 Rocío Naranjo1,2Paz González1,2Alejandro Lopez-Hurtado1,2Xosé M. Dopazo1,2Britt Mellström1,2
Rocío Naranjo1,2Paz González1,2Alejandro Lopez-Hurtado1,2Xosé M. Dopazo1,2Britt Mellström1,2 José R. Naranjo1,2*
José R. Naranjo1,2*- 1Spanish Network for Biomedical Research in Neurodegenerative Diseases (CIBERNED), ISCIII, Madrid, Spain
- 2National Biotechnology Center (CNB), CSIC, Madrid, Spain
Deregulated intracellular Ca2+ and protein homeostasis underlie synaptic dysfunction and are common features in neurodegenerative diseases. DREAM, also known as calsenilin or KChIP-3, is a multifunctional Ca2+ binding protein of the neuronal calcium sensor superfamily with specific functions through protein-DNA and protein-protein interactions. Small-molecules able to bind DREAM, like the anti-diabetic drug repaglinide, disrupt some of the interactions with other proteins and modulate DREAM activity on Kv4 channels or on the processing of activating transcription factor 6 (ATF6). Here, we show the interaction of endogenous DREAM and presenilin-2 (PS2) in mouse brain and, using DREAM deficient mice or transgenic mice overexpressing a dominant active DREAM (daDREAM) mutant in the brain, we provide genetic evidence of the role of DREAM in the endoproteolysis of endogenous PS2. We show that repaglinide disrupts the interaction between DREAM and the C-terminal PS2 fragment (Ct-PS2) by coimmunoprecipitation assays. Exposure to sub-micromolar concentrations of repaglinide reduces the levels of Ct-PS2 fragment in N2a neuroblastoma cells. These results suggest that the interaction between DREAM and PS2 may represent a new target for modulation of PS2 processing, which could have therapeutic potential in Alzheimer’s disease (AD) treatment.
Introduction
The neuronal calcium sensor DREAM, also known as calsenilin or KChIP-3, is a multifunctional Ca2+ binding protein with specific roles in different subcellular compartments through protein-DNA and/or protein-protein interactions. Thus, in the nucleus DREAM binds to DRE sites in the DNA and represses transcription of target genes (Carrión et al., 1999). In addition, DREAM interacts with other nucleoproteins, like CREB, CtBP1, nuclear receptors or TTF-1 and modifies their transcriptional function (Ledo et al., 2002; Rivas et al., 2004; Scsucova et al., 2005; Zaidi et al., 2006). Outside the nucleus, the DREAM interactome comprises a heterogeneous set of proteins encompassing ion channels, membrane receptors, GRK kinases and presenilins (PSs), among others (reviewed in Rivas et al., 2011; Burgoyne and Haynes, 2012). Binding of DREAM to these proteins modulates channel gating, response to agonist stimulation (An et al., 2000; Savignac et al., 2005; Rivas et al., 2009; Wu et al., 2010) or regulates DREAM activity through post-translational modifications including phosphorylation or sumoylation (Ruiz-Gomez et al., 2007; Palczewska et al., 2011). Binding to Ca2+ induces conformational changes in DREAM that prevents DREAM binding to DRE sites in the DNA (Carrión et al., 1999) and distinctly modifies the interaction with other proteins (Holmqvist et al., 2001; Naranjo et al., 2016). Furthermore, binding to arachidonic acid and other small-molecules, like repaglinide or Cl-888, also induces changes in DREAM that affect its activity on Kv4 channel gating or on the processing of activating transcription factor 6 (ATF6; Holmqvist et al., 2001; Okada et al., 2003; Bowlby et al., 2005; Naranjo et al., 2016).
PSs (PS1 and PS2) are the catalytic core of the γ-secretase complex, an enzymatic activity linked to the plasma membrane that cleaves multiple intramembrane substrates including the amyloid precursor protein (APP), cell surface receptors and adhesion molecules like Notch, E-cadherin and ErbB4 (McCarthy et al., 2009; Haapasalo and Kovacs, 2011). In addition, PS proteins participate in other γ-secretase independent cellular functions including Wnt/β-catenin signaling, calcium homeostasis, apoptosis, protein trafficking, lysosomal function and autophagy (reviewed in Jurisch-Yaksi et al., 2013; Duggan and McCarthy, 2016). Importantly, mutations in the PS genes are associated with inherited familial Alzheimer’s disease (AD), and they influence both γ-secretase dependent and independent cellular functions (Sherrington et al., 1995; Zhang et al., 1998; Lee et al., 2010; Supnet and Bezprozvanny, 2011).
Posttranslational modifications of the PSs include endoproteolysis, caspase cleavage, phosphorylation and ubiquitination (Duggan and McCarthy, 2016). PS endoproteolysis occurs in the large cytosolic loop of the protein generating N- and a C-terminal fragments that remain stably associated as a heterodimer, which constitutes the active core of the γ-secretase holoenzyme (Wolfe et al., 1999; Esler et al., 2000; Li et al., 2000; Spasic and Annaert, 2008; Lu et al., 2014; Sun et al., 2015). PS proteins are highly homologous, however, fragments generated from either PS1 or PS2 do not interact with each other, suggesting that the two PSs form independent complexes (Capell et al., 1998; Saura et al., 1999). Further supporting the idea of independent functions for PS proteins, PS1 knockout (KO) mice die shortly after birth displaying aberrant defects in the central nervous system and spinal ganglia (Shen et al., 1997; Wong et al., 1997). To date, the protease activity that mediates PS proteolysis is unknown, though the acidic protease inhibitor pepstatin A is the best known PS endoproteolysis inhibitor (Campbell et al., 2002). The involvement of other protease activities in PS cleavage like the proteasome (Liu et al., 2003; Kopan and Ilagan, 2004; Massey et al., 2005) or the γ-secretase complex (Takasugi et al., 2003; Xie et al., 2005; Fukumori et al., 2010; Honarnejad et al., 2013) has been suggested. Importantly, PS endoproteolysis is developmentally regulated (Hartmann et al., 1997) and the amount of PS fragments in the cell is tightly controlled (Steiner et al., 1998; Saura et al., 1999), though the mechanisms that regulate these activities are not well understood. Other posttranslational changes are also important for PS function. Thus, PS1 phosphorylation occurs at multiple sites by a variety of kinases and modulates γ-secretase activity (Kuo et al., 2008), γ-secretase independent functions (Walter et al., 1999; Uemura et al., 2007) and PS stability (Lau et al., 2002). Moreover, caspase cleavage of the C-terminal fragment of PS2 (Ct-PS2) has been associated with the role of PS2 in apoptosis (Alves da Costa et al., 2002, 2003, 2006) and PS ubiquitination has been related to the stability of the full-length holoproteins and the degradation of the PS fragments (Massey et al., 2005).
Yeast two-hybrid assays identified the interaction between Ct-PS2 and DREAM, and coimmunoprecipitation experiments after heterologous overexpression in COS-7 cells confirmed the binding of DREAM to PS1 and PS2 C-terminal fragments (Buxbaum et al., 1998). Recently, the structural determinants of the DREAM-Ct-PS1 interaction have been elegantly analyzed (Pham and Miksovska, 2016). Upon binding, DREAM regulates PS activity at different levels. First, overexpression of DREAM in human neuroglioma H4 cells increases PS2 cleavage and accumulation of Ct-PS2 (Buxbaum et al., 1998). Since mutations in the PS genes are associated with the development of familial type of AD (FAD), this finding linked DREAM/calsenilin and neurodegeneration in AD (Buxbaum et al., 1998). This mechanism, however, was questioned in a more recent study that showed no change or a decrease in Ct-PS2 in human neuroblastoma SH-5YSY cells overexpressing DREAM or Ca2+-insensitive DREAM mutants, respectively (Fedrizzi et al., 2008). Second, binding of PS2 to DREAM increases γ-secretase activity and the levels of Aβ peptides, one of the end-products of APP processing. In this regard, genetic ablation of DREAM leads to reduced APP processing and lower brain levels of Aβ peptides (Lilliehook et al., 2003). Third, PSs regulate calcium homeostasis through interaction with the Sarco/ER/Ca2+-ATPase (SERCA) pump, InsP3 and ryanodine receptors and/or forming leak channels for calcium ions in the endoplasmic reticulum membrane (Tu et al., 2006; Cheung et al., 2008; Green et al., 2008; Hayrapetyan et al., 2008; Nelson et al., 2011). DREAM overexpression enhances the depletion of calcium from the ER store (Fedrizzi et al., 2008) and reduction of PS endoproteolysis reduces calcium release from this subcellular compartment (Honarnejad et al., 2013). Finally, PSs have been associated with apoptosis through γ-secretase-dependent and -independent pathways (Alves da Costa et al., 2002, 2003, 2006; Wang et al., 2005; Zeng et al., 2015) and DREAM overexpression have been associated with cell death (Jo et al., 2001). Whether DREAM-induced Ct-PS2 fragment accumulation mediates this effect is currently not established.
Here, we show that endogenous DREAM interacts with PS2 and regulates PS2 endoproteolysis in the brain. Moreover, repaglinide, a DREAM binding molecule, blocks the DREAM/PS2 interaction and reduces PS2 processing in N2a mouse neuroblastoma cells.
Materials and Methods
Mice
Homozygous DREAM KO mice (Cheng et al., 2002) and transgenic mice expressing a dominant active mutant DREAM insensitive to calcium (Dierssen et al., 2012; Mellström et al., 2014) were used to assess the role of DREAM in PS2 endoproteolysis in the brain.
Cells
N2a mouse neuroblastoma and HEK293T cells were from ATCC. All cells were cultured in DMEM (with 10% FBS, penicillin/streptomycin, Glutamax; all from Invitrogen). Cell cultures were routinely checked for mycoplasma infection. To evaluate the effect of DREAM binding molecules on PS processing, neuroblastoma cells were exposed to increasing concentrations of repaglinide (30 nM to 3 μM, Sigma) and harvested 36 h later.
Coimmunoprecipitation
Endogenous DREAM and PS2 proteins were coimmunoprecipitated from whole cell extracts (200 μg) from mouse brain or N2a cells, using 5 μg affinity-purified mouse monoclonal anti-DREAM 1B1 (Ledo et al., 2002) Tagged proteins were coimmunoprecipitated from whole cell extracts (200 μg) from HEK293T cells cotransfected with plasmids encoding Myc-DREAM71–256 and Flag-Ct-PS2335–448, using 1 μg affinity-purified rabbit anti-Myc (ab9106, Abcam) in the presence of vehicle (DMSO) or repaglinide. Immunoprecipitated PS2 was detected by western blot with anti-PS2 (Cell Signaling Technology, #2192; endogenous) or mouse anti-Flag antibody (Sigma; overexpressed).
Western Blot
Mouse hippocampal and cortical extracts were prepared as described (López-Hurtado et al., 2018). Briefly, brain tissue was homogenized on ice in lysis buffer (RIPA, Cell Signaling Technology, #9806) supplemented with 1 mM PMSF. After 20 min incubation on ice, extracts were cleared by centrifugation (14,000× g, 20 min) and protein concentration assessed (Bradford, BioRad). For the analysis of the effect by DREAM binding molecules on PS2 processing, N2a cells were treated with repaglinide, or vehicle (DMSO) at the indicated concentrations for 36 h. Cells were pelleted and incubated on ice in lysis buffer for 45 min, with occasional vortexing. Samples (20 μg) were analyzed by SDS-PAGE and immunoblot. PVDF membranes were incubated overnight at 4°C with rabbit anti-PS2 (Cell Signaling Technology, #2192 or Abcam, EP1515Y, ab51249). Secondary antibody used was HRP-conjugated donkey anti-rabbit IgG (Jackson) and signals were detected with ECL Select (GE Healthcare). Control for equal loading were with total protein measured by Coomassie staining of the membrane after immunoblotting or HRP-conjugated β-actin (Sigma, A3854 clone AC-15). Lane and band intensity were quantified with ImageLab software (BioRad). We obtained similar results when we used as a primary antibody a rabbit monoclonal antibody raised against a C-terminal peptide of PS2 (Abcam Ab51249). This confirmed the specificity of the Ct-PS2 band detected by western blot.
Real-Time Quantitative PCR
RNA was isolated from tissue samples using TRIzol, treated with DNase (Ambion) and reverse transcribed using hexamer primer and Moloney murine leukemia virus reverse transcriptase. To confirm the absence of genomic DNA, each sample was processed in parallel without reverse transcriptase. Real-time quantitative PCR (qPCR) for endogenous PS2 was performed using the assay from Applied Biosystems (Mm00448405_m1) and expression was normalized using HPRT as reference with primers 5′ TTG GAT ACA GGC CAG ACT TTG TT 3′ and 5′ CTG AAG TAC TCA TTA TAG TCA AGG GCA TA 3′ and the MGB probe 5′ TTG AAA TTC CAG ACA AGT TT 3′.
Statistical Analysis
All data values shown are mean ± SEM. Differences were considered significant at P < 0.05. When possible, ordinary one-way ANOVA was used to analyze statistical differences among groups. For small or unequal sample size or non-Gaussian distribution, comparisons between groups were analyzed using t-test or the nonparametric ANOVA, Kruskal-Wallis test with Dunn’s multiple comparison between groups. Animal experiments were randomized. Sample size was not predetermined by statistical method. Prism 6.0 software (GraphPad) was used for statistical analysis.
Study Approval
Experiments with mice were conducted in accordance with standard ethical guidelines (European Communities Directive 86/609 EEC; National Institutes of Health 1995). The CNB-CSIC and Madrid Community ethical committees approved the experiments with mice (PROEX 28/05).
Results
DREAM Regulates the Processing of Presenilin-2 in Mouse Brain
Overexpression of DREAM increased PS2 processing and the levels of the Ct-PS2 in human H4 neuroglioma but not in SH-SY5Y neuroblastoma cells (Buxbaum et al., 1998; Fedrizzi et al., 2008). Overexpression conditions could be part of the controversy between these results. Thus, we aimed to investigate whether or not endogenous DREAM regulates PS2 processing in vivo in basal conditions in mouse brain. Using total extracts from mouse hippocampus or cerebral cortex, coimmunoprecipitation experiments showed the interaction of endogenous DREAM and Ct-PS2 both in the hippocampus and the cerebral cortex (Figure 1). Moreover, endogenous DREAM was shown to interact not only with the Ct-PS2 processed fragment but also with the full length PS2 holoprotein (Figure 1). Similar results were observed using whole cell extracts from mouse N2a neuroblastoma cells (Figure 1).
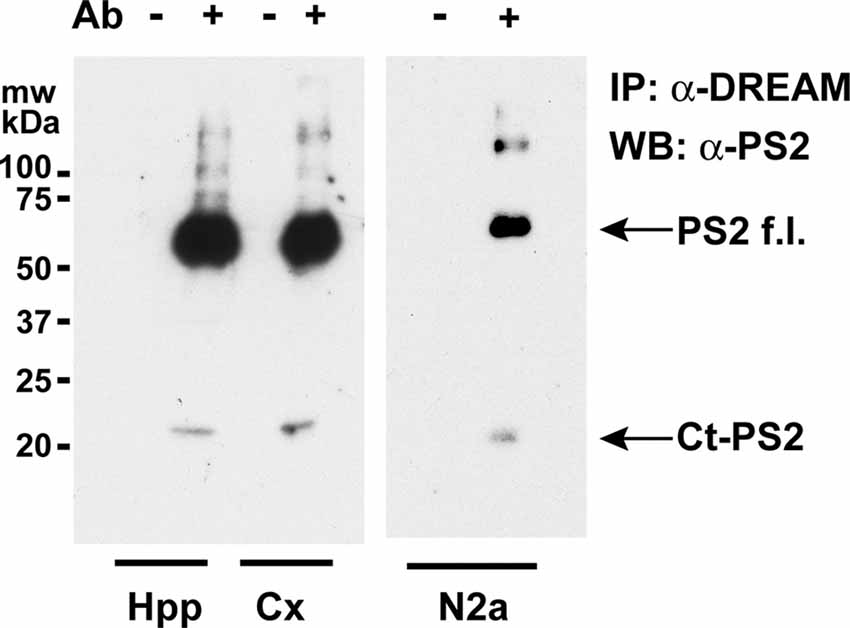
Figure 1. Coimmunoprecipitation of endogenous DREAM and presenilin-2 (PS2) proteins. Western blots of immunoprecipitated PS2 from mouse hippocampal (Hpp) and cortical (Cx) extracts (left) or from N2a whole cell extracts (right), with (+) or without (−) an anti-DREAM antibody (Ab), and developed with an antibody against PS2. Bands corresponding to the Ct-PS2 fragment and the full length PS2 holoprotein (PS2f.l.) are shown by arrows.
We then investigated whether PS2 processing is regulated by DREAM in mouse brain. For this, we compared Ct-PS2 basal brain levels in wild type (WT), DREAM deficient and transgenic mice overexpressing a dominant active DREAM (daDREAM) mutant in the brain. Absence of DREAM in DREAM deficient mice significantly reduced Ct-PS2 levels both in the cerebral cortex and the hippocampus (Figure 2A). Correspondingly, increased levels of DREAM protein in daDREAM transgenic mouse brain resulted in significant accumulation of Ct-PS2 in both brain areas (Figure 2A). Changes in Ct-PS2 levels in the cerebral cortex were not accompanied by significant changes in PS2 gene expression (Figure 2B). However, a slight but significant increase in PS2 mRNA level was observed in the hippocampus of daDREAM mice (Figure 2B). Taken together, these results indicate that endogenous DREAM interacts with endogenous PS2 and regulates PS2 endoproteolysis, most likely, without directly affecting PS2 gene expression.
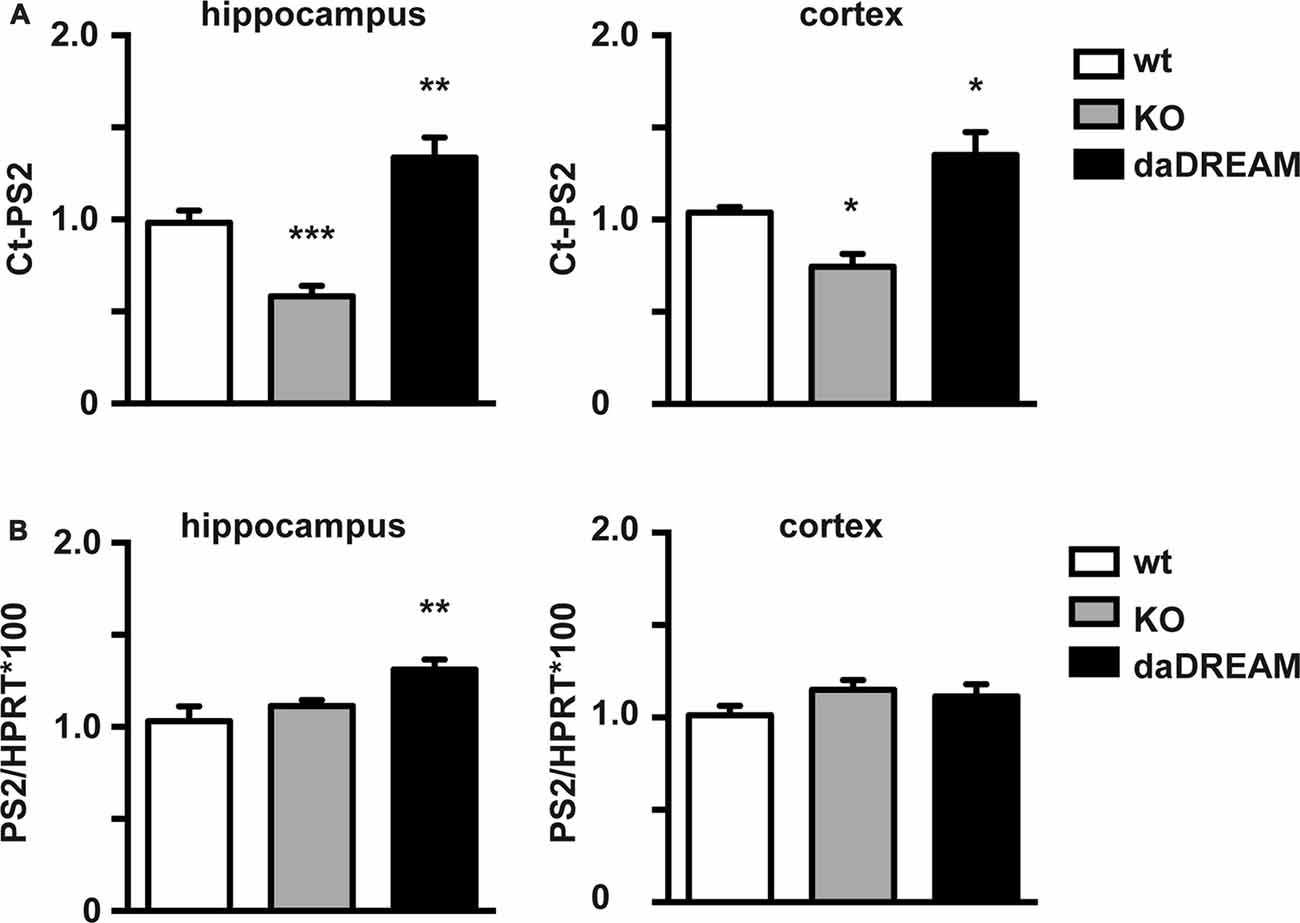
Figure 2. Effect of endogenous DREAM levels on PS2 endoproteolysis and PS2 gene expression. (A) Quantified Western blots with anti-PS2 antibody of hippocampal (Hpp) and cortical (Cx) extracts from wild-type (WT), DREAM deficient knockout (KO) and transgenic mice overexpressing dominant active DREAM (daDREAM). (B) Quantitative PCR (qPCR) analysis of PS2 mRNA levels in the hippocampus and the cerebral cortex from the same three genotypes as in (A). Ordinary one-way ANOVA with Holm-Sidak’s multiple comparison test (n = 11, for each genotype) *P < 0.05, **P < 0.01, ***P < 0.001.
Repaglinide Blocks the DREAM-PS2 Interaction
We next analyzed whether DREAM-binding molecules could affect the DREAM/PS2 interaction. For this, we reproduced previous coimmunoprecipitation experiments (Buxbaum et al., 1998) using HEK293T cells overexpressing tagged proteins. Combined overexpression of Myc-DREAM and Flag-Ct-PS2 resulted in coimmunoprecipitation of Flag-Ct-PS2 with an anti-Myc antibody (Figure 3A). Detection of the Flag-Ct-PS2 fragment with an anti-Flag antibody or an anti-Ct-PS2 antibody rendered identical results. As negative controls, non-transfected cells or cells transfected with only one of the expression plasmids did not show immunoprecipitation (Figure 3A). Inclusion in the immunoprecipitation reaction of 1 mM CaCl2 or up to 2 mM EGTA did not affect the interaction and did not modify the intensity of the immunoprecipitated band (Figure 3B). These results confirm the Ca2+-independent nature of this interaction, as previously described (Buxbaum et al., 1998; Fedrizzi et al., 2008). Then, we proceeded to investigate a potential blockage of the DREAM-PS2 interaction by the DREAM binding molecule repaglinide, known to affect the interaction of DREAM with other proteins including Kv4 potassium channels and ATF6 (Naranjo et al., 2016; López-Hurtado et al., 2018). In vitro exposure to repaglinide significantly reduced the Myc-DREAM/Flag-Ct-PS2 coimmunoprecipitation (Figure 3C). Inclusion of DMSO, the vehicle for repaglidine, as a negative control did not change the extent of the DREAM-PS2 interaction (Figure 3B).
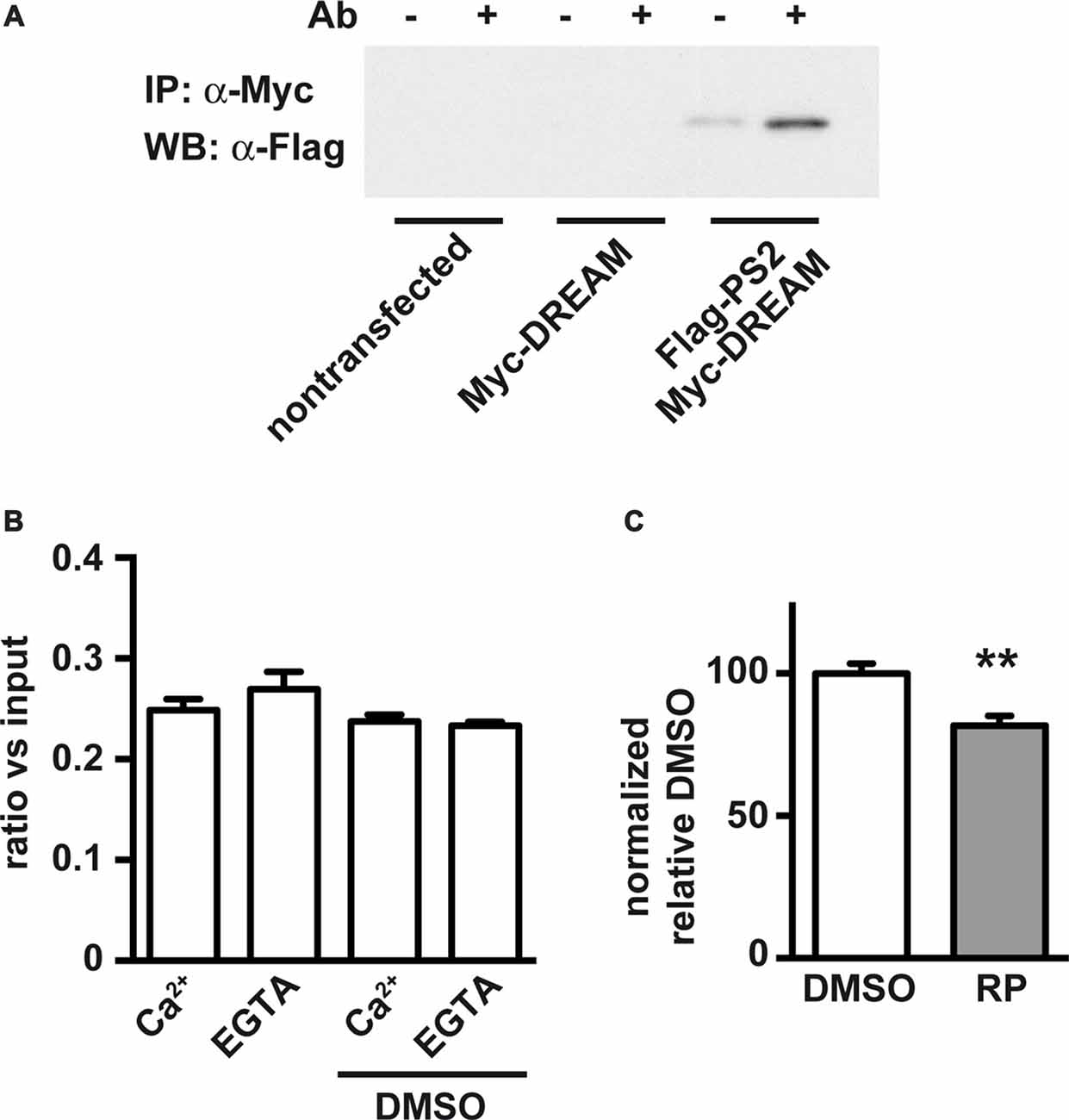
Figure 3. Coimmunoprecipitation of processed carboxy-terminal fragment of PS2 (Ct-PS2) and effect of calcium and the DREAM-binding molecule repaglinide. HEK293T cells were cotransfected with expression vectors for Myc-DREAM and Flag-Ct-PS2. (A) Immunoprecipitation with (+) or without (−) anti-Myc and immunoblot with anti-Flag antibody. (B) Immunoprecipitation in the presence or absence of calcium or DMSO, quantified after immunoblot with anti-Flag antibody. The experiment was repeated twice. No differences were observed among groups. (C) Immunoprecipitation in the presence of vehicle (DMSO) or repaglinide (RP), quantified after immunoblot with anti-Flag antibody. Unpaired two-tailed t-test (n = 10), **P < 0.01.
Repaglinide Blocks PS2 Endoproteolysis in N2a Cells
Finally, we analyzed whether partial block of the DREAM-PS2 interaction by repaglinide was translated in a noticeable effect on PS2 processing in an in vivo cell model. For this, we used N2a neuroblastoma cells, a cell model widely used to analyze PS2 endoproteolysis (Thinakaran et al., 1996). Exposure to repaglinide produced a concentration-dependent decrease in the levels of the Ct-PS2 fragment in N2a neuroblastoma cells with an estimated IC50 value of 0.76 μM (Figure 4).
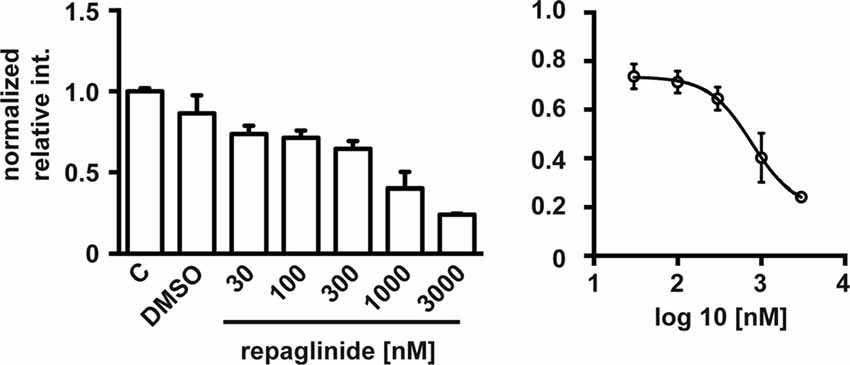
Figure 4. Repaglinide reduces endoproteolysis of endogenous PS2 in N2a cells. Cultures were untreated (C), treated with vehicle (DMSO) or with increasing concentrations of repaglinide. Whole cell extracts were analyzed by immunoblot with anti-PS2 antibody and band intensities quantified (left). The IC50 value (0.76 μM) was estimated after non-linear regression analysis (log(antagonist) vs. response (four parameters)) of the resulting dose-response curve (right; n = 2–5).
Discussion
In previous work, we have shown that reduction in DREAM protein levels or blockade of DREAM activity, using repaglinide, activates ATF6 processing which results in a neuroprotective effect in murine models of Huntington’s disease, delaying the onset and the progression of motor and cognitive decline in these mice (Naranjo et al., 2016; López-Hurtado et al., 2018). This finding opened a new avenue toward the search for effective HD treatments. Whether small-molecules binding to DREAM could have an effect on modifying the onset and/or the progression of other neurodegenerative pathologies is presently uncharacterized. DREAM has been associated with the progression of AD through its interaction with PSs (Buxbaum et al., 1998; Lilliehook et al., 2003). Therefore, we analyzed whether repaglinide has an effect on this interaction. Here, we show that blockade of the DREAM-PS2 interaction with repaglinide is directly translated in a decrease in PS2 endoproteolysis and in reduced levels of the Ct-PS2 fragment in N2a neuroblastoma cells. Whether this effect also occurs in vivo in mouse brain and the potential consequences for the onset and progression of AD remains to be investigated.
Repaglinide was developed as a potent insulinotropic agent for treatment of type-2 diabetes (Malaisse, 1995). Repaglinide binding to neuronal calcium sensors (NCS) was first reported in bovine brain and retinal extracts, which showed Ca2+-dependent binding respectively to neurocalcin and VILIP-1, or to recoverin (Okada et al., 2003). Repaglinide also binds to members of the DREAM/KChIP subfamily (Naranjo et al., 2016) which indicates that repaglinide binding is a characteristic of all proteins of the NCS superfamily but not to other Ca2+-binding proteins, including calmodulin or proteins of the S-100 superfamily (Okada et al., 2003). Nonetheless, using tagged proteins in the coimmunoprecipitation experiments here we show that repaglinide specifically blocks the DREAM-PS2 interaction. Furthermore, the interaction of PS2 with other NCS except DREAM has not been reported.
After binding, repaglinide interferes with the biological activity of the Ca2+ sensor, e.g., blockage of recoverin-mediated inhibition of rhodopsin kinase activity (Okada et al., 2003) DREAM-induced suppression of ATF6 processing (Naranjo et al., 2016) or, in this case, DREAM-induced activation of PS2 endoproteolysis. Work in progress, using single site mutations in DREAM aims to identify the binding pocket for repaglinide in the DREAM protein and whether these point mutations affect the interaction with PS2 and/or the induction of PS2 proteolysis.
The mechanisms that control PS endoproteolysis and finely keep a stoichiometry approximately 1:1 between N- and C-terminal fragments are poorly understood. To further complicate the picture, posttranslational mechanisms that potentially participate in these processes might be different for PS1 and PS2. Phosphorylation of PS1, but not of PS2, is known to occur at multiple sites within the cytosolic loop involving multiple kinases (Duggan and McCarthy, 2016). Early in vitro experiments described the interaction between DREAM and the C-terminal fragments of PS1 and PS2 as well as the increase in Ct-PS2 levels upon DREAM overexpression (Buxbaum et al., 1998). Our results confirm the DREAM-PS2 interaction in vivo and show the effect of DREAM inhibition on Ct-PS2 levels in N2a neuroblastoma cells. The functional consequences of the DREAM-PS1 interaction in terms of PS1 endoproteolysis and, the potential effect of DREAM interacting molecules in PS1 processing remains to be analyzed.
Overexpression of PS2 or DREAM has been associated with cell death (Janicki and Monteiro, 1997; Jo et al., 2001) and this effect is further enhanced by coexpression of DREAM and PS2 but not by a truncated form of PS2 (PS2/411stop) that lacks the C-terminal part (Jo et al., 2001). A parallel increase in Aβ1–42 levels after coexpression of DREAM with PS2, but not with PS2/411stop, indicated the involvement of γ-secretase activation in the cell death process, however, γ-secretase independent mechanisms can not be excluded. Interestingly, interaction between the calcium binding protein calmyrin and the cytosolic loop of PS2, and with lower affinity for the same domain in PS1, has been reported (Stabler et al., 1999). Like in the case of DREAM, coexpression of calmyrin and PS2 in HeLa cells modified the subcellular distribution of these proteins and increased cell death (Stabler et al., 1999). It was not analyzed, however, whether calmyrin modifies PS2 endoproteolysis, neither whether coexpression with PS1 also increases apoptosis (Stabler et al., 1999). The calmyrin-PS2 interaction requires and is regulated by Ca2+, suggesting that changes in cellular calcium homeostasis might control the functional output of this interaction (Stabler et al., 1999; Zhu et al., 2004). In contrast, the DREAM-PS2 interaction has been shown to be Ca2+-independent (Buxbaum et al., 1998; Fedrizzi et al., 2008). Our results support this view, however, titration curves for the binding of Apo-DREAM or Ca2+DREAM to peptides derived from the cytosolic loop of PS1 indicated that the interaction is stronger in the presence of Ca2+ (Pham and Miksovska, 2016). The different experimental conditions may account for this discrepancy.
With respect to the mechanism by which DREAM regulates PS2 endoproteolysis nothing has been reported. One possibility, among others, is that DREAM could compete with ubiquilin proteins for binding to the cytosolic loop region and/or to the C-terminal region of PS2, increasing the endoproteolysis of PS2 and/or the half-life of the Ct-PS2 fragment. It has been shown that ubiquilin-1 and -2 proteins bind to PSs and regulate PS stability by two different mechanisms (Mah et al., 2000). One, it has been shown that ubiquilin-1 and -2 regulate PS endoproteolysis and overexpression of ubiquilin proteins increases the amount of full length PS2 holoprotein and reduces the formation of Nt- and Ct-PS2 fragments (Mah et al., 2000; Massey et al., 2005). Though, a more recent study from the same group suggests that ubiquilin rather increases PSs biosynthesis (Ford and Monteiro, 2007). Two, binding of ubiquilin to the PS2 was initially associated with the ubiquitin-mediated degradation of PS2 through the proteosome pathway (Massey et al., 2004, 2005), however, binding of ubiquilin to Ct-PS2 does not require ubiquitination of critical lysine residues in this domain (Ford and Monteiro, 2007). Whether DREAM increases Ct-PS2 levels in vivo by competing the ubiquilin/PS2 interaction will need future experimental analysis.
Taken together, the effect of repaglinide blocking the DREAM-PS2 interaction and reducing PS2 endoproteolysis suggests that the interaction between DREAM and PS2 may represent a new target for modulation of PS2 processing, which could have therapeutic potential in AD treatment. Future studies, however, should analyze whether the reduction in PS2 endoproteolysis by DREAM binding molecules efficiently translates in a decrease in γ-secretase activity, in a reduction in β-amyloid accumulation and amyloid plaque formation and, finally, in an improvement in cognition.
Data Availability
All datasets generated for this study are included in the manuscript.
Author Contributions
RN, PG, AL-H and XD performed the experiments and analyzed the data. BM and JRN conceived the study, analyzed the data and wrote the article.
Funding
This work was funded by the Spanish Ministry of Economy, Industry and Competitivity (AEI-FEDER, EU grants): SAF2014-53412-R and SAF2017-89554-R (to JRN), the Instituto de Salud Carlos III biomedical research in neurodegenerative diseases (CIBERNED) program (to JRN) and the Madrid regional government/Neurodegmodels (to JRN).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Alves da Costa, C., Mattson, M. P., Ancolio, K., and Checler, F. (2003). The C-terminal fragment of presenilin 2 triggers p53-mediated staurosporine-induced apoptosis, a function independent of the presenilinase-derived N-terminal counterpart. J. Biol. Chem. 278, 12064–12069. doi: 10.1074/jbc.M212379200
Alves da Costa, C., Paitel, E., Mattson, M. P., Amson, R., Telerman, A., Ancolio, K., et al. (2002). Wild-type and mutated presenilins 2 trigger p53-dependent apoptosis and down-regulate presenilin 1 expression in HEK293 human cells and in murine neurons. Proc. Natl. Acad. Sci. U S A 99, 4043–4048. doi: 10.1073/pnas.062059899
Alves da Costa, C., Sunyach, C., Pardossi-Piquard, R., Sévalle, J., Vincent, B., Boyer, N., et al. (2006). Presenilin-dependent γ-secretase-mediated control of p53-associated cell death in Alzheimer’s disease. J. Neurosci. 26, 6377–6385. doi: 10.1523/JNEUROSCI.0651-06.2006
An, W. F., Bowlby, M. R., Betty, M., Cao, J., Ling, H.-P., Mendoza, G., et al. (2000). Modulation of A-type potassium channels by a family of calcium sensors. Nature 403, 553–556. doi: 10.1038/35000592
Bowlby, M. R., Chanda, P., Edris, W., Hinson, J., Jow, F., Katz, A. H., et al. (2005). Identification and characterization of small molecule modulators of KChIP/Kv4 function. Bioorg. Med. Chem. 13, 6112–6119. doi: 10.1016/j.bmc.2005.06.042
Burgoyne, R. D., and Haynes, L. P. (2012). Understanding the physiological roles of the neuronal calcium sensor proteins. Mol. Brain 5:2. doi: 10.1186/1756-6606-5-2
Buxbaum, J. D., Choi, E.-K., Luo, Y., Lilliehook, C., Crowley, A. C., Merriam, D. E., et al. (1998). Calsenilin: a calcium-binding protein that interacts with the presenilins and regulates the levels of a presenilin fragment. Nat. Med. 4, 1177–1181. doi: 10.1038/2673
Campbell, W. A., Iskandar, M. K., Reed, M. L., and Xia, W. (2002). Endoproteolysis of presenilin in vitro: inhibition by γ-secretase inhibitors. Biochemistry 41, 3372–3379. doi: 10.1021/bi015810h
Capell, A., Grunberg, J., Pesold, B., Diehlmann, A., Citron, M., Nixon, R., et al. (1998). The proteolytic fragments of the Alzheimer’s disease-associated presenilin-1 form heterodimers and occur as a 100–150-kDa molecular mass complex. J. Biol. Chem. 273, 3205–3211. doi: 10.1074/jbc.273.6.3205
Carrión, A. M., Link, W. A., Ledo, F., Mellström, B., and Naranjo, J. R. (1999). DREAM is a Ca2+-regulated transcriptional repressor. Nature 398, 80–84. doi: 10.1038/18044
Cheng, H. Y., Pitcher, G. M., Laviolette, S. R., Whishaw, I. Q., Tong, K. I., Kockeritz, L. K., et al. (2002). DREAM is a critical transcriptional repressor for pain modulation. Cell 108, 31–43. doi: 10.1016/s0092-8674(01)00629-8
Cheung, K. H., Shineman, D., Müller, M., Cárdenas, C., Mei, L., Yang, J., et al. (2008). Mechanism of Ca2+ disruption in Alzheimer’s disease by presenilin regulation of InsP3 receptor channel gating. Neuron 58, 871–883. doi: 10.1016/j.neuron.2008.04.015
Dierssen, M., Fedrizzi, L., Gomez-Villafuertes, R., de Lagran, M. M., Gutierrez-Adan, A., Sahun, I., et al. (2012). Reduced Mid1 expression and delayed neuromotor development in daDREAM transgenic mice. Front. Mol. Neurosci. 5:58. doi: 10.3389/fnmol.2012.00058
Duggan, S. P., and McCarthy, J. V. (2016). Beyond γ-secretase activity: the multifunctional nature of presenilins in cell signalling pathways. Cell. Signal. 28, 1–11. doi: 10.1016/j.cellsig.2015.10.006
Esler, W. P., Kimberly, W. T., Ostaszewski, B. L., Diehl, T. S., Moore, C. L., Tsai, J.-Y., et al. (2000). Transition-state analogue inhibitors of γ-secretase bind directly to presenilin-1. Nat. Cell Biol. 2, 428–434. doi: 10.1038/35017062
Fedrizzi, L., Lim, D., Carafoli, E., and Brini, M. (2008). Interplay of the Ca2+-binding protein DREAM with presenilin in neuronal Ca2+ signaling. J. Biol. Chem. 283, 27494–27503. doi: 10.1074/jbc.M804152200
Ford, D. L., and Monteiro, M. J. (2007). Studies of the role of ubiquitination in the interaction of ubiquilin with the loop and carboxyl terminal regions of presenilin-2. Biochemistry 46, 8827–8837. doi: 10.1021/bi700604q
Fukumori, A., Fluhrer, R., Steiner, H., and Haass, C. (2010). Three-amino acid spacing of presenilin endoproteolysis suggests a general stepwise cleavage of γ-secretase-mediated intramembrane proteolysis. J. Neurosci. 30, 7853–7862. doi: 10.1523/JNEUROSCI.1443-10.2010
Green, K. N., Demuro, A., Akbari, Y., Hitt, B. D., Smith, I. F., Parker, I., et al. (2008). SERCA pump activity is physiologically regulated by presenilin and regulates amyloid β production. J. Gen. Physiol. 132:i1. doi: 10.1085/jgp1322oia1
Haapasalo, A., and Kovacs, D. M. (2011). The many substrates of presenilin/γ-secretase. J. Alzheimers Dis. 25, 3–28. doi: 10.3233/jad-2011-101065
Hartmann, H., Busciglio, J., Baumann, K. H., Staufenbiel, M., and Yankner, B. A. (1997). Developmental regulation of presenilin-1 processing in the brain suggests a role in neuronal differentiation. J. Biol. Chem. 272, 14505–14508. doi: 10.1074/jbc.272.23.14505
Hayrapetyan, V., Rybalchenko, V., Rybalchenko, N., and Koulen, P. (2008). The N-terminus of presenilin-2 increases single channel activity of brain ryanodine receptors through direct protein-protein interaction. Cell Calcium 44, 507–518. doi: 10.1016/j.ceca.2008.03.004
Holmqvist, M. H., Cao, J., Knoppers, M. H., Jurman, M. E., Distefano, P. S., Rhodes, K. J., et al. (2001). Kinetic modulation of Kv4-mediated A-current by arachidonic acid is dependent on potassium channel interacting proteins. J. Neurosci. 21, 4154–4161. doi: 10.1523/JNEUROSCI.21-12-04154.2001
Honarnejad, K., Jung, C. K., Lammich, S., Arzberger, T., Kretzschmar, H., and Herms, J. (2013). Involvement of presenilin holoprotein upregulation in calcium dyshomeostasis of Alzheimer’s disease. J. Cell. Mol. Med. 17, 293–302. doi: 10.1111/jcmm.12008
Janicki, S., and Monteiro, M. J. (1997). Increased apoptosis arising from increased expression of the Alzheimer’s disease-associated presenilin-2 mutation (N141I). J. Cell Biol. 139, 485–495. doi: 10.1083/jcb.139.2.485
Jo, D.-G., Kim, M.-J., Choi, Y. H., Kim, I.-K., Song, Y.-H., Woo, H.-N., et al. (2001). Pro-apoptotic function of calsenilin/DREAM/KChIP3. FASEB J. 15, 589–591. doi: 10.1096/fj.00-0541fje
Jurisch-Yaksi, N., Sannerud, R., and Annaert, W. (2013). A fast growing spectrum of biological functions of γ-secretase in development and disease. Biochim. Biophys. Acta 1828, 2815–2827. doi: 10.1016/j.bbamem.2013.04.016
Kopan, R., and Ilagan, M. X. (2004). γ-secretase: proteasome of the membrane? Nat. Rev. Mol. Cell Biol. 5, 499–504. doi: 10.1038/nrm1406
Kuo, L.-H., Hu, M.-K., Hsu, W.-M., Tung, Y.-T., Wang, B.-J., Tsai, W.-W., et al. (2008). Tumor necrosis factor-α-elicited stimulation of γ-secretase is mediated by c-Jun N-terminal kinase-dependent phosphorylation of presenilin and nicastrin. Mol. Biol. Cell 19, 4201–4212. doi: 10.1091/mbc.E07-09-0987
Lau, K.-F., Howlett, D. R., Kesavapany, S., Standen, C. L., Dingwall, C., McLoughlin, D. M., et al. (2002). Cyclin-dependent kinase-5/p35 phosphorylates Presenilin 1 to regulate carboxy-terminal fragment stability. Mol. Cell. Neurosci. 20, 13–20. doi: 10.1006/mcne.2002.1108
Ledo, F., Kremer, L., Mellström, B., and Naranjo, J. R. (2002). Ca2+-dependent block of CREB-CBP transcription by repressor DREAM. EMBO J. 21, 4583–4592. doi: 10.1093/emboj/cdf440
Lee, J. H., Yu, W. H., Kumar, A., Lee, S., Mohan, P. S., Peterhoff, C. M., et al. (2010). Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 141, 1146–1158. doi: 10.1016/j.cell.2010.05.008
Li, Y. M., Xu, M., Lai, M. T., Huang, Q., Castro, J. L., DiMuzio-Mower, J., et al. (2000). Photoactivated γ-secretase inhibitors directed to the active site covalently label presenilin 1. Nature 405, 689–694. doi: 10.1038/35015085
Lilliehook, C., Bozdagi, O., Yao, J., Gomez-Ramirez, M., Zaidi, N. F., Wasco, W., et al. (2003). Altered Aβ formation and long-term potentiation in a calsenilin knock-out. J. Neurosci. 23, 9097–9106. doi: 10.1523/JNEUROSCI.23-27-09097.2003
Liu, C. W., Corboy, M. J., DeMartino, G. N., and Thomas, P. J. (2003). Endoproteolytic activity of the proteasome. Science 299, 408–411. doi: 10.1126/science.1079293
López-Hurtado, A., Burgos, D. F., González, P., Dopazo, X. M., González, V., Rábano, A., et al. (2018). Inhibition of DREAM-ATF6 interaction delays onset of cognition deficit in a mouse model of Huntington’s disease. Mol. Brain 11:13. doi: 10.1186/s13041-018-0359-6
Lu, P., Bai, X. C., Ma, D., Xie, T., Yan, C., Sun, L., et al. (2014). Three-dimensional structure of human γ-secretase. Nature 512, 166–170. doi: 10.1038/nature13567
Mah, A. L., Perry, G., Smith, M. A., and Monteiro, M. J. (2000). Identification of ubiquilin, a novel presenilin interactor that increases presenilin protein accumulation. J. Cell Biol. 151, 847–862. doi: 10.1016/s0197-4580(00)83116-4
Malaisse, W. J. (1995). Stimulation of insulin release by non-sulfonylurea hypoglycemic agents: the meglitinide family. Horm. Metab. Res. 27, 263–266. doi: 10.1055/s-2007-979955
Massey, L. K., Mah, A. L., Ford, D. L., Miller, J., Liang, J., Doong, H., et al. (2004). Overexpression of ubiquilin decreases ubiquitination and degradation of presenilin proteins. J. Alzheimers Dis. 6, 79–92. doi: 10.3233/jad-2004-6109
Massey, L. K., Mah, A. L., and Monteiro, M. J. (2005). Ubiquilin regulates presenilin endoproteolysis and modulates γ-secretase components, Pen-2 and nicastrin. Biochem. J. 391, 513–525. doi: 10.1042/bj20050491
McCarthy, J. V., Twomey, C., and Wujek, P. (2009). Presenilin-dependent regulated intramembrane proteolysis and γ-secretase activity. Cell. Mol. Life Sci. 66, 1534–1555. doi: 10.1007/s00018-009-8435-9
Mellström, B., Sahún, I., Ruiz-Nuño, A., Murtra, P., Gomez-Villafuertes, R., Savignac, M., et al. (2014). DREAM controls the on/off switch of specific activity-dependent transcription pathways. Mol. Cell. Biol. 34, 877–887. doi: 10.1128/mcb.00360-13
Naranjo, J. R., Zhang, H., Villar, D., González, P., Dopazo, X. M., Morón-Oset, J., et al. (2016). Activating transcription factor 6 derepression mediates neuroprotection in Huntington disease. J. Clin. Invest. 126, 627–638. doi: 10.1172/jci82670
Nelson, O., Supnet, C., Tolia, A., Horré, K., De Strooper, B., and Bezprozvanny, I. (2011). Mutagenesis mapping of the presenilin 1 calcium leak conductance pore. J. Biol. Chem. 286, 22339–22347. doi: 10.1074/jbc.M111.243063
Okada, M., Takezawa, D., Tachibanaki, S., Kawamura, S., Tokumitsu, H., and Kobayashi, R. (2003). Neuronal calcium sensor proteins are direct targets of the insulinotropic agent repaglinide. Biochem. J. 375, 87–97. doi: 10.1042/bj20030376
Palczewska, M., Casafont, I., Ghimire, K., Rojas, A. M., Valencia, A., Lafarga, M., et al. (2011). Sumoylation regulates nuclear localization of repressor DREAM. Biochim. Biophys. Acta 1813, 1050–1058. doi: 10.1016/j.bbamcr.2010.11.001
Pham, K., and Miksovska, J. (2016). Molecular insight of DREAM and presenilin 1 C-terminal fragment interactions. FEBS Lett. 590, 1114–1122. doi: 10.1002/1873-3468.12156
Rivas, M., Aurrekoetxea, K., Mellström, B., and Naranjo, J. R. (2011). Redox signaling regulates transcriptional activity of the Ca2+-dependent repressor DREAM. Antioxid Redox Signal. 14, 1237–1243. doi: 10.1089/ars.2010.3385
Rivas, M., Mellström, B., Naranjo, J. R., and Santisteban, P. (2004). Transcriptional repressor DREAM interacts with thyroid transcription factor-1 and regulates thyroglobulin gene expression. J. Biol. Chem. 279, 33114–33122. doi: 10.1074/jbc.M403526200
Rivas, M., Mellström, B., Torres, B., Cali, G., Ferrara, A. M., Terracciano, D., et al. (2009). The DREAM protein is associated with thyroid enlargement and nodular development. Mol. Endocrinol. 23, 862–870. doi: 10.1210/me.2008-0466
Ruiz-Gomez, A., Mellström, B., Tornero, D., Morato, E., Savignac, M., Holguín, H., et al. (2007). G protein-coupled receptor kinase 2-mediated phosphorylation of downstream regulatory element antagonist modulator regulates membrane trafficking of Kv4.2 potassium channel. J. Biol. Chem. 282, 1205–1215. doi: 10.1074/jbc.M607166200
Saura, C. A., Tomita, T., Davenport, F., Harris, C. L., Iwatsubo, T., and Thinakaran, G. (1999). Evidence that intramolecular associations between presenilin domains are obligatory for endoproteolytic processing. J. Biol. Chem. 274, 13818–13823. doi: 10.1074/jbc.274.20.13818
Savignac, M., Pintado, B., Gutierrez-Adan, A., Palczewska, M., Mellström, B., and Naranjo, J. R. (2005). Transcriptional repressor DREAM regulates T-lymphocyte proliferation and cytokine gene expression. EMBO J. 24, 3555–3564. doi: 10.1038/sj.emboj.7600810
Scsucova, S., Palacios, D., Savignac, M., Mellström, B., Naranjo, J. R., and Aranda, A. (2005). The repressor DREAM acts as a transcriptional activator on Vitamin D and retinoic acid response elements. Nucleic Acids Res. 33, 2269–2279. doi: 10.1093/nar/gki503
Shen, J., Bronson, R. T., Chen, D. F., Xia, W., Selkoe, D. J., and Tonegawa, S. (1997). Skeletal and CNS defects in Presenilin-1-deficient mice. Cell 89, 629–639. doi: 10.1016/s0092-8674(00)80244-5
Sherrington, R., Rogaev, E. I., Liang, Y., Rogaeva, E. A., Levesque, G., Ikeda, M., et al. (1995). Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature 375, 754–760. doi: 10.1038/375754a0
Spasic, D., and Annaert, W. (2008). Building γ-secretase: the bits and pieces. J. Cell Sci. 121, 413–420. doi: 10.1242/jcs.015255
Stabler, S. M., Ostrowski, L. L., Janicki, S. M., and Monteiro, M. J. (1999). A myristoylated calcium-binding protein that preferentially interacts with the Alzheimer’s disease presenilin 2 protein. J. Cell Biol. 145, 1277–1292. doi: 10.1083/jcb.145.6.1277
Steiner, H., Capell, A., Pesold, B., Citron, M., Kloetzel, P. M., Selkoe, D. J., et al. (1998). Expression of Alzheimer’s disease-associated presenilin-1 is controlled by proteolytic degradation and complex formation. J. Biol. Chem. 273, 32322–32331. doi: 10.1074/jbc.273.48.32322
Sun, L., Zhao, L., Yang, G., Yan, C., Zhou, R., Zhou, X., et al. (2015). Structural basis of human γ-secretase assembly. Proc. Natl. Acad. Sci. U S A 112, 6003–6008. doi: 10.1073/pnas.1506242112
Supnet, C., and Bezprozvanny, I. (2011). Presenilins function in ER calcium leak and Alzheimer’s disease pathogenesis. Cell Calcium 50, 303–309. doi: 10.1016/j.ceca.2011.05.013
Takasugi, N., Tomita, T., Hayashi, I., Tsuruoka, M., Niimura, M., Takahashi, Y., et al. (2003). The role of presenilin cofactors in the γ-secretase complex. Nature 422, 438–441. doi: 10.1038/nature01506
Thinakaran, G., Borchelt, D. R., Lee, M. K., Slunt, H. H., Spitzer, L., Kim, G., et al. (1996). Endoproteolysis of presenilin 1 and accumulation of processed derivatives in vivo. Neuron 17, 181–190. doi: 10.1016/s0896-6273(00)80291-3
Tu, H., Nelson, O., Bezprozvanny, A., Wang, Z., Lee, S. F., Hao, Y. H., et al. (2006). Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer’s disease-linked mutations. Cell 126, 981–993. doi: 10.1016/j.cell.2006.06.059
Uemura, K., Kuzuya, A., Shimozono, Y., Aoyagi, N., Ando, K., Shimohama, S., et al. (2007). GSK3β activity modifies the localization and function of presenilin 1. J. Biol. Chem. 282, 15823–15832. doi: 10.1074/jbc.M610708200
Walter, J., Schindzielorz, A., Grunberg, J., and Haass, C. (1999). Phosphorylation of presenilin-2 regulates its cleavage by caspases and retards progression of apoptosis. Proc. Natl. Acad. Sci. U S A 96, 1391–1396. doi: 10.1073/pnas.96.4.1391
Wang, H.-Q., Nakaya, Y., Du, Z., Yamane, T., Shirane, M., Kudo, T., et al. (2005). Interaction of presenilins with FKBP38 promotes apoptosis by reducing mitochondrial Bcl-2. Hum. Mol. Genet. 14, 1889–1902. doi: 10.1093/hmg/ddi195
Wolfe, M. S., Xia, W., Ostaszewski, B. L., Diehl, T. S., Kimberly, W. T., and Selkoe, D. J. (1999). Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and γ-secretase activity. Nature 398, 513–517. doi: 10.1038/19077
Wong, P. C., Zheng, H., Chen, H., Becher, M. W., Sirinathsinghji, D. J., Trumbauer, M. E., et al. (1997). Presenilin 1 is required for Notch 1 and DI1 expression in the paraxial mesoderm. Nature 387, 288–292. doi: 10.1038/387288a0
Wu, L. J., Mellström, B., Wang, H., Ren, M., Domingo, S., Kim, S. S., et al. (2010). DREAM (downstream regulatory element antagonist modulator) contributes to synaptic depression and contextual fear memory. Mol. Brain 3:3. doi: 10.1186/1756-6606-3-3
Xie, Z., Romano, D. M., and Tanzi, R. E. (2005). Effects of RNAi-mediated silencing of PEN-2, APH-1a, and nicastrin on wild-type vs. FAD mutant forms of presenilin 1. J. Mol. Neurosci. 25, 67–77. doi: 10.1385/jmn:25:1:067
Zaidi, N. F., Kuplast, K. G., Washicosky, K. J., Kajiwara, Y., Buxbaum, J. D., and Wasco, W. (2006). Calsenilin interacts with transcriptional co-repressor C-terminal binding protein(s). J. Neurochem. 98, 1290–1301. doi: 10.1111/j.1471-4159.2006.03972.x
Zeng, L., Hu, C., Zhang, F., Xu, D. C., Cui, M. Z., and Xu, X. (2015). Cellular FLICE-like inhibitory protein (c-FLIP) and PS1-associated protein (PSAP) mediate presenilin 1-induced γ-secretase-dependent and -independent apoptosis, respectively. J. Biol. Chem. 290, 18269–18280. doi: 10.1074/jbc.M115.640177
Zhang, Z., Hartmann, H., Do, V. M., Abramowski, D., Sturchler-Pierrat, C., Staufenbiel, M., et al. (1998). Destabilization of β-catenin by mutations in presenilin-1 potentiates neuronal apoptosis. Nature 395, 698–702. doi: 10.1038/27208
Keywords: presenilins, calcium, DREAM, neuronal calcium sensors, repaglinide
Citation: Naranjo R, González P, Lopez-Hurtado A, Dopazo XM, Mellström B and Naranjo JR (2018) Inhibition of the Neuronal Calcium Sensor DREAM Modulates Presenilin-2 Endoproteolysis. Front. Mol. Neurosci. 11:449. doi: 10.3389/fnmol.2018.00449
Received: 17 October 2018; Accepted: 21 November 2018;
Published: 03 December 2018.
Edited by:
Jaewon Ko, Daegu Gyeongbuk Institute of Science and Technology (DGIST), South KoreaReviewed by:
Homira Behbahani, Karolinska Institutet (KI), SwedenSe-Young Choi, Seoul National University, South Korea
Copyright © 2018 Naranjo, González, Lopez-Hurtado, Dopazo, Mellström and Naranjo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: José R. Naranjo, bmFyYW5qb0BjbmIuY3NpYy5lcw==