 Davide Pozzi1,2*
Davide Pozzi1,2* Elisabetta Menna2,3*
Elisabetta Menna2,3* Alice Canzi1
Alice Canzi1 Genni Desiato2,4
Genni Desiato2,4 Cristina Mantovani2
Cristina Mantovani2 Michela Matteoli2,3*
Michela Matteoli2,3*- 1Department of Biomedical Sciences, Humanitas University, Rozzano, Italy
- 2Humanitas Clinical and Research Center, Rozzano, Italy
- 3Istituto di Neuroscienze, Consiglio Nazionale delle Ricerche, Milan, Italy
- 4School of Medicine and Surgery, University of Milan-Bicocca, Milan, Italy
In the last 15 years, groundbreaking genetic progress has underlined a convergence onto coherent synaptic pathways for most psychiatric and neurodevelopmental disorders, which are now collectively called “synaptopathies.” However, the modest size of inheritance detected so far indicates a multifactorial etiology for these disorders, underlining the key contribution of environmental effects to them. Inflammation is known to influence the risk and/or severity of a variety of synaptopathies. In particular, pro-inflammatory cytokines, produced and released in the brain by activated astrocytes and microglia, may play a pivotal role in these pathologies. Although the link between immune system activation and defects in cognitive processes is nowadays clearly established, the knowledge of the molecular mechanisms by which inflammatory mediators specifically hit synaptic components implicated in synaptopathies is still in its infancy. This review summarizes recent evidence showing that the pro-inflammatory cytokine interleukin-1β (IL-1β) specifically targets synaptopathy molecular substrate, leading to memory defects and pathological processes. In particular, we describe three specific pathways through which IL-1β affects (1) synaptic maintenance/dendritic complexity, (2) spine morphology, and (3) the excitatory/inhibitory balance. We coin the term immune synaptopathies to identify this class of diseases.
Introduction
A large amount of evidence about the genetic architecture of psychiatric and neurodevelopmental diseases has progressively accumulated in the last 15 years or so. The identified pathways typically involve proteins chiefly affecting synapse formation and maintenance, a discovery which has led to the concept of “synaptopathies” (Grant, 2012). The initial excitement raised by these findings has been, however, tempered by the awareness that understanding a given synaptopathy at the level of its genetic, molecular, and synaptic dysfunction is typically insufficient to explain the disease onset, which depends indeed on additional genetic, epigenetic, and environmental factors (Beutner et al., 2007; Oh-Nishi et al., 2010; De Chiara et al., 2012; Millan, 2013; Giovanoli et al., 2016).
Inflammation is increasingly recognized as a key factor influencing physiology and pathology in the immature and mature brain, which can be exposed to inflammation in connection with viral or bacterial prenatal or postnatal infections or as a result of sterile CNS insults (Hagberg et al., 2015). Extensive research is providing evidence that inflammation has long-term consequences and could speculatively affect the risk and/or severity of a variety of brain diseases, including autism spectrum disorders (ASDs), schizophrenia, and intellectual disabilities (IDs), which represent recognized synaptopathies (Fan et al., 2007; Dantzer et al., 2008; Najjar et al., 2013). Accordingly, prenatal and early postnatal infections have been associated with increased risk for a number of neurodevelopmental disorders (Brown and Derkits, 2010; Hagberg et al., 2012; Lipina et al., 2013; Wischhof et al., 2015; Graham et al., 2018).
Pro-inflammatory cytokines, including interleukin-1β (IL-1β), interleukin-6 (IL-6), and tumor necrosis factor alpha (TNFα), appear to be at the forefront in the communication between the immune and the nervous system, playing dual roles in mediating physiological and neuroprotective roles in normal brain function (Kushima et al., 1992; Yamada and Hatanaka, 1994; Akaneya et al., 1995; Hirota et al., 1996; Wagner, 1996; Gadient and Otten, 1997; Parish et al., 2002; Nakanishi et al., 2007; Heese, 2017) or being detrimental and associated with brain diseases, especially when present at elevated concentrations (Yan et al., 1992; Katila et al., 1994; Gadient and Otten, 1997; Samuelsson et al., 2006; Garay and McAllister, 2010; Ashwood et al., 2011; Suzuki et al., 2011; Wei et al., 2011; Erta et al., 2012; Chase et al., 2016; O’Keeffe, 2017). In particular, experiments performed in rodents have unequivocally demonstrated that inflammation correlates with defective learning and memory paradigms. As an example, influenza infection associated to elevated pro-inflammatory cytokines was found to alter neuronal morphology leading to cognitive impairment in adult mice (Jurgens et al., 2012; Hosseini et al., 2018). Although the link between immune system activation and defects in cognition is solidly established, the molecular underpinnings of this correlation are not completely clear. In particular, the knowledge of whether inflammatory mediators specifically hit synaptic components, previously identified by genetic studies as implicated in synaptopathies, is poor and somehow fragmentary.
Excessive IL-1β Impairs Neuronal Plasticity and Memory
Interleukin-1β is a potent inflammatory cytokine and a fundamental component of the innate immune response (Dinarello, 1996). Besides affecting several organs during inflammatory processes, IL-1β also exerts a number of diverse actions in the central nervous system (CNS) as important mediator of neuronal injury. Twenty years of research have indeed indicated that IL-1β is involved in several brain diseases, including multiple sclerosis (Lin and Edelson, 2017), Alzheimer disease (Griffin et al., 2006), epilepsy (Iori et al., 2016), stroke (Murray et al., 2015), and even neurodevelopmental disorders such as schizophrenia and autism (Soderlund et al., 2009; Girard et al., 2010; Krakowiak et al., 2017).
Despite the heterogeneity of the diseases in which IL-1β is involved, a growing body of evidence points toward a shared physiological process hit by the cytokine: cognition. This concept originally emerged as the result of a series of pioneer experiments showing that the intraperitoneal or intrahippocampal injection of the cytokine results in learning and memory defects (Oitzl et al., 1993; Gibertini et al., 1995; Barrientos et al., 2002). The detrimental effects of IL-1β on cognition were later confirmed using transgenic mice expressing the cytokine in an inducible manner (Hein et al., 2010) or upon endogenous IL-1β elevations evoked by infections (Gibertini et al., 1995; Barrientos et al., 2006; Chen et al., 2008), with the memory deficits being prevented, in the latter case, by intraventricular infusion of the naturally occurring interleukin-1 receptor antagonist (IL-1ra; Goshen et al., 2007; Barrientos et al., 2009; Frank et al., 2010).
In line with the induction of cognitive defects, excessive IL-1β affects long-term potentiation (LTP), the synaptic process which underlies learning and memory. Indeed, elevated levels of IL-1β inhibit LTP in several regions of the hippocampus, including CA1 (Bellinger et al., 1993; Ross et al., 2003), CA3 (Katsuki et al., 1990), and dentate gyrus (Murray and Lynch, 1998; Kelly et al., 2003). Of note, the synaptic potentiation processes are not only affected in different pathological scenarios, but also during aging, when the overproduction of IL-1β and/or a synapse-specific IL-1 receptor subunit reconfiguration may produce specific deficits in consolidation of hippocampus-dependent memory (Patterson, 2015; Prieto et al., 2015) and also during stress-related conditions, a particular pathophysiological state which might deeply affect immature brain (Barron et al., 2017; Depino, 2017; Schiavone and Trabace, 2017).
IL-1β Affects Synapse Structure and Function
An extensive series of evidence accumulated over the last 10 years has pointed to the concept that IL-1β directly affects synapse structure and function. Indeed, early experiments performed in primary cultured neurons exposed to recombinant IL-1β revealed a significant decline in the levels of the synaptic vesicle protein synaptophysin (Li et al., 2003) and in the number of synaptic sites (Mishra et al., 2012). The occurrence of deleterious cytokine effects on the synapse structure has been reported in different non-neurological disorders associated to increases of IL-1β levels, like sepsis and obesity, where mice show memory impairment and reduced number of hippocampal and cortical excitatory synapses, through a mechanism fully prevented by the addition of IL-1ra (Erion et al., 2014; Moraes et al., 2015).
Different mechanisms have been called into question to shed light on the association between excessive IL-1β and synaptic alterations, including the modulation of the mitogen-activated protein kinase (MAPK) pathway and the modification of trophic factor production, such as brain-derived neurotrophic factor (BDNF). These processes have been thoroughly analyzed in a previous review (Patterson, 2015). Conversely, it is still unclear whether IL-1β may directly act on neuronal proteins involved in modulating the structure and function of dendritic spines and known to be the molecular targets of synaptopathies. In this review, we will illustrate some recently reported data showing that IL-1β may directly interfere with synaptic processes known to be at the root of neurodevelopmental diseases. The research was performed through PubMed. Inclusion criteria were based on the indicated keywords. Only peer-reviewed original articles and reviews were considered.
IL1RAPL1 and Synapse Stabilization
Interleukin-1-receptor accessory protein like 1 (IL1RAPL1) is a member of the interleukin-1 receptor family. It is selectively expressed in the brain (Carrie et al., 1999; Born et al., 2000), where it is mainly localized at excitatory synapses. Mutations in the gene encoding for IL1RAPL1 have been found in patients with cognitive impairments ranging from non-syndromic ID to ASD (Ramos-Brossier et al., 2015). Indeed, IL1RAPL1 KO mice display a reduction of spine density in the cortex (Yasumura et al., 2014) and in the CA1 region of the hippocampus (Pavlowsky et al., 2010; Yasumura et al., 2014) and are characterized by altered excitation/inhibition (E/I) balance in the cerebellum and amygdala (Gambino et al., 2009; Houbaert et al., 2013). These molecular and functional alterations are associated with memory deficits (Houbaert et al., 2013; Yasumura et al., 2014). In neurons, IL1RAPL1 plays a role in presynaptic differentiation, in spine formation and stabilization (Pavlowsky et al., 2010; Valnegri et al., 2011; Yoshida et al., 2011; Ramos-Brossier et al., 2015), and in dendritic morphology (Montani et al., 2017). Also IL1RAPL1 C-terminus interacts with the neuronal calcium sensor-1 which regulates voltage-gated calcium channel activity (Gambino et al., 2009). Thus, IL1RAPL1 may promote excitatory synapse formation through two main mechanisms: (i) a trans-synaptic signaling pathway involving the receptor tyrosine phosphatase δ (PTPδ) and RhoGAP2 (Valnegri et al., 2011) and (ii) the control of synaptic localization of PSD-95 through c-Jun N-terminal kinase (JNK) activity and PSD-95 phosphorylation (Pavlowsky et al., 2010).
IL1RAPL1 mediates some of the effects of IL-1β in neurons (Figure 1, pathway 1), controlling, in particular, the cytokine effects on dendritic morphology, possibly through the involvement of JNK pathway (Pavlowsky et al., 2010; Montani et al., 2017). IL1RAPL1 represents therefore the first identified synaptic target of IL-1β. The primary role of the protein in neuronal and synaptic development, together with the observation that IL1RAPL1 is a synaptopathy-related gene, identifies this protein as one of the possible key targets of the immune-to-neuron communication mediated by IL-1β. The future identification of the precise mechanisms by which IL1RAPL1 modulates the activity of IL-1β could offer the opportunity to specifically interfere with the vicious, harmful cycle leading to synapse dysfunction.
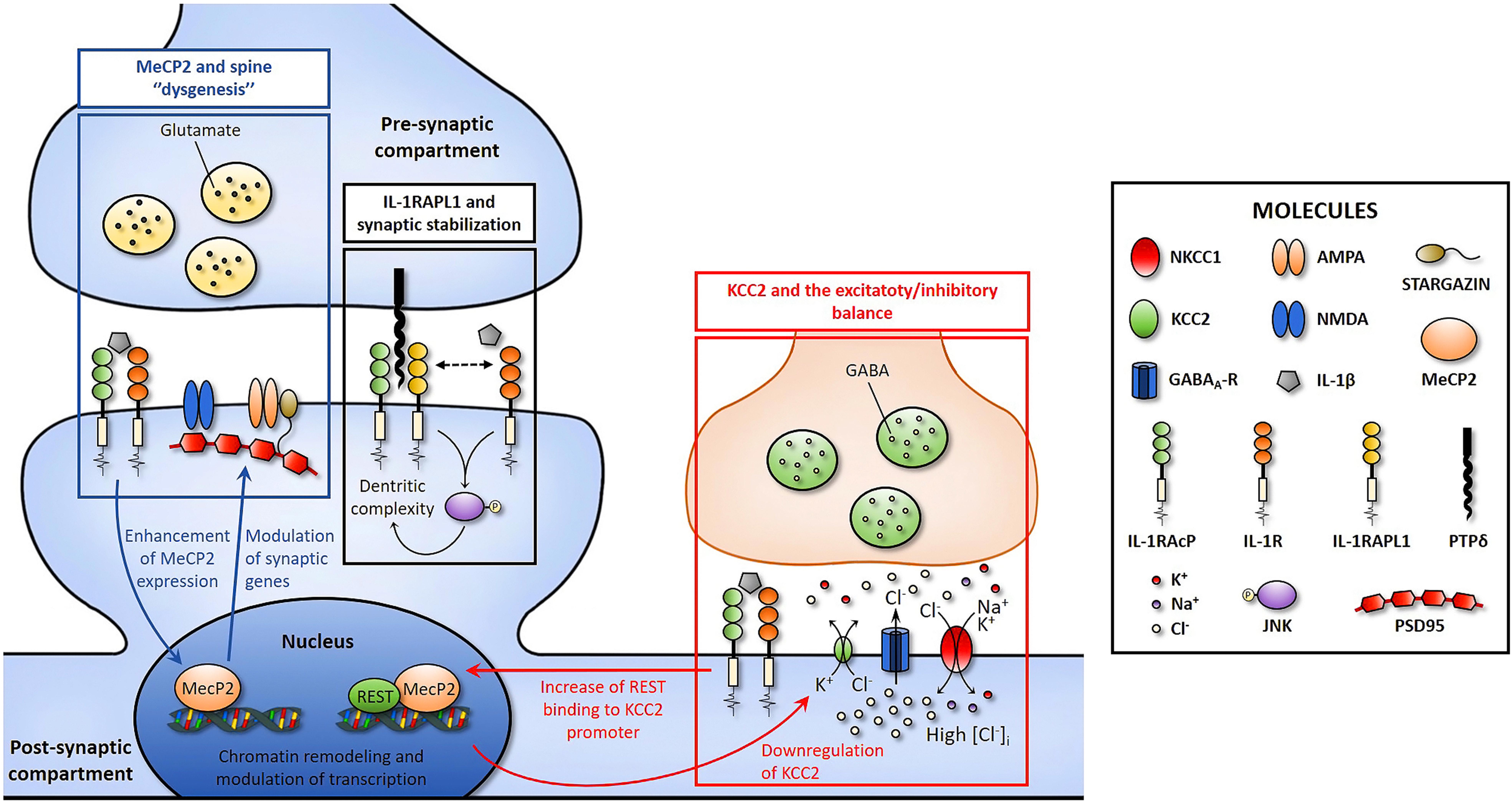
FIGURE 1. Cartoon depicting three possible pathways through which IL-1β might affect synaptic function and structure by acting on IL-R1 which is localized at synaptic site (Gardoni et al., 2011). Pathway 1: IL1RAPL1 mediates some of the effects of IL-1β in neurons, altering dendritic morphology. Pathway 2: IL-1β affects spine morphogenesis enhancing the expression of the transcription factor Mecp2, which in turn alters the expression of synaptic proteins via chromatin-remodeling mechanisms. Pathway3: IL-1β modulates the excitatory to inhibitory switch of GABA by reducing the expression of the chloride transporter KCC2, leading to E/I unbalance and higher susceptibility to seizures. Pathways 2 and 3 could be linked based on the REST/MeCP2 ability to bind KCC2 gene promoter and act as a repressor of the transporter transcription (see text for details).
MeCp2 and Spine “Dysgenesis”
Recent studies have exploited a genetic mouse model of IL-1β deregulation, the IL-1R8 KO mice, as a reliable and reproducible system for examining the effects of inflammation on synapse structure and function, also elucidating the molecular processes involved (Costello et al., 2011; Tomasoni et al., 2017). IL-1R8, also known as single Ig IL-1 related receptor (SIGIRR), reduces the activation of the Toll-like receptors (TLRs; protein sensors for microorganisms and tissue damage) and IL1R signaling pathways by intracellularly interfering with the association of adaptor molecules to the receptor complex including nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and JNK, thus leading to upregulation of IL-1 signaling. Mice lacking IL-1R8, previously shown to exhibit LTP deficits (Costello et al., 2011), have been used to investigate the mechanisms through which exaggerated inflammatory conditions impact synapse functions. IL-1R8KO neurons were found to display an increased number of immature, thin spines, and a decreased number of mature, mushroom spines, accompanied by reduction of PSD95 expression and impairment of synaptic plasticity (Tomasoni et al., 2017). The phenotype was rescued by IL-1ra, thus proving the direct involvement of IL-1β signaling (Tomasoni et al., 2017). The structural and functional alterations were found to be causally linked to upregulation of the mammalian target of rapamycin (mTOR) pathway and increased levels of the epigenetic regulator methyl CpG binding protein 2 (MeCP2), which were again normalized by IL-1Ra treatment (Tomasoni et al., 2017). The demonstration that enhanced IL-1β signaling increases the expression of MeCP2, thus negatively impacting synapse function, points to this factor as a potentially fundamental node linking inflammation and synaptic damage (Figure 1, pathway 2). Of note, alterations in the expression of MeCP2 are known to be responsible of neurodevelopmental diseases in humans: sporadic, loss-of-function mutations in the gene coding for MeCP2 result in Rett syndrome (Amir et al., 1999), while a double dosage of MeCP2 causes a severe developmental delay and ID, with even mild over-expression having a robust effect (Delobel et al., 1998; Van Esch et al., 2005). Interestingly, IL-1Ra reduces MeCP2 levels in wild-type neurons which, concomitantly, lose the ability to undergo LTP (Tomasoni et al., 2017). It appears therefore that the effects of IL-1β on neuronal plasticity follow a U-shaped dose–response curve, with levels in either excess or below the physiological range being deleterious to neuronal functions, which is exactly the case of MeCP2, whose levels need to be tightly regulated to guarantee a proper neuronal function (Cheng and Qiu, 2014; Lombardi et al., 2015). It will be important to assess whether the IL-1β-mediated modulation of MeCP2 also occurs in humans and to define the molecular mechanisms by which the cytokine modulates MeCP2 levels.
KCC2 and the Excitatory/Inhibitory Balance
Several lines of evidence have highlighted the capacity of IL-1β to promote an imbalance between excitation and inhibition, thus affecting neuronal network excitability. The first indications were obtained in the peripheral nervous system, where elevated levels of pro-inflammatory cytokines, including IL-1β, were found to alter neuronal excitability facilitating pain and hyperalgesia (Viviani et al., 2007; Schafers and Sorkin, 2008). Further studies conducted on the CNS have subsequently demonstrated that high levels of IL-1β, as occurring in neurological disorders such as multiple sclerosis and epilepsy, lead to an E/I imbalance, which might be responsible for – or contribute to – cognitive impairment (Vezzani et al., 2008; Rossi et al., 2012; Iori et al., 2016). Among the possible mechanisms, neuronal network hyperactivity may be the result of an altered excitatory-to-inhibitory switch of GABA signaling, leading to a reduced inhibitory action of GABA (Ganguly et al., 2001; Tyzio et al., 2007; Ben-Ari et al., 2012b), a process tightly controlled by the developmentally regulated expression of the two chloride co-transporters, potassium-chloride cotransporter 2 (KCC2) and neuronal Na–K–Cl cotransporter 1 (nKCC1) (Ben-Ari et al., 2012a; Watanabe and Fukuda, 2015; Raimondo et al., 2017). This evidence raised the question of whether an inflammatory event occurring during brain development may specifically affect this process. Such possibility has been called into question in a recent study, where an immune challenge – consisting in the injection of the viral mimicking molecule polyinosinic–polycytidylic acid (poly I:C) – was delivered to pregnant mice at early stages of embryo development (E9, corresponding to the mid/end of first trimester pregnancy in humans), a model of prenatal infection called maternal immune activation (MIA). The prenatal immune activation resulted in a delay of GABA switch in the offspring, resulting from a higher cortical and hippocampal expression of the transporter nKCC1 and a lower expression of KCC2 compared to controls. The alterations in nKCC1/KCC2 ratio resulted in GABA being excitatory and offspring more susceptible to seizures in the adult stage. The involvement of IL-1β in the process was indicated by the evidence that (i) IL1RKO embryos are protected from this detrimental effect and (ii) IL-1β delays the transition of GABA signaling in cultured neurons (Figure 1, pathway 3; Corradini et al., 2017). Among the transcriptional regulators of KCC2 expression, the neuronal repressor gene RE1-silencing transcription factor (REST) was found to modulate kcc2 gene expression (Yeo et al., 2009) and, in line with this evidence, its binding to KCC2 promoter was enhanced in the brain of offspring exposed to MIA (Corradini et al., 2017). However, given that MeCP2 also acts as a transcriptional repressor by binding to the KCC2 gene promoter (Tang et al., 2016), it will be interesting to define the interplay between these two factors and whether the increase of MeCP2 levels induced by IL-1β (Tomasoni et al., 2017) might contribute to KCC2 reduction.
The demonstration that a transient increase of IL-1β during neuronal development may induce long-lasting neuronal network hyperactivity by delaying GABA switch opens unexpected scenarios: impairment of the GABA developmental switch is indeed an important feature of several neurodevelopmental disorders, such as Down syndrome, Rett syndrome, and ASD (Lemonnier et al., 2012; He et al., 2014; Tyzio et al., 2014; Deidda et al., 2015; Inui et al., 2017), for which an important risk factor is represented by prenatal inflammatory conditions (Tarnow-Mordi et al., 2005; Missault et al., 2014).
An E/I imbalance has also been demonstrated in other neurological diseases characterized by excessive IL-1β levels, such as multiple sclerosis and epilepsy. In these cases, alternative molecular mechanisms were found to be involved. In a mouse model of multiple sclerosis, IL-1β-mediated inflammation was found to enhance glutamatergic transmission during the early phase of the disease (Centonze et al., 2009; Rossi et al., 2012, 2014; Mandolesi et al., 2013), through modulating vanilloid 1 channels in hippocampus (Rossi et al., 2012), downregulating the glutamate–aspartate transporter/excitatory amino acid transporter 1 (GLAST/EAAT1) in the cerebellum (Mandolesi et al., 2013, 2017), and by activating the apoptotic cascade through p53 activation (Rossi et al., 2014). An IL-1β-dependent enhancement of excitatory neurotransmission has also been reported in epilepsy (Iori et al., 2013) where danger signals such as High Mobility Group Box 1 (HMGB1) and Toll-like receptor 4 (TLR4) were proposed to mediate the IL-1β-dependent increase in neuronal excitability, through an Src kinase-mediated phosphorylation of the NR2B subunit of the N-methyl-D-aspartate (NMDA) receptor (Viviani et al., 2003; Maroso et al., 2010, 2011). Together these data indicate that IL-1β disrupts the correct E/I equilibrium acting on multiple molecular targets.
Conclusion and Perspectives
It is now clear that inflammation is an important contributor to defects in brain function, by affecting in particular cognitive processes (Rachal Pugh et al., 2001; Huang and Sheng, 2010; Yirmiya and Goshen, 2011) through concentration-dependent harmful effects induced by the pro-inflammatory cytokine IL-1β. Indeed, recent reports have highlighted the ability of IL-1β to selectively affect cell-to-cell communication in the brain, by targeting specific synaptic pathways (Mishra et al., 2012; Han et al., 2017) which are known to be altered in different synaptopathies (see Figures 1, 2). Among these, IL1RAPL1, MeCP2, and KCC2 are three central molecular players of neurodevelopmental disorders, whose functionality may be affected by IL-1β.
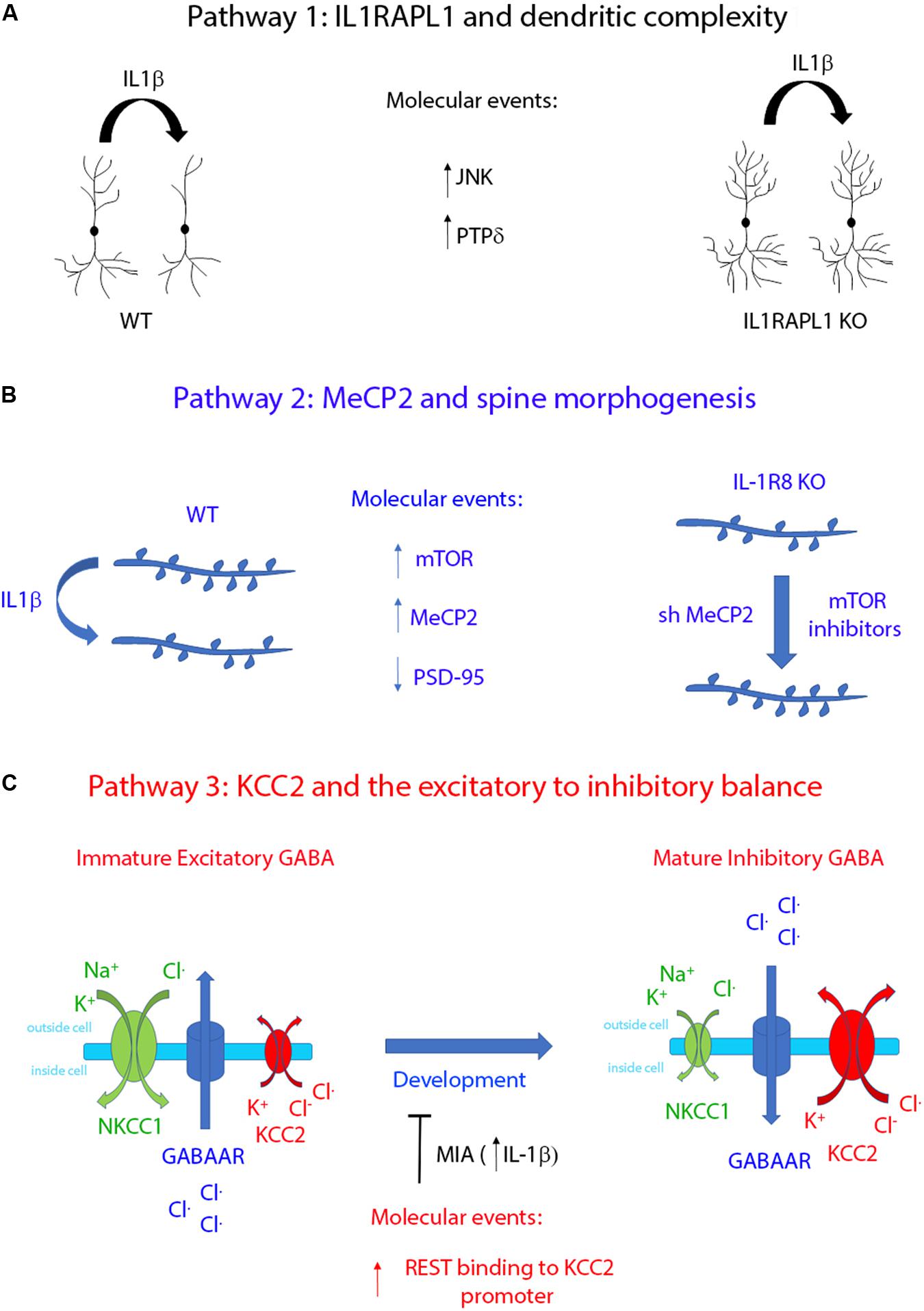
FIGURE 2. Schematic representation summarizing the functional effects of the three regulatory pathways controlled by IL-1β. (A) IL-1β affects dendritic complexity through IL1RAPL1-dependent mechanisms. This pathway activates JNK and PTPδ intracellular proteins (Montani et al., 2017). (B) IL-1β regulates dendritic spine morphology by upregulating the transcription factor MeCP2 in an mTOR-dependent manner (Tomasoni et al., 2017). (C) IL-1β leads to an excitatory/inhibitory unbalance by delaying the developmentally regulated switch of GABA signaling. This pathway involves the interplay between the two transcription factors REST and MeCP2, which in turn regulates the transcriptional level of KCC2 (Corradini et al., 2017).
These results may have important translational implications. As an example, the recognition that IL-1β modulates MeCP2 levels opens the challenging possibility to target the immune system for treating neurodevelopmental diseases characterized by altered levels of this transcription factor. Observational clinical data already support the possibility to treat cognitive symptoms by immunomodulatory drugs. Indeed, in patients affected by cryopyrin-associated periodic syndrome (CAPS), a group of rare genetic autoinflammatory diseases with levels of IL-1β being fivefold higher than in healthy individuals, symptoms of ID frequently occur. These cognitive defects are reversed following treatment with IL-1Ra (Bachove and Chang, 2014) or with specific neutralization of IL-1β with canakinumab (Kuemmerle-Deschner et al., 2011).
Although the modulation of IL-1β signaling in neurology or psychiatry is still in its infancy, the possibility of selectively inhibiting specific steps in the IL-1β cascade may represent a unique opportunity to treat immune synaptopathies (Box 1), i.e., synaptic dysfunctions resulting from deregulation of the immune system during brain development. A control of maternal inflammatory parameters during pregnancy may turn out to be a promising strategy to decrease the incidence of immune-mediated neurological or psychiatric illness in adulthood, as a consequence of maternal viral infections. Specific studies aimed at testing the efficacy of different IL-1β blockers that offer potential benefits to patients in individual disease states will be required in the next future.
Box 1. Synaptopathies and inflammation: the concept of immune-synaptopathies.
The term synaptopathy (from Greek συν, syn – together, απτειν, haptein – to clasp, and παθοϛ, pathos) refers to a class of neurological disorders characterized by alterations at the synaptic level (Brose et al., 2010). The concept of synaptopathy is based on several genetical studies which have identified the involvement, in different diseases, of specific genes whose products converge onto coherent biological pathways controlling various aspects of synaptic structure and function (see Table 1). Besides neurodevelopmental diseases, other brain diseases have been identified as synaptopathies, including neurodegenerative disorders, such as Alzheimer’s (Kerrigan and Randall, 2013) and Parkinson’s (Longhena et al., 2017) diseases. Synaptopathies are therefore defined as resulting from pathological events including synaptic loss (in which synaptic connectivity is compromised), alterations of synaptic functioning (in which the physiological activity of synapses is altered), or both (for a review, see Lepeta et al., 2016). We introduce the novel term immune-synaptopathy to underline the concept that the activation of the immune system, resulting in the formation of soluble immune mediators (i.e., cytokines), which are the main key effectors of the inflammatory response, directly impacts the physiological activity of the synapse producing a disease state (see text for details).
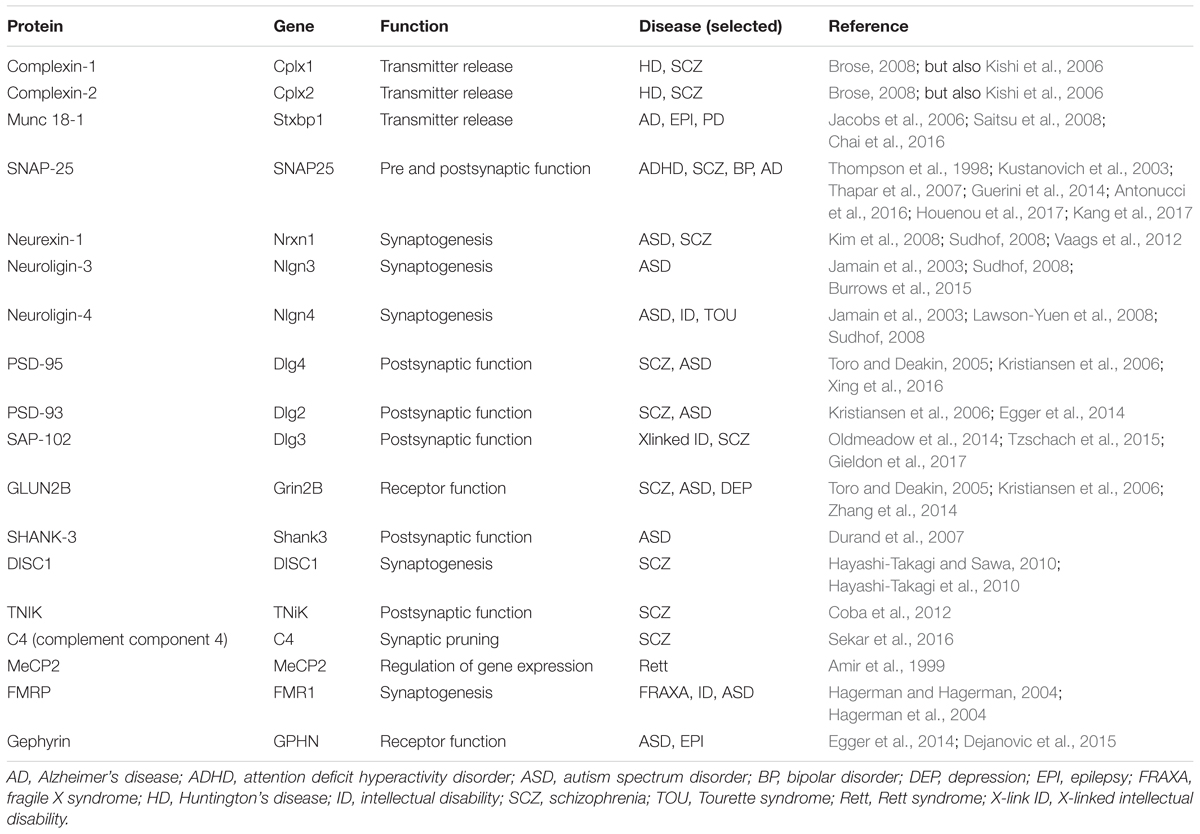
TABLE 1. List of selected synaptic and/or synaptic related gene disease.
Author Contributions
DP, EM, and MM designed the review outline. All the authors contributed to writing and designing the scheme. CM realized the figure.
Funding
The work in our laboratory was supported by Ministero della Salute GR-2011–02347377, Cariplo 2015–0594, AIRC, Project “AMANDA” CUP_B42F16000440005 from Regione Lombardia and CNR Research Project on Aging and Fondazione Veronesi to MM; Fondazione Vodafone to EM and MM; Cariplo no. 2017-0622 and Progetto Bandiera Interomics 2015–2017 to EM. Cariplo no. 2017-0886 to DP. We thank Monzino Foundation (Milano, Italy) for its generous gift of the Zeiss LSM800 confocal microscope to the section of Milan of the Institute of Neuroscience.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Akaneya, Y., Takahashi, M., and Hatanaka, H. (1995). Interleukin-1 beta enhances survival and interleukin-6 protects against MPP+ neurotoxicity in cultures of fetal rat dopaminergic neurons. Exp. Neurol. 136, 44–52. doi: 10.1006/exnr.1995.1082
Amir, R. E., Van Den Veyver, I. B., Wan, M., Tran, C. Q., Francke, U., and Zoghbi, H. Y. (1999). Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 23, 185–188. doi: 10.1038/13810
Antonucci, F., Corradini, I., Fossati, G., Tomasoni, R., Menna, E., and Matteoli, M. (2016). SNAP-25, a known presynaptic protein with emerging postsynaptic functions. Front. Synaptic Neurosci. 8:7. doi: 10.3389/fnsyn.2016.00007
Ashwood, P., Krakowiak, P., Hertz-Picciotto, I., Hansen, R., Pessah, I., and Van De Water, J. (2011). Elevated plasma cytokines in autism spectrum disorders provide evidence of immune dysfunction and are associated with impaired behavioral outcome. Brain Behav. Immun. 25, 40–45. doi: 10.1016/j.bbi.2010.08.003
Bachove, I., and Chang, C. (2014). Anakinra and related drugs targeting interleukin-1 in the treatment of cryopyrin-associated periodic syndromes. Open Access Rheumatol. 6, 15–25.
Barrientos, R. M., Frank, M. G., Hein, A. M., Higgins, E. A., Watkins, L. R., Rudy, J. W., et al. (2009). Time course of hippocampal IL-1 beta and memory consolidation impairments in aging rats following peripheral infection. Brain Behav. Immun. 23, 46–54. doi: 10.1016/j.bbi.2008.07.002
Barrientos, R. M., Higgins, E. A., Biedenkapp, J. C., Sprunger, D. B., Wright-Hardesty, K. J., Watkins, L. R., et al. (2006). Peripheral infection and aging interact to impair hippocampal memory consolidation. Neurobiol. Aging 27, 723–732. doi: 10.1016/j.neurobiolaging.2005.03.010
Barrientos, R. M., Higgins, E. A., Sprunger, D. B., Watkins, L. R., Rudy, J. W., and Maier, S. F. (2002). Memory for context is impaired by a post context exposure injection of interleukin-1 beta into dorsal hippocampus. Behav. Brain Res. 134, 291–298. doi: 10.1016/S0166-4328(02)00043-8
Barron, H., Hafizi, S., Andreazza, A. C., and Mizrahi, R. (2017). Neuroinflammation and oxidative stress in psychosis and psychosis risk. Int. J. Mol. Sci. 18:651. doi: 10.3390/ijms18030651
Bellinger, F. P., Madamba, S., and Siggins, G. R. (1993). Interleukin 1 beta inhibits synaptic strength and long-term potentiation in the rat CA1 hippocampus. Brain Res. 628, 227–234. doi: 10.1016/0006-8993(93)90959-Q
Ben-Ari, Y., Khalilov, I., Kahle, K. T., and Cherubini, E. (2012a). The GABA excitatory/inhibitory shift in brain maturation and neurological disorders. Neuroscientist 18, 467–486.
Ben-Ari, Y., Woodin, M. A., Sernagor, E., Cancedda, L., Vinay, L., Rivera, C., et al. (2012b). Refuting the challenges of the developmental shift of polarity of GABA actions: GABA more exciting than ever! Front. Cell. Neurosci. 6:35. doi: 10.3389/fncel.2012.00035
Beutner, D., Foerst, A., Lang-Roth, R., Von Wedel, H., and Walger, M. (2007). Risk factors for auditory neuropathy/auditory synaptopathy. ORL J. Otorhinolaryngol. Relat. Spec. 69, 239–244. doi: 10.1159/000101545
Born, T. L., Smith, D. E., Garka, K. E., Renshaw, B. R., Bertles, J. S., and Sims, J. E. (2000). Identification and characterization of two members of a novel class of the interleukin-1 receptor (IL-1R) family. Delineation Of a new class of IL-1R-related proteins based on signaling. J. Biol. Chem. 275, 29946–29954. doi: 10.1074/jbc.M004077200
Brose, N. (2008). Altered complexin expression in psychiatric and neurological disorders: cause or consequence? Mol. Cells 25, 7–19.
Brose, N., O’connor, V., and Skehel, P. (2010). Synaptopathy: dysfunction of synaptic function? Biochem. Soc. Trans. 38, 443–444. doi: 10.1042/BST0380443
Brown, A. S., and Derkits, E. J. (2010). Prenatal infection and schizophrenia: a review of epidemiologic and translational studies. Am. J. Psychiatry 167, 261–280. doi: 10.1176/appi.ajp.2009.09030361
Burrows, E. L., Laskaris, L., Koyama, L., Churilov, L., Bornstein, J. C., Hill-Yardin, E. L., et al. (2015). A neuroligin-3 mutation implicated in autism causes abnormal aggression and increases repetitive behavior in mice. Mol. Autism 6:62. doi: 10.1186/s13229-015-0055-7
Carrie, A., Jun, L., Bienvenu, T., Vinet, M. C., Mcdonell, N., Couvert, P., et al. (1999). A new member of the IL-1 receptor family highly expressed in hippocampus and involved in X-linked mental retardation. Nat. Genet. 23, 25–31. doi: 10.1038/12623
Centonze, D., Muzio, L., Rossi, S., Cavasinni, F., De Chiara, V., Bergami, A., et al. (2009). Inflammation triggers synaptic alteration and degeneration in experimental autoimmune encephalomyelitis. J. Neurosci. 29, 3442–3452. doi: 10.1523/JNEUROSCI.5804-08.2009
Chai, Y. J., Sierecki, E., Tomatis, V. M., Gormal, R. S., Giles, N., Morrow, I. C., et al. (2016). Munc18-1 is a molecular chaperone for α-synuclein, controlling its self-replicating aggregation. J. Cell Biol. 214, 705–718. doi: 10.1083/jcb.201512016
Chase, K. A., Cone, J. J., Rosen, C., and Sharma, R. P. (2016). The value of interleukin 6 as a peripheral diagnostic marker in schizophrenia. BMC Psychiatry 16:152. doi: 10.1186/s12888-016-0866-x
Chen, J., Buchanan, J. B., Sparkman, N. L., Godbout, J. P., Freund, G. G., and Johnson, R. W. (2008). Neuroinflammation and disruption in working memory in aged mice after acute stimulation of the peripheral innate immune system. Brain Behav. Immun. 22, 301–311. doi: 10.1016/j.bbi.2007.08.014
Cheng, T. L., and Qiu, Z. (2014). MeCP2: multifaceted roles in gene regulation and neural development. Neurosci. Bull. 30, 601–609. doi: 10.1007/s12264-014-1452-6
Coba, M. P., Komiyama, N. H., Nithianantharajah, J., Kopanitsa, M. V., Indersmitten, T., Skene, N. G., et al. (2012). TNiK is required for postsynaptic and nuclear signaling pathways and cognitive function. J. Neurosci. 32, 13987–13999. doi: 10.1523/JNEUROSCI.2433-12.2012
Corradini, I., Focchi, E., Rasile, M., Morini, R., Desiato, G., Tomasoni, R., et al. (2017). Maternal immune activation delays excitatory-to-inhibitory gamma-aminobutyric acid switch in offspring. Biol. Psychiatry 83, 680–691. doi: 10.1016/j.biopsych.2017.09.030
Costello, D. A., Watson, M. B., Cowley, T. R., Murphy, N., Murphy Royal, C., Garlanda, C., et al. (2011). Interleukin-1alpha and HMGB1 mediate hippocampal dysfunction in SIGIRR-deficient mice. J. Neurosci. 31, 3871–3879. doi: 10.1523/JNEUROSCI.6676-10.2011
Dantzer, R., O’connor, J. C., Freund, G. G., Johnson, R. W., and Kelley, K. W. (2008). From inflammation to sickness and depression: when the immune system subjugates the brain. Nat. Rev. Neurosci. 9, 46–56. doi: 10.1038/nrn2297
De Chiara, G., Marcocci, M. E., Sgarbanti, R., Civitelli, L., Ripoli, C., Piacentini, R., et al. (2012). Infectious agents and neurodegeneration. Mol. Neurobiol. 46, 614–638. doi: 10.1007/s12035-012-8320-7
Deidda, G., Parrini, M., Naskar, S., Bozarth, I. F., Contestabile, A., and Cancedda, L. (2015). Reversing excitatory GABAAR signaling restores synaptic plasticity and memory in a mouse model of Down syndrome. Nat. Med. 21, 318–326. doi: 10.1038/nm.3827
Dejanovic, B., Djemie, T., Grunewald, N., Suls, A., Kress, V., Hetsch, F., et al. (2015). Simultaneous impairment of neuronal and metabolic function of mutated gephyrin in a patient with epileptic encephalopathy. EMBO Mol. Med. 7, 1580–1594. doi: 10.15252/emmm.201505323
Delobel, B., Delannoy, V., Pini, G., Zapella, M., Tardieu, M., Vallee, L., et al. (1998). Identification and molecular characterization of a small 11q23.3 de novo duplication in a patient with Rett syndrome manifestations. Am. J. Med. Genet. 80, 273–280. doi: 10.1002/(SICI)1096-8628(19981116)80:3<273::AID-AJMG19>3.0.CO;2-6
Depino, A. M. (2017). Perinatal inflammation and adult psychopathology: from preclinical models to humans. Semin. Cell Dev. Biol. 77, 104–114. doi: 10.1016/j.semcdb.2017.09.010
Durand, C. M., Betancur, C., Boeckers, T. M., Bockmann, J., Chaste, P., Fauchereau, F., et al. (2007). Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat. Genet. 39, 25–27. doi: 10.1038/ng1933
Egger, G., Roetzer, K. M., Noor, A., Lionel, A. C., Mahmood, H., Schwarzbraun, T., et al. (2014). Identification of risk genes for autism spectrum disorder through copy number variation analysis in Austrian families. Neurogenetics 15, 117–127. doi: 10.1007/s10048-014-0394-0
Erion, J. R., Wosiski-Kuhn, M., Dey, A., Hao, S., Davis, C. L., Pollock, N. K., et al. (2014). Obesity elicits interleukin 1-mediated deficits in hippocampal synaptic plasticity. J. Neurosci. 34, 2618–2631. doi: 10.1523/JNEUROSCI.4200-13.2014
Erta, M., Quintana, A., and Hidalgo, J. (2012). Interleukin-6, a major cytokine in the central nervous system. Int. J. Biol. Sci. 8, 1254–1266. doi: 10.7150/ijbs.4679
Fan, X., Goff, D. C., and Henderson, D. C. (2007). Inflammation and schizophrenia. Expert Rev. Neurother. 7, 789–796. doi: 10.1586/14737175.7.7.789
Frank, M. G., Miguel, Z. D., Watkins, L. R., and Maier, S. F. (2010). Prior exposure to glucocorticoids sensitizes the neuroinflammatory and peripheral inflammatory responses to E. coli lipopolysaccharide. Brain Behav. Immun. 24, 19–30. doi: 10.1016/j.bbi.2009.07.008
Gadient, R. A., and Otten, U. H. (1997). Interleukin-6 (IL-6)–a molecule with both beneficial and destructive potentials. Prog. Neurobiol. 52, 379–390. doi: 10.1016/S0301-0082(97)00021-X
Gambino, F., Kneib, M., Pavlowsky, A., Skala, H., Heitz, S., Vitale, N., et al. (2009). IL1RAPL1 controls inhibitory networks during cerebellar development in mice. Eur. J. Neurosci. 30, 1476–1486. doi: 10.1111/j.1460-9568.2009.06975.x
Ganguly, K., Schinder, A. F., Wong, S. T., and Poo, M. (2001). Gaba itself promotes the developmental switch of neuronal GABAergic responses from excitation to inhibition. Cell 105, 521–532. doi: 10.1016/S0092-8674(01)00341-5
Garay, P. A., and McAllister, A. K. (2010). Novel roles for immune molecules in neural development: implications for neurodevelopmental disorders. Front. Synaptic Neurosci. 2:136. doi: 10.3389/fnsyn.2010.00136
Gardoni, F., Boraso, M., Zianni, E., Corsini, E., Galli, C. L., Cattabeni, F., et al. (2011). Distribution of interleukin-1 receptor complex at the synaptic membrane driven by interleukin-1beta and NMDA stimulation. J. Neuroinflammation 8:14. doi: 10.1186/1742-2094-8-14
Gibertini, M., Newton, C., Friedman, H., and Klein, T. W. (1995). Spatial learning impairment in mice infected with Legionella pneumophila or administered exogenous interleukin-1-beta. Brain Behav. Immun. 9, 113–128. doi: 10.1006/brbi.1995.1012
Gieldon, L., Mackenroth, L., Betcheva-Krajcir, E., Rump, A., Beck-Wodl, S., Schallner, J., et al. (2017). Skewed X-inactivation in a family with DLG3-associated X-linked intellectual disability. Am. J. Med. Genet. A 173, 2545–2550. doi: 10.1002/ajmg.a.38348
Giovanoli, S., Weber-Stadlbauer, U., Schedlowski, M., Meyer, U., and Engler, H. (2016). Prenatal immune activation causes hippocampal synaptic deficits in the absence of overt microglia anomalies. Brain Behav. Immun. 55, 25–38. doi: 10.1016/j.bbi.2015.09.015
Girard, S., Tremblay, L., Lepage, M., and Sebire, G. (2010). IL-1 receptor antagonist protects against placental and neurodevelopmental defects induced by maternal inflammation. J. Immunol. 184, 3997–4005. doi: 10.4049/jimmunol.0903349
Goshen, I., Kreisel, T., Ounallah-Saad, H., Renbaum, P., Zalzstein, Y., Ben-Hur, T., et al. (2007). A dual role for interleukin-1 in hippocampal-dependent memory processes. Psychoneuroendocrinology 32, 1106–1115. doi: 10.1016/j.psyneuen.2007.09.004
Graham, A. M., Rasmussen, J. M., Rudolph, M. D., Heim, C. M., Gilmore, J. H., Styner, M., et al. (2018). Maternal systemic interleukin-6 during pregnancy is associated with newborn amygdala phenotypes and subsequent behavior at 2 years of age. Biol. Psychiatry 83, 109–119. doi: 10.1016/j.biopsych.2017.05.027
Grant, S. G. (2012). Synaptopathies: diseases of the synaptome. Curr. Opin. Neurobiol. 22, 522–529. doi: 10.1016/j.conb.2012.02.002
Griffin, W. S., Liu, L., Li, Y., Mrak, R. E., and Barger, S. W. (2006). Interleukin-1 mediates Alzheimer and Lewy body pathologies. J. Neuroinflammation 3:5.
Guerini, F. R., Agliardi, C., Sironi, M., Arosio, B., Calabrese, E., Zanzottera, M., et al. (2014). Possible association between SNAP-25 single nucleotide polymorphisms and alterations of categorical fluency and functional MRI parameters in Alzheimer’s disease. J. Alzheimers Dis. 42, 1015–1028. doi: 10.3233/JAD-140057
Hagberg, H., Gressens, P., and Mallard, C. (2012). Inflammation during fetal and neonatal life: implications for neurologic and neuropsychiatric disease in children and adults. Ann. Neurol. 71, 444–457. doi: 10.1002/ana.22620
Hagberg, H., Mallard, C., Ferriero, D. M., Vannucci, S. J., Levison, S. W., Vexler, Z. S., et al. (2015). The role of inflammation in perinatal brain injury. Nat. Rev. Neurol. 11, 192–208. doi: 10.1038/nrneurol.2015.13
Hagerman, P. J., and Hagerman, R. J. (2004). Fragile X-associated tremor/ataxia syndrome (FXTAS). Ment. Retard. Dev. Disabil. Res. Rev. 10, 25–30. doi: 10.1002/mrdd.20005
Hagerman, R. J., Leavitt, B. R., Farzin, F., Jacquemont, S., Greco, C. M., Brunberg, J. A., et al. (2004). Fragile-X-associated tremor/ataxia syndrome (FXTAS) in females with the FMR1 premutation. Am. J. Hum. Genet. 74, 1051–1056. doi: 10.1086/420700
Han, Q., Lin, Q., Huang, P., Chen, M., Hu, X., Fu, H., et al. (2017). Microglia-derived IL-1β contributes to axon development disorders and synaptic deficit through p38-MAPK signal pathway in septic neonatal rats. J. Neuroinflammation 14:52. doi: 10.1186/s12974-017-0805-x
Hayashi-Takagi, A., and Sawa, A. (2010). Disturbed synaptic connectivity in schizophrenia: convergence of genetic risk factors during neurodevelopment. Brain Res. Bull. 83, 140–146. doi: 10.1016/j.brainresbull.2010.04.007
Hayashi-Takagi, A., Takaki, M., Graziane, N., Seshadri, S., Murdoch, H., Dunlop, A. J., et al. (2010). Disrupted-in-Schizophrenia 1 (DISC1) regulates spines of the glutamate synapse via Rac1. Nat. Neurosci. 13, 327–332. doi: 10.1038/nn.2487
He, Q., Nomura, T., Xu, J., and Contractor, A. (2014). The developmental switch in GABA polarity is delayed in fragile X mice. J. Neurosci. 34, 446–450. doi: 10.1523/JNEUROSCI.4447-13.2014
Heese, K. (2017). Functional repertoire of interleukin-6 in the central nervous system - a review. Restor. Neurol. Neurosci. 35, 693–701. doi: 10.3233/RNN-170772
Hein, A. M., Stasko, M. R., Matousek, S. B., Scott-Mckean, J. J., Maier, S. F., Olschowka, J. A., et al. (2010). Sustained hippocampal IL-1beta overexpression impairs contextual and spatial memory in transgenic mice. Brain Behav. Immun. 24, 243–253. doi: 10.1016/j.bbi.2009.10.002
Hirota, H., Kiyama, H., Kishimoto, T., and Taga, T. (1996). Accelerated nerve regeneration in mice by upregulated expression of interleukin (IL) 6 and IL-6 receptor after trauma. J. Exp. Med. 183, 2627–2634. doi: 10.1084/jem.183.6.2627
Hosseini, S., Wilk, E., Michaelsen-Preusse, K., Gerhauser, I., Baumgartner, W., Geffers, R., et al. (2018). Long-term neuroinflammation induced by influenza A virus infection and the impact on hippocampal neuron morphology and function. J. Neurosci. 38, 3060–3080. doi: 10.1523/JNEUROSCI.1740-17.2018
Houbaert, X., Zhang, C. L., Gambino, F., Lepleux, M., Deshors, M., Normand, E., et al. (2013). Target-specific vulnerability of excitatory synapses leads to deficits in associative memory in a model of intellectual disorder. J. Neurosci. 33, 13805–13819. doi: 10.1523/JNEUROSCI.1457-13.2013
Houenou, J., Boisgontier, J., Henrion, A., D’albis, M. A., Dumaine, A., Linke, J., et al. (2017). A multilevel functional study of a SNAP25 at-risk variant for bipolar disorder and schizophrenia. J. Neurosci. 37, 10389–10397. doi: 10.1523/JNEUROSCI.1040-17.2017
Huang, Z. B., and Sheng, G. Q. (2010). Interleukin-1beta with learning and memory. Neurosci. Bull. 26, 455–468. doi: 10.1007/s12264-010-6023-5
Inui, T., Kumagaya, S., and Myowa-Yamakoshi, M. (2017). Neurodevelopmental hypothesis about the etiology of autism spectrum disorders. Front. Hum. Neurosci. 11:354. doi: 10.3389/fnhum.2017.00354
Iori, V., Frigerio, F., and Vezzani, A. (2016). Modulation of neuronal excitability by immune mediators in epilepsy. Curr. Opin. Pharmacol. 26, 118–123. doi: 10.1016/j.coph.2015.11.002
Iori, V., Maroso, M., Rizzi, M., Iyer, A. M., Vertemara, R., Carli, M., et al. (2013). Receptor for advanced glycation endproducts is upregulated in temporal lobe epilepsy and contributes to experimental seizures. Neurobiol. Dis. 58, 102–114. doi: 10.1016/j.nbd.2013.03.006
Jacobs, E. H., Williams, R. J., and Francis, P. T. (2006). Cyclin-dependent kinase 5, Munc18a and Munc18-interacting protein 1/X11alpha protein up-regulation in Alzheimer’s disease. Neuroscience 138, 511–522. doi: 10.1016/j.neuroscience.2005.11.017
Jamain, S., Quach, H., Betancur, C., Rastam, M., Colineaux, C., Gillberg, I. C., et al. (2003). Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat. Genet. 34, 27–29. doi: 10.1038/ng1136
Jurgens, H. A., Amancherla, K., and Johnson, R. W. (2012). Influenza infection induces neuroinflammation, alters hippocampal neuron morphology, and impairs cognition in adult mice. J. Neurosci. 32, 3958–3968. doi: 10.1523/JNEUROSCI.6389-11.2012
Kang, Q., Chen, J., Yu, S., Yuan, A., Zhang, Y., Zhang, R., et al. (2017). Association of the 5-HT2A receptor gene promoter polymorphism-1438G/A with anorexia nervosa and psychopathological traits in the Chinese Han population: a preliminary study. Asia Pac. Psychiatry 9. doi: 10.1111/appy.12284
Katila, H., Appelberg, B., Hurme, M., and Rimon, R. (1994). Plasma levels of interleukin-1 beta and interleukin-6 in schizophrenia, other psychoses, and affective disorders. Schizophr. Res. 12, 29–34. doi: 10.1016/0920-9964(94)90081-7
Katsuki, H., Nakai, S., Hirai, Y., Akaji, K., Kiso, Y., and Satoh, M. (1990). Interleukin-1 beta inhibits long-term potentiation in the CA3 region of mouse hippocampal slices. Eur. J. Pharmacol. 181, 323–326. doi: 10.1016/0014-2999(90)90099-R
Kelly, A., Vereker, E., Nolan, Y., Brady, M., Barry, C., Loscher, C. E., et al. (2003). Activation of p38 plays a pivotal role in the inhibitory effect of lipopolysaccharide and interleukin-1 beta on long term potentiation in rat dentate gyrus. J. Biol. Chem. 278, 19453–19462. doi: 10.1074/jbc.M301938200
Kerrigan, T. L., and Randall, A. D. (2013). A new player in the “synaptopathy” of Alzheimer’s disease - arc/arg 3.1. Front. Neurol. 4:9. doi: 10.3389/fneur.2013.00009
Kim, H. G., Kishikawa, S., Higgins, A. W., Seong, I. S., Donovan, D. J., Shen, Y., et al. (2008). Disruption of neurexin 1 associated with autism spectrum disorder. Am. J. Hum. Genet. 82, 199–207. doi: 10.1016/j.ajhg.2007.09.011
Kishi, T., Ikeda, M., Suzuki, T., Kitajima, T., Yamanouchi, Y., Kinoshita, Y., et al. (2006). No association of complexin1 and complexin2 genes with schizophrenia in a Japanese population. Schizophr. Res. 82, 185–189. doi: 10.1016/j.schres.2005.12.842
Krakowiak, P., Goines, P. E., Tancredi, D. J., Ashwood, P., Hansen, R. L., Hertz-Picciotto, I., et al. (2017). Neonatal cytokine profiles associated with autism spectrum disorder. Biol. Psychiatry 81, 442–451. doi: 10.1016/j.biopsych.2015.08.007
Kristiansen, L. V., Beneyto, M., Haroutunian, V., and Meador-Woodruff, J. H. (2006). Changes in NMDA receptor subunits and interacting PSD proteins in dorsolateral prefrontal and anterior cingulate cortex indicate abnormal regional expression in schizophrenia. Mol. Psychiatry 11, 737–747. doi: 10.1038/sj.mp.4001844
Kuemmerle-Deschner, J. B., Ramos, E., Blank, N., Roesler, J., Felix, S. D., Jung, T., et al. (2011). Canakinumab (ACZ885, a fully human IgG1 anti-IL-1beta mAb) induces sustained remission in pediatric patients with cryopyrin-associated periodic syndrome (CAPS). Arthritis Res. Ther. 13:R34. doi: 10.1186/ar3266
Kushima, Y., Hama, T., and Hatanaka, H. (1992). Interleukin-6 as a neurotrophic factor for promoting the survival of cultured catecholaminergic neurons in a chemically defined medium from fetal and postnatal rat midbrains. Neurosci. Res. 13, 267–280. doi: 10.1016/0168-0102(92)90039-F
Kustanovich, V., Merriman, B., Mcgough, J., Mccracken, J. T., Smalley, S. L., and Nelson, S. F. (2003). Biased paternal transmission of SNAP-25 risk alleles in attention-deficit hyperactivity disorder. Mol. Psychiatry 8, 309–315. doi: 10.1038/sj.mp.4001247
Lawson-Yuen, A., Saldivar, J. S., Sommer, S., and Picker, J. (2008). Familial deletion within NLGN4 associated with autism and Tourette syndrome. Eur. J. Hum. Genet. 16, 614–618. doi: 10.1038/sj.ejhg.5202006
Lemonnier, E., Degrez, C., Phelep, M., Tyzio, R., Josse, F., Grandgeorge, M., et al. (2012). A randomised controlled trial of bumetanide in the treatment of autism in children. Transl. Psychiatry 2:e202. doi: 10.1038/tp.2012.124
Lepeta, K., Lourenco, M. V., Schweitzer, B. C., Martino Adami, P. V., Banerjee, P., Catuara-Solarz, S., et al. (2016). Synaptopathies: synaptic dysfunction in neurological disorders - A review from students to students. J. Neurochem. 138, 785–805. doi: 10.1111/jnc.13713
Li, Y., Liu, L., Barger, S. W., and Griffin, W. S. (2003). Interleukin-1 mediates pathological effects of microglia on tau phosphorylation and on synaptophysin synthesis in cortical neurons through a p38-MAPK pathway. J. Neurosci. 23, 1605–1611.
Lin, C. C., and Edelson, B. T. (2017). New insights into the role of IL-1beta in experimental autoimmune encephalomyelitis and multiple sclerosis. J. Immunol. 198, 4553–4560. doi: 10.4049/jimmunol.1700263
Lipina, T. V., Zai, C., Hlousek, D., Roder, J. C., and Wong, A. H. (2013). Maternal immune activation during gestation interacts with Disc1 point mutation to exacerbate schizophrenia-related behaviors in mice. J. Neurosci. 33, 7654–7666. doi: 10.1523/JNEUROSCI.0091-13.2013
Lombardi, L. M., Baker, S. A., and Zoghbi, H. Y. (2015). MECP2 disorders: from the clinic to mice and back. J. Clin. Invest. 125, 2914–2923. doi: 10.1172/JCI78167
Longhena, F., Faustini, G., Missale, C., Pizzi, M., Spano, P., and Bellucci, A. (2017). The contribution of alpha-synuclein spreading to Parkinson’s disease synaptopathy. Neural Plast. 2017:5012129. doi: 10.1155/2017/5012129
Mandolesi, G., De Vito, F., Musella, A., Gentile, A., Bullitta, S., Fresegna, D., et al. (2017). miR-142-3p is a key regulator of IL-1beta-dependent synaptopathy in neuroinflammation. J. Neurosci. 37, 546–561. doi: 10.1523/JNEUROSCI.0851-16.2016
Mandolesi, G., Musella, A., Gentile, A., Grasselli, G., Haji, N., Sepman, H., et al. (2013). Interleukin-1beta alters glutamate transmission at purkinje cell synapses in a mouse model of multiple sclerosis. J. Neurosci. 33, 12105–12121. doi: 10.1523/JNEUROSCI.5369-12.2013
Maroso, M., Balosso, S., Ravizza, T., Liu, J., Aronica, E., Iyer, A. M., et al. (2010). Toll-like receptor 4 and high-mobility group box-1 are involved in ictogenesis and can be targeted to reduce seizures. Nat. Med. 16, 413–419. doi: 10.1038/nm.2127
Maroso, M., Balosso, S., Ravizza, T., Liu, J., Bianchi, M. E., and Vezzani, A. (2011). Interleukin-1 type 1 receptor/toll-like receptor signalling in epilepsy: the importance of IL-1beta and high-mobility group box 1. J. Intern. Med. 270, 319–326. doi: 10.1111/j.1365-2796.2011.02431.x
Millan, M. J. (2013). An epigenetic framework for neurodevelopmental disorders: from pathogenesis to potential therapy. Neuropharmacology 68, 2–82. doi: 10.1016/j.neuropharm.2012.11.015
Mishra, A., Kim, H. J., Shin, A. H., and Thayer, S. A. (2012). Synapse loss induced by interleukin-1β requires pre- and post-synaptic mechanisms. J. Neuroimmune Pharmacol. 7, 571–578. doi: 10.1007/s11481-012-9342-7
Missault, S., Van Den Eynde, K., Vanden Berghe, W., Fransen, E., Weeren, A., Timmermans, J. P., et al. (2014). The risk for behavioural deficits is determined by the maternal immune response to prenatal immune challenge in a neurodevelopmental model. Brain Behav. Immun. 42, 138–146. doi: 10.1016/j.bbi.2014.06.013
Montani, C., Ramos-Brossier, M., Ponzoni, L., Gritti, L., Cwetsch, A. W., Braida, D., et al. (2017). The X-linked intellectual disability protein IL1RAPL1 regulates dendrite complexity. J. Neurosci. 37, 6606–6627. doi: 10.1523/JNEUROSCI.3775-16.2017
Moraes, C. A., Santos, G., De Sampaio e Spohr, T. C., D’avila, J. C., Lima, F. R., Benjamim, C. F., et al. (2015). Activated microglia-induced deficits in excitatory synapses through IL-1β: implications for cognitive impairment in sepsis. Mol. Neurobiol. 52, 653–663. doi: 10.1007/s12035-014-8868-5
Murray, C. A., and Lynch, M. A. (1998). Dietary supplementation with vitamin E reverses the age-related deficit in long term potentiation in dentate gyrus. J. Biol. Chem. 273, 12161–12168. doi: 10.1074/jbc.273.20.12161
Murray, K. N., Parry-Jones, A. R., and Allan, S. M. (2015). Interleukin-1 and acute brain injury. Front. Cell. Neurosci. 9:18. doi: 10.3389/fncel.2015.00018
Najjar, S., Pearlman, D. M., Alper, K., Najjar, A., and Devinsky, O. (2013). Neuroinflammation and psychiatric illness. J. Neuroinflammation 10:43. doi: 10.1186/1742-2094-10-43
Nakanishi, M., Niidome, T., Matsuda, S., Akaike, A., Kihara, T., and Sugimoto, H. (2007). Microglia-derived interleukin-6 and leukaemia inhibitory factor promote astrocytic differentiation of neural stem/progenitor cells. Eur. J. Neurosci. 25, 649–658. doi: 10.1111/j.1460-9568.2007.05309.x
Oh-Nishi, A., Obayashi, S., Sugihara, I., Minamimoto, T., and Suhara, T. (2010). Maternal immune activation by polyriboinosinic-polyribocytidilic acid injection produces synaptic dysfunction but not neuronal loss in the hippocampus of juvenile rat offspring. Brain Res. 1363, 170–179. doi: 10.1016/j.brainres.2010.09.054
Oitzl, M. S., Van Oers, H., Schobitz, B., and De Kloet, E. R. (1993). Interleukin-1 beta, but not interleukin-6, impairs spatial navigation learning. Brain Res. 613, 160–163. doi: 10.1016/0006-8993(93)90468-3
O’Keeffe, G. W. (2017). A new role for placental IL-6 signalling in determining neurodevelopmental outcome. Brain Behav. Immun. 62, 9–10. doi: 10.1016/j.bbi.2017.02.011
Oldmeadow, C., Mossman, D., Evans, T. J., Holliday, E. G., Tooney, P. A., Cairns, M. J., et al. (2014). Combined analysis of exon splicing and genome wide polymorphism data predict schizophrenia risk loci. J. Psychiatr. Res. 52, 44–49. doi: 10.1016/j.jpsychires.2014.01.011
Parish, C. L., Finkelstein, D. I., Tripanichkul, W., Satoskar, A. R., Drago, J., and Horne, M. K. (2002). The role of interleukin-1, interleukin-6, and glia in inducing growth of neuronal terminal arbors in mice. J. Neurosci. 22, 8034–8041.
Patterson, S. L. (2015). Immune dysregulation and cognitive vulnerability in the aging brain: interactions of microglia, IL-1β, BDNF and synaptic plasticity. Neuropharmacology 96, 11–18. doi: 10.1016/j.neuropharm.2014.12.020
Pavlowsky, A., Gianfelice, A., Pallotto, M., Zanchi, A., Vara, H., Khelfaoui, M., et al. (2010). A postsynaptic signaling pathway that may account for the cognitive defect due to IL1RAPL1 mutation. Curr. Biol. 20, 103–115. doi: 10.1016/j.cub.2009.12.030
Prieto, G. A., Snigdha, S., Baglietto-Vargas, D., Smith, E. D., Berchtold, N. C., Tong, L., et al. (2015). Synapse-specific IL-1 receptor subunit reconfiguration augments vulnerability to IL-1β in the aged hippocampus. Proc. Natl. Acad. Sci. U.S.A. 112, E5078–E5087. doi: 10.1073/pnas.1514486112
Rachal Pugh, C., Fleshner, M., Watkins, L. R., Maier, S. F., and Rudy, J. W. (2001). The immune system and memory consolidation: a role for the cytokine IL-1beta. Neurosci. Biobehav. Rev. 25, 29–41. doi: 10.1016/S0149-7634(00)00048-8
Raimondo, J. V., Richards, B. A., and Woodin, M. A. (2017). Neuronal chloride and excitability - the big impact of small changes. Curr. Opin. Neurobiol. 43, 35–42. doi: 10.1016/j.conb.2016.11.012
Ramos-Brossier, M., Montani, C., Lebrun, N., Gritti, L., Martin, C., Seminatore-Nole, C., et al. (2015). Novel IL1RAPL1 mutations associated with intellectual disability impair synaptogenesis. Hum. Mol. Genet. 24, 1106–1118. doi: 10.1093/hmg/ddu523
Ross, F. M., Allan, S. M., Rothwell, N. J., and Verkhratsky, A. (2003). A dual role for interleukin-1 in LTP in mouse hippocampal slices. J. Neuroimmunol. 144, 61–67. doi: 10.1016/j.jneuroim.2003.08.030
Rossi, S., Furlan, R., De Chiara, V., Motta, C., Studer, V., Mori, F., et al. (2012). Interleukin-1beta causes synaptic hyperexcitability in multiple sclerosis. Ann. Neurol. 71, 76–83. doi: 10.1002/ana.22512
Rossi, S., Motta, C., Studer, V., Macchiarulo, G., Volpe, E., Barbieri, F., et al. (2014). Interleukin-1β causes excitotoxic neurodegeneration and multiple sclerosis disease progression by activating the apoptotic protein p53. Mol. Neurodegener. 9:56. doi: 10.1186/1750-1326-9-56
Saitsu, H., Kato, M., Mizuguchi, T., Hamada, K., Osaka, H., Tohyama, J., et al. (2008). De novo mutations in the gene encoding STXBP1 (MUNC18-1) cause early infantile epileptic encephalopathy. Nat. Genet. 40, 782–788. doi: 10.1038/ng.150
Samuelsson, A. M., Jennische, E., Hansson, H. A., and Holmang, A. (2006). Prenatal exposure to interleukin-6 results in inflammatory neurodegeneration in hippocampus with NMDA/GABA(A) dysregulation and impaired spatial learning. Am. J. Physiol. Regul. Integr. Comp. Physiol. 290, R1345–R1356.
Schafers, M., and Sorkin, L. (2008). Effect of cytokines on neuronal excitability. Neurosci. Lett. 437, 188–193. doi: 10.1016/j.neulet.2008.03.052
Schiavone, S., and Trabace, L. (2017). Inflammation, stress response, and redox dysregulation biomarkers: clinical outcomes and pharmacological implications for psychosis. Front. Psychiatry 8:203. doi: 10.3389/fpsyt.2017.00203
Sekar, A., Bialas, A. R., De Rivera, H., Davis, A., Hammond, T. R., Kamitaki, N., et al. (2016). Schizophrenia risk from complex variation of complement component 4. Nature 530, 177–183. doi: 10.1038/nature16549
Soderlund, J., Schroder, J., Nordin, C., Samuelsson, M., Walther-Jallow, L., Karlsson, H., et al. (2009). Activation of brain interleukin-1beta in schizophrenia. Mol. Psychiatry 14, 1069–1071. doi: 10.1038/mp.2009.52
Sudhof, T. C. (2008). Neuroligins and neurexins link synaptic function to cognitive disease. Nature 455, 903–911. doi: 10.1038/nature07456
Suzuki, K., Matsuzaki, H., Iwata, K., Kameno, Y., Shimmura, C., Kawai, S., et al. (2011). Plasma cytokine profiles in subjects with high-functioning autism spectrum disorders. PLoS One 6:e20470. doi: 10.1371/journal.pone.0020470
Tang, X., Kim, J., Zhou, L., Wengert, E., Zhang, L., Wu, Z., et al. (2016). KCC2 rescues functional deficits in human neurons derived from patients with Rett syndrome. Proc. Natl. Acad. Sci. U.S.A. 113, 751–756. doi: 10.1073/pnas.1524013113
Tarnow-Mordi, W., Isaacs, D., Smart, D. H., Stenson, B., and Haque, K. (2005). Neurodevelopmental impairment and neonatal infections. JAMA 293, 932; author reply 932.
Thapar, A., Langley, K., Owen, M. J., and O’donovan, M. C. (2007). Advances in genetic findings on attention deficit hyperactivity disorder. Psychol. Med. 37, 1681–1692. doi: 10.1017/S0033291707000773
Thompson, P. M., Sower, A. C., and Perrone-Bizzozero, N. I. (1998). Altered levels of the synaptosomal associated protein SNAP-25 in schizophrenia. Biol. Psychiatry 43, 239–243. doi: 10.1016/S0006-3223(97)00204-7
Tomasoni, R., Morini, R., Lopez-Atalaya, J. P., Corradini, I., Canzi, A., Rasile, M., et al. (2017). Lack of IL-1R8 in neurons causes hyperactivation of IL-1 receptor pathway and induces MECP2-dependent synaptic defects. Elife 6:e21735. doi: 10.7554/eLife.21735
Toro, C., and Deakin, J. F. (2005). NMDA receptor subunit NRI and postsynaptic protein PSD-95 in hippocampus and orbitofrontal cortex in schizophrenia and mood disorder. Schizophr. Res. 80, 323–330. doi: 10.1016/j.schres.2005.07.003
Tyzio, R., Holmes, G. L., Ben-Ari, Y., and Khazipov, R. (2007). Timing of the developmental switch in GABA(A) mediated signaling from excitation to inhibition in CA3 rat hippocampus using gramicidin perforated patch and extracellular recordings. Epilepsia 48(Suppl. 5), 96–105. doi: 10.1111/j.1528-1167.2007.01295.x
Tyzio, R., Nardou, R., Ferrari, D. C., Tsintsadze, T., Shahrokhi, A., Eftekhari, S., et al. (2014). Oxytocin-mediated GABA inhibition during delivery attenuates autism pathogenesis in rodent offspring. Science 343, 675–679. doi: 10.1126/science.1247190
Tzschach, A., Grasshoff, U., Beck-Woedl, S., Dufke, C., Bauer, C., Kehrer, M., et al. (2015). Next-generation sequencing in X-linked intellectual disability. Eur. J. Hum. Genet. 23, 1513–1518. doi: 10.1038/ejhg.2015.5
Vaags, A. K., Lionel, A. C., Sato, D., Goodenberger, M., Stein, Q. P., Curran, S., et al. (2012). Rare deletions at the neurexin 3 locus in autism spectrum disorder. Am. J. Hum. Genet. 90, 133–141. doi: 10.1016/j.ajhg.2011.11.025
Valnegri, P., Montrasio, C., Brambilla, D., Ko, J., Passafaro, M., and Sala, C. (2011). The X-linked intellectual disability protein IL1RAPL1 regulates excitatory synapse formation by binding PTPdelta and RhoGAP2. Hum. Mol. Genet. 20, 4797–4809. doi: 10.1093/hmg/ddr418
Van Esch, H., Bauters, M., Ignatius, J., Jansen, M., Raynaud, M., Hollanders, K., et al. (2005). Duplication of the MECP2 region is a frequent cause of severe mental retardation and progressive neurological symptoms in males. Am. J. Hum. Genet. 77, 442–453. doi: 10.1086/444549
Vezzani, A., Balosso, S., and Ravizza, T. (2008). The role of cytokines in the pathophysiology of epilepsy. Brain Behav. Immun. 22, 797–803. doi: 10.1016/j.bbi.2008.03.009
Viviani, B., Bartesaghi, S., Gardoni, F., Vezzani, A., Behrens, M. M., Bartfai, T., et al. (2003). Interleukin-1beta enhances NMDA receptor-mediated intracellular calcium increase through activation of the Src family of kinases. J. Neurosci. 23, 8692–8700.
Viviani, B., Gardoni, F., and Marinovich, M. (2007). Cytokines and neuronal ion channels in health and disease. Int. Rev. Neurobiol. 82, 247–263. doi: 10.1016/S0074-7742(07)82013-7
Wagner, J. A. (1996). Is IL-6 both a cytokine and a neurotrophic factor? J. Exp. Med. 183, 2417–2419. doi: 10.1084/jem.183.6.2417
Watanabe, M., and Fukuda, A. (2015). Development and regulation of chloride homeostasis in the central nervous system. Front. Cell. Neurosci. 9:371. doi: 10.3389/fncel.2015.00371
Wei, H., Zou, H., Sheikh, A. M., Malik, M., Dobkin, C., Brown, W. T., et al. (2011). IL-6 is increased in the cerebellum of autistic brain and alters neural cell adhesion, migration and synaptic formation. J. Neuroinflammation 8:52. doi: 10.1186/1742-2094-8-52
Wischhof, L., Irrsack, E., Osorio, C., and Koch, M. (2015). Prenatal LPS-exposure–a neurodevelopmental rat model of schizophrenia–differentially affects cognitive functions, myelination and parvalbumin expression in male and female offspring. Prog. Neuropsychopharmacol. Biol. Psychiatry 57, 17–30. doi: 10.1016/j.pnpbp.2014.10.004
Xing, J., Kimura, H., Wang, C., Ishizuka, K., Kushima, I., Arioka, Y., et al. (2016). Resequencing and association analysis of six PSD-95-related genes as possible susceptibility genes for schizophrenia and autism spectrum disorders. Sci. Rep. 6:27491. doi: 10.1038/srep27491
Yamada, M., and Hatanaka, H. (1994). Interleukin-6 protects cultured rat hippocampal neurons against glutamate-induced cell death. Brain Res. 643, 173–180. doi: 10.1016/0006-8993(94)90023-X
Yan, H. Q., Banos, M. A., Herregodts, P., Hooghe, R., and Hooghe-Peters, E. L. (1992). Expression of interleukin (IL)-1 beta, IL-6 and their respective receptors in the normal rat brain and after injury. Eur. J. Immunol. 22, 2963–2971. doi: 10.1002/eji.1830221131
Yasumura, M., Yoshida, T., Yamazaki, M., Abe, M., Natsume, R., Kanno, K., et al. (2014). IL1RAPL1 knockout mice show spine density decrease, learning deficiency, hyperactivity and reduced anxiety-like behaviours. Sci. Rep. 4:6613. doi: 10.1038/srep06613
Yeo, M., Berglund, K., Augustine, G., and Liedtke, W. (2009). Novel repression of Kcc2 transcription by REST-RE-1 controls developmental switch in neuronal chloride. J. Neurosci. 29, 14652–14662. doi: 10.1523/JNEUROSCI.2934-09.2009
Yirmiya, R., and Goshen, I. (2011). Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain Behav. Immun. 25, 181–213. doi: 10.1016/j.bbi.2010.10.015
Yoshida, T., Yasumura, M., Uemura, T., Lee, S. J., Ra, M., Taguchi, R., et al. (2011). IL-1 receptor accessory protein-like 1 associated with mental retardation and autism mediates synapse formation by trans-synaptic interaction with protein tyrosine phosphatase delta. J. Neurosci. 31, 13485–13499. doi: 10.1523/JNEUROSCI.2136-11.2011
Keywords: synaptopathies, inflammation, cytokines, IL-1β, IL1RAPL1, neurodevelopmental diseases, KCC2, MeCP2
Citation: Pozzi D, Menna E, Canzi A, Desiato G, Mantovani C and Matteoli M (2018) The Communication Between the Immune and Nervous Systems: The Role of IL-1β in Synaptopathies. Front. Mol. Neurosci. 11:111. doi: 10.3389/fnmol.2018.00111
Received: 14 January 2018; Accepted: 20 March 2018;
Published: 05 April 2018.
Edited by:
Andras Bilkei-Gorzo, Universität Bonn, GermanyReviewed by:
Luigia Trabace, University of Foggia, ItalyRommy Von Bernhardi, Pontificia Universidad Católica de Chile, Chile
Copyright © 2018 Pozzi, Menna, Canzi, Desiato, Mantovani and Matteoli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Davide Pozzi, RGF2aWRlLnBvenppQGh1bWFuaXRhc3Jlc2VhcmNoLml0 Elisabetta Menna, ZS5tZW5uYUBpbi5jbnIuaXQ= Michela Matteoli, bS5tYXR0ZW9saUBpbi5jbnIuaXQ=