Usdin, K. (2003). The fragile X syndrome repeats form RNA hairpins that do not activate the interferon-inducible protein kinase, PKR, but are cut by Dicer. Nucleic Acids Res. 31, 6243–6248. doi: 10.1093/nar/gkg818
Handt, M., Epplen, A., Hoffjan, S., Mese, K., Epplen, J. T., and Dekomien, G. (2014). Point mutation frequency in the FMR1 gene as revealed by fragile X syndrome screening. Mol. Cell. Probes 28, 279–283. doi: 10.1016/j.mcp.2014.08.003
Hannan, A. J. (2012). Tandem repeat polymorphisms: mediators of genetic plasticity, modulators of biological diversity and dynamic sources of disease susceptibility. Adv. Exp. Med. Biol. 769, 1–9. doi: 10.1007/978-1-4614-5434-2_1
Hansen, B. C., Gografe, S., Pritt, S., Jen, K. C., McWhirter, C. A., Barman, S. M., et al. (2017). Ensuring due process in the IACUC and animal welfare setting: considerations in developing noncompliance policies and procedures for institutional animal care and use committees and institutional officials. FASEB J. 31, 4216–4225. doi: 10.1096/fj.201601250R
Hansen, R. S., Gartler, S. M., Scott, C. R., Chen, S. H., and Laird, C. D. (1992). Methylation analysis of CGG sites in the CpG island of the human FMR1 gene. Hum. Mol. Genet. 1, 571–578. doi: 10.1093/hmg/1.8.571
Hanson, D. M., Jackson, A. W. III., and Hagerman, R. J. (1986). Speech disturbances (cluttering) in mildly impaired males with the Martin-Bell/fragile X syndrome. Am. J. Med. Genet. 23, 195–206. doi: 10.1002/ajmg.1320230114
Harlow, E. G., Till, S. M., Russell, T. A., Wijetunge, L. S., Kind, P., and Contractor, A. (2010). Critical period plasticity is disrupted in the barrel cortex of FMR1 knockout mice. Neuron 65, 385–398. doi: 10.1016/j.neuron.2010.01.024
Harris, E. H., Washburn, D. A., Beran, M. J., and Sevcik, R. A. (2007). Rhesus monkeys (Macaca mulatta) select Arabic numerals or visual quantities corresponding to a number of sequentially completed maze trials. Learn. Behav. 35, 53–59. doi: 10.3758/bf03196074
Hartley, S. L., Seltzer, M. M., Raspa, M., Olmstead, M., Bishop, E., and Bailey, D. B. (2011). Exploring the adult life of men and women with fragile X syndrome: results from a national survey. Am. J. Intellect. Dev. Disabil. 116, 16–35. doi: 10.1352/1944-7558-116.1.16
Hashem, V., Galloway, J. N., Mori, M., Willemsen, R., Oostra, B. A., Paylor, R., et al. (2009). Ectopic expression of CGG containing mRNA is neurotoxic in mammals. Hum. Mol. Genet. 18, 2443–2451. doi: 10.1093/hmg/ddp182
Hayashi, M. L., Rao, B. S., Seo, J. S., Choi, H. S., Dolan, B. M., Choi, S. Y., et al. (2007). Inhibition of p21-activated kinase rescues symptoms of fragile X syndrome in mice. Proc. Natl. Acad. Sci. U S A 104, 11489–11494. doi: 10.1073/pnas.0705003104
Hébert, B., Pietropaolo, S., Même, S., Laudier, B., Laugeray, A., Doisne, N., et al. (2014). Rescue of fragile X syndrome phenotypes in Fmr1 KO mice by a BKCa channel opener molecule. Orphanet J. Rare Dis. 9:124. doi: 10.1186/s13023-014-0124-6
Heitz, D., Devys, D., Imbert, G., Kretz, C., and Mandel, J. L. (1992). Inheritance of the fragile X syndrome: size of the fragile X premutation is a major determinant of the transition to full mutation. J. Med. Genet. 29, 794–801. doi: 10.1136/jmg.29.11.794
Henderson, G. I., Chen, J. J., and Schenker, S. (1999). Ethanol, oxidative stress, reactive aldehydes, and the fetus. Front. Biosci. 15, D541–D550. doi: 10.2741/henderson
Henderson, C., Wijetunge, L., Kinoshita, M. N., Shumway, M., Hammond, R. S., Postma, F. R., et al. (2012). Reversal of disease-related pathologies in the fragile X mouse model by selective activation of GABAB receptors with arbaclofen. Sci. Transl. Med. 4:152ra128. doi: 10.1126/scitranslmed.3004218
Herculano-Houzel, S. (2009). The human brain in numbers: a linearly scaled-up primate brain. Front. Hum. Neurosci. 3:31. doi: 10.3389/neuro.09.031.2009
Hernández, J. A., López-Sánchez, R. C., and Rendón-Ramírez, A. (2016). Lipids and oxidative stress associated with ethanol-induced neurological damage. Oxid. Med. Cell. Longev. 2016:1543809. doi: 10.1155/2016/1543809
Hernando-Herraez, I., Heyn, H., Fernandez-Callejo, M., Vidal, E., Fernandez-Bellon, H., Prado-Martinez, J., et al. (2015). The interplay between DNA methylation and sequence divergence in recent human evolution. Nucleic Acids Res. 43, 8204–8214. doi: 10.1093/nar/gkv693
Hernando-Herraez, I., Prado-Martinez, J., Garg, P., Fernandez-Callejo, M., Heyn, H., Hvilsom, C., et al. (2013). Dynamics of DNA methylation in recent human and great ape evolution. PLoS Genet. 9:e1003763. doi: 10.1371/journal.pgen.1003763
Herzog, H. (2002). Ethical aspects of relationships between humans and research animals. ILAR J. 43, 27–32. doi: 10.1093/ilar.43.1.27
Heulens, I., D’Hulst, C., Van Dam, D., De Deyn, P. P., and Kooy, R. F. (2012). Pharmacological treatment of fragile X syndrome with GABAergic drugs in a knockout mouse model. Behav. Brain Res. 229, 244–249. doi: 10.1016/j.bbr.2012.01.031
Heyer, D. B., and Meredith, R. M. (2017). Environmental toxicology: sensitive periods of development and neurodevelopmental disorders. Neurotoxicology 58, 23–41. doi: 10.1016/j.neuro.2016.10.017
Higashimori, H., Schin, C. S., Chiang, M. S., Morel, L., Shoneye, T. A., Nelson, D. L., et al. (2016). Selective deletion of astroglial FMRP dysregulates glutamate transporter GLT1 and contributes to fragile X syndrome phenotypes in vivo. J. Neurosci. 36, 7079–7094. doi: 10.1523/JNEUROSCI.1069-16.2016
Hill, J., Inder, T., Neil, J., Dierker, D., Harwell, J., and Van Essen, D. (2010). Similar patterns of cortical expansion during human development and evolution. Proc. Natl. Acad. Sci. U S A 107, 13135–13140. doi: 10.1073/pnas.1001229107
Hinds, H. L., Ashley, C. T., Sutcliffe, J. S., Nelson, D. L., Warren, S. T., Housman, D. E., et al. (1993). Tissue specific expression of FMR-1 provides evidence for a functional role in fragile X syndrome. Nat. Genet. 3, 36–43. doi: 10.1038/ng0193-36
Hinton, V. J., Brown, W. T., Wisniewski, K., and Rudelli, R. D. (1991). Analysis of neocortex in three males with the fragile X syndrome. Am. J. Med. Genet. 41, 289–294. doi: 10.1002/ajmg.1320410306
Hirst, M. C., and White, P. J. (1998). Cloned human FMR1 trinucleotide repeats exhibit a length- and orientation-dependent instability suggestive of in vivo lagging strand secondary structure. Nucleic Acids Res. 26, 2353–2358. doi: 10.1093/nar/26.10.2353
Hoeffer, C. A., Sanchez, E., Hagerman, R. J., Mu, Y., Nguyen, D. V., Wong, H., et al. (2012). Altered mTOR signaling and enhanced CYFIP2 expression levels in subjects with fragile X syndrome. Genes Brain Behav. 11, 332–341. doi: 10.1111/j.1601-183x.2012.00768.x
Hoehndorf, R., Hiebert, T., Hardy, N. W., Schofield, P. N., Gkoutos, G. V., and Dumontier, M. (2014). Mouse model phenotypes provide information about human drug targets. Bioinformatics 30, 719–725. doi: 10.1093/bioinformatics/btt613
Holsen, L. M., Dalton, K. M., Johnstone, T., and Davidson, R. J. (2008). Prefrontal social cognition network dysfunction underlying face encoding and social anxiety in fragile X syndrome. Neuroimage 43, 592–604. doi: 10.1016/j.neuroimage.2008.08.009
Hopkins, W. D. (2013). Neuroanatomical asymmetries and handedness in chimpanzees (Pan troglodytes): a case for continuity in the evolution of hemispheric specialization. Ann. N Y Acad. Sci. 1288, 17–35. doi: 10.1111/nyas.12109
Horner, A. E., Heath, C. J., Hvoslef-Eide, M., Kent, B. A., Kim, C. H., Nilsson, S. R., et al. (2013). The touchscreen operant platform for testing learning and memory in rats and mice. Nat. Protoc. 8, 1961–1984. doi: 10.1038/nprot.2013.122
Hornstra, I. K., Nelson, D. L., Warren, S. T., and Yang, T. P. (1993). High resolution methylation analysis of the FMR1 gene trinucleotide repeat region in fragile X syndrome. Hum. Mol. Genet. 2, 1659–1665. doi: 10.1093/hmg/2.10.1659
Hou, L., Antion, M. D., Hu, D., Spencer, C. M., Paylor, R., and Klann, E. (2006). Dynamic translational and proteasomal regulation of fragile X mental retardation protein controls mGluR-dependent long-term depression. Neuron 51, 441–454. doi: 10.1016/j.neuron.2006.07.005
Hu, H., Qin, Y., Bochorishvili, G., Zhu, Y., van Aelst, L., and Zhu, J. J. (2008). Ras signaling mechanisms underlying impaired GluR1-dependent plasticity associated with fragile X syndrome. J. Neurosci. 28, 7847–7862. doi: 10.1523/JNEUROSCI.1496-08.2008
Huang, J., Wang, Y., and Zhao, J. (2017). CRISPR editing in biological and biomedical investigation. J. Cell. Physiol. doi: 10.1002/jcp.26141 [Epub ahead of print].
Huber, K. M., Gallagher, S. M., Warren, S. T., and Bear, M. F. (2002). Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc. Natl. Acad. Sci. U S A 99, 7746–7750. doi: 10.1073/pnas.122205699
Huddleston, L. B., Visootsak, J., and Sherman, S. L. (2014). Cognitive aspects of fragile X syndrome. Wiley Interdiscip. Rev. Cogn. Sci. 5, 501–508. doi: 10.1002/wcs.1296
Hukema, R. K., Buijsen, R. A., Raske, C., Severijnen, L. A., Nieuwenhuizen-Bakker, I., Minneboo, M., et al. (2014). Induced expression of expanded CGG RNA causes mitochondrial dysfunction in vivo. Cell Cycle 13, 2600–2608. doi: 10.4161/15384101.2014.943112
Hulbert, A. J. (2005). On the importance of fatty acid composition of membranes for aging. J. Theor. Biol. 234, 277–288. doi: 10.1016/j.jtbi.2004.11.024
Hunsaker, M. R., Goodrich-Hunsaker, N. J., Willemsen, R., and Berman, R. F. (2010). Temporal ordering deficits in female CGG KI mice heterozygous for the fragile X premutation. Behav. Brain Res. 213, 263–268. doi: 10.1016/j.bbr.2010.05.010
Hunsaker, M. R., von Leden, R. E., Ta, B. T., Goodrich-Hunsaker, N. J., Arque, G., Kim, K., et al. (2011). Motor deficits on a ladder rung task in male and female adolescent and adult CGG knock-in mice. Behav. Brain Res. 222, 117–121. doi: 10.1016/j.bbr.2011.03.039
Hunter, J., Rivero-Arias, O., Angelov, A., Kim, E., Fotheringham, I., and Leal, J. (2014). Epidemiology of fragile X syndrome: a systematic review and meta-analysis. Am. J. Med. Genet. A 164A, 1648–1658. doi: 10.1002/ajmg.a.36511
Hvoslef-Eide, M., Nilsson, S. R., Saksida, L. M., and Bussey, T. J. (2016). Cognitive translation using the rodent touchscreen testing approach. Curr. Top. Behav. Neurosci. 28, 423–447. doi: 10.1007/7854_2015_5007
Iida, T., Nakahori, Y., Tsutsumi, O., Taketani, Y., and Nakagome, Y. (1994). The CpG island of the FMR-1 gene is methylated differently among embryonic tissues: implication for prenatal diagnosis. Hum. Reprod. 9, 1471–1473. doi: 10.1093/oxfordjournals.humrep.a138732
Illingworth, R. S., Gruenewald-Schneider, U., Webb, S., Kerr, A. R., James, K. D., Turner, D. J., et al. (2010). Orphan CpG islands identify numerous conserved promoters in the mammalian genome. PLoS Genet. 6:1001134. doi: 10.1371/journal.pgen.1001134
Irwin, S. A., Patel, B., Idupulapati, M., Harris, J. B., Crisostomo, R. A., Larsen, B. P., et al. (2001). Abnormal dendritic spine characteristics in the temporal and visual cortices of patients with fragile-X syndrome: a quantitative examination. Am. J. Med. Genet. 98, 161–167. doi: 10.1002/1096-8628(20010115)98:2<161::aid-ajmg1025>3.0.co;2-b
Jacobs, J. (2013). Hippocampal theta oscillations are slower in humans than in rodents: implications for models of spatial navigation and memory. Philos. Trans. R. Soc. Lond. B Biol. Sci. 369:20130304. doi: 10.1098/rstb.2013.0304
Jacobs, S., Cheng, C., and Doering, L. C. (2016). Hippocampal neuronal subtypes develop abnormal dendritic arbors in the presence of Fragile X astrocytes. Neuroscience 324, 202–217. doi: 10.1016/j.neuroscience.2016.03.011
Jacobs, R. A., Díaz, V., Meinild, A.-K., Gassmann, M., and Lundby, C. (2013). The C57Bl/6 mouse serves as a suitable model of human skeletal muscle mitochondrial function. Exp. Physiol. 98, 908–921. doi: 10.1113/expphysiol.2012.070037
Jacobs, S., and Doering, L. C. (2010). Astrocytes prevent abnormal neuronal development in the fragile x mouse. J. Neurosci. 30, 4508–4514. doi: 10.1523/JNEUROSCI.5027-09.2010
Jacobs, S., Nathwani, M., and Doering, L. C. (2010). Fragile X astrocytes induce developmental delays in dendrite maturation and synaptic protein expression. BMC Neurosci. 11:132. doi: 10.1186/1471-2202-11-132
Jacquemont, S., Curie, A., Des Portes, V., Torrioli, M. G., Berry-Kravis, E., Hagerman, R. J., et al. (2011). Epigenetic modification of the FMR1 gene in fragile X syndrome is associated with differential response to the mGluR5 antagonist AFQ056. Sci. Transl. Med. 3:64ra1. doi: 10.1126/scitranslmed.3001708
Jarem, D. A., Huckaby, L. V., and Delaney, S. (2010). AGG interruptions in (CGG)n DNA repeat tracts modulate the structure and thermodynamics of non-B conformations in vitro. Biochemistry 49, 6826–6837. doi: 10.1021/bi1007782
Joly, O., Pallier, C., Ramus, F., Pressnitzer, D., Vanduffel, W., and Orban, G. A. (2012). Processing of vocalizations in humans and monkeys: a comparative fMRI study. Neuroimage 62, 1376–1389. doi: 10.1016/j.neuroimage.2012.05.070
Jones, O. D. (2017). Do group I metabotropic glutamate receptors mediate LTD? Neurobiol. Learn. Mem. 138, 85–97. doi: 10.1016/j.nlm.2016.08.010
Jones, S. M., Cantlon, J. F., Merritt, D. J., and Brannon, E. M. (2010). Context affects the numerical semantic congruity effect in rhesus monkeys (Macaca mulatta). Behav. Processes 83, 191–196. doi: 10.1016/j.beproc.2009.12.009
Kaas, J. H. (1989). Why does the brain have so many visual areas? J. Cogn. Neurosci. 1, 121–135. doi: 10.1162/jocn.1989.1.2.121
Kaas, J. H. (2011). Reconstructing the areal organization of the neocortex of the first mammals. Brain Behav. Evol. 78, 7–21. doi: 10.1159/000327316
Kalinowska, M., Castillo, C., and Francesconi, A. (2015). Quantitative profiling of brain lipid raft proteome in a mouse model of fragile X syndrome. PLoS One 10:e0121464. doi: 10.1371/journal.pone.0121464
Kandel, E. R. (2001). The molecular biology of memory storage: a dialogue between genes and synapses. Science 294, 1030–1038. doi: 10.1126/science.1067020
Kandel, E. R. (2009). The biology of memory: a forty-year perspective. J. Neurosci. 29, 12748–12756. doi: 10.1523/JNEUROSCI.3958-09.2009
Kang, J. Y., Chadchankar, J., Vien, T. N., Mighdoll, M. I., Hyde, T. M., Mather, R. J., et al. (2017). Deficits in the activity of presynaptic γ-aminobutyric acid type B receptors contribute to altered neuronal excitability in fragile X syndrome. J. Biol. Chem. 292, 6621–6632. doi: 10.1074/jbc.M116.772541
Kang, S. J., and Kaang, B. K. (2016). Metabotropic glutamate receptor dependent long-term depression in the cortex. Korean J. Physiol. Pharmacol. 20, 557–564. doi: 10.4196/kjpp.2016.20.6.557
Kangas, B. D., and Bergman, J. (2017). Touchscreen technology in the study of cognition-related behavior. Behav. Pharmacol. 28, 623–629. doi: 10.1097/FBP.0000000000000356
Kashi, Y., and King, D. G. (2006). Simple sequence repeats as advantageous mutators in evolution. Trends Genet. 22, 253–259. doi: 10.1016/j.tig.2006.03.005
Kato, Y., Gokan, H., Oh-Nishi, A., Suhara, T., Watanabe, S., and Minamimoto, T. (2014). Vocalizations associated with anxiety and fear in the common marmoset (Callithrix jacchus). Behav. Brain Res. 275, 43–52. doi: 10.1016/j.bbr.2014.08.047
Kaufmann, W. E., Abrams, M. T., Chen, W., and Reiss, A. L. (1999). Genotype, molecular phenotype and cognitive phenotype: correlations in fragile X syndrome. Am. J. Med. Genet. 83, 286–295. doi: 10.1002/(sici)1096-8628(19990402)83:4<286::aid-ajmg10>3.3.co;2-8
Kazdoba, T. M., Leach, P. T., Silverman, J. L., and Crawley, J. N. (2014). Modeling fragile X syndrome in the Fmr1 knockout mouse. Intractable Rare Dis. Res. 3, 118–133. doi: 10.5582/irdr.2014.01024
Kemp, C., and Kaplan, G. (2013). Facial expressions in common marmosets (Callithrix jacchus) and their use by conspecifics. Anim. Cogn. 16, 773–788. doi: 10.1007/s10071-013-0611-5
Kenkel, W. M., Yee, J. R., Moore, K., Madularu, D., Kulkarni, P., Gamber, K., et al. (2016). Functional magnetic resonance imaging in awake transgenic fragile X rats: evidence of dysregulation in reward processing in the mesolimbic/habenular neural circuit. Transl. Psychiatry 22:e763. doi: 10.1038/tp.2016.15
Kenneson, A., Zhang, F., Hagedorn, C. H., and Warren, S. T. (2001). Reduced FMRP and increased FMR1 transcription is proportionally associated with CGG repeat number in intermediate-length and premutation carriers. Hum. Mol. Genet. 10, 1449–1454. doi: 10.1093/hmg/10.14.1449
Kenny, P., and Ceman, S. (2016). RNA secondary structure modulates FMRP’s bi-functional role in the MicroRNA pathway. Int. J. Mol. Sci. 17:E985. doi: 10.3390/ijms17060985
Kettani, A., Kumara, R. A., and Patel, J. D. (1995). Solution structure of a DNA quadruplex containing the fragile X syndrome triplet repeat. J. Mol. Biol. 254, 638–656. doi: 10.1006/jmbi.1995.0644
Keysers, C., Kaas, J. H., and Gazzola, V. (2010). Somatosensation in social perception. Nat. Rev. Neurosci. 11, 417–428. doi: 10.1038/nrn2833
Khalfallah, O., Jarjat, M., Davidovic, L., Nottet, N., Cestele, S., Mantegazza, M., et al. (2017). Depletion of the fragile X mental retardation protein in embryonic stem cells alters the kinetics of neurogenesis. Stem Cells 35, 374–385. doi: 10.1002/stem.2505
Khateb, S., Weisman-Shomer, P., Hershco-Shani, I., Ludwig, A. L., and Fry, M. (2007). The tetraplex (CGG)n destabilizing proteins hnRNP A2 and CBF-A enhance the in vivo translation of fragile X premutation mRNA. Nucleic Acids Res. 35, 5775–5788. doi: 10.1093/nar/gkm636
Kim, M., Bellini, M., and Ceman, S. (2009). Fragile X mental retardation protein FMRP binds mRNAs in the nucleus. Mol. Cell. Biol. 29, 214–228. doi: 10.1128/MCB.01377-08
Kim, S. Y., Burris, J., Bassal, F., Koldewyn, K., Chattarji, S., Tassone, F., et al. (2014). Fear-specific amygdala function in children and adolescents on the fragile X spectrum: a dosage response of the FMR1 gene. Cereb. Cortex 24, 600–613. doi: 10.1093/cercor/bhs341
Kim, H., Gibboni, R., Kirkhart, C., and Bao, S. (2013). Impaired critical period plasticity in primary auditory cortex of fragile X model mice. J. Neurosci. 33, 15686–15692. doi: 10.1523/JNEUROSCI.3246-12.2013
Kim, S. H., Markham, J. A., Weiler, I. J., and Greenough, W. T. (2008). Aberrant early-phase ERK inactivation impedes neuronal function in fragile X syndrome. Proc. Natl. Acad. Sci. U S A 105, 4429–4434. doi: 10.1073/pnas.0800257105
Klein, E. D., Evans, T. A., Schultz, N. B., and Beran, M. J. (2013). Learning how to “make a deal”: human (Homo sapiens) and monkey (Macaca mulatta) performance when repeatedly faced with the Monty Hall Dilemma. J. Comp. Psychol. 127, 103–108. doi: 10.1037/a0029057
Klusek, J., Martin, G. E., and Losh, M. (2014). A comparison of pragmatic language in boys with autism and fragile X syndrome. J. Speech Lang. Hear. Res. 57, 1692–1707. doi: 10.1044/2014_JSLHR-L-13-0064
Koekkoek, S. K., Yamaguchi, K., Milojkovic, B. A., Dortland, B. R., Ruigrok, T. J., Maex, R., et al. (2005). Deletion of FMR1 in Purkinje cells enhances parallel fiber LTD, enlarges spines and attenuates cerebellar eyelid conditioning in Fragile X syndrome. Neuron 47, 339–352. doi: 10.1016/j.neuron.2005.07.005
Koga, K., Liu, M. G., Qiu, S., Song, Q., O’Den, G., Chen, T., et al. (2015). Impaired presynaptic long-term potentiation in the anterior cingulate cortex of Fmr1 knock-out mice. J. Neurosci. 35, 2033–2043. doi: 10.1523/JNEUROSCI.2644-14.2015
Kogan, C. S., Turk, J., Hagerman, R. J., and Cornish, K. M. (2008). Impact of the Fragile X mental retardation 1 (FMR1) gene premutation on neuropsychiatric functioning in adult males without fragile X-associated Tremor/Ataxia syndrome: a controlled study. Am. J. Med. Genet. B Neuropsychiatr. Genet. 5, 859–872. doi: 10.1002/ajmg.b.30685
Korb, E., Herre, M., Zucker-Scharff, I., Gresack, J., Allis, C. D., and Darnell, R. B. (2017). Excess translation of epigenetic regulators contributes to fragile X syndrome and is alleviated by Brd4 inhibition. Cell 170, 1209.e20–1223.e20. doi: 10.1016/j.cell.2017.07.033
Korzhevskii, D. E., Otellin, V. A., and Grigor’ev, I. P. (2005). Glial fibrillary acidic protein in astrocytes in the human neocortex. Neurosci. Behav. Physiol. 35, 789–792. doi: 10.1007/s11055-005-0125-y
Koski, S. E., and Burkart, J. M. (2015). Common marmosets show social plasticity and group-level similarity in personality. Sci. Rep. 5:8878. doi: 10.1038/srep08878
Kotani, M., Shimono, K., Yoneyama, T., Nakako, T., Matsumoto, K., Ogi, Y., et al. (2017). An eye tracking system for monitoring face scanning patterns reveals the enhancing effect of oxytocin on eye contact in common marmosets. Psychoneuroendocrinology 83, 42–48. doi: 10.1016/j.psyneuen.2017.05.009
Ku, H.-H., Brunk, U. T., and Sohal, R. S. (1993). Relationship between mitochondrial superoxide and hydrogen peroxide production and longevity of mammalian species. Free Radic. Biol. Med. 15, 621–627. doi: 10.1016/0891-5849(93)90165-q
Kumari, D., Bhattacharya, A., Nadel, J., Moulton, K., Zeak, N. M., Glicksman, A., et al. (2014). Identification of fragile X syndrome specific molecular markers in human fibroblasts: a useful model to test the efficacy of therapeutic drugs. Hum. Mutat. 35, 1485–1494. doi: 10.1002/humu.22699
Kumari, D., and Usdin, K. (2010). The distribution of repressive histone modifications on silenced FMR1 alleles provides clues to the mechanism of gene silencing in fragile X syndrome. Hum. Mol. Genet. 19, 4634–4642. doi: 10.1093/hmg/ddq394
Kumari, D., and Usdin, K. (2016). Sustained expression of FMR1 mRNA from reactivated fragile X syndrome alleles after treatment with small molecules that prevent trimethylation of H3K27. Hum. Mol. Genet. 25, 3689–3698. doi: 10.1093/hmg/ddw215
Kuo, A. Y., Reiss, A. L., Freund, L. S., and Huffman, L. C. (2002). Family environment and cognitive abilities in girls with fragile-X syndrome. J. Intellect. Disabil. Res. 46, 328–339. doi: 10.1046/j.1365-2788.2002.00396.x
Kwon, H., Menon, V., Eliez, S., Warsofsky, I. S., White, C. D., Dyer-Friedman, J., et al. (2001). Functional neuroanatomy of visuospatial working memory in fragile X syndrome: relation to behavioral and molecular measures. Am. J. Psychiatry 158, 1040–1051. doi: 10.1176/appi.ajp.158.7.1040
La Fata, G., Gärtner, A., Domínguez-Iturza, N., Dresselaers, T., Dawitz, J., Poorthuis, R. B., et al. (2014). FMRP regulates multipolar to bipolar transition affecting neuronal migration and cortical circuitry. Nat. Neurosci. 17, 1693–1700. doi: 10.1038/nn.3870
Lai, Y., Xu, M., Zhang, Z., and Liu, Y. (2013). Instability of CTG repeats is governed by the position of a DNA base lesion through base excision repair. PLoS One 8:e56960. doi: 10.1371/journal.pone.0056960
Lalanne, L., Weiner, L., Trojak, B., Berna, F., and Bertschy, G. (2015). Substance-use disorder in high-functioning autism: clinical and neurocognitive insights from two case reports. BMC Psychiatry 15:149. doi: 10.1186/s12888-015-0541-7
Landrigan, P. J. (2010). What causes autism? Exploring the environmental contribution. Curr. Opin. Pediatr. 22, 219–225. doi: 10.1097/mop.0b013e328336eb9a
Larsen, L. A., Armstrong, J. S., Grønskov, K., Hjalgrim, H., Macpherson, J. N., Brøndum-Nielsen, K., et al. (2000). Haplotype and AGG-interspersion analysis of FMR1 (CGG)n alleles in the Danish population: implications for multiple mutational pathways towards fragile X alleles. Am. J. Med. Genet. 93, 99–106. doi: 10.1002/1096-8628(20000717)93:2<99::aid-ajmg4>3.0.co;2-w
Larsen, H., van der Zwaluw, C. S., Overbeek, G., Granic, I., Franke, B., and Engels, R. C. (2010). A variable-number-of-tandem-repeats polymorphism in the dopamine D4 receptor gene affects social adaptation of alcohol use: investigation of a gene-environment interaction. Psychol. Sci. 21, 1064–1068. doi: 10.1177/0956797610376654
Larson, J., Jessen, R. E., Kim, D., Fine, A. K., and Du Hoffmann, J. (2005). Age-dependent and selective impairment of long-term potentiation in the anterior piriform cortex of mice lacking the fragile X mental retardation protein. J. Neurosci. 25, 9460–9469. doi: 10.1523/JNEUROSCI.2638-05.2005
Lauterborn, J. C., Rex, C. S., Kramár, E., Chen, L. Y., Pandyarajan, V., Lynch, G., et al. (2007). Brain-derived neurotrophic factor rescues synaptic plasticity in a mouse model of fragile X syndrome. J. Neurosci. 27, 10685–10694. doi: 10.1523/JNEUROSCI.2624-07.2007
Lavedan, C. N., Garrett, L., and Nussbaum, R. L. (1997). Trinucleotide repeats (CGG)22TGG(CGG)43TGG(CGG)21 from the fragile X gene remain stable in transgenic mice. Hum. Genet. 100, 407–414. doi: 10.1007/s004390050525
Lavedan, C., Grabczyk, E., Usdin, K., and Nussbaum, R. L. (1998). Long uninterrupted CGG repeats within the first exon of the human FMR1 gene are not intrinsically unstable in transgenic mice. Genomics 50, 229–240. doi: 10.1006/geno.1998.5299
Lazaro-Perea, C., de Fátima Arruda, M., and Snowdon, C. T. (2004). Grooming as a reward? Social function of grooming between females in cooperatively breeding marmosets. Anim. Behav. 67, 627–636. doi: 10.1016/j.anbehav.2003.06.004
Leach, P. T., Hayes, J., Pride, M., Silverman, J. L., and Crawley, J. N. (2016). Normal performance of Fmr1 mice on a touchscreen delayed nonmatching to position working memory task. eNeuro 3:ENEURO.0143-15.2016. doi: 10.1523/ENEURO.0143-15.2016
Lee, H. Y., Ge, W. P., Huang, W., He, Y., Wang, G. X., Rowson-Baldwin, A., et al. (2011). Bidirectional regulation of dendritic voltage-gated potassium channels by the fragile X mental retardation protein. Neuron 72, 630–642. doi: 10.1016/j.neuron.2011.09.033
Lee, E., Lee, J., and Kim, E. (2017). Excitation/inhibition imbalance in animal models of autism spectrum disorders. Biol. Psychiatry 81, 838–847. doi: 10.1016/j.biopsych.2016.05.011
Levenga, J., Hayashi, S., de Vrij, F. M., Koekkoek, S. K., van der Linde, H. C., Nieuwenhuizen, I., et al. (2011). AFQ056, a new mGluR5 antagonist for treatment of fragile X syndrome. Neurobiol. Dis. 42, 311–317. doi: 10.1016/j.nbd.2011.01.022
Lewis, P., Abbeduto, L., Murphy, M., Richmond, E., Giles, N., Bruno, L., et al. (2006). Cognitive, language and social-cognitive skills of individuals with fragile X syndrome with and without autism. J. Intellect. Disabil. Res. 50, 532–545. doi: 10.1111/j.1365-2788.2006.00803.x
Li, J., Pelletier, M. R., Perez Velazquez, J.-L., and Carlen, P. L. (2002). Reduced cortical synaptic plasticity and GluR1 expression associated with fragile X mental retardation protein deficiency. Mol. Cell. Neurosci. 19, 138–151. doi: 10.1006/mcne.2001.1085
Li, Y., Stockton, M. E., Bhuiyan, I., Eisinger, B. E., Gao, Y., Miller, J. L., et al. (2016). MDM2 inhibition rescues neurogenic and cognitive deficits in a mouse model of fragile X syndrome. Sci. Transl. Med. 8:336ra61. doi: 10.1126/scitranslmed.aad9370
Liang, K. C., Tseng, J. T., Tsai, S. J., and Sun, H. S. (2015). Characterization and distribution of repetitive elements in association with genes in the human genome. Comput. Biol. Chem. 57, 29–38. doi: 10.1016/j.compbiolchem.2015.02.007
Liang, P., Xu, Y., Zhang, X., Ding, C., Huang, R., Zhang, Z., et al. (2015). CRISPR/Cas9-mediated gene editing in human tripronuclear zygotes. Protein Cell 6, 363–372. doi: 10.1007/s13238-015-0153-5
Liao, L. J., Park, S. K., Xu, T., Vanderklish, P., and Yates, J. R. (2008). Quantitative proteomic analysis of primary neurons reveals diverse changes in synaptic protein content in fmr1 knockout mice. Proc. Natl. Acad. Sci. U S A 105, 15281–15286. doi: 10.1073/pnas.0804678105
Ligsay, A., and Hagerman, R. J. (2016). Review of targeted treatments in fragile X syndrome. Intractable Rare Dis. Res. 5, 158–167. doi: 10.5582/irdr.2016.01045
Lim, C. S., Hoang, E. T., Viar, K. E., Stornetta, R. L., Scott, M. M., and Zhu, J. J. (2014). Pharmacological rescue of Ras signaling, GluA1-dependent synaptic plasticity and learning deficits in a fragile X model. Genes Dev. 28, 273–289. doi: 10.1101/gad.232470.113
Lin, Y., Tang, C., He, H., and Duan, R. (2013). Activation of mTOR ameliorates fragile X premutation rCGG repeat-mediated neurodegeneration. PLoS One 8:e62572. doi: 10.1371/journal.pone.0062572
Linderkamp, O., Janus, L., Linder, R., and Skoruppa, D. B. (2009). Time table of normal foetal brain development. Int. J. Prenat. Perinat. Psychol. Med. 21, 4–16.
Liu, Z. H., Chuang, D. M., and Smith, C. B. (2011). Lithium ameliorates phenotypic deficits in a mouse model of fragile X syndrome. Int. J. Neuropsychopharmacol. 14, 618–630. doi: 10.1017/s1461145710000520
Liu, Z. H., Huang, T., and Smith, C. B. (2012). Lithium reverses increased rates of cerebral protein synthesis in a mouse model of fragile X syndrome. Neurobiol. Dis. 45, 1145–1152. doi: 10.1016/j.nbd.2011.12.037
Liu, Z., Li, X., Zhang, J. T., Cai, Y. J., Cheng, T. L., Cheng, C., et al. (2016). Autism-like behaviours and germline transmission in transgenic monkeys overexpressing MeCP2. Nature 530, 98–102. doi: 10.1038/nature16533
Liu, H., Yin, J., Xiao, M., Gao, C., Mason, A. S., Zhao, Z., et al. (2012). Characterization and evolution of 5’ and 3’ untranslated regions in eukaryotes. Gene 507, 106–111. doi: 10.1016/j.gene.2012.07.034
Lloyd, B. A., Hake, H. S., Ishiwata, T., Farmer, C. E., Loetz, E. C., Fleshner, M., et al. (2017). Exercise increases mTOR signaling in brain regions involved in cognition and emotional behavior. Behav. Brain Res. 323, 56–67. doi: 10.1016/j.bbr.2017.01.033
Loesch, D. Z., Huggins, R. M., Bui, Q. M., Taylor, A. K., Pratt, C., Epstein, J., et al. (2003). Effect of fragile X status categories and FMRP deficits on cognitive profiles estimated by robust pedigree analysis. Am. J. Med. Genet. A 15, 13–23. doi: 10.1002/ajmg.a.20214
Loesch, D. Z., Huggins, R. M., and Hagerman, R. J. (2004). Phenotypic variation and FMRP levels in fragile X. Ment. Retard. Dev. Disabil. Res. Rev. 10, 31–41. doi: 10.1002/mrdd.20006
Loesch, D. Z., Sherwell, S., Kinsella, G., Tassone, F., Taylor, A., Amor, D., et al. (2012). Fragile X-associated tremor/ataxia phenotype in a male carrier of unmethylated full mutation in the FMR1 gene. Clin. Genet. 82, 88–92. doi: 10.1111/j.1399-0004.2011.01675.x
Long, H. K., Sims, D., Heger, A., Blackledge, N. P., Kutter, C., Wright, M. L., et al. (2013). Epigenetic conservation at gene regulatory elements revealed by non-methylated DNA profiling in seven vertebrates. Elife 26:e00348. doi: 10.7554/eLife.00348
Loomis, E. W., Sanz, L. A., Chedin, F., and Hagerman, P. J. (2014). Transcription-associated R-loop formation across the human FMR1 CGG-repeat region. PLoS Genet. 10:e1004294. doi: 10.1371/journal.pgen.1004294
Lovelace, J. W., Wen, T. H., Reinhard, S., Hsu, M. S., Sidhu, H., Ethell, I. M., et al. (2016). Matrix metalloproteinase-9 deletion rescues auditory evoked potential habituation deficit in a mouse model of Fragile X Syndrome. Neurobiol. Dis. 89, 126–135. doi: 10.1016/j.nbd.2016.02.002
Lowenstine, L. J., McManamon, R., and Terio, K. A. (2016). Comparative pathology of aging great apes: bonobos, chimpanzees, gorillas and orangutans. Vet. Pathol. 53, 250–276. doi: 10.1177/0300985815612154
Lubojemska, A., Borejko, M., Czapiewski, P., Dziadziuszko, R., and Biernat, W. (2016). Of mice and men: olfactory neuroblastoma among animals and humans. Vet. Comp. Oncol. 14, e70–e82. doi: 10.1111/vco.12102
Lucarelli, M., Visalberghi, E., Adriani, W., Addessi, E., Pierandrei, S., Manciocco, A., et al. (2017). Polymorphism of the 3’-UTR of the dopamine transporter gene (DAT) in New World monkeys. Primates 58, 169–178. doi: 10.1007/s10329-016-0560-0
Ludwig, A. L., Espinal, G. M., Pretto, D. I., Jamal, A. L., Arque, G., Tassone, F., et al. (2014). CNS expression of murine fragile X protein (FMRP) as a function of CGG-repeat size. Hum. Mol. Genet. 23, 3228–3238. doi: 10.1093/hmg/ddu032
Luecke, R. H., Wosilait, W. D., and Young, J. F. (1999). Mathematical modeling of human embryonic and fetal growth rates. Growth Dev. Aging 63, 49–59.
Luque, M. A., Beltran-Matas, P., Marin, M. C., Torres, B., and Herrero, L. (2017). Excitability is increased in hippocampal CA1 pyramidal cells of Fmr1 knockout mice. PLoS One 12:e0185067. doi: 10.1371/journal.pone.0185067
Luy, J. (2007). Ethical and legal aspects of animal experiments on non-human primates. Dtsch. Tierarztl. Wochenschr. 114, 81–85.
Macdonald, D. W. (1981). Dwindling resources and the social behaviour of Capybaras, (Hydrochoerus hydrochaeris) (Mammalia). J. Zool. 194, 371–391. doi: 10.1111/j.1469-7998.1981.tb04588.x
Machado, C. J., and Nelson, E. E. (2011). Eye-tracking with nonhuman primates is now more accessible than ever before. Am. J. Primatol. 73, 562–569. doi: 10.1002/ajp.20928
MacLeod, L. S., Kogan, C. S., Collin, C. A., Berry-Kravis, E., Messier, C., and Gandhi, R. (2010). A comparative study of the performance of individuals with fragile X syndrome and Fmr1 knockout mice on Hebb-Williams mazes. Genes Brain Behav. 9, 53–64. doi: 10.1111/j.1601-183x.2009.00534.x
Maes, B., Fryns, J. P., Van Walleghem, M., and Van Den Berghe, H. (1994). Cognitive functioning and information processing of adult mentally retarded men with fragile-X syndrome. Am. J. Med. Genet. 50, 190–200. doi: 10.1002/ajmg.1320500211
Malgowska, M., Gudanis, D., Kierzek, R., Wyszko, E., Gabelica, V., and Gdaniec, Z. (2014). Distinctive structural motifs of RNA G-quadruplexes composed of AGG, CGG and UGG trinucleotide repeats. Nucleic Acids Res. 42, 10196–10207. doi: 10.1093/nar/gku710
Margaret Elowson, A., Snowdon, C. T., and Lazaro-Perea, C. (1998). ‘Babbling’ and social context in infant monkeys: parallels to human infants. Trends Cogn. Sci. 2, 31–37. doi: 10.1016/s1364-6613(97)01115-7
Margulies, D. S. (2017). Unraveling the complex tapestry of association networks. Neuron 95, 239–241. doi: 10.1016/j.neuron.2017.07.006
Markram, K., and Markram, H. (2010). The intense world theory–a unifying theory of the neurobiology of autism. Front. Hum. Neurosci. 4:224. doi: 10.3389/fnhum.2010.00224
Martin, B. S., Corbin, J. G., and Huntsman, M. M. (2014). Deficient tonic GABAergic conductance and synaptic balance in the fragile X syndrome amygdala. J. Neurophysiol. 112, 890–902. doi: 10.1152/jn.00597.2013
Martin, H. G. S., Lassalle, O., Brown, J. T., and Manzoni, O. J. (2016). Age-dependent long-term potentiation deficits in the prefrontal cortex of the Fmr1 knockout mouse model of fragile X syndrome. Cereb. Cortex 26, 2084–2092. doi: 10.1093/cercor/bhv031
Martin, B. S., Martinez-Botella, G., Loya, C. M., Salituro, F. G., Robichaud, A. J., Huntsman, M. M., et al. (2016). Rescue of deficient amygdala tonic γ-aminobutyric acidergic currents in the Fmr-/y mouse model of fragile X syndrome by a novel γ-aminobutyric acid type A receptor-positive allosteric modulator. J. Neurosci. Res. 94, 568–578. doi: 10.1002/jnr.23632
Martin, D. I., Singer, M., Dhahbi, J., Mao, G., Zhang, L., Schroth, G. P., et al. (2011). Phyloepigenomic comparison of great apes reveals a correlation between somatic and germline methylation states. Genome Res. 21, 2049–2057. doi: 10.1101/gr.122721.111
Martinez, L. A., and Tejada-Simon, M. V. (2017). Pharmacological rescue of hippocampal fear learning deficits in fragile X syndrome. Mol. Neurobiol. doi: 10.1007/s12035-017-0819-5 [Epub ahead of print].
Massen, J. J., Šlipogor, V., and Gallup, A. C. (2016). An observational investigation of behavioral contagion in common marmosets (Callithrix jacchus): indications for contagious scent-marking. Front. Psychol. 7:1190. doi: 10.3389/fpsyg.2016.01190
Matic, K., Eninger, T., Bardoni, B., Davidovic, L., and Macek, B. (2014). Quantitative phosphoproteomics of murine Fmr1-KO cell lines provides new insights into FMRP-dependent signal transduction mechanisms. J. Proteome Res. 13, 4388–4397. doi: 10.1021/pr5006372
Matsuo, K., Clay, O., Takahashi, T., Silke, J., and Schaffner, W. (1993). Evidence for erosion of mouse CpG islands during mammalian evolution. Somat. Cell Mol. Genet. 19, 543–555. doi: 10.1007/bf01233381
Mazzocco, M. M., Pennington, B. F., and Hagerman, R. J. (1994). Social cognition skills among females with fragile X. J. Autism Dev. Disord. 24, 473–485. doi: 10.1007/bf02172129
Meredith, R. M., de Jong, R., and Mansvelder, H. D. (2011). Functional rescue of excitatory synaptic transmission in the developing hippocampus in Fmr1-KO mouse. Neurobiol. Dis. 41, 104–110. doi: 10.1016/j.nbd.2010.08.026
Mestas, J., and Hughes, C. C. (2004). Of mice and not men: differences between mouse and human immunology. J. Immunol. 172, 2731–2738. doi: 10.4049/jimmunol.172.5.2731
Michalon, A., Sidorov, M., Ballard, T. M., Ozmen, L., Spooren, W., Wettstein, J. G., et al. (2012). Chronic pharmacological mGlu5 inhibition corrects fragile X in adult mice. Neuron 74, 49–56. doi: 10.1016/j.neuron.2012.03.009
Mientjes, E. J., Nieuwenhuizen, I., Kirkpatrick, L., Zu, T., Hoogeveen-Westerveld, M., Severijnen, L., et al. (2006). The generation of a conditional Fmr1 knock out mouse model to study Fmrp function in vivo. Neurobiol. Dis. 21, 549–555. doi: 10.1016/j.nbd.2005.08.019
Miller, C. T., Mandel, K., and Wang, X. (2010). The communicative content of the common marmoset phee call during antiphonal calling. Am. J. Primatol. 72, 974–980. doi: 10.1002/ajp.20854
Miller, C. T., and Wang, X. (2006). Sensory-motor interactions modulate a primate vocal behavior: antiphonal calling in common marmosets. J. Comp. Physiol. A Neuroethol. Sens. Neural Behav. Physiol. 192, 27–38. doi: 10.1007/s00359-005-0043-z
Ming, X., Johnson, W. G., Stenroos, E. S., Mars, A., Lambert, G. H., and Buyske, S. (2010). Genetic variant of glutathione peroxidase 1 in autism. Brain Dev. 32, 105–109. doi: 10.1016/j.braindev.2008.12.017
Mitas, M., Yu, A., Dill, J., and Haworth, S. I. (1995). The trinucleotide repeat sequence d(CGG)15 forms a heat-stable hairpin containing Gsyn. Ganti base pairs. Biochemistry 34, 12803–12811. doi: 10.1021/bi00039a041
Miyashiro, K. Y., Beckel-Mitchener, A., Purk, T. P., Becker, K. G., Barret, T., Liu, L., et al. (2003). RNA cargoes associating with FMRP reveal deficits in cellular functioning in Fmr1 null mice. Neuron 37, 417–431. doi: 10.1016/s0896-6273(03)00034-5
Mohammadparast, S., Bayat, H., Biglarian, A., and Ohadi, M. (2014). Exceptional expansion and conservation of a CT-repeat complex in the core promoter of PAXBP1 in primates. Am. J. Primatol. 76, 747–756. doi: 10.1002/ajp.22266
Molaro, A., Hodges, E., Fang, F., Song, Q., Mccombie, W. R., Hannon, G. J., et al. (2011). Sperm methylation profiles reveal features of epigenetic inheritance and evolution in primates. Cell 146, 1029–1041. doi: 10.1016/j.cell.2011.08.016
Moore, T. L., Killiany, R. J., Herndon, J. G., Rosene, D. L., and Moss, M. B. (2003). Impairment in abstraction and set shifting in aged rhesus monkeys. Neurobiol. Aging 24, 125–134. doi: 10.1016/s0197-4580(02)00054-4
Moore, T. L., Killiany, R. J., Herndon, J. G., Rosene, D. L., and Moss, M. B. (2005). A non-human primate test of abstraction and set shifting: an automated adaptation of the Wisconsin Card Sorting Test. J. Neurosci. Methods 146, 165–173. doi: 10.1016/j.jneumeth.2005.02.005
Moore, T. L., Killiany, R. J., Herndon, J. G., Rosene, D. L., and Moss, M. B. (2006). Executive system dysfunction occurs as early as middle-age in the rhesus monkey. Neurobiol. Aging 27, 1484–1493. doi: 10.1016/j.neurobiolaging.2005.08.004
Morecraft, R. J., Stilwell-Morecraft, K. S., Ge, J., Cipolloni, P. B., and Pandya, D. N. (2015). Cytoarchitecture and cortical connections of the anterior insula and adjacent frontal motor fields in the rhesus monkey. Brain Res. Bull. 119, 52–72. doi: 10.1016/j.brainresbull.2015.10.004
Mornet, E., Chateau, C., Taillandier, A., Simon-Bouy, B., and Serre, J. L. (1996). Recurrent and unexpected segregation of the FMR1 CGG repeat in a family with fragile X syndrome. Hum. Genet. 97, 512–515. doi: 10.1007/s004390050083
Morrill, R. J., Paukner, A., Ferrari, P. F., and Ghazanfar, A. A. (2012). Monkey lipsmacking develops like the human speech rhythm. Dev. Sci. 15, 557–568. doi: 10.1111/j.1467-7687.2012.01149.x
Morrill, R. J., Thomas, A. W., Schiel, N., Souto, A., and Miller, C. T. (2013). The effect of habitat acoustics on common marmoset vocal signal transmission. Am. J. Primatol. 75, 904–916. doi: 10.1002/ajp.22152
Mor-Shaked, H., and Eiges, R. (2016). Modeling fragile X syndrome using human pluripotent stem cells. Genes 7:E77. doi: 10.3390/genes7100077
Mor-Shaked, H., and Eiges, R. (2018). Reevaluation of FMR1 hypermethylation timing in fragile X syndrome. Front. Mol. Neurosci. 11:31. doi: 10.3389/fnmol.2018.00031
Moser, E. I., Kropff, E., and Moser, M. B. (2008). Place cells, grid cells and the brain’s spatial representation system. Annu. Rev. Neurosci. 31, 69–89. doi: 10.1146/annurev.neuro.31.061307.090723
Mulvihill, D. J., Nichol Edamura, K., Hagerman, K. A., Pearson, C. E., and Wang, Y. H. (2005). Effect of CAT or AGG interruptions and CpG methylation on nucleosome assembly upon trinucleotide repeats on spinocerebellar ataxia, type 1 and fragile X syndrome. J. Biol. Chem. 280, 4498–4503. doi: 10.1074/jbc.M413239200
Munir, F., Cornish, K. M., and Wilding, J. (2000). A neuropsychological profile of attention deficits in young males with fragile X syndrome. Neuropsychologia 38, 1261–1270. doi: 10.1016/s0028-3932(00)00036-1
Murphy, M. M., and Abbeduto, L. (2007). Gender differences in repetitive language in fragile X syndrome. J. Intellect. Disabil. Res. 51, 387–400. doi: 10.1111/j.1365-2788.2006.00888.x
Muzar, Z., Adams, P. E., Schneider, A., Hagerman, R. J., and Lozano, R. (2014). Addictive substances may induce a rapid neurological deterioration in fragile X-associated tremor ataxia syndrome: a report of two cases. Intractable Rare Dis. Res. 3, 162–165. doi: 10.5582/irdr.2014.01023
Myrick, L. K., Deng, P. Y., Hashimoto, H., Oh, Y. M., Cho, Y., Poidevin, M. J., et al. (2015). Independent role for presynaptic FMRP revealed by an FMR1 missense mutation associated with intellectual disability and seizures. Proc. Natl. Acad. Sci. U S A 112, 949–956. doi: 10.1073/pnas.1423094112
Myrick, L. K., Nakamoto-Kinoshita, M., Lindor, N. M., Kirmani, S., Cheng, X., and Warren, S. T. (2014). Fragile X syndrome due to a missense mutation. Eur. J. Hum. Genet. 22, 1185–1189. doi: 10.1038/ejhg.2013.311
Nadel, Y., Weisman-Shomer, P., and Fry, M. (1995). The fragile X syndrome single strand d(CGG)n nucleotide repeats readily fold back to form unimolecular hairpin structures. J. Biol. Chem. 270, 28970–28977. doi: 10.1074/jbc.270.48.28970
Nakamoto, M., Nalavadi, V., Epstein, M. P., Narayanan, U., Bassell, G. J., and Warren, S. T. (2007). Fragile X mental retardation protein deficiency leads to excessive mGluR5-dependent internalization of AMPA receptors. Proc. Natl. Acad. Sci. U S A 104, 15537–15542. doi: 10.1073/pnas.0707484104
Namdar-Aligoodarzi, P., Mohammadparast, S., Zaker-Kandjani, B., Talebi Kakroodi, S., Jafari Vesiehsari, M., and Ohadi, M. (2015). Exceptionally long 5’ UTR short tandem repeats specifically linked to primates. Gene 569, 88–94. doi: 10.1016/j.gene.2015.05.053
Napierala, M., Michalowski, D., de Mezer, M., and Krzyzosiak, W. J. (2005). Facile FMR1 mRNA structure regulation by interruptions in CGG repeats. Nucleic Acids Res. 33, 451–463. doi: 10.1093/nar/gki186
Narayanan, U., Nalavadi, V., Nakamoto, M., Thomas, G., Ceman, S., Bassell, G. J., et al. (2008). S6K1 phosphorylates and regulates fragile X mental retardation protein (FMRP) with the neuronal protein synthesis-dependent mammalian target of rapamycin (mTOR) signaling cascade. J. Biol. Chem. 283, 18478–18482. doi: 10.1074/jbc.C800055200
Nelson, D. L., Orr, H. T., and Warren, S. T. (2013). The unstable repeats–three evolving faces of neurological disease. Neuron 77, 825–843. doi: 10.1016/j.neuron.2013.02.022
Neubert, F. X., Mars, R. B., Thomas, A. G., Sallet, J., and Rushworth, M. F. (2014). Comparison of human ventral frontal cortex areas for cognitive control and language with areas in monkey frontal cortex. Neuron 81, 700–713. doi: 10.1016/j.neuron.2013.11.012
Neves, G., Cooke, S. F., and Bliss, T. V. (2008). Synaptic plasticity, memory and the hippocampus: a neural network approach to causality. Nat. Rev. Neurosci. 9, 65–75. doi: 10.1038/nrn2303
Neyman, S., and Manahan-Vaughan, D. (2008). Metabotropic glutamate receptor 1 (mGluR1) and 5 (mGluR5) regulate late phases of LTP and LTD in the hippocampal CA1 region in vitro. Eur. J. Neurosci. 27, 1345–1352. doi: 10.1111/j.1460-9568.2008.06109.x
Nichol Edamura, K., Leonard, M. R., and Pearson, C. E. (2005). Role of replication and CpG methylation in fragile X syndrome CGG deletions in primate cells. Am. J. Hum. Genet. 76, 302–311. doi: 10.1086/427928
Niere, F., Wilkerson, J. R., and Huber, K. M. (2012). Evidence for a fragile X mental retardation protein-mediated translational switch in metabotropic glutamate receptor-triggered Arc translation and long-term depression. J. Neurosci. 32, 5924–5936. doi: 10.1523/JNEUROSCI.4650-11.2012
Niu, Y., Shen, B., Cui, Y., Chen, Y., Wang, J., Wang, L., et al. (2014). Generation of gene-modified cynomolgus monkey via Cas9/RNA-mediated gene targeting in one-cell embryos. Cell 156, 836–843. doi: 10.1016/j.cell.2014.01.027
Nolin, S. L., Brown, W. T., Glicksman, A., Houck, G. E. Jr., Gargano, A. D., Sullivan, A., et al. (2003). Expansion of the fragile X CGG repeat in females with premutation or intermediate alleles. Am. J. Hum. Genet. 72, 454–464. doi: 10.1086/367713
Nolin, S. L., Glicksman, A., Ersalesi, N., Dobkin, C., Brown, W. T., Cao, R., et al. (2015). Fragile X full mutation expansions are inhibited by one or more AGG interruptions in premutation carriers. Genet. Med. 17, 358–364. doi: 10.1038/gim.2014.106
Nolin, S. L., Houck, G. E. Jr., Gargano, A. D., Blumstein, H., Dobkin, C. S., and Brown, W. T. (1999). FMR1 CGG-repeat instability in single sperm and lymphocytes of fragile-X premutation males. Am. J. Hum. Genet. 65, 680–688. doi: 10.1086/302543
Nolin, S. L., Lewis, F. A. III., Ye, L. L., Houck, G. E. Jr., Glicksman, A. E., Limprasert, P., et al. (1996). Familial transmission of the FMR1 CGG repeat. Am. J. Hum. Genet. 59, 1252–1261.
Nolin, S. L., Sah, S., Glicksman, A., Sherman, S. L., Allen, E., Berry-Kravis, E., et al. (2013). Fragile X AGG analysis provides new risk predictions for 45–69 repeat alleles. Am. J. Med. Genet. A 4, 771–778. doi: 10.1002/ajmg.a.35833
Nosyreva, E. D., and Huber, K. M. (2006). Metabotropic receptor-dependent long-term depression persists in the absence of protein synthesis in the mouse model of fragile X syndrome. J. Neurophysiol. 95, 3291–3295. doi: 10.1152/jn.01316.2005
Oberheim, N. A., Takano, T., Han, X., He, W., Lin, J. H., Wang, F., et al. (2009). Uniquely hominid features of adult human astrocytes. J. Neurosci. 29, 3276–3287. doi: 10.1523/JNEUROSCI.4707-08.2009
Oberlé, I., Rousseau, F., Heitz, D., Kretz, C., Devys, D., Hanauer, A., et al. (1991). Instability of a 550-base pair DNA segment and abnormal methylation in fragile X syndrome. Science 252, 1097–1102. doi: 10.1126/science.252.5009.1097
Oberman, L., Ifert-Miller, F., Najib, U., Bashir, S., Woollacott, I., Gonzalez-Heydrich, J., et al. (2010). Transcranial magnetic stimulation provides means to assess cortical plasticity and excitability in humans with fragile x syndrome and autism spectrum disorder. Front. Synaptic Neurosci. 2:26. doi: 10.3389/fnsyn.2010.00026
Ohadi, M., Valipour, E., Ghadimi-Haddadan, S., Namdar-Aligoodarzi, P., Bagheri, A., Kowsari, A., et al. (2015). Core promoter short tandem repeats as evolutionary switch codes for primate speciation. Am. J. Primatol. 77, 34–43. doi: 10.1002/ajp.22308
Oikonomidis, L., Santangelo, A. M., Shiba, Y., Clarke, F. H., Robbins, T. W., and Roberts, A. C. (2017). A dimensional approach to modeling symptoms of neuropsychiatric disorders in the marmoset monkey. Dev. Neurobiol. 77, 328–353. doi: 10.1002/dneu.22446
Olmos-Serrano, J. L., Corbin, J. G., and Burns, M. P. (2011). The GABAA receptor agonist THIP ameliorates specific behavioral deficits in the mouse model of fragile X syndrome. Dev. Neurosci. 33, 395–403. doi: 10.1159/000332884
Olmos-Serrano, J. L., Paluszkiewicz, S. M., Martin, B. S., Kaufmann, W. E., Corbin, J. G., and Huntsman, M. M. (2010). Defective GABAergic neurotransmission and pharmacological rescue of neuronal hyperexcitability in the amygdala in a mouse model of fragile X syndrome. J. Neurosci. 30, 9929–9938. doi: 10.1523/JNEUROSCI.1714-10.2010
Oostra, B. A. (1998). “Instability of triplet repeats: factors and mechanisms,” in Trinucleotide Diseases and Instability, eds R. D. Wells, A. Bacolla and R. P. Bowater (Berlin, Heidelberg: Springer-Verlag), 133–165.
Orban, G. A., Claeys, K., Nelissen, K., Smans, R., Sunaert, S., Todd, J. T., et al. (2006). Mapping the parietal cortex of human and non-human primates. Neuropsychologia 44, 2647–2667. doi: 10.1016/j.neuropsychologia.2005.11.001
Orekhova, E. V., Stroganova, T. A., Prokofyev, A. O., Nygren, G., Gillberg, C., and Elam, M. (2008). Sensory gating in young children with autism: relation to age, IQ and EEG γ oscillations. Neurosci. Lett. 434, 218–223. doi: 10.1016/j.neulet.2008.01.066
Ornstein, P. A., Schaaf, J. M., Hooper, S. R., Hatton, D. D., Mirrett, P., and Bailey, D. B. Jr. (2008). Memory skills of boys with fragile X syndrome. Am. J. Ment. Retard. 113, 453–465. doi: 10.1352/2008.113:453-465
Osterweil, E. K., Chuang, S. C., Chubykin, A. A., Sidorov, M., Bianchi, R., Wong, R. K., et al. (2013). Lovastatin corrects excess protein synthesis and prevents epileptogenesis in a mouse model of fragile X syndrome. Neuron 77, 243–250. doi: 10.1016/j.neuron.2012.01.034
Osterweil, E. K., Krueger, D. D., Reinhold, K., and Bear, M. F. (2010). Hypersensitivity to mGluR5 and ERK1/2 leads to excessive protein synthesis in the hippocampus of a mouse model of fragile X syndrome. J. Neurosci. 30, 15616–15627. doi: 10.1523/JNEUROSCI.3888-10.2010
Pacey, L. K., Doss, L., Cifelli, C., van der Kooy, D., Heximer, S. P., and Hampson, D. R. (2011a). Genetic deletion of regulator of G-protein signaling 4 (RGS4) rescues a subset of fragile X related phenotypes in the FMR1 knockout mouse. Mol. Cell. Neurosci. 46, 563–572. doi: 10.1016/j.mcn.2010.12.005
Pacey, L. K., Tharmalingam, S., and Hampson, D. R. (2011b). Subchronic administration and combination metabotropic glutamate and GABAB receptor drug therapy in fragile X syndrome. J. Pharmacol. Exp. Ther. 338, 897–905. doi: 10.1124/jpet.111.183327
Padmashri, R., Reiner, B. C., Suresh, A., Spartz, E., and Dunaevsky, A. (2013). Altered structural and functional synaptic plasticity with motor skill learning in a mouse model of fragile X syndrome. J. Neurosci. 33, 19715–19723. doi: 10.1523/JNEUROSCI.2514-13.2013
Pai, A. A., Bell, J. T., Marioni, J. C., Pritchard, J. K., and Gilad, Y. (2011). A genome-wide study of DNA methylation patterns and gene expression levels in multiple human and chimpanzee tissues. PLoS Genet. 7:24. doi: 10.1371/journal.pgen.1001316
Paigen, K. (2003). One hundred years of mouse genetics: an intellectual history. I. The classical period (1902–1980). Genetics 163, 1–7.
Paluszkiewicz, S. M., Olmos-Serrano, J. L., Corbin, J. G., and Huntsman, M. M. (2011). Impaired inhibitory control of cortical synchronization in fragile X syndrome. J. Neurophysiol. 106, 2264–2272. doi: 10.1152/jn.00421.2011
Paoloni-Giacobino, A., D’Aiuto, L., Cirio, M. C., Reinhart, B., and Chaillet, J. R. (2007). Conserved features of imprinted differentially methylated domains. Gene 399, 33–45. doi: 10.1016/j.gene.2007.04.028
Paradee, W., Melikian, H. E., Rasmussen, D. L., Kenneson, A., Conn, P. J., and Warren, S. T. (1999). Fragile X mouse: strain effects of knockout phenotype and evidence suggesting deficient amygdala function. Neuroscience 94, 185–192. doi: 10.1016/s0306-4522(99)00285-7
Pardo, M., Beurel, E., and Jope, R. S. (2017). Cotinine administration improves impaired cognition in the mouse model of Fragile X syndrome. Eur. J. Neurosci. 45, 490–498. doi: 10.1111/ejn.13446
Park, C. Y., Halevy, T., Lee, D. R., Sung, J. J., Lee, J. S., Yanuka, O., et al. (2015). Reversion of FMR1 methylation and silencing by editing the triplet repeats in fragile X iPSC-derived neurons. Cell Rep. 13, 234–241. doi: 10.1016/j.celrep.2015.08.084
Park, S., Park, J. M., Kim, S., Kim, J. A., Shepherd, J. D., Smith-Hicks, C. L., et al. (2008). Elongation factor 2 and fragile X mental retardation protein control the dynamic translation of Arc/Arg3.1 essential for mGluR-LTD. Neuron 59, 70–83. doi: 10.1016/j.neuron.2008.05.023
Park, J. E., Zhang, X. F., Choi, S. H., Okahara, J., Sasaki, E., and Silva, A. C. (2016). Generation of transgenic marmosets expressing genetically encoded calcium indicators. Sci. Rep. 6:34931. doi: 10.1038/srep34931
Patel, P. K., Bhavesh, N. S., and Hosur, R. V. (2000). Cation-dependent conformational switches in d-TGGCGGC containing two triplet repeats of Fragile X Syndrome: NMR observations. Biochem. Biophys. Res. Commun. 278, 833–838. doi: 10.1006/bbrc.2000.3878
Patel, A. B., Hays, S. A., Bureau, I., Huber, K. M., and Gibson, J. R. (2013). A target cell-specific role for presynaptic Fmr1 in regulating glutamate release onto neocortical fast-spiking inhibitory neurons. J. Neurosci. 33, 2593–2604. doi: 10.1523/JNEUROSCI.2447-12.2013
Paul, K., Venkitaramani, D. V., and Cox, C. L. (2013). Dampened dopamine-mediated neuromodulation in prefrontal cortex of fragile X mice. J. Physiol. 591, 1133–1143. doi: 10.1113/jphysiol.2012.241067
Peier, A. M., and Nelson, D. L. (2002). Instability of a premutation-sized CGG repeat in FMR1 YAC transgenic mice. Genomics 80, 423–432. doi: 10.1006/geno.2002.6849
Pellerin, D., Çaku, A., Fradet, M., Bouvier, P., Dubé, J., and Corbin, F. (2016). Lovastatin corrects ERK pathway hyperactivation in fragile X syndrome: potential of platelet’s signaling cascades as new outcome measures in clinical trials. Biomarkers 21, 497–508. doi: 10.3109/1354750x.2016.1160289
Pereira, S., Veeraraghavan, P., Ghosh, S., and Gandhi, M. (2004). Animal experimentation and ethics in India: the CPCSEA makes a difference. Altern. Lab. Anim. 32, 411–415.
Petersen, B. (2017). Basics of genome editing technology and its application in livestock species. Reprod. Domest. Anim. 3, 4–13. doi: 10.1111/rda.13012
Pezer, Z., Brajković, J., Feliciello, I., and Ugarkovć, D. (2012). Satellite DNA-mediated effects on genome regulation. Genome Dyn. 7, 153–169. doi: 10.1159/000337116
Phillips, K. A., Bales, K. L., Capitanio, J. P., Conley, A., Czoty, P. W., ‘t Hart, B. A., et al. (2014). Why primate models matter. Am. J. Primatol. 76, 801–827. doi: 10.1002/ajp.22281
Pieretti, M., Zhang, F. P., Fu, Y. H., Warren, S. T., Oostra, B. A., Caskey, C. T., et al. (1991). Absence of expression of the FMR-1 gene in fragile X syndrome. Cell 66, 817–822. doi: 10.1016/0092-8674(91)90125-i
Pietrobono, R., Tabolacci, E., Zalfa, F., Zito, I., Terracciano, A., Moscato, U., et al. (2005). Molecular dissection of the events leading to inactivation of the FMR1 gene. Hum. Mol. Genet. 14, 267–277. doi: 10.1093/hmg/ddi024
Pietropaolo, S., Goubran, M. G., Joffre, C., Aubert, A., Lemaire-Mayo, V., Crusio, W. E., et al. (2014). Dietary supplementation of omega-3 fatty acids rescues fragile X phenotypes in Fmr1-Ko mice. Psychoneuroendocrinology 49, 119–129. doi: 10.1016/j.psyneuen.2014.07.002
Pistorio, A. L., Vintch, B., and Wang, X. (2006). Acoustic analysis of vocal development in a New World primate, the common marmoset (Callithrix jacchus). J. Acoust. Soc. Am. 120, 1655–1670. doi: 10.1121/1.2225899
Plohl, M., Meštrović, N., and Mravinac, B. (2012). Satellite DNA evolution. Genome Dyn. 7, 126–152. doi: 10.1159/000337122
Powell, W. T., Coulson, R. L., Gonzales, M. L., Crary, F. K., Wong, S. S., Adams, S., et al. (2013). R-loop formation at Snord116 mediates topotecan inhibition of Ube3a-antisense and allele-specific chromatin decondensation. Proc. Natl. Acad. Sci. U S A 110, 13938–13943. doi: 10.1073/pnas.1305426110
Pretto, D., Yrigollen, C. M., Tang, H. T., Williamson, J., Espinal, G., Iwahashi, C. K., et al. (2014). Clinical and molecular implications of mosaicism in FMR1 full mutations. Front. Genet. 5:318. doi: 10.3389/fgene.2014.00318
Preuss, T. M. (2011). The human brain: rewired and running hot. Ann. N Y Acad. Sci. 1225, E182–E191. doi: 10.1111/j.1749-6632.2011.06001.x
Primerano, B., Tassone, F., Hagerman, R. J., Hagerman, P., Amaldi, F., and Bagni, C. (2002). Reduced FMR1 mRNA translation efficiency in fragile X patients with premutations. RNA 8, 1482–1488.
Quartier, A., Poquet, H., Gilbert-Dussardier, B., Rossi, M., Casteleyn, A. S., Portes, V. D., et al. (2017). Intragenic FMR1 disease-causing variants: a significant mutational mechanism leading to Fragile-X syndrome. Eur. J. Hum. Genet. 25, 423–431. doi: 10.1038/ejhg.2016.204
Quilez, J., Guilmatre, A., Garg, P., Highnam, G., Gymrek, M., Erlich, Y., et al. (2016). Polymorphic tandem repeats within gene promoters act as modifiers of gene expression and DNA methylation in humans. Nucleic Acids Res. 44, 3750–3762. doi: 10.1093/nar/gkw219
Radwan, B., Dvorak, D., and Fenton, A. A. (2016). Impaired cognitive discrimination and discoordination of coupled theta-γ oscillations in Fmr1 knockout mice. Neurobiol. Dis. 88, 125–138. doi: 10.1016/j.nbd.2016.01.003
Rajan-Babu, I. S., and Chong, S. S. (2016). Molecular correlates and recent advancements in the diagnosis and screening of FMR1-related disorders. Genes (Basel) 7:E87. doi: 10.3390/genes7100087
Ranganath, C., and Ritchey, M. (2012). Two cortical systems for memory-guided behaviour. Nat. Rev. Neurosci. 13, 713–726. doi: 10.1038/nrn3338
Reisin, H. D., and Colombo, J. A. (2002). Astroglial interlaminar processes in human cerebral cortex: variations in cytoskeletal profiles. Brain Res. 937, 51–57. doi: 10.1016/s0006-8993(02)02464-2
Reyniers, E., Martin, J.-J., Cras, P., Van Marck, E., Handig, I., Jorens, H. Z. J., et al. (1999). Postmortem examination of two fragile X brothers with an FMR1 full mutation. Am. J. Med. Genet. 84, 245–249. doi: 10.1002/(sici)1096-8628(19990528)84:3<245::aid-ajmg16>3.0.co;2-u
Reyniers, E., Vits, L., De Boulle, K., Roy, B. V., Velzen, D. V., de Graaff, E., et al. (1993). The full mutation in the FMR-1 gene of male fragile X patients is absent in their sperm. Nat. Genet. 4, 143–146. doi: 10.1038/ng0693-143
Rezazadeh, M., Gharesouran, J., Mirabzadeh, A., Khorram Khorshid, H. R., Biglarian, A., and Ohadi, M. (2015). A primate-specific functional GTTT-repeat in the core promoter of CYTH4 is linked to bipolar disorder in human. Prog. Neuropsychopharmacol. Biol. Psychiatry 56, 161–167. doi: 10.1016/j.pnpbp.2014.09.001
Riedel, G., and Reymann, K. G. (1996). Metabotropic glutamate receptors in hippocampal long-term potentiation and learning and memory. Acta Physiol. Scand. 157, 1–19. doi: 10.1046/j.1365-201x.1996.484231000.x
Riley, D. E., and Krieger, J. N. (2009). Embryonic nervous system genes predominate in searches for dinucleotide simple sequence repeats flanked by conserved sequences. Gene 429, 74–79. doi: 10.1016/j.gene.2008.09.025
Roberts, J. E., Hennon, E. A., Price, J. R., Dear, E., Anderson, K., and Vandergrift, N. A. (2007). Expressive language during conversational speech in boys with fragile X syndrome. Am. J. Ment. Retard. 112, 1–17. doi: 10.1352/0895-8017(2007)112[1:eldcsi]2.0.CO;2
Robertson, C. E., Ratai, E. M., and Kanwisher, N. (2016). Reduced GABAergic action in the autistic brain. Curr. Biol. 26, 80–85. doi: 10.1016/j.cub.2015.11.019
Robinson, A. N., Roberts, J. E., Brady, N. C., McQuillin, S. D., and Warren, S. F. (2016). Physiological correlates of maternal responsivity in mothers of preschoolers with fragile X syndrome. Am. J. Intellect. Dev. Disabil. 121, 111–120. doi: 10.1352/1944-7558-121.2.111
Ronesi, J. A., Collins, K. A., Hays, S. A., Tsai, N.-P., Guo, W., Birnbaum, S. G., et al. (2012). Disrupted mGluR5-Homer scaffolds mediate abnormal mGluR5 signaling, circuit function and behavior in a mouse model of Fragile X Syndrome. Nat. Neurosci. 15:S431.
Ronesi, J. A., and Huber, K. M. (2008). Homer interactions are necessary for metabotropic glutamate receptor-induced long-term depression and translational activation. J. Neurosci. 28, 543–547. doi: 10.1523/JNEUROSCI.5019-07.2008
Rosati, A. G., Arre, A. M., Platt, M. L., and Santos, L. R. (2016). Rhesus monkeys show human-like changes in gaze following across the lifespan. Proc. Biol. Sci. 283:20160376. doi: 10.1098/rspb.2016.0376
Rotschafer, S., and Razak, K. (2013). Altered auditory processing in a mouse model of fragile X syndrome. Brain Res. 19, 12–24. doi: 10.1016/j.brainres.2013.02.038
Routh, B. N., Rathour, R. K., Baumgardner, M. E., Kalmbach, B. E., Johnston, D., and Brager, D. H. (2017). Increased transient Na+ conductance and action potential output in layer 2/3 prefrontal cortex neurons of the fmr1-/y mouse. J. Physiol. 595, 4431–4448. doi: 10.1113/JP274258
Sabanov, V., Braat, S., D’Andrea, L., Willemsen, R., Zeidler, S., Rooms, L., et al. (2016). Impaired GABAergic inhibition in the hippocampus of Fmr1 knockout mice. Neuropharmacology 116, 71–81. doi: 10.1016/j.neuropharm.2016.12.010
Sabaratnam, M., Vroegop, P. G., and Gangadharan, S. K. (2001). Epilepsy and EEG findings in 18 males with fragile X syndrome. Seizure 10, 60–63. doi: 10.1053/seiz.2000.0492
Sabino, F. C., Ribeiro, A. O., Tufik, S., Torres, L. B., Oliveira, J. A., Mello, L. E., et al. (2014). Evolutionary history of the PER3 variable number of tandem repeats (VNTR): idiosyncratic aspect of primate molecular circadian clock. PLoS One 9:e107198. doi: 10.1371/journal.pone.0107198
Saffary, R., and Xie, Z. (2011). FMRP regulates the transition from radial glial cells to intermediate progenitor cells during neocortical development. J. Neurosci. 31, 1427–1439. doi: 10.1523/JNEUROSCI.4854-10.2011
Saito, A. (2015). The marmoset as a model for the study of primate parental behavior. Neurosci. Res. 93, 99–109. doi: 10.1016/j.neures.2014.12.011
Salcedo-Arellano, M. J., Lozano, R., Tassone, F., Hagerman, R. J., and Saldarriaga, W. (2016). Alcohol use dependence in fragile X syndrome. Intractable Rare Dis. Res. 5, 207–213. doi: 10.5582/irdr.2016.01046
Saldarriaga, W., Tassone, F., González-Teshima, L. Y., Forero-Forero, J. V., Ayala-Zapata, S., and Hagerman, R. (2014). Fragile X syndrome. Colomb. Med. 45, 190–198.
Santa Maria, L., Pugin, A., Alliende, M. A., Aliaga, S., Curotto, B., Aravena, T., et al. (2014). FXTAS in an unmethylated mosaic male with fragile X syndrome from Chile. Clin. Genet. 86, 378–382. doi: 10.1111/cge.12278
Saré, R. M., Song, A., Loutaev, I., Cook, A., Maita, I., Lemons, A., et al. (2018). Negative effects of chronic rapamycin treatment on behavior in a mouse model of fragile X syndrome. Front. Mol. Neurosci. 10:452. doi: 10.3389/fnmol.2017.00452
Sasaki, E., Suemizu, H., Shimada, A., Hanazawa, K., Oiwa, R., Kamioka, M., et al. (2009). Generation of transgenic non-human primates with germline transmission. Nature 459, 523–527. doi: 10.1038/nature08090
Sato, K., Oiwa, R., Kumita, W., Henry, R., Sakuma, T., Ito, R., et al. (2016). Generation of a nonhuman primate model of severe combined immunodeficiency using highly efficient genome editing. Cell Stem Cell 19, 127–138. doi: 10.1016/j.stem.2016.06.003
Sawaya, S., Bagshaw, A., Buschiazzo, E., Kumar, P., Chowdhury, S., Black, M. A., et al. (2013). Microsatellite tandem repeats are abundant in human promoters and are associated with regulatory elements. PLoS One 8:e54710. doi: 10.1371/journal.pone.0054710
Sawaya, S. M., Bagshaw, A. T., Buschiazzo, E., and Gemmell, N. J. (2012). Promoter microsatellites as modulators of human gene expression. Adv. Exp. Med. Biol. 769, 41–54. doi: 10.1007/978-1-4614-5434-2_4
Sawicka, K., Pyronneau, A., Chao, M., Bennett, M. V., and Zukin, R. S. (2016). Elevated ERK/p90 ribosomal S6 kinase activity underlies audiogenic seizure susceptibility in fragile X mice. Proc. Natl. Acad. Sci. U S A 113, E6290–E6297. doi: 10.1073/pnas.1610812113
Scharf, S. H., Jaeschke, G., Wettstein, J. G., and Lindemann, L. (2015). Metabotropic glutamate receptor 5 as drug target for Fragile X syndrome. Curr. Opin. Pharmacol. 20, 124–134. doi: 10.1016/j.coph.2014.11.004
Schibler, L., Roig, A., Mahe, M. F., Laurent, P., Hayes, H., Rodolphe, F., et al. (2006). High-resolution comparative mapping among man, cattle and mouse suggests a role for repeat sequences in mammalian genome evolution. BMC Genomics 7:194. doi: 10.1186/1471-2164-7-194
Schiel, N., and Souto, A. (2017). The common marmoset: an overview of its natural history, ecology and behavior. Dev. Neurobiol. 77, 244–262. doi: 10.1002/dneu.22458
Schmidt, A., Morales-Prieto, D. M., Pastuschek, J., Fröhlich, K., and Markert, U. R. (2015). Only humans have human placentas: molecular differences between mice and humans. J. Reprod. Immunol. 108, 65–71. doi: 10.1016/j.jri.2015.03.001
Selby, L., Zhang, C., and Sun, Q. Q. (2007). Major defects in neocortical GABAergic inhibitory circuits in mice lacking the fragile X mental retardation protein. Neurosci. Lett. 412, 227–232. doi: 10.1016/j.neulet.2006.11.062
Semendeferi, K., Lu, A., Schenker, N., and Damasio, H. (2002). Humans and great apes share a large frontal cortex. Nat. Neurosci. 5, 272–276. doi: 10.1038/nn814
Semendeferi, K., Teffer, K., Buxhoeveden, D. P., Park, M. S., Bludau, S., Amunts, K., et al. (2011). Spatial organization of neurons in the frontal pole sets humans apart from great apes. Cereb. Cortex 21, 1485–1497. doi: 10.1093/cercor/bhQ211
Shang, Y., Wang, H., Mercaldo, V., Li, X., Chen, T., and Zhuo, M. (2009). Fragile X mental retardation protein is required for chemically-induced long-term potentiation of the hippocampus in adult mice. J. Neurochem. 111, 635–646. doi: 10.1111/j.1471-4159.2009.06314.x
Sharma, A., Hoeffer, C. A., Takayasu, Y., Miyawaki, T., McBride, S. M., Klann, E., et al. (2010). Dysregulation of mTOR signaling in fragile X syndrome. J. Neurosci. 30, 694–702. doi: 10.1523/JNEUROSCI.3696-09.2010
Sheridan, S. D., Theriault, K. M., Reis, S. A., Zhou, F., Madison, J. M., Daheron, L., et al. (2011). Epigenetic characterization of the FMR1 gene and aberrant neurodevelopment in human induced pluripotent stem cell models of fragile X syndrome. PLoS One 6:e26203. doi: 10.1371/journal.pone.0026203
Sidhu, H., Dansie, L. E., Hickmott, P. W., Ethell, D. W., and Ethell, I. M. (2014). Genetic removal of matrix metalloproteinase 9 rescues the symptoms of fragile X syndrome in a mouse model. J. Neurosci. 34, 9867–9879. doi: 10.1523/JNEUROSCI.1162-14.2014
Sigurdsson, T., and Duvarci, S. (2016). Hippocampal-prefrontal interactions in cognition, behavior and psychiatric disease. Front. Syst. Neurosci. 9:190. doi: 10.3389/fnsys.2015.00190
Simmons, Z. L., and Roney, J. R. (2011). Variation in CAG repeat length of the androgen receptor gene predicts variables associated with intrasexual competitiveness in human males. Horm. Behav. 60, 306–312. doi: 10.1016/j.yhbeh.2011.06.006
Siomi, H., Choi, M., Siomi, M. C., Nussbaum, R. L., and Dreyfuss, G. (1994). Essential role for KH domains in RNA binding: impaired RNA binding by a mutation in the KH domain of FMR1 that causes fragile X syndrome. Cell 77, 33–39. doi: 10.1016/0092-8674(94)90232-1
Smeets, H. J., Smits, A. P., Verheij, C. E., Theelen, J. P., Willemsen, R., van de Burgt, I., et al. (1995). Normal phenotype in two brothers with a full FMR1 mutation. Hum. Mol. Genet. 4, 2103–2108. doi: 10.1093/hmg/4.11.2103
Smith, L. E., Hong, J., Greenberg, J. S., and Mailick, M. R. (2016). Change in the behavioral phenotype of adolescents and adults with FXS: role of the family environment. J. Autism Dev. Disord. 46, 1824–1833. doi: 10.1007/s10803-016-2714-8
Smrzka, O. W., Faé, I., Stöger, R., Kurzbauer, R., Fischer, G. F., Henn, T., et al. (1995). Conservation of a maternal-specific methylation signal at the human IGF2R locus. Hum. Mol. Genet. 4, 1945–1952. doi: 10.1093/hmg/4.10.1945
Sousa, M. B., Albuquerque, A. C., Albuquerque Fda, S., Araujo, A., Yamamoto, M. E., and Arruda Mde, F. (2005). Behavioral strategies and hormonal profiles of dominant and subordinate common marmoset (Callithrix jacchus) females in wild monogamous groups. Am. J. Primatol. 67, 37–50. doi: 10.1002/ajp.20168
Spinelli, S., Pennanen, L., Dettling, A. C., Feldon, J., Higgins, G. A., and Pryce, C. R. (2004). Performance of the marmoset monkey on computerized tasks of attention and working memory. Cogn. Brain Res. 19, 123–137. doi: 10.1016/j.cogbrainres.2003.11.007
Street, S. E., Navarrete, A. F., Reader, S. M., and Laland, K. N. (2017). Coevolution of cultural intelligence, extended life history, sociality and brain size in primates. Proc. Natl. Acad. Sci. U S A 24:1620734114. doi: 10.1073/pnas.1620734114
Stuber, E. F., Baumgartner, C., Dingemanse, N. J., Kempenaers, B., and Mueller, J. C. (2016). Genetic correlates of individual differences in sleep behavior of free-living great tits (Parus major). G3 (Bethesda) 6, 599–607. doi: 10.1534/g3.115.024216
Su, T., Fan, H. X., Jiang, T., Sun, W. W., Den, W. Y., Gao, M. M., et al. (2011). Early continuous inhibition of group 1 mGlu signaling partially rescues dendritic spine abnormalities in the Fmr1 knockout mouse model for fragile X syndrome. Psychopharmacology 215, 291–300. doi: 10.1007/s00213-010-2130-2
Subramanian, P. S., Nelson, D. L., and Chinault, A. C. (1996). Large domains of apparent delayed replication timing associated with triplet repeat expansion at FRAXA and FRAXE. Am. J. Hum. Genet. 59, 407–416.
Sun, M. K., Hongpaisan, J., and Alkon, D. L. (2016). Rescue of synaptic phenotypes and spatial memory in young fragile X mice. J. Pharmacol. Exp. Ther. 357, 300–310. doi: 10.1124/jpet.115.231100
Sun, M. K., Hongpaisan, J., Lim, C. S., and Alkon, D. L. (2014). Bryostatin-1 restores hippocampal synapses and spatial learning and memory in adult fragile x mice. J. Pharmacol. Exp. Ther. 349, 393–401. doi: 10.1124/jpet.114.214098
Sutcliffe, J. S., Nelson, D. L., Zhang, F., Pieretti, M., Caskey, C. T., Saxe, D., et al. (1992). DNA methylation represses FMR-1 transcription in fragile X syndrome. Hum. Mol. Genet. 1, 397–400. doi: 10.1093/hmg/1.6.397
Suvrathan, A., Hoeffer, C. A., Wong, H., Klann, E., and Chattarji, S. (2010). Characterization and reversal of synaptic defects in the amygdala in a mouse model of fragile X syndrome. Proc. Natl. Acad. Sci. U S A 107, 11591–11596. doi: 10.1073/pnas.1002262107
Suzumori, K., Yamauchi, M., Seki, N., Kondo, I., and Hori, T. (1993). Prenatal diagnosis of a hypermethylated full fragile X mutation in chorionic villi of a male fetus. J. Med. Genet. 30, 785–787. doi: 10.1136/jmg.30.9.785
Symonds, E. L., Peiris, M., Page, A. J., Chia, B., Dogra, H., Masding, A., et al. (2015). Mechanisms of activation of mouse and human enteroendocrine cells by nutrients. Gut 64, 618–626. doi: 10.1136/gutjnl-2014-306834
Tabata, H., Yoshinaga, S., and Nakajima, K. (2012). Cytoarchitecture of mouse and human subventricular zone in developing cerebral neocortex. Exp. Brain Res. 216, 161–168. doi: 10.1007/s00221-011-2933-3
Tabolacci, E., Mancano, G., Lanni, S., Palumbo, F., Goracci, M., Ferre, F., et al. (2016a). Genome-wide methylation analysis demonstrates that 5-aza-2-deoxycytidine treatment does not cause random DNA demethylation in fragile X syndrome cells. Epigenetics Chromatin 9, 016–0060. doi: 10.1186/s13072-016-0060-x
Tabolacci, E., Palumbo, F., Nobile, V., and Neri, G. (2016b). Transcriptional reactivation of the FMR1 gene. A possible approach to the treatment of the fragile X syndrome. Genes 7:E49. doi: 10.3390/genes7080049
Tabolacci, E., Moscato, U., Zalfa, F., Bagni, C., Chiurazzi, P., and Neri, G. (2008). Epigenetic analysis reveals a euchromatic configuration in the FMR1 unmethylated full mutations. Eur. J. Hum. Genet. 16, 1487–1498. doi: 10.1038/ejhg.2008.130
Tabolacci, E., Pietrobono, R., Moscato, U., Oostra, B. A., Chiurazzi, P., and Neri, G. (2005). Differential epigenetic modifications in the FMR1 gene of the fragile X syndrome after reactivating pharmacological treatments. Eur. J. Hum. Genet. 13, 641–648. doi: 10.1038/sj.ejhg.5201393
Takahashi, D. Y., Fenley, A. R., and Ghazanfar, A. A. (2016). Early development of turn-taking with parents shapes vocal acoustics in infant marmoset monkeys. Philos. Trans. R. Soc. Lond. B Biol. Sci. 371:20150370. doi: 10.1098/rstb.2015.0370
Takahashi, D. Y., Fenley, A. R., Teramoto, Y., Narayanan, D. Z., Borjon, J. I., Holmes, P., et al. (2015). LANGUAGE DEVELOPMENT. The developmental dynamics of marmoset monkey vocal production. Science 349, 734–738. doi: 10.1126/science.aab1058
Takahashi, D. Y., Liao, D. A., and Ghazanfar, A. A. (2017). Vocal learning via social reinforcement by infant marmoset monkeys. Curr. Biol. 27, 1844–1852. doi: 10.1016/j.cub.2017.05.004
Takahashi, D. Y., Narayanan, D. Z., and Ghazanfar, A. A. (2013). Coupled oscillator dynamics of vocal turn-taking in monkeys. Curr. Biol. 23, 2162–2168. doi: 10.1016/j.cub.2013.09.005
Takahashi, M., Ueno, Y., and Fujita, K. (2015). Inference in a social context: a comparative study of capuchin monkeys (Cebus apella), tree shrews (Tupaia belangeri), hamsters (Mesocricetus auratus), and rats (Rattus norvegicus). J. Comp. Psychol. 129, 402–411. doi: 10.1037/a0039732
Takemoto, A., Izumi, A., Miwa, M., and Nakamura, K. (2011). Development of a compact and general-purpose experimental apparatus with a touch-sensitive screen for use in evaluating cognitive functions in common marmosets. J. Neurosci. Methods 199, 82–86. doi: 10.1016/j.jneumeth.2011.04.029
Tan, W., Schauder, C., Naryshkina, T., Minakhina, S., and Steward, R. (2016). Zfrp8 forms a complex with fragile-X mental retardation protein and regulates its localization and function. Dev. Biol. 410, 202–212. doi: 10.1016/j.ydbio.2015.12.008
Tang, A. H., and Alger, B. E. (2015). Homer protein-metabotropic glutamate receptor binding regulates endocannabinoid signaling and affects hyperexcitability in a mouse model of fragile X syndrome. J. Neurosci. 35, 3938–3945. doi: 10.1523/JNEUROSCI.4499-14.2015
Tang, B., Wang, T., Wan, H., Han, L., Qin, X., Zhang, Y., et al. (2015). Fmr1 deficiency promotes age-dependent alterations in the cortical synaptic proteome. Proc. Natl. Acad. Sci. U S A 112, E4697–E4706. doi: 10.1073/pnas.1502258112
Tang, L., Zeng, Y., Du, H., Gong, M., Peng, J., Zhang, B., et al. (2017). CRISPR/Cas9-mediated gene editing in human zygotes using Cas9 protein. Mol. Genet. Genomics 292, 525–533. doi: 10.1007/s00438-017-1299-z
Taqi, M. M., Wärmländer, S. K. T. S., Yamskova, O., Madani, F., Bazov, I., Luo, J., et al. (2012). Conformation effects of CpG methylation on single-stranded DNA oligonucleotides: analysis of the opioid peptide dynorphin-coding sequences. PLoS One 7:e39605. doi: 10.1371/journal.pone.0039605
Tardif, S. D., Coleman, K., Hobbs, T. R., and Lutz, C. (2013). IACUC review of nonhuman primate research. ILAR J. 54, 234–245. doi: 10.1093/ilar/ilt040
Tassone, F., Hagerman, R. J., Loesch, D. Z., Lachiewicz, A., Taylor, A. K., and Hagerman, P. J. (2000a). Fragile X males with unmethylated, full mutation trinucleotide repeat expansions have elevated levels of FMR1 messenger RNA. Am. J. Med. Genet. 94, 232–236. doi: 10.1002/1096-8628(20000918)94:3<232::aid-ajmg9>3.0.co;2-h
Tassone, F., Hagerman, R. J., Taylor, A. K., Gane, L. W., Godfrey, T. E., and Hagerman, P. J. (2000b). Elevated levels of FMR1 mRNA in carrier males: a new mechanism of involvement in the fragile-X syndrome. Am. J. Hum. Genet. 66, 6–15. doi: 10.1086/302720
Tassone, F., Hagerman, R. J., Taylor, A. K., and Hagerman, P. J. (2001). A majority of fragile X males with methylated, full mutation alleles have significant levels of FMR1 messenger RNA. J. Med. Genet. 38, 453–456. doi: 10.1136/jmg.38.7.453
Taylor, A. K., Tassone, F., Dyer, P. N., Hersch, S. M., Harris, J. B., Greenough, W. T., et al. (1999). Tissue heterogeneity of the FMR1 mutation in a high-functioning male with fragile X syndrome. Am. J. Med. Genet. 84, 233–239. doi: 10.1002/(sici)1096-8628(19990528)84:3<233::aid-ajmg14>3.3.co;2-y
Tebartz van Elst, L., Maier, S., Fangmeier, T., Endres, D., Mueller, G. T., Nickel, K., et al. (2014). Disturbed cingulate glutamate metabolism in adults with high-functioning autism spectrum disorder: evidence in support of the excitatory/inhibitory imbalance hypothesis. Mol. Psychiatry 19, 1314–1325. doi: 10.1038/mp.2014.62
Tecott, L. H. (2003). The genes and brains of mice and men. Am. J. Psychiatry 160, 646–656. doi: 10.1176/appi.ajp.160.4.646
Telias, M., Segal, M., and Ben-Yosef, D. (2013). Neural differentiation of Fragile X human Embryonic Stem Cells reveals abnormal patterns of development despite successful neurogenesis. Dev. Biol. 374, 32–45. doi: 10.1016/j.ydbio.2012.11.031
Thomas, A. M., Bui, N., Graham, D., Perkins, J. R., Yuva-Paylor, L. A., and Paylor, R. (2011). Genetic reduction of group 1 metabotropic glutamate receptors alters select behaviors in a mouse model for fragile X syndrome. Behav. Brain Res. 223, 310–321. doi: 10.1016/j.bbr.2011.04.049
Thomson, S. R., Seo, S. S., Barnes, S. A., Louros, S. R., Muscas, M., Dando, O., et al. (2017). Cell-type-specific translation profiling reveals a novel strategy for treating fragile X syndrome. Neuron 95, 550–563. doi: 10.1016/j.neuron.2017.07.013
Tian, M., Zeng, Y., Hu, Y., Yuan, X., Liu, S., Li, J., et al. (2015). 7, 8-Dihydroxyflavone induces synapse expression of AMPA GluA1 and ameliorates cognitive and spine abnormalities in a mouse model of fragile X syndrome. Neuropharmacology 89, 43–53. doi: 10.1016/j.neuropharm.2014.09.006
Tian, Y., Yang, C., Shang, S., Cai, Y., Deng, X., Zhang, J., et al. (2017). Loss of FMRP impaired hippocampal long-term plasticity and spatial learning in rats. Front. Mol. Neurosci. 10:269. doi: 10.3389/fnmol.2017.00269
Till, S. M., Asiminas, A., Jackson, A. D., Katsanevaki, D., Barnes, S. A., Osterweil, E. K., et al. (2015). Conserved hippocampal cellular pathophysiology but distinct behavioural deficits in a new rat model of FXS. Hum. Mol. Genet. 24, 5977–5984. doi: 10.1093/hmg/ddv299
Todd, P. K., Oh, S. Y., Krans, A., He, F., Sellier, C., Frazer, M., et al. (2013). CGG repeat-associated translation mediates neurodegeneration in fragile X tremor ataxia syndrome. Neuron 78, 440–455. doi: 10.1016/j.neuron.2013.03.026
Toft, A. K., Lundbye, C. J., and Banke, T. G. (2016). Dysregulated NMDA-receptor signaling inhibits long-term depression in a mouse model of fragile X syndrome. J. Neurosci. 36, 9817–9827. doi: 10.1523/JNEUROSCI.3038-15.2016
Tokuno, H., Watson, C., Roberts, A., Sasaki, E., and Okano, H. (2015). Marmoset neuroscience. Neurosci. Res. 93, 1–2. doi: 10.1016/j.neures.2015.03.001
Truszkowski, T. L., James, E. J., Hasan, M., Wishard, T. J., Liu, Z., Pratt, K. G., et al. (2016). Fragile X mental retardation protein knockdown in the developing Xenopus tadpole optic tectum results in enhanced feedforward inhibition and behavioral deficits. Neural. Dev. 11:14. doi: 10.1186/s13064-016-0069-7
Tu, Z., Yang, W., Yan, S., Yin, A., Gao, J., Liu, X., et al. (2017). Promoting Cas9 degradation reduces mosaic mutations in non-human primate embryos. Sci. Rep. 7:42081. doi: 10.1038/srep42081
Tzschach, A., and Ropers, H.-H. (2007). Genetik der mentalen Retardierung. Dtsch Arztebl Int. 104, 1400–1405. Available online at: https://www.aerzteblatt.de/archiv/55715/Genetik-der-mentalen-Retardierung
Udagawa, T., Farny, N. G., Jakovcevski, M., Kaphzan, H., Alarcon, J. M., Anilkumar, S., et al. (2013). Genetic and acute CPEB1 depletion ameliorate fragile X pathophysiology. Nat. Med. 19, 1473–1477. doi: 10.1038/nm.3353
Urbach, A., Bar-Nur, O., Daley, G. Q., and Benvenisty, N. (2010). Differential modeling of fragile X syndrome by human embryonic stem cells and induced pluripotent stem cells. Cell Stem Cell 6, 407–411. doi: 10.1016/j.stem.2010.04.005
Usdin, K., and Kumari, D. (2015). Repeat-mediated epigenetic dysregulation of the FMR1 gene in the fragile X-related disorders. Front. Genet. 6:192. doi: 10.3389/fgene.2015.00192
Usdin, K., and Woodford, K. J. (1995). CGG repeats associated with DNA instability and chromosome fragility form structures that block DNA synthesis in vitro. Nucleic Acids Res. 23, 4202–4209. doi: 10.1093/nar/23.20.4202
Uzunova, G., Pallanti, S., and Hollander, E. (2016). Excitatory/inhibitory imbalance in autism spectrum disorders: implications for interventions and therapeutics. World J. Biol. Psychiatry 17, 174–186. doi: 10.3109/15622975.2015.1085597
Valomon, A., Holst, S. C., Bachmann, V., Viola, A. U., Schmidt, C., Zürcher, J., et al. (2014). Genetic polymorphisms of DAT1 and COMT differentially associate with actigraphy-derived sleep-wake cycles in young adults. Chronobiol. Int. 31, 705–714. doi: 10.3109/07420528.2014.896376
Van Dam, D., D’Hooge, R., Hauben, E., Reyniers, E., Gantois, I., Bakker, C. E., et al. (2000). Spatial learning, contextual fear conditioning and conditioned emotional response in Fmr1 knockout mice. Behav. Brain Res. 117, 127–136. doi: 10.1016/s0166-4328(00)00296-5
Van Dam, D., Errijgers, V., Kooy, R. F., Willemsen, R., Mientjes, E., Oostra, B. A., et al. (2005). Cognitive decline, neuromotor and behavioural disturbances in a mouse model for fragile-X-associated tremor/ataxia syndrome (FXTAS). Behav. Brain Res. 162, 233–239. doi: 10.1016/j.bbr.2005.03.007
Van der Molen, M. J., Huizinga, M., Huizenga, H. M., Ridderinkhof, K. R., Van der Molen, M. W., Hamel, B. J., et al. (2010). Profiling Fragile X Syndrome in males: strengths and weaknesses in cognitive abilities. Res. Dev. Disabil. 31, 426–439. doi: 10.1016/j.ridd.2009.10.013
Van der Molen, M. J., Stam, C. J., and Van der Molen, M. W. (2014). Resting-state EEG oscillatory dynamics in fragile X syndrome: abnormal functional connectivity and brain network organization. PLoS One 9:e88451. doi: 10.1371/journal.pone.0088451
Van der Molen, M. J., and Van der Molen, M. W. (2013). Reduced α and exaggerated theta power during the resting-state EEG in fragile X syndrome. Biol. Psychol. 92, 216–219. doi: 10.1016/j.biopsycho.2012.11.013
Van der Weyden, L., and Adams, D. J. (2013). Cancer of mice and men: old twists and new tails. J. Pathol. 230, 4–16. doi: 10.1002/path.4184
Vandamme, T. F. (2014). Use of rodents as models of human diseases. J. Pharm. Bioallied Sci. 6, 2–9. doi: 10.4103/0975-7406.124301
Vasile, F., Dossi, E., and Rouach, N. (2017). Human astrocytes: structure and functions in the healthy brain. Brain Struct. Funct. 222, 2017–2029. doi: 10.1007/s00429-017-1383-5
Veeraragavan, S., Bui, N., Perkins, J. R., Yuva-Paylor, L. A., and Paylor, R. (2011). The modulation of fragile X behaviors by the muscarinic M4 antagonist, tropicamide. Behav. Neurosci. 125, 783–790. doi: 10.1037/a0025202
Verkerk, A. J. M. H., Pieretti, M., Sutcliffe, J. S., Fu, Y.-H., Kuhl, D. P. A., Pizzuti, A., et al. (1991). Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 65, 905–914. doi: 10.1016/0092-8674(91)90397-H
Verrico, C. D., Liu, S., Asafu-Adjei, J. K., Sampson, A. R., Bradberry, C. W., and Lewis, D. A. (2011). Acquisition and baseline performance of working memory tasks by adolescent rhesus monkeys. Brain Res. 10, 91–104. doi: 10.1016/j.brainres.2010.12.081
Vinces, M. D., Legendre, M., Caldara, M., Hagihara, M., and Verstrepen, K. J. (2009). Unstable tandem repeats in promoters confer transcriptional evolvability. Science 324, 1213–1216. doi: 10.1126/science.1170097
Vinueza Veloz, M. F., Buijsen, R. A., Willemsen, R., Cupido, A., Bosman, L. W., Koekkoek, S. K., et al. (2012). The effect of an mGluR5 inhibitor on procedural memory and avoidance discrimination impairments in Fmr1 KO mice. Genes Brain Behav. 11, 325–331. doi: 10.1111/j.1601-183X.2012.00763.x
Vislay, R. L., Martin, B. S., Olmos-Serrano, J. L., Kratovac, S., Nelson, D. L., Corbin, J. G., et al. (2013). Homeostatic responses fail to correct defective amygdala inhibitory circuit maturation in fragile X syndrome. J. Neurosci. 33, 7548–7558. doi: 10.1523/JNEUROSCI.2764-12.2013
Voelkl, B., and Huber, L. (2007). Imitation as faithful copying of a novel technique in marmoset monkeys. PLoS One 2:e611. doi: 10.1371/journal.pone.0000611
Volk, L. J., Pfeiffer, B. E., Gibson, J. R., and Huber, K. M. (2007). Multiple Gq-coupled receptors converge on a common protein synthesis-dependent long-term depression that is affected in fragile X syndrome mental retardation. J. Neurosci. 27, 11624–11634. doi: 10.1523/JNEUROSCI.2266-07.2007
Volle, C. B., and Delaney, S. (2013). AGG/CCT interruptions affect nucleosome formation and positioning of healthy-length CGG/CCG triplet repeats. BMC Biochem. 14:33. doi: 10.1186/1471-2091-14-33
Votinov, M., Pripfl, J., Windischberger, C., Moser, E., Sailer, U., and Lamm, C. (2015). A functional polymorphism in the prodynorphin gene affects cognitive flexibility and brain activation during reversal learning. Front. Behav. Neurosci. 9:172. doi: 10.3389/fnbeh.2015.00172
Wahlstrom-Helgren, S., and Klyachko, V. A. (2015). GABAB receptor-mediated feed-forward circuit dysfunction in the mouse model of fragile X syndrome. J. Physiol. 593, 5009–5024. doi: 10.1113/JP271190
Wallingford, J., Scott, A. L., Rodrigues, K., and Doering, L. C. (2017). Altered developmental expression of the astrocyte-secreted factors hevin and sparc in the fragile X mouse model. Front. Mol. Neurosci. 10:268. doi: 10.3389/fnmol.2017.00268
Wang, J., Ethridge, L. E., Mosconi, M. W., White, S. P., Binder, D. K., Pedapati, E. V., et al. (2017). A resting EEG study of neocortical hyperexcitability and altered functional connectivity in fragile X syndrome. J. Neurodev. Disord. 9:11. doi: 10.1186/s11689-017-9191-z
Wang, J. Y., Hessl, D., Iwahashi, C., Cheung, K., Schneider, A., Hagerman, R. J., et al. (2013). Influence of the fragile X mental retardation (FMR1) gene on the brain and working memory in men with normal FMR1 alleles. Neuroimage 65, 288–298. doi: 10.1016/j.neuroimage.2012.09.075
Wang, J. M., Koldewyn, K., Hashimoto, R., Schneider, A., Le, L., Tassone, F., et al. (2012). Male carriers of the FMR1 premutation show altered hippocampal-prefrontal function during memory encoding. Front. Hum. Neurosci. 6:297. doi: 10.3389/fnhum.2012.00297
Wang, H., Ku, L., Osterhout, D. J., Li, W., Ahmadian, A., Liang, Z., et al. (2004). Developmentally-programmed FMRP expression in oligodendrocytes: a potential role of FMRP in regulating translation in oligodendroglia progenitors. Hum. Mol. Genet. 13, 79–89. doi: 10.1093/hmg/ddh009
Wang, X., Snape, M., Klann, E., Stone, J. G., Singh, A., Petersen, R. B., et al. (2012). Activation of the extracellular signal-regulated kinase pathway contributes to the behavioral deficit of fragile X-syndrome. J. Neurochem. 121, 672–679. doi: 10.1111/j.1471-4159.2012.07722.x
Waterston, R. H., Lindblad-Toh, K., Birney, E., Rogers, J., Abril, J. F., Agarwal, P., et al. (2002). Initial sequencing and comparative analysis of the mouse genome. Nature 420, 520–562. doi: 10.1038/nature01262
Watson, C. F., and Caldwell, C. A. (2010). Neighbor effects in marmosets: social contagion of agonism and affiliation in captive Callithrix jacchus. Am. J. Primatol. 72, 549–558. doi: 10.1002/ajp.20805
Weiler, I. J., Irwin, S. A., Klintsova, A. Y., Spencer, C. M., Brazelton, A. D., Miyashiro, K., et al. (1997). Fragile X mental retardation protein is translated near synapses in response to neurotransmitter activation. Proc. Natl. Acad. Sci. U S A 94, 5395–5400. doi: 10.1073/pnas.94.10.5395
Weisman-Shomer, P., Cohen, E., and Fry, M. (2000). Interruption of the fragile X syndrome expanded sequence d(CGG)(n) by interspersed d(AGG) trinucleotides diminishes the formation and stability of d(CGG)(n) tetrahelical structures. Nucleic Acids Res. 28, 1535–1541. doi: 10.1093/nar/28.7.1535
Weiss, D., and Hampshire, V. (2015). Primate wellness exams. Lab Anim. (NY) 44, 342–344. doi: 10.1038/laban.835
Weiss, K., Orr-Urtreger, A., Kaplan Ber, I., Naiman, T., Shomrat, R., Bardugu, E., et al. (2014). Ethnic effect on FMR1 carrier rate and AGG repeat interruptions among Ashkenazi women. Genet. Med. 16, 940–944. doi: 10.1038/gim.2014.64
Weitzmann, M. N., Woodford, K. J., and Usdin, K. (1997). DNA secondary structures and the evolution of hypervariable tandem arrays. J. Biol. Chem. 272, 9517–9523. doi: 10.1074/jbc.272.14.9517
Wells, P. G., Bhatia, S., Drake, D. M., and Miller-Pinsler, L. (2016). Fetal oxidative stress mechanisms of neurodevelopmental deficits and exacerbation by ethanol and methamphetamine. Birth Defects Res. C Embryo Today 108, 108–130. doi: 10.1002/bdrc.21134
Weng, N., Weiler, I., Sumis, A., Berry-Kravis, E., and Greenough, W. (2008). Early-phase ERK activation as a biomarker for metabolic status in fragile X syndrome. Am. J. Med. Genet. B Neuropsychiatr. Genet. 147B, 1253–1257. doi: 10.1002/ajmg.b.30765
Wenzel, H. J., Hunsaker, M. R., Greco, C. M., Willemsen, R., and Berman, R. F. (2010). Ubiquitin-positive intranuclear inclusions in neuronal and glial cells in a mouse model of the fragile X premutation. Brain Res. 8, 155–166. doi: 10.1016/j.brainres.2009.12.077
Westmark, C. J. (2017). Commentary: depletion of the fragile X mental retardation protein in embryonic stem cells alters the kinetics of neurogenesis. Front. Mol. Neurosci. 10:29. doi: 10.3389/fnmol.2017.00029
Westmark, C. J., Chuang, S. C., Hays, S. A., Filon, M. J., Ray, B. C., Westmark, P. R., et al. (2016). APP causes hyperexcitability in fragile X mice. Front. Mol. Neurosci. 9:147. doi: 10.3389/fnmol.2016.00147
Westmark, C. J., Westmark, P. R., O’Riordan, K. J., Ray, B. C., Hervey, C. M., Salamat, M. S., et al. (2011). Reversal of fragile X phenotypes by manipulation of AβPP/Aβ levels in Fmr1KO mice. PLoS One 6:e26549. doi: 10.1371/journal.pone.0026549
Willemsen, R., Bontekoe, C. J., Severijnen, L. A., and Oostra, B. A. (2002). Timing of the absence of FMR1 expression in full mutation chorionic villi. Hum. Genet. 110, 601–605. doi: 10.1007/s00439-002-0723-5
Willemsen, R., Hoogeveen-Westerveld, M., Reis, S., Holstege, J., Severijnen, L. A., Nieuwenhuizen, I. M., et al. (2003). The FMR1 CGG repeat mouse displays ubiquitin-positive intranuclear neuronal inclusions; implications for the cerebellar tremor/ataxia syndrome. Hum. Mol. Genet. 12, 949–959. doi: 10.1093/hmg/ddg114
Wilson, B. M., and Cox, C. L. (2007). Absence of metabotropic glutamate receptor-mediated plasticity in the neocortex of fragile X mice. Proc. Natl. Acad. Sci. U S A 104, 2454–2459. doi: 10.1073/pnas.0610875104
Wilson, B., Kikuchi, Y., Sun, L., Hunter, D., Dick, F., Smith, K., et al. (2015a). Auditory sequence processing reveals evolutionarily conserved regions of frontal cortex in macaques and humans. Nat. Commun. 6:8901. doi: 10.1038/ncomms9901
Wilson, B., Smith, K., and Petkov, C. I. (2015b). Mixed-complexity artificial grammar learning in humans and macaque monkeys: evaluating learning strategies. Eur. J. Neurosci. 41, 568–578. doi: 10.1111/ejn.12834
Wilson, B., Slater, H., Kikuchi, Y., Milne, A. E., Marslen-Wilson, W. D., Smith, K., et al. (2013). Auditory artificial grammar learning in macaque and marmoset monkeys. J. Neurosci. 33, 18825–18835. doi: 10.1523/jneurosci.2414-13.2013
Wittkopp, P. J., and Kalay, G. (2011). Cis-regulatory elements: molecular mechanisms and evolutionary processes underlying divergence. Nat. Rev. Genet. 13, 59–69. doi: 10.1038/nrg3095
Wöhrle, D., Hennig, I., Vogel, W., and Steinbach, P. (1993). Mitotic stability of fragile X mutations in differentiated cells indicates early post-conceptional trinucleotide repeat expansion. Nat. Genet. 4, 140–142. doi: 10.1038/ng0693-140
Wöhrle, D., Salat, U., Hameister, H., Vogel, W., and Steinbach, P. (2001). Demethylation, reactivation and destabilization of human fragile X full-mutation alleles in mouse embryocarcinoma cells. Am. J. Hum. Genet. 69, 504–515. doi: 10.1086/322739
Wu, C. L., Huang, L. Y., and Chang, C. L. (2017). Linking arsenite- and cadmium-generated oxidative stress to microsatellite instability in vitro and in vivo. Free Radic. Biol. Med. 112, 12–23. doi: 10.1016/j.freeradbiomed.2017.07.006
Wu, D., Zhai, Q., and Shi, X. (2006). Alcohol-induced oxidative stress and cell responses. J. Gastroenterol. Hepatol. 21, S26–S29. doi: 10.1111/j.1440-1746.2006.04589.x
Xu, H., Rosales-Reynoso, M. A., Barros-Núñez, P., and Peprah, E. (2013). DNA repair/replication transcripts are down regulated in patients with fragile X syndrome. BMC Res. Notes 6:90. doi: 10.1186/1756-0500-6-90
Xu, Z. H., Yang, Q., Feng, B., Liu, S. B., Zhang, N., Xing, J. H., et al. (2012a). Group I mGluR antagonist rescues the deficit of D1-induced LTP in a mouse model of fragile X syndrome. Mol. Neurodegener. 7:24. doi: 10.1186/1750-1326-7-24
Xu, Z. H., Yang, Q., Ma, L., Liu, S. B., Chen, G. S., Wu, Y. M., et al. (2012b). Deficits in LTP induction by 5-HT2A receptor antagonist in a mouse model for fragile X syndrome. PLoS One 7:e48741. doi: 10.1371/journal.pone.0048741
Yamamoto, M. E., Araujo, A., Arruda Mde, F., Lima, A. K., Siqueira Jde, O., and Hattori, W. T. (2014). Male and female breeding strategies in a cooperative primate. Behav. Processes 109, 27–33. doi: 10.1016/j.beproc.2014.06.009
Yamazaki, Y., Iriki, A., and Watanabe, S. (2011). Modulation of physical understanding by common marmosets (Callithrix jacchus). Anim. Cogn. 14, 175–186. doi: 10.1007/s10071-010-0351-8
Yamazaki, Y., Saiki, M., Inada, M., Iriki, A., and Watanabe, S. (2014). Transposition and its generalization in common marmosets. J. Exp. Psychol. Anim. Learn. Cogn. 40, 317–326. doi: 10.1037/xan0000027
Yan, Q. J., Asafo-Adjei, P. K., Arnold, H. M., Brown, R. E., and Bauchwitz, R. P. (2004). A phenotypic and molecular characterization of the fmr1-tm1Cgr fragile X mouse. Genes Brain Behav. 3, 337–359. doi: 10.1111/j.1601-183x.2004.00087.x
Yan, Q. J., Rammal, M., Tranfaglia, M., and Bauchwitz, R. P. (2005). Suppression of two major Fragile X Syndrome mouse model phenotypes by the mGluR5 antagonist MPEP. Neuropharmacology 49, 1053–1066. doi: 10.1016/j.neuropharm.2005.06.004
Yang, Q., Yang, L., Zhang, K., Guo, Y. Y., Liu, S. B., Wu, Y. M., et al. (2015). Increased coupling of caveolin-1 and estrogen receptor α contributes to the fragile X syndrome. Ann. Neurol. 77, 618–636. doi: 10.1002/ana.24358
Yang, S., Park, J. S., Kirkwood, A., and Bao, S. (2014). Failed stabilization for long-term potentiation in the auditory cortex of FMR1 knockout mice. PLoS One 9:e104691. doi: 10.1371/journal.pone.0104691
Yau, S. Y., Bostrom, C. A., Chiu, J., Fontaine, C. J., Sawchuk, S., Meconi, A., et al. (2016). Impaired bidirectional NMDA receptor dependent synaptic plasticity in the dentate gyrus of adult female Fmr1 heterozygous knockout mice. Neurobiol. Dis. 96, 261–270. doi: 10.1016/j.nbd.2016.09.012
Yrigollen, C. M., Durbin-Johnson, B., Gane, L., Nelson, D. L., Hagerman, R., Hagerman, P. J., et al. (2012). AGG interruptions within the maternal FMR1 gene reduce the risk of offspring with fragile X syndrome. Genet. Med. 14, 729–736. doi: 10.1038/gim.2012.34
Yrigollen, C. M., Martorell, L., Durbin-Johnson, B., Naudo, M., Genoves, J., Murgia, A., et al. (2014). AGG interruptions and maternal age affect FMR1 CGG repeat allele stability during transmission. J. Neurodev. Disord. 6:24. doi: 10.1186/1866-1955-6-24
Yrigollen, C. M., Mendoza-Morales, G., Hagerman, R., and Tassone, F. (2013). Transmission of an FMR1 premutation allele in a large family identified through newborn screening: the role of AGG interruptions. J. Hum. Genet. 58, 553–559. doi: 10.1038/jhg.2013.50
Yrigollen, C. M., Pacini, L., Nobile, V., Lozano, R., Hagerman, R. J., Bagni, C., et al. (2016). Clinical and molecular assessment in a female with fragile X syndrome and tuberous sclerosis. J. Genet. Disord. Genet. Rep. 5:139. doi: 10.4172/2327-5790.1000139
Yue, F., Cheng, Y., Breschi, A., Vierstra, J., Wu, W., Ryba, T., et al. (2014). A comparative encyclopedia of DNA elements in the mouse genome. Nature 515, 355–364. doi: 10.1038/nature13992
Yu, S., Mulley, J., Loesch, D., Turner, G., Donnelly, A., Gedeon, A., et al. (1992). Fragile-X syndrome: unique genetics of the heritable unstable element. Am. J. Hum. Genet. 50, 968–980.
Yuskaitis, C. J., Mines, M. A., King, M. K., Sweatt, J. D., Miller, C. A., and Jope, R. S. (2010). Lithium ameliorates altered glycogen synthase kinase-3 and behavior in a mouse model of fragile X syndrome. Biochem. Pharmacol. 79, 632–646. doi: 10.1016/j.bcp.2009.09.023
Zalfa, F., Giorgi, M., Primerano, B., Moro, A., Di Penta, A., Reis, S., et al. (2003). The fragile X syndrome protein FMRP associates with BC1 RNA and regulates the translation of specific mRNAs at synapses. Cell 112, 317–327. doi: 10.1016/s0092-8674(03)00079-5
Zang, J. B., Nosyreva, E. D., Spencer, C. M., Volk, L. J., Musunuru, K., Zhong, R., et al. (2009). A mouse model of the human fragile X syndrome I304N mutation. PLoS Genet. 5:e1000758. doi: 10.1371/journal.pgen.1000758
Zeier, Z., Kumar, A., Bodhinathan, K., Feller, J. A., Foster, T. C., and Bloom, D. C. (2009). Fragile X mental retardation protein replacement restores hippocampal synaptic function in a mouse model of fragile X syndrome. Gene Ther. 16, 1122–1129. doi: 10.1038/gt.2009.83
Zeng, J., Konopka, G., Hunt, B. G., Preuss, T. M., Geschwind, D., and Yi, S. V. (2012). Divergent whole-genome methylation maps of human and chimpanzee brains reveal epigenetic basis of human regulatory evolution. Am. J. Hum. Genet. 91, 455–465. doi: 10.1016/j.ajhg.2012.07.024
Zhang, N., and Ashizawa, T. (2017). RNA toxicity and foci formation in microsatellite expansion diseases. Curr. Opin. Genet. Dev. 44, 17–29. doi: 10.1016/j.gde.2017.01.005
Zhang, Y., Bonnan, A., Bony, G., Ferezou, I., Pietropaolo, S., Ginger, M., et al. (2014). Dendritic channelopathies contribute to neocortical and sensory hyperexcitability in Fmr1−/y mice. Nat. Neurosci. 17, 1701–1709. doi: 10.1038/nn.3864
Zhang, J., Hou, L., Klann, E., and Nelson, D. L. (2009). Altered hippocampal synaptic plasticity in the FMR1 gene family knockout mouse models. J. Neurophysiol. 101, 2572–2580. doi: 10.1152/jn.90558.2008
Zhang, Y. W., Luo, H. R., Ryder, O. A., and Zhang, Y. P. (2004). Evolution of the tandem repeats in thymidylate synthase enhancer region (TSER) in primates. Gene 338, 47–54. doi: 10.1016/j.gene.2004.05.018
Zhang, X. L., Pang, W., Hu, X. T., Li, J. L., Yao, Y. G., and Zheng, Y. T. (2014). Experimental primates and non-human primate (NHP) models of human diseases in China: current status and progress. Dongwuxue Yanjiu 35, 447–464. doi: 10.13918/j.issn.2095-8137.2014.6.447
Zhao, W., Chuang, S. C., Young, S. R., Bianchi, R., and Wong, R. K. (2015). Extracellular glutamate exposure facilitates group I mGluR-mediated epileptogenesis in the hippocampus. J. Neurosci. 35, 308–315. doi: 10.1523/jneurosci.1944-14.2015
Zhao, X. N., Kumari, D., Gupta, S., Wu, D., Evanitsky, M., Yang, W., et al. (2015). Mutsbeta generates both expansions and contractions in a mouse model of the Fragile X-associated disorders. Hum. Mol. Genet. 24, 7087–7096. doi: 10.1093/hmg/ddv408
Zhao, M. G., Toyoda, H., Ko, S. W., Ding, H. K., Wu, L. J., and Zhuo, M. (2005). Deficits in trace fear memory and long-term potentiation in a mouse model for fragile X syndrome. J. Neurosci. 25, 7385–7392. doi: 10.1523/jneurosci.1520-05.2005
Zhao, H., Tu, Z., Xu, H., Yan, S., Yan, H., Zheng, Y., et al. (2017). Altered neurogenesis and disrupted expression of synaptic proteins in prefrontal cortex of SHANK3-deficient non-human primate. Cell Res. 27, 1293–1297. doi: 10.1038/cr.2017.95
Zhao, X. N., and Usdin, K. (2014). Gender and cell-type-specific effects of the transcription-coupled repair protein, ERCC6/CSB, on repeat expansion in a mouse model of the fragile X-related disorders. Hum. Mutat. 35, 341–349. doi: 10.1002/humu.22495
Zhong, J., Chuang, S. C., Bianchi, R., Zhao, W., Lee, H., Fenton, A. A., et al. (2009). BC1 regulation of metabotropic glutamate receptor-mediated neuronal excitability. J. Neurosci. 29, 9977–9986. doi: 10.1523/jneurosci.3893-08.2009
Zhou, Q. (2014). Balancing the welfare: the use of non-human primates in research. Trends Genet. 30, 476–478. doi: 10.1016/j.tig.2014.09.005
Zhou, Y., Kumari, D., Sciascia, N., and Usdin, K. (2016). CGG-repeat dynamics and FMR1 gene silencing in fragile X syndrome stem cells and stem cell-derived neurons. Mol. Autism 7:42. doi: 10.1186/s13229-016-0105-9
Zumwalt, M., Ludwig, A., Hagerman, P. J., and Dieckmann, T. (2007). Secondary structure and dynamics of the r(CGG) repeat in the mRNA of the fragile X mental retardation 1 (FMR1) gene. RNA Biol. 4, 93–100. doi: 10.4161/rna.4.2.5039
Zuo, E., Cai, Y. J., Li, K., Wei, Y., Wang, B. A., Sun, Y., et al. (2017). One-step generation of complete gene knockout mice and monkeys by CRISPR/Cas9-mediated gene editing with multiple sgRNAs. Cell Res. 27, 933–945. doi: 10.1038/cr.2017.81
Keywords: Fragile X Syndrome, mouse model, FMR1, microsatellite instability, E/I balance, behavior and cognition, primates, autism spectrum disorders
Citation: Dahlhaus R (2018) Of Men and Mice: Modeling the Fragile X Syndrome. Front. Mol. Neurosci. 11:41. doi: 10.3389/fnmol.2018.00041
Received: 23 November 2017; Accepted: 31 January 2018;
Published: 15 March 2018.
Edited by:
Rolf Sprengel, Max Planck Institute for Medical Research (MPG), GermanyReviewed by:
Maria Vincenza Catania, Institute of Neurological Sciences, ItalyBarbara Bardoni, UMR7275 Institut de Pharmacologie Moléculaire et Cellulaire (IPMC), France
Zhengping Jia, Hospital for Sick Children, Canada
Copyright © 2018 Dahlhaus. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Regina Dahlhaus, cmVnaW5hLmRhaGxoYXVzQGgtYnJzLmRl
†Present address: Regina Dahlhaus, Hochschule Bonn Rhein Sieg, Rheinbach, Germany