 Andrea Ruetenik1,2
Andrea Ruetenik1,2 Antonio Barrientos1,2,3*
Antonio Barrientos1,2,3*- 1Department of Neurology, School of Medicine, University of Miami Miller School of Medicine, Miami, FL, United States
- 2Neuroscience Graduate Program, School of Medicine, University of Miami Miller School of Medicine, Miami, FL, United States
- 3Department of Biochemistry, School of Medicine, University of Miami Miller School of Medicine, Miami, FL, United States
Over the last few decades, the budding yeast Saccharomyces cerevisiae has been extensively used as a valuable organism to explore mechanisms of aging and human age-associated neurodegenerative disorders. Yeast models can be used to study loss of function of disease-related conserved genes and to investigate gain of function activities, frequently proteotoxicity, exerted by non-conserved human mutant proteins responsible for neurodegeneration. Most published models of proteotoxicity have used rapidly dividing cells and suffer from a high level of protein expression resulting in acute growth arrest or cell death. This contrasts with the slow development of neurodegenerative proteotoxicity during aging and the characteristic post-mitotic state of the affected cell type, the neuron. Here, we will review the efforts to create and characterize yeast models of neurodegeneration using the chronological life span model of aging, and the specific information they can provide regarding the chronology of physiological events leading to neurotoxic proteotoxicity-induced cell death and the identification of new pathways involved.
Neurodegenerative Diseases Modeled in Yeast
The unicellular yeast Saccharomyces cerevisiae, known as baker’s yeast or brewer’s yeast, has been extensively used in the areas of biotechnology and biomedicine. Over the last century, S. cerevisiae has been used as a valuable organism for studying the principles of microbiology, characterizing biochemical pathways and understanding the biology of more complex eukaryotic organisms (Botstein, 1991). A multiplicity of basic cellular activities are conserved from yeast to humans, including DNA replication, recombination and repair, RNA transcription and translation, intracellular trafficking, enzymatic activities of general metabolism and mitochondrial biogenesis, protein quality control pathways, nutrient sensing, and stress resistance pathways (reviewed in Barrientos, 2003). Therefore, knowledge gained in yeast has been fundamental to understanding the physiology of human cells and the pathophysiology of human diseases.
Over the last two decades, yeast has been used to model the human aging process and complex neurodegenerative disorders, including amyotrophic lateral sclerosis (ALS), Parkinson’s disease (PD), and Huntington’s disease (HD) (reviewed in Miller-Fleming et al., 2008). In humans, these neurodegenerative disorders are characterized by the progressive, selective loss of neurons in different areas of the brain associated with the misfolding of disease-specific proteins. Although yeast cells are less complex than human neurons, basic metabolic pathways involved in neurodegeneration are well-conserved in S. cerevisiae, as mentioned earlier.
Constructing a yeast model of a human neurodegenerative disorder does not present major technical difficulties per se but requires a carefully designed multistep plan (Figure 1). A major goal is that the yeast model of a particular disease must recapitulate the crucial events preceding cell death that are manifested during the course of the human disorder.
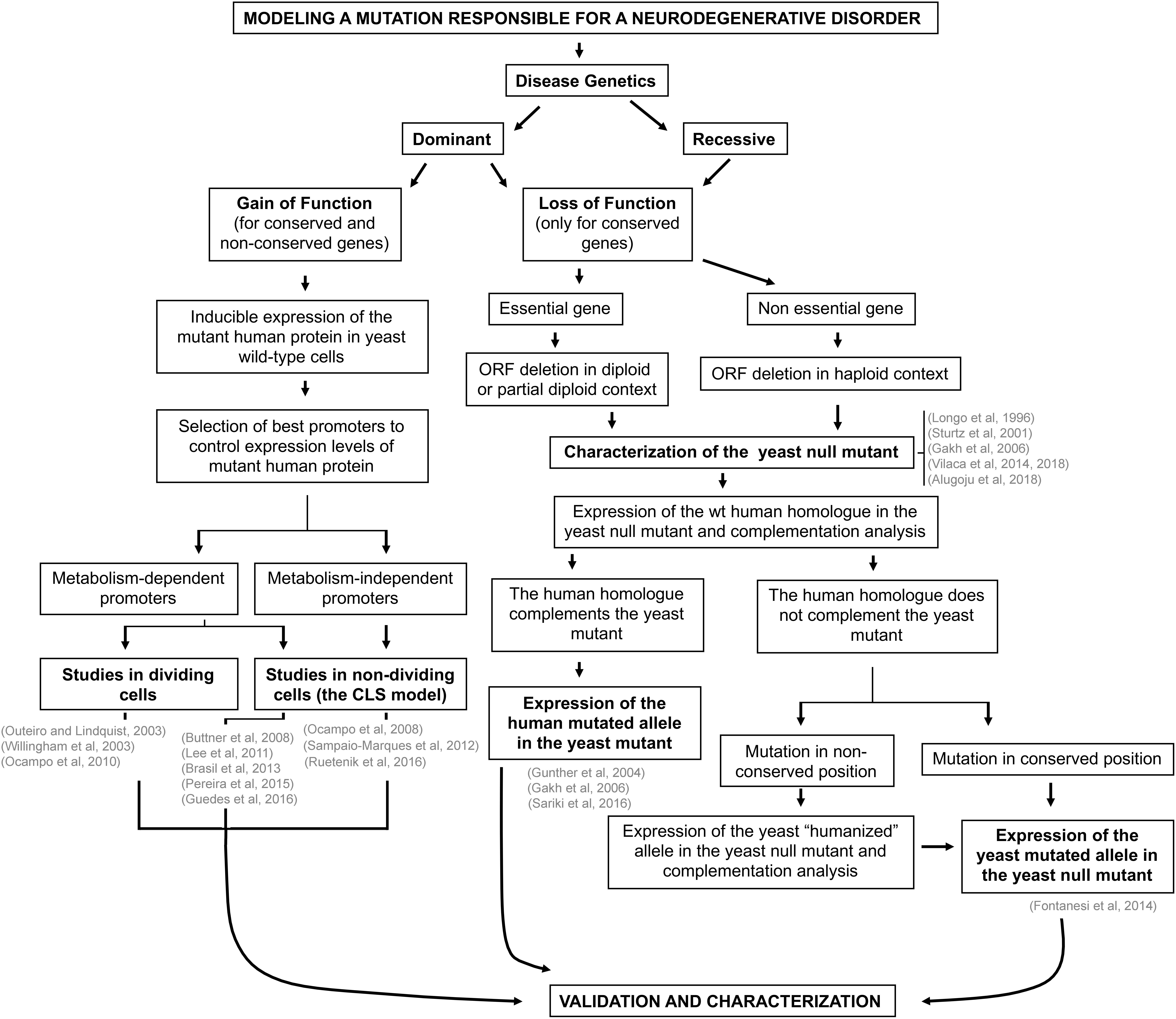
FIGURE 1. Scheme depicting the strategic planning for the creation of yeast models of neurodegenerative disorders. The strategies used for the construction of yeast models of human monogenic neurodegenerative diseases depend on genetic and pathophysiological constraints. Whether the disease is dominant or recessive, whether the phenotype results from a gain or a loss of function of the protein involved, and whether the gene is functionally conserved or not from yeast to humans are determinants of the kind of yeast model that can be generated. References located in the relevant text boxes provide actual examples in the literature of yeast models of neurodegenerative disorders. See full explanation in the text. Figure modified from Fontanesi et al. (2009).
The strategies that are usually followed in the construction of yeast models of human neurodegenerative diseases depend on genetic and pathophysiological constraints. In some cases, human disorders result from a loss of function of the disease gene encoded protein. In these cases, when the human disease gene is conserved from yeast to humans, functional complementation studies will allow determining whether the human disease gene product partially or fully replaces the function of the yeast gene product. If complementation occurs, human disease gene mutant alleles are expressed in yeast and tested for functionality as for mutations in the Cu–Zn superoxide dismutase gene responsible for ALS (Gunther et al., 2004). If complementation does not occur, the disease mutations, frequently involving conserved protein residues, are alternatively introduced in the yeast protein and subsequently analyzed as it has been reported for mutations in the adenine nucleotide translocator (ANT1) responsible for cases of external progressive ophthalmoplegia (Fontanesi et al., 2004).
In age-associated neurodegenerative disorders such as Alzheimer’s disease (AD), PD, or HD, the human disease genes are restricted to vertebrates. In these diseases, however, a gain of function of the disease mutant proteins greatly contributed to pathogenicity. Mutant forms of the proteins huntingtin (htt) and α-synuclein, responsible for HD and some familiar forms of PD, respectively, undergo misfolding and damage several cellular structures thus leading to cell death. Yeast models of these disorders are constructed by expressing the human disease gene in yeast thus providing paradigms where the toxic effect of the misfolded protein on the cellular physiology and metabolism can be conveniently studied (Outeiro and Lindquist, 2003; Willingham et al., 2003; Ocampo et al., 2010).
The Yeast Chronological Life Span Assay: a Better System to Model Neurodegeneration?
Yeast Aging
The ideal yeast models of age-associated neurodegeneration should incorporate the concept of cellular aging. Yeast has two life spans; a replicative life span (RLS), defined as the number of daughters produced by each dividing mother cell, and a chronological life span (CLS), defined as the capacity of stationary (Go) cultures to maintain viability over time. The CLS assay, initially established by Valter Longo (University of Southern California) (Longo et al., 1996, 2012; Fabrizio et al., 2001; Fabrizio and Longo, 2007), has been proposed to reflect aging in post-mitotic mammalian cells, such as neurons (Chen et al., 2005; Braun et al., 2009). Despite the different level of complexity between yeast and human post-mitotic cells, yeast models of aging have been instrumental for the identification of essential conserved pathways that influence healthspan and life span. For example, studies aiming to understand the molecular mechanisms of calorie-restriction (CR)-mediated longevity, allowed for the identification of several longevity genes (reviewed in Ruetenik et al., 2016). In yeast, CR down-regulates the conserved Ras/cAMP/PKA, TOR, and Sch9 signaling pathways that integrate the nutrient and other environmental cues to regulate cell growth, division and life span (Wei et al., 2008). Deletion of RAS2, TOR1, or SCH9 enhances cellular protection against thermal and oxidative stresses and extends yeast CLS (Longo and Finch, 2003). Inhibition of these pathways converges on the activation of stress resistance transcription factors that will induce the expression of cell protection systems (e.g., catalase and superoxide dismutase -SOD2) and accumulation of stored nutrients (trehalose and glycogen). The key components of these pathways also regulate stress resistance and life span in higher eukaryotes (Fontana et al., 2010). For example, both Akt and S6K, homologs of yeast SCH9, regulate life span in higher eukaryotes and inhibition of Tor/S6K signaling extends life span in worms, flies, and mice (Paradis et al., 1999; Hertweck et al., 2004; Kapahi et al., 2004; Selman et al., 2009). Also, mice deficient in elements of the Ras pathway have extended health and life span (Yan et al., 2007; Enns et al., 2009). The longevity pathways play an essential role in the regulation of mitochondrial biogenesis and function, crucial for the management of neuronal life and death, in yeast and higher eukaryotes including mammals (Bonawitz et al., 2007; Anderson and Prolla, 2009). For example, deletion of the TOR1 gene extends yeast CLS in part by increasing mitochondrial mass and respiration (Bonawitz et al., 2007) and by promoting adaptive mitochondrial reactive oxygen species (ROS) signaling (Pan et al., 2011). The Ras/cAMP/PKA pathway senses excessive ROS to signal to the Hap2,3,4,5 transcriptional system and down-regulate mitochondrial biogenesis (Dejean et al., 2002; Chevtzoff et al., 2009). Also in mammals, modulation of mitochondrial biogenesis and metabolism through the Tor, Akt1, and Ras pathways involves the transcriptional co-activator PGC-1α (Anderson and Prolla, 2009). PGC-1α transcriptional activity appears to be induced in the oxidative stress response and CR through a shared mechanism, suggesting that in mammals, regulation of mitochondrial function is a critical element in both cell survival and longevity (Anderson et al., 2008).
Therefore, the yeast CLS model of aging is expected to be an informative paradigm regarding connections between aging and neurodegeneration, at least for those diseases originating from proteotoxic stress.
The CLS Assay
Chronological life span determination must be conducted in exact conditions to ensure reproducibility. CLS is classically determined in cells grown in liquid synthetic complete media containing 2% glucose (SDC) supplemented with standard amounts of amino acids and nucleotide bases as previously described (Sherman, 1991; Ocampo and Barrientos, 2011). The assay is depicted in Figure 2 and briefly explained in the figure legend. During CLS, multiple approaches can be implemented to analyze the progression or decline of physiological features over time, how they are influenced by the expression of toxic proteins, and how they correlate with survival. As an example, an array of assays that can be implemented to analyze mitochondrial function and ROS production is presented in Figure 3.
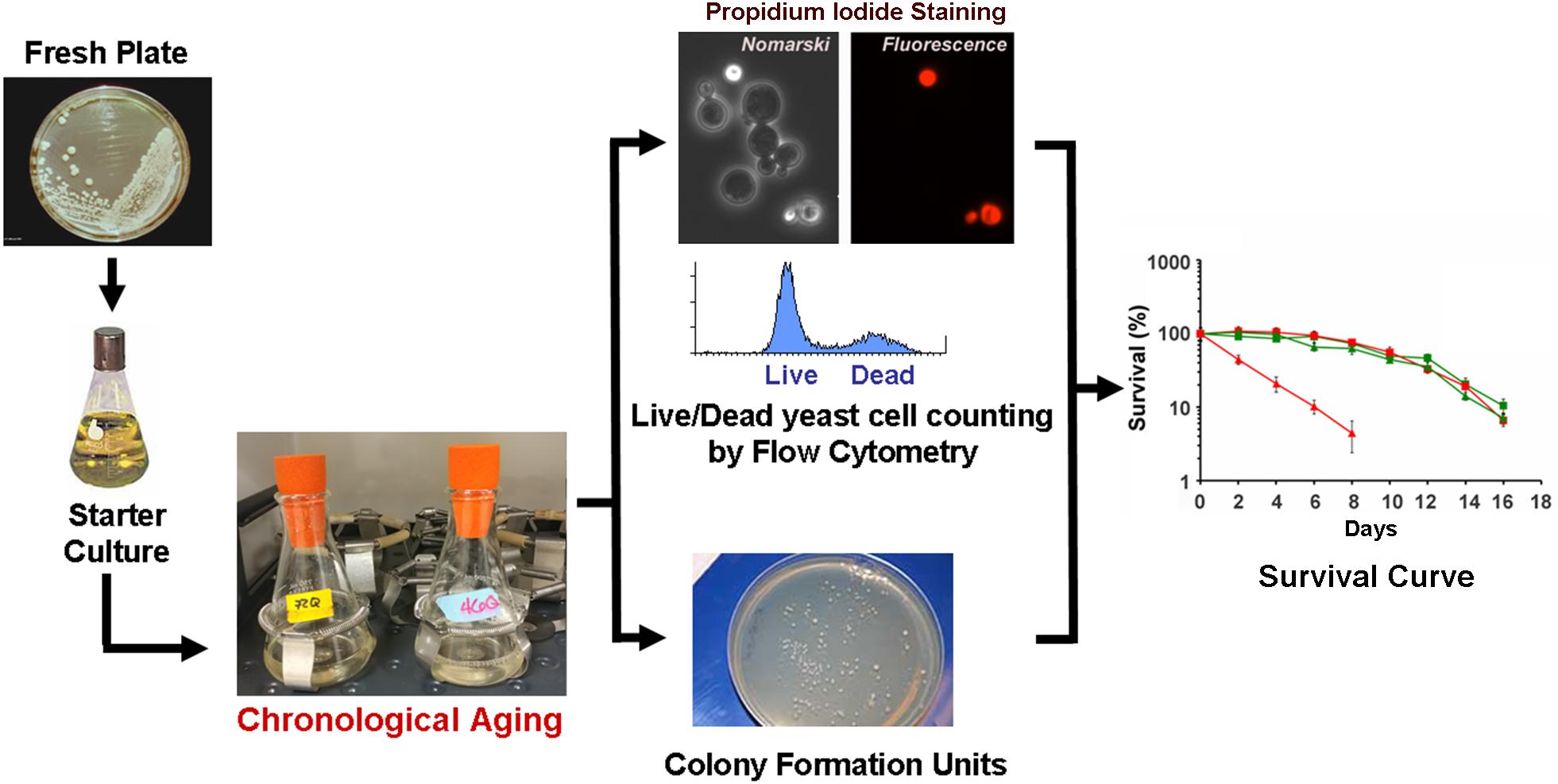
FIGURE 2. The chronological life span (CLS) assay. In a typical CLS assay, yeast strains from frozen stocks (–80°C) are patched onto YPD agar plates (2% glucose) and incubated at 30°C. The following day, cells are inoculated into 10 ml of SDC media and grown overnight. After 24 h, cells are inoculated into 50 ml of synthetic (SDC) media in 250-ml flasks to an optical density at 600 nm (OD600) of 0.250. Cultures are then grown with shaking (250 rpm) at 30°C. We recommend that all flasks are capped using Bio-Silico plugs that ensure sterility and maximize airflow (Hirschmann, Louisville, KY, United States). Maximum cell density is normally reached after 48 h of growth in SDC, therefore we consider 3 days after inoculation as Day 0 of CLS. Subsequently, cellular viability is determined every other day by either a clonogenic approach using the colony formation unit (CFU) assay of propidium iodide staining and flow cytometry analyses (PI-FCM) as described (Ocampo and Barrientos, 2012; Ocampo et al., 2012). The data obtained from viability analysis is then used to construct survival curves.
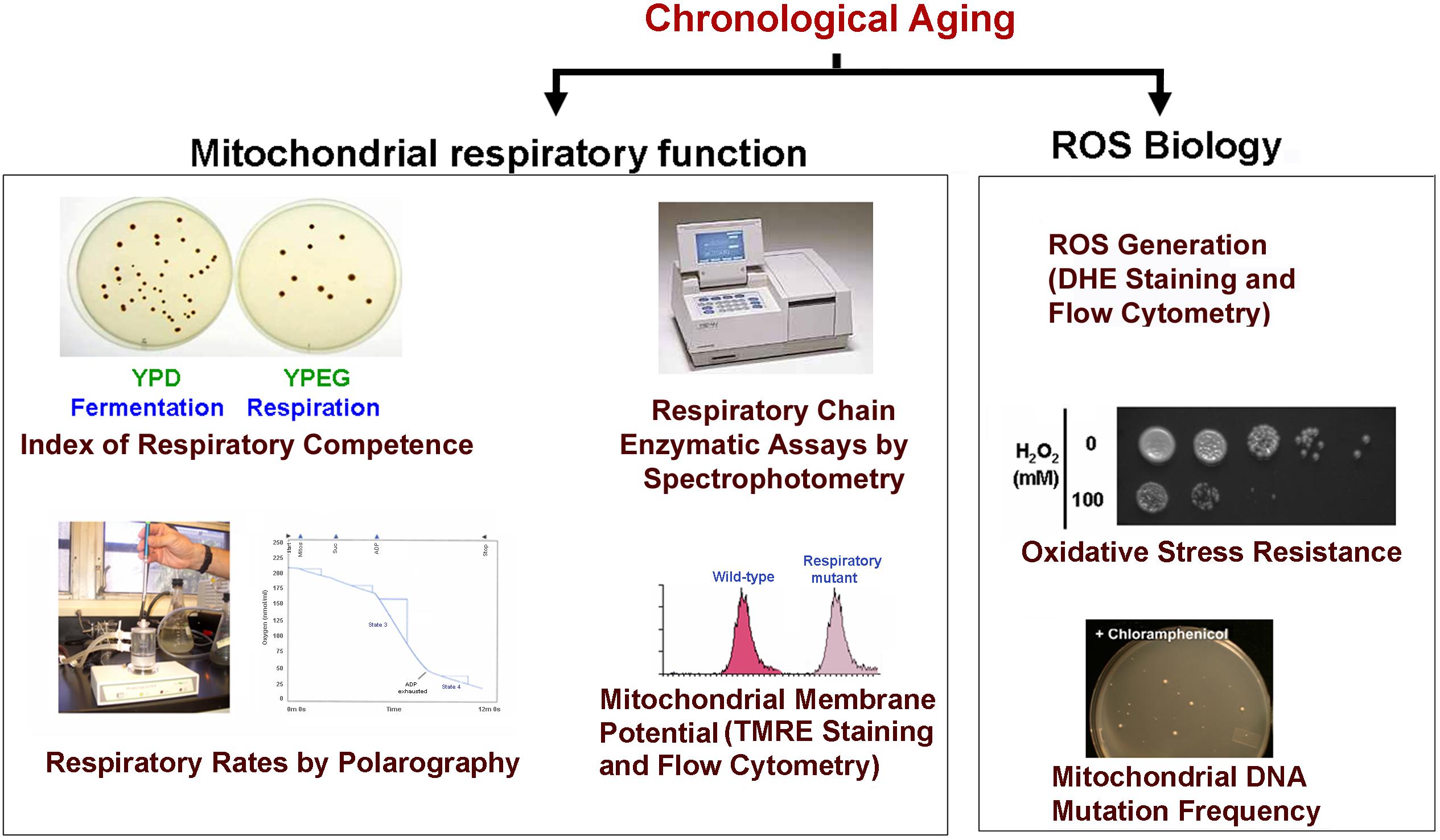
FIGURE 3. Scheme of some of the experiments used to monitor mitochondrial damage during CLS. During CLS of wild-type yeast cultures expressing or not the toxic proteins are collected at different time points and analyzed for markers of mitochondrial function including: (1) The index of respiratory competence (Parrella and Longo, 2008) or proportion of cells that are able to form colonies in media containing respiratory (glycerol) versus fermentable (glucose) carbon sources. (2) Endogenous cell respiration measured polarographically and mitochondrial respiratory chain (MRC) enzymatic activities measured spectrophotometrically (Barrientos et al., 2009). (3) Mitochondrial membrane potential estimated in vivo by flow cytometry using the fluorescent probe tetramethylrhodamine ethyl ester (TMRE) (Ocampo et al., 2010). (4) Oxidative stress and damage: mitochondrial and cellular reactive oxygen species (ROS) production can be measured in whole cells by using the oxidant-sensitive fluorescent probes, MitoSOX Red and dihydroethidium (DHE) as described (Ocampo et al., 2012). Additionally, resistance to oxidative stress can be determined by exposing cells to high H2O2 concentrations (up to 100 mM) for 1 h prior to testing growth in complete solid media. Also, mtDNA point mutation frequency can be estimated by counting the proportion of cells that acquire resistance to specific antibiotics (chloramphenicol or erythromycin). Cells are plated in respiratory media supplemented with the drugs as described (O’Rourke et al., 2002).
Strain Background
When preparing yeast models of aging and neurodegeneration it is important to consider the strain background. Several laboratory strains commonly used in aging research are genetically and physiologically heterogeneous (Mortimer and Johnston, 1986; van Dijken et al., 2000), which is reflected in some manner in their CLS (Fabrizio and Longo, 2007; Ocampo and Barrientos, 2011; Ocampo et al., 2012). For example, two different yeast genetic backgrounds commonly used in this field are the short-lived strains BY4741 (Powers et al., 2006; Murakami et al., 2008; Burtner et al., 2009) and DBY2006 (Bonawitz et al., 2007; Pan and Shadel, 2009). Many commonly used strains, including BY4741, CEN.PK113-7D and DBY2006, are S288c derivatives that carry a mutation affecting Hap1, a heme-dependent regulator of a number of genes involved in electron-transfer reactions (Gaisne et al., 1999). This contributes to their reduced ability to respire compared with strains carrying a wild-type HAP1 gene such as W303, although other genetic differences between the strains may also contribute (Ocampo et al., 2012). As a consequence, when the cells are grown in synthetic medium containing 2% glucose, the CLS of BY4741 (∼8 days), CEN.PK113-7D (∼9 days), and DBY2006 (∼3 days) is shorter than of W303 yeast (∼10.5 days), and the effect of mutations affecting nutrient-sensing and stress-resistance pathways are not exactly equal (for a comparison of CLS in different yeast strains, see, Ocampo and Barrientos, 2011; Ocampo et al., 2012; Chen and Petranovic, 2015). Therefore, the use of at least two different strain backgrounds is always advised to obtain interpretable results.
External Modulators of Yeast CLS
Various external conditions have been found to shorten or extend yeast CLS in wild-type cells. Those need to be taken into account when designing the CLS assays with yeast models of neurodegenerative diseases and when interpreting the results obtained. For example, the CLS on the type of medium in which cells are initially grown. Yeast grown in synthetic medium containing dextrose survive for a few days (from 6 to 11 depending on the yeast strain), which is a very short, high-metabolic post-diauxic phase (Sinclair et al., 1998; Longo, 1999). In contrast, wild-type yeast grown in YPD, and maintained on expired YPD medium, can survive for several weeks, and thus have a prolonged period of hypometabolism in stationary phase (Sinclair et al., 1998; Longo, 1999 and reviewed in Chen et al., 2005). Yeast cultures even have slightly longer survival if they are pre-grown in YPEG (respiratory media) (MacLean et al., 2001). Additional modulators of CLS include: pH of the growth culture medium (Burtner et al., 2009; Longo et al., 2012; Ocampo et al., 2012), amino acid content (Johnson and Johnson, 2014; Maruyama et al., 2016), nitrogen source (Hess et al., 2006; Santos et al., 2015), osmolarity (Kaeberlein et al., 2002; Smith et al., 2007; Murakami et al., 2008), and temperature (Smith et al., 2007). All these factors need to be carefully controlled and maintained constant through experimentation since they are significant sources of experimental variability.
CLS Studies Using Yeast Models of Neurodegenerative Disease
Although, the CLS assay has been used extensively to study and identify modifiers of normal yeast aging, proportionately few studies have employed the CLS assay to study yeast models of neurodegenerative disease. This represents a vast missed opportunity. A summary of studies conducted to date using the CLS assay to evaluate the toxicity of disease-related proteins in a wild-type yeast background as well as studies that attempted to find toxicity suppressors in these models using the CLS assay are listed in Table 1. In this section, we will briefly introduce the main findings of these studies, the study limitations, and our advice for future work. As seen in Table 1, the majority of these studies also quantified yeast viability throughout the CLS assay using only the colony formation unit (CFU) assay method, which, as explained previously may affect the strength of the interpretations that can be made from these studies if not combined with the parallel use of the PI–FCM method.
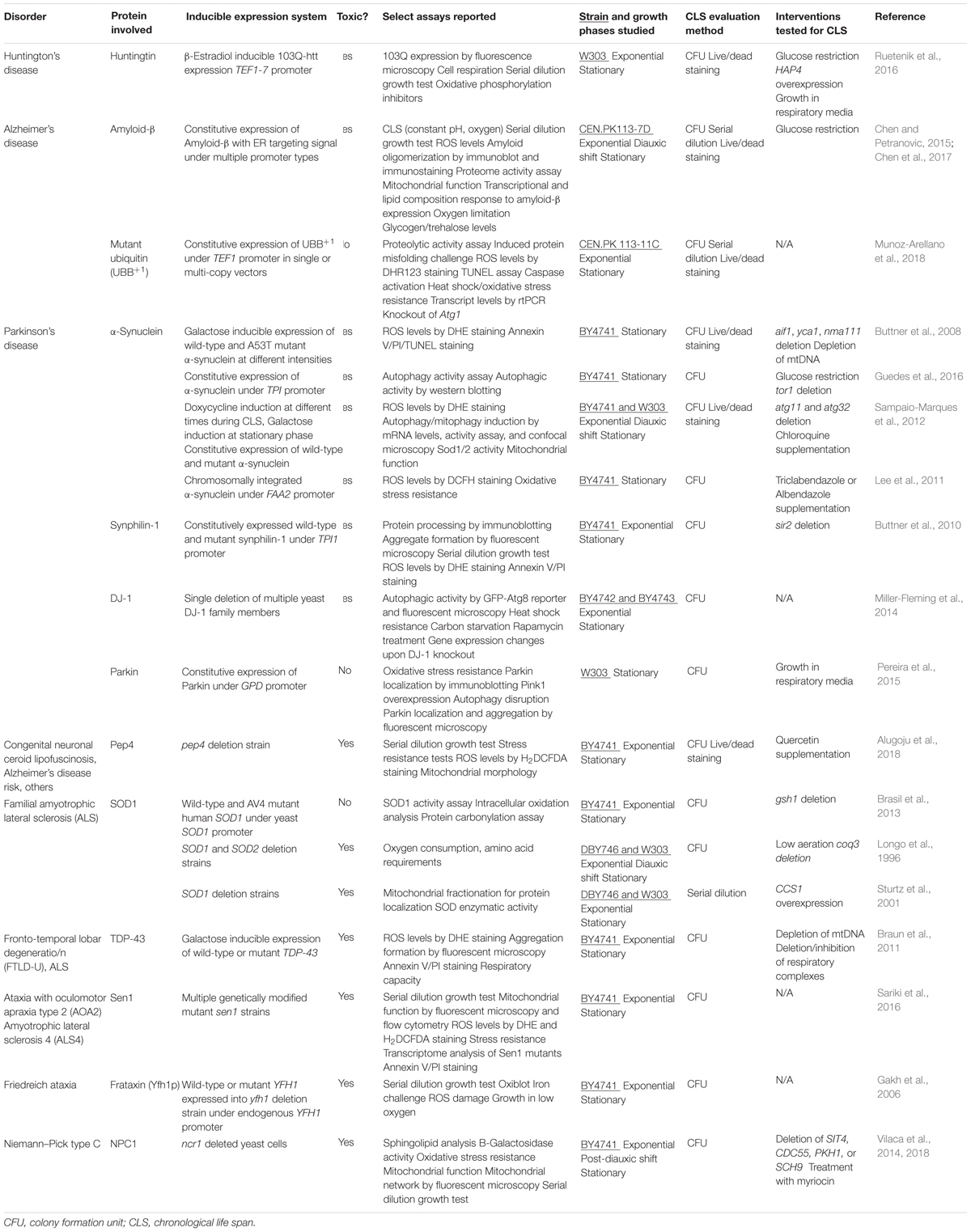
TABLE 1. Yeast models of neurodegenerative disorders established in the context of the chronological life span model of aging.
Huntington’s Disease (HD) and Other Polyglutamine (PolyQ) Disorders
Polyglutamine (PolyQ) diseases are caused by a CAG codon repeat expansion in disease-specific genes resulting in the expression of misfolding/aggregation-prone proteins with expanded polyQ stretches. These include HD, characterized by intranuclear and cytoplasmic htt inclusions, and six types of spinocerebellar ataxias (Shao and Diamond, 2007). Mitochondrial dysfunction, altered mitochondrial integrity and dynamics and impaired axonal trafficking have been associated with the pathogenesis of polyQ diseases in human patients and several research model organisms (Panov et al., 2002; Trushina et al., 2004; Cui et al., 2006; Lin and Beal, 2006; Wang et al., 2009). Mutant htt may also damage neurons directly by inducing mitochondrial depolarization and altering calcium homeostasis in patients and in mouse models (Panov et al., 2002). Additionally, mutant htt has been shown to alter mitochondrial function indirectly by inhibiting the expression of the transcriptional co-activator PGC-1α, which regulates mitochondrial biogenesis and respiration (Cui et al., 2006).
Yeast Models in Dividing Cells
In yeast, expression of htt exon I fragments comprising the polyQ stretches faithfully recapitulates htt misfolding/aggregation in a polyQ length-dependent manner, as shown by the pioneering work of Dr. Susan Lindquist (Krobitsch and Lindquist, 2000). These models were constructed by placing polyQ expression under the control of a galactose-inducible promoter. Cells were transferred from glucose (or raffinose) to galactose-containing media to induce strong polyQ expression and acute cytotoxicity. Upon polyQ toxicity, several pathways, including ER stress, cytoskeletal disturbances, oxidative stress, and mitochondrial dysfunction were found to contribute to growth arrest and cell death (Meriin et al., 2002; Muchowski et al., 2002; Solans et al., 2006; Duennwald and Lindquist, 2008; Ocampo et al., 2010). These models have been used extensively to search for suppressors of polyQ toxicity. Toxicity can be reduced by enhancing the clearance pathways for the removal of cytoplasmic aggregate-prone proteins by increasing autophagy (Sarkar et al., 2009), activating the ubiquitin–proteasome system (Bauer and Nukina, 2009) or repairing the endoplasmic reticulum-associated degradation (ERAD) pathway (Duennwald and Lindquist, 2008). Toxicity is ameliorated by modulating the chaperone systems involved in protein refolding, aggregation and disaggregation, thus shifting the balance toward the non-toxic species (Krobitsch and Lindquist, 2000; Vacher et al., 2005). Suppression of polyQ toxicity is also achieved by protecting the cells against events downstream from polyQ misfolding and oligomerization, including suppression of cytoskeletal instability (Kaminosono et al., 2008), suppression of the kynurenine pathway to limit the accumulation of toxic metabolites that increase ROS generation (Giorgini et al., 2005) and protection of mitochondrial integrity and function (Ocampo et al., 2010). These yeast models have been also used for genome-wide screens to identify genes that enhance polyQ toxicity. The identified genes clustered in the functionally related cellular processes of response to stress, protein folding, and ubiquitin-dependent protein catabolism (Willingham et al., 2003).
Yeast Models in Non-dividing Cells
To ascertain how polyQ toxicity modulates aging, we have studied suppressors of mutant htt toxicity using the CLS assay in a yeast model of HD by expressing 103Q starting at day 0 of the stationary phase using an inducible β-estradiol expression system (Figure 4) (Ruetenik et al., 2016). In the CLS assay, in standard synthetic media growth conditions, we observed that overexpression of this 103Q htt fragment through induction with 50 nM β-estradiol resulted in a severe shortening of CLS compared to control cells (Ruetenik et al., 2016) (Figure 4).
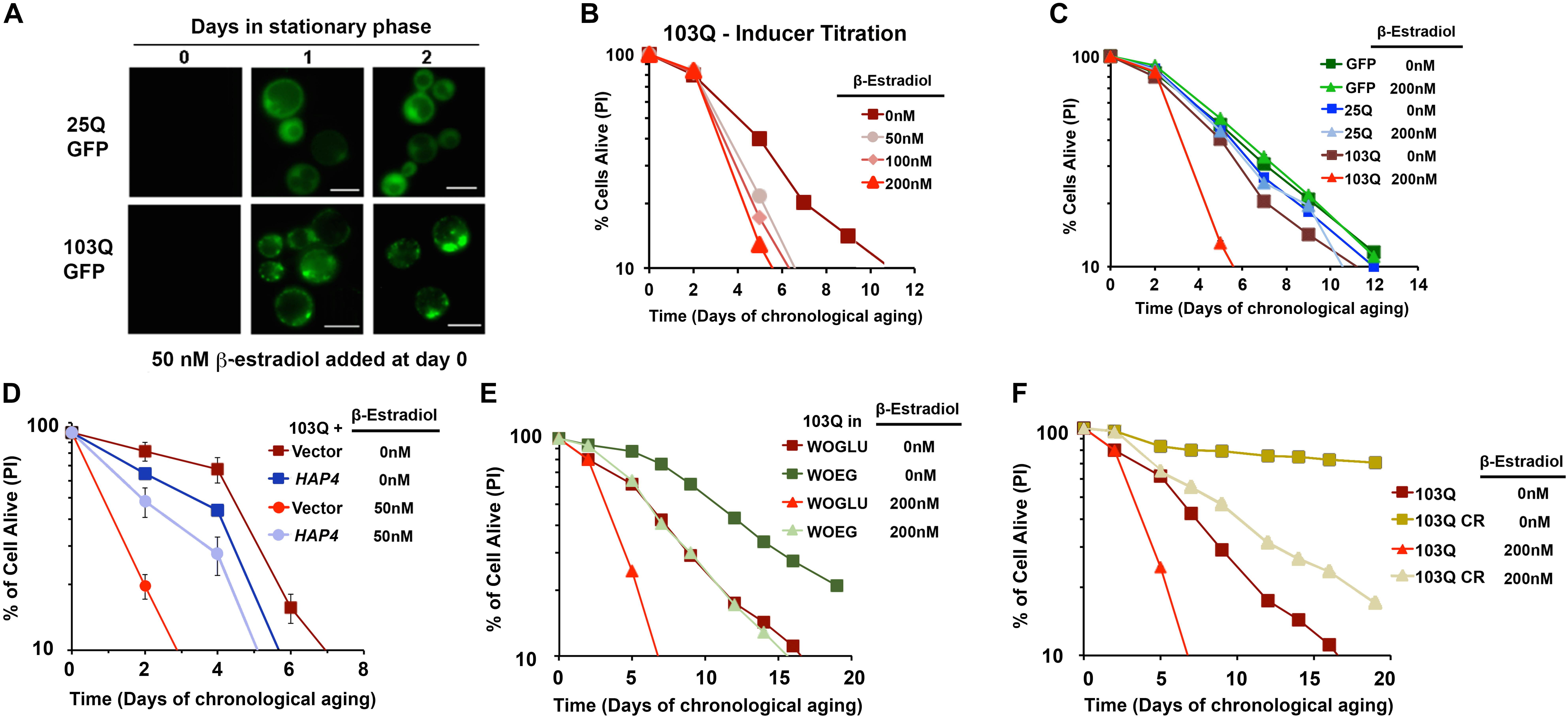
FIGURE 4. β-Estradiol inducible yeast models of polyglutamine disorders. (A) Chronology of polyQ-GFP protein accumulation, followed by fluorescence microscopy, in cells induced with 50 nM β-estradiol. The bar is 5 μm. (B,C) Yeast CLS. Survival of wild-type cells expressing 25Q or 103Q from a β-estradiol-inducible promoter activated with the indicated amounts of inducer or supplemented with the solvent (ethanol) was estimated by propidium iodide (PI) staining and flow cytometry analysis of 10,000 cells. Data are average of three samples in % of cells alive at day 0. In (C) a β-estradiol titration was performed. (D) Effect of increased mitochondrial biogenesis by HAP4 overexpression on CLS of yeast expressing 103Q from day 0 in the stationary phase. Error bars represent SEM for three independent experiments. (E) Effect of growth in synthetic medium containing ethanol and glycerol as non-fermentable (respiratory) carbon sources (WOEG) on 103Q yeast CLS compared to synthetic medium containing glucose as fermentable carbon source (WOGLU). SD <1 for all samples, n = 3. (F) Effect of calorie restriction (CR) modeled by growing the cells in the presence of 0.5% glucose vs. non-CR (2% glucose) in 103Q yeast CLS. SD <1 for all samples, n = 3. This figure was constructed using panels previously published in Ruetenik et al. (2016) with permission since they were published under the terms of the Creative Commons Attribution (CC BY) license.
In previous studies with this yeast model, using exponentially-growing cells, we had discovered that the 103Q mutant htt fragment associates with the mitochondrial membrane and disrupts several key mitochondrial functions (Solans et al., 2004; Ocampo et al., 2010). In an independent study, aiming to understand the determinants of CLS in wild-type yeast, we reported that there are mitochondrial respiratory thresholds during the exponential phase of growth, below which yeast CLS is shortened (Ocampo et al., 2012). We were therefore interested in assessing whether increasing the mitochondrial respiratory capacity in our inducible yeast model of HD from culture initiation could partially or entirely suppress the toxicity of 103Q overexpression in the CLS assay. To test this hypothesis, we explored three separate interventions that had previously been shown to increase mitochondrial respiration during exponential growth and to increase overall CLS. The first intervention, was the constitutive overexpression of HAP4, the catalytic subunit of the master transcriptional regulator complex that controls the expression of nuclear genes coding for mitochondrial proteins, which we had previously shown increases mitochondrial respiration in dividing cells when overexpressed (Ocampo et al., 2010). We next tested two nutritional modulators of yeast CLS that increase mitochondrial respiration: growth in media containing non-fermentable carbon sources, and glucose restriction. As expected, each of these three interventions did increase mitochondrial respiration exponential growth phase compared to cells, and these interventions did not decrease 103Q expression compared to controls. Importantly, all three of these interventions tested robustly extended the CLS of yeast overexpressing 103Q beyond that of cells overexpressing 103Q without the interventions (Ruetenik et al., 2016) (Figure 4). Therefore, the results in our HD yeast model suggest that treatments targeting mitochondrial biogenesis, and specifically mitochondrial respiration, may be beneficial in the treatment of HD patients to delay neuronal death.
Alzheimer’s Disease (AD)
Chen and Petranovic (2015) employed the CLS assay to study yeast models of AD and how overexpression of amyloid β peptides affects yeast CLS. In these experiments, the authors studied two different amyloid precursor protein cleavage products, Aβ40 and Aβ42. Importantly, Aβ42 has been found to be more prone to aggregation in previous studies by other groups, and it has been hypothesized that mutations that lead to an increase in Aβ42 production may be one of the primary causes of familial AD (Hampel et al., 2010). The experiments by Chen and Petranovic (2015) explored the effects of the constitutive overexpression, at various levels, of either Aβ40 or Aβ42, both with targeting signals directing the peptides to the endoplasmic reticulum (ER) secretory pathway. CLS studies were conducted at a constant pH of 5.0 and dissolved oxygen levels were kept above 30%, all other conditions being standard. In these conditions, overexpression of ER-targeted Aβ40 or Aβ42 both shortened CLS compared to cells without Aβ expression, with Aβ42 overexpression causing the most robust CLS shortening. Propidium iodide live/dead staining further confirmed these findings, revealing that overexpression of the ER-targeted Aβ42 peptide significantly increased the proportion of dead cells throughout the stationary phase compared to control cells (Chen and Petranovic, 2015). In a second publication by this group, a glucose restriction from the standard 2% concentration to 1% glucose at culture initiation was found to robustly increase CLS in the same model system, measured through propidium iodide staining (Chen et al., 2017).
Munoz-Arellano et al. (2018) have also recently studied the consequences of the expression of the loss-of-function ubiquitin variant UBB+1 in yeast using the CLS model. This UBB+1 variant, found to be neurotoxic in high levels, specifically accumulate and co-aggregate with the tangles and plaques of AD, as well as in HD and some other diseases of protein misfolding, while, in non-diseased cells the UBB+1 variant is degraded albeit inefficiently. UBB+1 has a high affinity for the proteasome and thus can make it inefficient to degrading other ubiquitinated moieties. To examine the effects of UBB+1 on aging cell health, Munoz-Arellano et al. (2018) expressed UBB+1 both at low and high levels. Whereas high expression of UBB+1 had no effect on yeast CLS, low UBB+1 levels greatly extended CLS, through a mechanism that included reduction of ROS levels and attenuation of apoptosis markers compared to control and high UBB+1 expressing cells. Altering the ubiquitin–proteasome system capacity by low UBB+1 expression was also found to protect cells against the adverse effects of protein misfolding induced through amino acid analogs (Munoz-Arellano et al., 2018). Further exploitation of this UBB+1 yeast model might help to identify additional factors that together with UBB+1 may contribute to the neurotoxic proteinotaphy disease initiation and/or progression.
Parkinson’s Disease (PD) and Other α-Synucleopathies
α-Synuclein (α-syn) is a presynaptic brain protein. Mainly cytosolic, α-synuclein can bind membranes and participate in vesicle trafficking. Misfolded/aggregated α-synuclein is the major constituent of cytoplasmic inclusions called Lewy bodies, a pathological hallmark of α-synucleinopathies (Uversky, 2008). This group of diseases includes PD, multiple system atrophy, and dementia. Mutant α-synuclein, as well as increased α-synuclein levels due to gene duplication, can cause PD. OXPHOS dysfunction and excessive ROS generation have been linked to PD (Schapira et al., 1990; Bender et al., 2006; Kraytsberg et al., 2006; Henchcliffe and Beal, 2008).
Yeast Models in Dividing Cells
Yeast models of α-synucleinopathies consist of overexpression of mutant or wild-type forms of α-synuclein (Outeiro and Lindquist, 2003). α-Synuclein misfolding-aggregation cause growth arrest and induces toxicity and cell death in an expression level-dependent manner (Outeiro and Lindquist, 2003). α-Synuclein toxicity in yeast is characterized by altered lipid metabolism, disrupted vesicular trafficking, ER stress, oxidative stress, impaired protein degradation, and mitochondrion-dependent cell death (Outeiro and Lindquist, 2003; Willingham et al., 2003; Kritzer et al., 2009; Yeger-Lotem et al., 2009; Auluck et al., 2010; Donmez et al., 2012). These yeast models have been also used for genome-wide screens to identify genes that enhance α-synuclein toxicity. Different from what it was found in yeast models of polyQ toxicity, the identified genes clustered in the processes of lipid metabolism and vesicle-mediated transport (Willingham et al., 2003).
Yeast Models in Non-dividing Cells
Whereas studies in growing cells have provided valuable mechanistic and physiological insight into α-synuclein toxicity, they need to be complemented with studies in non-dividing yeast cells to explore how α-synuclein toxicity modulates aging and vice versa. Of the neurodegenerative diseases, PD has been the most extensively studied using yeast models and the CLS assay. Of these studies, four have explored the effects of α-synuclein overexpression using different expression systems. The earliest of these studies, by Buttner et al. (2008), used a galactose-inducible system in which wild-type α-synuclein or α-synuclein with the toxic A53T point mutation, a cause of hereditary PD, was highly expressed in yeast at the beginning of the stationary phase. In this system, the overexpression of each form of α-synuclein was highly toxic in the CLS assay, with only about 20% of cells expressing either protein remaining alive by day 3 of the stationary phase, compared to about 70% of cells alive in the empty vector control cells. There was no significant difference seen in cell death between the expression of wild-type or A53T α-synuclein. Deletion of three proteins of interest involved in the apoptotic pathway was found to have no effect on either wild-type or A53T α-synuclein toxicity in the CLS assay. Finally, the authors expressed wild-type or A53T α-synuclein in yeast cells lacking mitochondrial DNA in the CLS assay. Surprisingly, ablation of mitochondrial DNA eliminated the toxic effects of α-synuclein overexpression in yeast cells in the stationary phase, indicating that the α-synuclein toxicity seen in the CLS assay is dependent on functional mitochondria (Buttner et al., 2008).
In another study, Sampaio-Marques et al. (2012) explored α-synuclein-induced toxicity in the CLS assay using a Tet-On system to induce wild-type α-synuclein expression during exponential growth, the diauxic shift, or the stationary phase of the CLS, and then continued following the yeast life span. Overexpression of α-synuclein was found to be toxic when induced at all stages of growth, but CLS, evaluated through CFU, was shortened most significantly when α-synuclein was induced starting in the stationary phase. These results indicate that aged cells may be particularly vulnerable to high levels of α-synuclein. Interestingly, when α-synuclein was induced at day 0 of the stationary phase, post-exponential phase and diauxic shift, and autophagy was inhibited using the pharmacological drug chloroquine, yeast CLS was greatly extended past that of cells overexpressing α-synuclein alone. However, a chloroquine-only treatment group without α-synuclein expression was not included, making this result difficult to interpret, as autophagy may play a significant role in the survival of wild-type cells in the CLS assay as well. As found by Buttner et al. (2008), when expressed from a high-overexpression galactose-inducible system, both wild-type or A53T α-synuclein expression were highly toxic to cells in stationary growth, reducing the number of viable cells to only 10% of the initial amount after 30 h of expression, compared to 100% remaining viable in cells with empty vector or overexpressing an α-synuclein A30P mutant protein (Sampaio-Marques et al., 2012). Under moderate expression throughout cell life, controlled by a constitutive TPI1 promoter, expression of all three forms of α-synuclein showed toxicity compared to vector control cells, with A53T α-synuclein shortening CLS the most, and A30P moderately reducing yeast CLS. Unexpectedly, when the three forms of α-synuclein were expressed in cells lacking Atg11, which encodes a protein involved in autophagy coordination, under the TPI1 constitutive promoter, only the A30P mutant, thought to be the less toxic form of α-synuclein from previous experiments, was found to have a shortened CLS. No toxicity was seen through overexpression of any form of α-synuclein in a mitophagy-deficient atg32 deletion strain, indicating that mitophagy is specifically essential for α-synuclein induced CLS shortening (Sampaio-Marques et al., 2012).
Two more recent studies have explored exogenous modulators on α-synuclein toxicity. First, a small study by Lee et al. (2011), presented a screen of the Prestwick and NIH chemical libraries for drugs that protect S. cerevisiae from sugar-induced cell death (SICD). SICD is triggered when stationary-phase yeast cells are transferred from spent rich medium into water with 2% glucose and no other nutrients, a paradigm that induces the generation of large amounts of ROS. The authors found that triclabendazole, a drug commonly used to treat liver fluke infections in cattle and humans, and approved for veterinary use in the United States, partially protects against SICD. Further tests found that treatment from culture initiation with triclabendazole, also suppresses the toxicity induced by constitutively expressed α-synuclein. Treatment with triclabendazole increased the average CLS, measured by CFU, of cells overexpressing α-synuclein to 194% of the CLS of cells overexpressing α-synuclein with DMSO control (Lee et al., 2011). Triclabendazole is believed to be effective against liver fluke infections by inhibiting β-tubulin. However, triclabenzadole was found to extend the life span of wild-type yeast cells over a DMSO control at concentrations that did not affect microtubule morphology (Lee et al., 2011), consistently with the fact that two drugs known to target microtubules in yeast, benomyl and nocodazole did not extend yeast life span (Lee et al., 2011). Because triclabenzadole effectively protected stationary-phase yeast cells from hydrogen peroxide-induced toxicity, the authors concluded that its protection of yeast cells from α-synuclein-induced cell could involve oxidative stress attenuation (Lee et al., 2011).
Most recently, a study by Guedes et al. (2017), reported that glucose restriction to 0.5% extends CLS in yeast overexpressing wild-type α-synuclein from the constitutive TPI1 promoter. A similar effect was observed upon deletion of Tor1, the target of rapamycin, and both interventions were associated with decreased autophagy, which was maintained at homeostatic levels. Maintenance of autophagy in α-synuclein-expressing cells under CR or upon tor1 deletion was proposed to be achieved by decreasing levels and activity of the yeast sirtuin Sir2 (Guedes et al., 2017).
Synphilin-1
In addition to α-synuclein, additional proteins that have been implicated in PD have been studied using the CLS assay. One of these proteins is Synphilin-1, a known interactor of α-synuclein that was first found using a yeast two-hybrid screen.
Interestingly, while studying this protein, Buttner et al. (2010) observed that whereas overexpression of α-synuclein is much more toxic to yeast than synphilin-1 when overexpressed during exponential growth (dividing cells), expression of synphilin-1 or expression of α-synuclein through the constitutive TPI1 promoter shortened CLS to a similar extent. Furthermore, co-expression of synphilin-1 along with α-synuclein showed induced higher toxicity to yeast in the stationary phase, shortening their CLS more that expression of either of the proteins alone, thus highlighting the relevance of CLS studies to model neurodegeneration. When synphilin-1 and α-synuclein were expressed in a yeast strain lacking sir2, a gene implicated in the process of yeast replicative aging, the toxicity and CLS-shortening induced by expression of the proteins individually or in combination was decreased significantly, although toxicity was still higher when synphilin-1 and α-synuclein were expressed together.
Human DJ-1 Protein
It is also known as PD protein 7 and a member of the DJ-1 superfamily of proteins. DJ-1 has been previously shown to inhibit the aggregation of α-synuclein (Shendelman et al., 2004) and mutations in human DJ-1 have been implicated in a form of autosomal recessive early-onset parkinsonism (Bonifati et al., 2003). DJ-1 plays some role in protection from oxidative stress, but how it functions is still unclear. Since yeast contains homologs of proteins belonging to the DJ-1 superfamily, Miller-Fleming et al. (2014) studied how deleting these proteins in yeast would affect cell viability during the CLS assay. In wild-type yeast cells, without the expression of α-synuclein, single deletion of multiple DJ-1 family member homologs in yeast shortened CLS (Miller-Fleming et al., 2014). However, the synergistic effect that deletion of these DJ-1 family members may have on α-synuclein toxicity was not studied in this model.
Mutations in PRKN or PARK2
The coding for the protein Parkin, have been also associated with an autosomal recessive form of juvenile PD (Kitada et al., 1998). Parkin is an E3 ubiquitin ligase, which plays essential roles in mitochondrial quality control and turnover. A yeast model to study the function of the Parkin protein was recently developed, in which wild-type Parkin was constitutively expressed in wild-type yeast cells and grown either in standard glucose media or non-fermentable media containing ethanol and glycerol (Pereira et al., 2015). In standard glucose media, no difference between CLS length was seen between cells with or without Parkin expression. However, when grown in non-fermentable media, cells overexpressing Parkin displayed an extended CLS (Pereira et al., 2015). The effects of Parkin overexpression concomitantly with α-synuclein expression are yet to be studied in the yeast CLS assay.
Familial Amyotrophic Lateral Sclerosis (ALS)
Mutations in the Gene SOD1
The coding for the mostly cytosolic form of the Cu–Zn superoxide dismutase, was one of the first genetic causes of familial ALS to be discovered, thus the effects of the deletion and overexpression of the SOD1 protein and SOD1 mutants have been a general research focus and have also been studied using the CLS assay. Across species, mitochondria contain a small fraction of SOD1 in the intermembrane space (Sturtz et al., 2001) and a matrix-located Mn-SOD known as SOD2 (Longo et al., 1996). In the original study by Longo et al. (1996) exploring the role of SOD1 and SOD2 in superoxide detoxification and its effect on CLS, single and double deletion yeast were grown in standard conditions and then transferred to sterile water upon entrance into the stationary phase. In these conditions, the Δsod2 strain showed slightly shortened CLS compared to wild-type cells, the Δsod1 strain showed a moderately shortened CLS, and Δsod1Δsod2 yeast displayed a severely reduced CLS. Interestingly, in low aeration conditions in sterile water, the Δsod1 strain showed a decreased level of toxicity compared to normoxia, while the Δsod2 mutant displayed enhanced toxicity compared to normoxia. The CLS shortening observed from the Δsod1Δsod2 strain remained the most extreme. Deletion of COQ3, a required gene in the synthesis of coenzyme Q, eliminated the respiratory capacity of the yeast cells, and appeared protective against the deletions of sod1 and sod2, however, the cells quickly died during the diauxic shift of the CLS assay, as respiration is required for entrance into the stationary phase (Longo et al., 1996).
As mentioned earlier, although SOD1 is synthesized in the cytosol, a ∼5% fraction of SOD1 and its copper chaperone Ccs1 are transported into the mitochondria to mature within the mitochondrial intermembrane space (Sturtz et al., 2001). Overexpression of the copper chaperone for SOD1, CCS, was found to increase the mitochondrial localization of the SOD1 protein, and notably, yeast cells in which mitochondrial SOD1 was enriched had extended CLS compared to yeast in which SOD1 was expressed without this mitochondrial enrichment (Sturtz et al., 2001). These results suggest that minimizing mitochondrial ROS generation is essential to maintain wild-type life span, although because multiple reports have also documented ROS signaling during exponential growth phase that promotes stress resistance that subsequently extends CLS (Pan et al., 2011; Ocampo et al., 2012), a critical ROS homeostasis needs to be maintained for optimal chronological aging.
Brasil et al. (2013) also studied the effects of SOD1 or SOD1 mutant overexpression in wild-type yeast cells. This group specifically studied the differences between overexpression of wild-type SOD1 and the SOD1 AV4 mutant, the mutation that has been most commonly associated with familial forms of ALS. As glutathione levels within the cell have also been found to be associated with mutant SOD1 toxicity, and overexpression has been previously seen to reduce mutant SOD1 aggregation, the authors also overexpressed both the wild-type and AV4 SOD1 variants in cells lacking the protein that acts as the first step in glutathione synthesis. In the CLS assay, when cells were transferred to sterile water and kept at 37°C during the stationary phase, overexpression of either wild-type or the AV4 SOD1 mutant significantly extended CLS, though the extension by the AV4 mutant was significantly shorter than that of wild-type SOD1. However, when overexpressed in the strain lacking glutathione synthesis, the protective effects of SOD1 overexpression were abolished entirely, and overexpression of the AV4 mutant shortened CLS (Brasil et al., 2013).
Senataxin (SETX)
SETX, which plays a vital role in maintaining RNA transcriptome homeostasis, is an RNA helicase implicated in two neurodegenerative disorders. Dominantly inherited mutations were identified in rare juvenile-onset, motor neuron disease pedigrees in a familial form of ALS (ALS4), whereas recessive mutations were found to cause a severe early-onset ataxia with oculomotor apraxia (AOA2) that is the second most common recessive ataxia after Freidreich’s ataxia (Bennett and La Spada, 2015). Senataxin dysfunction has been studied through the CLS assay by targeting its yeast homolog, Sen1. Specifically, a yeast strain missing the N-terminal region of the Sen1 protein was seen to have a severely shortened CLS in standard conditions compared to wild-type cells. This N-terminal region was previously seen to be necessary for protein–protein interactions (Sariki et al., 2016).
TAR DNA-Binding Protein 43 (TDP-43)
It is associated with a spectrum of neurodegenerative diseases, including ALS. TDP-43 has been shown to bind both DNA and RNA and have multiple functions in transcriptional repression, pre-mRNA splicing and translational regulation (Sephton et al., 2011). Yeast overexpressing human TDP-43 protein has been used to model dementia and motor neuron disorders, including ALS, in which this protein has been discovered in inclusion bodies. In this yeast model, expression of TDP-43 or TDP-43 mutants was driven by a galactose-inducible promoter on a low-copy-number plasmid, resulting in an intermediate level of expression when induced. In dividing cells, it has been shown that TDP-43 turnover and toxicity depend in part upon the endocytosis pathway (Liu et al., 2017). As seen in ALS patient tissues, TDP-43 inhibits endocytosis, and co-localizes strongly with endocytic proteins. Furthermore, impairing endocytosis increases TDP-43 toxicity, aggregation, and protein levels, whereas enhancing endocytosis reverses these phenotypes (Liu et al., 2017; Leibiger et al., 2018). In non-dividing cells, induction of TDP-43 expression (wild-type or the TDP-43 Q331K variant implicated in ALS) at the beginning of the stationary phase, shortened CLS compared to vector-carrying control cells. Curtailed CLS was similar for both TDP-43 variants by the third day of stationary phase, although TDP-43 Q331K resulted in a much faster decrease in cell viability at early time points. Conversely, when a high-copy-number plasmid was used, induction of wild-type TDP-43 resulted in a high degree of toxicity that quickly reduced cell viability to ∼20% 12 h after induction compared to control cells that remained near 100% viable at this time (Braun et al., 2011). When mitochondrial DNA was depleted, this severe toxic phenotype from wild-type TDP-43 overexpression was partially suppressed (Braun et al., 2011), as previously reported for yeast cells expressing α-synuclein (Buttner et al., 2008). Therefore, mitochondrial respiration has been proposed to play a major role in TDP-43- and α-synuclein-induced cytotoxicities in stationary-phase yeast cells. These results are interesting and would fit with the reported mitochondrial metabolic abnormalities often seen in patients suffering from neurodegenerative disorders. They are also intriguing because although respiratory defects in yeast (even absence of respiration) are less detrimental when produced experimentally only in the stationary phase, once yeast have accumulated nutrient stores during growth and undergone their metabolic remodeling during the diauxic shift, a minimum 40%-residual cell respiration during exponential growth is required for normal CLS (Ocampo et al., 2012).
Other Neurological Disorders
A handful of other neurological disorders have also been modeled in yeast and studied using the CLS assay.
Friedreich Ataxia
Friedreich ataxia is a hereditary autosomal recessive disease that causes severe neurological dysfunction (Rotig et al., 1997). The disease is caused by lowered expression of the human mitochondrial protein frataxin, which is conserved in yeast. In all organisms, frataxin plays an essential role in iron homeostasis and therefore in critical mitochondrial functions such as heme biosynthesis and iron–sulfur cluster assembly (Rotig et al., 1997). Gakh et al. (2006) studied how different mutations in yeast frataxin affected CLS. Their results showed that mutations that resulted in the inability of frataxin to oxidize iron or the inability to mineralize iron, both significantly reduced yeast CLS with similar severity (Gakh et al., 2006). Thus, both these functions seem equally important to preserving cell viability during CLS.
Niemann–Pick Type C (NPC)
Loss of function mutations in human NPC1 cause the rare, but very severe neurodegenerative disorder called Niemann–Pick type C (NPC). Human NPC1 and its yeast homolog Ncr1 are sphingolipid transporters that localize to vacuole membrane and to the ER. The yeast ncr1 deletion strain has been studied in the CLS assay. Yeast lacking Ncr1 display a premature aging phenotype (cells grown in standard synthetic media at 26°C) and higher sensitivity to oxidative stress associated with mitochondrial dysfunction and accumulation of long-chain bases (Vilaca et al., 2014). Importantly, deletion of the ceramide-activated protein phosphatases Pkh1, Sit4 and its activator cdc55 suppressed ncr1-deletion phenotypes but downregulation of de novo sphingolipid biosynthesis had no protective effect, suggesting that long-chain bases accumulation and shorten CLS may result from an increased turnover of complex sphingolipids (Vilaca et al., 2014, 2018).
Cathepsin D-Related Diseases
The loss of human protein cathepsin D has been linked to several neurodegenerative disorders. Cathepsin D is a protease required for efficient lysosomal protein breakdown, calcineurin signaling, and endosomal sorting (Aufschnaiter et al., 2017). The yeast homolog of cathepsin D is the vacuolar aspartyl protease (proteinase A) named Pep4. A pep4 deletion yeast strain was found to accumulate ROS and to have shorter CLS in standard conditions. Pointing toward a deleterious oxidative stress in the deletion strain, supplementation of the culture media with quercetin, a dietary flavonoid with antioxidant properties, found in a variety of variety of fruits and vegetables, increased CLS in the Δpep4 strain although a positive effect was also observed in wild-type yeast (Alugoju et al., 2018).
Gene Expression Systems and CLS Models
Inducible Gene Expression Systems: Advantages and Limitations
To date, most inducible yeast models of (gain of function) neurodegenerative diseases have been created by heterologous expression of human genes under the control of the strong GAL1 promoter, which is activated by galactose and repressed by glucose. Although these models have provided a significant amount of information, the lack of regulation of expression levels, generally resulting in high levels of expression, often leads to acute toxicity. These acute effects can be advantageous when screening for drugs or genetic suppressors of cytotoxicity but are not the ideal system for analyzing metabolic or physiological disturbances leading to cytotoxicity. Additionally, gene expression under the control of a galactose-inducible promoter introduces a metabolic constraint, since gene expression is induced upon transferring the cells to media containing 2% galactose, which is a fermentable carbon source. This system would prevent studies in non-fermentable, respiratory conditions that can be relevant to the study. For example, the use of metabolism-independent inducers would allow for the study of cell toxicities in situations in which the cells are forced to exclusively respire, creating a better model of the highly oxidative neuronal metabolism. Besides, the use of metabolism-independent inducers allows studies in non-dividing post-mitotic cells using the yeast stationary phase model of aging or CLS (Sinclair et al., 1998), the focus of this manuscript. As explained in the previous sections, in this post-mitotic state, energetic dependence on mitochondrial respiration and concomitant ROS production highly resemble the situation in which neuronal cells age.
To create refined inducible yeast models of neurodegenerative disorders, researchers have tested several systems (Table 2). Sampaio-Marques and colleagues used a Tet-On inducible system to study how age traits potentiate the cytotoxic effects of α-synuclein. In these models, α-synuclein expression was induced from a Tet-On promoter in different phases of yeast growth (exponential, diauxic, or stationary) and CLS was then determined (Sampaio-Marques et al., 2012). The Tet-on system is highly regulatable although tend to be leaky. Importantly, even at low concentrations, the inducer of the system, tetracyclines, are known inhibitors of mitochondrial translation and can therefore provoke mitochondrial proteotoxic stress, leading to changes in nuclear gene expression and altered mitochondrial dynamics and function, which will introduce a confounding variable in experimental settings (Moullan et al., 2015). The use of newly developed tetracycline-based systems that are more sensitive could be an alternative to minimize the dose of the antibiotic that needs to be used; however, even if no apparent mitochondrial toxicity is detected, widespread gene expression changes may sensitize cells to the intended tetracycline-controlled loss or gain of function, still confounding the results (Chatzispyrou et al., 2015).
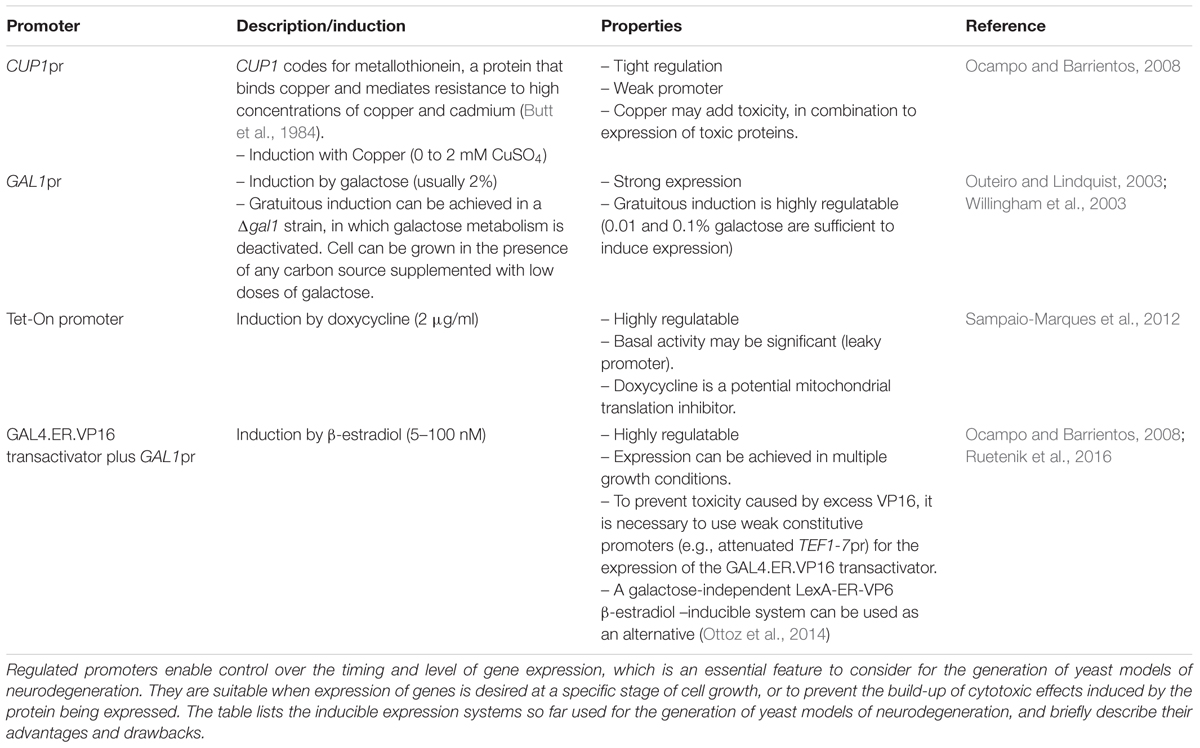
TABLE 2. Inducible gene expression systems used for the construction of yeast models of neurodegeneration.
In our laboratory, we have focused on modeling HD, based on the expression of polyQ domains of normal and pathological length under the control of different promoters, to test for their advantages and drawbacks. We tested the CUP1 promoter, two different β-estradiol-inducible GAL1 promoter systems and the GAL1 promoter in a Δgal1 mutant background (Ocampo and Barrientos, 2008, 2012) (Table 2). Evaluation and comparison of the different systems was performed to assess their tightness, regulation of expression levels, self-toxicity, and effects on metabolism. The use of copper is not recommended since it can promote the generation of ROS. Models created using the GAL1 promoter in cells in which their galactose metabolism is disabled by disrupting the endogenous GAL1 gene are a good choice, since they are highly regulatable, metabolically gratuitous, and non-toxic. We have opted to more extensively use β-estradiol inducible models, which are strongly regulated and are based on the constitutive expression of a transactivator fusion protein GAL4.ER.VP16, which is formed by a GAL4 DNA binding domain, a β-estradiol receptor domain and a VP16 (virus protein 16) transcriptional activator that can activate transcription of a gene placed under the control of a galactose-inducible promoter (GAL1pr). These models have been further optimized to eliminate VP16 toxicity by using a weak constitutive promoter (an attenuated version of the TEF1 promoter) for the expression of the GAL4.ER.VP16 transactivator (Ruetenik et al., 2016). In the absence of the β-estradiol hormone, the fusion protein in this system is repressed by the yeast chaperones from the Hsp90 family (Louvion et al., 1993). Upon media supplementation with β-estradiol, the fusion protein is de-repressed, binds to GAL1pr through the GAL4 DNA binding domain and the VP16 recruits the transcriptional machinery to start transcription of the gene of interest. This system has been already used successfully to create models of neurotoxic proteotoxicity expressing polyglutamine domains (Ruetenik et al., 2016) in the context of chronological aging. Using an alternative galactose-independent transactivator could further improve the β-estradiol inducible models. In this line, the recently developed LexA-ER-AD β-estradiol -inducible system, based on a heterologous DNA-binding domain (LexA) and non-toxic activation domains, can be used as an alternative (Ottoz et al., 2014).
Alternative Culture Models for the Study of Chronological Life Span
In the classical CLS assays, the age-dependent viability of non-dividing cells is estimated in conditions that involved starvation of exogenous nutrients and reliance on storage carbohydrates, glycogen and trehalose (Cao et al., 2016). However, because many terminally differentiated human cell types rarely starve and some, such as neurons, can consume a large amount of energy (Bourre, 2006), it has been considered imperative to develop a yeast model to study CLS, in which nutrients are not limited, but cell division stops and metabolism remains highly active. With this goal in mind, uncoupling metabolism from cell division, Nagarajan et al. (2014) have promoted the study of CLS in immobilized cells using bioreactors, a technology that has been extensively used in brewing and bioethanol production (Verbelen et al., 2006). To model CLS under nutrient-replete conditions, the authors encapsulated cells in a matrix made of calcium alginate to form beads that were packed into bioreactors and fed ad libitum (Nagarajan et al., 2014). In these conditions, cells stopped dividing, retained high glycolytic flux but decreased expression of genes in the tricarboxylic acid cycle, stored large amounts of glycogen, had enhanced expression of stress resistance genes and maintained >95% viability over prolonged culture (17 days). Therefore, yeast metabolism in bioreactors is very different from metabolism in batch cultures, where yeast cells depend on respiration to survive the diauxic shift and reach the stationary phase (Ocampo et al., 2012). Respiratory-competent cells grown in batch liquid cultures in the presence of 2% glucose use both fermentation of stored carbohydrates and respiration for survival once they reach the stationary phase (Ocampo et al., 2012). However, when yeast is grown in liquid cultures containing only 0.5% glucose (what is considered as a model of calorie restriction), their stress resistance is enhanced, accumulation of stored carbohydrates is increased and CLS is extended by 3–4 fold (above 1 month) while their requirement of oxidative metabolism for survival is abolished (Ocampo et al., 2012). Immobilized yeast under calorie-unrestricted bioreactor conditions also exhibits long CLS in the absence of respiration, which is precluded by glucose repression and low O2 tension (Nagarajan et al., 2014). In conclusion, the pattern of gene expression and the metabolism of immobilized cells grown in bioreactors is similar to that of stationary phase liquid cultures initially grown in the presence of low glucose concentrations. Despite the value of immobilized yeast cultures, the classical liquid cultures may be easier to implement in most research laboratories and offer the possibility to modulate media composition to allow aging cells to use fermentative and respiratory metabolisms.
Concluding Remarks
Yeast models of neurodegenerative disorders have traditionally used rapidly dividing cells and expression of human disease genes. Although some disease genes are conserved along evolution, some of those responsible for age-associated neurodegenerative disorders are restricted to vertebrates. While many features of the disease cannot be modeled in yeast (e.g., neuron type specificity or loss of function activities), yeast models have proven to provide a robust paradigm for the elucidation of pathways leading to cell death upon deletion or overexpression of conserved genes, or upon heterologous expression of human disease genes. The refinement of yeast models by the incorporation of neurotoxic gene expression controlled by metabolism-independent promoters has allowed the study of neurotoxic proteotoxicity in non-dividing yeast using the CLS model of aging. Although relatively few studies using the yeast CLS model are found so far in the literature, they are expected to exponentially increase as they can be a source of information regarding the chronology of physiological events leading to neurotoxic proteotoxicity-induced cell death and the identification of new pathways involved. Importantly, the yeast CLS model has been instrumental for the identification of pathways that modulate life span in yeast and higher organisms. Therefore, inducible yeast models of neurodegeneration in the context of CLS will allow testing whether and how life span modulators (e.g., nutrient-sensing and stress-resistance pathways) postpone the physiological effects and cell death induced by neurotoxic proteins.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported by a Research grant from The Army Research Office (ARO) # W911NF-16-1-0311 (to AB), a Merit award from the Veterans Administration (VA-) Biomedical Laboratory Research and Development 1I01BX003303-01 (to AB) and an HD Human Biology Project Fellowship from the Huntington’s Disease Society of America (HDSA) # GR009606 (to AR).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Julia Denissova for her help with the construction of Table 1.
References
Alugoju, P., Janardhanshetty, S. S., Subaramanian, S., Periyasamy, L., and Dyavaiah, M. (2018). Quercetin protects yeast Saccharomyces cerevisiae pep4 mutant from oxidative and apoptotic stress and extends chronological lifespan. Curr. Microbiol. 75, 519–530. doi: 10.1007/s00284-017-1412-x
Anderson, R., and Prolla, T. (2009). PGC-1alpha in aging and anti-aging interventions. Biochim. Biophys. Acta 1790, 1059–1066. doi: 10.1016/j.bbagen.2009.04.005
Anderson, R. M., Barger, J. L., Edwards, M. G., Braun, K. H., O’connor, C. E., Prolla, T. A., et al. (2008). Dynamic regulation of PGC-1alpha localization and turnover implicates mitochondrial adaptation in calorie restriction and the stress response. Aging Cell 7, 101–111. doi: 10.1111/j.1474-9726.2007.00357.x
Aufschnaiter, A., Kohler, V., and Buttner, S. (2017). Taking out the garbage: cathepsin D and calcineurin in neurodegeneration. Neural Regen. Res. 12, 1776–1779. doi: 10.4103/1673-5374.219031
Auluck, P. K., Caraveo, G., and Lindquist, S. (2010). alpha-Synuclein: membrane interactions and toxicity in Parkinson’s disease. Annu. Rev. Cell Dev. Biol. 26, 211–233. doi: 10.1146/annurev.cellbio.042308.113313
Barrientos, A. (2003). Yeast models of human mitochondrial diseases. IUBMB Life 55, 83–95. doi: 10.1002/tbmb.718540876
Barrientos, A., Fontanesi, F., and Diaz, F. (2009). Evaluation of the mitochondrial respiratory chain and oxidative phosphorylation system using polarography and spectrophotometric enzyme assays. Curr. Protoc. Hum. Genet. 63, 19.3.1–19.3.14. doi: 10.1002/0471142905.hg1903s63
Bauer, P. O., and Nukina, N. (2009). Enhanced degradation of mutant huntingtin by rho kinase inhibition is mediated through activation of proteasome and macroautophagy. Autophagy 5, 747–748. doi: 10.4161/auto.5.5.8704
Bender, A., Krishnan, K. J., Morris, C. M., Taylor, G. A., Reeve, A. K., Perry, R. H., et al. (2006). High levels of mitochondrial DNA deletions in Substantia nigra neurons in aging and Parkinson disease. Nat. Genet. 38, 515–517. doi: 10.1038/ng1769
Bennett, C. L., and La Spada, A. R. (2015). Unwinding the role of senataxin in neurodegeneration. Discov. Med. 19, 127–136.
Bonawitz, N. D., Chatenay-Lapointe, M., Pan, Y., and Shadel, G. S. (2007). Reduced TOR signaling extends chronological life span via increased respiration and upregulation of mitochondrial gene expression. Cell Metab. 5, 265–277. doi: 10.1016/j.cmet.2007.02.009
Bonifati, V., Rizzu, P., Van Baren, M. J., Schaap, O., Breedveld, G. J., Krieger, E., et al. (2003). Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 299, 256–259. doi: 10.1126/science.1077209
Bourre, J. M. (2006). Effects of nutrients (in food) on the structure and function of the nervous system: update on dietary requirements for brain. Part 2: macronutrients. J. Nutr. Health Aging 10, 386–399.
Brasil, A. A., Belati, A., Mannarino, S. C., Panek, A. D., Eleutherio, E. C., and Pereira, M. D. (2013). The involvement of GSH in the activation of human Sod1 linked to FALS in chronologically aged yeast cells. FEMS Yeast Res. 13, 433–440. doi: 10.1111/1567-1364.12045
Braun, R. J., Buttner, S., Ring, J., Kroemer, G., and Madeo, F. (2009). Nervous yeast: modeling neurotoxic cell death. Trends Biochem. Sci. 35, 135–144. doi: 10.1016/j.tibs.2009.10.005
Braun, R. J., Sommer, C., Carmona-Gutierrez, D., Khoury, C. M., Ring, J., Buttner, S., et al. (2011). Neurotoxic 43-kDa TAR DNA-binding protein (TDP-43) triggers mitochondrion-dependent programmed cell death in yeast. J. Biol. Chem. 286, 19958–19972. doi: 10.1074/jbc.M110.194852
Burtner, C. R., Murakami, C. J., Kennedy, B. K., and Kaeberlein, M. (2009). A molecular mechanism of chronological aging in yeast. Cell Cycle 8, 1256–1270. doi: 10.4161/cc.8.8.8287
Butt, T. R., Sternberg, E. J., Gorman, J. A., Clark, P., Hamer, D., Rosenberg, M., et al. (1984). Copper metallothionein of yeast, structure of the gene, and regulation of expression. Proc. Natl. Acad. Sci. U.S.A. 81, 3332–3336. doi: 10.1073/pnas.81.11.3332
Buttner, S., Bitto, A., Ring, J., Augsten, M., Zabrocki, P., Eisenberg, T., et al. (2008). Functional mitochondria are required for alpha-synuclein toxicity in aging yeast. J. Biol. Chem. 283, 7554–7560. doi: 10.1074/jbc.M708477200
Buttner, S., Delay, C., Franssens, V., Bammens, T., Ruli, D., Zaunschirm, S., et al. (2010). Synphilin-1 enhances alpha-synuclein aggregation in yeast and contributes to cellular stress and cell death in a Sir2-dependent manner. PLoS One 5:e13700. doi: 10.1371/journal.pone.0013700
Cao, L., Tang, Y., Quan, Z., Zhang, Z., Oliver, S. G., and Zhang, N. (2016). Chronological Llfespan in yeast is dependent on the accumulation of storage carbohydrates mediated by Yak1, Mck1 and Rim15 Kinases. PLoS Genet. 12:e1006458. doi: 10.1371/journal.pgen.1006458
Chatzispyrou, I. A., Held, N. M., Mouchiroud, L., Auwerx, J., and Houtkooper, R. H. (2015). Tetracycline antibiotics impair mitochondrial function and its experimental use confounds research. Cancer Res. 75, 4446–4449. doi: 10.1158/0008-5472.CAN-15-1626
Chen, Q., Ding, Q., and Keller, J. N. (2005). The stationary phase model of aging in yeast for the study of oxidative stress and age-related neurodegeneration. Biogerontology 6, 1–13. doi: 10.1007/s10522-004-7379-6
Chen, X., Bisschops, M. M. M., Agarwal, N. R., Ji, B., Shanmugavel, K. P., and Petranovic, D. (2017). Interplay of energetics and ER stress exacerbates Alzheimer’s amyloid-beta (Abeta) toxicity in yeast. Front. Mol. Neurosci. 10:232. doi: 10.3389/fnmol.2017.00232
Chen, X., and Petranovic, D. (2015). Amyloid-beta peptide-induced cytotoxicity and mitochondrial dysfunction in yeast. FEMS Yeast Res. 15:fov061. doi: 10.1093/femsyr/fov061
Chevtzoff, C., Yoboue, E. D., Galinier, A., Casteilla, L., Daignan-Fornier, B., Rigoulet, M., et al. (2009). Reactive oxygen species mediated regulation of mitochondrial biogenesis in the yeast Saccharomyces cerevisiae. J. Biol. Chem. 285, 1733–1742. doi: 10.1074/jbc.M109.019570
Cui, L., Jeong, H., Borovecki, F., Parkhurst, C. N., Tanese, N., and Krainc, D. (2006). Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell 127, 59–69. doi: 10.1016/j.cell.2006.09.015
Dejean, L., Beauvoit, B., Bunoust, O., Guerin, B., and Rigoulet, M. (2002). Activation of Ras cascade increases the mitochondrial enzyme content of respiratory competent yeast. Biochem. Biophys. Res. Commun. 293, 1383–1388. doi: 10.1016/S0006-291X(02)00391-1
Donmez, G., Arun, A., Chung, C. Y., Mclean, P. J., Lindquist, S., and Guarente, L. (2012). SIRT1 protects against alpha-synuclein aggregation by activating molecular chaperones. J. Neurosci. 32, 124–132. doi: 10.1523/JNEUROSCI.3442-11.2012
Duennwald, M. L., and Lindquist, S. (2008). Impaired ERAD and ER stress are early and specific events in polyglutamine toxicity. Genes Dev. 22, 3308–3319. doi: 10.1101/gad.1673408
Enns, L. C., Morton, J. F., Treuting, P. R., Emond, M. J., Wolf, N. S., Dai, D. F., et al. (2009). Disruption of protein kinase A in mice enhances healthy aging. PLoS One 4:e5963. doi: 10.1371/journal.pone.0005963
Fabrizio, P., and Longo, V. D. (2007). The chronological life span of Saccharomyces cerevisiae. Methods Mol. Biol. 371, 89–95. doi: 10.1007/978-1-59745-361-5_8
Fabrizio, P., Pozza, F., Pletcher, S. D., Gendron, C. M., and Longo, V. D. (2001). Regulation of longevity and stress resistance by Sch9 in yeast. Science 292, 288–290. doi: 10.1126/science.1059497
Fontana, L., Partridge, L., and Longo, V. D. (2010). Extending healthy life span–from yeast to humans. Science 328, 321–326. doi: 10.1126/science.1172539
Fontanesi, F., Diaz, F., and Barrientos, A. (2009). Evaluation of the mitochondrial respiratory chain and oxidative phosphorylation system using yeast models of OXPHOS deficiencies. Curr. Protoc. Hum. Genet. 63, 19.5.1–19.5.20. doi: 10.1002/0471142905.hg1905s63
Fontanesi, F., Palmieri, L., Scarcia, P., Lodi, T., Donnini, C., Limongelli, A., et al. (2004). Mutations in AAC2, equivalent to human adPEO-associated ANT1 mutations, lead to defective oxidative phosphorylation in Saccharomyces cerevisiae and affect mitochondrial DNA stability. Hum. Mol. Genet. 13, 923–934. doi: 10.1093/hmg/ddh108
Gaisne, M., Becam, A. M., Verdiere, J., and Herbert, C. J. (1999). A ‘natural’ mutation in Saccharomyces cerevisiae strains derived from S288c affects the complex regulatory gene HAP1 (CYP1). Curr. Genet. 36, 195–200. doi: 10.1007/s002940050490
Gakh, O., Park, S., Liu, G., Macomber, L., Imlay, J. A., Ferreira, G. C., et al. (2006). Mitochondrial iron detoxification is a primary function of frataxin that limits oxidative damage and preserves cell longevity. Hum. Mol. Genet. 15, 467–479. doi: 10.1093/hmg/ddi461
Giorgini, F., Guidetti, P., Nguyen, Q., Bennett, S. C., and Muchowski, P. J. (2005). A genomic screen in yeast implicates kynurenine 3-monooxygenase as a therapeutic target for Huntington disease. Nat. Genet. 37, 526–531. doi: 10.1038/ng1542
Guedes, A., Ludovico, P., and Sampaio-Marques, B. (2016). Caloric restriction alleviates alpha-synuclein toxicity in aged yeast cells by controlling the opposite roles of Tor1 and Sir2 on autophagy. Mech. Ageing Dev. 161(Pt B), 270–276. doi: 10.1016/j.mad.2016.04.006
Guedes, A., Ludovico, P., and Sampaio-Marques, B. (2017). Caloric restriction alleviates alpha-synuclein toxicity in aged yeast cells by controlling the opposite roles of Tor1 and Sir2 on autophagy. Mech. Ageing Dev. 161, 270–276. doi: 10.1016/j.mad.2016.04.006
Gunther, M. R., Vangilder, R., Fang, J., and Beattie, D. S. (2004). Expression of a familial amyotrophic lateral sclerosis-associated mutant human superoxide dismutase in yeast leads to decreased mitochondrial electron transport. Arch. Biochem. Biophys. 431, 207–214. doi: 10.1016/j.abb.2004.08.009
Hampel, H., Shen, Y., Walsh, D. M., Aisen, P., Shaw, L. M., Zetterberg, H., et al. (2010). Biological markers of amyloid beta-related mechanisms in Alzheimer’s disease. Exp. Neurol. 223, 334–346. doi: 10.1016/j.expneurol.2009.09.024
Henchcliffe, C., and Beal, M. F. (2008). Mitochondrial biology and oxidative stress in Parkinson disease pathogenesis. Nat. Clin. Pract. Neurol. 4, 600–609. doi: 10.1038/ncpneuro0924
Hertweck, M., Gobel, C., and Baumeister, R. (2004). C. elegans SGK-1 is the critical component in the Akt/PKB kinase complex to control stress response and life span. Dev. Cell 6, 577–588. doi: 10.1016/S1534-5807(04)00095-4
Hess, D. C., Lu, W., Rabinowitz, J. D., and Botstein, D. (2006). Ammonium toxicity and potassium limitation in yeast. PLoS Biol. 4:e351. doi: 10.1371/journal.pbio.0040351
Johnson, J. E., and Johnson, F. B. (2014). Methionine restriction activates the retrograde response and confers both stress tolerance and lifespan extension to yeast, mouse and human cells. PLoS One 9:e97729. doi: 10.1371/journal.pone.0097729
Kaeberlein, M., Andalis, A. A., Fink, G. R., and Guarente, L. (2002). High osmolarity extends life span in Saccharomyces cerevisiae by a mechanism related to calorie restriction. Mol. Cell. Biol. 22, 8056–8066. doi: 10.1128/MCB.22.22.8056-8066.2002
Kaminosono, S., Saito, T., Oyama, F., Ohshima, T., Asada, A., Nagai, Y., et al. (2008). Suppression of mutant Huntingtin aggregate formation by Cdk5/p35 through the effect on microtubule stability. J. Neurosci. 28, 8747–8755. doi: 10.1523/JNEUROSCI.0973-08.2008
Kapahi, P., Zid, B. M., Harper, T., Koslover, D., Sapin, V., and Benzer, S. (2004). Regulation of lifespan in Drosophila by modulation of genes in the TOR signaling pathway. Curr. Biol. 14, 885–890. doi: 10.1016/j.cub.2004.03.059
Kitada, T., Asakawa, S., Hattori, N., Matsumine, H., Yamamura, Y., Minoshima, S., et al. (1998). Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392, 605–608. doi: 10.1038/33416
Kraytsberg, Y., Kudryavtseva, E., Mckee, A. C., Geula, C., Kowall, N. W., and Khrapko, K. (2006). Mitochondrial DNA deletions are abundant and cause functional impairment in aged human Substantia nigra neurons. Nat. Genet. 38, 518–520. doi: 10.1038/ng1778
Kritzer, J. A., Hamamichi, S., Mccaffery, J. M., Santagata, S., Naumann, T. A., Caldwell, K. A., et al. (2009). Rapid selection of cyclic peptides that reduce alpha-synuclein toxicity in yeast and animal models. Nat. Chem. Biol. 5, 655–663. doi: 10.1038/nchembio.193
Krobitsch, S., and Lindquist, S. (2000). Aggregation of huntingtin in yeast varies with the length of the polyglutamine expansion and the expression of chaperone proteins. Proc. Natl. Acad. Sci. U.S.A. 97, 1589–1594. doi: 10.1073/pnas.97.4.1589
Lee, Y. J., Burlet, E., Wang, S., Xu, B., Huang, S., Galiano, F. J., et al. (2011). Triclabendazole protects yeast and mammalian cells from oxidative stress: identification of a potential neuroprotective compound. Biochem. Biophys. Res. Commun. 414, 205–208. doi: 10.1016/j.bbrc.2011.09.057
Leibiger, C., Deisel, J., Aufschnaiter, A., Ambros, S., Tereshchenko, M., Verheijen, B. M., et al. (2018). TDP-43 controls lysosomal pathways thereby determining its own clearance and cytotoxicity. Hum. Mol. Genet. 27, 1593–1607. doi: 10.1093/hmg/ddy066
Lin, M. T., and Beal, M. F. (2006). Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443, 787–795. doi: 10.1038/nature05292
Liu, G., Coyne, A. N., Pei, F., Vaughan, S., Chaung, M., Zarnescu, D. C., et al. (2017). Endocytosis regulates TDP-43 toxicity and turnover. Nat. Commun. 8:2092. doi: 10.1038/s41467-017-02017-x
Longo, V. D. (1999). Mutations in signal transduction proteins increase stress resistance and longevity in yeast, nematodes, fruit flies, and mammalian neuronal cells. Neurobiol. Aging 20, 479–486. doi: 10.1016/S0197-4580(99)00089-5
Longo, V. D., and Finch, C. E. (2003). Evolutionary medicine: from dwarf model systems to healthy centenarians? Science 299, 1342–1346.
Longo, V. D., Gralla, E. B., and Valentine, J. S. (1996). Superoxide dismutase activity is essential for stationary phase survival in Saccharomyces cerevisiae. Mitochondrial production of toxic oxygen species in vivo. J. Biol. Chem. 271, 12275–12280. doi: 10.1074/jbc.271.21.12275
Longo, V. D., Shadel, G. S., Kaeberlein, M., and Kennedy, B. (2012). Replicative and chronological aging in Saccharomyces cerevisiae. Cell 16, 18–31. doi: 10.1016/j.cmet.2012.06.002
Louvion, J. F., Havaux-Copf, B., and Picard, D. (1993). Fusion of GAL4-VP16 to a steroid-binding domain provides a tool for gratuitous induction of galactose-responsive genes in yeast. Gene 131, 129–134. doi: 10.1016/0378-1119(93)90681-R
MacLean, M., Harris, N., and Piper, P. W. (2001). Chronological lifespan of stationary phase yeast cells; a model for investigating the factors that might influence the ageing of postmitotic tissues in higher organisms. Yeast 18, 499–509. doi: 10.1002/yea.701
Maruyama, Y., Ito, T., Kodama, H., and Matsuura, A. (2016). Availability of amino acids extends chronological lifespan by suppressing hyper-acidification of the environment in Saccharomyces cerevisiae. PLoS One 11:e0151894. doi: 10.1371/journal.pone.0151894
Meriin, A. B., Zhang, X., He, X., Newnam, G. P., Chernoff, Y. O., and Sherman, M. Y. (2002). Huntington toxicity in yeast model depends on polyglutamine aggregation mediated by a prion-like protein Rnq1. J. Cell Biol. 157, 997–1004. doi: 10.1083/jcb.200112104
Miller-Fleming, L., Antas, P., Pais, T. F., Smalley, J. L., Giorgini, F., and Outeiro, T. F. (2014). Yeast DJ-1 superfamily members are required for diauxic-shift reprogramming and cell survival in stationary phase. Proc. Natl. Acad. Sci. U.S.A. 111, 7012–7017. doi: 10.1073/pnas.1319221111
Miller-Fleming, L., Giorgini, F., and Outeiro, T. F. (2008). Yeast as a model for studying human neurodegenerative disorders. Biotechnol. J. 3, 325–338. doi: 10.1002/biot.200700217
Mortimer, R. K., and Johnston, J. R. (1986). Genealogy of principal strains of the yeast genetic stock center. Genetics 113, 35–43.
Moullan, N., Mouchiroud, L., Wang, X., Ryu, D., Williams, E. G., Mottis, A., et al. (2015). Tetracyclines disturb mitochondrial function across eukaryotic models: a call for caution in biomedical research. Cell Rep. 10, 00180–00181. doi: 10.1016/j.celrep.2015.02.034
Muchowski, P. J., Ning, K., D’souza-Schorey, C., and Fields, S. (2002). Requirement of an intact microtubule cytoskeleton for aggregation and inclusion body formation by a mutant huntingtin fragment. Proc. Natl. Acad. Sci. U.S.A. 99, 727–732. doi: 10.1073/pnas.022628699
Munoz-Arellano, A. J., Chen, X., Molt, A., Meza, E., and Petranovic, D. (2018). Different expression levels of human mutant ubiquitin B(+ 1) (UBB(+ 1)) can modify chronological lifespan or stress resistance of Saccharomyces cerevisiae. Front. Mol. Neurosci. 11:200. doi: 10.3389/fnmol.2018.00200
Murakami, C. J., Burtner, C. R., Kennedy, B. K., and Kaeberlein, M. (2008). A method for high-throughput quantitative analysis of yeast chronological life span. J. Gerontol. A Biol. Sci. Med. Sci. 63, 113–121. doi: 10.1093/gerona/63.2.113
Nagarajan, S., Kruckeberg, A. L., Schmidt, K. H., Kroll, E., Hamilton, M., Mcinnerney, K., et al. (2014). Uncoupling reproduction from metabolism extends chronological lifespan in yeast. Proc. Natl. Acad. Sci. U.S.A. 111, E1538–E1547. doi: 10.1073/pnas.1323918111
Ocampo, A., and Barrientos, A. (2008). From the bakery to the brain business: developing inducible yeast models of human neurodegenerative disorders. Biotechniques 45, vii–xiv. doi: 10.2144/000112746
Ocampo, A., and Barrientos, A. (2011). Quick and reliable assessment of chronological life span in yeast cell populations by flow cytometry. Mech. Ageing Dev. 132, 315–323. doi: 10.1016/j.mad.2011.06.007
Ocampo, A., and Barrientos, A. (2012). Developing yeast models of human neurodegenerative disorders. Methods Mol. Biol. 793, 113–127. doi: 10.1007/978-1-61779-328-8_8
Ocampo, A., Liu, J., Schroeder, E. A., Shadel, G. S., and Barrientos, A. (2012). Mitochondrial respiratory thresholds regulate yeast chronological life span and its extension by caloric restriction. Cell Metab. 16, 55–67. doi: 10.1016/j.cmet.2012.05.013
Ocampo, A., Zambrano, A., and Barrientos, A. (2010). Suppression of polyglutamine-induced cytotoxicity in Saccharomyces cerevisiae by enhancement of mitochondrial biogenesis. FASEB J. 24, 1431–1441. doi: 10.1096/fj.09-148601
O’Rourke, T. W., Doudican, N. A., Mackereth, M. D., Doetsch, P. W., and Shadel, G. S. (2002). Mitochondrial dysfunction due to oxidative mitochondrial DNA damage is reduced through cooperative actions of diverse proteins. Mol. Cell Biol. 22, 4086–4093. doi: 10.1128/MCB.22.12.4086-4093.2002
Ottoz, D. S., Rudolf, F., and Stelling, J. (2014). Inducible, tightly regulated and growth condition-independent transcription factor in Saccharomyces cerevisiae. Nucleic Acids Res. 42:e130. doi: 10.1093/nar/gku616
Outeiro, T. F., and Lindquist, S. (2003). Yeast cells provide insight into alpha-synuclein biology and pathobiology. Science 302, 1772–1775. doi: 10.1126/science.1090439
Pan, Y., Schroeder, E. A., Ocampo, A., Barrientos, A., and Shadel, G. S. (2011). Regulation of yeast chronological life span by TORC1 via adaptive mitochondrial ROS signaling. Cell Metab. 13, 668–678. doi: 10.1016/j.cmet.2011.03.018
Pan, Y., and Shadel, G. S. (2009). Extension of chronological life span by reduced TOR signaling requires down-regulation of Sch9p and involves increased mitochondrial OXPHOS complex density. Aging 1, 131–145. doi: 10.18632/aging.100016
Panov, A. V., Gutekunst, C. A., Leavitt, B. R., Hayden, M. R., Burke, J. R., Strittmatter, W. J., et al. (2002). Early mitochondrial calcium defects in Huntington’s disease are a direct effect of polyglutamines. Nat. Neurosci. 5, 731–736. doi: 10.1038/nn884
Paradis, S., Ailion, M., Toker, A., Thomas, J. H., and Ruvkun, G. (1999). A PDK1 homolog is necessary and sufficient to transduce AGE-1 PI3 kinase signals that regulate diapause in Caenorhabditis elegans. Genes Dev. 13, 1438–1452. doi: 10.1101/gad.13.11.1438
Parrella, E., and Longo, V. D. (2008). The chronological life span of Saccharomyces cerevisiae to study mitochondrial dysfunction and disease. Methods 46, 256–262. doi: 10.1016/j.ymeth.2008.10.004
Pereira, C., Costa, V., Martins, L. M., and Saraiva, L. (2015). A yeast model of the Parkinson’s disease-associated protein Parkin. Exp. Cell Res. 333, 73–79. doi: 10.1016/j.yexcr.2015.02.018
Powers, R. W. III, Kaeberlein, M., Caldwell, S. D., Kennedy, B. K., and Fields, S. (2006). Extension of chronological life span in yeast by decreased TOR pathway signaling. Genes Dev. 20, 174–184. doi: 10.1101/gad.1381406
Rotig, A., De Lonlay, P., Chretien, D., Foury, F., Koenig, M., Sidi, D., et al. (1997). Aconitase and mitochondrial iron-sulphur protein deficiency in Friedreich ataxia. Nat. Genet. 17, 215–217. doi: 10.1038/ng1097-215
Ruetenik, A. L., Ocampo, A., Ruan, K., Zhu, Y., Li, C., Zhai, R. G., et al. (2016). Attenuation of polyglutamine-induced toxicity by enhancement of mitochondrial OXPHOS in yeast and fly models of aging. Microb. Cell 3, 338–351. doi: 10.15698/mic2016.08.518
Sampaio-Marques, B., Felgueiras, C., Silva, A., Rodrigues, M., Tenreiro, S., Franssens, V., et al. (2012). SNCA (alpha-synuclein)-induced toxicity in yeast cells is dependent on sirtuin 2 (Sir2)-mediated mitophagy. Autophagy 8, 1494–1509. doi: 10.4161/auto.21275
Santos, J., Leitao-Correia, F., Sousa, M. J., and Leao, C. (2015). Ammonium is a key determinant on the dietary restriction of yeast chronological aging in culture medium. Oncotarget 6, 6511–6523. doi: 10.18632/oncotarget.2989
Sariki, S. K., Sahu, P. K., Golla, U., Singh, V., Azad, G. K., and Tomar, R. S. (2016). Sen1, the homolog of human Senataxin, is critical for cell survival through regulation of redox homeostasis, mitochondrial function, and the TOR pathway in Saccharomyces cerevisiae. FEBS J. 283, 4056–4083. doi: 10.1111/febs.13917
Sarkar, S., Ravikumar, B., Floto, R. A., and Rubinsztein, D. C. (2009). Rapamycin and mTOR-independent autophagy inducers ameliorate toxicity of polyglutamine-expanded huntingtin and related proteinopathies. Cell Death Differ. 16, 46–56. doi: 10.1038/cdd.2008.110
Schapira, A. H., Cooper, J. M., Dexter, D., Clark, J. B., Jenner, P., and Marsden, C. D. (1990). Mitochondrial complex I deficiency in Parkinson’s disease. J. Neurochem. 54, 823–827. doi: 10.1111/j.1471-4159.1990.tb02325.x
Selman, C., Tullet, J. M., Wieser, D., Irvine, E., Lingard, S. J., Choudhury, A. I., et al. (2009). Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science 326, 140–144. doi: 10.1126/science.1177221
Sephton, C. F., Cenik, C., Kucukural, A., Dammer, E. B., Cenik, B., Han, Y., et al. (2011). Identification of neuronal RNA targets of TDP-43-containing ribonucleoprotein complexes. J. Biol. Chem. 286, 1204–1215. doi: 10.1074/jbc.M110.190884
Shao, J., and Diamond, M. I. (2007). Polyglutamine diseases: emerging concepts in pathogenesis and therapy. Hum. Mol. Genet. 16, R115–R123. doi: 10.1093/hmg/ddm213
Shendelman, S., Jonason, A., Martinat, C., Leete, T., and Abeliovich, A. (2004). DJ-1 is a redox-dependent molecular chaperone that inhibits alpha-synuclein aggregate formation. PLoS Biol. 2:e362. doi: 10.1371/journal.pbio.0020362
Sherman, F. (1991). Getting started with yeast. Methods Enzymol. 194, 3–21. doi: 10.1016/0076-6879(91)94004-V
Sinclair, D., Mills, K., and Guarente, L. (1998). Aging in Saccharomyces cerevisiae. Annu. Rev. Microbiol. 52, 533–560. doi: 10.1146/annurev.micro.52.1.533
Smith, D. L. Jr., Mcclure, J. M., Matecic, M., and Smith, J. S. (2007). Calorie restriction extends the chronological lifespan of Saccharomyces cerevisiae independently of the Sirtuins. Aging Cell 6, 649–662. doi: 10.1111/j.1474-9726.2007.00326.x
Solans, A., Zambrano, A., and Barrientos, A. (2004). Cytochrome c oxidase deficiency: from yeast to human. Preclinica 2, 336–348.
Solans, A., Zambrano, A., Rodriguez, M., and Barrientos, A. (2006). Cytotoxicity of a mutant huntingtin fragment in yeast involves early alterations in mitochondrial OXPHOS complexes II and III. Hum. Mol. Genet. 15, 3063–3081. doi: 10.1093/hmg/ddl248
Sturtz, L. A., Diekert, K., Jensen, L. T., Lill, R., and Culotta, V. C. (2001). A fraction of yeast Cu, Zn-superoxide dismutase and its metallochaperone, CCS, localize to the intermembrane space of mitochondria. A physiological role for SOD1 in guarding against mitochondrial oxidative damage. J. Biol. Chem. 276, 38084–38089.
Trushina, E., Dyer, R. B., Badger, J. D. II, Ure, D., Eide, L., Tran, D. D., et al. (2004). Mutant huntingtin impairs axonal trafficking in mammalian neurons in vivo and in vitro. Mol. Cell Biol. 24, 8195–8209. doi: 10.1128/MCB.24.18.8195-8209.2004
Uversky, V. N. (2008). Alpha-synuclein misfolding and neurodegenerative diseases. Curr. Protein Pept. Sci. 9, 507–540. doi: 10.2174/138920308785915218
Vacher, C., Garcia-Oroz, L., and Rubinsztein, D. C. (2005). Overexpression of yeast hsp104 reduces polyglutamine aggregation and prolongs survival of a transgenic mouse model of Huntington’s disease. Hum. Mol. Genet. 14, 3425–3433. doi: 10.1093/hmg/ddi372
van Dijken, J. P., Bauer, J., Brambilla, L., Duboc, P., Francois, J. M., Gancedo, C., et al. (2000). An interlaboratory comparison of physiological and genetic properties of four Saccharomyces cerevisiae strains. Enzyme Microb. Technol. 26, 706–714. doi: 10.1016/S0141-0229(00)00162-9
Verbelen, P. J., De Schutter, D. P., Delvaux, F., Verstrepen, K. J., and Delvaux, F. R. (2006). Immobilized yeast cell systems for continuous fermentation applications. Biotechnol. Lett. 28, 1515–1525. doi: 10.1007/s10529-006-9132-5
Vilaca, R., Barros, I., Matmati, N., Silva, E., Martins, T., Teixeira, V., et al. (2018). The ceramide activated protein phosphatase Sit4 impairs sphingolipid dynamics, mitochondrial function and lifespan in a yeast model of Niemann-Pick type C1. Biochim. Biophys. Acta 1864, 79–88. doi: 10.1016/j.bbadis.2017.10.010
Vilaca, R., Silva, E., Nadais, A., Teixeira, V., Matmati, N., Gaifem, J., et al. (2014). Sphingolipid signalling mediates mitochondrial dysfunctions and reduced chronological lifespan in the yeast model of Niemann-Pick type C1. Mol. Microbiol. 91, 438–451. doi: 10.1111/mmi.12470
Wang, H., Lim, P. J., Karbowski, M., and Monteiro, M. J. (2009). Effects of overexpression of huntingtin proteins on mitochondrial integrity. Hum. Mol. Genet. 18, 737–752. doi: 10.1093/hmg/ddn404
Wei, M., Fabrizio, P., Hu, J., Ge, H., Cheng, C., Li, L., et al. (2008). Life span extension by calorie restriction depends on Rim15 and transcription factors downstream of Ras/PKA, Tor, and Sch9. PLoS Genet. 4:e13. doi: 10.1371/journal.pgen.0040013
Willingham, S., Outeiro, T. F., Devit, M. J., Lindquist, S. L., and Muchowski, P. J. (2003). Yeast genes that enhance the toxicity of a mutant huntingtin fragment or alpha-synuclein. Science 302, 1769–1772. doi: 10.1126/science.1090389
Yan, L., Vatner, D. E., O’connor, J. P., Ivessa, A., Ge, H., Chen, W., et al. (2007). Type 5 adenylyl cyclase disruption increases longevity and protects against stress. Cell 130, 247–258. doi: 10.1016/j.cell.2007.05.038
Keywords: inducible yeast model, Saccharomyces cerevisiae, chronological life span, neurodegenerative disorder, mitochondria, proteotoxicity, reactive oxygen species
Citation: Ruetenik A and Barrientos A (2018) Exploiting Post-mitotic Yeast Cultures to Model Neurodegeneration. Front. Mol. Neurosci. 11:400. doi: 10.3389/fnmol.2018.00400
Received: 08 June 2018; Accepted: 12 October 2018;
Published: 02 November 2018.
Edited by:
Ralf J. Braun, University of Bayreuth, GermanyReviewed by:
Per Olov Ingvar Widlund, University of Gothenburg, SwedenDina Petranovic, Chalmers University of Technology, Sweden
Copyright © 2018 Ruetenik and Barrientos. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Antonio Barrientos, YWJhcnJpZW50b3NAbWVkLm1pYW1pLmVkdQ==