 Isabelle Néant1
Isabelle Néant1 Jacques Haiech2
Jacques Haiech2 Marie-Claude Kilhoffer2
Marie-Claude Kilhoffer2 Francisco J. Aulestia3Marc Moreau1
Francisco J. Aulestia3Marc Moreau1 Catherine Leclerc1*
Catherine Leclerc1*- 1Centre de Biologie du Développement (CBD), Centre de Biologie Intégrative (CBI), CNRS, UPS, Université de Toulouse, Toulouse, France
- 2Laboratoire d’Excellence Medalis, CNRS, LIT UMR 7200, Université de Strasbourg, Strasbourg, France
- 3Department of Basic Science and Craniofacial Biology, NYU College of Dentistry, New York, NY, United States
Glioblastomas (GBMs) are the most aggressive and lethal primary astrocytic tumors in adults, with very poor prognosis. Recurrence in GBM is attributed to glioblastoma stem-like cells (GSLCs). The behavior of the tumor, including proliferation, progression, invasion, and significant resistance to therapies, is a consequence of the self-renewing properties of the GSLCs, and their high resistance to chemotherapies have been attributed to their capacity to enter quiescence. Thus, targeting GSLCs may constitute one of the possible therapeutic challenges to significantly improve anti-cancer treatment regimens for GBM. Ca2+ signaling is an important regulator of tumorigenesis in GBM, and the transition from proliferation to quiescence involves the modification of the kinetics of Ca2+ influx through store-operated channels due to an increased capacity of the mitochondria of quiescent GSLC to capture Ca2+. Therefore, the identification of new therapeutic targets requires the analysis of the calcium-regulated elements at transcriptional levels. In this review, we focus onto the direct regulation of gene expression by KCNIP proteins (KCNIP1–4). These proteins constitute the class E of Ca2+ sensor family with four EF-hand Ca2+-binding motifs and control gene transcription directly by binding, via a Ca2+-dependent mechanism, to specific DNA sites on target genes, called downstream regulatory element (DRE). The presence of putative DRE sites on genes associated with unfavorable outcome for GBM patients suggests that KCNIP proteins may contribute to the alteration of the expression of these prognosis genes. Indeed, in GBM, KCNIP2 expression appears to be significantly linked to the overall survival of patients. In this review, we summarize the current knowledge regarding the quiescent GSLCs with respect to Ca2+ signaling and discuss how Ca2+ via KCNIP proteins may affect prognosis genes expression in GBM. This original mechanism may constitute the basis of the development of new therapeutic strategies.
Introduction
Among tumors of the central nervous system, glioblastomas (GBMs) are the most aggressive and lethal primary astrocytic tumors in adults, with very poor prognosis (Louis et al., 2016; Lapointe et al., 2018). More than 90% of the patients show recurrence after therapies combining surgical resection, radiotherapy, and temozolomide (TMZ)-based chemotherapy, and the mean survival period rarely exceeds 2 years (Stupp et al., 2005). According to the cancer stem cell model, recurrence in GBM is attributed to a small sub-population of tumor cells called glioblastoma stem-like cells (GSLCs). These GSLCs have stem-like properties and are responsible for the initiation and the growth of the tumors (Visvader and Lindeman, 2008). Indeed, the GSLCs provide all the subtypes of cells that comprise the tumor including some pseudo-endothelial cells (Ricci-Vitiani et al., 2010). GSLCs are characterized by a molecular signature which combines markers of neural and/or embryonic stem cells and of mesenchymal cells. Numerous studies support the proposal that the behavior of the tumor, including proliferation, progression, invasion, and significant resistance to therapies, is determined by the self-renewing properties of the GSLCs (Stupp et al., 2005; Bao et al., 2006; Hegi et al., 2006; Stupp and Hegi, 2007; Murat et al., 2008). More importantly, this high resistance capacity to TMZ treatment have been attributed to slow cycling or relatively quiescent GSLCs (Pistollato et al., 2010; Deleyrolle et al., 2011). Quiescent GSLCs have been identified in vivo in a mouse model of GBM (Chen et al., 2012) and in human GBM tumors (Ishii et al., 2016). Thus, targeting GSLCs and their stem cell-like properties may constitute one of the possible therapeutic challenges to significantly improve anti-cancer treatment regimens for GBM.
Ca2+ is a crucial second messenger (Carafoli and Krebs, 2016) that controls a wide variety of cell functions from cell proliferation and apoptosis to organogenesis (Berridge et al., 2000; Machaca, 2011; Moreau et al., 2016). Thus, the intracellular Ca2+ concentration ([Ca2+]i) is tightly regulated and involves Ca2+ channels, pumps, and exchangers both at the plasma membrane and at the membrane of endoplasmic reticulum, mitochondria, or Golgi apparatus (Bootman, 2012; Humeau et al., 2018). In addition, changes in [Ca2+]i do not proceed in a stereotypical manner. The Ca2+ signal can be described by its amplitude (variations of [Ca2+]i levels) and by its spatial (sources of Ca2+; organelles where changes occur) and time-dependent (duration, frequency) components (Berridge, 1992; Haiech et al., 2011; Smedler and Uhlén, 2014; Monteith et al., 2017). The remodeling of Ca2+ signaling contributes also to cancer hallmarks such as excessive proliferation, survival, or resistance to cell death (Roderick and Cook, 2008; Prevarskaya et al., 2014) and accumulating evidence suggests that Ca2+ is also an important positive regulator of tumorigenesis in GBM (Robil et al., 2015; Leclerc et al., 2016). Interestingly, screening of the Prestwick Chemical library identified bisacodyl, an organic compound used as a stimulant laxative drug, with cytotoxic effect on quiescent GSLCs (Zeniou et al., 2015). Bisacodyl inhibits Ca2+ release from inositol 1,4,5-triphosphate-dependent Ca2+ stores without affecting the store-operated Ca2+ entry (SOCE) (Dong et al., 2017). These data exemplify the fact that Ca2+ channels, pumps, and exchangers may represent potential therapeutic targets. In this review, we will summarize the current knowledge regarding the quiescent GSLCs with respect to Ca2+ signaling and describe an original mechanism by which Ca2+ can activate some genes involved in the prognosis of GBM in order to propose new strategies to explore the molecular basis of GBM development for therapeutic issues.
Transition from Proliferation to Quiescence and Ca2+ Signaling
Quiescent cells are non-proliferative cells, arrested in a specific phase of the cell cycle called G0 (Coller et al., 2006). Quiescence is not a prolonged G1 phase and in contrary to the cell-cycle arrest observed in differentiation or senescence, it is reversible. Transcriptional profiling data reveal that quiescent stem cells are characterized by a common set of genes which are either downregulated, these are genes associated with cell-cycle progression (i.e., CCNA2, CCNB1, and CCNE2), or upregulated and classified as tumor suppressors, including the cyclin-dependent kinase inhibitor p21 (CDKN1A) and the G0/G1 switch gene 2 (G0S2) (Yamada et al., 2012; Cheung and Rando, 2013). Quiescence represents a strategy for GSLCs to evade killing. It is thus vital to better characterize the quiescent GSLCs and to understand the mechanisms involved in the transition from a proliferative to a quiescence state. Quiescence is actively regulated by signals provided by the stem cell microenvironment. In GBM, quiescent cells are found close to necrotic tissues, in specific niches characterized by a hypoxic (Pistollato et al., 2010; Persano et al., 2011; Ishii et al., 2016) and acidic microenvironment (Garcia-Martin et al., 2006; Honasoge et al., 2014).
A recent study suggests that Ca2+ is an important regulator of the balance between quiescence and proliferation in hematopoietic stem cell (HSC) (Umemoto et al., 2018). In HSCs, re-entry into cell-cycle requires Ca2+ influx through Cav1 voltage-dependent Ca2+ channel and the resultant activation of mitochondria. Recent findings in our group showed that Ca2+ signaling is also required for GBM stem cells quiescence. On GSLCs lines, established from surgical resections of primary GBMs, we showed that change in Ca2+ homeostasis is an important actor of the transition from proliferation to quiescence. In order to analyze the signals underlying this switch, we modified the culture condition by lowering the extracellular pH from pH 7.5 to 6.5. GSLCs kept in such conditions for 5 days enter G0. This simple protocol allowed to reversibly maintain GSLCs in a proliferating or in quiescent state (Zeniou et al., 2015; Aulestia et al., 2018). A RNAseq analysis, focusing on the Ca2+ toolkit genes (Robil et al., 2015), established the transcriptional profiles of these proliferative and quiescent GSLCs and revealed that genes regulating plasma membrane Ca2+ channels (CACNA2D1 and ORAI2) and mitochondrial Ca2+-uptake (MCU, MICU1, MICU2, and VDAC1) are downregulated in quiescence while others are upregulated (CACNB1, CAPS, and SLC8B1). A functional analysis through a bioluminescent Ca2+ imaging approach showed that quiescence in GSLCs does not involve Cav1 channels like in HSCs, but is rather due to the modification of the kinetics of the store-operated Ca2+ entry (SOCE), mediated by plasma membrane ORAI channels associated with the ER membrane protein STIM1. The inhibition of store-operated channels (SOC) by SKF96365 triggers quiescence, further supporting the crucial role of SOC in quiescence in GSLCs. Interestingly, the use of bioluminescent Ca2+ reporter targeted to mitochondria revealed that this change in SOCE kinetics is due to an increased capacity of quiescent GSLCs’ mitochondria to capture Ca2+ and not to the modification of the SOCE mechanism itself (Aulestia et al., 2018). These data highlight the importance of mitochondria as regulator of Ca2+ homeostasis.
Over the past decade, many studies have identified changes in the expression levels of proteins involved in Ca2+ homeostasis such as Ca2+ channels, pumps, and exchangers and established that some of these proteins contribute to tumorigenesis through regulation of proliferation, migration, or apoptosis (Monteith et al., 2012; Leclerc et al., 2016). As a second messenger, Ca2+ is also an important regulator of gene expression. This occurs either indirectly, via changes in the transactivating properties of transcription factors following the activation of Ca2+-dependent kinases and/or phosphatases (Dolmetsch, 2001; West et al., 2001; Kornhauser et al., 2002; Spotts et al., 2002), or directly via EF hand Ca2+-binding proteins which belongs to a group of four proteins (KCNIP1–4) (Mellström et al., 2008). The identification of new therapeutic targets now requires not only to target the identified proteins but also to analyze the molecular mechanisms responsible for the changes in gene expression observed in cancer cells. In this review, we choose to focus on the direct mode of action of Ca2+ on transcription with the implication of KCNIPs in GBM.
The Family of Neuronal Ca2+ Sensors: KCNIPs
Potassium channel-interacting proteins (KCNIPs), which constitute the class E of Ca2+ sensor family, are globular proteins of 217–270 amino acids in size, with variable N- and C-termini and a conserved core domain containing four EF-hand Ca2+-binding motifs (EF-1, EF-2, EF-3, and EF-4). Among the four EF hands, EF-1 is not able to bind Ca2+ (Buxbaum et al., 1998; Carrión et al., 1999; An et al., 2000). Drosophila melanogaster has a single Kcnip, whereas mammals have four KCNIPs (KCNIP1–4) and several alternatively spliced variants (Burgoyne, 2007). In mammals, the four KCNIPs are predominantly expressed in adult brain, with specific or overlapping patterns according to the tissues (Rhodes, 2004; Xiong et al., 2004; Pruunsild and Timmusk, 2005). KCNIP3, also called calsenilin, KChIP3, and DREAM [i.e., Downstream Regulatory Element (DRE) Antagonist Modulator] is also found in the thyroid gland (Dandrea et al., 2005; Rivas et al., 2009) and in the hematopoietic progenitor cells (Sanz, 2001). KCNIP2 and KCNIP3 are found in T and B lymphocytes (Savignac et al., 2005, 2010). During mouse development, Kcnip3 transcript first occurs at E10.5 (Spreafico et al., 2001) and Kcnip1, 2, and 4 are not detected before E13 (Pruunsild and Timmusk, 2005). In the fish Danio rerio, the embryonic expressions of kcnip1b and kcnip3 are not detectable before somitogenesis (Stetsyuk et al., 2007) and in the amphibian Xenopus laevis among the four kcnips, only kcnip1 is expressed at all developmental stages, from fertilized egg to the tadpole stages. By contrast, the transcripts for kcnip2, kcnip3, and kcnip4 are expressed at later stages, after the specification of neural territories (Néant et al., 2015).
KCNIP proteins are known to control gene transcription directly by binding, via a Ca2+-dependent mechanism, to specific DNA sites, called DRE, of target genes. DRE sites are localized in the proximal 5′ sequence of the gene, downstream of the TATA box and upstream of the start codon, with the sequence GTCA forming the central core of the DRE site (Carrión et al., 1999; Ledo et al., 2000). This mechanism has been particularly well described for KCNIP3 (DREAM). When the intracellular Ca2+ level is low, KCNIP3 is bound as tetramer to the DRE sites, acting mainly as a transcriptional repressor. An increase in intracellular Ca2+ leads to dissociation of the KCNIP3 tetramer from its DRE site, thus allowing transcription (Carrión et al., 1999). KCNIP3 can affect transcription by acting either as a transcriptional repressor (Carrión et al., 1999; Link, 2004) or activator (Scsucova, 2005; Cebolla et al., 2008). In a more recent study, KCNIP3 has been shown to be required for human embryonic stem cells (hESCs) survival and to maintain hESCs pluripotency (Fontán-Lozano et al., 2016). KCNIP3 was initially the only Ca2+ sensor known to bind to DRE sites and to directly regulate transcription in a Ca2+-dependent manner (Mellström and Naranjo, 2001). However, all the four KCNIPs exhibit DRE-binding site affinity as homo or heterotetramers and act as Ca2+-dependent transcriptional regulators (Osawa et al., 2001; Craig et al., 2002; Link, 2004), allowing functional redundancy. KCNIP2 and KCNIP3 interactions are indirectly evidenced by two-hybrid and immunoprecipitation experiments (Savignac et al., 2005) and by the fact that KCNIP3 and KCNIP2 are both able to physically interact with EF-hand mutated KCNIP3 and that such associations still inhibit DRE-dependent gene expression (Gomez-Villafuertes, 2005; Savignac et al., 2005). In vivo studies also argue for the existence of compensatory mechanisms and the formation of functional KCNIP heterotetramers. Particularly, while in cortico-hippocampal neurons from Kcnip3 knockdown mice, the expression levels of KCNIP3 target genes such as Npas4 and c-fos are not significantly modified, the additional invalidation of Kcnip2 with an antisense lentiviral vector (in this Kcnip3 KO context) results in a significant increase in the expression of these KCNIP3-dependent target genes (Mellström et al., 2014). In amphibian embryos, we demonstrated that Kcnip1 binds DRE sites in a Ca2+-dependent manner. Kcnip1 is the earliest kcnip gene expressed in X. laevis embryo. Its transcripts are timely and spatially present in the presumptive neural territories. In this in vivo model, loss of function experiments indicate that Kcnip1 is a Ca2+-dependent transcriptional repressor that controls the size of the neural plate by regulating the proliferation of neural progenitors (Néant et al., 2015).
KCNIP Proteins in Glioblastoma
To the best of our knowledge, no published work has analyzed the expression of KCNIPs in GSLCs or more generally in cancer stem cells. Using the UALCAN server (Chandrashekar et al., 2017), it was possible to compare gene expression in normal brain tissues versus GBM multiform. KCNIP1–4 are expressed in normal tissues at comparable levels. Interestingly, in GBM tissues while KCNIP1 is significantly upregulated compared to its levels in normal brain tissues, KCNIP2 and KCNIP3 are strongly downregulated (Table 1). Although KCNIP4 expression appears downregulated in GBM, the results are not statistically significant. This is probably due to large variability of KCNIP4 expression in normal brain tissues and the small number of samples analyzed. In terms of survival, only KCNIP2 expression is relevant. Among GBM patients, those with high KCNIP2 expression appear to have a significant reduction in their overall survival time (UALCAN analysis). A recent study incidentally provides additional information on KCNIP expression in BT189 GSLC (Wang et al., 2018). Wang and coworkers analyzed the function of ING5, an epigenetic regulator overexpressed in GBM, and showed that ING5 promotes GSLCs self-renewal capabilities. Using the fluorescent Ca2+ probe fluo3, these authors showed that [Ca2+]i increases in cells overexpressing ING5. This increase in the resting Ca2+ level is required to maintain GSLCs’ self-renewal. Conversely, ING5 knockdown results in a strong reduction of the resting [Ca2+]i. To decipher further how ING5 is acting, they performed the transcriptomic analysis of GSLC cells where ING5 is knockdown. Among the differentially expressed genes, several Ca2+ channels were identified as upregulated by ING5, including some subunits of L and P/Q types of voltage-gated Ca2+ channels (CACNA1F, 1S, 1D, and 1C and CACN1A, respectively) and of transient receptor potential cation channels (TRPC3, C5, C4, and M1). Of note, close examination of this list revealed that KCNIP1–4 are indeed expressed in the BT189 GSLCs, although with different expression levels, and that KCNIP2 is upregulated by ING5 in this GLSC (see Supplementary Table S1 in Wang et al., 2018).

Table 1. KCNIP genes expression in glioblastoma multiform.
These data suggest a role of KCNIP proteins in stemness maintenance and dormant status of the GSLCs. The importance of KCNIPs in GBM is further emphasized by the presence of potential DRE sites within the proximal promoter of MCU and MICU2, two genes downregulated in quiescent GSLCs (Aulestia et al., 2018) and within the proximal promoters of TRPC5, TRPC4, and TRPM1, genes from the TRP family upregulated by the epigenetic factor ING5 in BT189 GSLC (Wang et al., 2018; Table 2).
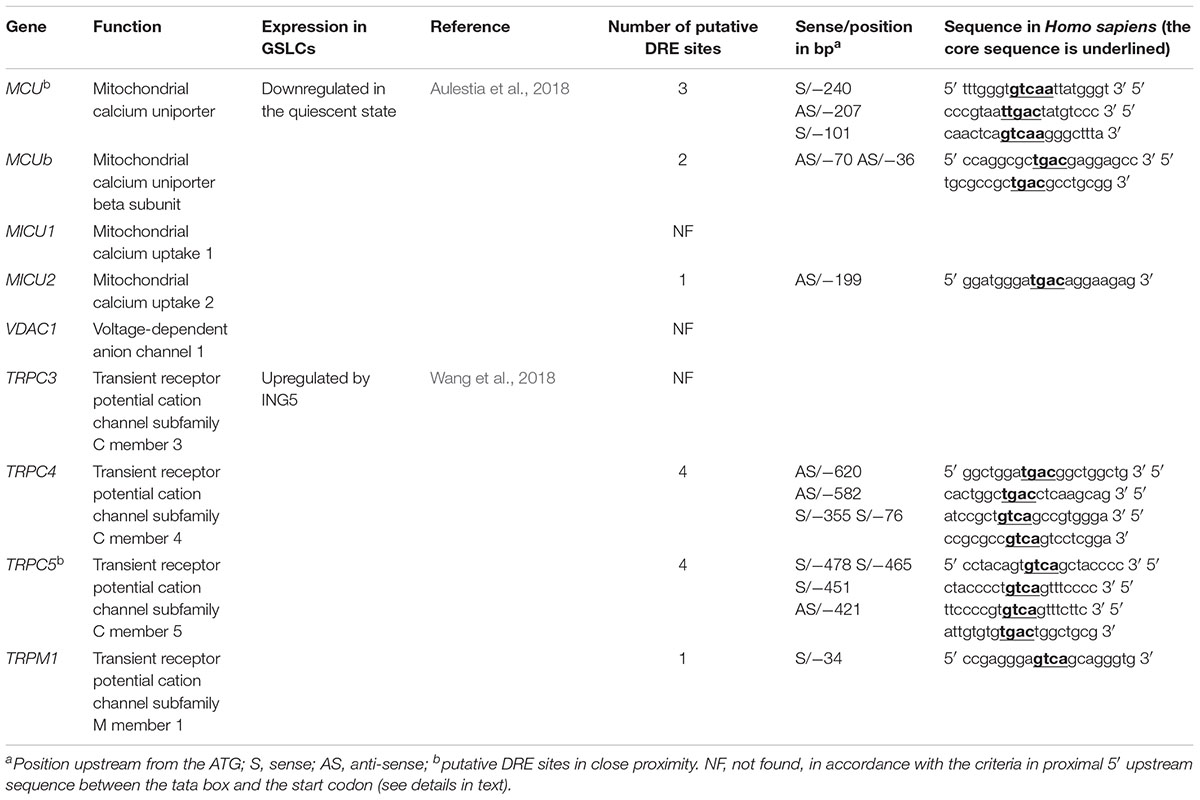
Table 2. Candidate genes with putative DRE site and expressed in GSLCs.
Regulation of GBM Prognosis Genes by KCNIP Proteins
Ion channels are now considered as important actors in cancers. Recent studies using microarray datasets of glioma samples obtained from the CGGA (Chinese Glioma Genome Atlas) and the TCGA (The Cancer Genome Atlas) identified genes belonging to the Ca2+ signaling machinery as new candidate genes that can predict GBM patients with high risk of unfavorable outcome (Wang et al., 2016; Zhang et al., 2017, 2018). These genes, listed in Table 3, are ion channels genes namely CACNA1H, a T-type Ca2+ channel (Cav3.2); KCNN4, a potassium Ca2+-activated channel (KCa3.1); KCNB1, a voltage-gated potassium channel (Kv2.1); KCNJ10, a potassium Ca2+-activated channel (Kir4.1); and classified as Ca2+-binding protein; PRKCG, Ca2+-dependent serine/threonine protein kinase Cγ (PKCγ); PRKCB, Ca2+-dependent serine/threonine protein kinase Cβ (PKCβ); and CAMK2A, the Ca2+-calmodulin-dependent protein kinase IIα. KCNIP proteins are known to control gene transcription directly by binding to DRE sites. To test whether KCNIP proteins may be involved in the regulation of the expression of these selected prognosis genes, we searched for the presence of DRE sites within their proximal promoters. The CACNA1H and PRKCB genes present both one DRE potential site in their proximal promoter and KCNB1 presents two DRE-binding sites (Table 3). More exciting are the three and four putative DRE sequences exhibited by CAMK2A and KCNN4, respectively, ideally positioned between the TATA box and the start codon, within the highly conserved sequence of proximal promoter in primates (Figure 1). The CAMK2A proximal promoter is also particularly conserved in mouse compared to human (87%), their DRE sites respect orientation and repartition, even for tandem organization. This promising observation has to be tested for KCNIP binding efficiency. Recent evidence argues for the existence of functional DRE sites within the CAMK2A proximal promoter. KCNIP3 mutants with two amino acids substitution in the EF-hands two, three, and four are unable to respond to Ca2+ and function as a constitutively dominant active (daDREAM) transcriptional repressor (Savignac et al., 2005). In transgenic mice with neuronal expression of this daDREAM, the CAMK2A mRNA level is reduced by 1.7-fold compared to wild type (Benedet et al., 2017). Mouse promoter for KCNN4 is conserved (79%), but in a lesser extend concerning DRE sequences. These sequence alignments for proximal promoters let guess a putative regulatory role of KNCIPs in the expression of some prognosis genes in GBM. Of note, not all of these prognosis genes exhibit DRE-like sites, as no DRE putative sequence was detected for KCNJ10 or PRKCG (Table 3), suggesting that KCNIPs are not the only transcriptional regulators directly implicated in the regulation of these prognosis genes, but the hypothesis of their contribution remains attractive. It is noteworthy that the previous results were obtained using transcriptomic data issued from DNA chips. When using the portal UALCAN (Chandrashekar et al., 2017) interfaced with the TCGA data base of transcriptomic cancer profiles obtained by RNA-seq techniques, only CACNA1H and KCNN4 expression levels are correlated with significant differences in survival curves. It is noticeable that these two genes present one and three DRE sites, respectively. Anyhow, the presence of these putative DRE sites on prognosis genes, suggests that remodeling of Ca2+ homeostasis in GBM stem cells may contribute to the alteration of the expression of these prognosis genes. These preliminary observations urge for a more complete analysis taking into account the high level of false negatives when using the transcriptomic signatures built from DNA chip data.
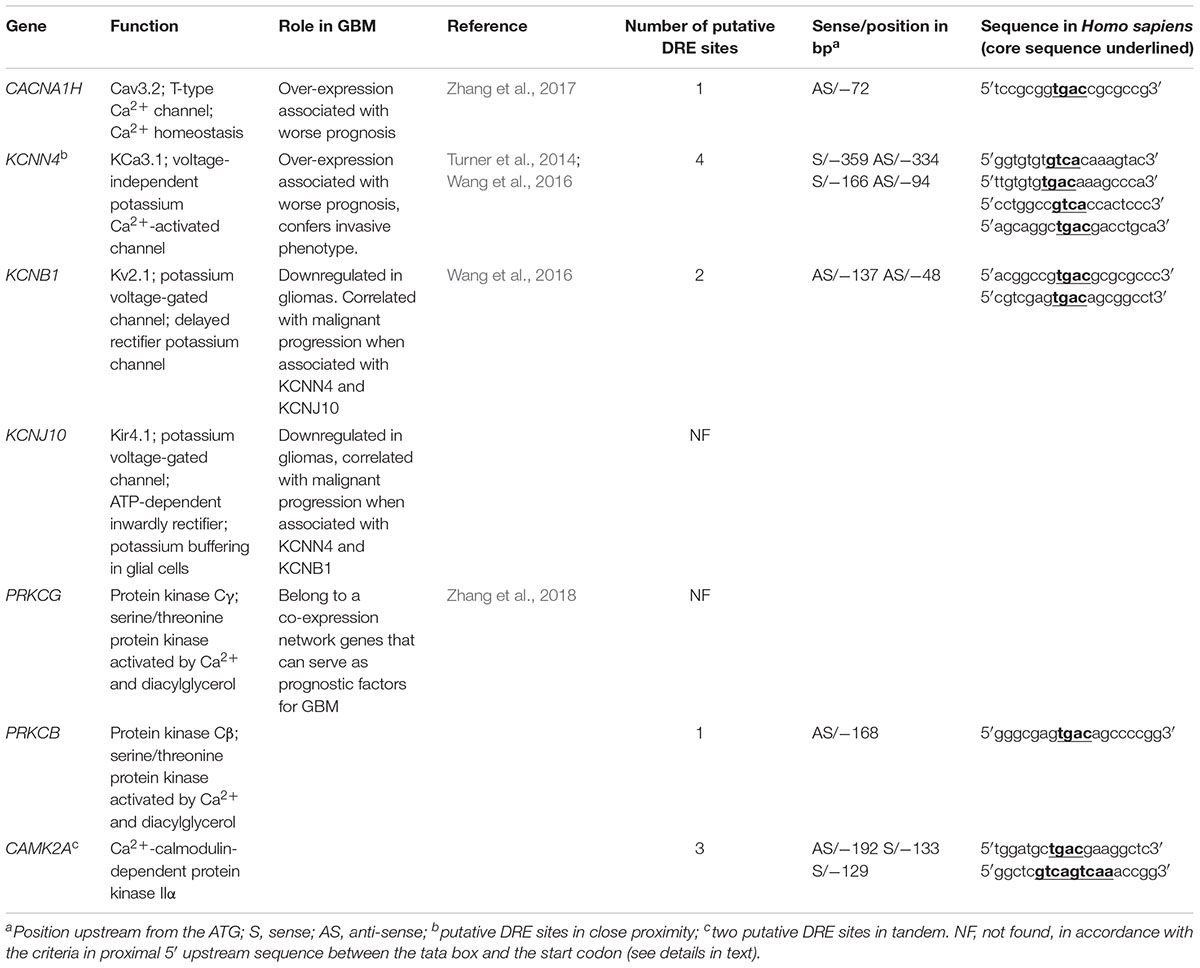
Table 3. Candidate genes associated with unfavorable prognosis in GBM.
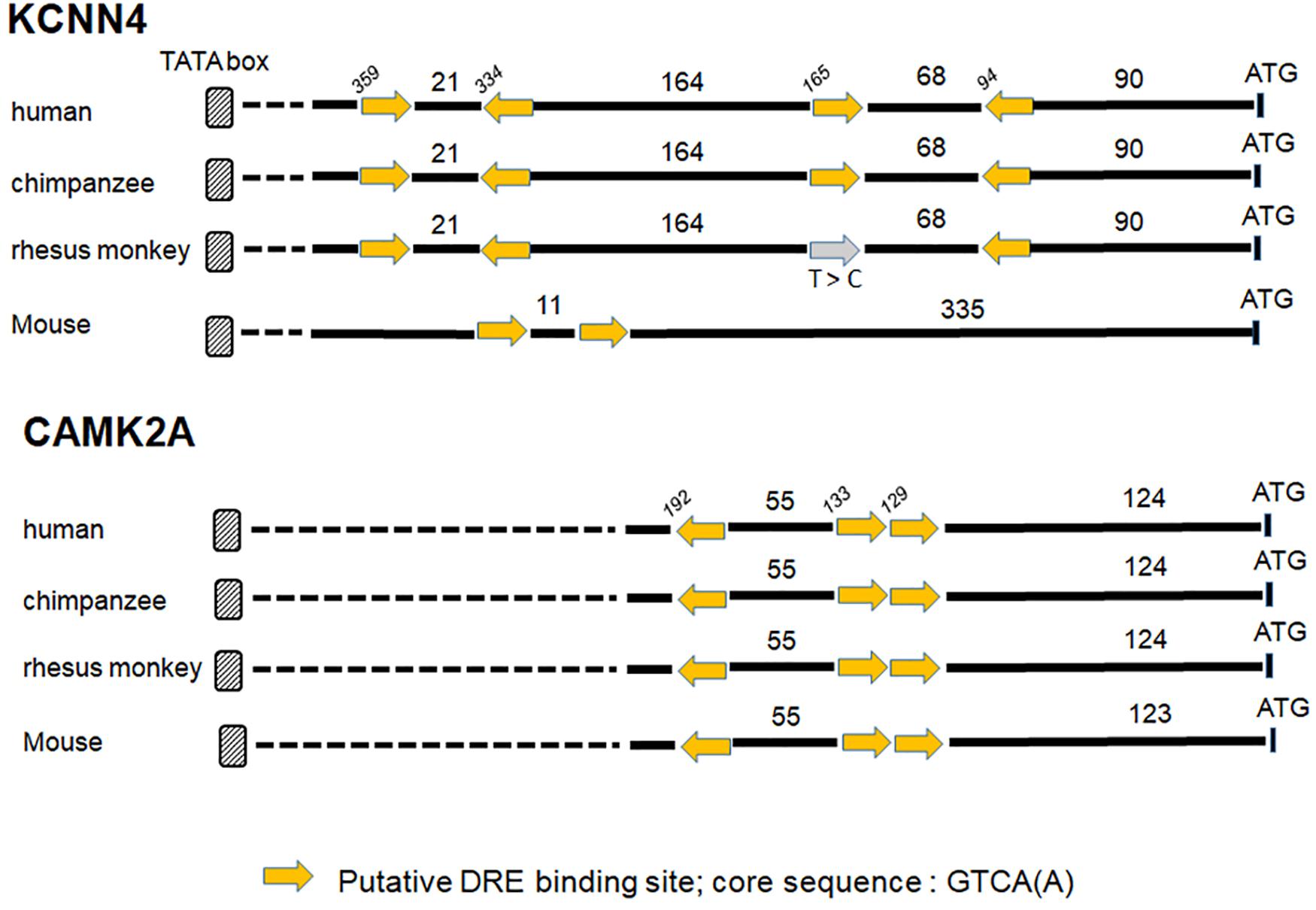
Figure 1. Putative DRE sites for two prognosis genes KCNN4 and CAMK2A. The proximal promoters of primates (human, chimpanzee, and rhesus monkey) and mouse are highly conserved for KCNN4 and CAMK2A genes, the putative DRE-binding sites within these proximal promoters are positioned regarding to ATG (numbers in italic), the lengths of the 5′ fragment sequences are expressed in bp, and the yellow arrow gives the sequence orientation (see Table 3 for DRE sequence details). A point mutation (T to C) in a “sense” DRE site within rhesus monkey sequence is mentioned by the gray arrow.
Perspectives/Prospect on KCNIPs in GBM
Although no specific data are available for KCNIPs’ function in GBM or even cancers, one can speculate taking into account published functions of KCNIP in other cell types. KCNIPs are in fact multifunctional EF hand Ca2+-binding proteins and according to their interaction partners and subcellular localization one can discriminate at least three main functions: (1) regulation of cellular excitability, (2) regulation of intracellular signaling, and (3) control of transcription.
Control of Cellular Excitability
The control of cellular excitability which involves the formation of a macromolecular signaling complex between KCNIP1 or 2, the A-type Kv4 potassium channel, and the T-type Ca2+ channel Cav3 (Anderson et al., 2010a,b) is unlikely to occur in GSLCs. Indeed, investigation of the electrophysiological properties of glioma cells revealed the absence of A-type potassium channels in these cells (Bordey and Sontheimer, 1998). Therefore, only the two other functions of KCNIP may be relevant to GBM physiology.
Regulation of Intracellular Ca2+ Signaling
In cardiomyocytes, KCNIP2 participates in the modulation of Ca2+ release through ryanodine receptors (RyR) by interacting with the ryanodine modulator, presenilin (Nassal et al., 2017). The presenilin/KCNIP3 complex has also been shown to modulate IP3-mediated Ca2+ release (Leissring et al., 2000). We have already shown that the unique drug able to kill quiescent GSLCs acts through a modulation of IP3 signaling (Dong et al., 2017).
Control of Transcription
As mentioned above, all KCNIPs can bind to DRE sites on DNA and directly control transcription. KCNIP3 (DREAM) can also interact with other transcription factors such as CREB and therefore affects transcription of genes that do not contain DRE sites (review in Rivas et al., 2011). Interestingly, in cardiomyocytes, it has been shown that the complex Ca2+/CAMK2 regulates nuclear translocation of KCNIP3 (Ronkainen et al., 2011). As CAMK2A has been identified as a prognosis gene in GBM (Table 3), such a mechanism is likely to occur in GBM.
In conclusion, since no experimental data exists for the moment in the literature concerning the function of KCNIP family in GBM, this opens a new field of research. In other models, KCNIPs have pleiotropic effects. Their well-known role as transcriptional repressors, and the presence of DRE sites in the promoter region of some GBM prognosis genes argue for a transcriptional function of KCNIPs in GBM. However, non-transcriptional roles have also to be considered more closely in the future.
Author Contributions
IN, JH, M-CK, FA, MM, and CL designed the experiments. IN, FA, and CL performed and analyzed the experiments. IN, JH, M-CK, MM, and CL wrote the manuscript. JH, MM, and CL analyzed the data, provided financial support, and the final approval of manuscript. All authors reviewed the manuscript.
Funding
This work was supported by the Centre National de la Recherche Scientifique (CNRS), Université de Strasbourg, Université Toulouse 3, by a joint grant from the Agence Nationale de la Recherche (ANR) given between France and Hong Kong to CL, JH, and MM (CalciumGlioStem ANR-13-ISV1-0004 and A-HKUST601/13), SATT Conectus (M-CK), and has been performed within the LABEX ANR-10-LABX-0034_Medalis and received a financial support from French Government managed by “Agence Nationale de la Recherche” under “Programme d’investissement d’avenir.” FA was supported by a grant from the ANR CalciumGlioStem.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
An, W. F., Bowlby, M. R., Betty, M., Cao, J., Ling, H.-P., Mendoza, G., et al. (2000). Modulation of A-type potassium channels by a family of calcium sensors. Nature 403, 553–556. doi: 10.1038/35000592
Anderson, D., Mehaffey, W. H., Iftinca, M., Rehak, R., Engbers, J. D. T., Hameed, S., et al. (2010a). Regulation of neuronal activity by Cav3-Kv4 channel signaling complexes. Nat. Neurosci. 13, 333–337. doi: 10.1038/nn.2493
Anderson, D., Rehak, R., Hameed, S., Mehaffey, W. H., Zamponi, G. W., and Turner, R. W. (2010b). Regulation of the K V 4.2 complex by Ca V 3.1 calcium channels. Channels 4, 163–167. doi: 10.4161/chan.4.3.11955
Aulestia, F. J., Néant, I., Dong, J., Haiech, J., Kilhoffer, M.-C., Moreau, M., et al. (2018). Quiescence status of glioblastoma stem-like cells involves remodelling of Ca 2+ signalling and mitochondrial shape. Sci. Rep. 8:9731. doi: 10.1038/s41598-018-28157-8
Bao, S., Wu, Q., McLendon, R. E., Hao, Y., Shi, Q., Hjelmeland, A. B., et al. (2006). Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 444, 756–760. doi: 10.1038/nature05236
Benedet, T., Gonzalez, P., Oliveros, J. C., Dopazo, J. M., Ghimire, K., Palczewska, M., et al. (2017). Transcriptional repressor DREAM regulates trigeminal noxious perception. J. Neurochem. 141, 544–552. doi: 10.1111/jnc.13584
Berridge, M. J. (1992). Spatiotemporal aspects of calcium signalling. Jpn. J. Pharmacol. 58(Suppl. 2), 142–149.
Berridge, M. J., Lipp, P., and Bootman, M. D. (2000). The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 1, 11–21. doi: 10.1038/35036035
Bootman, M. D. (2012). Calcium Signaling. Cold Spring Harb. Perspect. Biol. 4:a011171. doi: 10.1101/cshperspect.a011171
Bordey, A., and Sontheimer, H. (1998). Electrophysiological properties of human astrocytic tumor cells In situ: enigma of spiking glial cells. J. Neurophysiol. 79, 2782–2793. doi: 10.1152/jn.1998.79.5.2782
Burgoyne, R. D. (2007). Neuronal calcium sensor proteins: generating diversity in neuronal Ca2+ signalling. Nat. Rev. Neurosci. 8, 182–193. doi: 10.1038/nrn2093
Buxbaum, J. D., Choi, E.-K., Luo, Y., Lilliehook, C., Crowley, A. C., Merriam, D. E., et al. (1998). Calsenilin: a calcium-binding protein that interacts with the presenilins and regulates the levels of a presenilin fragment. Nat. Med. 4, 1177–1181. doi: 10.1038/2673
Carafoli, E., and Krebs, J. (2016). Why calcium? How calcium became the best communicator. J. Biol. Chem. 291, 20849–20857. doi: 10.1074/jbc.R116.735894
Carrión, A. M., Link, W. A., Ledo, F., Mellström, B., and Naranjo, J. R. (1999). DREAM is a Ca2+-regulated transcriptional repressor. Nature 398, 80–84. doi: 10.1038/18044
Cebolla, B., Fernandez-Perez, A., Perea, G., Araque, A., and Vallejo, M. (2008). DREAM Mediates cAMP-dependent, Ca2+-induced stimulation of GFAP gene expression and regulates cortical astrogliogenesis. J. Neurosci. 28, 6703–6713. doi: 10.1523/JNEUROSCI.0215-08.2008
Chandrashekar, D. S., Bashel, B., Balasubramanya, S. A. H., Creighton, C. J., Ponce-Rodriguez, I., Chakravarthi, B. V. S. K., et al. (2017). UALCAN: a portal for facilitating tumor subgroup gene expression and survival analyses. Neoplasia 19, 649–658. doi: 10.1016/j.neo.2017.05.002
Chen, J., Li, Y., Yu, T.-S., McKay, R. M., Burns, D. K., Kernie, S. G., et al. (2012). A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 488, 522–526. doi: 10.1038/nature11287
Cheung, T. H., and Rando, T. A. (2013). Molecular regulation of stem cell quiescence. Nat. Rev. Mol. Cell Biol. 14, 329–340. doi: 10.1038/nrm3591
Coller, H. A., Sang, L., and Roberts, J. M. (2006). A new description of cellular quiescence. PLoS Biol. 4:e83. doi: 10.1371/journal.pbio.0040083
Craig, T. A., Benson, L. M., Venyaminov, S. Y., Klimtchuk, E. S., Bajzer, Z., Prendergast, F. G., et al. (2002). The metal-binding properties of DREAM: evidence for calcium-mediated changes in DREAM structure. J. Biol. Chem. 277, 10955–10966. doi: 10.1074/jbc.M109660200
Dandrea, B., Dipalma, T., Mascia, A., Motti, M., Viglietto, G., Nitsch, L., et al. (2005). The transcriptional repressor DREAM is involved in thyroid gene expression. Exp. Cell Res. 305, 166–178. doi: 10.1016/j.yexcr.2004.12.012
Deleyrolle, L. P., Harding, A., Cato, K., Siebzehnrubl, F. A., Rahman, M., Azari, H., et al. (2011). Evidence for label-retaining tumour-initiating cells in human glioblastoma. Brain 134, 1331–1343. doi: 10.1093/brain/awr081
Dolmetsch, R. E. (2001). Signaling to the nucleus by an L-type calcium channel-calmodulin complex through the MAP kinase pathway. Science 294, 333–339. doi: 10.1126/science.1063395
Dong, J., Aulestia, F. J., Assad Kahn, S., Zeniou, M., Dubois, L. G., El-Habr, E. A., et al. (2017). Bisacodyl and its cytotoxic activity on human glioblastoma stem-like cells. Implication of inositol 1,4,5-triphosphate receptor dependent calcium signaling. Biochim. Biophys. Acta 1864, 1018–1027. doi: 10.1016/j.bbamcr.2017.01.010
Fontán-Lozano, A., Capilla-Gonzalez, V., Aguilera, Y., Mellado, N., Carrión, A. M., Soria, B., et al. (2016). Impact of transient down-regulation of DREAM in human embryonic stem cell pluripotency. Stem Cell Res. 16, 568–578. doi: 10.1016/j.scr.2016.03.001
Garcia-Martin, M. L., Martinez, G. V., Raghunand, N., Sherry, A. D., Zhang, S., and Gillies, R. J. (2006). High resolution pHe imaging of rat glioma using pH-dependent relaxivity. Magn. Reson. Med. 55, 309–315. doi: 10.1002/mrm.20773
Gomez-Villafuertes, R. (2005). Downstream regulatory element antagonist modulator regulates Ca2+ homeostasis and viability in cerebellar neurons. J. Neurosci. 25, 10822–10830. doi: 10.1523/JNEUROSCI.3912-05.2005
Haiech, J., Audran, E., Fève, M., Ranjeva, R., and Kilhoffer, M.-C. (2011). Revisiting intracellular calcium signaling semantics. Biochimie 93, 2029–2037. doi: 10.1016/j.biochi.2011.05.003
Hegi, M. E., Murat, A., Lambiv, W. L., and Stupp, R. (2006). Brain tumors: molecular biology and targeted therapies. Ann. Oncol. 17, x191–x197. doi: 10.1093/annonc/mdl259
Honasoge, A., Shelton, K. A., and Sontheimer, H. (2014). Autocrine regulation of glioma cell proliferation via pH e -sensitive K + channels. Am. J. Physiol. Cell Physiol. 306, C493–C505. doi: 10.1152/ajpcell.00097.2013
Humeau, J., Bravo-San Pedro, J. M., Vitale, I., Nuñez, L., Villalobos, C., Kroemer, G., et al. (2018). Calcium signaling and cell cycle: progression or death. Cell Calcium 70, 3–15. doi: 10.1016/j.ceca.2017.07.006
Ishii, A., Kimura, T., Sadahiro, H., Kawano, H., Takubo, K., Suzuki, M., et al. (2016). Histological characterization of the tumorigenic “peri-necrotic niche” harboring quiescent stem-like tumor cells in glioblastoma. PLoS One 11:e0147366. doi: 10.1371/journal.pone.0147366
Kornhauser, J. M., Cowan, C. W., Shaywitz, A. J., Dolmetsch, R. E., Griffith, E. C., Hu, L. S., et al. (2002). CREB transcriptional activity in neurons is regulated by multiple, calcium-specific phosphorylation events. Neuron 34, 221–233. doi: 10.1016/S0896-6273(02)00655-4
Lapointe, S., Perry, A., and Butowski, N. A. (2018). Primary brain tumours in adults. Lancet 392, 432–446. doi: 10.1016/S0140-6736(18)30990-5
Leclerc, C., Haeich, J., Aulestia, F. J., Kilhoffer, M.-C., Miller, A. L., Néant, I., et al. (2016). Calcium signaling orchestrates glioblastoma development: facts and conjunctures. Biochim. Biophys. Acta 1863, 1447–1459. doi: 10.1016/j.bbamcr.2016.01.018
Ledo, F., Link, W. A., Carrión, A. M., Echeverria, V., Mellström, B., and Naranjo, J. R. (2000). The DREAM–DRE interaction: key nucleotides and dominant negative mutants. Biochim. Biophys. Acta 1498, 162–168. doi: 10.1016/S0167-4889(00)00092-6
Leissring, M. A., Yamasaki, T. R., Wasco, W., Buxbaum, J. D., Parker, I., and LaFerla, F. M. (2000). Calsenilin reverses presenilin-mediated enhancement of calcium signaling. Proc. Natl. Acad. Sci. U.S.A. 97, 8590–8593. doi: 10.1073/pnas.97.15.8590
Link, W. A. (2004). Day-night changes in downstream regulatory element antagonist modulator/potassium channel interacting protein activity contribute to circadian gene expression in pineal gland. J. Neurosci. 24, 5346–5355. doi: 10.1523/JNEUROSCI.1460-04.2004
Louis, D. N., Perry, A., Reifenberger, G., von Deimling, A., Figarella-Branger, D., Cavenee, W. K., et al. (2016). The 2016 world health organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 131, 803–820. doi: 10.1007/s00401-016-1545-1
Machaca, K. (2011). Ca2+ signaling, genes and the cell cycle. Cell Calcium 49, 323–330. doi: 10.1016/j.ceca.2011.05.004
Mellström, B., and Naranjo, J. R. (2001). Ca2+-dependent transcriptional repression and derepression: DREAM, a direct effector. Semin. Cell Dev. Biol. 12, 59–63. doi: 10.1006/scdb.2000.0218
Mellström, B., Sahún, I., Ruiz-Nuño, A., Murtra, P., Gomez-Villafuertes, R., Savignac, M., et al. (2014). DREAM controls the on/off switch of specific activity-dependent transcription pathways. Mol. Cell. Biol. 34, 877–887. doi: 10.1128/MCB.00360-13
Mellström, B., Savignac, M., Gomez-Villafuertes, R., and Naranjo, J. R. (2008). Ca 2+ -operated transcriptional networks: molecular mechanisms and in vivo models. Physiol. Rev. 88, 421–449. doi: 10.1152/physrev.00041.2005
Monteith, G. R., Davis, F. M., and Roberts-Thomson, S. J. (2012). Calcium channels and pumps in cancer: changes and consequences. J. Biol. Chem. 287, 31666–31673. doi: 10.1074/jbc.R112.343061
Monteith, G. R., Prevarskaya, N., and Roberts-Thomson, S. J. (2017). The calcium–cancer signalling nexus. Nat. Rev. Cancer 17, 367–380. doi: 10.1038/nrc.2017.18
Moreau, M., Néant, I., Webb, S. E., Miller, A. L., Riou, J.-F., and Leclerc, C. (2016). Ca2+ coding and decoding strategies for the specification of neural and renal precursor cells during development. Cell Calcium 59, 75–83. doi: 10.1016/j.ceca.2015.12.003
Murat, A., Migliavacca, E., Gorlia, T., Lambiv, W. L., Shay, T., Hamou, M.-F., et al. (2008). Stem cell–related “self-renewal” signature and high epidermal growth factor receptor expression associated with resistance to concomitant chemoradiotherapy in glioblastoma. J. Clin. Oncol. 26, 3015–3024. doi: 10.1200/JCO.2007.15.7164
Nassal, D. M., Wan, X., Liu, H., Laurita, K. R., and Deschênes, I. (2017). KChIP2 regulates the cardiac Ca2+ transient and myocyte contractility by targeting ryanodine receptor activity. PLoS One 12:e0175221. doi: 10.1371/journal.pone.0175221
Néant, I., Mellström, B., Gonzalez, P., Naranjo, J. R., Moreau, M., and Leclerc, C. (2015). Kcnip1 a Ca2+-dependent transcriptional repressor regulates the size of the neural plate in Xenopus. Biochim. Biophys. Acta 1853, 2077–2085. doi: 10.1016/j.bbamcr.2014.12.007
Osawa, M., Tong, K. I., Lilliehook, C., Wasco, W., Buxbaum, J. D., Cheng, H.-Y. M., et al. (2001). Calcium-regulated DNA binding and oligomerization of the neuronal calcium-sensing protein, calsenilin/DREAM/KChIP3. J. Biol. Chem. 276, 41005–41013. doi: 10.1074/jbc.M105842200
Persano, L., Rampazzo, E., Della Puppa, A., Pistollato, F., and Basso, G. (2011). The three-layer concentric model of glioblastoma: cancer stem cells, microenvironmental regulation, and therapeutic implications. ScientificWorldJournal 11, 1829–1841. doi: 10.1100/2011/736480
Pistollato, F., Abbadi, S., Rampazzo, E., Persano, L., Puppa, A. D., Frasson, C., et al. (2010). Intratumoral hypoxic gradient drives stem cells distribution and MGMT expression in glioblastoma. Stem Cells 28, 851–862. doi: 10.1002/stem.415
Prevarskaya, N., Ouadid-Ahidouch, H., Skryma, R., and Shuba, Y. (2014). Remodelling of Ca2+ transport in cancer: how it contributes to cancer hallmarks? Philos. Trans. R. Soc. Lond. B Biol. Sci. 369:20130097. doi: 10.1098/rstb.2013.0097
Pruunsild, P., and Timmusk, T. (2005). Structure, alternative splicing, and expression of the human and mouse KCNIP gene family. Genomics 86, 581–593. doi: 10.1016/j.ygeno.2005.07.001
Rhodes, K. J. (2004). KChIPs and Kv4 subunits as integral components of A-Type potassium channels in mammalian brain. J. Neurosci. 24, 7903–7915. doi: 10.1523/JNEUROSCI.0776-04.2004
Ricci-Vitiani, L., Pallini, R., Biffoni, M., Todaro, M., Invernici, G., Cenci, T., et al. (2010). Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature 468, 824–828. doi: 10.1038/nature09557
Rivas, M., Mellström, B., Torres, B., Cali, G., Ferrara, A. M., Terracciano, D., et al. (2009). The DREAM protein is associated with thyroid enlargement and nodular development. Mol. Endocrinol. 23, 862–870. doi: 10.1210/me.2008-0466
Rivas, M., Villar, D., González, P., Dopazo, X. M., Mellstrom, B., and Naranjo, J. R. (2011). Building the DREAM interactome. Sci. China Life Sci. 54, 786–792. doi: 10.1007/s11427-011-4196-4
Robil, N., Petel, F., Kilhoffer, M.-C., and Haiech, J. (2015). Glioblastoma and calcium signaling - analysis of calcium toolbox expression. Int. J. Dev. Biol. 59, 407–415. doi: 10.1387/ijdb.150200jh
Roderick, H. L., and Cook, S. J. (2008). Ca2+ signalling checkpoints in cancer: remodelling Ca2+ for cancer cell proliferation and survival. Nat. Rev. Cancer 8, 361–375. doi: 10.1038/nrc2374
Ronkainen, J. J., Hänninen, S. L., Korhonen, T., Koivumäki, J. T., Skoumal, R., Rautio, S., et al. (2011). Ca 2+ -calmodulin-dependent protein kinase II represses cardiac transcription of the L-type calcium channel α 1C -subunit gene (Cacna1c) by DREAM translocation: CaMKII represses Cacna1c by DREAM translocation. J. Physiol. 589, 2669–2686. doi: 10.1113/jphysiol.2010.201400
Sanz, C. (2001). Interleukin 3-dependent activation of DREAM is involved in transcriptional silencing of the apoptotic hrk gene in hematopoietic progenitor cells. EMBO J. 20, 2286–2292. doi: 10.1093/emboj/20.9.2286
Savignac, M., Mellstrom, B., Bebin, A.-G., Oliveros, J. C., Delpy, L., Pinaud, E., et al. (2010). Increased B cell proliferation and reduced Ig production in DREAM transgenic mice. J. Immunol. 185, 7527–7536. doi: 10.4049/jimmunol.1000152
Savignac, M., Pintado, B., Gutierrez-Adan, A., Palczewska, M., Mellström, B., and Naranjo, J. R. (2005). Transcriptional repressor DREAM regulates T-lymphocyte proliferation and cytokine gene expression. EMBO J. 24, 3555–3564. doi: 10.1038/sj.emboj.7600810
Scsucova, S. (2005). The repressor DREAM acts as a transcriptional activator on Vitamin D and retinoic acid response elements. Nucleic Acids Res. 33, 2269–2279. doi: 10.1093/nar/gki503
Smedler, E., and Uhlén, P. (2014). Frequency decoding of calcium oscillations. Biochim. Biophys. Acta 1840, 964–969. doi: 10.1016/j.bbagen.2013.11.015
Spotts, J. M., Dolmetsch, R. E., and Greenberg, M. E. (2002). Time-lapse imaging of a dynamic phosphorylation-dependent protein-protein interaction in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 99, 15142–15147. doi: 10.1073/pnas.232565699
Spreafico, F., Barski, J. J., Farina, C., and Meyer, M. (2001). Mouse DREAM/Calsenilin/KChIP3: gene structure, coding potential, and expression. Mol. Cell. Neurosci. 17, 1–16. doi: 10.1006/mcne.2000.0913
Stetsyuk, V., Peers, B., Mavropoulos, A., Verbruggen, V., Thisse, B., Thisse, C., et al. (2007). Calsenilin is required for endocrine pancreas development in zebrafish. Dev. Dyn. 236, 1517–1525. doi: 10.1002/dvdy.21149
Stupp, R., and Hegi, M. E. (2007). Targeting brain-tumor stem cells. Nat. Biotechnol. 25, 193–194. doi: 10.1038/nbt0207-193
Stupp, R., Mason, W. P., van den Bent, M. J., Weller, M., Fisher, B., Taphoorn, M. J. B., et al. (2005). Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 352, 987–996. doi: 10.1056/NEJMoa043330
Turner, K. L., Honasoge, A., Robert, S. M., McFerrin, M. M., and Sontheimer, H. (2014). A proinvasive role for the Ca 2+ -activated K + channel KCa3.1 in malignant glioma: KCa3.1 inhibition slows glioma migration. Glia 62, 971–981. doi: 10.1002/glia.22655
Umemoto, T., Hashimoto, M., Matsumura, T., Nakamura-Ishizu, A., and Suda, T. (2018). Ca 2+ –mitochondria axis drives cell division in hematopoietic stem cells. J. Exp. Med. 215, 2097–2113. doi: 10.1084/jem.20180421
Visvader, J. E., and Lindeman, G. J. (2008). Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat. Rev. Cancer 8, 755–768. doi: 10.1038/nrc2499
Wang, F., Wang, A. Y., Chesnelong, C., Yang, Y., Nabbi, A., Thalappilly, S., et al. (2018). ING5 activity in self-renewal of glioblastoma stem cells via calcium and follicle stimulating hormone pathways. Oncogene 37, 286–301. doi: 10.1038/onc.2017.324
Wang, H.-Y., Li, J.-Y., Liu, X., Yan, X.-Y., Wang, W., Wu, F., et al. (2016). A three ion channel genes-based signature predicts prognosis of primary glioblastoma patients and reveals a chemotherapy sensitive subtype. Oncotarget 7, 74895–74903. doi: 10.18632/oncotarget.12462
West, A. E., Chen, W. G., Dalva, M. B., Dolmetsch, R. E., Kornhauser, J. M., Shaywitz, A. J., et al. (2001). Calcium regulation of neuronal gene expression. Proc. Natl. Acad. Sci. U.S.A. 98, 11024–11031. doi: 10.1073/pnas.191352298
Xiong, H., Kovacs, I., and Zhang, Z. (2004). Differential distribution of KChIPs mRNAs in adult mouse brain. Mol. Brain Res. 128, 103–111. doi: 10.1016/j.molbrainres.2004.06.024
Yamada, T., Park, C. S., Burns, A., Nakada, D., and Lacorazza, H. D. (2012). The cytosolic protein G0S2 maintains quiescence in hematopoietic stem cells. PLoS One 7:e38280. doi: 10.1371/journal.pone.0038280
Zeniou, M., Fève, M., Mameri, S., Dong, J., Salomé, C., Chen, W., et al. (2015). Chemical library screening and structure-function relationship studies identify bisacodyl as a potent and selective cytotoxic agent towards quiescent human glioblastoma tumor stem-like cells. PLoS One 10:e0134793. doi: 10.1371/journal.pone.0134793
Zhang, Y., Cruickshanks, N., Yuan, F., Wang, B., Pahuski, M., Wulfkuhle, J., et al. (2017). Targetable T-type calcium channels drive glioblastoma. Cancer Res. 77, 3479–3490. doi: 10.1158/0008-5472.CAN-16-2347
Keywords: Ca2+ signaling, neuronal Ca2+ sensors, KCNIP, glioblastoma multiform, cancer stem cells (CSC), quiescence
Citation: Néant I, Haiech J, Kilhoffer M-C, Aulestia FJ, Moreau M and Leclerc C (2018) Ca2+-Dependent Transcriptional Repressors KCNIP and Regulation of Prognosis Genes in Glioblastoma. Front. Mol. Neurosci. 11:472. doi: 10.3389/fnmol.2018.00472
Received: 03 October 2018; Accepted: 04 December 2018;
Published: 18 December 2018.
Edited by:
Jose R. Naranjo, Spanish National Research Council (CSIC), SpainReviewed by:
Ana Cristina Calvo, Universidad de Zaragoza, SpainMarisa Brini, Università degli Studi di Padova, Italy
Mario Vallejo, Instituto de Investigaciones Biomédicas Alberto Sols (IIBM), Spain
Copyright © 2018 Néant, Haiech, Kilhoffer, Aulestia, Moreau and Leclerc. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Catherine Leclerc, Y2F0aGVyaW5lLmxlY2xlcmNAdW5pdi10bHNlMy5mcg==