 Christelle Lasbleiz1
Christelle Lasbleiz1 Nadine Mestre-Francés1
Nadine Mestre-Francés1 Gina Devau1
Gina Devau1 Maria-Rosario Luquin2
Maria-Rosario Luquin2 Liliane Tenenbaum3
Liliane Tenenbaum3 Eric J. Kremer4
Eric J. Kremer4 Jean-Michel Verdier1*
Jean-Michel Verdier1*- 1MMDN, University of Montpellier, EPHE, INSERM, U1198, PSL University, Montpellier, France
- 2Department of Neurology, Clinica Universidad de Navarra, Pamplona, Spain
- 3Laboratory of Molecular Neurotherapies and NeuroModulation, Clinical Neuroscience Department, Lausanne University Hospital, Lausanne, Switzerland
- 4Institut de Génétique Moléculaire de Montpellier, University of Montpellier, CNRS, Montpellier, France
Parkinson’s disease (PD) is a progressive CNS disorder that is primarily associated with impaired movement. PD develops over decades and is linked to the gradual loss of dopamine delivery to the striatum, via the loss of dopaminergic (DA) neurons in the substantia nigra pars compacta (SNpc). While the administration of L-dopa and deep brain stimulation are potent therapies, their costs, side effects and gradual loss of efficacy underlines the need to develop other approaches. Unfortunately, the lack of pertinent animal models that reproduce DA neuron loss and behavior deficits—in a timeline that mimics PD progression—has hindered the identification of alternative therapies. A complementary approach to transgenic animals is the use of nonhuman primates (NHPs) combined with the overexpression of disease-related genes using viral vectors. This approach may induce phenotypes that are not influenced by developmental compensation mechanisms, and that take into account the personality of animals. In this review article, we discuss the combination of gene transfer and NHPs to develop “genetic” models of PD that are suitable for testing therapeutic approaches.
Introduction
Parkinson’s disease (PD) is a disorder of the CNS primarily due to the degeneration of nigro-striatal dopaminergic (DA) neurons. Early symptoms are movement-related, including shaking, rigidity, slowness of movement, postural instability and difficulty with walking. Collectively, these symptoms are called “parkinsonism”. However, non-motor symptoms, such as depression and apathy, which are attributed to the degeneration of the mesolimbic mesocortical DA pathway [neurons of the ventral tegmental area (VTA) projecting to the nucleus accumbens, Lewis et al., 2003; Carriere et al., 2014; Dujardin and Lopes, 2014; Dujardin et al., 2014], frequently appear before the motor symptoms (Figure 1). In addition to fine-tuning of motor function, these pathways are also involved in reward (motivation), pleasure, compulsion and perseveration. Later, sensory, sleep, emotional problems, depression and dementia may arise in the late stages. Regarding the latter, the degeneration of non-DA neurons (e.g., serotoninergic) are thought to contribute to depression (Tan et al., 2011). These early neuropsychiatric manifestations often precede motor symptoms, which appear when approximately 70% of the substantia nigra pars compacta (SNpc) DA neurons are lost or are unable to deliver dopamine to the striatum. This implies that PD is, in part, an axonopathy (O’Keeffe and Sullivan, 2018). Finally, cognitive symptoms like dementia and hallucinations tend to appear in the late phases of the disease and are related to perturbation of the mesocortial pathway, which connects the VTA to the prefrontal cortex. Eventually, deficits in the noradrenergic, serotoninergic and acetylcholinergic systems also appear. Clearly, early diagnosis will be of utmost importance for disease prevention/reversal.
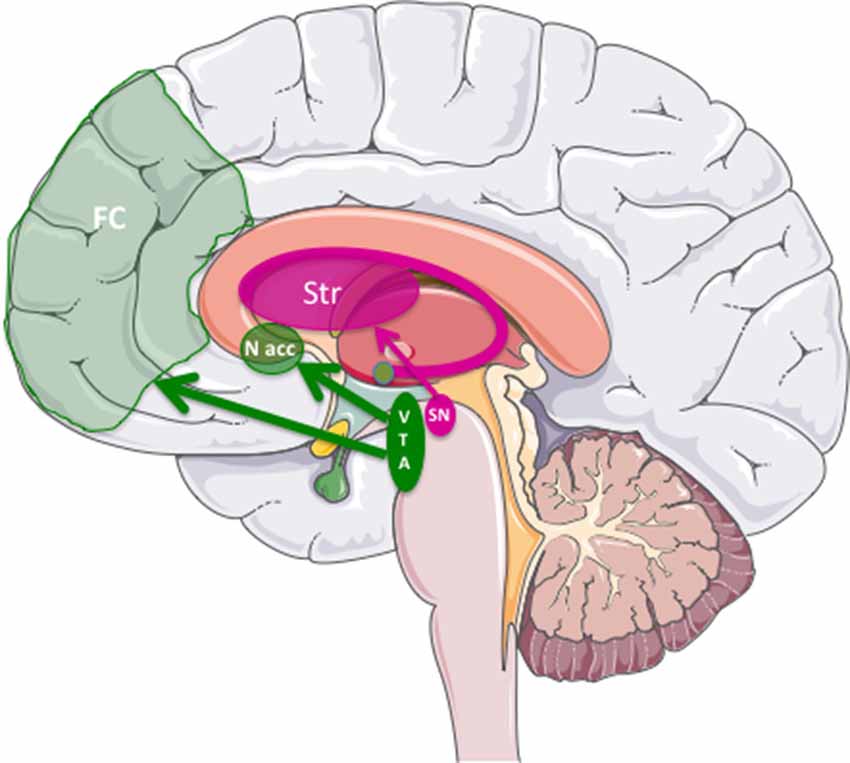
Figure 1. The three major dopaminergic (DA) pathways in the brain linked to Parkinson’s Disease (PD). The nigrostriatal pathway was DA cells from substantia nigra pars compacta (SNpc) project into the striatum (in dark pink). The mesolimbic/mesocortical pathway, which corresponds to the projection of the midbrain ventral tegmental area (VTA) to the nucleus accumbens (N. Acc) in the limbic areas, and to the frontal cortex (FC), respectively.
A handful of genes are involved in monogenic (recessive or dominant) forms of PD. Together these genes account for around 30% of familial forms and 3%–5% of sporadic cases (Klein and Westenberger, 2012). Among these PD-related genes, α-synuclein (SNCA) and leucine-rich repeat kinase 2 (LRRK2 or PARK8) or are the best characterized because over-expression and/or mutations in SNCA and LRRK2 are responsible for autosomal-dominant PD forms. A mutation in SCNA that causes an A53T change was identified in four families (Polymeropoulos et al., 1997). Since then, other mutations, duplication and triplication of this gene have been linked to PD (Deng and Yuan, 2014). LRRK2G2019S is the most common mutation in familial and sporadic PD. LRRK2 mutations are also found in sporadic cases further supporting the prominent role of this gene in PD aetiology. Finally, disease evolution of patients with LRRK2 mutations, including the accumulation of Lewy bodies, are clinically indistinguishable from those with idiopathic PD (Gasser, 2009). Familial forms of PD are slowly providing clues to underlying mechanisms of neurodegeneration.
While some drugs have markedly improved parkinsonism, their efficacy often declines as PD progresses. To date, there are no long-term disease-modifying treatments available for the 10 million people worldwide suffering from PD. Therefore, using pertinent models that allow the scientific community to develop new approaches are of utmost importance to combat PD.
Acute and Chronic Models of PD
One bottleneck associated with identifying therapeutic options for PD is the lack of a robust and pertinent animal model. While many models give potential results on a given aspect of parkinsonism, none fully recapitulate the pathognomonic lesions of PD (Dawson et al., 2010). Two broad categories of models are being used: neurotoxin-based (acute) and genetic-based (chronic) models. Neurotoxin models are the most popular. They can be produced by the use of the toxin 6-hydroxydopamine (6-OHDA), which preferentially kills DA neurons by production of free radicals (Przedborski et al., 1995), or 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine (MPTP); Schober, 2004), which interferes with the mitochondrial metabolism, also producing free radicals (Petroske et al., 2001) and strong neuroinflammation (Luchtman et al., 2009, 2012). In addition to the loss of dopamine in the nigrostriatal DA system, they also reach extrastriatal regions, such as the subcortex and brainstem (Bezard et al., 2013). While we have learned much from toxin-induced PD models, their use in the development of disease-modifying therapies is challenging. A well-recognized caveat of toxin-induced PD models is that they mainly cause the degeneration of the nigrostriatal DA pathway, which induces robust motor symptoms, but poorly recapitulate symptoms related to most, but not all, pathways (Brown et al., 2012). These models, although very useful to test motor deficits or L-dopa responsiveness (Dawson et al., 2002), remain acute models where the progressive DA cell death is absent, and poorly mimic PD progression over time (Hattori and Sato, 2007). To circumvent these drawbacks many labs have opted for the creation of transgenic mice, widely based on SNCA and LRRK2 mutants. However, transgenic SNCA mice, based on missenses mutations A30P, E46K or A53T, have led to limited parkinsonism (Deng and Yuan, 2014), especially in terms of nigrostriatal degeneration (Chesselet, 2008; Dawson et al., 2010). On the other side, the current cohort of LRRK2 transgenic mice induce mild, if any, degeneration of nigrostriatal DA neurons, Lewy body formation, or behavior effects (Ramonet et al., 2011; Blesa and Przedborski, 2014). Of note though, LRRK2 overexpression does accelerate the pathological consequences of SNCAA53T in double transgenic mice (Lin et al., 2009). The latter study also suggests that the LRRK2 protein affects the intracellular trafficking and accumulation of SNCA protein. A transgenic rat overexpressing the G2019S mutation impaired dopamine uptake but did not show any nigral DA cell loss and striatal dopamine contents in aged rats (Zhou et al., 2011). Interestingly, transgene expression in adult animals using viral vectors can induce pronounced phenotypes in rodents, presumably by circumventing developmental compensatory effects, and by producing high level of transgene expression. In particular, vector-mediated expression of native or mutant SNCA can lead to DA neuron cell death and motor symptoms. Cognitive symptoms such as spatial learning and memory deficits (Hall et al., 2013), depression (Caudal et al., 2015) and emotional memory impairment, are influenced by VTA neurons (Alvarsson et al., 2016) in rats. In conclusion, animal models that recapitulate the early and late, motor and non-motor symptoms, within a time frame suitable to evaluate PD-modifying treatments, are still needed.
Viral Vector-Mediated PD Effects in Nonhuman Primates
Nonhuman primates (NHPs) are particularly relevant in preclinical research because they share several genetic, physiological and anatomical similarities with humans. NHPs display complex cognitive functions, complex motor skills and a highly developed cerebral cortex (Verdier et al., 2015). Equally important, NHPs can be studied under controlled and humane experimental conditions. Interestingly, aged rhesus monkeys can naturally display a significant loss of tyrosine-hydroxylase and dopamine-transporter immunoreactivity correlated with motor impairments (Emborg et al., 1998). Furthermore, aged-related SNCA increase in rhesus monkeys has been observed in the nigral pathway (Chu and Kordower, 2007). These observations led to the idea that aged NHPs are at the threshold to develop a PD and, as a consequence, constitute a model of choice. Several viral vectors have been used to drive the development of PD in NHPs. Vector-mediated transgenesis is also versatile and transposable between species. Taking nothing away from the ground-breaking work performed in rodents, we believe that NHPs are needed to unravel PD induction, progression, therapeutic strategies (Emborg, 2007), and understand long-term pathophysiological, biochemical and behavioral anomalies. To develop these models, intracerebral injection of viral vectors bearing mutated SNCA or LRRK2 have been tested for modeling “genetic” PD. Monkeys overexpressing simian or human SNCA coding for a protein with the A53T change via adeno-associated virus (AAV) vectors exhibit motor impairment and neuropathological features of PD including but not limited to: head position bias, loss of TH- and VMAT2-positive innervation throughout caudate nucleus and putamen, dystrophic neurites and swollen axons, SNCA-positive inclusions (Kirik et al., 2003). AAV vectors have been successfully used for expression of human SNCAA53T in cynomolgus macaque SN, and led to a 50% loss of nigral DA neurons (Koprich et al., 2016). Lentivirus vectors expression of SNCAA53T into the SN of rhesus monkeys resulted in more neuronal pathology and chronicity in monkey brains than in mouse brains (Yang et al., 2015). Of note, NHPs are also responsive to dopamine replacement therapies, and show complications resulting from long-term use such as dyskinesia and motor fluctuations when the medication is not working well.
In contrast to the small (~16 kDa) SNCA protein, the LRRK2 protein is ~250 kDa with at least seven different functional domains (Taymans and Greggio, 2016). The G2019S change located in the kinase domain, leads to a hyperkinase activity. LRRK2 was recently shown to be involved in the endoplasmic reticulum to Golgi export. Interestingly, this function is altered in the PD-related LRRK2R1441C mutation located in the GTPase domain (Cho et al., 2014).
Helper-Dependent Canine Adenovirus for Developing New PD Models
The LRRK2 cDNA is about 8 kb and therefore a vector with an appropriately large cloning capacity is needed, and precludes its efficient expression in AAV and lentivirus vectors. A handful of attempts have been made to develop animal models expressing LRRK2G2019S via viral vectors. To date, three vector platforms have been used to deliver the LRRK2G2019S cDNA: human adenovirus type 5 (HAd5; Dusonchet et al., 2011; Tsika et al., 2014) were used in rats, herpes simplex virus (HSV) were used in mice. The HAd5-LRRK2G2019S vector was injected into the striatum of rodents and due to its preferential transduction of glia cells and poor retrograde transport, the direct effect of LRRK2G2019S on DA neurons in the nigra could not be addressed. Using a HSV vectors expressing LRRK2 or LRRK2G2019S, Lee et al. (2010) showed that the hyperkinase activity of LRRK2G2019S was responsible for the PD phenotype, and that LRRK2 kinase inhibitors provide a potential neuroprotective treatment for PD. Interestingly, they also showed that overexpression of wild type LRRK2 caused neurite shortening in vitro.
Clearly, the ability to efficiently and simultaneously deliver expression cassettes to multiple regions of the brain could be a notable plus for PD. Taking nothing away from the encouraging results when using HSV vectors (Goverdhana et al., 2005; Lee et al., 2010), we believe that we can improve PD modeling by using helper-dependent (HD) canine adenovirus (CAV-2; Junyent and Kremer, 2015; Figure 2). HD CAV-2 vectors have a unique combination of characteristics that make it ideally suited for PD modeling: CAV-2 vectors preferentially transduce neurons in rodent and NHPs, have no long-term impact on adult of newborn neuron homeostasis, have a 30 kb cloning capacity. Following injection in the rodent and NHP striatum, CAV-2 efficiently transits into afferent (axonal projections into the striatum) structures and is ≥100-fold more efficient than HAd5 vectors. This is particularly pertinent for PD modeling because efficient and stable gene transfer to DA neurons via injections into the SN is pernicious because DA neurons are particularly sensitive to stress (Albert et al., 2017). Due to the efficient retrograde transport of CAV-2 vectors in DA neurons (Soudais et al., 2001b; Schwarz et al., 2015), HD-LRRK2 vector can be delivered in the striatum, thus bypassing the potential damage incurred by SN injections.
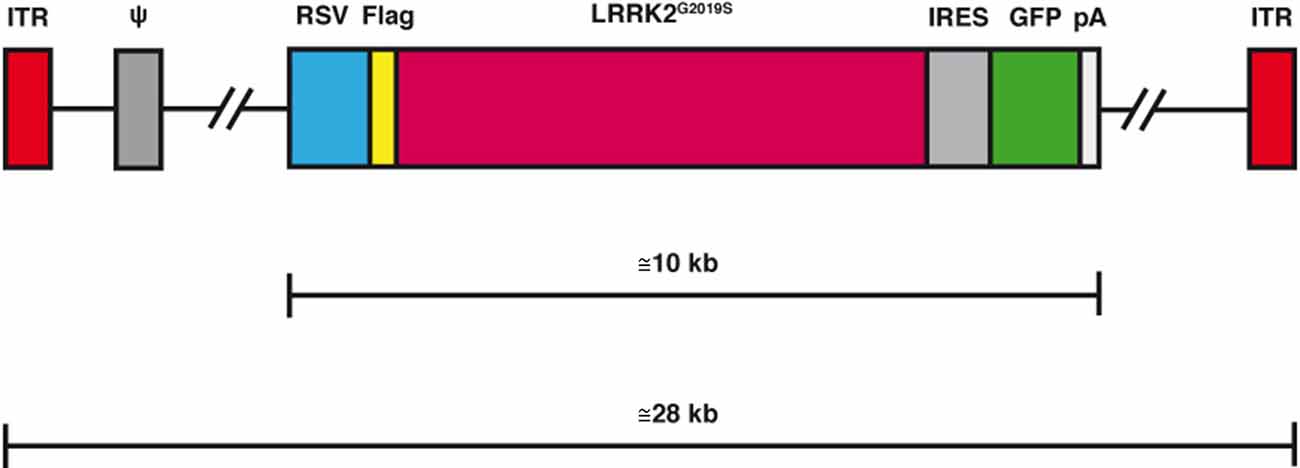
Figure 2. Schematic representation of the helper-dependent (HD) canine adenovirus-2 (CAV-2) vector expressing leucine-rich repeat kinase 2 (LRRK2)G2019S. While the HD genome is devoid of all viral coding sequences, it still retains the 200 bp inverted terminal repeat (ITR) at each end and the 150 bp packaging signal (ψ) at the left end of the genome. To create a stable capsid the genome must fill the interior of the capsid which therefore requires it to be 95%-105% of the 32 kbp wild type genome. Depending on the size of the expression cassette [here it contains the 600 bp Rous sarcoma virus early promoter (RSV), an internal ribosome entry signal (IRES), a green fluorescent protein cDNa 5GF] the LRRK2 cDNA and a 250 polyA signal (pA), the remaining sequence is made up of noncoding intronic sequence from the human genome.
HD CAV-2 vectors lead to long-term expression in the brain: transgene expression was stable for >1 year post-transduction (Soudais et al., 2001b) and cellular protein expression showed no change. These data demonstrate the low immunogenicity of HD CAV-2. Finally, scalable high titre production is also possible (Junyent and Kremer, 2015). We have invested significantly in optimizing CAV-2 vector cloning, creation and production parameters (Kremer et al., 2000; Soudais et al., 2001a; Fernandes et al., 2013, 2015a,b; Ibanes and Kremer, 2013), understanding CAV-2 uptake and trafficking (Soudais et al., 2000, 2001b; Chillon and Kremer, 2001; Martin-Touaux et al., 2002; Salinas et al., 2009), the physiological role of CAV-2’s receptor (Seiradake et al., 2006; Schoehn et al., 2008; Salinas et al., 2010; Rademacher et al., 2012; Piersanti et al., 2013; Kremer and Nemerow, 2015; Loustalot et al., 2015), and the in vivo use of the vectors [Junyent and Kremer, 2015 and del Rio et al. (in this Research Topics issue)]. HD CAV-2 vectors are therefore ideal for LRRK2 cDNA delivery.
New NHP Models of PD
LRRK2G2019S Expression in the Lemurian Primate Microcebus murinus
By complementing and extending the work performed in rodents, NHPs can help unravel PD induction, progression, therapeutic strategies (Emborg, 2007), and understand long-term pathophysiological, biochemical and behavioral anomalies. Although several type of NHPs are used to study neurodegenerative diseases (Verdier et al., 2015) the use of NHPs is time demanding, expensive, and the number of available animals for research is limited. To circumvent these issues, the Microcebus murinus (or gray mouse lemur) has the notable advantages to be small and efficiently bred in captivity. M. murinus are ideally for cerebral ageing and neurodegenerative studies because they develops complex behavioral (emotional), cognitive, and motor tests [(Joly et al., 2004, 2006, 2014; Picq, 2007; Trouche et al., 2010; Picq et al., 2012); for a review article, see also (Languille et al., 2012)], and also through transcriptomic studies (Abdel Rassoul et al., 2010) and transmissibility of neurodegenerative diseases (Mestre-Francés et al., 2012) or for gene transfer (Alba et al., 2012). In addition, its brain structure is similar to that of the humans, with a relative proportion of each region.
Recently, we generated HD CAV-2 vectors containing a LRRK2G2019S expression cassette (HD-LRRK2G2019S) that we injected unilaterally into the putamen of M. murinus. We found preferential transduction of neurons at the injection site, and in numerous areas harboring neurons that project into the striatum. The long-term expression leads to the progressive unilateral loss of DA cells in the SNpc accounting for up to 30%–40% a decreased of the DA fibers, dystrophic neurites and swollen axons, characteristic of neurodegeneration. This neurodegeneration was accompanied by dopamine loss in the striatum, and PD-like motor symptoms (bradykinesia, rigidity, and difficulty in prehension; Mestre-Francés et al., 2018).
LRRK2G2019S Expression in Macaques
The promising outcomes obtained in the M. murinus model, prompted us to determine if CAV-2 vectors were also effective gene transfer tools in the Macaca fascicularis brain, and if CAV-2–mediated expression of LRRK2G2019S in the SN neurons could induce pathological features associated with PD in a more complex NHP model. Our study demonstrated the neuronal tropism, retrograde transport, biodistribution, and efficacy of CAV-2 vectors expressing GFP in the M. fascicularis brain (Di Caudo et al., in preparation). Furthermore, we also demonstrated that CAV-2-mediated HD-LRRK2G2019S expression in the SN leads to the loss of DA cells, neurite dystrophy, axon swelling and mitochondrial abnormalities. Unfortunately, animals injected with HD-LRRK2G2019S into the striatum did not develop clear parkinsonian features, but they exhibited a significant reduction of striatal F-dopa uptake, indicating that they represent an early stage of the disease.
Together, these data demonstrate that robust PD NHP models can be generated using HD CAV-2 vectors and in turn could allow detailed evaluation of the therapeutic options for PD motor, emotional, and cognitive deficits.
Viral Vectors for Developing Disease-Modifying Treatments
While currently available treatments can temporarily relieve the symptoms, they have little influence on the neurodegenerative process.
Neurotropic factors (NFs), which mediate pro-survival effects on neurons, potentially constitute a disease-modifying option. However, the results obtained with NFs are controversial, and largely depend on the model. For instance, in an AAV-SNCA-injected rat, the delivery of AAV-GDNF (glial cell line-derived neurotrophic factor), 2–3 weeks before AAV-SNCA injection, failed to demonstrate a neuroprotective effect (Decressac et al., 2012). In this case, SNCA overexpression resulted in Ret downregulation and disruption of GDNF signaling. However, a recent study demonstrated that Ret is not downregulated in PD patients (Su et al., 2017). In other studies, the therapeutic potential of NFs was demonstrated in toxin injected (6-OHDA and MPTP) rodents and NHPs, in which NFs reduced motor symptoms (Bilang-Bleuel et al., 1997; Kirik et al., 2000; Kordower et al., 2000, 2006; Eslamboli et al., 2003; Su et al., 2009). Following these encouraging results, clinical trials were conducted using AAV2-GDNF (still ongoing) or AAV2-neurturin (NRTN; Marks et al., 2010; Warren Olanow et al., 2015). Although the AAV2-NRTN trials demonstrated acceptable tolerance, after a 1-year follow-up, no significant improvement was observed in the “Unified PD Rating Scale” (UPDRS). However, post hoc analyses suggested that a subgroup of patients had beneficial effects (Marks et al., 2010). Post-mortem analysis of four patients showed that although surviving DA neurons were still present in the SN, very few co-stained with NRTN. These observations suggested that retrograde transport was inefficient (Bartus et al., 2011, 2015). Strikingly, these results were not predicted by the pre-clinical animal models used to establish the clinical protocol (MPTP-treated NHPs and 6-OHDA-injected rats) in which the surviving nigro-striatal DA neurons still had functional projections proficient for retrograde transport and could be efficiently rescued. In a subsequent study (Kordower et al., 2013), analysis of brains from untreated PD patients at different stages, showed that the putamen innervation had almost totally disappeared at 4 years post-diagnosis, whereas numerous DA neurons cell bodies were still present in the SNpc. Because most of the patients enrolled in the AAV2-NRTN trial were more than 5 years post-diagnosis, it is likely that their putaminal DA innervation had been lost or was dysfunctional. Although unsuccessful, the AAV2-NTRN trial was informative since it allowed: (i) to identify the limitations of the toxin-induced models; (ii) to suggest that disease-interfering treatments should be administered before disappearance the DA fibers; and (iii) supported enrolment of patients at earlier disease stages in gene therapy trials.
Therefore, disease-modifying treatments will need animal models that more faithfully recapitulate the mechanisms underlying the progression of PD, and should be administered at the earliest possible stage.
Concluding Remarks
No animal model manifests all the characteristics of PD in humans, i.e., SNCA aggregation, DA reduction, progressive DA cell death, motor and non-motor symptoms. If transgenic models offer tremendous advantages over toxin-induced models, the overexpression of human disease causing mutated genes should be kept within the range of physiological levels. Viral vector-mediated local transgenesis offers the advantage to allow adjusting the transgene copy number to avoid confounding effects of a non-physiological overdosage. Clearly though it is difficult, if not impossible, to argue that NHP will not be the most informative path towards testing PD therapies. As underlined by Blesa and Przedborski (2014), models are just models, and should answer the question asked, not all the questions. Because NHPs have their own personality each animal can produce different emotional, motor, or cognitive behavior (that is why they are their own control). In addition, NHPs allow us to monitor early phases of the disease and follow-up. The combination of gene therapy and the use of NHPs should open new route to a disease-modifying treatment of PD.
Data Availability
The datasets generated for this study are available on request to the corresponding author.
Author Contributions
CL, M-RL, LT, EK, and J-MV wrote this review article. NM-F and GD edited and revised it.
Funding
This work was supported in part by France Parkinson’s (EK), BrainCAV (EC FP7 contract #292222; EK, J-MV, NM-F), BrainVector (EC FP7 contract #286071; EK), Université de Montpellier (EK, J-MV), the Region Languedoc Roussillon (EK), IGMM (EK), and LabEx EpiGenMed, an Investissements d’Avenir Program (PIA), ANR-10-LABX-12-01 (EK), MMDN (J-MV), Instituto de Salud Carlos III (PI15/01816; M-RL), INSERM and Swiss National Foundation (Contract #31003A_179527; LT), Institut National de la Santé et de la Recherche Médicale (INSERM; NM-F, J-MV).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abdel Rassoul, R., Alves, S., Pantesco, V., De Vos, J., Michel, B., Perret, M., et al. (2010). Distinct transcriptome expression of the temporal cortex of the primate Microcebus murinus during brain aging versus Alzheimer’s disease-like pathology. PLoS One 5:e12770. doi: 10.1371/journal.pone.0012770
Alba, R., Bradshaw, A. C., Mestre-Francés, N., Verdier, J.-M., Henaff, D., and Baker, A. H. (2012). Coagulation factor X mediates adenovirus type 5 liver gene transfer in non-human primates (Microcebus murinus). Gene Ther 19, 109–113. doi: 10.1038/gt.2011.87
Albert, K., Voutilainen, M. H., Domanskyi, A., and Airavaara, M. (2017). AAV vector-mediated gene delivery to substantia nigra dopamine neurons: implications for gene therapy and disease models. Genes Basel. 8:63. doi: 10.3390/genes8020063
Alvarsson, A., Caudal, D., Björklund, A., and Svenningsson, P. (2016). Emotional memory impairments induced by AAV-mediated overexpression of human α-synuclein in dopaminergic neurons of the ventral tegmental area. Behav. Brain Res. 296, 129–133. doi: 10.1016/j.bbr.2015.08.034
Bartus, R. T., Herzog, C. D., Chu, Y., Wilson, A., Brown, L., Siffert, J., et al. (2011). Bioactivity of AAV2-neurturin gene therapy (CERE-120): differences between Parkinson’s disease and nonhuman primate brains. Mov. Disord. 26, 27–36. doi: 10.1002/mds.23442
Bartus, R. T., Kordower, J. H., Johnson, E. M. Jr., Brown, L., Kruegel, B. R., Chu, Y., et al. (2015). Post-mortem assessment of the short and long-term effects of the trophic factor neurturin in patients with α-synucleinopathies. Neurobiol. Dis. 78, 162–171. doi: 10.1016/j.nbd.2015.03.023
Bezard, E., Yue, Z., Kirik, D., and Spillantini, M. G. (2013). Animal models of Parkinson’s disease: limits and relevance to neuroprotection studies. Mov. Disord. 28, 61–70. doi: 10.1002/mds.25108
Bilang-Bleuel, A., Revah, F., Colin, P., Locquet, I., Robert, J.-J., Mallet, J., et al. (1997). Intrastriatal injection of an adenoviral vector expressing glial-cell-line-derived neurotrophic factor prevents dopaminergic neuron degeneration and behavioral impairment in a rat model of Parkinson disease. Proc. Natl. Acad. Sci. U S A 94, 8818–8823. doi: 10.1073/pnas.94.16.8818
Blesa, J., and Przedborski, S. (2014). Parkinson’s disease: animal models and dopaminergic cell vulnerability. Front. Neuroanat. 8:155. doi: 10.3389/fnana.2014.00155
Brown, C. A., Campbell, M. C., Karimi, M., Tabbal, S. D., Loftin, S. K., Tian, L. L., et al. (2012). Dopamine pathway loss in nucleus accumbens and ventral tegmental area predicts apathetic behavior in MPTP-lesioned monkeys. Exp. Neurol. 236, 190–197. doi: 10.1016/j.expneurol.2012.04.025
Carriere, N., Besson, P., Dujardin, K., Duhamel, A., Defebvre, L., Delmaire, C., et al. (2014). Apathy in Parkinson’s disease is associated with nucleus accumbens atrophy: a magnetic resonance imaging shape analysis. Mov. Disord. 29, 897–903. doi: 10.1002/mds.25904
Caudal, D., Alvarsson, A., Björklund, A., and Svenningsson, P. (2015). Depressive-like phenotype induced by AAV-mediated overexpression of human α-synuclein in midbrain dopaminergic neurons. Exp. Neurol 273, 243–252. doi: 10.1016/j.expneurol.2015.09.002
Chesselet, M.-F. (2008). In vivo alpha-synuclein overexpression in rodents: a useful model of Parkinson’s disease? Exp. Neurol. 209, 22–27. doi: 10.1016/j.expneurol.2007.08.006
Chillon, M., and Kremer, E. J. (2001). Trafficking and propagation of canine adenovirus vectors lacking a known integrin-interacting motif. Hum. Gene Ther. 12, 1815–1823. doi: 10.1089/104303401750476302
Cho, H. J., Yu, J., Xie, C., Rudrabhatla, P., Chen, X., Wu, J., et al. (2014). Leucine-rich repeat kinase 2 regulates Sec16A at ER exit sites to allow ER-Golgi export. EMBO J. 33, 2314–2331. doi: 10.15252/embj.201487807
Chu, Y., and Kordower, J. H. (2007). Age-associated increases of α-synuclein in monkeys and humans are associated with nigrostriatal dopamine depletion: is this the target for Parkinson’s disease? Neurobiol. Dis. 25, 134–149. doi: 10.1016/j.nbd.2006.08.021
Dawson, T. M., Ko, H. S., and Dawson, V. L. (2010). Genetic animal models of Parkinson’s disease. Neuron 66, 646–661. doi: 10.1016/j.neuron.2010.04.034
Dawson, T., Mandir, A., and Lee, M. (2002). Animal models of PD: pieces of the same puzzle? Neuron 35, 219–222. doi: 10.1016/S0896-6273(02)00780-8
Decressac, M., Kadkhodaei, B., Mattsson, B., Laguna, A., Perlmann, T., and Bjorklund, A. (2012). α-Synuclein-induced down-regulation of Nurr1 disrupts GDNF signaling in nigral dopamine neurons. Sci. Transl. Med. 4:163ra156. doi: 10.1126/scitranslmed.3004676
Deng, H., and Yuan, L. (2014). Genetic variants and animal models in SNCA and Parkinson disease. Ageing Res. Rev. 15, 161–176. doi: 10.1016/j.arr.2014.04.002
Dujardin, K., and Lopes, R. (2014). Apathy and impaired recognition of emotion: are they related in Parkinson’s disease? J. Neurol. Neurosurg. Psychiatry 85:1061. doi: 10.1136/jnnp-2013-307224
Dujardin, K., Langlois, C., Plomhause, L., Carette, A.-S., Delliaux, M., Duhamel, A., et al. (2014). Apathy in untreated early-stage Parkinson disease: relationship with other non-motor symptoms. Mov. Disord. 29, 1796–1801. doi: 10.1002/mds.26058
Dusonchet, J., Kochubey, O., Stafa, K., Young, S. M. Jr., Zufferey, R., Moore, D. J., et al. (2011). A rat model of progressive nigral neurodegeneration induced by the Parkinson’s disease-associated G2019S mutation in LRRK2. J. Neurosci. 31, 907–912. doi: 10.1523/JNEUROSCI.5092-10.2011
Emborg, M. E. (2007). Nonhuman primate models of Parkinson’s disease. ILAR J. 48, 339–355. doi: 10.1093/ilar.48.4.339
Emborg, M. E., Ma, S. Y., Mufson, E. J., Levey, A. I., Taylor, M. D., Brown, W. D., et al. (1998). Age-related declines in nigral neuronal function correlate with motor impairments in rhesus monkeys. J. Comp. Neurol. 401, 253–265. doi: 10.1002/(SICI)1096-9861(19981116)401:2<253::AID-CNE7>3.0.CO;2-X
Eslamboli, A., Cummings, R. M., Ridley, R. M., Baker, H. F., Muzyczka, N., Burger, C., et al. (2003). Recombinant adeno-associated viral vector (rAAV) delivery of GDNF provides protection against 6-OHDA lesion in the common marmoset monkey (Callithrix jacchus). Exp. Neurol. 184, 536–548. doi: 10.1016/j.expneurol.2003.08.007
Fernandes, P., Almeida, A. I., Kremer, E. J., Alves, P. M., and Coroadinha, A. S. (2015a). Canine helper-dependent vectors production: implications of Cre activity and co-infection on adenovirus propagation. Sci. Rep. 5:9135. doi: 10.1038/srep09135
Fernandes, P., Santiago, V. M., Rodrigues, A. F., Tomás, H., Kremer, E. J., Alves, P. M., et al. (2013). Impact of E1 and Cre on adenovirus vector amplification: developing MDCK CAV-2-E1 and E1-Cre transcomplementing cell lines. PLoS ONE 8:e60342. doi: 10.1371/journal.pone.0060342
Fernandes, P., Simão, D., Guerreiro, M. R., Kremer, E. J., Coroadinha, A. S., and Alves, P. M. (2015b). Impact of adenovirus life cycle progression on the generation of canine helper-dependent vectors. Gene Ther. 22, 40–49. doi: 10.1038/gt.2014.92
Gasser, T. (2009). Molecular pathogenesis of Parkinson disease: insights from genetic studies. Expert Rev. Mol. Med. 11:e22. doi: 10.1017/s1462399409001148
Goverdhana, S., Puntel, M., Xiong, W., Zirger, J. M., Barcia, C., Curtin, J. F., et al. (2005). Regulatable gene expression systems for gene therapy applications: progress and future challenges. Mol. Ther. 12, 189–211. doi: 10.1016/j.ymthe.2005.03.022
Hall, H., Jewett, M., Landeck, N., Nilsson, N., SchagerlÖf, U., Leanza, G., et al. (2013). Characterization of cognitive deficits in rats overexpressing human alpha-synuclein in the ventral tegmental area and medial septum using recombinant adeno-associated viral vectors. PLoS One 8:e64844. doi: 10.1371/journal.pone.0064844
Hattori, N., and Sato, S. (2007). Animal models of Parkinson’s disease: similarities and differences between the disease and models. Neuropathology 27, 479–483. doi: 10.1111/j.1440-1789.2007.00842.x
Ibanes, S., and Kremer, E. J. (2013). Canine adenovirus type 2 vector generation via I-Sce1-mediated intracellular genome release. PLoS One 8:e71032. doi: 10.1371/journal.pone.0071032
Joly, M., Ammersdörfer, S., Schmidtke, D., and Zimmermann, E. (2014). Touchscreen-Based Cognitive Tasks Reveal Age-Related Impairment in a Primate Aging Model, the Grey Mouse Lemur (Microcebus murinus). PLoS One 9:e109393. doi: 10.1371/journal.pone.0109393
Joly, M., Deputte, B., and Verdier, J. M. (2006). Age effect on olfactory discrimination in a non-human primate, Microcebus murinus. Neurobiol. Aging 27, 1045–1049. doi: 10.1016/j.neurobiolaging.2005.05.001
Joly, M., Michel, B., Deputte, B., and Verdier, J.-M. (2004). Odor discrimination assessment with an automated olfactometric method in a prosimian primate, Microcebus murinus. Physiol. Behav. 82, 325–329. doi: 10.1016/j.physbeh.2004.03.019
Junyent, F., and Kremer, E. J. (2015). CAV-2—why a canine virus is a neurobiologist’s best friend. Curr. Opin. Pharmacol. 24, 86–93. doi: 10.1016/j.coph.2015.08.004
Kirik, D., Annett, L. E., Burger, C., Muzyczka, N., Mandel, R. J., and Björklund, A. (2003). Nigrostriatal α-synucleinopathy induced by viral vector-mediated overexpression of human α-synuclein: a new primate model of Parkinson’s disease. Proc. Natl. Acad. Sci. U S A 100, 2884–2889. doi: 10.1073/pnas.0536383100
Kirik, D., Rosenblad, C., Bjorklund, A., and Mandel, R. J. (2000). Long-term rAAV-mediated gene transfer of GDNF in the rat Parkinson’s model: intrastriatal but not intranigral transduction promotes functional regeneration in the lesioned nigrostriatal system. J. Neurosci. 20, 4686–4700. doi: 10.1523/JNEUROSCI.20-12-04686.2000
Klein, C., and Westenberger, A. (2012). Genetics of Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2:a008888. doi: 10.1101/cshperspect.a008888
Koprich, J. B., Johnston, T. H., Reyes, G., Omana, V., and Brotchie, J. M. (2016). Towards a Non-Human Primate Model of Alpha-Synucleinopathy for Development of Therapeutics for Parkinson’s Disease: Optimization of AAV1/2 Delivery Parameters to Drive Sustained Expression of Alpha Synuclein and Dopaminergic Degeneration in Macaque. PLoS ONE 11:e0167235. doi: 10.1371/journal.pone.0167235
Kordower, J. H., Emborg, M. E., Bloch, J., Ma, S. Y., Chu, Y., Leventhal, L., et al. (2000). Neurodegeneration prevented by lentiviral vector delivery of GDNF in primate models of Parkinson’s disease. Science 290, 767–773. doi: 10.1126/science.290.5492.767
Kordower, J. H., Herzog, C. D., Dass, B., Bakay, R. A., Stansell, J. 3rd, Gasmi, M., et al. (2006). Delivery of neurturin by AAV2 (CERE-120)-mediated gene transfer provides structural and functional neuroprotection and neurorestoration in MPTP-treated monkeys. Ann. Neurol. 60, 706–715. doi: 10.1002/ana.21032
Kordower, J. H., Olanow, C. W., Dodiya, H. B., Chu, Y., Beach, T. G., Adler, C. H., et al. (2013). Disease duration and the integrity of the nigrostriatal system in Parkinson’s disease. Brain J. Neurol. 136, 2419–2431. doi: 10.1093/brain/awt192
Kremer, E. J., and Nemerow, G. R. (2015). Adenovirus tales: from the cell surface to the nuclear pore complex. PLoS Pathog 11:e1004821. doi: 10.1371/journal.ppat.1004821
Kremer, E. J., Boutin, S., Chillon, M., and Danos, O. (2000). Canine adenovirus vectors: an alternative for adenovirus mediated gene transfer. J. Virol. 74, 505–512. doi: 10.1128/jvi.74.1.505-512.2000
Languille, S., Blanc, S., Blin, O., Canale, C. I., Dal-Pan, A., Devau, G., et al. (2012). The grey mouse lemur: a non-human primate model for ageing studies. Ageing Res. Rev. 11, 150–162. doi: 10.1016/j.arr.2011.07.001
Lee, B. D., Shin, J.-H., VanKampen, J., Petrucelli, L., West, A. B., Ko, H. S., et al. (2010). Inhibitors of leucine-rich repeat kinase-2 protect against models of Parkinson’s disease. Nat. Med. 16, 998–1000. doi: 10.1038/nm.2199
Lewis, S. J., Dove, A., Robbins, T. W., Barker, R. A., and Owen, A. M. (2003). Cognitive impairments in early Parkinson’s disease are accompanied by reductions in activity in frontostriatal neural circuitry. J. Neurosci 23, 6351–6356. doi: 10.1523/JNEUROSCI.23-15-06351.2003
Lin, X., Parisiadou, L., Gu, X. L., Wang, L., Shim, H., Sun, L., et al. (2009). Leucine-rich repeat kinase 2 regulates the progression of neuropathology induced by Parkinson’s-disease-related mutant alpha-synuclein. Neuron 64, 807–827. doi: 10.1016/j.neuron.2009.11.006
Loustalot, F., Kremer, E. J., and Salinas, S. (2015). The intracellular domain of the coxsackievirus and adenovirus receptor differentially influences adenovirus entry. J. Virol 89, 9417–9426. doi: 10.1128/jvi.01488-15
Luchtman, D. W., Meng, Q., and Song, C. (2012). Ethyl-eicosapentaenoate (E-EPA) attenuates motor impairments and inflammation in the MPTP-probenecid mouse model of Parkinson’s disease. Behav. Brain Res. 226, 386–396. doi: 10.1016/j.bbr.2011.09.033
Luchtman, D. W., Shao, D., and Song, C. (2009). Behavior, neurotransmitters and inflammation in three regimens of the MPTP mouse model of Parkinson’s disease. Physiol. Behav. 98, 130–138. doi: 10.1016/j.physbeh.2009.04.021
Marks, W. J. Jr., Bartus, R. T., Siffert, J., Davis, C. S., Lozano, A., Boulis, N., et al. (2010). Gene delivery of AAV2-neurturin for Parkinson’s disease: a double-blind, randomised, controlled trial. Lancet Neurol. 9, 1164–1172. doi: 10.1016/S1474-4422(10)70254-4
Martin-Touaux, E., Puech, J. P., Château, D., Emiliani, C., Kremer, E. J., Raben, N., et al. (2002). Muscle as a putative producer of acid alpha-glucosidase for glycogenosis type II gene therapy. Hum. Mol. Genet 11, 1637–1645. doi: 10.1093/hmg/11.14.1637
Mestre-Francés, N., Nicot, S., Rouland, S., Biacabe, A. G., Quadrio, I., Perret-Liaudet, A., et al. (2012). Oral transmission of L-type bovine spongiform encephalopathy in primate model. Emerging Infect. Dis. 18, 142–145. doi: 10.3201/eid1801.111092
Mestre-Francés, N., Serratrice, N., Gennetier, A., Devau, G., Cobo, S., Trouche, S. G., et al. (2018). Exogenous LRRK2G2019S induces parkinsonian-like pathology in a nonhuman primate. JCI Insight 3:e98202. doi: 10.1172/jci.insight.98202
O’Keeffe, G. W., and Sullivan, A. M. (2018). Evidence for dopaminergic axonal degeneration as an early pathological process in Parkinson’s disease. Parkinsonism Relat. Disord. 56, 9–15. doi: 10.1016/j.parkreldis.2018.06.025
Petroske, E., Meredith, G. E., Callen, S., Totterdell, S., and Lau, Y. S. (2001). Mouse model of Parkinsonism: a comparison between subacute MPTP and chronic MPTP/probenecid treatment. Neuroscience 106, 589–601. doi: 10.1016/s0306-4522(01)00295-0
Picq, J. L. (2007). Aging affects executive functions and memory in mouse lemur primates. Exp. Gerontol. 42, 223–232. doi: 10.1016/j.exger.2006.09.013
Picq, J. L., Aujard, F., Volk, A., and Dhenain, M. (2012). Age-related cerebral atrophy in nonhuman primates predicts cognitive impairments. Neurobiol. Aging 33, 1096–1109. doi: 10.1016/j.neurobiolaging.2010.09.009
Piersanti, S., Astrologo, L., Licursi, V., Costa, R., Roncaglia, E., Gennetier, A., et al. (2013). Differentiated neuroprogenitor cells incubated with human or canine adenovirus, or lentiviral vectors have distinct transcriptome profiles. PLoS One 8:e69808. doi: 10.1371/journal.pone.0069808
Polymeropoulos, M. H., Lavedan, C., Leroy, E., Ide, S. E., Dehejia, A., Dutra, A., et al. (1997). Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276, 2045–2047. doi: 10.1126/science.276.5321.2045
Przedborski, S., Levivier, M., Jiang, H., Ferreira, M., Jackson-Lewis, V., Donaldson, D., et al. (1995). Dose-dependent lesions of the dopaminergic nigrostriatal pathway induced by intrastriatal injection of 6-hydroxydopamine. Neuroscience 67, 631–647. doi: 10.1016/0306-4522(95)00066-r
Rademacher, C., Bru, T., McBride, R., Robison, E., Nycholat, C. M., Kremer, E. J., et al. (2012). A Siglec-like sialic-acid-binding motif revealed in an adenovirus capsid protein. Glycobiology 22, 1086–1091. doi: 10.1093/glycob/cws073
Ramonet, D., Daher, J. P., Lin, B. M., Stafa, K., Kim, J., Banerjee, R., et al. (2011). Dopaminergic neuronal loss, reduced neurite complexity and autophagic abnormalities in transgenic mice expressing G2019S mutant LRRK2. PLoS ONE 6:e18568. doi: 10.1371/journal.pone.0018568
Salinas, S., Bilsland, L. G., Henaff, D., Weston, A. E., Keriel, A., Schiavo, G., et al. (2009). CAR-associated vesicular transport of an adenovirus in motor neuron axons. PLoS Pathog 5:e1000442. doi: 10.3410/f.1162577.624132
Salinas, S., Schiavo, G., and Kremer, E. J. (2010). A hitchhiker’s guide to the nervous system: the complex journey of viruses and toxins. Nat. Rev. Microbiol. 8, 645–655. doi: 10.1038/nrmicro2395
Schober, A. (2004). Classic toxin-induced animal models of Parkinson’s disease: 6-OHDA and MPTP. Cell Tissue Res. 318, 215–224. doi: 10.1007/s00441-004-0938-y
Schoehn, G., El Bakkouri, M., Fabry, C. M., Billet, O., Estrozi, L. F., Le, L., et al. (2008). Three-dimensional structure of canine adenovirus serotype 2 capsid. J. Virol. 82, 3192–3203. doi: 10.1128/jvi.02393-07
Schwarz, L. A., Miyamichi, K., Gao, X. J., Beier, K. T., Weissbourd, B., DeLoach, K. E., et al. (2015). Viral-genetic tracing of the input-output organization of a central noradrenaline circuit. Nature 524, 88–92. doi: 10.1038/nature14600
Seiradake, E., Lortat-Jacob, H., Billet, O., Kremer, E. J., and Cusack, S. (2006). Structural and mutational analysis of human Ad37 and canine adenovirus 2 fiber heads in complex with the D1 domain of coxsackie and adenovirus receptor. J. Biol. Chem. 281, 33704–33716. doi: 10.1074/jbc.m605316200
Soudais, C., Boutin, S., and Kremer, E. J. (2001a). Characterization of cis-acting sequences involved in canine adenovirus packaging. Mol. Ther. 3, 631–640. doi: 10.1006/mthe.2001.0263
Soudais, C., Boutin, S., Hong, S. S., Chillon, M., Danos, O., Bergelson, J. M., et al. (2000). Canine adenovirus type 2 attachment and internalization: Coxsackievirus-adenovirus receptor, alternative receptors and an RGD-independent pathway. J. Virol. 74, 10639–10649. doi: 10.1128/jvi.74.22.10639-10649.2000
Soudais, C., Laplace-Builhe, C., Kissa, K., and Kremer, E. J. (2001b). Preferential transduction of neurons by canine adenovirus vectors and their efficient retrograde transport in vivo. FASEB J 15, 2283–2285. doi: 10.1096/fj.01-0321fje
Su, X., Fischer, D. L., Li, X., Bankiewicz, K., Sortwell, C. E., and Federoff, H. J. (2017). Alpha-Synuclein mRNA is not increased in sporadic PD and alpha-synuclein accumulation does not block GDNF signaling in Parkinson’s disease and disease models. Mol. Ther. J. Am. Soc. Gene Ther. 25, 2231–2235. doi: 10.1016/j.ymthe.2017.04.018
Su, X., Kells, A. P., Huang, E. J., Lee, H. S., Hadaczek, P., Beyer, J., et al. (2009). Safety evaluation of AAV2-GDNF gene transfer into the dopaminergic nigrostriatal pathway in aged and parkinsonian rhesus monkeys. Hum. Gene Ther. 20, 1627–1640. doi: 10.1089/hum.2009.103
Tan, S. K., Hartung, H., Sharp, T., and Temel, Y. (2011). Serotonin-dependent depression in Parkinson’s Disease: a role for the subthalamic nucleus? Neuropharmacology 61, 387–399. doi: 10.1016/j.neuropharm.2011.01.006
Taymans, J.-M., and Greggio, E. (2016). LRRK2 kinase inhibition as a therapeutic strategy for Parkinson’s Disease, where do we stand? Curr. Neuropharmacol. 14, 214–225. doi: 10.2174/1570159X13666151030102847
Trouche, S. G., Maurice, T., Rouland, S., Verdier, J. M., and Mestre-Francés, N. (2010). The three-panel runway maze adapted to Microcebus murinus reveals age-related differences in memory and perseverance performances. Neurobiol. Learn. Mem. 94, 100–106. doi: 10.1016/j.nlm.2010.04.006
Tsika, E., Kannan, M., Foo, C. S., Dikeman, D., Glauser, L., Gellhaar, S., et al. (2014). Conditional expression of Parkinson’s disease-related R1441C LRRK2 in midbrain dopaminergic neurons of mice causes nuclear abnormalities without neurodegeneration. Neurobiol. Dis. 71, 345–358. doi: 10.1016/j.nbd.2014.08.027
Verdier, J. M., Acquatella, I., Lautier, C., Devau, G., Trouche, S. G., Lasbleiz, C., et al. (2015). Lessons from the analysis of nonhuman primates for understanding human aging and neurodegenerative diseases. Front. Neurosci. 9:64. doi: 10.3389/fnins.2015.00064
Warren Olanow, C., Bartus, R. T., Baumann, T. L., Factor, S., Boulis, N., Stacy, M., et al. (2015). Gene delivery of neurturin to putamen and substantia nigra in Parkinson disease: A double-blind, randomized, controlled trial. Ann. Neurol. 78, 248–257. doi: 10.1002/ana.24436
Yang, W., Wang, G., Wang, C.-E., Guo, X., Yin, P., Gao, J., et al. (2015). Mutant alpha-synuclein causes age-dependent neuropathology in monkey brain. J. Neurosci. 35, 8345–8358. doi: 10.1523/jneurosci.2727-15.2015
Keywords: Parkinson’s disease, primate, CAV vectors, gene transfer, dopaminergic neurons
Citation: Lasbleiz C, Mestre-Francés N, Devau G, Luquin M-R, Tenenbaum L, Kremer EJ and Verdier J-M (2019) Combining Gene Transfer and Nonhuman Primates to Better Understand and Treat Parkinson’s Disease. Front. Mol. Neurosci. 12:10. doi: 10.3389/fnmol.2019.00010
Received: 02 November 2018; Accepted: 14 January 2019;
Published: 11 February 2019.
Edited by:
Jean-Marc Taymans, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceReviewed by:
Masahiko Takada, Kyoto University, JapanHui-Yun Chang, National Tsing Hua University, Taiwan
Copyright © 2019 Lasbleiz, Mestre-Francés, Devau, Luquin, Tenenbaum, Kremer and Verdier. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jean-Michel Verdier, amVhbi1taWNoZWwudmVyZGllckBlcGhlLnBzbC5ldQ==