 Xue Yang1†
Xue Yang1† Yong Wang1†
Yong Wang1† Qiyan Li1
Qiyan Li1 Yaxian Zhong1
Yaxian Zhong1 Liangpei Chen1
Liangpei Chen1 Yajun Du1
Yajun Du1 Jing He1
Jing He1 Lvshuang Liao2
Lvshuang Liao2 Kun Xiong2
Kun Xiong2 Chun-xia Yi3
Chun-xia Yi3 Jie Yan1*
Jie Yan1*- 1Department of Forensic Science, School of Basic Medical Sciences, Central South University, Changsha, China
- 2Department of Anatomy and Neurobiology, School of Basic Medical Science, Central South University, Changsha, China
- 3Department of Endocrinology and Metabolism, Academic Medical Center, University of Amsterdam, Amsterdam, Netherlands
Methamphetamine (METH) is a popular new-type psychostimulant drug with complicated neurotoxicity. In spite of mounting evidence on METH-induced damage of neural cell, the accurate mechanism of toxic effect of the drug on central nervous system (CNS) has not yet been completely deciphered. Besides, effective treatment strategies toward METH neurotoxicity remain scarce and more efficacious drugs are to be developed. In this review, we summarize cellular and molecular bases that might contribute to METH-elicited neurotoxicity, which mainly include oxidative stress, excitotoxicity, and neuroinflammation. We also discuss some drugs that protect neural cells suffering from METH-induced neurotoxic consequences. We hope more in-depth investigations of exact details that how METH produces toxicity in CNS could be carried out in future and the development of new drugs as natural compounds and immunotherapies, including clinic trials, are expected.
Introduction
Methamphetamine (METH), also known as “ice” or “crystal,” is an addictive pharmacologic psychostimulant with strong neurotoxic effects on the central nervous system (CNS). It has been abused by >33 million people worldwide and seen a steady increase in use over the last few decades. Such use is associated with deleterious effects on families, loss of productivity, major public-health concerns, and a consumption of substantial resources for medical intervention (Courtney and Ray, 2014; Moratalla et al., 2017). A recent editorial in the Lancet stated that the shift in public-health priorities to opioids in the last few years in the United States has enabled the METH market to flourish; as a result, this market is primed for resurgence. Accordingly, drug control may be more challenging than anticipated as a second “METH wave” begins (Lancet, 2018).
Methamphetamine belongs to a class of synthetic drugs known as amphetamine-type stimulants, which includes amphetamine, METH, methylenedioxy-methamphetamine, and other designer drugs (Chomchai and Chomchai, 2015). METH is similar to amphetamine with regard to pharmacodynamic effects; however, users are more likely to become addicted to METH because of its better penetration into the CNS and longer duration of action (Won et al., 2013). Long-term abuse of METH causes serious physical and mental damage. Overall, METH abuse is associated with an increased risk of infection by the human immunodeficiency virus, hepatitis viruses, as well as dangerously high body temperature, periodontal disease, pulmonary hypertension, adrenergic storm, cerebrovascular events, stroke, circulatory collapse, and kidney failure (Ho et al., 2009; Schep et al., 2010; Moratalla et al., 2017). METH abusers are more likely to develop Parkinson’s disease, depression, schizophrenia, psychosis, and other neuropsychiatric and cognitive sequelae (Rawson and Condon, 2007; Forray and Sofuoglu, 2014; Hsieh et al., 2014); these are mostly attributed to METH-induced neurotoxicity. The neurotoxic effects of METH are of strong concern, and exploration of the mechanisms underlying this neurotoxicity has become a research hotspot in recent years (Xiong et al., 2016; Ashok et al., 2017; Xie et al., 2018).
In general, neurotoxicity is defined as physical damage to neurons. In a broader sense, neurotoxicity may refer to a permanent or reversible adverse effect of a substance on neuronal structure/function that induces disruption of neuronal components, collapse of entire neurons, histologic signs of neuronal injury, and/or behavioral abnormalities (Moszczynska and Callan, 2017). METH-induced neurotoxic effects include damage to dopaminergic and serotonergic terminals, neuronal apoptosis, as well as activated astroglia and microglia that lead to a neuroinflammatory response within the brain (Cadet and Krasnova, 2009; Panenka et al., 2013; Moratalla et al., 2017).
In clinical trials, psychological therapies have been shown to generate small-to-moderate reductions in METH use; however, these have not yet been translated into clinical practice (Colfax et al., 2010; Carroll, 2014). Poor outcomes of psychosocial interventions may be related to METH-produced neurotoxicity. Further, psychosocial treatments must be utilized clinically in conjunction with other strategies as pharmacotherapies (Aharonovich et al., 2006).
In this review, we discuss briefly some of the principal mechanisms underlying the neurotoxicity induced by METH and summarize targeted pharmacologic treatments. We anticipate that more efficacious intervention strategies that protect neural cells against METH-induced neurotoxic consequences may be implemented in the future.
Mechanisms Underlying METH-Induced Neurotoxicity
Oxidative Stress
The neurotoxic mechanism of METH is complex and involves multiple pathways. Oxidative stress has been demonstrated to be a significant factor contributing to cellular toxicity. METH induces the considerable production of reactive oxygen species (ROS), such as hydroxyl radicals (OH-), hydrogen peroxide (H2O2), and the superoxide anion (O2-), by increasing the oxidation of dopamine (DA) (Hansen, 2002). METH passes through the blood–brain barrier and penetrates the brain readily due to its high lipid solubility (Nordahl et al., 2003). Then, it enters dopaminergic terminals via the dopamine transporter (DAT) because of its similarity to DA (Shin et al., 2017), as well as by passive diffusion (Moszczynska and Callan, 2017). METH enhances DA concentration in the cytosol and synaptic cleft significantly by impairing vesicle monoamine transporter 2 (VMAT2) function and promoting DA release; this process may represent the main mechanism underlying the neurotoxic effect of METH in the brain (Baumann et al., 2002). Within dopaminergic terminals and in synaptic clefts, excess DA is autoxidized to quinone or semi-quinone (LaVoie and Hastings, 1999) to generate large amounts of H2O2, OH-, and O2- (Baumann et al., 2002). Further, a small proportion of DA metabolism mediated by monoamine oxidase (MAO) or catechol-O-methyltransferase (COMT) produces H2O2 as a byproduct (Cadet and Brannock, 1998; Olanow and Tatton, 1999). H2O2 reacts with transition-metal ions to produce highly toxic OH-. Eventually, abundant ROS promote a series of oxidative stress reactions, such as lipid peroxidation and activation of proteases, which trigger the cell-death cascade. Furthermore, highly toxic peroxynitrite ions (ONOO-) produced via O2- react with nitric oxide (NO) to damage proteins, nucleic acids, and phospholipids in cells by circumventing antioxidative enzymes (Cadet and Brannock, 1998; Figure 1).
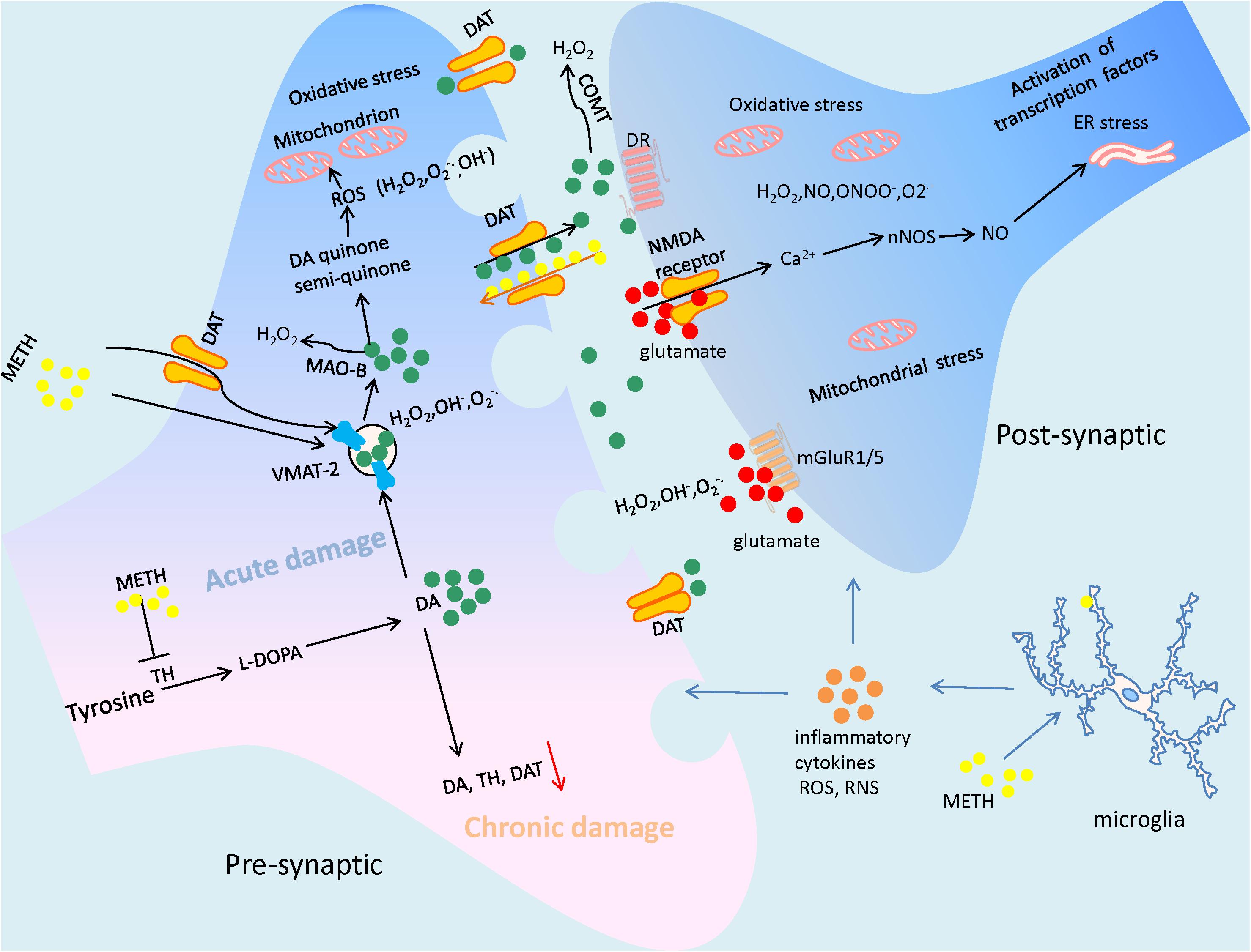
FIGURE 1. The illustration summarizes the main mechanisms of Methamphetamine (METH)-elicited neurotoxic effects, which include DA oxidation, excessive glutamate production, generating a large amount of reactive oxygen species (ROS) and reactive nitrogen species (RNS), and subsequently leading to mitochondrial dysfunction and ER stress. The neuroinflammation mediated by microglial cells also contribute to the neuronal damage by attacking it with inflammatory cytokines. As a result of the suffering from METH, the neuronal cells may undergo terminal degeneration or apoptosis. In particular, due to the neurotoxicity of the drug, long time abuse of METH often cause the decrease of dopaminergic markers such as dopamine (DA), tyrosine hydroxylase (TH), and dopamine transporter (DAT).
Mitochondria represent a major site of METH-induced ROS production within neural cells (Dawson and Dawson, 2017). Mitochondria, which are intracellular organelles composed of two bilayers, act as the energy generators of cells through oxidative phosphorylation and adenosine triphosphate (ATP) production. Defects in mitochondrial respiration have been implicated in neuronal death and several neurodegenerative diseases (Dawson and Dawson, 2017). Several studies have suggested that dysfunction of mitochondrial metabolism plays a critical role in METH-induced dopaminergic neurotoxicity through inhibition of the Krebs cycle and electron transport chain (ETC) as well as by promotion of oxidative stress; these effects result in imbalance between oxidation and antioxidation in neural cells (Annepu and Ravindranath, 2000; Beer et al., 2004; Shin et al., 2017). ROS and reactive nitrogen species (RNS) generated by DA oxidation inhibit several key enzymes directly as complexes I, II, III, and IV of the ETC, causing mitochondrial dysfunction and damage to DNA structure as well as loss of genetic information (Burrows et al., 2000; Brown et al., 2005; Bachmann et al., 2009; Moratalla et al., 2017; Moszczynska and Callan, 2017). In turn, inhibition of ETC components by METH enhances O2- production due to electron leakage. This positive-feedback loop aggravates disturbance of mitochondrial energy metabolism and neurotoxicity.
The changes in mitochondrial enzymes in response to METH remain controversial. Klongpanichapak et al. (2006) found significant inhibition of expression of striatal complex I following repeated METH treatment in mice. In accordance with this finding, Thrash et al. (2010) and Thrash-Williams et al. (2013) showed that intraperitoneal administration of METH decreased the activity of striatal complex I significantly but had no significant effect on the activity of complex IV in vivo or in vitro. Several researchers have found that acute exposure to METH induces a decrease in glutathione (GSH) levels and an increase in levels of oxidized glutathione (GSSG) in the striatum; this leads to a reduction in the GSH/GSSG ratio, which is essential for the inhibition of striatal complex I activity (Annepu and Ravindranath, 2000; Beer et al., 2004). In contrast, a regimen of rapid (<1 h) binge administration of METH decreased complex II activity in the striatal brain regions, but did not decrease the activity of complex I in vivo (Brown et al., 2005). Killinger et al. (2014) also found that a binge regimen of METH every 2 h, via four successive intraperitoneal injections, did not alter the levels of mitochondrial complex I in striatal synaptosomes in vivo. An in vitro study showed no significant alterations in the protein content of mitochondrial respiratory complex I; however, METH treatment caused time-dependent reductions in the protein contents of complex IV (Wu et al., 2007). Although the reactions of the mitochondrial ETC differ according to the route of drug delivery and modes of METH administration, these studies suggest that METH inhibits the activity of the mitochondrial respiratory chain complex; this effect is considered to play a crucial role in the pathogenesis of several psychiatric disorders such as depression, bipolar disorder and schizophrenia (Manji et al., 2012).
In recent years, dynamic disorders of mitochondria have been reported to result from mitochondrial dysfunction triggered by METH (Lenzi et al., 2012; Lin et al., 2012). In general, mitochondrial biogenesis coupled with dynamic fusion and fission maintain healthy mitochondria, whereas damaged mitochondria are degraded by mitophagy (Michel et al., 2012). The key molecules of mitochondrial biogenesis are peroxisome proliferator-activated receptor gamma coactivator-1α (PGC-1α), nuclear respiratory factors (NRFs), and mitochondrial transcription factor A (TFAM) (Lee and Wei, 2005). PGC-1α regulates and coordinates the activity of NRF and TFAM to serve as a nutrient-sensing system that increases mitochondrial biogenesis (Nervina et al., 2006). Using a repeated escalating METH regimen in rats, Valian et al. (2017) detected an increase in expression of PGC1α and TFAM in the substantia nigra, but not the striatum. However, Beirami et al. (2018) revealed a significant decrease in expression of PGC-1α, NRF, and TFAM in the hippocampus of rats with a different repeated METH administration regimen. These inconsistent results suggest that the indicators of mitochondrial biogenesis appear to be expressed aberrantly in various brain regions or according to the route of drug administration.
The regulation of mitochondrial fusion is primarily through mitofusin-1, mitofusin-2, and optic atrophy protein 1 (Opa1), whereas mitochondrial fission is regulated by mitochondrial fission 1 protein (Fis1) and dynamin-related protein 1 (Drp1) (Ding et al., 2012). In in vitro studies, Parameyong et al. (2013, 2015) revealed that METH decreased cell viability significantly and increased the levels of Fis1 and Drp1 in isolated mitochondria, whereas Drp1 expression in the cytosol of METH-treated cells showed no significant differences compared with the control group. Interestingly, Borgmann and Ghorpade (2018) reported a larger and more rod-shaped morphology and dysfunction of mitochondria in astrocytes during prolonged exposure to low levels of METH. This may have been mediated by inhibition of phosphorylation of Drp1 and an increase in mitofusin levels, implying an overall increase in the number of mitochondria in astrocytes (Borgmann and Ghorpade, 2018). Considering the difference of dose and time points of METH used in these studies, the regimen of drug administration may be the main cause of the alteration in mitochondrial dynamics. Nonetheless, the mechanisms of mitochondrial impairment induced by METH have not been elucidated to date.
Oxidative stress occurs primarily in mitochondria and leads to mitochondrial dysfunction by attacking mitochondria in the CNS (Thrash-Williams et al., 2016). Accordingly, METH-induced mitochondrial damage may contribute to dopaminergic toxicity by enhancing susceptibility to oxidative stress and promoting the apoptosis of neural cells. This phenomenon is of clinical relevance as it eventually results in devastating neuropathological effects in the brain due to mitochondrial impairment, subsequent caspase activation, and apoptotic neuronal death following METH administration (Nguyen et al., 2015; Chamorro et al., 2016; Xiong et al., 2017). For instance, METH exposure has been shown to increase expression of the pro-apoptotic proteins Bax, Bad, and Bid (Jayanthi et al., 2001; Bachmann et al., 2009; Beauvais et al., 2011; Raineri et al., 2012) and decrease the expression of the anti-apoptotic proteins Bcl-2 and Bcl-xL (Jayanthi et al., 2001; Beauvais et al., 2011; Raineri et al., 2012). The decrease in the Bcl-2/Bax ratio in mitochondrial fractions has been shown to promote cytochrome c (Cyt c) release from mitochondria (Qiao et al., 2014; Nam et al., 2015). Subsequently, Cyt c becomes part of the apoptosome with apoptotic peptidase activating factor-1 (Apaf-1) and induced sequential activation of the apoptosis executioners caspase-3, -6, and -7 (Shin et al., 2017).
Several studies have reported that multiple molecules are involved in the apoptotic death of neurons induced by METH. For instance, Kim et al. (1999) found that inhibition of protein kinase C delta (PKCδ) or overexpression of a cleavage-resistant PKCδ mutant protected against METH-elicited apoptosis in mesencephalic dopaminergic cell cultures in vitro (Lin et al., 2012). In follow-up studies, they showed that PKCδ inhibition may rescue METH-elicited mitochondrial burden, pro-apoptosis, and dopaminergic degeneration, implying that PKCδ is an important gene involved in METH-induced mitochondrial dysfunction and apoptosis in dopaminergic neuronal cells (Nam et al., 2015; Nguyen et al., 2015; Shin et al., 2016; Mai et al., 2018).
Recently, Chen et al. (2016) reported that p53-upregulated modulator of apoptosis (PUMA) was involved in the mitochondrial apoptotic pathway induced by METH in PC12 cells and SH-SY5Y cells. They suggested that PUMA interacts with Bax and Bcl-2 in mitochondrial membranes to drive Cyt c relocation from mitochondria to the cytoplasm, causing activation of caspase-3, poly-ADP-ribose polymerase (PARP) and apoptosis (Chen et al., 2016). Furthermore, through microinjection of anti-micro (mi)R143 into the hippocampi of mice, Zhang et al. (2016) revealed that miR143-dependent PUMA upregulation reversed the METH-induced decrease in microglial survival via regulation of apoptosis and autophagy. In addition to miRNAs, long non-coding RNAs (lncRNAs) appear to participate in METH-induced neuronal apoptosis by regulating the coding genes of neurons. In our recent investigation, we reported that several lncRNAs were expressed differentially in primary cultured prefrontal cortical neurons treated with METH. Further, using bioinformatics, we hypothesized that lncRNA GAS5 modulates downstream molecules involved in p53-mediated neuronal apoptosis, although more direct evidence from in vivo and in vitro studies is needed (Figure 2; Xiong et al., 2017).
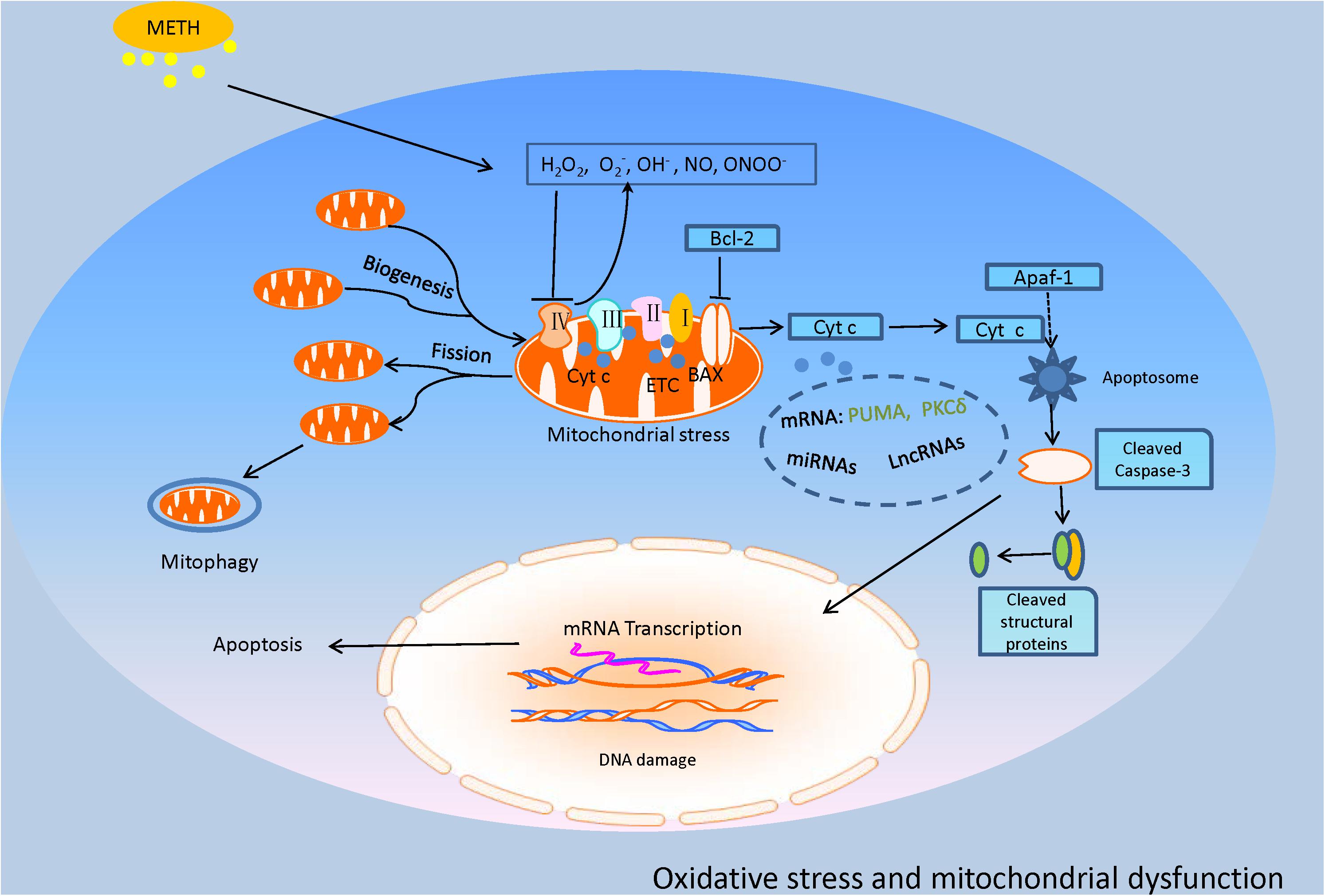
FIGURE 2. This model illustrates the oxidative stress and mitochondrial dysfunction involved in METH-induced neurotoxic consequences. METH exposure produced a considerable amount of ROS and RNS, named OH-, H2O2, O2-, NO, and ONOO-. The excessive oxidative stress inhibits the key enzymes of the ETC, causing mitochondrial dysfunction that leads to mitochondrial fission and mitophagy. Particularly, the impaired mitochondria trigger the increase of Bax and decrease of Bcl-2 and sequential cytochrome c (Cyt c) release, inducing activation of executioner caspases-3 and apoptosis which might be regulated by some molecules such as p53-upregulated modulator of apoptosis (PUMA), protein kinase C delta (PKCδ), miRNAs, and long non-coding RNAs (lncRNAs) which are reviewed in the text.
Excitotoxicity
Glutamate (Glu), which is the main excitatory neurotransmitter in the brain, has been reported to have an important role in METH-induced excitotoxicity (Moratalla et al., 2017). Glu accumulation over-activates various downstream signaling pathways, mostly involving a surge in Ca2+ influx, to trigger an increase in intracellular Ca2+ concentration (Chamorro et al., 2016). Specifically, excessive Glu activates N-methyl-D-aspartate receptors (NMDARs) and metabotropic glutamate receptors (mGluRs) (Tseng et al., 2010). Activation of mGluRs induces protein kinase C (PKC) phosphorylation and upregulates NMDAR function, leading to an increase in Ca2+ influx, which acts as a pervasive and pluripotent second messenger (Bahar et al., 2016). Excess intracellular Ca2+ triggers a cascade of reactions within cells to activate protein kinases, phosphatases, and nitric oxide synthase (NOS). The latter subsequently promotes NO production (Tseng et al., 2010), which causes endoplasmic reticulum (ER) stress, activation of apoptotic pathways and, eventually, METH-produced neurotoxic sequelae (Moratalla et al., 2017).
In particular, ER stress occurs under various toxic stimuli along with accumulation of misfolded proteins and activation of unfolded protein response (UPR), which removes unfolded and/or misfolded proteins in the ER, thereby recovering ER homeostasis. ER stress leads to the activation of three ER-resident transmembrane proteins: activating transcription factor-6 (ATF6), inositol requiring protein-1 (IRE1) and protein kinase RNA-like ER kinase (PERK) (Szegezdi et al., 2006; Hetz, 2012). The roles of these three signaling pathways involve a reduction in protein synthesis and expression of specific genes to cope with proteotoxic stress (Wongprayoon and Govitrapong, 2017). During prolonged ER stress, IRE1, PERK, and ATF6 may induce pro-apoptotic signaling through activation of C/EBP homologous protein (CHOP), which subsequently leads to the initiation of ER stress-mediated apoptosis through regulation of Bcl-2 family members (Bahar et al., 2016). ER stress leads to apoptosis, including various mechanisms involving activation of death receptors and participation of the mitochondria-dependent cell death pathway (Sano and Reed, 2013). It has been shown that neurotoxic doses of METH induce expression of several ER stress genes, including those that encode the 78-kDa glucose-regulated protein (GRP-78), CHOP, and ATF4, in the rat striatum (Jayanthi et al., 2004; Beauvais et al., 2011). The ER stress induced by METH appears to be associated with dopaminergic toxicity and activation of the DA D1 receptor (Beauvais et al., 2011). Recently, Wongprayoon and Govitrapong (2017) suggested that METH-induced apoptotic death is mediated (at least in part) through an ER-dependent mechanism involving CHOP, spliced X-box binding protein 1 (XBP1), caspase-12, and caspase-3 in vitro. In addition, exposure to relatively high-dose METH increases nuclear protein 1 (Nupr1) expression, which promotes dopaminergic neuronal apoptosis and autophagy through a Nupr1/CHOP pathway (Figure 3; Xu X. et al., 2017).
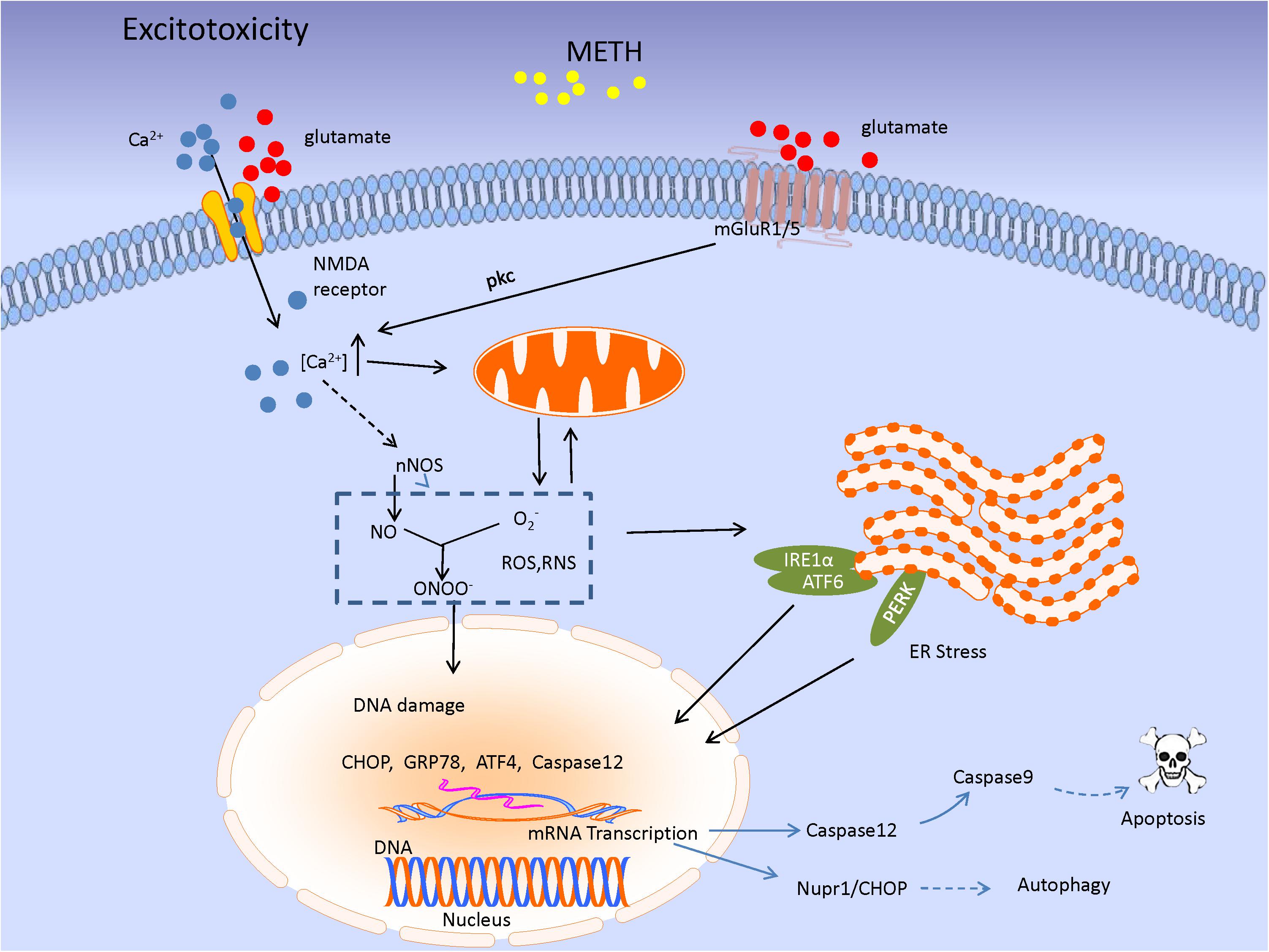
FIGURE 3. The image shows the excitatory toxicity model of METH. METH-mediated increase in extracellular glutamate level leads to stimulation of mGluR1/5 or N-methyl-D-aspartate receptors (NMDARs). mGluR1/5-induced protein kinase C (PKC) activation phosphorylates and upregulates NMDAR function, leading to Ca2+ influx. The signaling results in enhancement of cytosolic Ca2+ level associated with nNOS activity, leading to NO production. NO acts as an ER stressor, and then, UPR signaling pathway would be initiated in response to ER stress through three ER transmembrane mediators [inositol requiring protein-1 (IRE1)α, activating transcription factor-6 (ATF6), and protein kinase RNA-like ER kinase (PERK)]. Subsequently, the mediators lead to special genes transcription as CHOP, GRP78, and Caspase 12 which triggering a series of cascade involving apoptosis and autophagy.
Neuroinflammation
Reactive neurogliocytes are considered to be sensitive markers of nerve damage, which is a common response to CNS injury (Ares-Santos et al., 2013). The neuroinflammation caused by METH shows a close correlation with microglial activity as it is activated rapidly after METH administration in DA-innervated areas. Microglial activation following METH exposure may result from the neuronal release of damage-associated molecular patterns (DAMPs) (Xu E. et al., 2017). For instance, high-mobility group box-1 (HMGB1) expression was found up-regulated in response to METH treatment and shown to mediate the neuroin?ammatory response in the nucleus accumbens, ventral tegmental, and prefrontal cortex of the brain (Frank et al., 2016). Thomas and colleagues suggested that METH-induced microglial activation was regulated by DA-quinone (DAQ), a metabolite of DA, because it activated microglia dose- and time-dependently, and because inhibition of DAQ formation blocked (at least partially) microgliosis (Thomas et al., 2008). The vital role of DA in neuroinflammation was also evidenced by the observation that excessive DA released into the synaptic cleft may stimulate regional microglia directly and trigger a neurotoxic signal cascade (Thomas et al., 2008). The underlying mechanism of microglial activation induced by METH is related to the Toll-like receptor 4 (TLR4) located on microglia, which is involved in the immune surveillance of pathogens and exogenous small molecules (Bachtell et al., 2015). Activation of microglia may also be mediated through the sigma-1 receptor, which involves ROS generation and activation of the mitogen-activated protein kinase (MAPK) and PI3K/Akt pathways in the neurotoxicity of METH (Chao et al., 2017).
Activated microglial cells secrete not only neurotrophic factors to prolong neuronal survival, but also cytotoxic mediators and cytokines that induce inflammation and neurotoxicity (Zhang et al., 2016). Studies have suggested that METH activates nuclear factor-kappa B (NF-κB), inducing its transfer to the nucleus and promoting the transcription of pro-inflammatory cytokines in microglia (Ojaniemi et al., 2003; Shah et al., 2012; Snider et al., 2013). This results in the release of various pro-inflammatory factors such as interleukin 6 (IL-6), interleukin 1β (IL-1β), tumor necrosis factor-α (TNF-α), monocyte chemo-attractant protein 1 (MCP-1), and cellular adhesion molecule (ICAM-1) (Yamaguchi et al., 1991; Nakajima et al., 2004; Goncalves et al., 2008; Snider et al., 2012), which are thought to play key roles in METH-induced neuroinflammation (Figure 4). Interestingly, astrocytes (which protect neurons and promote sprouting) are activated because glial fibrillary acidic protein (GFAP) immunoreactivity increases in the striatum and indusium griseum upon METH treatment, thereby implying complex mechanisms involving guidance molecules and cytotoxic mediators in the neurotoxic consequences of METH (Ares-Santos et al., 2013; Moratalla et al., 2017).
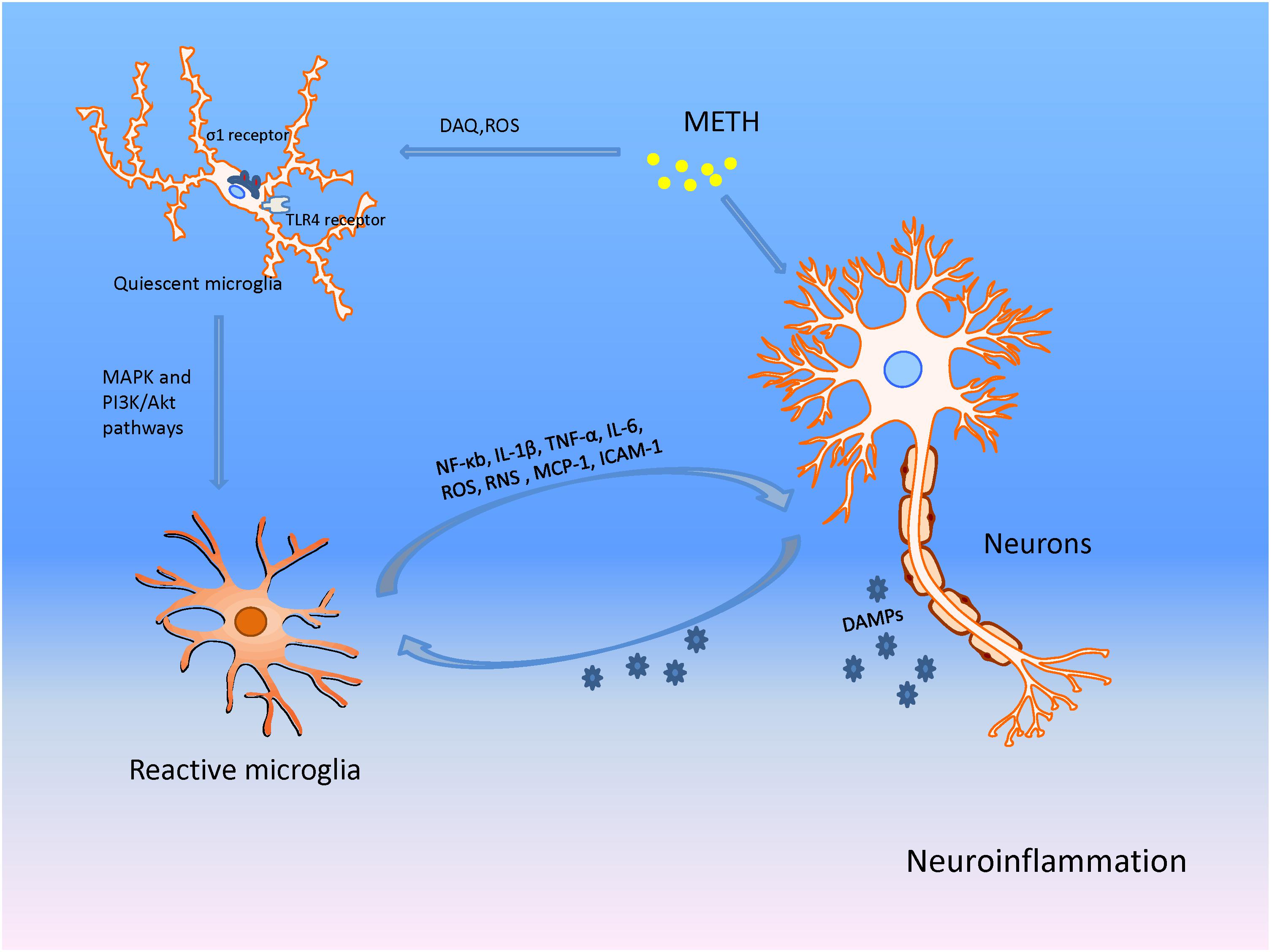
FIGURE 4. METH produces neuronal damage through microglia associated neuroinflammation in addition to direct actions on neurons. METH damages presynaptic terminals of neurons causing the production of DA-quinone (DAQ) and sequential ROS; these facilitate microglial activation. The activated microglia then increase production of nuclear factor-kappa B (NF-κB), tumor necrosis factor-α (TNF-α), interleukin 6 (IL-6), interleukin 1β (IL-1β), monocyte chemo-attractant protein 1 (MCP-1), ICAM-1, ROS, and RNS, promoting neuroinflammation and neuronal injury. The damaged neurons release DAMPs that act on microglia, and aggravate the inflammation and eventual neurotoxicity through the positive feedback mechanism.
Long-Term Neurotoxicity
The effects of long-term exposure to METH are different from those of acute injury. The former is characterized by impairment of expression of tyrosine, tyrosine hydroxylase (TH), DAT, and serotonin transporters (SERT), as well as DA depletion, decrease in the density of DA D2 receptors, hypo-dopaminergic status, and neuronal degeneration (Barr et al., 2002). In Wilson et al. (1996) observed decreased expression of DA, TH, and DAT in the striatum of chronic METH abusers post mortem, which might explain the dysphoric effects of the drug and dose escalation observed in some METH users (Wilson et al., 1996). The decrease in levels of dopaminergic markers in the striatum was shown by other studies, and could last for months to years after METH abstinence in human abusers of METH (Volkow et al., 2001a,b; Kitamura et al., 2007). Besides the striatum, a long-lasting decrease in DAT levels was also observed in the nucleus accumbens and prefrontal cortex of the brain (Davidson et al., 2001; Sekine et al., 2001). In accordance with this study, several previous reports have demonstrated that the density of DAT, VMAT2, SERT, and DA D2 receptors is significantly lower than that in healthy controls according to positron emission tomography (PET) scans of the brains of METH abusers (Sekine et al., 2001; Volkow et al., 2001b, 2015; Kish et al., 2009; Boileau et al., 2016). Similar to that observed in METH abusers, significant and sustained reductions in the levels of DA, TH, and DAT were shown following single, high-dose, or binge administration of METH in animal models (Figure 1; Hassan et al., 1973; McConnell et al., 2015; Nguyen et al., 2015). These results suggest persistent DA deficits as well as structural and metabolic dysfunctions in specific brain regions that correlate with several types of behavioral neurologic sequelae induced by METH (Moszczynska and Callan, 2017).
When using a regimen that involves gradual increases in METH administration to rats to mimic progressively larger doses of the drug used by some human abusers of METH, METH preconditioning protects against DA depletion caused by binge METH challenge in the brain (Cadet et al., 2011; Shen et al., 2016; Li et al., 2017). Further, chronic injections of METH activate programs that prevent DA toxicity without influencing drug-induced pathologic changes in serotoninergic systems (Cadet et al., 2009). The plethora of exciting applications of preconditioning in neuroprotection (Shen et al., 2016; Li et al., 2017) reflect differences in the mechanisms involved in METH toxicity on monoaminergic systems between the various routes of METH administration. For instance, Cadet et al. (2011) reported differential METH-induced gene expression in the striatum, including brain-derived neurotrophic factor (BDNF), heme oxygenase-1 (HO-1) and heat shock protein 27 (Hsp27), in the absence and presence of METH preconditioning and between various brain regions. This result implies that the brain cannot be considered as a homogeneous structure when assessing the molecular effects of METH preconditioning (Cadet et al., 2011; Table 1).
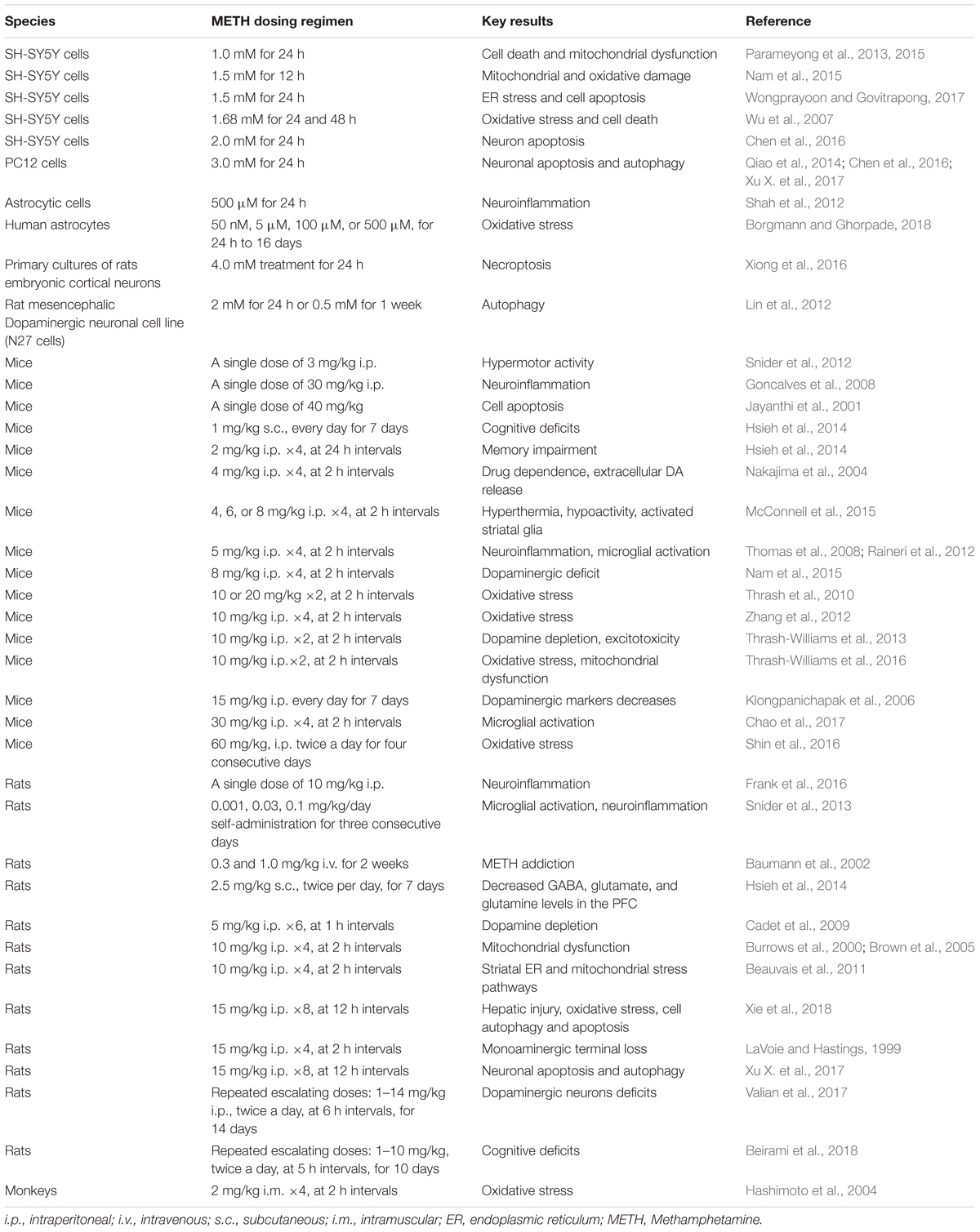
TABLE 1. The summary of common METH treatment protocols in vivo or in vitro.
Treatment of METH-Induced Neurotoxicity
Targeting Oxidative Stress and Mitochondrial Toxicity
Preclinical and clinical investigations have been applied to seek effective and efficacious pharmacologic strategies for treatment of METH neurotoxicity. As stated above, METH interferes with DA reuptake and leads to DA oxidation; this leads to the production of ROS and RNS, which trigger degeneration of dopaminergic terminals and neuronal apoptosis. This pathway demonstrates that oxidative stress is one of the main mechanisms through which METH injures the CNS. Accordingly, antioxidant pharmacotherapies have been applied to explore efficacious strategies for the protection of neural cells suffering from oxidative stress generated by METH.
For example, vitamin C (Vit.C) reduces the production of free radicals, maintains GSH homeostasis, and induces the expression of HO-1, which is critical in limiting cellular damage by maintaining redox homeostasis within the brain (Rice, 2000; Hediger, 2002). Huang et al. (2012) found that pretreatment with Vit.C enhanced METH-elicited HO-1 expression and attenuated METH-induced ROS production in neuronal/glial co-cultures. Conversely, pharmacologic inhibition of HO-1 activity abolished the suppressive effects of Vit.C (Huang et al., 2012). Recently, Huang et al. (2017) found that treating cells with Vit.C before METH exposure attenuated production of ROS and Beclin 1 time-dependently, suggesting that the protective effect of Vit.C against METH toxicity is achieved via the attenuation of ROS production and apoptosis, and that HO-1 induction by Vit.C serves as a strategy for alleviation of METH neurotoxicity. However, an in vivo study to test the neuroprotective action of Vit.C in terms of METH exposure has not been conducted.
Selenium is an essential mineral found naturally in water, soil, and food. It is often used as an antioxidant and dietary supplement (Wang et al., 2017). In an in vivo study, Kim et al. (1999) found that selenium supplementation for 13 weeks significantly blocked a METH-induced increase in the lipid-peroxidation marker malondialdehyde (MDA), decreased the ratio of GSH/GSSG, and appeared to attenuate the loss of DA in the striatum and substantia nigra. Consistently, Imam et al. (1999) demonstrated that selenium pretreatment in drinking water for 1 week prevented (at least in part) the depletion of striatal DA induced by METH exposure in rodent brains. Further in vitro research supported their findings by showing that the increased oxidative stress induced by METH is reversed upon the treatment of SH-SY5Y neuronal cells with selenium, possibly through a reduction in glutathione peroxidase (GPx) levels. This effects is attributed to the incorporation of selenium into the amino acid selenocysteine by GPx1 and GPx4, which have antioxidant functions (Barayuga et al., 2013). However, in view of the narrow range between the therapeutic and toxic doses of selenium, as well as the dependence of the effect on the applied formulation, dose, and method of treatment, supplementation should be undertaken with appropriate precautions and avoidance of the side-effects of selenium (Ungvari et al., 2014; Ghosh et al., 2015; Kielczykowska et al., 2018).
Considering that mitochondrial dysfunction plays an important part in METH-induced neurotoxic insult, improvement of mitochondrial function might ameliorate the oxidative stress and neural damage associated with this disorder. The mood stabilizers lithium and valproate attenuate a series of METH-induced changes, such as reduction of mitochondrial Cyt c, the mitochondrial anti-apoptotic Bcl-2/Bax ratio, activity of mitochondrial Cyt c oxidase, and the expression of several proteins related to the apoptotic pathway. These phenomena illustrate that lithium and valproate enhance mitochondrial function and protect against the mitochondria-mediated toxicity of METH (Bachmann et al., 2009). Feier et al. (2013) found that lithium and valproate attenuated the effects of METH on the activity of enzymes in the Krebs cycle, thereby alleviating the impairment of respiratory chain complex activity (complexes I, II, III, and IV). In their study, the effects of lithium and valproate on some enzymes and in some brain areas were not always identical. For instance, interventions using lithium and valproate reversed the decrease in complex II activity in the hippocampus and striatum of rats; however, in the amygdala and prefrontal cortex, valproate (but not lithium) increased complex II activity, notably in the METH group. This region-specific effect on oxidative stress is in accordance with the previously shown heterogeneity of oxidative-stress parameters across brain regions and treatment regimens (Musavi and Kakkar, 2003).
Nicotinamide, which is a co-factor in the ETC, has been reported to be an efficacious treatment for mitochondrial encephalopathies, possibly through energy repletion (Penn et al., 1992). Huang et al. (1997) revealed that nicotinamide treatment before METH injection attenuated reductions in striatal DA and ATP content 5 days later in vivo. These findings suggest a close relationship between METH-induced perturbations of energy metabolism and dopaminergic neurotoxicity, and that potential therapeutic strategies might bypass bioenergetic defects if defective mitochondrial energy metabolism has a role in neurotoxicity. Inconsistently, Stephans et al. (1998) found that nicotinamide perfusion during METH administration had no effect on the long-term toxicity induced by METH. Given the localized effect of nicotinamide on nerve terminals (i.e., striatum), it is likely to produce consequences vastly different from those produced by systemic administration of METH. The distinct routes of administration in the two studies, as well as altered pharmacokinetics and bioavailability, might account for this discrepancy.
It has been reported that METH users experience a higher risk of developing Parkinson’s disease despite the different brain areas affected by METH and Parkinson’s disease (Callaghan et al., 2010, 2012; Curtin et al., 2015). Talipexole is used as an antiparkinsonian agent in Japan. It was shown to react strongly with OH-, with similar reaction kinetics against METH neurotoxicity in vitro and in vivo, indicating the potency of the neuroprotective action of talipexole due to its scavenging of OH- (Mizuno et al., 1993; Kondo et al., 1998). In addition, Kish et al. (2017) suggested that, in principle, the striatal DA deficiency caused by METH may be corrected by DA substitution if safety and patient selection could be resolved. We hope that these findings will prompt researchers to investigate the potential of antiparkinsonian drugs and therapies targeted at specific brain regions for amelioration of the effects of METH on the brain.
Targeting Excitotoxicity
Excitatory Receptor Antagonists
As discussed above, the excitatory toxicity of METH is closely related to the high release of Glu, which activates NMDARs and GluRs, leading to the influx of excess Ca2+ and triggering a series of intracellular cascade reactions. Therefore, several drugs targeting these receptors have been explored in recent years.
For example, melatonin is a potent protector against oxidative damage and regulates the movement of free Ca2+ intracellularly in the CNS (Suwanjang et al., 2016; Xu et al., 2016). Using a melatonin preparation, Singhakumar et al. (2015) demonstrated that the suppression of neuronal nitrogen-activated protein kinase and alteration of the NMDAR subunits NR2A and NR2B induced by METH were attenuated in the hippocampus of mice. Ekthuwapranee et al. (2015) suggested that melatonin reduced the METH-elicited inflammation, autophagy, and death of hippocampal progenitor cells in vitro. These findings are of interest to clinicians as melatonin protects against mitochondrial dysfunction, apoptosis, and dopaminergic degeneration, which are considered to contribute to several psychosomatic manifestations in METH abusers.
N-acetylcysteine (NAC) is a pro-glutamatergic compound. It is of considerable interest because NAC attenuates the excitatory toxicity of Glu by standardizing extracellular Glu levels in the nucleus accumbens via stimulation of the cystine–glutamate antiporter (Mcketin et al., 2017). Other actions of NAC include antioxidant activity, modulation of DA release, improvement of mitochondrial dysfunction, and reductions in levels of pro-inflammatory cytokines (Scofield and Kalivas, 2014). Several investigators have examined the effects of NAC in METH-exposure models owing to its broad efficacy against neuropsychiatric disorders involving schizophrenia, bipolar disorder, depression, memory impairments, and cognitive sequelae (Berk et al., 2012, 2014; Rapado-Castro et al., 2017). They found that pre-treatment with NAC significantly improved the reduction in density of DA transporters induced by high-dose METH treatment in mice, rats, and monkeys using PET (Hashimoto et al., 2004; Zhang et al., 2012). These results suggest that NAC reduces neurotoxic damage and possibly alleviates associated neuropsychiatric symptoms in METH abusers; these data were supported by clinical findings (Gray et al., 2012; Prado et al., 2015). Nonetheless, full-scale clinical trials to establish definitively if NAC has a therapeutic benefit (and the nature of this benefit) are needed.
Other chemical compounds targeting excitatory receptors have also shown therapeutic potential against METH. Baldwin et al. (1993) found that dizocilpine, a non-competitive NMDAR antagonist, acted as a potent anti-convulsant and protected dopaminergic neurons in the striatum against METH-induced neurotoxicity in vivo. Ma et al. (2013) reported that topiramate showed considerable potential in reducing the excitatory toxicity of METH. They hypothesized that topiramate has a complex mechanism of action that includes antagonism of several Glu receptors, blockade of voltage-dependent Na+ channels, and inhibition of carbonic anhydrase (Ma et al., 2013). In addition, Baptista et al. (2012) suggested that neuropeptide Y, with receptors Y1, Y2, and Y5, reduce Glu release to protect neurons from METH excitotoxicity. Activation of Y1 or Y2 receptors prevents METH-induced cell death, and the Y1 subtype is responsible for blocking the decrease in neuronal differentiation induced by METH (Baptista et al., 2012).
Neuronal (n)NOS Inhibitors
The excitability of METH has been shown to be closely related to ONOO- generation, which can be protected selectively by nNOS inhibitors, ONOO- scavengers, or decomposition catalysts (Imam et al., 2000). nNOS inhibitors, such as 7-nitroindazole (7-NI) (Di Monte et al., 1996; Virmani et al., 2003), AR-R17477AR (Sanchez et al., 2003), S-Methylthiocitrulline, 3-bromo-7-nitroindazole (Itzhak et al., 2000), and the new nitrone-based radical scavenger S34176 (Lockhart et al., 2005), have shown significant protective effects and been used in investigations of METH-evoked neurotoxicity. Callahan and Ricaurte (1998) found that 7-NI generated hypothermic effects and afforded total protection against the DA depletions elicited by METH in the striatum of mice at 20°C. However, at 28°C, 7-NI produced minimal effects on body temperature and failed to alleviate METH-induced DA reductions. This findings suggest that the neuroprotective action of 7-NI was likely related to its ability to induce hypothermia (Callahan and Ricaurte, 1998). Itzhak et al. (2000) revealed that pretreatment with 7-NI before METH injection afforded protection against the depletion of dopaminergic markers induced by METH, but did not affect persistent hyperthermia at a low dose. These data suggest that diminished production of NO by nNOS inhibitors, rather than thermoregulation, might prevent METH-produced neurotoxicity (Itzhak et al., 2000). However, the detailed mechanism of action, such as the possible role of NO regulation by 7-NI, has not been elucidated.
Anti-neuroinflammation
The inflammatory response induced by activated microglia plays a crucial role in the neurotoxicity of METH. Hence, blockade of microglial activation seems to be a promising method for the suppression of METH-induced neurotoxic effects (Abdul Muneer et al., 2011). The antibiotic minocycline has anti-inflammatory and neuroprotective effects in the CNS that are thought to be mediated by the inhibition of microglial activation (Zhang et al., 2006). Zhang et al. (2006) observed that the reduction of DA and DAT immunoreactivity after repeated administration of METH was attenuated dose-dependently if minocycline was used in the striatum of mice. They suggested that minocycline may (at least in part) protect against METH-induced neurotoxicity via inhibition of microglial activation in vivo (Zhang et al., 2006). The phosphodiesterase-4 inhibitor ibudilast increases brain levels of glial-derived neurotrophic factor and reduces microglial activation and production of pro-inflammatory cytokines (Charntikov et al., 2015). Clinical trials have shown that ibudilast can improve METH-induced acute injury (Bates et al., 2006). Further, ibudilast reverses the decrease in levels of synaptic signaling protein produced by chronic intake of METH (Charntikov et al., 2015). Although further clinical trials and animal experiments are needed, minocycline and ibudilast may have therapeutic potential for METH neurotoxicity through modulation of neuroimmune signaling. In addition, Raineri et al. (2012) revealed that modafinil (a cognitive enhancer with weak stimulant-like behavior used for the treatment of sleep apnea, narcolepsy, and shift work-induced sleep disorders) prevented METH-induced microglial and astroglial activation in the human brain, thus avoiding the induction of inflammatory processes.
Vaccine Immunotherapies
Immunotherapies reduce the amount of drug in the CNS by triggering the production of antibodies binding the drug molecule after systemic absorption of METH (Ballester et al., 2017). Miller et al. (2013) found that, in rats receiving a KLH-conjugated METH-like hapten vaccine (MH6-KLH) and the vaccine succinyl MA (SMA–KLH), higher antibody titer-dependent METH serum concentrations, yet lower METH concentrations, in the CNS were observed, which suggested reduced METH concentrations in the brain. In addition, it was shown that anti-METH/AMP mAb4G9 therapy protected maternal and fetal rat brains from METH-induced damage (Gentry et al., 2006; White et al., 2014). Another study reported that a human–mouse monoclonal antibody binding to methylphenidate, named anti-METH antibody (mAB7F9), elicited a significant reduction in the METH concentration (Ballester et al., 2017). In particular, a combined approach using monoclonal and polyclonal antibodies was more effective in reducing the METH concentration in the brain (Hambuchen et al., 2015). However, the limitations of a vaccine remain; these include incomplete blockade of drug effects, prolonged delay in the production of sufficient circulating antibodies, and considerable variation in the antibody titer (Baracz and Cornish, 2016). Furthermore, given that anti-METH antibodies cannot cross the blood–brain barrier, this therapy may be too expensive because antibodies must be administered repeatedly to maintain an effective level (Chen et al., 2013).
Drugs for Chronic Damage
Long-term exposure to METH leads to DA depletion, impairment of dopaminergic markers, and neuronal degeneration (Barr et al., 2002; Ballester et al., 2017). Therefore, extensive investigations, including those of several drugs in clinical trials, have been performed to explore efficacious pharmaceutical interventions for recovery from neural injury resulting from METH use. For instance, Reiner et al. (2014) reported that 9 cis-retinoic acid (9cRA), a biologically active derivative of Vit.A, reversed the METH-induced decrease in immunoreactivity and apoptosis of T-helper cells via inhibition of the export of nuclear receptor 77 from the nucleus to the cytosol in primary cultured dopaminergic neurons. Their in vivo experiments showed that 9cRA (delivered via the intracerebroventricular route) also antagonized the immunoreactivity of T-helper cells and locomotor activity in the striatum of rats indirectly through a signaling mechanism involving bone morphogenetic protein. This result is similar to that of a study by Yin et al. (2012) showing that 9cRA produced neuroprotective effects in a rodent model of Parkinson’s disease involving reduced rotational behavior and loss of T-helper cells in the substantia nigra, but increased DA release in the striatum. In the report by Yin et al. (2012), 9cRA was repeatedly administered through the intra-nasal route as intracerebral delivery is not feasible for repeated drug administration and may require chronic cannulation, whereas small molecules bypass the blood–brain barrier and reach brain parenchyma non-invasively if delivered via the intranasal route.
Cognition-enhancing as well as antidepressant and neuroprotective effects are conferred by 7, 8-Dihydroxyflavone (7, 8-DHF), a high-affinity tropomyosin receptor kinase B (TrkB) agonist that activates downstream signaling (Devi and Ohno, 2012; Hashimoto, 2013). Ren et al. (2014) observed that 7, 8-DHF significantly prevented the reduction of DAT and microglial activation in the striatum of mice after repeated METH administration. In their study, it was investigated whether 7, 8-DHF prevents neurotoxicity through a signaling pathway upstream of DA terminals, and if reduction of microglial activation occurs as a consequence (rather than a cause) of METH-induced neurotoxicity. In view of the vital role of microglial activation in neurotoxicity, this should be explored in further detail. Endogenous hormones have shown therapeutic potential in METH treatment. For instance, Cholecystokinin-8 (CCK-8) pretreatment has been shown to attenuate the decrease in expression of TH and DAT in the striatum (Gou et al., 2015). Overall, these drugs have shown the potential for amelioration of hypo-dopaminergic status and neuron degeneration; however, their precise mechanism of action is not known (Table 2).
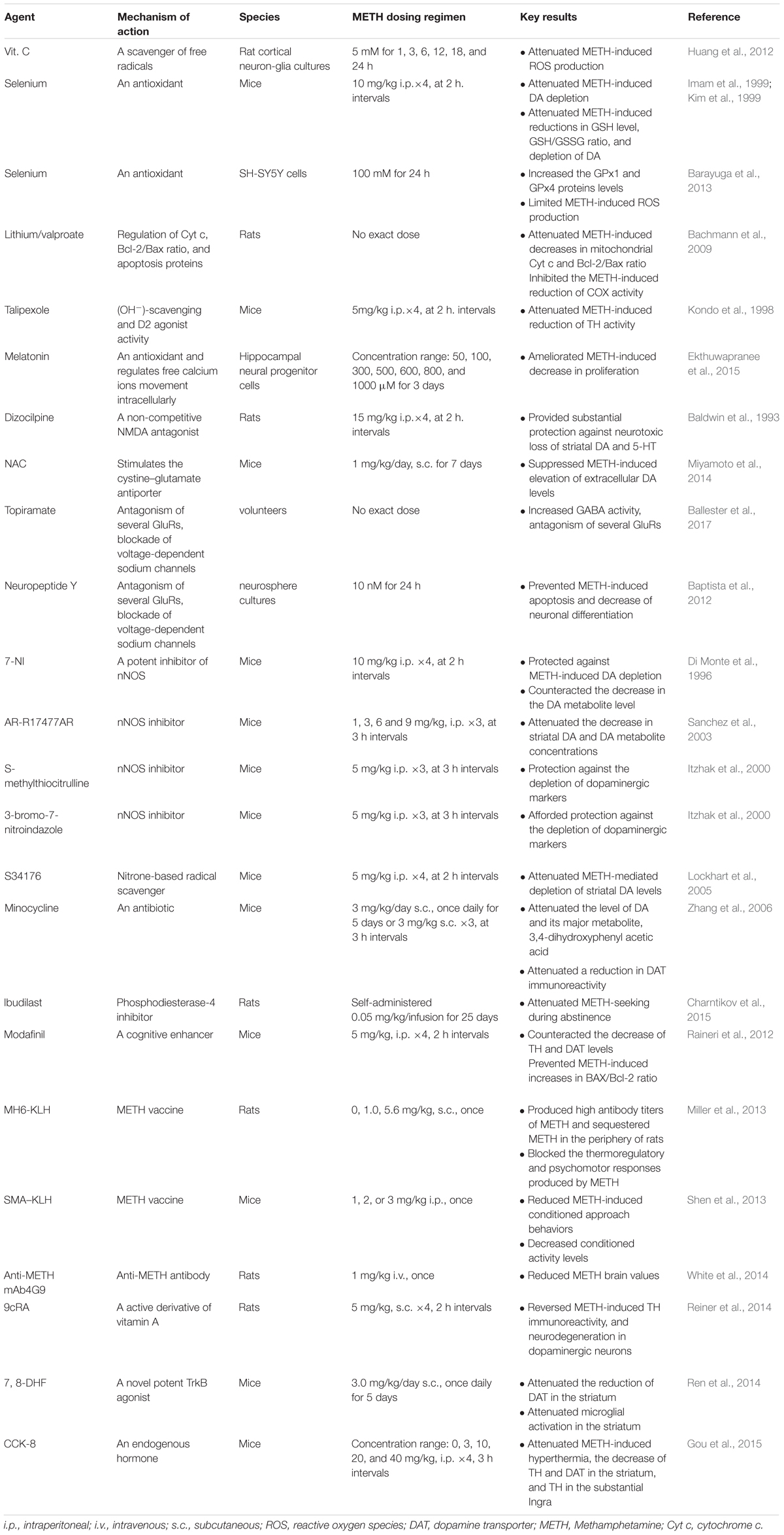
TABLE 2. The summary of pharmacotherapy approaches in METH-induced neurotoxicity in vivo or in vitro.
Future Perspectives
The complex mechanisms underlying METH-evoked neurotoxicity affect sub-regions of the CNS in various ways. Important advances have been made regarding the basic neurobiology of METH-produced neurotoxicity; however, these findings have not resulted in the development of efficacious drug therapy. Nevertheless, in view of the great harm elicited by METH to the CNS, exploration of toxic mechanisms and pharmacotherapies are anticipated in preclinical/clinical investigations. Natural compounds such as 1-methyl-l, 2, 3, 4-tetrahydroisoquinoline (1MeTIQ), resveratrol, curcumin, and gingko biloba have garnered considerable inhibition owing to their ability to scavenge excess free radicals to protect neurons. Accordingly, some researchers have shifted focus to natural compounds to make major breakthroughs. Wasik and Antkiewicz-Michaluk (2017) suggested that 1MeTIQ can apply a brake to several processes related to neurotoxicity (e.g., elimination of free radicals, inhibition of MAO, and Glu-dependent excitotoxicity) without side-effects. Therefore, the use of natural compounds may represent a promising strategy for preventing METH neurotoxicity. In a recent study, the therapeutic effect of carbon nanotubes, which enabled oxidation of METH-enhanced extracellular DA in the striatum of mice, was demonstrated (Xue et al., 2016). Considering the favorable properties (e.g., large surface areas, superior bundle strength, and highly electrostatic attraction to neurotransmitters such as DA) of nanotubes, their use merits further research as a new approach for treatment of METH abuse-induced neurotoxicity.
Author Contributions
XY and JY wrote the manuscript. YW, QL, YZ, LC, and YD completed the figures and the tables. LL, KX, and C-xY provided advice. All authors reviewed the manuscript.
Funding
This research was supported by National Natural Science Foundation of China (81772024) and Natural Science Foundation of Hunan Province (2017JJ2339).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Mrs. Tang Qiu for the kind support and constructive suggestions.
References
Abdul Muneer, P. M., Alikunju, S., Szlachetka, A. M., and Haorah, J. (2011). Methamphetamine inhibits the glucose uptake by human neurons and astrocytes: stabilization by acetyl-L-carnitine. PLoS One 6:e19258. doi: 10.1371/journal.pone.0019258
Aharonovich, E., Hasin, D. S., Brooks, A. C., Liu, X., Bisaga, A., and Nunes, E. V. (2006). Cognitive deficits predict low treatment retention in cocaine dependent patients. Drug Alcohol Depend. 81, 313–322. doi: 10.1016/j.drugalcdep.2005.08.003
Annepu, J., and Ravindranath, V. (2000). 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced complex I inhibition is reversed by disulfide reductant, dithiothreitol in mouse brain. Neurosci. Lett. 289, 209–212. doi: 10.1016/S0304-3940(00)01300-8
Ares-Santos, S., Granado, N., and Moratalla, R. (2013). The role of dopamine receptors in the neurotoxicity of methamphetamine. J. Intern. Med. 273, 437–453. doi: 10.1111/joim.12049
Ashok, A. H., Mizuno, Y., Volkow, N. D., and Howes, O. D. (2017). Association of stimulant use with dopaminergic alterations in users of cocaine, amphetamine, or methamphetamine a systematic review and meta-analysis. JAMA Psychiatry 74, 511–519. doi: 10.1001/jamapsychiatry.2017.0135
Bachmann, R. F., Wang, Y., Yuan, P., Zhou, R., Li, X., Alesci, S., et al. (2009). Common effects of lithium and valproate on mitochondrial functions: protection against methamphetamine-induced mitochondrial damage. Int. J. Neuropsychopharmacol. 12, 805–822. doi: 10.1017/S1461145708009802
Bachtell, R., Hutchinson, M. R., Wang, X., Rice, K. C., Maier, S. F., and Watkins, L. R. (2015). Targeting the toll of drug abuse: the translational potential of toll-like receptor 4. CNS Neurol. Disord. Drug Targets 14, 692–699. doi: 10.2174/1871527314666150529132503
Bahar, E., Kim, H., and Yoon, H. (2016). ER Stress-mediated signaling: action potential and Ca(2+) as key players. Int. J. Mol. Sci. 17:E1558. doi: 10.3390/ijms17091558
Baldwin, H. A., Colado, M. I., Murray, T. K., and Green, A. R. (1993). Striatal dopamine release in vivo following neurotoxic doses of methamphetamine and effect of the neuroprotective drugs, chlormethiazole and dizocilpine. Br. J. Pharmacol. 1108, 1590–1596. doi: 10.1111/j.1476-5381.1993.tb12847.x
Ballester, J., Valentine, G., and Sofuoglu, M. (2017). Pharmacological treatments for methamphetamine addiction: current status and future directions. Expert Rev. Clin. Pharmacol. 10, 305–314. doi: 10.1080/17512433.2017.1268916
Baptista, S., Bento, A. R., Goncalves, J., Bernardino, L., Summavielle, T., Lobo, A., et al. (2012). Neuropeptide Y promotes neurogenesis and protection against methamphetamine-induced toxicity in mouse dentate gyrus-derived neurosphere cultures. Neuropharmacology 62, 2413–2423. doi: 10.1016/j.neuropharm.2012.02.015
Baracz, S. J., and Cornish, J. L. (2016). The neurocircuitry involved in oxytocin modulation of methamphetamine addiction. Front. Neuroendocrinol. 43, 1–18. doi: 10.1016/j.yfrne.2016.08.001
Barayuga, S. M., Pang, X., Andres, M. A., Panee, J., and Bellinger, F. P. (2013). Methamphetamine decreases levels of glutathione peroxidases 1 and 4 in SH-SY5Y neuronal cells: protective effects of selenium. Neurotoxicology 37, 240–246. doi: 10.1016/j.neuro.2013.05.009
Barr, A. M., Markou, A., and Phillips, A. G. (2002). A ‘crash’ course on psychostimulant withdrawal as a model of depression. Trends Pharmacol. Sci. 23, 475–482. doi: 10.1016/S0165-6147(02)02086-2
Bates, M. E., Pawlak, A. P., Tonigan, J. S., and Buckman, J. F. (2006). Cognitive impairment influences drinking outcome by altering therapeutic mechanisms of change. Psychol. Addict Behav. 20, 241–253. doi: 10.1037/0893-164X.20.3.241
Baumann, M. H., Ayestas, M. A., Sharpe, L. G., Lewis, D. B., Rice, K. C., and Rothman, R. B. (2002). Persistent antagonism of methamphetamine-induced dopamine release in rats pretreated with GBR12909 decanoate. J. Pharmacol. Exp. Ther. 301, 1190–1197. doi: 10.1124/jpet.301.3.1190
Beauvais, G., Atwell, K., Jayanthi, S., Ladenheim, B., and Cadet, J. L. (2011). Involvement of dopamine receptors in binge methamphetamine-induced activation of endoplasmic reticulum and mitochondrial stress pathways. PLoS One 6:e28946. doi: 10.1371/journal.pone.0028946
Beer, S. M., Taylor, E. R., Brown, S. E., Dahm, C. C., Costa, N. J., Runswick, M. J., et al. (2004). Glutaredoxin 2 catalyzes the reversible oxidation and glutathionylation of mitochondrial membrane thiol proteins: implications for mitochondrial redox regulation and antioxidant DEFENSE. J. Biol. Chem. 279, 47939–47951. doi: 10.1074/jbc.M408011200
Beirami, E., Oryan, S., Seyedhosseini Tamijani, S. M., Ahmadiani, A., and Dargahi, L. (2018). Intranasal insulin treatment restores cognitive deficits and insulin signaling impairment induced by repeated methamphetamine exposure. J. Cell. Biochem. 119, 2345–2355. doi: 10.1002/jcb.26398
Berk, M., Dean, O. M., Cotton, S. M., Gama, C. S., Kapczinski, F., Fernandes, B., et al. (2012). Maintenance N-acetyl cysteine treatment for bipolar disorder: a double-blind randomized placebo controlled trial. BMC Med. 10:91. doi: 10.1186/1741-7015-10-91
Berk, M., Dean, O. M., Cotton, S. M., Jeavons, S., Tanious, M., Kohlmann, K., et al. (2014). The efficacy of adjunctive N-acetylcysteine in major depressive disorder: a double-blind, randomized, placebo-controlled trial. J. Clin. Psychiatry 75, 628–636. doi: 10.4088/JCP.13m08454
Boileau, I., McCluskey, T., Tong, J., Furukawa, Y., Houle, S., and Kish, S. J. (2016). Rapid recovery of vesicular dopamine levels in methamphetamine users in early abstinence. Neuropsychopharmacology 41, 1179–1187. doi: 10.1038/npp.2015.267
Borgmann, K., and Ghorpade, A. (2018). Methamphetamine augments concurrent astrocyte mitochondrial stress, oxidative burden, and antioxidant capacity: tipping the balance in HIV-Associated neurodegeneration. Neurotox. Res. 33, 433–447. doi: 10.1007/s12640-017-9812-z
Brown, J. M., Quinton, M. S., and Yamamoto, B. K. (2005). Methamphetamine-induced inhibition of mitochondrial complex II: roles of glutamate and peroxynitrite. J. Neurochem. 95, 429–436. doi: 10.1111/j.1471-4159.2005.03379.x
Burrows, K. B., Gudelsky, G., and Yamamoto, B. K. (2000). Rapid and transient inhibition of mitochondrial function following methamphetamine or 3,4-methylenedioxymethamphetamine administration. Eur. J. Pharmacol. 398, 11–18. doi: 10.1016/S0014-2999(00)00264-8
Cadet, J. L., and Brannock, C. (1998). Free radicals and the pathobiology of brain dopamine systems. Neurochem. Int. 32, 117–131. doi: 10.1016/S0197-0186(97)00031-4
Cadet, J. L., Brannock, C., Ladenheim, B., McCoy, M. T., Beauvais, G., Hodges, A. B., et al. (2011). Methamphetamine preconditioning causes differential changes in striatal transcriptional responses to large doses of the drug. Dose Response 9, 165–181. doi: 10.2203/dose-response.10-011.Cadet
Cadet, J. L., and Krasnova, I. N. (2009). Molecular bases of methamphetamine-induced neurodegeneration. Int. Rev. Neurobiol. 88, 101–119. doi: 10.1016/S0074-7742(09)88005-7
Cadet, J. L., Krasnova, I. N., Ladenheim, B., Cai, N. S., McCoy, M. T., and Atianjoh, F. E. (2009). Methamphetamine preconditioning: differential protective effects on monoaminergic systems in the rat brain. Neurotox. Res. 15, 252–259. doi: 10.1007/s12640-009-9026-0
Callaghan, R. C., Cunningham, J. K., Sajeev, G., and Kish, S. J. (2010). Incidence of Parkinson’s disease among hospital patients with methamphetamine-use disorders. Mov. Disord. 25, 2333–2339. doi: 10.1002/mds.23263
Callaghan, R. C., Cunningham, J. K., Sykes, J., and Kish, S. J. (2012). Increased risk of Parkinson’s disease in individuals hospitalized with conditions related to the use of methamphetamine or other amphetamine-type drugs. Drug Alcohol Depend. 120, 35–40. doi: 10.1016/j.drugalcdep.2011.06.013
Callahan, B. T., and Ricaurte, G. A. (1998). Effect of 7-nitroindazole on body temperature and methamphetamine-induced dopamine toxicity. Neuroreport 9, 2691–2695. doi: 10.1097/00001756-199808240-00001
Carroll, K. M. (2014). Lost in translation? Moving contingency management and cognitive behavioral therapy into clinical practice. Ann. N. Y. Acad. Sci. 1327, 94–111. doi: 10.1111/nyas.12501
Chamorro, A., Dirnagl, U., Urra, X., and Planas, A. M. (2016). Neuroprotection in acute stroke: targeting excitotoxicity, oxidative and nitrosative stress, and inflammation. Lancet Neurol. 15, 869–881. doi: 10.1016/S1474-4422(16)00114-9
Chao, J., Zhang, Y., Du, L., Zhou, R., Wu, X., Shen, K., et al. (2017). Molecular mechanisms underlying the involvement of the sigma-1 receptor in methamphetamine-mediated microglial polarization. Sci. Rep. 7:11540. doi: 10.1038/s41598-017-11065-8
Charntikov, S., Pittenger, S. T., Thapa, I., Bastola, D. R., Bevins, R. A., and Pendyala, G. (2015). Ibudilast reverses the decrease in the synaptic signaling protein phosphatidylethanolamine-binding protein 1 (PEBP1) produced by chronic methamphetamine intake in rats. Drug Alcohol Depend. 152, 15–23. doi: 10.1016/j.drugalcdep.2015.04.012
Chen, C., Qincao, L., Xu, J., Du, S., Huang, E., Liu, C., et al. (2016). Role of PUMA in methamphetamine-induced neuronal apoptosis. Toxicol. Lett. 240, 149–160. doi: 10.1016/j.toxlet.2015.10.020
Chen, Y. H., Wu, K. L., Tsai, H. M., and Chen, C. H. (2013). Treatment of methamphetamine abuse: an antibody-based immunotherapy approach. J. Food Drug Anal. 21, S82–S86. doi: 10.1016/j.jfda.2013.09.040
Chomchai, C., and Chomchai, S. (2015). Global patterns of methamphetamine use. Curr. Opin. Psychiatry 28, 269–274. doi: 10.1097/YCO.0000000000000168
Colfax, G., Santos, G. M., Chu, P., Vittinghoff, E., Pluddemann, A., Kumar, S., et al. (2010). Amphetamine-group substances and HIV. Lancet 376, 458–474. doi: 10.1016/S0140-6736(10)60753-2
Courtney, K. E., and Ray, L. A. (2014). Methamphetamine: an update on epidemiology, pharmacology, clinical phenomenology, and treatment literature. Drug Alcohol Depend. 143, 11–21. doi: 10.1016/j.drugalcdep.2014.08.003
Curtin, K., Fleckenstein, A. E., Robison, R. J., Crookston, M. J., Smith, K. R., and Hanson, G. R. (2015). Methamphetamine/amphetamine abuse and risk of Parkinson’s disease in Utah: a population-based assessment. Drug Alcohol Depend. 146, 30–38. doi: 10.1016/j.drugalcdep.2014.10.027
Davidson, C., Gow, A. J., Lee, T. H., and Ellinwood, E. H. (2001). Methamphetamine neurotoxicity: necrotic and apoptotic mechanisms and relevance to human abuse and treatment. Brain Res. Brain Res. Rev. 36, 1–22. doi: 10.1016/S0165-0173(01)00054-6
Dawson, T. M., and Dawson, V. L. (2017). Mitochondrial mechanisms of neuronal cell death: potential therapeutics. Annu. Rev. Pharmacol. Toxicol. 57, 437–454. doi: 10.1146/annurev-pharmtox-010716-105001
Devi, L., and Ohno, M. (2012). 7,8-dihydroxyflavone, a small-molecule TrkB agonist, reverses memory deficits and BACE1 elevation in a mouse model of Alzheimer’s disease. Neuropsychopharmacology 37, 434–444. doi: 10.1038/npp.2011.191
Di Monte, D. A., Royland, E. J., and Jakowec, M. W. (1996). Role of nitric oxide in methamphetamine neurotoxicity :protection by 7-Nitroindazole, an inhibitor of neuronal nitric oxide synthase. J. Neurochem. 67, 2443–2450. doi: 10.1046/j.1471-4159.1996.67062443.x
Ding, W. X., Guo, F., Ni, H. M., Bockus, A., Manley, S., Stolz, D. B., et al. (2012). Parkin and mitofusins reciprocally regulate mitophagy and mitochondrial spheroid formation. J. Biol. Chem. 287, 42379–42388. doi: 10.1074/jbc.M112.413682
Ekthuwapranee, K., Sotthibundhu, A., and Govitrapong, P. (2015). Melatonin attenuates methamphetamine-induced inhibition of proliferation of adult rat hippocampal progenitor cells in vitro. J. Pineal Res. 58, 418–428. doi: 10.1111/jpi.12225
Feier, G., Valvassori, S. S., Varela, R. B., Resende, W. R., Bavaresco, D. V., Morais, M. O., et al. (2013). Lithium and valproate modulate energy metabolism in an animal model of mania induced by methamphetamine. Pharmacol. Biochem. Behav. 103, 589–596. doi: 10.1016/j.pbb.2012.09.010
Forray, A., and Sofuoglu, M. (2014). Future pharmacological treatments for substance use disorders. Br. J. Clin. Pharmacol. 77, 382–400. doi: 10.1111/j.1365-2125.2012.04474.x
Frank, M. G., Adhikary, S., Sobesky, J. L., Weber, M. D., Watkins, L. R., and Maier, S. F. (2016). The danger-associated molecular pattern HMGB1 mediates the neuroinflammatory effects of methamphetamine. Brain Behav. Immun. 51, 99–108. doi: 10.1016/j.bbi.2015.08.001
Gentry, W. B., Laurenzana, E. M., Williams, D. K., West, J. R., Berg, R. J., Terlea, T., et al. (2006). Safety and efficiency of an anti-(+)-methamphetamine monoclonal antibody in the protection against cardiovascular and central nervous system effects of (+)-methamphetamine in rats. Int. Immunopharmacol. 6, 968–977. doi: 10.1016/j.intimp.2006.01.008
Ghosh, P., Bhattacharjee, A., Basu, A., Singha Roy, S., and Bhattacharya, S. (2015). Attenuation of cyclophosphamide-induced pulmonary toxicity in Swiss albino mice by naphthalimide-based organoselenium compound 2-(5-selenocyanatopentyl)-benzo[de]isoquinoline 1,3-dione. Pharm. Biol. 53, 524–532. doi: 10.3109/13880209.2014.931440
Goncalves, J., Martins, T., Ferreira, R., Milhazes, N., Borges, F., Ribeiro, C. F., et al. (2008). Methamphetamine-induced early increase of IL-6 and TNF-alpha mRNA expression in the mouse brain. Ann. N. Y. Acad. Sci. 1139, 103–111. doi: 10.1196/annals.1432.043
Gou, H., Wen, D., Ma, C., Li, M., Li, Y., Zhang, W., et al. (2015). Protective effects of cholecystokinin-8 on methamphetamine-induced behavioral changes and dopaminergic neurodegeneration in mice. Behav. Brain Res. 283, 87–96. doi: 10.1016/j.bbr.2015.01.028
Gray, K. M., Carpenter, M. J., Baker, N. L., DeSantis, S. M., Kryway, E., Hartwell, K. J., et al. (2012). A double-blind randomized controlled trial of N-acetylcysteine in cannabis-dependent adolescents. Am. J. Psychiatry 169, 805–812. doi: 10.1176/appi.ajp.2012.12010055
Hambuchen, M. D., Carroll, F. I., Ruedi-Bettschen, D., Hendrickson, H. P., Hennings, L. J., Blough, B. E., et al. (2015). Combining active immunization with monoclonal antibody therapy to facilitate early initiation of a long-acting anti-methamphetamine antibody response. J. Med. Chem. 58, 4665–4677. doi: 10.1021/acs.jmedchem.5b00220
Hansen, J. P. (2002). Ten keys to successful NCQA accreditation: a health plan perspective. J. Healthc. Qual. 24, 39–42. doi: 10.1111/j.1945-1474.2002.tb00459.x
Hashimoto, K. (2013). Sigma-1 receptor chaperone and brain-derived neurotrophic factor: emerging links between cardiovascular disease and depression. Prog. Neurobiol. 100, 15–29. doi: 10.1016/j.pneurobio.2012.09.001
Hashimoto, K., Tsukada, H., Nishiyama, S., Fukumoto, D., Kakiuchi, T., Shimizu, E., et al. (2004). Effects of N-acetyl-L-cysteine on the reduction of brain dopamine transporters in monkey treated with methamphetamine. Ann. N. Y. Acad. Sci. 1025, 231–235. doi: 10.1196/annals.1316.028
Hassan, A., Erian, M., Said, F., Sorensen, K., and Sippel, J. (1973). Comparison of co-trimoxazole and chloramphenicol in enteric fever. Br. Med. J. 3:108. doi: 10.1136/bmj.3.5871.108-a
Hetz, C. (2012). The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 13, 89–102. doi: 10.1038/nrm3270
Ho, E. L., Josephson, S. A., Lee, H. S., and Smith, W. S. (2009). Cerebrovascular complications of methamphetamine abuse. Neurocrit. Care 10, 295–305. doi: 10.1007/s12028-008-9177-5
Hsieh, J. H., Stein, D. J., and Howells, F. M. (2014). The neurobiology of methamphetamine induced psychosis. Front. Hum. Neurosci. 8:537. doi: 10.3389/fnhum.2014.00537
Huang, N. K., Wan, F. J., Tseng, C. J., and Tung, C. S. (1997). Nicotinamide attenuates methamphetamine-induced striatal dopamine depletion in rats. Neuroreport 8, 1883–1885. doi: 10.1097/00001756-199705260-00018
Huang, Y. N., Wang, J. Y., Lee, C. T., Lin, C. H., Lai, C. C., and Wang, J. Y. (2012). L-ascorbate attenuates methamphetamine neurotoxicity through enhancing the induction of endogenous heme oxygenase-1. Toxicol. Appl. Pharmacol. 265, 241–252. doi: 10.1016/j.taap.2012.08.036
Huang, Y. N., Yang, L. Y., Wang, J. Y., Lai, C. C., Chiu, C. T., and Wang, J. Y. (2017). L-Ascorbate protects against methamphetamine-induced neurotoxicity of cortical cells via inhibiting oxidative stress. Autophagy, and Apoptosis. Mol. Neurobiol. 54, 125–136. doi: 10.1007/s12035-015-9561-z
Imam, S. Z., Islam, F., Itzhak, Y., Slikker, W., and Ali, S. F. (2000). Prevention of dopaminergic neurotoxicity by targeting nitric oxide and peroxynitrite: implications for the prevention of methamphetamine-induced neurotoxic damage. Ann. N. Y. Acad. Sci. 914, 157–171. doi: 10.1111/j.1749-6632.2000.tb05193.x
Imam, S. Z., Newport, G. D., Islam, F., Slikker, W. Jr., and Ali, S. F. (1999). Selenium, an antioxidant, protects against methamphetamine-induced dopaminergic neurotoxicity. Brain Res. 818, 575–578. doi: 10.1016/S0006-8993(98)01311-0
Itzhak, Y., Martin, J. L., and Ail, S. F. (2000). nNOS inhibitors attenuate methamphetamine-induced dopaminergic neurotoxicity but not hyperthermia in mice. Neuroreport 11, 2943–2946. doi: 10.1097/00001756-200009110-00022
Jayanthi, S., Deng, X., Bordelon, M., McCoy, M. T., and Cadet, J. L. (2001). Methamphetamine causes differential regulation of pro-death and anti-death Bcl-2 genes in the mouse neocortex. FASEB J. 15, 1745–1752. doi: 10.1096/fj.01-0025com
Jayanthi, S., Deng, X., Noailles, P. A., Ladenheim, B., and Cadet, J. L. (2004). Methamphetamine induces neuronal apoptosis via cross-talks between endoplasmic reticulum and mitochondria-dependent death cascades. FASEB J. 18, 238–251. doi: 10.1096/fj.03-0295com
Kielczykowska, M., Kocot, J., Pazdzior, M., and Musik, I. (2018). Selenium - a fascinating antioxidant of protective properties. Adv. Clin. Exp. Med. 27, 245–255. doi: 10.17219/acem/67222
Killinger, B., Shah, M., and Moszczynska, A. (2014). Co-administration of betulinic acid and methamphetamine causes toxicity to dopaminergic and serotonergic nerve terminals in the striatum of late adolescent rats. J. Neurochem. 128, 764–775. doi: 10.1111/jnc.12496
Kim, H. C., Jhoo, W. K., Choi, D. Y., Im, D. H., Shin, E. J., Suh, J. H., et al. (1999). Protection of methamphetamine nigrostriatal toxicity by dietary selenium. Brain Res. 851, 76–86. doi: 10.1016/S0006-8993(99)02122-8
Kish, S. J., Boileau, I., Callaghan, R. C., and Tong, J. (2017). Brain dopamine neurone ‘damage’: methamphetamine users vs. Parkinson’s disease - a critical assessment of the evidence. Eur. J. Neurosci. 45, 58–66. doi: 10.1111/ejn.13363
Kish, S. J., Fitzmaurice, P. S., Boileau, I., Schmunk, G. A., Ang, L. C., Furukawa, Y., et al. (2009). Brain serotonin transporter in human methamphetamine users. Psychopharmacology 202, 649–661. doi: 10.1007/s00213-008-1346-x
Kitamura, O., Tokunaga, I., Gotohda, T., and Kubo, S. (2007). Immunohistochemical investigation of dopaminergic terminal markers and caspase-3 activation in the striatum of human methamphetamine users. Int. J. Legal Med. 121, 163–168. doi: 10.1007/s00414-006-0087-9
Klongpanichapak, S., Govitrapong, P., Sharma, S. K., and Ebadi, M. (2006). Attenuation of cocaine and methamphetamine neurotoxicity by coenzyme Q10. Neurochem. Res. 31, 303–311. doi: 10.1007/s11064-005-9025-3
Kondo, T., Shimada, H., Hatori, K., Sugita, Y., and Mizuno, Y. (1998). Talipexole protects dopaminergic neurons from methamphetamine toxicity in C57BL/6N mouse. Neurosci. Lett. 247, 143–146. doi: 10.1016/S0304-3940(98)00300-0
Lancet (2018). Opioids and methamphetamine: a tale of two crises. Lancet 391:713. doi: 10.1016/S0140-6736(18)30319-2
LaVoie, M. J., and Hastings, T. G. (1999). Dopamine quinone formation and protein modification associated with the striatal neurotoxicity of methamphetamine: evidence against a role for extracellular dopamine. J. Neurosci. 19, 1484–1491. doi: 10.1523/JNEUROSCI.19-04-01484.1999
Lee, H. C., and Wei, Y. H. (2005). Mitochondrial biogenesis and mitochondrial DNA maintenance of mammalian cells under oxidative stress. Int. J. Biochem. Cell Biol. 37, 822–834. doi: 10.1016/j.biocel.2004.09.010
Lenzi, P., Marongiu, R., Falleni, A., Gelmetti, V., Busceti, C. L., Michiorri, S., et al. (2012). A subcellular analysis of genetic modulation of PINK1 on mitochondrial alterations, autophagy and cell death. Arch. Ital. Biol. 150, 194–217.
Li, S., Hafeez, A., Noorulla, F., Geng, X., Shao, G., Ren, C., et al. (2017). Preconditioning in neuroprotection: from hypoxia to ischemia. Prog. Neurobiol. 157, 79–91. doi: 10.1016/j.pneurobio.2017.01.001
Lin, M., Chandramani-Shivalingappa, P., Jin, H., Ghosh, A., Anantharam, V., Ali, S., et al. (2012). Methamphetamine-induced neurotoxicity linked to ubiquitin-proteasome system dysfunction and autophagy-related changes that can be modulated by protein kinase C delta in dopaminergic neuronal cells. Neuroscience 210, 308–332. doi: 10.1016/j.neuroscience.2012.03.004
Lockhart, B., Roger, A., Bonhomme, N., Goldstein, S., and Lestage, P. (2005). In vivo neuroprotective effects of the novel imidazolyl nitrone free-radical scavenger (Z)-alpha-[2-thiazol-2-yl)imidazol-4-yl]-N-tert-butylnitrone (S34176). Eur. J. Pharmacol. 511, 127–136. doi: 10.1016/j.ejphar.2005.01.043
Ma, J. Z., Johnson, B. A., Yu, E., Weiss, D., McSherry, F., Saadvandi, J., et al. (2013). Fine-grain analysis of the treatment effect of topiramate on methamphetamine addiction with latent variable analysis. Drug Alcohol Depend. 130, 45–51. doi: 10.1016/j.drugalcdep.2012.10.009
Mai, H. N., Sharma, N., Shin, E. J., Nguyen, B. T., Nguyen, P. T., Jeong, J. H., et al. (2018). Exposure to far-infrared ray attenuates methamphetamine-induced impairment in recognition memory through inhibition of protein kinase C delta in male mice: comparison with the antipsychotic clozapine. J. Neurosci. Res. doi: 10.1002/jnr.24228 [Epub ahead of print].
Manji, H., Kato, T., Di Prospero, N. A., Ness, S., Beal, M. F., Krams, M., et al. (2012). Impaired mitochondrial function in psychiatric disorders. Nat. Rev. Neurosci. 13, 293–307. doi: 10.1038/nrn3229
McConnell, S. E., O’Banion, M. K., Cory-Slechta, D. A., Olschowka, J. A., and Opanashuk, L. A. (2015). Characterization of binge-dosed methamphetamine-induced neurotoxicity and neuroinflammation. Neurotoxicology 50, 131–141. doi: 10.1016/j.neuro.2015.08.006
Mcketin, R., Dean, O. M., Baker, A. L., Carter, G., Turner, A., Kelly, P. J., et al. (2017). A potential role for N-acetylcysteine in the management of methamphetamine dependence. Drug Alcohol Rev. 36, 153–159. doi: 10.1111/dar.12414
Michel, S., Wanet, A., De Pauw, A., Rommelaere, G., Arnould, T., and Renard, P. (2012). Crosstalk between mitochondrial (dys)function and mitochondrial abundance. J. Cell. Physiol. 227, 2297–2310. doi: 10.1002/jcp.23021
Miller, M. L., Moreno, A. Y., Aarde, S. M., Creehan, K. M., Vandewater, S. A., Vaillancourt, B. D., et al. (2013). A methamphetamine vaccine attenuates methamphetamine-induced disruptions in thermoregulation and activity in rats. Biol. Psychiatry 73, 721–728. doi: 10.1016/j.biopsych.2012.09.010
Miyamoto, Y., Ishikawa, Y., Iegaki, N., Sumi, K., Fu, K., Sato, K., et al. (2014). Overexpression of Shati/Nat8l, an nacetyltransferase, in the nucleus accumbens attenuates the response to methamphetamine via activation of group II mGluRs in mice. Int. J. Neuropsychopharmacol. 17, 1283–1294. doi: 10.1017/S146114571400011X
Mizuno, Y., Kowa, H., Nakanishi, T., and Yanagisawa, N. (1993). Preliminary study of B-HT 920, a Novel dopamine agonist, for the treatment of Parkinson’s Disease. Drug Invest. 5, 186–192. doi: 10.1007/BF03258445
Moratalla, R., Khairnar, A., Simola, N., Granado, N., Garcia-Montes, J. R., Porceddu, P. F., et al. (2017). Amphetamine-related drugs neurotoxicity in humans and in experimental animals: main mechanisms. Prog. Neurobiol. 155, 149–170. doi: 10.1016/j.pneurobio.2015.09.011
Moszczynska, A., and Callan, S. P. (2017). Molecular, behavioral, and physiological consequences of methamphetamine neurotoxicity: implications for treatment. J. Pharmacol. Exp. Ther. 362, 474–488. doi: 10.1124/jpet.116.238501
Musavi, S., and Kakkar, P. (2003). Effect of diazepam treatment and its withdrawal on pro/antioxidative processes in rat brain. Mol. Cell. Biochem. 245, 51–56. doi: 10.1023/A:1022857508987
Nakajima, A., Yamada, K., Nagai, T., Uchiyama, T., Miyamoto, Y., Mamiya, T., et al. (2004). Role of tumor necrosis factor-alpha in methamphetamine-induced drug dependence and neurotoxicity. J. Neurosci. 24, 2212–2225. doi: 10.1523/JNEUROSCI.4847-03.2004
Nam, Y., Wie, M. B., Shin, E. J., Nguyen, T. T., Nah, S. Y., Ko, S. K., et al. (2015). Ginsenoside Re protects methamphetamine-induced mitochondrial burdens and proapoptosis via genetic inhibition of protein kinase C delta in human neuroblastoma dopaminergic SH-SY5Y cell lines. J. Appl. Toxicol. 35, 927–944. doi: 10.1002/jat.3093
Nervina, J. M., Magyar, C. E., Pirih, F. Q., and Tetradis, S. (2006). PGC-1alpha is induced by parathyroid hormone and coactivates Nurr1-mediated promoter activity in osteoblasts. Bone 39, 1018–1025. doi: 10.1016/j.bone.2006.04.023
Nguyen, X. K., Lee, J., Shin, E. J., Dang, D. K., Jeong, J. H., Nguyen, T. T., et al. (2015). Liposomal melatonin rescues methamphetamine-elicited mitochondrial burdens, pro-apoptosis, and dopaminergic degeneration through the inhibition PKCdelta gene. J. Pineal Res. 58, 86–106. doi: 10.1111/jpi.12195
Nordahl, T. E., Salo, R., and Leamon, M. (2003). Neuropsychological effects of chronic methamphetamine use on neurotransmitters and cognition: a review. J. Neuropsychiatry Clin. Neurosci. 15, 317–325. doi: 10.1176/jnp.15.3.317
Ojaniemi, M., Glumoff, V., Harju, K., Liljeroos, M., Vuori, K., and Hallman, M. (2003). Phosphatidylinositol 3-kinase is involved in Toll-like receptor 4-mediated cytokine expression in mouse macrophages. Eur. J. Immunol. 33, 597–605. doi: 10.1002/eji.200323376
Olanow, C. W., and Tatton, W. G. (1999). Etiology and pathogenesis of Parkinson’s disease. Annu. Rev. Neurosci. 22, 123–144. doi: 10.1146/annurev.neuro.22.1.123
Panenka, W. J., Procyshyn, R. M., Lecomte, T., MacEwan, G. W., Flynn, S. W., Honer, W. G., et al. (2013). Methamphetamine use: a comprehensive review of molecular, preclinical and clinical findings. Drug Alcohol Depend. 129, 167–179. doi: 10.1016/j.drugalcdep.2012.11.016
Parameyong, A., Charngkaew, K., Govitrapong, P., and Chetsawang, B. (2013). Melatonin attenuates methamphetamine-induced disturbances in mitochondrial dynamics and degeneration in neuroblastoma SH-SY5Y cells. J. Pineal Res. 55, 313–323. doi: 10.1111/jpi.12078
Parameyong, A., Govitrapong, P., and Chetsawang, B. (2015). Melatonin attenuates the mitochondrial translocation of mitochondrial fission proteins and Bax, cytosolic calcium overload and cell death in methamphetamine-induced toxicity in neuroblastoma SH-SY5Y cells. Mitochondrion 24, 1–8. doi: 10.1016/j.mito.2015.07.004
Penn, A. M., Lee, J. W., Thuillier, P., Wagner, M., Maclure, K. M., Menard, M. R., et al. (1992). Melas syndrome with mitochondrial tRNA(Leu)(UUR) mutation: correlation of clinical state, nerve conduction, and muscle 31P magnetic resonance spectroscopy during treatment with nicotinamide and riboflavin. Neurology 42, 2147–2152. doi: 10.1212/WNL.42.11.2147
Prado, E., Maes, M., Piccoli, L. G., Baracat, M., Barbosa, D. S., Franco, O., et al. (2015). N-acetylcysteine for therapy-resistant tobacco use disorder: a pilot study. Redox Rep. 20, 215–222. doi: 10.1179/1351000215Y.0000000004
Qiao, D., Xu, J., Le, C., Huang, E., Liu, C., Qiu, P., et al. (2014). Insulin-like growth factor binding protein 5 (IGFBP5) mediates methamphetamine-induced dopaminergic neuron apoptosis. Toxicol. Lett. 230, 444–453. doi: 10.1016/j.toxlet.2014.08.010
Raineri, M., Gonzalez, B., Goitia, B., Garcia-Rill, E., Krasnova, I. N., Cadet, J. L., et al. (2012). Modafinil abrogates methamphetamine-induced neuroinflammation and apoptotic effects in the mouse striatum. PLoS One 7:e46599. doi: 10.1371/journal.pone.0046599
Rapado-Castro, M., Dodd, S., Bush, A. I., Malhi, G. S., Skvarc, D. R., On, Z. X., et al. (2017). Cognitive effects of adjunctive N-acetyl cysteine in psychosis. Psychol. Med. 47, 866–876. doi: 10.1017/S0033291716002932
Rawson, R. A., and Condon, T. P. (2007). Why do we need an Addiction supplement focused on methamphetamine? Addiction 102(Suppl. 1), 1–4. doi: 10.1111/j.1360-0443.2006.01781.x
Reiner, D. J., Yu, S. J., Shen, H., He, Y., Bae, E., and Wang, Y. (2014). 9-Cis retinoic acid protects against methamphetamine-induced neurotoxicity in nigrostriatal dopamine neurons. Neurotox. Res. 25, 248–261. doi: 10.1007/s12640-013-9413-4
Ren, Q., Zhang, J. C., Ma, M., Fujita, Y., Wu, J., and Hashimoto, K. (2014). 7,8-Dihydroxyflavone, a TrkB agonist, attenuates behavioral abnormalities and neurotoxicity in mice after administration of methamphetamine. Psychopharmacology 231, 159–166. doi: 10.1007/s00213-013-3221-7
Rice, M. E. (2000). Ascorbate regulation and its neuroprotective role in the brain. Trends Neurosci. 23, 209–216. doi: 10.1016/S0166-2236(99)01543-X
Sanchez, V., Zeini, M., Camarero, J., O’Shea, E., Bosca, L., Green, A. R., et al. (2003). The nNOS inhibitor, AR-R17477AR, prevents the loss of NF68 immunoreactivity induced by methamphetamine in the mouse striatum. J. Neurochem. 85, 515–524. doi: 10.1046/j.1471-4159.2003.01714.x
Sano, R., and Reed, J. C. (2013). ER stress-induced cell death mechanisms. Biochim. Biophys. Acta 1833, 3460–3470. doi: 10.1016/j.bbamcr.2013.06.028
Schep, L. J., Slaughter, R. J., and Beasley, D. M. (2010). The clinical toxicology of metamfetamine. Clin. Toxicol. 48, 675–694. doi: 10.3109/15563650.2010.516752
Scofield, M. D., and Kalivas, P. W. (2014). Astrocytic dysfunction and addiction: consequences of impaired glutamate homeostasis. Neuroscientist 20, 610–622. doi: 10.1177/1073858413520347
Sekine, Y., Iyo, M., Ouchi, Y., Matsunaga, T., Tsukada, H., Okada, H., et al. (2001). Methamphetamine-related psychiatric symptoms and reduced brain dopamine transporters studied with PET. Am. J. Psychiatry 158, 1206–1214. doi: 10.1176/appi.ajp.158.8.1206
Shah, A., Silverstein, P. S., Singh, D. P., and Kumar, A. (2012). Involvement of metabotropic glutamate receptor 5, AKT/PI3K signaling and NF-kappaB pathway in methamphetamine-mediated increase in IL-6 and IL-8 expression in astrocytes. J. Neuroinflammation 9:52. doi: 10.1186/1742-2094-9-52
Shen, P. P., Hou, S., Ma, D., Zhao, M. M., Zhu, M. Q., Zhang, J. D., et al. (2016). Cortical spreading depression-induced preconditioning in the brain. Neural Regen. Res. 11, 1857–1864.
Shen, X. Y., Kosten, T. A., Lopez, A. Y., Kinsey, B. M., Kosten, T. R., and Orson, F. M. (2013). A vaccine against methamphetamine attenuates its behavioral effects in mice. Drug Alcohol Depend. 129, 41–48. doi: 10.1016/j.drugalcdep.2012.09.007
Shin, E. J., Dang, D. K., Tran, H. Q., Nam, Y., Jeong, J. H., Lee, Y. H., et al. (2016). PKCdelta knockout mice are protected from para-methoxymethamphetamine-induced mitochondrial stress and associated neurotoxicity in the striatum of mice. Neurochem. Int. 100, 146–158. doi: 10.1016/j.neuint.2016.09.008
Shin, E. J., Tran, H. Q., Nguyen, P. T., Jeong, J. H., Nah, S. Y., Jang, C. G., et al. (2017). Role of mitochondria in methamphetamine-induced dopaminergic neurotoxicity: involvement in oxidative stress, neuroinflammation, and pro-apoptosis-a review. Neurochem. Res. 43, 57–69. doi: 10.1007/s11064-017-2318-5
Singhakumar, R., Boontem, P., Ekthuwapranee, K., Sotthibundhu, A., Mukda, S., Chetsawang, B., et al. (2015). Melatonin attenuates methamphetamine-induced inhibition of neurogenesis in the adult mouse hippocampus: an in vivo study. Neurosci. Lett. 606, 209–214. doi: 10.1016/j.neulet.2015.09.011
Snider, S. E., Hendrick, E. S., and Beardsley, P. M. (2013). Glial cell modulators attenuate methamphetamine self-administration in the rat. Eur. J. Pharmacol. 701, 124–130. doi: 10.1016/j.ejphar.2013.01.016
Snider, S. E., Vunck, S. A., van den Oord, E. J., Adkins, D. E., McClay, J. L., and Beardsley, P. M. (2012). The glial cell modulators, ibudilast and its amino analog, AV1013, attenuate methamphetamine locomotor activity and its sensitization in mice. Eur. J. Pharmacol. 679, 75–80. doi: 10.1016/j.ejphar.2012.01.013
Stephans, S. E., Whittingham, T. S., Douglas, A. J., Lust, W. D., and Yamamoto, B. K. (1998). Substrates of energy metabolism attenuate methamphetamine-induced neurotoxicity in striatum. J. Neurochem. 71, 613–621. doi: 10.1046/j.1471-4159.1998.71020613.x
Suwanjang, W., Abramov, A. Y., Charngkaew, K., Govitrapong, P., and Chetsawang, B. (2016). Melatonin prevents cytosolic calcium overload, mitochondrial damage and cell death due to toxically high doses of dexamethasone-induced oxidative stress in human neuroblastoma SH-SY5Y cells. Neurochem. Int. 97, 34–41. doi: 10.1016/j.neuint.2016.05.003
Szegezdi, E., Logue, S. E., Gorman, A. M., and Samali, A. (2006). Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 7, 880–885. doi: 10.1038/sj.embor.7400779
Thomas, D. M., Francescutti-Verbeem, D. M., and Kuhn, D. M. (2008). The newly synthesized pool of dopamine determines the severity of methamphetamine-induced neurotoxicity. J. Neurochem. 105, 605–616. doi: 10.1111/j.1471-4159.2007.05155.x
Thrash, B., Karuppagounder, S. S., Uthayathas, S., Suppiramaniam, V., and Dhanasekaran, M. (2010). Neurotoxic effects of methamphetamine. Neurochem. Res. 35, 171–179. doi: 10.1007/s11064-009-0042-5
Thrash-Williams, B., Ahuja, M., Karuppagounder, S. S., Uthayathas, S., Suppiramaniam, V., and Dhanasekaran, M. (2013). Assessment of therapeutic potential of amantadine in methamphetamine induced neurotoxicity. Neurochem. Res. 38, 2084–2094. doi: 10.1007/s11064-013-1117-x
Thrash-Williams, B., Karuppagounder, S. S., Bhattacharya, D., Ahuja, M., Suppiramaniam, V., and Dhanasekaran, M. (2016). Methamphetamine-induced dopaminergic toxicity prevented owing to the neuroprotective effects of salicylic acid. Life Sci. 154, 24–29. doi: 10.1016/j.lfs.2016.02.072
Tseng, E. E., Brock, M. V., Lange, M. S., Troncoso, J. C., Blue, M. E., Lowenstein, C. J., et al. (2010). Glutamate excitotoxicity mediates neuronal apoptosis after hypothermic circulatory arrest. Ann. Thorac. Surg. 89, 440–445. doi: 10.1016/j.athoracsur.2009.10.059
Ungvari, E., Monori, I., Megyeri, A., Csiki, Z., Prokisch, J., Sztrik, A., et al. (2014). Protective effects of meat from lambs on selenium nanoparticle supplemented diet in a mouse model of polycyclic aromatic hydrocarbon-induced immunotoxicity. Food Chem. Toxicol. 64, 298–306. doi: 10.1016/j.fct.2013.12.004
Valian, N., Ahmadiani, A., and Dargahi, L. (2017). Escalating methamphetamine regimen induces compensatory mechanisms, mitochondrial biogenesis, and GDNF expression, in Substantia Nigra. J. Cell. Biochem. 118, 1369–1378. doi: 10.1002/jcb.25795
Virmani, A., Gaetani, F., Imam, S., Binienda, Z., and Ali, S. (2003). Possible mechanism for the neuroprotective effects of L-carnitine on methamphetamine-evoked neurotoxicity. Ann. N. Y. Acad. Sci. 993, 197–207; discussion 287–198. doi: 10.1111/j.1749-6632.2003.tb07530.x
Volkow, N. D., Chang, L., Wang, G. J., Fowler, J. S., Franceschi, D., Sedler, M., et al. (2001a). Loss of dopamine transporters in methamphetamine abusers recovers with protracted abstinence. J. Neurosci. 21, 9414–9418. doi: 10.1523/JNEUROSCI.21-23-09414.2001
Volkow, N. D., Chang, L., Wang, G. J., Fowler, J. S., Leonido-Yee, M., Franceschi, D., et al. (2001b). Association of dopamine transporter reduction with psychomotor impairment in methamphetamine abusers. Am. J. Psychiatry 158, 377–382. doi: 10.1176/appi.ajp.158.3.377
Volkow, N. D., Wang, G. J., Smith, L., Fowler, J. S., Telang, F., Logan, J., et al. (2015). Recovery of dopamine transporters with methamphetamine detoxification is not linked to changes in dopamine release. Neuroimage 121, 20–28. doi: 10.1016/j.neuroimage.2015.07.035
Wang, N., Tan, H. Y., Li, S., Xu, Y., Guo, W., and Feng, Y. (2017). Supplementation of micronutrient selenium in metabolic diseases: its role as an antioxidant. Oxid. Med. Cell Longev. 2017:7478523. doi: 10.1155/2017/7478523
Wasik, A., and Antkiewicz-Michaluk, L. (2017). The mechanism of neuroprotective action of natural compounds. Pharmacol. Rep. 69, 851–860. doi: 10.1016/j.pharep.2017.03.018
White, S. J., Hendrickson, H. P., Atchley, W. T., Laurenzana, E. M., Gentry, W. B., Williams, D. K., et al. (2014). Treatment with a monoclonal antibody against methamphetamine and amphetamine reduces maternal and fetal rat brain concentrations in late pregnancy. Drug Metab. Dispos. 42, 1285–1291. doi: 10.1124/dmd.114.056879
Wilson, J. M., Kalasinsky, K. S., Levey, A. I., Bergeron, C., Reiber, G., Anthony, R. M., et al. (1996). Striatal dopamine nerve terminal markers in human, chronic methamphetamine users. Nat. Med. 2, 699–703. doi: 10.1038/nm0696-699
Won, S., Hong, R. A., Shohet, R. V., Seto, T. B., and Parikh, N. I. (2013). Methamphetamine-associated cardiomyopathy. Clin. Cardiol. 36, 737–742. doi: 10.1002/clc.22195
Wongprayoon, P., and Govitrapong, P. (2017). Melatonin Protects SH-SY5Y Neuronal Cells Against methamphetamine-induced endoplasmic reticulum stress and apoptotic cell death. Neurotox. Res. 31, 1–10. doi: 10.1007/s12640-016-9647-z
Wu, C. W., Ping, Y. H., Yen, J. C., Chang, C. Y., Wang, S. F., Yeh, C. L., et al. (2007). Enhanced oxidative stress and aberrant mitochondrial biogenesis in human neuroblastoma SH-SY5Y cells during methamphetamine induced apoptosis. Toxicol. Appl. Pharmacol. 220, 243–251. doi: 10.1016/j.taap.2007.01.011
Xie, X. L., He, J. T., Wang, Z. T., Xiao, H. Q., Zhou, W. T., Du, S. H., et al. (2018). Lactulose attenuates METH-induced neurotoxicity by alleviating the impaired autophagy, stabilizing the perturbed antioxidant system and suppressing apoptosis in rat striatum. Toxicol. Lett. 289, 107–113. doi: 10.1016/j.toxlet.2018.03.015
Xiong, K., Liao, H., Long, L., Ding, Y., Huang, J., and Yan, J. (2016). Necroptosis contributes to methamphetamine-induced cytotoxicity in rat cortical neurons. Toxicol. In Vitro 35, 163–168. doi: 10.1016/j.tiv.2016.06.002
Xiong, K., Long, L., Zhang, X., Qu, H., Deng, H., Ding, Y., et al. (2017). Overview of long non-coding RNA and mRNA expression in response to methamphetamine treatment in vitro. Toxicol. In Vitro 44, 1–10. doi: 10.1016/j.tiv.2017.06.009
Xu, E., Liu, J., Liu, H., Wang, X., and Xiong, H. (2017). Role of microglia in methamphetamine-induced neurotoxicity. Int. J. Physiol. Pathophysiol. Pharmacol. 9, 84–100.
Xu, S., Pi, H., Zhang, L., Zhang, N., Li, Y., Zhang, H., et al. (2016). Melatonin prevents abnormal mitochondrial dynamics resulting from the neurotoxicity of cadmium by blocking calcium-dependent translocation of Drp1 to the mitochondria. J. Pineal Res. 60, 291–302. doi: 10.1111/jpi.12310
Xu, X., Huang, E., Tai, Y., Zhao, X., Chen, X., Chen, C., et al. (2017). Nupr1 modulates methamphetamine-induced dopaminergic neuronal apoptosis and autophagy through CHOP-Trib3-Mediated endoplasmic reticulum stress signaling pathway. Front. Mol. Neurosci. 10:203. doi: 10.3389/fnmol.2017.00203
Xue, X., Yang, J. Y., He, Y., Wang, L. R., Liu, P., Yu, L. S., et al. (2016). Aggregated single-walled carbon nanotubes attenuate the behavioural and neurochemical effects of methamphetamine in mice. Nat. Nanotechnol. 11, 613–620. doi: 10.1038/nnano.2016.23
Yamaguchi, T., Kuraishi, Y., Minami, M., Nakai, S., Hirai, Y., and Satoh, M. (1991). Methamphetamine-induced expression of interleukin-1 beta mRNA in the rat hypothalamus. Neurosci. Lett. 128, 90–92. doi: 10.1016/0304-3940(91)90766-M
Yin, L. H., Shen, H., Diaz-Ruiz, O., Backman, C. M., Bae, E., Yu, S. J., et al. (2012). Early post-treatment with 9-cis retinoic acid reduces neurodegeneration of dopaminergic neurons in a rat model of Parkinson’s disease. BMC Neurosci. 13:120. doi: 10.1186/1471-2202-13-120
Zhang, L., Kitaichi, K., Fujimoto, Y., Nakayama, H., Shimizu, E., Iyo, M., et al. (2006). Protective effects of minocycline on behavioral changes and neurotoxicity in mice after administration of methamphetamine. Prog. Neuropsychopharmacol. Biol. Psychiatry 30, 1381–1393. doi: 10.1016/j.pnpbp.2006.05.015
Zhang, X., Tobwala, S., and Ercal, N. (2012). N-acetylcysteine amide protects against methamphetamine-induced tissue damage in CD-1 mice. Hum. Exp. Toxicol. 31, 931–944. doi: 10.1177/0960327112438287
Keywords: methamphetamine, neurotoxicity, oxidative stress, excitotoxicity, neuroinflammation, immunotherapy
Citation: Yang X, Wang Y, Li Q, Zhong Y, Chen L, Du Y, He J, Liao L, Xiong K, Yi C-x and Yan J (2018) The Main Molecular Mechanisms Underlying Methamphetamine- Induced Neurotoxicity and Implications for Pharmacological Treatment. Front. Mol. Neurosci. 11:186. doi: 10.3389/fnmol.2018.00186
Received: 05 February 2018; Accepted: 14 May 2018;
Published: 04 June 2018.
Edited by:
Ildikó Rácz, Universitätsklinikum Bonn, GermanyReviewed by:
Rozi Andretic Waldowski, University of Rijeka, CroatiaJana Ruda-Kucerova, Masaryk University, Czechia
Copyright © 2018 Yang, Wang, Li, Zhong, Chen, Du, He, Liao, Xiong, Yi and Yan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jie Yan, d2lsbHMyMTIxNTZAY3N1LmVkdS5jbg==
†These authors have contributed equally to this work.