 Eftychia Vasili
Eftychia Vasili Antonio Dominguez-Meijide
Antonio Dominguez-Meijide Tiago Fleming Outeiro
Tiago Fleming Outeiro- 1Department of Experimental Neurodegeneration, Center for Nanoscale Microscopy and Molecular Physiology of the Brain, Center for Biostructural Imaging of Neurodegeneration, University Medical Center Goettingen, Goettingen, Germany
- 2Max Planck Institute for Experimental Medicine, Goettingen, Germany
- 3The Medical School, Institute of Neuroscience, Newcastle University, Newcastle Upon Tyne, United Kingdom
Alzheimer's disease (AD) and Parkinson's disease (PD) are age-associated neurodegenerative disorders characterized by the misfolding and aggregation of alpha-synuclein (aSyn) and tau, respectively. The coexistence of aSyn and tau aggregates suggests a strong overlap between tauopathies and synucleinopathies. Interestingly, misfolded forms of aSyn and tau can propagate from cell to cell, and throughout the brain, thereby templating the misfolding of native forms of the proteins. The exact mechanisms involved in the propagation of the two proteins show similarities, and are reminiscent of the spreading characteristic of prion diseases. Recently, several models were developed to study the spreading of aSyn and tau. Here, we discuss the mechanisms involved, the similarities and differences between the spreading of the two proteins and that of the prion protein, and the different cell and animal models used for studying these processes. Ultimately, a deeper understanding of the molecular mechanisms involved may lead to the identification of novel targets for therapeutic intervention in a variety of devastating neurodegenerative diseases.
Introduction
Alzheimer's disease (AD) and Parkinson's disease (PD) are progressive, age-associated neurodegenerative disorders. Recent epidemiological studies revealed that around 50 million people worldwide are living with AD, and more than 10 million people above 60 years old with PD, respectively (Karlawish et al., 2017; Tysnes and Storstein, 2017). The prevalence of both diseases is increased in the highest age groups and the number will escalate rapidly the coming years (Karlawish et al., 2017; Tysnes and Storstein, 2017). While the clinical features are quite distinct between the two diseases, at the molecular level they are characterized by the misfolding, aggregation, and deposition of proteins in characteristic types of inclusions (Brion et al., 1985; Kosik et al., 1986; Iwai et al., 1995; Spillantini et al., 1997). Accumulation of aggregated tau is a hallmark of AD and related tauopathies and the accumulation of alpha-synuclein (aSyn) aggregates is the hallmark of PD and related synucleinopathies (Brion et al., 1985, 1986; Kosik et al., 1986; Wood et al., 1986; Wischik et al., 1988; Spillantini et al., 1997; Baba et al., 1998; Bayer et al., 1999). aSyn and tau are abundant brain proteins, both known as intrinsically disordered proteins (IDPs) with prion-like properties, as they can misfold, seed, and spread the misfolded conformation to normal monomeric forms of each protein (Uversky and Fink, 2004; Eliezer, 2009; Bartels et al., 2010; Wu and Baum, 2010; Coelho-Cerqueira et al., 2013).
Different strains of aSyn and tau display different cell binding and penetration properties, resulting in transmission of pathology between cells (Clavaguera et al., 2009; Guo and Lee, 2011; Hansen et al., 2011; Angot et al., 2012; Kfoury et al., 2012; Masuda-Suzukake et al., 2013; Wu et al., 2013; Recasens and Dehay, 2014; Sanders et al., 2014; Grozdanov and Danzer, 2018). It is currently thought that distinct protein conformations account for differences in seeding potency. Interestingly, several studies revealed the accumulation of abnormal tau aggregates in numerous cases of aSyn deposition, and vice versa (Brion et al., 1985; Kosik et al., 1986). The coexistence of aSyn and tau aggregates suggests a strong cross-talk between tauopathies and synucleinopathies, and raises the hypothesis that cross-seeding might take place, thereby contributing to disease progression (Kosik et al., 1986; Spillantini et al., 1997; Cabrales Fontela et al., 2017). Furthermore, the interaction between aSyn and tau appear to promote the oligomerization and solubility of each other in vitro and in vivo, thereby disrupting cytoskeletal organization, impairing axonal transport, and compromising synaptic organization (Masliah et al., 2001; Giasson et al., 2003; Kotzbauer et al., 2004; Bellani et al., 2010; Cabrales Fontela et al., 2017; Sotiropoulos et al., 2017; Biswas and Kalil, 2018; Ordonez et al., 2018; Prots et al., 2018; Tuerde et al., 2018; Yuan et al., 2018). However, the exact molecular mechanisms involved in the cross-talk between the two proteins, and in the propagation of pathology, are still obscure. Here, we discuss the current knowledge about the mechanisms involved in transmission of both proteins, focusing on similarities and differences between the different spreading mechanisms.
aSyn Structure and Function
Alpha-synuclein (aSyn) is a 14.5 kDa acidic protein of 140 amino acid residues, encoded by the SNCA gene (Chen et al., 1995), and is strongly implicated in PD. aSyn belongs to the synuclein family, together with beta- and gamma-synuclein.
aSyn was first isolated from the synaptic vesicles and nuclei of the electric organ of Torpedo californica (Maroteaux et al., 1988). In 1997, aSyn was identified as the major protein component of Lewy bodies (LBs) and Lewy neurites (LNs), the pathognomonic deposits in PD (Spillantini et al., 1997). In the same year, the first point mutation in the SNCA gene was associated with autosomal-dominant forms of PD, demonstrating the role of genetics in the disease (Polymeropoulos et al., 1997). Furthermore, the identification of families with duplications and triplications of the SNCA locus confirmed that increased levels of aSyn can cause disease (Singleton et al., 2003). These findings, along with a plethora of in vitro and in vivo studies, suggest that aSyn is a central player in a group of neurodegenerative disorders known as synucleinopathies.
aSyn is classified as an intrinsically disordered protein (IDP) as it lacks defined secondary structure (Uversky, 2003, 2011a,b; Bernado et al., 2005; Breydo et al., 2012). Although the precise physiological function of aSyn is still unclear, several studies suggest that aSyn is involved in the regulation of synaptic membrane processes and in neurotransmitter release through interactions with members of the SNARE family (Tsigelny et al., 2012; Bellucci et al., 2016). Surprisingly, studies in aSyn knockout mice revealed that aSyn is not essential for synapse formation and cell survival (Bisaglia et al., 2009).
The primary sequence of aSyn can be divided in three distinct domains: the amino-terminal domain (N-terminal, residues 1–60), the central domain (residues 61–95) and the carboxy-terminal domain (C-terminal domain, residues 96–140). The N-terminal domain includes four repeats of the 11 amino acid alpha-helical lipid-binding motif (KTKEGV) (Figure 1Ai, R1–4), enabling the formation of amphipathic α-helical structures upon interaction with lipid membranes (Jao et al., 2004, 2008; Georgieva et al., 2008). The lipid composition of membranes is critical for aSyn binding. aSyn specifically prefers the binding in membranes characterized by high concentrations in cholesterol and sphingolipids, known also as lipid rafts. It seems that lipid rafts serve as a platform, which promotes aSyn binding and oligomerization (Davidson et al., 1998; Jo et al., 2000; Fortin et al., 2004; Zabrocki et al., 2008; Middleton and Rhoades, 2010; Fabelo et al., 2011; Hellstrand et al., 2013).
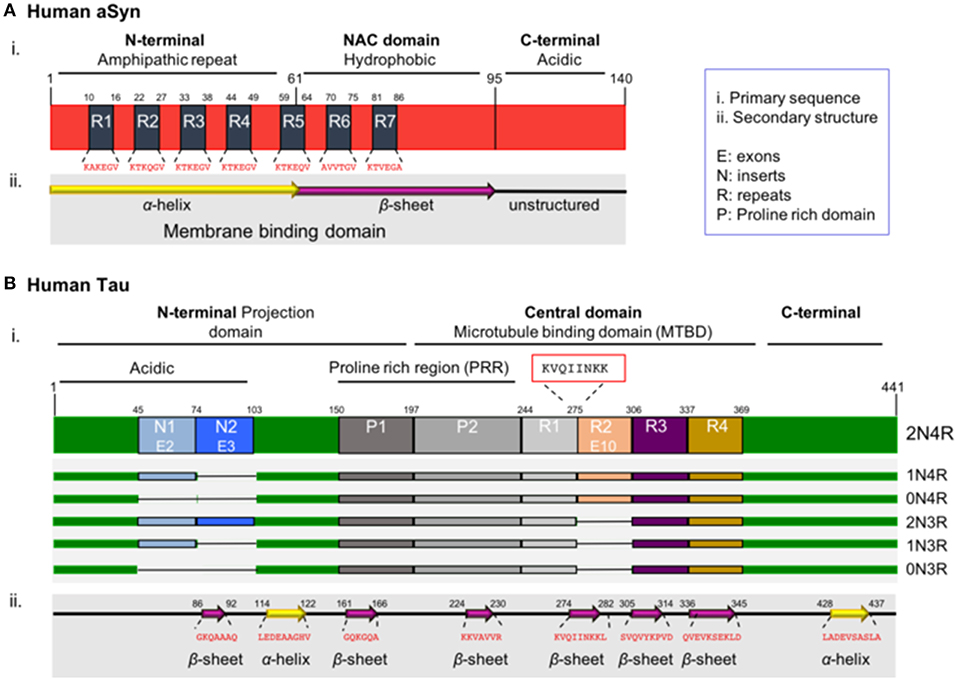
Figure 1. Schematic illustration of aSyn and tau proteins. (A) aSyn is encoded by the SNCA gene. The primary sequence of aSyn can be divided in three distinct domains: the amino-terminal domain (N-terminal, residues 1–60), the central domain also known as NAC domain (residues 61–95), and the carboxy-terminal domain (C-terminal domain, residues 96–140). The N-terminal domain includes four repeats (R1–R4) of the 11 amino acid alpha-helical lipid-binding motif (KTKEGV). This region has propensity to form amphipathic α-helical structures upon interacting with lipid membranes. The NAC domain (non-amyloid-β component), contains three additional repeats (R5–R7) of the lipid-binding motif, is enriched in hydrophobic residues, leading to the formation of cylindrical β-sheets and amyloid-β fibrils. Both the N-terminal and NAC domain are characterized part of the membrane binding domain. The C-terminal domain is rich in acidic residues (15 acidic amino acids: 10 Glu and 5 Asp residues) and lacks defined secondary structure. (B) Tau is encoded by the MAPT gene. Alternative splicing of the MAPT gene results in six isoforms known as 2N/4R, 1N/4R, 0N/4R, 2N/3R, 1N/3R, and 0N/3R, depending on the presence or absence of exon 10 (4R or 3R) and on the numbers of amino-terminal inserts (0N, 1N, and 2N) encoded by exons 2 and 3. The primary sequence of the full-length human tau isoform can be divided in the N-terminal domain also known as projection domain, the central domain which is the microtubule binding domain (MTBD) and the C-terminal tail. The N-terminal consists of the acidic part encoded by exons 2 and 3 (E2-3) called inserts 1 and 2 (N1-2), followed by the proline-rich region (PRR). The MTBD in the longest isoform contains four repeats (R1-4). The region with the strongest propensity for microtubule polymerization is the oligopeptide “KVQIINKK” (residues 274–281), located in the sub-region between the R1–R2 repeats (red box). The C-terminal tail is enriched in positively charged residues. Notably, the region with the strongest propensity for microtubule binding is located in the sub-region between the R1–R2 repeats and, more specifically, is the oligopeptide “KVQIINKK” (residues 274–281), which is included only in the 4R tau isoform, providing a stronger binding affinity when compared to the 3R isoforms (Brandt and Lee, 1993; Goode and Feinstein, 1994; Sergeant et al., 2005).
Importantly, all the known mutations associated with familial forms of PD are clustered within the N-terminal region of aSyn (Polymeropoulos et al., 1997; Kruger et al., 1998; Zarranz et al., 2004; Appel-Cresswell et al., 2013; Lesage et al., 2013; Proukakis et al., 2013; Pasanen et al., 2014), reinforcing the hypothesis that changes in the lipid binding domain may be linked to aSyn pathology. Interestingly, aSyn was reported to be acetylated at the N-terminus in cells, an essential modification that protects its native conformation against pathological aggregation (Iyer et al., 2016; Bu et al., 2017). The central domain, also known as NAC domain (non-amyloid-β component) (Figure 1Ai), is enriched in hydrophobic residues and is involved in the pathologic aggregation of the protein due to conformational changes (El-Agnaf et al., 1998; Giasson et al., 2001; Bellucci et al., 2012). Interestingly, one phosphorylation site is present in the NAC domain—the S87 residue. S87 phosphorylation is increased in synucleinopathies, leading to inhibition of aSyn oligomerization which influences synuclein-membrane interactions (Paleologou et al., 2010). The carboxy-terminal domain (C-terminal domain) is characterized by a non-defined structure (Bisaglia et al., 2009) and incorporates most of the posttranslational modification sites (PTMs), including the most common phosphorylation at S129 (Fujiwara et al., 2002; Oueslati, 2016). The importance of phosphorylation is emphasized by a study showing that in DLB brains, approximately 90% of insoluble aSyn is phosphorylated at S129, compared with only 4% in soluble cytosolic aSyn (Anderson et al., 2006). This suggests the implication of phosphorylation in the aggregation propensity. These PTMs may act by modulating the structure, the physiological functions and the toxicity of aSyn. Furthermore, they can modulate protein-protein interactions, interaction with metal ions (Paik et al., 1999; Brown, 2007; Bisaglia et al., 2009), including Ca2+ binding (Nielsen et al., 2001), polyamine complexes binding, modulation of phospholipid-binding (Paleologou et al., 2008; Visanji et al., 2011), affecting the aggregation propensity of the protein. Moreover, the region plays a protective role against aggregation, due to the presence of all the five proline (Pro) residues of the protein (Meuvis et al., 2010). Changes in the charge or the hydrophobicity by residue substitution as well as deletion of the C-terminal lead to accelerated aggregation of aSyn in vitro (Hoyer et al., 2004). As mentioned above, aSyn has the ability to bind to acidic membranes. This binding is mediated by the amphipathic α-helix in the N-terminal domain. Under physiological conditions aSyn exists in a dynamic equilibrium between the unfolded cytosolic and the membrane–bound state (Burre et al., 2014). In contrast, under pathological conditions, aSyn adopts a β-sheet–rich amyloid conformation, which leads to the fibril formation and subsequently aSyn deposition into LBs (Pineda and Burre, 2017). Importantly, the α-helical part is responsible for the formation of the different types of oligomers, the species currently considered to be most toxic. However, the exact nature of those toxic species remains unknown, and is still unclear whether aggregation initiates from its lipid-bound part or from the unstructured cytosolic protein (Trexler and Rhoades, 2012; Chen et al., 2015; Ghosh et al., 2015; Gallea et al., 2018).
Tau Structure and Function
Tau was first discovered associated with microtubules, together with other microtubule-associated proteins (Weingarten et al., 1975; Kolarova et al., 2012). For this reason, it was included in the family of microtubule-associated proteins (MAPs). There are six different isoforms of tau in the central nervous system, generated from the MAPT gene, as a result of alternative splicing. These isoforms range from 352 to 441 amino acids (Neve et al., 1986). Each of the six tau isoforms differs in their primary structure due to the content of three (3R) or four repeats (4R) of the microtubule binding domains in the C-terminal region, in combination with the presence or absence of one (N1) or two (N2) amino acid inserts in the N-terminal part of the protein. The six isoforms are known as 0N/3R (352 residues, 60 kDa), 1N/3R (381 residues, 64 kDa), 2N/3R (410 residues, 69 kDa), 0N/4R (383 residues, 64 kDa), 1N/4R (412 residues, 69 kDa), and 2N/4R (441 residues, 74 kDa) (Figure 1 Bi), all showing higher apparent molecular masses than the predicted ones. Since the isoforms are differentially expressed in the brain during development, and stimulate microtubule assembly with different efficiencies, possibly possess particular physiological roles and implicated at different biological activities (Utton et al., 2001; Stanford et al., 2003). The shortest isoform is also known as “fetal tau isoform” because it is expressed also in the fetal brain, while all of them are detected in the human adult brain (Kosik et al., 1989; Stanford et al., 2003).
Importantly, in neurodegenerative diseases such as AD and PD, modified proportions of the different tau isoforms have been observed (Bre and Karsenti, 1990; Avila et al., 2004). Tau stabilizes the polymerization of microtubules through the three or four MTBR repeats in case of the longest isoform (Drubin and Kirschner, 1986; Maccioni et al., 1989). Under physiological conditions, in mature neurons, all tau protein is likely to be microtubule bound (Ackmann et al., 2000), and it is considered a dipole protein since the two ends of the protein have opposite charges (Sergeant et al., 2008).
The primary sequence of tau consists of the N-terminal domain, half of which is enriched in acidic residues, followed by a proline-rich region and the positively charged C-terminal tail.
Tau, like aSyn, is an intrinsically disordered protein, since it contains regions without defined secondary structure that are inserted between very short β-sheets and α-helices (Figure 1Bii). The protein can also undergo different types of PTMs like phosphorylation, ubiquitination, acetylation, glycation, methylation, truncation of the N- or C-terminal regions or nitration (Avila et al., 2004; Garcia-Sierra et al., 2008; Avila, 2009; Morris et al., 2015; Huang et al., 2016; Iqbal et al., 2016), that likely modulate its normal function and lead to pathological features. Notably, tau contains a high number of potential phosphorylation sites (80 serines/threonines and 5 tyrosines) (Grundke-Iqbal et al., 1986b; Bancher et al., 1989; Wang and Mandelkow, 2016). Most of these sites are located within the proline-rich region in close proximity to the MTBR domains and in the C-terminal tail (Figure 1Bi) (Buee et al., 2000; Sergeant et al., 2008). The phosphorylation state of the protein affects the secondary structure and, subsequently, regulates all the normal and abnormal functions like development, interaction with different protein partners such as microtubules, localization, aggregation, and spreading (Camero et al., 2014; Multhaup et al., 2015; Wang and Mandelkow, 2016). In principle, a normal and strictly controlled level of phosphorylation is required for the appropriate function of the protein, while the pathological state is characterized by hyperphosphorylation that leads the tau to lose its biological activity (Kopke et al., 1993).
The deposition of hyperphosphorylated tau in insoluble filaments in the brain is a pathological hallmark not only of AD but also of related neurodegenerative diseases, known as tauopathies, including frontotemporal dementias (FTD) like Pick's Disease and argyrophilic grain disease, progressive supranuclear palsy, and corticobasal degeneration (Grundke-Iqbal et al., 1986b). Major differences between tauopathies are the deposition of different isoforms of tau (Rademakers et al., 2004) and the occurrence of different structures of tau aggregates (Gerson et al., 2014; Dujardin et al., 2018). In AD, all the six-tau isoforms are hyperphosphorylated and aggregated into paired helical filaments (PHF) (Grundke-Iqbal et al., 1986a,b). In Pick's disease 3R isoforms are predominant, and the arrangement of tau is different than that in inclusions found in AD (Falcon et al., 2018). On the other hand, in the other tauopathies only the 4R isoform is present in the filaments (Goedert, 2015). In AD brains, the abnormally hyperphosphorylated tau is presented in the cytosol inhibiting the assembly of tubulin and disrupting microtubules. Furthermore, as a result of self-assembly is accumulated into neurofibrillary deposits in neurons and glial cells (Iqbal et al., 2010). In sporadic and familial FTD, several mutations have been identified in the tau gene. Some of these mutations are thought to disrupt the normal binding of tau to tubulin resulting in pathological deposits of hyperphosphorylated tau (Rademakers et al., 2004), or makes tau more vulnerable to self-aggregation (S422E, ΔK280, R5L, P301L, and R406W) (Haase et al., 2004; van Swieten et al., 2007; Mutreja et al., 2019). As with aSyn, it is believed that in a variety of tauopathies, the most toxic species are oligomeric, but the controversy is still not fully resolved. These oligomeric species are non-fibrillar, multimeric, soluble forms of the protein (Haase et al., 2004; Ghag et al., 2018). Examples of tauopathies where oligomers have been proposed as the toxic species include AD, corticobasal degeneration, Pick's disease, and progressive supranuclear palsy (Maeda et al., 2006; Patterson et al., 2011; Gerson et al., 2014).
Prions and Prion-like Spreading of Pathology
Prion diseases are infectious diseases that can be transmitted horizontally between individuals of the same or even different species (Costanzo and Zurzolo, 2013; Kizhakke et al., 2017). In these diseases, PrPC misfolds and converts into the pathogenic form PrPSc. PrPSc then acts as a template, converting endogenous PrPC into additional PrPSc, thereby spreading pathology in the brain (Brandner et al., 1996). Other proteins may manifest prion-like behavior. The prion-like behavior of amyloid-β has been broadly studied (Walker et al., 2016; Ruiz-Riquelme et al., 2018; Sarnataro, 2018). Alterations in amyloid-β conformation lead to aggregation and the formation of plaques, and it has been reported that amyloid-β can reach the brain form outside the CNS (Eisele et al., 2014).
The stable propagation of different misfolded protein conformations was established as a defining feature of the prion paradigm and of the prion-like spreading of pathology (Jucker and Walker, 2013). Importantly, both aSyn and tau appear to spread in a prion-like manner (Holmes et al., 2013). However, different structural features may affect the way they propagate. Each protein has a characteristic core that undergoes conformational changes and may lead to aggregation. In particular, these are the NAC region in aSyn and the MTBR (together with the final part of the poliproline region) domain in tau (El-Agnaf et al., 1998; Ackmann et al., 2000; Giasson et al., 2001). Differences in these regions may lead to differences in the aggregated species formed. Whether aSyn and tau spreading have an infectious nature like that of the prion protein remains unclear. In general, protein infectivity depends on several factors, such as irreversibility of misfolded protein assemblies, the efficiency by which precursor polypeptides are recruited into aggregates, the clearance of the aggregates, and the efficiency of the spreading of misfolded protein proteins (Brundin et al., 2010).
Spreading of aSyn Pathology
aSyn neuropathology typically progresses in a predictable manner throughout the brain. Post mortem analysis of human brains revealed progression of neuropathology in a series of stages (Braak et al., 2003a). Initially, the lesions start in the olfactory bulb, anterior olfactory nucleus, and dorsal motor nucleus of the vagus (Ordonez et al., 2018) in what is considered the first stage. During the second stage, pathology spreads to the lower raphe nuclei, the magnocellular portions of the reticular formation and the locus coeruleus (Prots et al., 2018). In the third stage, the pathology reaches the midbrain, affecting fundamentally the substantia nigra pars compacta (Braak et al., 2003a). Pathology spreads then to the cortex during the fourth stage. In this stage the mesocortex is affected whilst the neocortex is unaffected (Braak et al., 2003a,b). In the last two stages, pathology reaches the neocortex. Initially affecting the prefrontal neocortex and then moving to the premotor areas, the primary sensory areas and the primary motor field (Braak et al., 2003a).
aSyn can cross the blood-brain barrier (Peelaerts et al., 2015) and was shown to reach the central nervous system (CNS) after gastrointestinal administration (Holmqvist et al., 2014). It has been found in the choroid plexus, where it may be produced by the choroid cells that participate in its transport between the blood and cerebrospinal fluid (Bellani et al., 2010).
Additional studies showed the spreading of aSyn from diseased to healthy tissue. Several PD patients underwent embryonic neuronal cell transplantation developed the disease years after the surgery. The postmortem analysis of the tissue showed signs of PD, including the presence of LB and LN, in the grafted tissue. Interestingly, in these studies, the presence of cytosolic aSyn phosphorylated at S129 was also shown (Kordower et al., 2008; Li et al., 2008).
These studies, together with the Braak staging hypothesis, were considered strong evidence in favor of the prion-like spreading of aSyn pathology in the brain (Olanow and Prusiner, 2009).
Following these findings, several new studies revealed that aSyn can propagate from host to grafted tissue (Desplats et al., 2009; Angot et al., 2012; Reyes et al., 2014). A different set of studies showed how the administration of brain lysates from multiple system atrophy (MSA) patients into TgM83 mice brain leads to transmission in a way that is reminiscent of the transmission of the prion protein in chimpanzee brains in a model of Kuru (Gajdusek et al., 1966; Watts et al., 2013). This process was proposed to happen through cell-to-cell transmission following not only cell connectivity, and may reach parts of the CNS away from the injection site (Luk et al., 2012). Furthermore, 9 months after the inoculation of pathological aSyn from sarkosyl-insoluble fractions from cortical brain tissue from MSA patients, aSyn aggregates were found in the side contralateral to the administration (Bernis et al., 2015). Interestingly, in all these experiments human material leads to disease in different species. The transmission among different species is one of the main characteristics of prion proteins. Another argument in favor of the prion-like behavior of aSyn is that, under certain conditions, aSyn can assemble aberrantly forming prion strains (Guo et al., 2013; Peelaerts et al., 2015). When aSyn fibrils are inoculated in Wistar rats they act as seeds imprinting their intrinsic structures, turning monomeric aSyn into fibrils. When aSyn ribbons were injected, endogenous aSyn in Wistar rats acquired this specific conformation (Breydo et al., 2012). Furthermore, when two different strains of aSyn pre formed fibrils were inoculated in mice, endogenous aSyn acquired the structure of the strain inoculated (Peelaerts et al., 2015). Also, assemblies such as fibrils and ribbons can cross the blood-brain barrier and reach the CNS after intravenous injection (Uversky, 2011b).
Several mechanisms have been put forward to explain the spreading of aSyn between cells. These include membrane pores (Stockl et al., 2013), passive diffusion (Ahn et al., 2006; Grozdanov and Danzer, 2018), receptor mediated endocytosis (Mao et al., 2016), through exo- and endocytosis (Lee et al., 2005), exosomal transport (Emmanouilidou et al., 2010), tunneling nanotubes (Abounit et al., 2016a,b; Dieriks et al., 2017), and the possibility of transport through carrier proteins (Sung et al., 2001; Yang et al., 2017) (Figure 2).
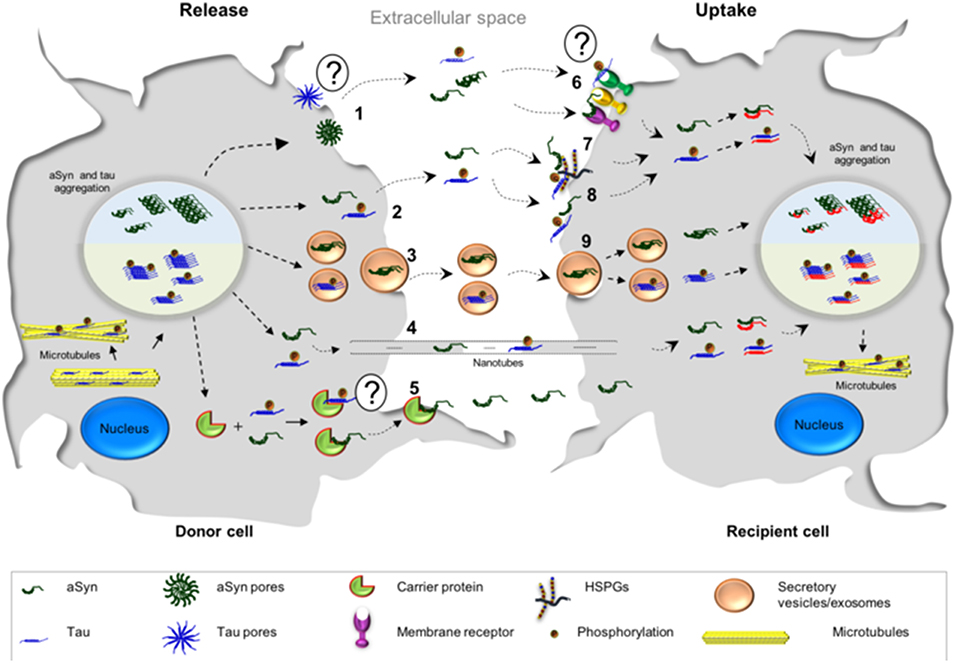
Figure 2. Possible mechanisms associated with the cell-to-cell transmission of aSyn and tau. The release of tau and aSyn is thought to take place via different mechanisms: (1) aSyn oligomers may form pore-like structures that penetrate the plasma membrane. These structures may act as non-selective channels, leading to the release of aSyn. At the present moment, there is less evidence in support of such mechanism for tau; (2, 8) Direct penetration of the plasma membrane may lead to protein release through passive diffusion and, consequently, passive uptake from the extracellular space, a common mechanism for both proteins; (3) aSyn and tau monomers and oligomers may be released in exosomes/secretory vesicles; (4) transmission of aSyn and tau may also occur via tunneling nanotubes which are membrane bridges between the cells composed by F-actin; (5) another possibility is that aSyn interacts with a possible carrier protein, which mediates the transfer in the plasma membrane and subsequently the release—it is still not known if tau could be released in this manner; (6) the release of both proteins may also take place from dying cells and the uptake of aSyn and tau could be mediated by cell surface receptors; (7) Heparan sulfate proteoglycans (HSPGs) may facilitate the internalization of aSyn and tau. Both proteins (monomers or fibrils) bind HSPGs at the cell surface, and then get internalized; (9) both proteins may be taken up by endocytosis.
aSyn is present in extracellular fluids, as it has been found in human plasma and cerebrospinal fluid and in medium from cultured human M17 neuroblastoma cell line (El-Agnaf et al., 2003). The release of aSyn has also been shown in other cell lines such as SH-SY5Y cells, H4, MES cells, and in primary neurons (Lee et al., 2005; Danzer et al., 2011; Yamada and Iwatsubo, 2018). In these cells, aSyn is released through a calcium-dependent non-conventional pathway. Treatment with compounds that interfere with the normal function of endosomes result to significant changes in the extracellular levels of aSyn. Thus, the release pathway of aSyn is dependent on the integrity of the endosomal compartment (Emmanouilidou et al., 2010; Alvarez-Erviti et al., 2011; Emmanouilidou and Vekrellis, 2016). Release can also happen through exosomes. Exosomes are vesicles of <100 nm of diameter that facilitate intercellular communication transporting proteins or RNA (Jansen et al., 2017; Mutreja and Gamblin, 2017). They can be released through budding forming a small vesicle or formed inside the multivesicular body, which fuses then with the cell membrane releasing its vesicles into the extracellular space (Beaudoin and Grondin, 1991; Denzer et al., 2000; Rustom et al., 2004). Membrane carrier proteins like the secretory carrier membrane protein 5 can also participate in the release of aSyn through exosomes (Yang et al., 2017). These release processes can happen from either the cell soma or the synaptic button, as aSyn is also transmitted trans-synaptically (Danzer et al., 2011; Freundt et al., 2012; Yamada and Iwatsubo, 2018), in processes that may or may not require axon-dendrite contacts (Freundt et al., 2012). Interestingly, mutant forms of aSyn such as the H50Q and the G51D are more prone to be released via exosomes and other types of extracellular vesicles than the wild type protein (Falcon et al., 2018). In addition, aSyn can also reach the extracellular space, not only by active mechanisms, but also passively by leakage through damaged cell membranes or by cell impairments. This process can be exacerbated by aSyn itself, as its interactions and fibrillization may disrupt cell membrane integrity (Volles and Lansbury, 2002; Chaudhary et al., 2014).
Internalization can then happen through the aforementioned mechanisms. During pinocytosis, aSyn is internalized in a dynamin-dependent process which seems to be more relevant for monomeric than for aggregated aSyn (Hansen et al., 2011). Furthermore, the endocytic process of aSyn internalization is a dynamin-dependent process, but not clathrin-dependent (Uversky, 2011a).
Tunneling nanotubes are F-actin containing membranous bridges that connect the cytoplasm of remote cells, first described in PC12 cells (Abounit and Zurzolo, 2012). Fibrillar aSyn can spread from cell to cell in a prion-like way through tunneling nanotubes by mechanisms such as intercellular trafficking of lysosomes (Abounit et al., 2016a; Dieriks et al., 2017). This happens not only among neuronal cells, but also in pericytes and astrocytes (Dieriks et al., 2017; Rostami et al., 2017), and from one type of cell to another (Sun et al., 2012).
Changes in aggregation propensity like the ones mentioned above lead to changes in the internalization and clearance of aSyn (Lee et al., 2008).
It has been proposed that aSyn can cross the cell membrane through pore-like structures such as the β-barrel voltage-dependent anion channel (Hoogerheide et al., 2017). These pores can be formed by aSyn itself in its oligomeric form, as it crosses the cell membrane leading to the formation of octameric ring structures, being the mutant A53T more prone to do it (Ma et al., 2017). The formation of these pores by aSyn is related with its binding properties to membranes and lipid layers. The association with these membranes leads to changes in membrane conductance which result to changes in its pore activity formation (Tosatto et al., 2012). The association with membranes is dependent on the presence of the KTKEGV repeat motif (Jao et al., 2004). In fact, binding of aSyn to the cell membrane causes permeabilization of the cell membrane by decreasing the lipid order (Stockl et al., 2013). This process also facilitates passive diffusion of the protein. Passive diffusion is a mechanism that allows exclusively the internalization of monomers (Lee et al., 2008). It is noteworthy that changes in the amphipatic N-terminus lead to an altered binding of aSyn to membranes. This results in abnormal vesicle interactions and changes the conformation of the vesicles, affecting aSyn spreading (Taneva et al., 2012; Dettmer et al., 2017). Out of the three known membrane proteins described to interact with aSyn, only the LAG3 and PrPC mediate in its internalization through endocytosis (Chen et al., 2015; Mao et al., 2016; De Cecco and Legname, 2018). Activation of N-Methyl-D-Aspartate receptor (NMDAR) leads to the clathrin-mediated internalization of the receptor (Chen et al., 2017). Interestingly, activation of PrPc receptor by extracellular aSyn oligomers also leads to the phosphorylation of Fyn kinase activating the NMDAR receptor in a process that is independent of pore formation and impairs hippocampal long term potentiation leading to cognitive impairment (Diogenes et al., 2012; Chen et al., 2015).
Spreading of Tau Pathology
Tau also progresses in a predictable manner throughout the CNS. In AD, tau starts its propagation in the transentorhinal region and progresses through the hippocampus, cortex and the superior temporal gyrus, finally reaching the neocortex (Uversky, 2003; Braak et al., 2006). Nonetheless, particularly in FTD cases with Pick's disease type of tau pathology, atrophy progression starts in the frontal lobe and the hippocampus, then the temporal lobe and the insula, and finally pathology reaches areas of the parietal lobe (Broe et al., 2003; Gallea et al., 2018).
In mice, tau isoforms 2N4R, 2N3R, and 0N4R were found to readily and bidirectionally cross the blood-brain barrier (Ghosh et al., 2015).
Tau propagates trans-synaptically, mostly based on connectivity and not on proximity (Ahmed et al., 2014). This propagation happens mostly through afferent connections (Iba et al., 2015) and is mediated by trans-synaptic mechanisms (Dujardin et al., 2014), where transmission was proposed to take place through exosomes (Wang et al., 2017). Although propagation happens mainly through afferent connections, small tau species can be transported anterogradely and retrogradely in neurons (Wu et al., 2013). In P301S transgenic mice, tau pre-formed fibrils lead to templated misfolding of tau in a prion-like manner along neuronal connections (Stancu et al., 2015). When injected into the brain, aggregated P301S tau present in brain homogenates from P301S mice induces the spreading of filamentous tau pathology in ALZ17 mice (Clavaguera et al., 2009). Interestingly, the P301S mutant spreads at least five times faster than the wild type (WT) tau (Kundel et al., 2018).
In normal conditions, purified human tau does not form protein assemblies (Crowther et al., 1994). Polyanionic substances, especially glycosaminoglycans such as heparin or heparan sulfate proteoglicans (HSPGs), promote tau aggregation, thereby accelerating the formation of amyloid tau fibrils (Montejo de Garcini et al., 1986; Friedhoff et al., 1998). All six tau isoforms are able to aggregate even though, in vitro, the 4R isoforms are more prone to aggregation than the 3R isoforms (Zhong et al., 2012). Once the protein forms assemblies, these can act as seeds for the generation of new assemblies, even when they are applied extracellularly, spreading subsequently to other cells (Goedert and Spillantini, 2017).
Tau spreads from cell to cell through several different putative mechanisms, similar to those proposed for aSyn (Guo and Lee, 2011; Tardivel et al., 2016; Katsinelos et al., 2018; Polanco et al., 2018) (Figure 2). A mechanism that was proposed to be more frequent for tau than for aSyn, is the internalization of the protein via macropinocytosis (Lee et al., 2008; Holmes et al., 2013) (Figure 2). Macropinocytosis is an endocytic process driven by actin and involves the formation of the macropinosome in response to the direct actions of cargo/receptor molecules that coordinate the activity and recruitment of specific effector molecules, and subsequently fuse with degradative compartments of the cell (Kirkham and Parton, 2005; Kerr and Teasdale, 2009). Different types of HSPGs have been described to facilitate cellular internalization of aSyn and tau in vitro and in vivo and blocking their expression diminishes the internalization of tau and aSyn monomer and aggregates (Holmes et al., 2013; Gerson et al., 2014; Dujardin et al., 2018). Oligomers and short fibrils which bind to membranes can be internalized through receptor-independent mechanisms. On the other hand, monomers, long fibrils, or long filaments, are more dependent on receptor-mediated mechanisms (Wu et al., 2013). Extracellular tau can also affect the accumulation of endogenous tau in a way that is dependent on the tau isoform found in the extracellular space. In addition, it has been shown that oligomeric 0N4R tau induces the accumulation of endogenous tau to a small extent, while oligomeric 0N3R, 1N3R, and 1N4R tau do not stimulate accumulation of intracellular tau (Swanson et al., 2017).
Detachment of tau from microtubules, e.g., due to hyperphosphorylation, increases the levels of free intracellular protein, which can then cross the membrane through translocation mechanisms (Katsinelos et al., 2018). Certain extracellular forms of tau, especially soluble forms composed mostly of monomers and small oligomers, appear to be cytotoxic through muscarinic receptor activation, involving the tissue non-specific alkaline phosphatase (Sebastian-Serrano et al., 2018). Extracellular tau, mainly truncated forms, can also contribute to synaptic dysfunction (Brandt et al., 1995; Sebastian-Serrano et al., 2018). In fact, at least 75% of tau in the synapse of AD patients is C-terminally truncated, and can be released from cortical synapses and affect its physiological role (Sokolow et al., 2015). Interestingly, these truncated forms have been reported to undergo truncation from the C-terminus to inner regions and have been linked to the pathogenesis of AD (Basurto-Islas et al., 2008; Garcia-Sierra et al., 2008). So far, two major sites of truncation have been studied, especially truncation at residues E391 and D421. Interestingly, the truncation at D421 is not only found in AD brains, but also in Pick's disease brains, where C-terminal truncated tau has been proposed to be the main isoform found in exosomes (Mena et al., 1995; Gamblin et al., 2003; Basurto-Islas et al., 2008; Mondragon-Rodriguez et al., 2008; Kanmert et al., 2015). Furthermore, overexpression of the projection domain of tau suppresses neuronal growth factor-induced neurite formation, contributing to synaptic dysfunction (Brandt et al., 1995).
Cell Models for Studying the Spreading of aSyn and Tau
The mechanisms by which aSyn and tau spread through the central nervous system are of utmost importance to understand the progression of PD and AD, respectively. To study these mechanisms, several cell models of aSyn and tau spreading have been used in the last years.
Different cell lines have been used to study specific mechanisms underlying the spreading of aSyn and tau pathology. For instance, the mouse neuroblastoma N2a cell line has been extensively used to study cell-to-cell spreading through exosomes (Wang et al., 2017). Other cell lines, such as the HEK293, SH-SY5Y, and B103, or primary neuronal cultures, were also used to study exosomal-mediated release, but less frequently (Alvarez-Erviti et al., 2011; Polanco et al., 2016; Ngolab et al., 2017). The cell-to-cell transmission through tunneling nanotubes has been studied in the cathecolaminergic CAD cell line (Abounit et al., 2016a). The C17.2 neural precursor cell line was used to study macropinocytosis (Holmes et al., 2013) (Tables 1, 2).
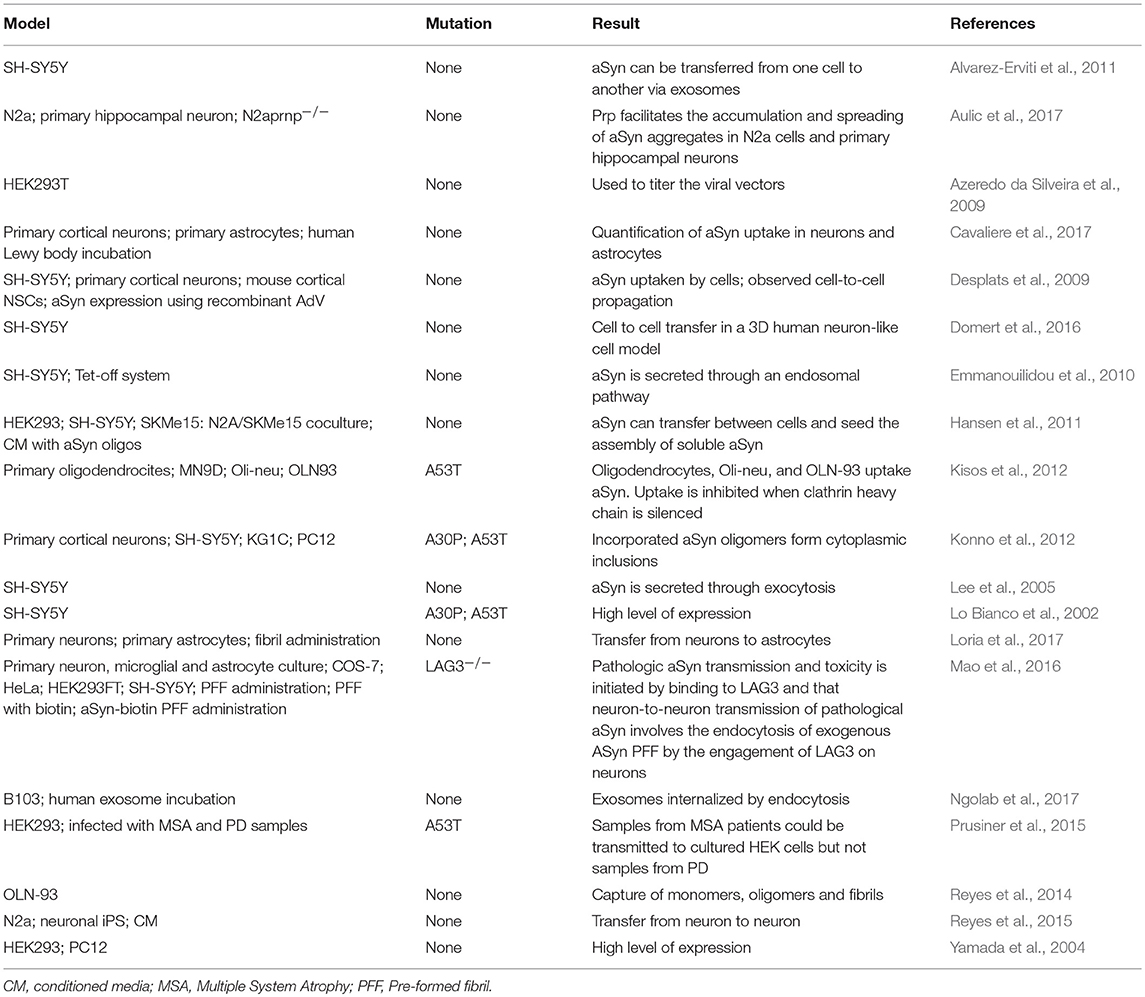
Table 1. Cell models used to study aSyn spreading.
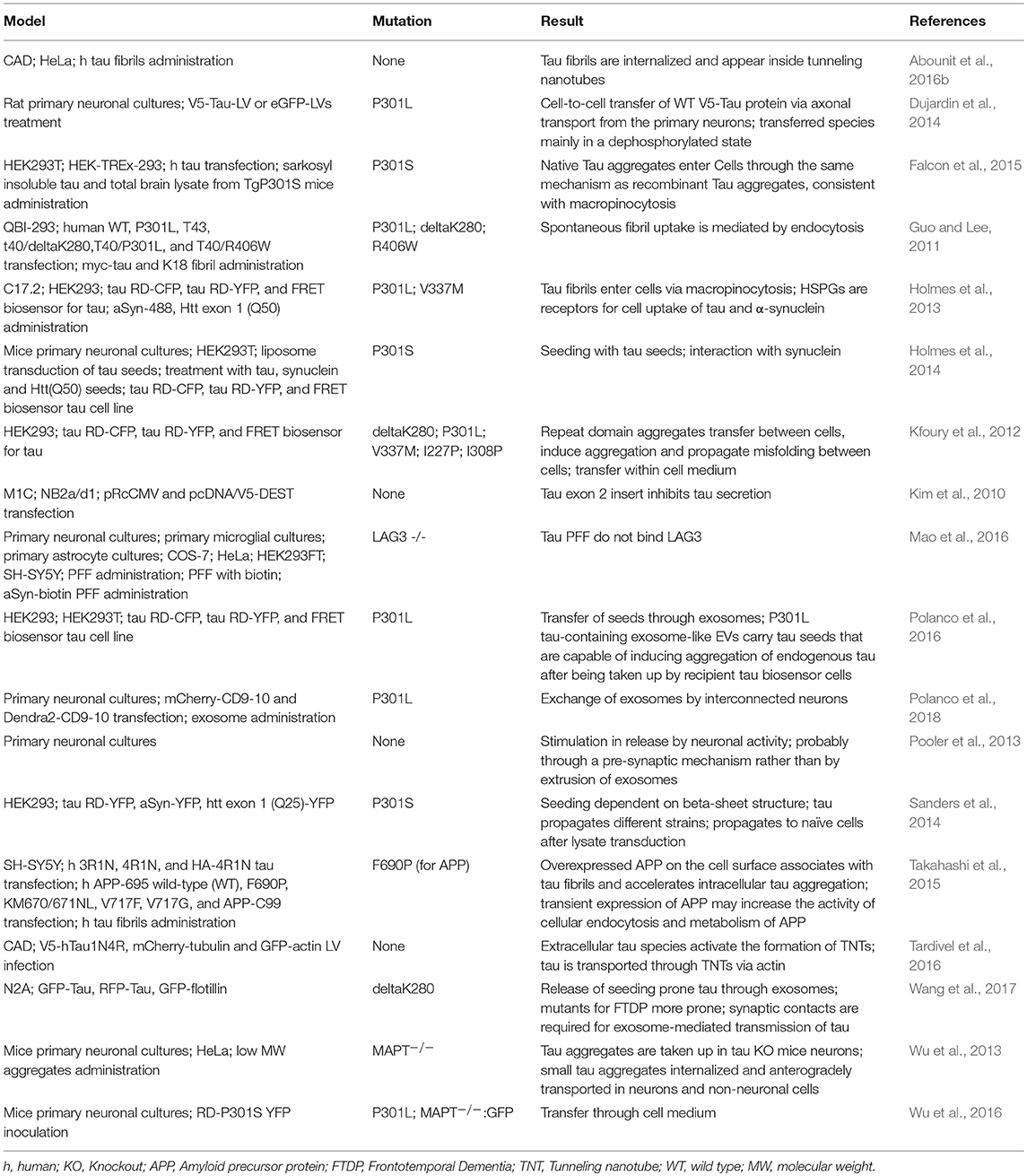
Table 2. Cell models used to study tau spreading.
Animal Models for Studying the Spreading of aSyn and Tau
In recent years, animal models based in the administration of different forms of aSyn and tau in specific brain and peripheral areas have been used (Table 3). The use of animal models enables us to address specific aspects of the spreading of aSyn or tau pathology, in particular those related to neuronal connectivity, or to the spreading between different organs (Braak and Braak, 1991; Delacourte et al., 1999; Zaccai et al., 2008). For aSyn, both mice and rats have been extensively used (Hansen et al., 2011; Masuda-Suzukake et al., 2013; Holmqvist et al., 2014; Recasens and Dehay, 2014), while in the case of tau most studies employed mice (Dujardin et al., 2014).
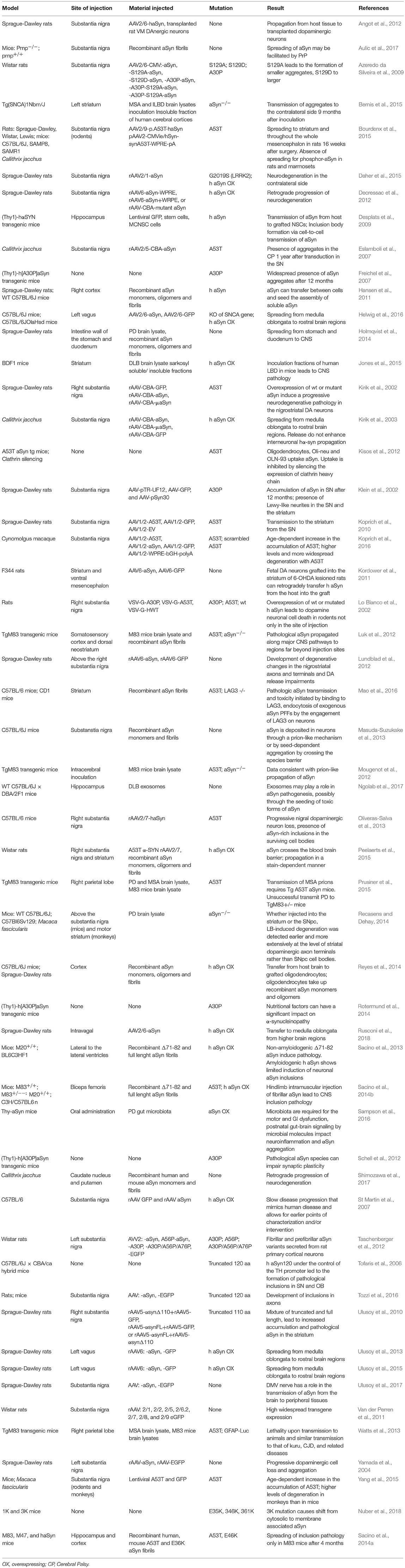
Table 3. Animal models used to study aSyn spreading.
Non-human primates have already been used to study the spreading of aSyn pathology (Kirik et al., 2003; Eslamboli et al., 2007; Shimozawa et al., 2017), but have not been reported thus far for the study of tau spreading. Interestingly, a lamprey model has been used to study the effects of several tau mutations in the progression of neurodegeneration (Lee et al., 2009).
Transgenic mice expressing mutant forms of aSyn, such as A53T and A30P, have also been used, and are proving useful for studying other synucleinopathies including MSA (Giasson et al., 2002; Prusiner et al., 2015). For tau, the rTg4510 mouse line, harboring the P301L mutation, has been widely used (Barghorn et al., 2000).
Additionally, injections of viral vectors have been also extensively used to induce aSyn and tau overexpression. In the case of aSyn, several studies reported the mimicking of relevant PD features, the progressive nature, and the spreading of pathology, in mice, rats, and non-human primates (Kirik et al., 2002, 2003; St Martin et al., 2007; Low and Aebischer, 2012). In the case of tau there are also models based on the use of viral vectors (Klein et al., 2008), including models for the study of rapid tau propagation (Asai et al., 2015) (Tables 3, 4).
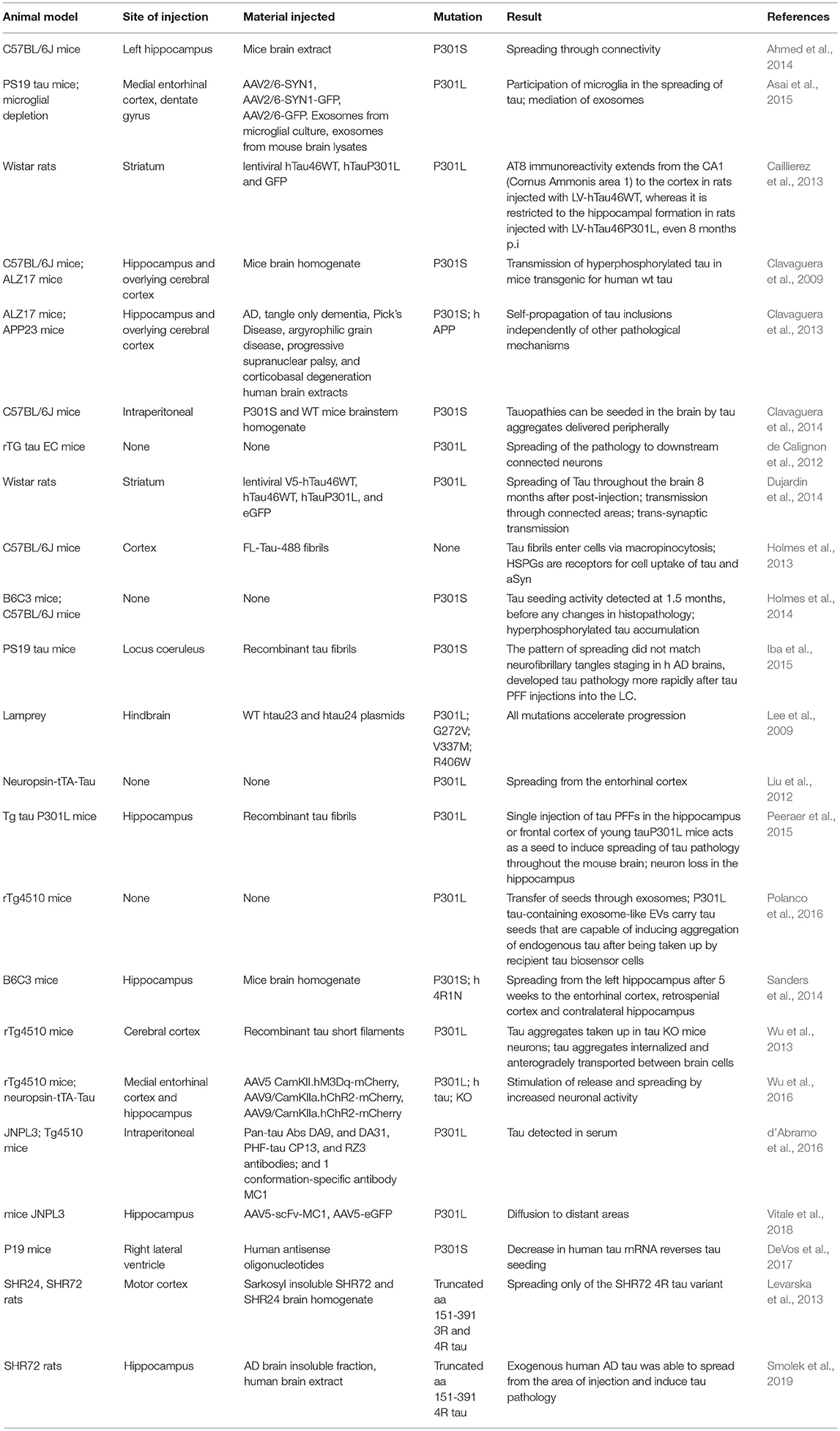
Table 4. Animal models used to study tau spreading.
Conclusions
Tau and aSyn pathologies spread in a manner that is reminiscent of the process of prion spreading in prion diseases, whereby misfolded forms of the proteins can act as templates and induce the misfolding of normally structured proteins. The altered proteins can spread from cell to cell throughout the CNS, and possibly also between different organs through neuronal connections.
In contrast to prion diseases, in AD and PD there is still no definitive evidence for horizontal transmission of tau or aSyn pathologies, as these proteins have not been shown to be infectious. Additionally, the spreading of pathology in synucleinopathies and tauopathies seems to be slower than that observed in prion diseases, but the reason for this is still unknown (Yekhlef et al., 2003; Peden et al., 2004; Schofield et al., 2005; Desplats et al., 2009; Angot et al., 2012; Reyes et al., 2014; Mabbott, 2017).
The putative mechanisms involved in the spreading of both tau and aSyn are thought to be similar. One difference is that tau appears to have greater propensity to be internalized through macropinocytosis. Another difference is that, thus far, no putative direct receptors have been involved in mediating tau internalization, whereas for aSyn several putative “receptors” have been reported.
From an anatomical point of view, aSyn and tau appear to spread from and to different locations, in patterns that are relatively predictable (McCann et al., 2016; Hoenig et al., 2018; Schwarz et al., 2018).
In conclusion, through the use of different model systems, it is becoming evident that the spreading of aSyn and tau pathologies share similar mechanisms, and there is hope that a deeper understanding of those mechanisms may lead to the identification of novel targets for therapeutic intervention in various neurodegenerative diseases.
Author Contributions
EV and AD-M reviewed the literature and contributed equally to the design and writing of the manuscript. TO reviewed the final manuscript.
Funding
TO is supported by the DFG Center for Nanoscale Microscopy and Molecular Physiology of the Brain (CNMPB) and by a grant from Fundación La Marato de TV3 (Ref. 20144330). This work has received support from the EU/EFPIA/Innovative Medicines Initiative [2] Joint Undertaking (IMPRIND grant n° 116060). AD-M is supported by a postdoctoral fellowship from the Galician Government (Programa de axuda á etapa posdoutoral, XUGA, GAIN, ED481B 2017/053).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abounit, S., Bousset, L., Loria, F., Zhu, S., de Chaumont, F., Pieri, L., et al. (2016a). Tunneling nanotubes spread fibrillar alpha-synuclein by intercellular trafficking of lysosomes. EMBO J. 35, 2120–2138. doi: 10.15252/embj.201593411
Abounit, S., Wu, J. W., Duff, K., Victoria, G. S., and Zurzolo, C. (2016b). Tunneling nanotubes: a possible highway in the spreading of tau and other prion-like proteins in neurodegenerative diseases. Prion 10, 344–351. doi: 10.1080/19336896.2016.1223003
Abounit, S., and Zurzolo, C. (2012). Wiring through tunneling nanotubes–from electrical signals to organelle transfer. J. Cell Sci. 125, 1089–1098. doi: 10.1242/jcs.083279
Ackmann, M., Wiech, H., and Mandelkow, E. (2000). Nonsaturable binding indicates clustering of tau on the microtubule surface in a paired helical filament-like conformation. J. Biol. Chem. 275, 30335–30343. doi: 10.1074/jbc.M002590200
Ahmed, Z., Cooper, J., Murray, T. K., Garn, K., McNaughton, E., Clarke, H., et al. (2014). A novel in vivo model of tau propagation with rapid and progressive neurofibrillary tangle pathology: the pattern of spread is determined by connectivity, not proximity. Acta Neuropathol. 127, 667–683. doi: 10.1007/s00401-014-1254-6
Ahn, K. J., Paik, S. R., Chung, K. C., and Kim, J. (2006). Amino acid sequence motifs and mechanistic features of the membrane translocation of alpha-synuclein. J. Neurochem. 97, 265–279. doi: 10.1111/j.1471-4159.2006.03731.x
Alvarez-Erviti, L., Seow, Y., Schapira, A. H., Gardiner, C., Sargent, I. L., Wood, M. J., et al. (2011). Lysosomal dysfunction increases exosome-mediated alpha-synuclein release and transmission. Neurobiol. Dis. 42, 360–367. doi: 10.1016/j.nbd.2011.01.029
Anderson, J. P., Walker, D. E., Goldstein, J. M., de Laat, R., Banducci, K., Caccavello, R. J., et al. (2006). Phosphorylation of Ser-129 is the dominant pathological modification of alpha-synuclein in familial and sporadic Lewy body disease. J. Biol. Chem. 281, 29739–29752. doi: 10.1074/jbc.M600933200
Angot, E., Steiner, J. A., Lema Tome, C. M., Ekstrom, P., Mattsson, B., Bjorklund, A., et al. (2012). Alpha-synuclein cell-to-cell transfer and seeding in grafted dopaminergic neurons in vivo. PLoS ONE 7:e39465. doi: 10.1371/journal.pone.0039465
Appel-Cresswell, S., Vilarino-Guell, C., Encarnacion, M., Sherman, H., Yu, I., Shah, B., et al. (2013). Alpha-synuclein p.H50Q, a novel pathogenic mutation for Parkinson's disease. Mov. Disord. 28, 811–813. doi: 10.1002/mds.25421
Asai, H., Ikezu, S., Tsunoda, S., Medalla, M., Luebke, J., Haydar, T., et al. (2015). Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci. 18, 1584–1593. doi: 10.1038/nn.4132
Aulic, S., Masperone, L., Narkiewicz, J., Isopi, E., Bistaffa, E., Ambrosetti, E., et al. (2017). alpha-synuclein amyloids hijack prion protein to gain cell entry, facilitate cell-to-cell spreading and block prion replication. Sci. Rep. 7:10050. doi: 10.1038/s41598-017-10236-x
Avila, J., Lucas, J. J., Perez, M., and Hernandez, F. (2004). Role of tau protein in both physiological and pathological conditions. Physiol. Rev. 84, 361–384. doi: 10.1152/physrev.00024.2003
Azeredo da Silveira, S., Schneider, B. L., Cifuentes-Diaz, C., Sage, D., Abbas-Terki, T., Iwatsubo, T., et al. (2009). Phosphorylation does not prompt, nor prevent, the formation of alpha-synuclein toxic species in a rat model of Parkinson's disease. Hum. Mol. Genet. 18, 872–887. doi: 10.1093/hmg/ddn417
Baba, M., Nakajo, S., Tu, P. H., Tomita, T., Nakaya, K., Lee, V. M., et al. (1998). Aggregation of alpha-synuclein in Lewy bodies of sporadic Parkinson's disease and dementia with Lewy bodies. Am. J. Pathol. 152, 879–884.
Bancher, C., Brunner, C., Lassmann, H., Budka, H., Jellinger, K., Wiche, G., et al. (1989). Accumulation of abnormally phosphorylated tau precedes the formation of neurofibrillary tangles in Alzheimer's disease. Brain Res. 477, 90–99. doi: 10.1016/0006-8993(89)91396-6
Barghorn, S., Zheng-Fischhofer, Q., Ackmann, M., Biernat, J., von Bergen, M., Mandelkow, E. M., et al. (2000). Structure, microtubule interactions, and paired helical filament aggregation by tau mutants of frontotemporal dementias. Biochemistry 39, 11714–11721. doi: 10.1021/bi000850r
Bartels, T., Ahlstrom, L. S., Leftin, A., Kamp, F., Haass, C., Brown, M. F., et al. (2010). The N-terminus of the intrinsically disordered protein alpha-synuclein triggers membrane binding and helix folding. Biophys. J. 99, 2116–2124. doi: 10.1016/j.bpj.2010.06.035
Basurto-Islas, G., Luna-Munoz, J., Guillozet-Bongaarts, A. L., Binder, L. I., Mena, R., and Garcia-Sierra, F. (2008). Accumulation of aspartic acid421- and glutamic acid391-cleaved tau in neurofibrillary tangles correlates with progression in Alzheimer disease. J. Neuropathol. Exp. Neurol. 67, 470–483. doi: 10.1097/NEN.0b013e31817275c7
Bayer, T. A., Jakala, P., Hartmann, T., Havas, L., McLean, C., Culvenor, J. G., et al. (1999). Alpha-synuclein accumulates in Lewy bodies in Parkinson's disease and dementia with Lewy bodies but not in Alzheimer's disease beta-amyloid plaque cores. Neurosci. Lett. 266, 213–216. doi: 10.1016/S0304-3940(99)00311-0
Beaudoin, A. R., and Grondin, G. (1991). Shedding of vesicular material from the cell surface of eukaryotic cells: different cellular phenomena. Biochim. Biophys. Acta 1071, 203–219. doi: 10.1016/0304-4157(91)90014-N
Bellani, S., Sousa, V. L., Ronzitti, G., Valtorta, F., Meldolesi, J., and Chieregatti, E. (2010). The regulation of synaptic function by alpha-synuclein. Commun. Integr. Biol. 3, 106–109. doi: 10.4161/cib.3.2.10964
Bellucci, A., Mercuri, N. B., Venneri, A., Faustini, G., Longhena, F., Pizzi, M., et al. (2016). Review: Parkinson's disease: from synaptic loss to connectome dysfunction. Neuropathol. Appl. Neurobiol. 42, 77–94. doi: 10.1111/nan.12297
Bellucci, A., Zaltieri, M., Navarria, L., Grigoletto, J., Missale, C., and Spano, P. (2012). From alpha-synuclein to synaptic dysfunctions: new insights into the pathophysiology of Parkinson's disease. Brain Res. 1476, 183–202. doi: 10.1016/j.brainres.2012.04.014
Bernado, P., Bertoncini, C. W., Griesinger, C., Zweckstetter, M., and Blackledge, M. (2005). Defining long-range order and local disorder in native alpha-synuclein using residual dipolar couplings. J. Am. Chem. Soc. 127, 17968–17969. doi: 10.1021/ja055538p
Bernis, M. E., Babila, J. T., Breid, S., Wusten, K. A., Wullner, U., and Tamguney, G. (2015). Prion-like propagation of human brain-derived alpha-synuclein in transgenic mice expressing human wild-type alpha-synuclein. Acta Neuropathol. Commun. 3:75. doi: 10.1186/s40478-015-0254-7
Bisaglia, M., Mammi, S., and Bubacco, L. (2009). Structural insights on physiological functions and pathological effects of alpha-synuclein. FASEB J. 23, 329–340. doi: 10.1096/fj.08-119784
Biswas, S., and Kalil, K. (2018). The microtubule-associated protein tau mediates the organization of microtubules and their dynamic exploration of actin-rich lamellipodia and filopodia of cortical growth cones. J. Neurosci. 38, 291–307. doi: 10.1523/JNEUROSCI.2281-17.2017
Bourdenx, M., Dovero, S., Engeln, M., Bido, S., Bastide, M. F., Dutheil, N., et al. (2015). Lack of additive role of ageing in nigrostriatal neurodegeneration triggered by alpha-synuclein overexpression. Acta Neuropathol. Commun. 3:46. doi: 10.1186/s40478-015-0222-2
Braak, H., Alafuzoff, I., Arzberger, T., Kretzschmar, H., and Del Tredici, K. (2006). Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 112, 389–404. doi: 10.1007/s00401-006-0127-z
Braak, H., and Braak, E. (1991). Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 82, 239–259. doi: 10.1007/BF00308809
Braak, H., Del Tredici, K., Rub, U., de Vos, R. A., Jansen Steur, E. N., and Braak, E. (2003a). Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol. Aging 24, 197–211. doi: 10.1016/S0197-4580(02)00065-9
Braak, H., Rub, U., Gai, W. P., and Del Tredici, K. (2003b). Idiopathic Parkinson's disease: possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J. Neural. Transm. 110, 517–536. doi: 10.1007/s00702-002-0808-2
Brandner, S., Raeber, A., Sailer, A., Blattler, T., Fischer, M., Weissmann, C., et al. (1996). Normal host prion protein (PrPC) is required for scrapie spread within the central nervous system. Proc. Natl. Acad. Sci. U.S.A. 93, 13148–13151. doi: 10.1073/pnas.93.23.13148
Brandt, R., and Lee, G. (1993). Functional organization of microtubule-associated protein tau. Identification of regions which affect microtubule growth, nucleation, and bundle formation in vitro. J. Biol. Chem. 268, 3414–3419.
Brandt, R., Leger, J., and Lee, G. (1995). Interaction of tau with the neural plasma membrane mediated by tau's amino-terminal projection domain. J. Cell Biol. 131, 1327–1340. doi: 10.1083/jcb.131.5.1327
Bre, M. H., and Karsenti, E. (1990). Effects of brain microtubule-associated proteins on microtubule dynamics and the nucleating activity of centrosomes. Cell Motil. Cytoskeleton 15, 88–98. doi: 10.1002/cm.970150205
Breydo, L., Wu, J. W., and Uversky, V. N. (2012). Alpha-synuclein misfolding and Parkinson's disease. Biochim. Biophys. Acta 1822, 261–285. doi: 10.1016/j.bbadis.2011.10.002
Brion, J. P., Couck, A. M., Passareiro, E., and Flament-Durand, J. (1985). Neurofibrillary tangles of Alzheimer's disease: an immunohistochemical study. J. Submicrosc. Cytol. 17, 89–96.
Brion, J. P., Flament-Durand, J., and Dustin, P. (1986). Alzheimer's disease and tau proteins. Lancet 2:1098. doi: 10.1016/S0140-6736(86)90495-2
Broe, M., Hodges, J. R., Schofield, E., Shepherd, C. E., Kril, J. J., and Halliday, G. M. (2003). Staging disease severity in pathologically confirmed cases of frontotemporal dementia. Neurology 60, 1005–1011. doi: 10.1212/01.WNL.0000052685.09194.39
Brown, D. R. (2007). Interactions between metals and alpha-synuclein–function or artefact? FEBS J. 274, 3766–3774. doi: 10.1111/j.1742-4658.2007.05917.x
Brundin, P., Melki, R., and Kopito, R. (2010). Prion-like transmission of protein aggregates in neurodegenerative diseases. Nat. Rev. Mol. Cell Biol. 11, 301–307. doi: 10.1038/nrm2873
Bu, B., Tong, X., Li, D., Hu, Y., He, W., Zhao, C., et al. (2017). N-Terminal acetylation preserves alpha-synuclein from oligomerization by blocking intermolecular hydrogen bonds. ACS Chem. Neurosci. 8, 2145–2151. doi: 10.1021/acschemneuro.7b00250
Buee, L., Bussiere, T., Buee-Scherrer, V., Delacourte, A., and Hof, P. R. (2000). Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res. Brain Res. Rev. 33, 95–130. doi: 10.1016/S0165-0173(00)00019-9
Burre, J., Sharma, M., and Sudhof, T. C. (2014). alpha-Synuclein assembles into higher-order multimers upon membrane binding to promote SNARE complex formation. Proc. Natl. Acad. Sci. U.S.A. 111, E4274–E4283. doi: 10.1073/pnas.1416598111
Cabrales Fontela, Y., Kadavath, H., Biernat, J., Riedel, D., Mandelkow, E., and Zweckstetter, M. (2017). Multivalent cross-linking of actin filaments and microtubules through the microtubule-associated protein Tau. Nat. Commun. 8:1981. doi: 10.1038/s41467-017-02230-8
Caillierez, R., Begard, S., Lecolle, K., Deramecourt, V., Zommer, N., Dujardin, S., et al. (2013). Lentiviral delivery of the human wild-type tau protein mediates a slow and progressive neurodegenerative tau pathology in the rat brain. Mol. Ther. 21, 1358–1368. doi: 10.1038/mt.2013.66
Camero, S., Benitez, M. J., Cuadros, R., Hernandez, F., Avila, J., and Jimenez, J. S. (2014). Thermodynamics of the interaction between Alzheimer's disease related tau protein and DNA. PLoS ONE 9:e104690. doi: 10.1371/journal.pone.0104690
Cavaliere, F., Cerf, L., Dehay, B., Ramos-Gonzalez, P., De Giorgi, F., Bourdenx, M., et al. (2017). In vitro alpha-synuclein neurotoxicity and spreading among neurons and astrocytes using Lewy body extracts from Parkinson disease brains. Neurobiol. Dis. 103, 101–112. doi: 10.1016/j.nbd.2017.04.011
Chaudhary, H., Stefanovic, A. N., Subramaniam, V., and Claessens, M. M. (2014). Membrane interactions and fibrillization of alpha-synuclein play an essential role in membrane disruption. FEBS Lett. 588, 4457–4463. doi: 10.1016/j.febslet.2014.10.016
Chen, P. H., Bendris, N., Hsiao, Y. J., Reis, C. R., Mettlen, M., Chen, H. Y., et al. (2017). Crosstalk between CLCb/dyn1-mediated adaptive clathrin-mediated endocytosis and epidermal growth factor receptor signaling increases metastasis. Dev. Cell 40, 278–288.e275. doi: 10.1016/j.devcel.2017.01.007
Chen, S. W., Drakulic, S., Deas, E., Ouberai, M., Aprile, F. A., Arranz, R., et al. (2015). Structural characterization of toxic oligomers that are kinetically trapped during alpha-synuclein fibril formation. Proc. Natl. Acad. Sci. U.S.A. 112, E1994–E2003. doi: 10.1073/pnas.1421204112
Chen, X., de Silva, H. A., Pettenati, M. J., Rao, P. N., St George-Hyslop, P., Roses, A. D., et al. (1995). The human NACP/alpha-synuclein gene: chromosome assignment to 4q21.3-q22 and TaqI RFLP analysis. Genomics 26, 425–427. doi: 10.1016/0888-7543(95)80237-G
Clavaguera, F., Akatsu, H., Fraser, G., Crowther, R. A., Frank, S., Hench, J., et al. (2013). Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc. Natl. Acad. Sci. U.S.A. 110, 9535–9540. doi: 10.1073/pnas.1301175110
Clavaguera, F., Bolmont, T., Crowther, R. A., Abramowski, D., Frank, S., Probst, A., et al. (2009). Transmission and spreading of tauopathy in transgenic mouse brain. Nat. Cell Biol. 11, 909–913. doi: 10.1038/ncb1901
Clavaguera, F., Hench, J., Lavenir, I., Schweighauser, G., Frank, S., Goedert, M., et al. (2014). Peripheral administration of tau aggregates triggers intracerebral tauopathy in transgenic mice. Acta Neuropathol. 127, 299–301. doi: 10.1007/s00401-013-1231-5
Coelho-Cerqueira, E., Carmo-Goncalves, P., Pinheiro, A. S., Cortines, J., and Follmer, C. (2013). alpha-Synuclein as an intrinsically disordered monomer–fact or artefact? FEBS J. 280, 4915–4927. doi: 10.1111/febs.12471
Costanzo, M., and Zurzolo, C. (2013). The cell biology of prion-like spread of protein aggregates: mechanisms and implication in neurodegeneration. Biochem. J. 452, 1–17. doi: 10.1042/BJ20121898
Crowther, R. A., Olesen, O. F., Smith, M. J., Jakes, R., and Goedert, M. (1994). Assembly of Alzheimer-like filaments from full-length tau protein. FEBS Lett. 337, 135–138. doi: 10.1016/0014-5793(94)80260-2
d'Abramo, C., Acker, C. M., Schachter, J. B., Terracina, G., Wang, X., Forest, S. K., et al. (2016). Detecting tau in serum of transgenic animal models after tau immunotherapy treatment. Neurobiol. Aging 37, 58–65. doi: 10.1016/j.neurobiolaging.2015.09.017
Daher, J. P., Abdelmotilib, H. A., Hu, X., Volpicelli-Daley, L. A., Moehle, M. S., Fraser, K. B., et al. (2015). Leucine-rich Repeat Kinase 2 (LRRK2) pharmacological inhibition abates alpha-synuclein gene-induced neurodegeneration. J. Biol. Chem. 290, 19433–19444. doi: 10.1074/jbc.M115.660001
Danzer, K. M., Ruf, W. P., Putcha, P., Joyner, D., Hashimoto, T., Glabe, C., et al. (2011). Heat-shock protein 70 modulates toxic extracellular alpha-synuclein oligomers and rescues trans-synaptic toxicity. FASEB J. 25, 326–336. doi: 10.1096/fj.10-164624
Davidson, W. S., Jonas, A., Clayton, D. F., and George, J. M. (1998). Stabilization of alpha-synuclein secondary structure upon binding to synthetic membranes. J. Biol. Chem. 273, 9443–9449. doi: 10.1074/jbc.273.16.9443
de Calignon, A., Polydoro, M., Suarez-Calvet, M., William, C., Adamowicz, D. H., Kopeikina, K. J., et al. (2012). Propagation of tau pathology in a model of early Alzheimer's disease. Neuron 73, 685–697. doi: 10.1016/j.neuron.2011.11.033
De Cecco, E., and Legname, G. (2018). The role of the prion protein in the internalization of alpha-synuclein amyloids. Prion 12, 23–27. doi: 10.1080/19336896.2017.1423186
Decressac, M., Mattsson, B., Lundblad, M., Weikop, P., and Bjorklund, A. (2012). Progressive neurodegenerative and behavioural changes induced by AAV-mediated overexpression of alpha-synuclein in midbrain dopamine neurons. Neurobiol. Dis. 45, 939–953. doi: 10.1016/j.nbd.2011.12.013
Delacourte, A., David, J. P., Sergeant, N., Buee, L., Wattez, A., Vermersch, P., et al. (1999). The biochemical pathway of neurofibrillary degeneration in aging and Alzheimer's disease. Neurology 52, 1158–1165. doi: 10.1212/WNL.52.6.1158
Denzer, K., Kleijmeer, M. J., Heijnen, H. F., Stoorvogel, W., and Geuze, H. J. (2000). Exosome: from internal vesicle of the multivesicular body to intercellular signaling device. J. Cell Sci. 113(Pt 19), 3365–3374.
Desplats, P., Lee, H. J., Bae, E. J., Patrick, C., Rockenstein, E., Crews, L., et al. (2009). Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc. Natl. Acad. Sci. U.S.A. 106, 13010–13015. doi: 10.1073/pnas.0903691106
Dettmer, U., Ramalingam, N., von Saucken, V. E., Kim, T. E., Newman, A. J., Terry-Kantor, E., et al. (2017). Loss of native alpha-synuclein multimerization by strategically mutating its amphipathic helix causes abnormal vesicle interactions in neuronal cells. Hum. Mol. Genet. 26, 3466–3481. doi: 10.1093/hmg/ddx227
DeVos, S. L., Miller, R. L., Schoch, K. M., Holmes, B. B., Kebodeaux, C. S., Wegener, A. J., et al. (2017). Tau reduction prevents neuronal loss and reverses pathological tau deposition and seeding in mice with tauopathy. Sci. Transl. Med. 9:eaag0481. doi: 10.1126/scitranslmed.aag0481
Dieriks, B. V., Park, T. I., Fourie, C., Faull, R. L., Dragunow, M., and Curtis, M. A. (2017). alpha-synuclein transfer through tunneling nanotubes occurs in SH-SY5Y cells and primary brain pericytes from Parkinson's disease patients. Sci. Rep. 7:42984. doi: 10.1038/srep42984
Diogenes, M. J., Dias, R. B., Rombo, D. M., Vicente Miranda, H., Maiolino, F., Guerreiro, P., et al. (2012). Extracellular alpha-synuclein oligomers modulate synaptic transmission and impair LTP via NMDA-receptor activation. J. Neurosci. 32, 11750–11762. doi: 10.1523/JNEUROSCI.0234-12.2012
Domert, J., Sackmann, C., Severinsson, E., Agholme, L., Bergstrom, J., Ingelsson, M., et al. (2016). Aggregated alpha-synuclein transfer efficiently between cultured human neuron-like cells and localize to lysosomes. PLoS ONE 11:e0168700. doi: 10.1371/journal.pone.0168700
Drubin, D. G., and Kirschner, M. W. (1986). Tau protein function in living cells. J. Cell Biol. 103, 2739–2746. doi: 10.1083/jcb.103.6.2739
Dujardin, S., Begard, S., Caillierez, R., Lachaud, C., Carrier, S., Lieger, S., et al. (2018). Different tau species lead to heterogeneous tau pathology propagation and misfolding. Acta Neuropathol. Commun. 6:132. doi: 10.1186/s40478-018-0637-7
Dujardin, S., Lecolle, K., Caillierez, R., Begard, S., Zommer, N., Lachaud, C., et al. (2014). Neuron-to-neuron wild-type Tau protein transfer through a trans-synaptic mechanism: relevance to sporadic tauopathies. Acta Neuropathol. Commun. 2:14. doi: 10.1186/2051-5960-2-14
Eisele, Y. S., Fritschi, S. K., Hamaguchi, T., Obermuller, U., Fuger, P., Skodras, A., et al. (2014). Multiple factors contribute to the peripheral induction of cerebral beta-amyloidosis. J. Neurosci. 34, 10264–10273. doi: 10.1523/JNEUROSCI.1608-14.2014
El-Agnaf, O. M., Jakes, R., Curran, M. D., Middleton, D., Ingenito, R., Bianchi, E., et al. (1998). Aggregates from mutant and wild-type alpha-synuclein proteins and NAC peptide induce apoptotic cell death in human neuroblastoma cells by formation of beta-sheet and amyloid-like filaments. FEBS Lett. 440, 71–75. doi: 10.1016/S0014-5793(98)01418-5
El-Agnaf, O. M., Salem, S. A., Paleologou, K. E., Cooper, L. J., Fullwood, N. J., Gibson, M. J., et al. (2003). Alpha-synuclein implicated in Parkinson's disease is present in extracellular biological fluids, including human plasma. FASEB J. 17, 1945–1947. doi: 10.1096/fj.03-0098fje
Eliezer, D. (2009). Biophysical characterization of intrinsically disordered proteins. Curr. Opin. Struct. Biol. 19, 23–30. doi: 10.1016/j.sbi.2008.12.004
Emmanouilidou, E., Melachroinou, K., Roumeliotis, T., Garbis, S. D., Ntzouni, M., Margaritis, L. H., et al. (2010). Cell-produced alpha-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J. Neurosci. 30, 6838–6851. doi: 10.1523/JNEUROSCI.5699-09.2010
Emmanouilidou, E., and Vekrellis, K. (2016). Exocytosis and spreading of normal and aberrant alpha-synuclein. Brain Pathol. 26, 398–403. doi: 10.1111/bpa.12373
Eslamboli, A., Romero-Ramos, M., Burger, C., Bjorklund, T., Muzyczka, N., Mandel, R. J., et al. (2007). Long-term consequences of human alpha-synuclein overexpression in the primate ventral midbrain. Brain 130, 799–815. doi: 10.1093/brain/awl382
Fabelo, N., Martin, V., Santpere, G., Marin, R., Torrent, L., Ferrer, I., et al. (2011). Severe alterations in lipid composition of frontal cortex lipid rafts from Parkinson's disease and incidental Parkinson's disease. Mol. Med. 17, 1107–1118. doi: 10.2119/molmed.2011.00119
Falcon, B., Cavallini, A., Angers, R., Glover, S., Murray, T. K., Barnham, L., et al. (2015). Conformation determines the seeding potencies of native and recombinant Tau aggregates. J. Biol. Chem. 290, 1049–1065. doi: 10.1074/jbc.M114.589309
Falcon, B., Zhang, W., Murzin, A. G., Murshudov, G., Garringer, H. J., Vidal, R., et al. (2018). Structures of filaments from Pick's disease reveal a novel tau protein fold. Nature 561, 137–140. doi: 10.1038/s41586-018-0454-y
Fortin, D. L., Troyer, M. D., Nakamura, K., Kubo, S., Anthony, M. D., and Edwards, R. H. (2004). Lipid rafts mediate the synaptic localization of alpha-synuclein. J. Neurosci. 24, 6715–6723. doi: 10.1523/JNEUROSCI.1594-04.2004
Freichel, C., Neumann, M., Ballard, T., Muller, V., Woolley, M., Ozmen, L., et al. (2007). Age-dependent cognitive decline and amygdala pathology in alpha-synuclein transgenic mice. Neurobiol. Aging 28, 1421–1435. doi: 10.1016/j.neurobiolaging.2006.06.013
Freundt, E. C., Maynard, N., Clancy, E. K., Roy, S., Bousset, L., Sourigues, Y., et al. (2012). Neuron-to-neuron transmission of alpha-synuclein fibrils through axonal transport. Ann. Neurol. 72, 517–524. doi: 10.1002/ana.23747
Friedhoff, P., von Bergen, M., Mandelkow, E. M., Davies, P., and Mandelkow, E. (1998). A nucleated assembly mechanism of Alzheimer paired helical filaments. Proc. Natl. Acad. Sci. U.S.A. 95, 15712–15717. doi: 10.1073/pnas.95.26.15712
Fujiwara, H., Hasegawa, M., Dohmae, N., Kawashima, A., Masliah, E., Goldberg, M. S., et al. (2002). alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell Biol. 4, 160–164. doi: 10.1038/ncb748
Gajdusek, D. C., Gibbs, C. J., and Alpers, M. (1966). Experimental transmission of a Kuru-like syndrome to chimpanzees. Nature 209, 794–796. doi: 10.1038/209794a0
Gallea, J. I., Ambroggio, E. E., Vilcaes, A. A., James, N. G., Jameson, D. M., and Celej, M. S. (2018). Amyloid oligomerization of the Parkinson's disease related protein alpha-synuclein impacts on its curvature-membrane sensitivity. J. Neurochem. 147, 541–556. doi: 10.1111/jnc.14573
Gamblin, T. C., Chen, F., Zambrano, A., Abraha, A., Lagalwar, S., Guillozet, A. L., et al. (2003). Caspase cleavage of tau: linking amyloid and neurofibrillary tangles in Alzheimer's disease. Proc. Natl. Acad. Sci. U.S.A. 100, 10032–10037. doi: 10.1073/pnas.1630428100
Garcia-Sierra, F., Mondragon-Rodriguez, S., and Basurto-Islas, G. (2008). Truncation of tau protein and its pathological significance in Alzheimer's disease. J. Alzheimers. Dis. 14, 401–409. doi: 10.3233/JAD-2008-14407
Georgieva, E. R., Ramlall, T. F., Borbat, P. P., Freed, J. H., and Eliezer, D. (2008). Membrane-bound alpha-synuclein forms an extended helix: long-distance pulsed ESR measurements using vesicles, bicelles, and rodlike micelles. J. Am. Chem. Soc. 130, 12856–12857. doi: 10.1021/ja804517m
Gerson, J. E., Sengupta, U., Lasagna-Reeves, C. A., Guerrero-Munoz, M. J., Troncoso, J., and Kayed, R. (2014). Characterization of tau oligomeric seeds in progressive supranuclear palsy. Acta Neuropathol. Commun. 2:73. doi: 10.1186/2051-5960-2-73
Ghag, G., Bhatt, N., Cantu, D. V., Guerrero-Munoz, M. J., Ellsworth, A., Sengupta, U., et al. (2018). Soluble tau aggregates, not large fibrils, are the toxic species that display seeding and cross-seeding behavior. Protein Sci. 27, 1901–1909. doi: 10.1002/pro.3499
Ghosh, D., Singh, P. K., Sahay, S., Jha, N. N., Jacob, R. S., Sen, S., et al. (2015). Structure based aggregation studies reveal the presence of helix-rich intermediate during alpha-Synuclein aggregation. Sci. Rep. 5:9228. doi: 10.1038/srep09228
Giasson, B. I., Duda, J. E., Quinn, S. M., Zhang, B., Trojanowski, J. Q., and Lee, V. M. (2002). Neuronal alpha-synucleinopathy with severe movement disorder in mice expressing A53T human alpha-synuclein. Neuron 34, 521–533. doi: 10.1016/S0896-6273(02)00682-7
Giasson, B. I., Forman, M. S., Higuchi, M., Golbe, L. I., Graves, C. L., Kotzbauer, P. T., et al. (2003). Initiation and synergistic fibrillization of tau and alpha-synuclein. Science 300, 636–640. doi: 10.1126/science.1082324
Giasson, B. I., Murray, I. V., Trojanowski, J. Q., and Lee, V. M. (2001). A hydrophobic stretch of 12 amino acid residues in the middle of alpha-synuclein is essential for filament assembly. J. Biol. Chem. 276, 2380–2386. doi: 10.1074/jbc.M008919200
Goedert, M. (2015). NEURODEGENERATION. Alzheimer's and Parkinson's diseases: the prion concept in relation to assembled Abeta, tau, and alpha-synuclein. Science 349:1255555. doi: 10.1126/science.1255555
Goedert, M., and Spillantini, M. G. (2017). Propagation of Tau aggregates. Mol. Brain 10:18. doi: 10.1186/s13041-017-0298-7
Goode, B. L., and Feinstein, S. C. (1994). Identification of a novel microtubule binding and assembly domain in the developmentally regulated inter-repeat region of tau. J. Cell Biol. 124, 769–782. doi: 10.1083/jcb.124.5.769
Grozdanov, V., and Danzer, K. M. (2018). Release and uptake of pathologic alpha-synuclein. Cell Tissue Res. 373, 175–182. doi: 10.1007/s00441-017-2775-9
Grundke-Iqbal, I., Iqbal, K., Quinlan, M., Tung, Y. C., Zaidi, M. S., and Wisniewski, H. M. (1986b). Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J. Biol. Chem. 261, 6084–6089.
Grundke-Iqbal, I., Iqbal, K., Tung, Y. C., Quinlan, M., Wisniewski, H. M., and Binder, L. I. (1986a). Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc. Natl. Acad. Sci. U.S.A. 83, 4913–4917. doi: 10.1073/pnas.83.13.4913
Guo, J. L., Covell, D. J., Daniels, J. P., Iba, M., Stieber, A., Zhang, B., et al. (2013). Distinct alpha-synuclein strains differentially promote tau inclusions in neurons. Cell 154, 103–117. doi: 10.1016/j.cell.2013.05.057
Guo, J. L., and Lee, V. M. (2011). Seeding of normal Tau by pathological Tau conformers drives pathogenesis of Alzheimer-like tangles. J. Biol. Chem. 286, 15317–15331. doi: 10.1074/jbc.M110.209296
Haase, C., Stieler, J. T., Arendt, T., and Holzer, M. (2004). Pseudophosphorylation of tau protein alters its ability for self-aggregation. J. Neurochem. 88, 1509–1520. doi: 10.1046/j.1471-4159.2003.02287.x
Hansen, C., Angot, E., Bergstrom, A. L., Steiner, J. A., Pieri, L., Paul, G., et al. (2011). alpha-Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. J. Clin. Invest. 121, 715–725. doi: 10.1172/JCI43366
Hellstrand, E., Nowacka, A., Topgaard, D., Linse, S., and Sparr, E. (2013). Membrane lipid co-aggregation with alpha-synuclein fibrils. PLoS ONE 8:e77235. doi: 10.1371/journal.pone.0077235
Helwig, M., Klinkenberg, M., Rusconi, R., Musgrove, R. E., Majbour, N. K., El-Agnaf, O. M., et al. (2016). Brain propagation of transduced alpha-synuclein involves non-fibrillar protein species and is enhanced in alpha-synuclein null mice. Brain 139, 856–870. doi: 10.1093/brain/awv376
Hoenig, M. C., Bischof, G. N., Seemiller, J., Hammes, J., Kukolja, J., Onur, O. A., et al. (2018). Networks of tau distribution in Alzheimer's disease. Brain 141, 568–581. doi: 10.1093/brain/awx353
Holmes, B. B., DeVos, S. L., Kfoury, N., Li, M., Jacks, R., Yanamandra, K., et al. (2013). Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc. Natl. Acad. Sci. U.S.A. 110, E3138–E3147. doi: 10.1073/pnas.1301440110
Holmes, B. B., Furman, J. L., Mahan, T. E., Yamasaki, T. R., Mirbaha, H., Eades, W. C., et al. (2014). Proteopathic tau seeding predicts tauopathy in vivo. Proc. Natl. Acad. Sci. U.S.A. 111, E4376–E4385. doi: 10.1073/pnas.1411649111
Holmqvist, S., Chutna, O., Bousset, L., Aldrin-Kirk, P., Li, W., Bjorklund, T., et al. (2014). Direct evidence of Parkinson pathology spread from the gastrointestinal tract to the brain in rats. Acta Neuropathol. 128, 805–820. doi: 10.1007/s00401-014-1343-6
Hoogerheide, D. P., Gurnev, P. A., Rostovtseva, T. K., and Bezrukov, S. M. (2017). Mechanism of alpha-synuclein translocation through a VDAC nanopore revealed by energy landscape modeling of escape time distributions. Nanoscale 9, 183–192. doi: 10.1039/C6NR08145B
Hoyer, W., Cherny, D., Subramaniam, V., and Jovin, T. M. (2004). Rapid self-assembly of alpha-synuclein observed by in situ atomic force microscopy. J. Mol. Biol. 340, 127–139. doi: 10.1016/j.jmb.2004.04.051
Huang, Y., Wu, Z., and Zhou, B. (2016). Behind the curtain of tauopathy: a show of multiple players orchestrating tau toxicity. Cell. Mol. Life Sci. 73, 1–21. doi: 10.1007/s00018-015-2042-8
Iba, M., McBride, J. D., Guo, J. L., Zhang, B., Trojanowski, J. Q., and Lee, V. M. (2015). Tau pathology spread in PS19 tau transgenic mice following locus coeruleus (LC) injections of synthetic tau fibrils is determined by the LC's afferent and efferent connections. Acta Neuropathol. 130, 349–362. doi: 10.1007/s00401-015-1458-4
Iqbal, K., Liu, F., and Gong, C. X. (2016). Tau and neurodegenerative disease: the story so far. Nat. Rev. Neurol. 12, 15–27. doi: 10.1038/nrneurol.2015.225
Iqbal, K., Liu, F., Gong, C. X., and Grundke-Iqbal, I. (2010). Tau in Alzheimer disease and related tauopathies. Curr. Alzheimer Res. 7, 656–664. doi: 10.2174/156720510793611592
Iwai, A., Masliah, E., Yoshimoto, M., Ge, N., Flanagan, L., de Silva, H. A., et al. (1995). The precursor protein of non-A beta component of Alzheimer's disease amyloid is a presynaptic protein of the central nervous system. Neuron 14, 467–475. doi: 10.1016/0896-6273(95)90302-X
Iyer, A., Roeters, S. J., Schilderink, N., Hommersom, B., Heeren, R. M., Woutersen, S., et al. (2016). The impact of N-terminal acetylation of alpha-synuclein on phospholipid membrane binding and fibril structure. J. Biol. Chem. 291, 21110–21122. doi: 10.1074/jbc.M116.726612
Jansen, S., Melkova, K., Trosanova, Z., Hanakova, K., Zachrdla, M., Novacek, J., et al. (2017). Quantitative mapping of microtubule-associated protein 2c (MAP2c) phosphorylation and regulatory protein 14-3-3zeta-binding sites reveals key differences between MAP2c and its homolog Tau. J. Biol. Chem. 292, 6715–6727. doi: 10.1074/jbc.A116.771097
Jao, C. C., Der-Sarkissian, A., Chen, J., and Langen, R. (2004). Structure of membrane-bound alpha-synuclein studied by site-directed spin labeling. Proc. Natl. Acad. Sci. U.S.A. 101, 8331–8336. doi: 10.1073/pnas.0400553101
Jao, C. C., Hegde, B. G., Chen, J., Haworth, I. S., and Langen, R. (2008). Structure of membrane-bound alpha-synuclein from site-directed spin labeling and computational refinement. Proc. Natl. Acad. Sci. U.S.A. 105, 19666–19671. doi: 10.1073/pnas.0807826105
Jo, E., McLaurin, J., Yip, C. M., St George-Hyslop, P., and Fraser, P. E. (2000). alpha-Synuclein membrane interactions and lipid specificity. J. Biol. Chem. 275, 34328–34334. doi: 10.1074/jbc.M004345200
Jones, D. R., Delenclos, M., Baine, A. T., DeTure, M., Murray, M. E., Dickson, D. W., et al. (2015). Transmission of soluble and insoluble alpha-synuclein to mice. J. Neuropathol. Exp. Neurol. 74, 1158–1169. doi: 10.1097/NEN.0000000000000262
Jucker, M., and Walker, L. C. (2013). Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 501, 45–51. doi: 10.1038/nature12481
Kanmert, D., Cantlon, A., Muratore, C. R., Jin, M., O'Malley, T. T., Lee, G., et al. (2015). C-terminally truncated forms of tau, but not full-length tau or its C-terminal fragments, are released from neurons independently of cell death. J. Neurosci. 35, 10851–10865. doi: 10.1523/JNEUROSCI.0387-15.2015
Karlawish, J., Jack, C. R. Jr., Rocca, W. A., Snyder, H. M., and Carrillo, M. C. (2017). Alzheimer's disease: the next frontier-special report 2017. Alzheimers Dement. 13, 374–380. doi: 10.1016/j.jalz.2017.02.006
Katsinelos, T., Zeitler, M., Dimou, E., Karakatsani, A., Muller, H. M., Nachman, E., et al. (2018). Unconventional secretion mediates the trans-cellular spreading of tau. Cell Rep. 23, 2039–2055. doi: 10.1016/j.celrep.2018.04.056
Kerr, M. C., and Teasdale, R. D. (2009). Defining macropinocytosis. Traffic 10, 364–371. doi: 10.1111/j.1600-0854.2009.00878.x
Kfoury, N., Holmes, B. B., Jiang, H., Holtzman, D. M., and Diamond, M. I. (2012). Trans-cellular propagation of Tau aggregation by fibrillar species. J. Biol. Chem. 287, 19440–19451. doi: 10.1074/jbc.M112.346072
Kim, W., Lee, S., and Hall, G. F. (2010). Secretion of human tau fragments resembling CSF-tau in Alzheimer's disease is modulated by the presence of the exon 2 insert. FEBS Lett. 584, 3085–3088. doi: 10.1016/j.febslet.2010.05.042
Kirik, D., Annett, L. E., Burger, C., Muzyczka, N., Mandel, R. J., and Bjorklund, A. (2003). Nigrostriatal alpha-synucleinopathy induced by viral vector-mediated overexpression of human alpha-synuclein: a new primate model of Parkinson's disease. Proc. Natl. Acad. Sci. U.S.A. 100, 2884–2889. doi: 10.1073/pnas.0536383100
Kirik, D., Rosenblad, C., Burger, C., Lundberg, C., Johansen, T. E., Muzyczka, N., et al. (2002). Parkinson-like neurodegeneration induced by targeted overexpression of alpha-synuclein in the nigrostriatal system. J. Neurosci. 22, 2780–2791. doi: 10.1523/JNEUROSCI.22-07-02780.2002
Kirkham, M., and Parton, R. G. (2005). Clathrin-independent endocytosis: new insights into caveolae and non-caveolar lipid raft carriers. Biochim. Biophys. Acta 1746, 349–363. doi: 10.1016/j.bbamcr.2005.11.005
Kisos, H., Pukass, K., Ben-Hur, T., Richter-Landsberg, C., and Sharon, R. (2012). Increased neuronal alpha-synuclein pathology associates with its accumulation in oligodendrocytes in mice modeling alpha-synucleinopathies. PLoS ONE 7:e46817. doi: 10.1371/journal.pone.0046817
Kizhakke, P. A., Olakkaran, S., Antony, A., Tilagul, K. S., and Hunasanahally, P. G. (2017). Convolvulus pluricaulis (Shankhapushpi) ameliorates human microtubule-associated protein tau (hMAPtau) induced neurotoxicity in Alzheimer's disease Drosophila model. J Chem Neuroanat. 95, 115–122. doi: 10.1016/j.jchemneu.2017.10.002
Klein, R. L., Dayton, R. D., Tatom, J. B., Henderson, K. M., and Henning, P. P. (2008). AAV8, 9, Rh10, Rh43 vector gene transfer in the rat brain: effects of serotype, promoter and purification method. Mol. Ther. 16, 89–96. doi: 10.1038/sj.mt.6300331
Klein, R. L., King, M. A., Hamby, M. E., and Meyer, E. M. (2002). Dopaminergic cell loss induced by human A30P alpha-synuclein gene transfer to the rat substantia nigra. Hum. Gene Ther. 13, 605–612. doi: 10.1089/10430340252837206
Kolarova, M., Garcia-Sierra, F., Bartos, A., Ricny, J., and Ripova, D. (2012). Structure and pathology of tau protein in Alzheimer disease. Int. J. Alzheimers. Dis. 2012:731526. doi: 10.1155/2012/731526
Konno, M., Hasegawa, T., Baba, T., Miura, E., Sugeno, N., Kikuchi, A., et al. (2012). Suppression of dynamin GTPase decreases alpha-synuclein uptake by neuronal and oligodendroglial cells: a potent therapeutic target for synucleinopathy. Mol. Neurodegener. 7:38. doi: 10.1186/1750-1326-7-38
Kopke, E., Tung, Y. C., Shaikh, S., Alonso, A. C., Iqbal, K., and Grundke-Iqbal, I. (1993). Microtubule-associated protein tau. Abnormal phosphorylation of a non-paired helical filament pool in Alzheimer disease. J. Biol. Chem. 268, 24374–24384.
Koprich, J. B., Johnston, T. H., Reyes, G., Omana, V., and Brotchie, J. M. (2016). Towards a non-human primate model of alpha-synucleinopathy for development of therapeutics for Parkinson's disease: optimization of AAV1/2 delivery parameters to drive sustained expression of alpha synuclein and dopaminergic degeneration in Macaque. PLoS ONE 11:e0167235. doi: 10.1371/journal.pone.0167235
Koprich, J. B., Johnston, T. H., Reyes, M. G., Sun, X., and Brotchie, J. M. (2010). Expression of human A53T alpha-synuclein in the rat substantia nigra using a novel AAV1/2 vector produces a rapidly evolving pathology with protein aggregation, dystrophic neurite architecture and nigrostriatal degeneration with potential to model the pathology of Parkinson's disease. Mol. Neurodegener. 5:43. doi: 10.1186/1750-1326-5-43
Kordower, J. H., Chu, Y., Hauser, R. A., Freeman, T. B., and Olanow, C. W. (2008). Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson's disease. Nat. Med. 14, 504–506. doi: 10.1038/nm1747
Kordower, J. H., Dodiya, H. B., Kordower, A. M., Terpstra, B., Paumier, K., Madhavan, L., et al. (2011). Transfer of host-derived alpha synuclein to grafted dopaminergic neurons in rat. Neurobiol. Dis. 43, 552–557. doi: 10.1016/j.nbd.2011.05.001
Kosik, K. S., Joachim, C. L., and Selkoe, D. J. (1986). Microtubule-associated protein tau (tau) is a major antigenic component of paired helical filaments in Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 83, 4044–4048. doi: 10.1073/pnas.83.11.4044
Kosik, K. S., Orecchio, L. D., Bakalis, S., and Neve, R. L. (1989). Developmentally regulated expression of specific tau sequences. Neuron 2, 1389–1397. doi: 10.1016/0896-6273(89)90077-9
Kotzbauer, P. T., Giasson, B. I., Kravitz, A. V., Golbe, L. I., Mark, M. H., Trojanowski, J. Q., et al. (2004). Fibrillization of alpha-synuclein and tau in familial Parkinson's disease caused by the A53T alpha-synuclein mutation. Exp. Neurol. 187, 279–288. doi: 10.1016/j.expneurol.2004.01.007
Kruger, R., Kuhn, W., Muller, T., Woitalla, D., Graeber, M., Kosel, S., et al. (1998). Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson's disease. Nat. Genet. 18, 106–108. doi: 10.1038/ng0298-106
Kundel, F., Hong, L., Falcon, B., McEwan, W. A., Michaels, T. C. T., Meisl, G., et al. (2018). Measurement of tau filament fragmentation provides insights into prion-like spreading. ACS Chem. Neurosci. 9, 1276–1282. doi: 10.1021/acschemneuro.8b00094
Lee, H. J., Patel, S., and Lee, S. J. (2005). Intravesicular localization and exocytosis of alpha-synuclein and its aggregates. J. Neurosci. 25, 6016–6024. doi: 10.1523/JNEUROSCI.0692-05.2005
Lee, H. J., Suk, J. E., Bae, E. J., Lee, J. H., Paik, S. R., and Lee, S. J. (2008). Assembly-dependent endocytosis and clearance of extracellular alpha-synuclein. Int. J. Biochem. Cell Biol. 40, 1835–1849. doi: 10.1016/j.biocel.2008.01.017
Lee, S., Jung, C., Lee, G., and Hall, G. F. (2009). Exonic point mutations of human tau enhance its toxicity and cause characteristic changes in neuronal morphology, tau distribution and tau phosphorylation in the lamprey cellular model of tauopathy. J. Alzheimers. Dis. 16, 99–111. doi: 10.3233/JAD-2009-0954
Lesage, S., Anheim, M., Letournel, F., Bousset, L., Honore, A., Rozas, N., et al. (2013). G51D alpha-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann. Neurol. 73, 459–471. doi: 10.1002/ana.23894
Levarska, L., Zilka, N., Jadhav, S., Neradil, P., and Novak, M. (2013). Of rodents and men: the mysterious interneuronal pilgrimage of misfolded protein tau in Alzheimer's disease. J. Alzheimers. Dis. 37, 569–577. doi: 10.3233/JAD-131106
Li, J. Y., Englund, E., Holton, J. L., Soulet, D., Hagell, P., Lees, A. J., et al. (2008). Lewy bodies in grafted neurons in subjects with Parkinson's disease suggest host-to-graft disease propagation. Nat. Med. 14, 501–503. doi: 10.1038/nm1746
Liu, L., Drouet, V., Wu, J. W., Witter, M. P., Small, S. A., Clelland, C., et al. (2012). Trans-synaptic spread of tau pathology in vivo. PLoS ONE 7:e31302. doi: 10.1371/journal.pone.0031302
Lo Bianco, C., Ridet, J. L., Schneider, B. L., Deglon, N., and Aebischer, P. (2002). alpha -Synucleinopathy and selective dopaminergic neuron loss in a rat lentiviral-based model of Parkinson's disease. Proc. Natl. Acad. Sci. U.S.A. 99, 10813–10818. doi: 10.1073/pnas.152339799
Loria, F., Vargas, J. Y., Bousset, L., Syan, S., Salles, A., Melki, R., et al. (2017). alpha-Synuclein transfer between neurons and astrocytes indicates that astrocytes play a role in degradation rather than in spreading. Acta Neuropathol. 134, 789–808. doi: 10.1007/s00401-017-1746-2
Low, K., and Aebischer, P. (2012). Use of viral vectors to create animal models for Parkinson's disease. Neurobiol. Dis. 48, 189–201. doi: 10.1016/j.nbd.2011.12.038
Luk, K. C., Kehm, V. M., Zhang, B., O'Brien, P., Trojanowski, J. Q., and Lee, V. M. (2012). Intracerebral inoculation of pathological alpha-synuclein initiates a rapidly progressive neurodegenerative alpha-synucleinopathy in mice. J. Exp. Med. 209, 975–986. doi: 10.1084/jem.20112457
Lundblad, M., Decressac, M., Mattsson, B., and Bjorklund, A. (2012). Impaired neurotransmission caused by overexpression of alpha-synuclein in nigral dopamine neurons. Proc. Natl. Acad. Sci. U.S.A. 109, 3213–3219. doi: 10.1073/pnas.1200575109
Ma, R. H., Zhang, Y., Hong, X. Y., Zhang, J. F., Wang, J. Z., and Liu, G. P. (2017). Role of microtubule-associated protein tau phosphorylation in Alzheimer's disease. J. Huazhong Univ. Sci. Technol. Med. Sci. 37, 307–312. doi: 10.1007/s11596-017-1732-x
Mabbott, N. A. (2017). How do PrP(Sc) prions spread between host species, and within hosts? Pathogens 6:60. doi: 10.3390/pathogens6040060
Maccioni, R. B., Vera, J. C., Dominguez, J., and Avila, J. (1989). A discrete repeated sequence defines a tubulin binding domain on microtubule-associated protein tau. Arch. Biochem. Biophys. 275, 568–579. doi: 10.1016/0003-9861(89)90403-7
Maeda, S., Sahara, N., Saito, Y., Murayama, S., Ikai, A., and Takashima, A. (2006). Increased levels of granular tau oligomers: an early sign of brain aging and Alzheimer's disease. Neurosci. Res. 54, 197–201. doi: 10.1016/j.neures.2005.11.009
Mao, X., Ou, M. T., Karuppagounder, S. S., Kam, T. I., Yin, X., Xiong, Y., et al. (2016). Pathological α-synuclein transmission initiated by binding lymphocyte-activation gene 3. Science 353:aah3374. doi: 10.1126/science.aah3374
Maroteaux, L., Campanelli, J. T., and Scheller, R. H. (1988). Synuclein: a neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J. Neurosci. 8, 2804–2815. doi: 10.1523/JNEUROSCI.08-08-02804.1988
Masliah, E., Rockenstein, E., Veinbergs, I., Sagara, Y., Mallory, M., Hashimoto, M., et al. (2001). beta-amyloid peptides enhance alpha-synuclein accumulation and neuronal deficits in a transgenic mouse model linking Alzheimer's disease and Parkinson's disease. Proc. Natl. Acad. Sci. U.S.A. 98, 12245–12250. doi: 10.1073/pnas.211412398
Masuda-Suzukake, M., Nonaka, T., Hosokawa, M., Oikawa, T., Arai, T., Akiyama, H., et al. (2013). Prion-like spreading of pathological alpha-synuclein in brain. Brain 136, 1128–1138. doi: 10.1093/brain/awt037
McCann, H., Cartwright, H., and Halliday, G. M. (2016). Neuropathology of alpha-synuclein propagation and braak hypothesis. Mov. Disord. 31, 152–160. doi: 10.1002/mds.26421
Mena, R., Edwards, P., Perez-Olvera, O., and Wischik, C. M. (1995). Monitoring pathological assembly of tau and beta-amyloid proteins in Alzheimer's disease. Acta Neuropathol. 89, 50–56. doi: 10.1007/BF00294259
Meuvis, J., Gerard, M., Desender, L., Baekelandt, V., and Engelborghs, Y. (2010). The conformation and the aggregation kinetics of alpha-synuclein depend on the proline residues in its C-terminal region. Biochemistry 49, 9345–9352. doi: 10.1021/bi1010927
Middleton, E. R., and Rhoades, E. (2010). Effects of curvature and composition on alpha-synuclein binding to lipid vesicles. Biophys. J. 99, 2279–2288. doi: 10.1016/j.bpj.2010.07.056
Mondragon-Rodriguez, S., Mena, R., Binder, L. I., Smith, M. A., Perry, G., and Garcia-Sierra, F. (2008). Conformational changes and cleavage of tau in Pick bodies parallel the early processing of tau found in Alzheimer pathology. Neuropathol. Appl. Neurobiol. 34, 62–75. doi: 10.1111/j.1365-2990.2007.00853.x
Montejo de Garcini, E., Serrano, L., and Avila, J. (1986). Self assembly of microtubule associated protein tau into filaments resembling those found in Alzheimer disease. Biochem. Biophys. Res. Commun. 141, 790–796. doi: 10.1016/S0006-291X(86)80242-X
Morris, M., Knudsen, G. M., Maeda, S., Trinidad, J. C., Ioanoviciu, A., Burlingame, A. L., et al. (2015). Tau post-translational modifications in wild-type and human amyloid precursor protein transgenic mice. Nat. Neurosci. 18, 1183–1189. doi: 10.1038/nn.4067
Mougenot, A. L., Nicot, S., Bencsik, A., Morignat, E., Verchere, J., Lakhdar, L., et al. (2012). Prion-like acceleration of a synucleinopathy in a transgenic mouse model. Neurobiol. Aging 33, 2225–2228. doi: 10.1016/j.neurobiolaging.2011.06.022
Multhaup, G., Huber, O., Buee, L., and Galas, M. C. (2015). Amyloid Precursor Protein (APP) Metabolites APP Intracellular Fragment (AICD), Abeta42, and Tau in Nuclear Roles. J. Biol. Chem. 290, 23515–23522. doi: 10.1074/jbc.R115.677211
Mutreja, Y., Combs, B., and Gamblin, T. C. (2019). FTDP-17 mutations alter the aggregation and microtubule stabilization propensity of tau in an isoform-specific fashion. Biochemistry 58, 742–754. doi: 10.1021/acs.biochem.8b01039
Mutreja, Y., and Gamblin, T. C. (2017). Optimization of in vitro conditions to study the arachidonic acid induction of 4R isoforms of the microtubule-associated protein tau. Methods Cell Biol. 141, 65–88. doi: 10.1016/bs.mcb.2017.06.007
Neve, R. L., Harris, P., Kosik, K. S., Kurnit, D. M., and Donlon, T. A. (1986). Identification of cDNA clones for the human microtubule-associated protein tau and chromosomal localization of the genes for tau and microtubule-associated protein 2. Brain Res. 387, 271–280. doi: 10.1016/0169-328X(86)90033-1
Ngolab, J., Trinh, I., Rockenstein, E., Mante, M., Florio, J., Trejo, M., et al. (2017). Brain-derived exosomes from dementia with Lewy bodies propagate alpha-synuclein pathology. Acta Neuropathol. Commun. 5:46. doi: 10.1186/s40478-017-0445-5
Nielsen, L., Khurana, R., Coats, A., Frokjaer, S., Brange, J., Vyas, S., et al. (2001). Effect of environmental factors on the kinetics of insulin fibril formation: elucidation of the molecular mechanism. Biochemistry 40, 6036–6046. doi: 10.1021/bi002555c
Nuber, S., Rajsombath, M., Minakaki, G., Winkler, J., Muller, C. P., Ericsson, M., et al. (2018). Abrogating native alpha-synuclein tetramers in mice causes a L-DOPA-responsive motor syndrome closely resembling Parkinson's disease. Neuron 100, 75–90.e75. doi: 10.1016/j.neuron.2018.09.014
Olanow, C. W., and Prusiner, S. B. (2009). Is Parkinson's disease a prion disorder? Proc. Natl. Acad. Sci. U.S.A. 106, 12571–12572. doi: 10.1073/pnas.0906759106
Oliveras-Salva, M., Van der Perren, A., Casadei, N., Stroobants, S., Nuber, S., D'Hooge, R., et al. (2013). rAAV2/7 vector-mediated overexpression of alpha-synuclein in mouse substantia nigra induces protein aggregation and progressive dose-dependent neurodegeneration. Mol. Neurodegener. 8:44. doi: 10.1186/1750-1326-8-44
Ordonez, D. G., Lee, M. K., and Feany, M. B. (2018). α-synuclein induces mitochondrial dysfunction through spectrin and the actin cytoskeleton. Neuron 97, 108–124.e106. doi: 10.1016/j.neuron.2017.11.036
Oueslati, A. (2016). Implication of alpha-synuclein phosphorylation at S129 in synucleinopathies: what have we learned in the last decade? J. Parkinsons. Dis. 6, 39–51. doi: 10.3233/JPD-160779
Paik, S. R., Shin, H. J., Lee, J. H., Chang, C. S., and Kim, J. (1999). Copper(II)-induced self-oligomerization of alpha-synuclein. Biochem. J. 340(Pt 3), 821–828.
Paleologou, K. E., Oueslati, A., Shakked, G., Rospigliosi, C. C., Kim, H. Y., Lamberto, G. R., et al. (2010). Phosphorylation at S87 is enhanced in synucleinopathies, inhibits alpha-synuclein oligomerization, and influences synuclein-membrane interactions. J. Neurosci. 30, 3184–3198. doi: 10.1523/JNEUROSCI.5922-09.2010
Paleologou, K. E., Schmid, A. W., Rospigliosi, C. C., Kim, H. Y., Lamberto, G. R., Fredenburg, R. A., et al. (2008). Phosphorylation at Ser-129 but not the phosphomimics S129E/D inhibits the fibrillation of alpha-synuclein. J. Biol. Chem. 283, 16895–16905. doi: 10.1074/jbc.M800747200
Pasanen, P., Myllykangas, L., Siitonen, M., Raunio, A., Kaakkola, S., Lyytinen, J., et al. (2014). Novel alpha-synuclein mutation A53E associated with atypical multiple system atrophy and Parkinson's disease-type pathology. Neurobiol. Aging 35, 2180.e2181–2180.e2185. doi: 10.1016/j.neurobiolaging.2014.03.024
Patterson, K. R., Remmers, C., Fu, Y., Brooker, S., Kanaan, N. M., Vana, L., et al. (2011). Characterization of prefibrillar Tau oligomers in vitro and in Alzheimer disease. J. Biol. Chem. 286, 23063–23076. doi: 10.1074/jbc.M111.237974
Peden, A. H., Head, M. W., Ritchie, D. L., Bell, J. E., and Ironside, J. W. (2004). Preclinical vCJD after blood transfusion in a PRNP codon 129 heterozygous patient. Lancet 364, 527–529. doi: 10.1016/S0140-6736(04)16811-6
Peelaerts, W., Bousset, L., Van der Perren, A., Moskalyuk, A., Pulizzi, R., Giugliano, M., et al. (2015). alpha-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature 522, 340–344. doi: 10.1038/nature14547
Peeraer, E., Bottelbergs, A., Van Kolen, K., Stancu, I. C., Vasconcelos, B., Mahieu, M., et al. (2015). Intracerebral injection of preformed synthetic tau fibrils initiates widespread tauopathy and neuronal loss in the brains of tau transgenic mice. Neurobiol. Dis. 73, 83–95. doi: 10.1016/j.nbd.2014.08.032
Pineda, A., and Burre, J. (2017). Modulating membrane binding of alpha-synuclein as a therapeutic strategy. Proc. Natl. Acad. Sci. U.S.A. 114, 1223–1225. doi: 10.1073/pnas.1620159114
Polanco, J. C., Li, C., Durisic, N., Sullivan, R., and Gotz, J. (2018). Exosomes taken up by neurons hijack the endosomal pathway to spread to interconnected neurons. Acta Neuropathol. Commun. 6:10. doi: 10.1186/s40478-018-0514-4
Polanco, J. C., Scicluna, B. J., Hill, A. F., and Gotz, J. (2016). Extracellular vesicles isolated from the brains of rTg4510 mice seed tau protein aggregation in a threshold-dependent manner. J. Biol. Chem. 291, 12445–12466. doi: 10.1074/jbc.M115.709485
Polymeropoulos, M. H., Lavedan, C., Leroy, E., Ide, S. E., Dehejia, A., Dutra, A., et al. (1997). Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science 276, 2045–2047. doi: 10.1126/science.276.5321.2045
Pooler, A. M., Phillips, E. C., Lau, D. H., Noble, W., and Hanger, D. P. (2013). Physiological release of endogenous tau is stimulated by neuronal activity. EMBO Rep. 14, 389–394. doi: 10.1038/embor.2013.15
Prots, I., Grosch, J., Brazdis, R. M., Simmnacher, K., Veber, V., Havlicek, S., et al. (2018). α-Synuclein oligomers induce early axonal dysfunction in human iPSC-based models of synucleinopathies. Proc. Natl. Acad. Sci. U.S.A. 115, 7813–7818. doi: 10.1073/pnas.1713129115
Proukakis, C., Dudzik, C. G., Brier, T., MacKay, D. S., Cooper, J. M., Millhauser, G. L., et al. (2013). A novel alpha-synuclein missense mutation in Parkinson disease. Neurology 80, 1062–1064. doi: 10.1212/WNL.0b013e31828727ba
Prusiner, S. B., Woerman, A. L., Mordes, D. A., Watts, J. C., Rampersaud, R., Berry, D. B., et al. (2015). Evidence for alpha-synuclein prions causing multiple system atrophy in humans with parkinsonism. Proc. Natl. Acad. Sci. U.S.A. 112, E5308–E5317. doi: 10.1073/pnas.1514475112
Rademakers, R., Cruts, M., and van Broeckhoven, C. (2004). The role of tau (MAPT) in frontotemporal dementia and related tauopathies. Hum. Mutat. 24, 277–295. doi: 10.1002/humu.20086
Recasens, A., and Dehay, B. (2014). Alpha-synuclein spreading in Parkinson's disease. Front. Neuroanat. 8:159. doi: 10.3389/fnana.2014.00159
Reyes, J. F., Olsson, T. T., Lamberts, J. T., Devine, M. J., Kunath, T., and Brundin, P. (2015). A cell culture model for monitoring alpha-synuclein cell-to-cell transfer. Neurobiol. Dis. 77, 266–275. doi: 10.1016/j.nbd.2014.07.003
Reyes, J. F., Rey, N. L., Bousset, L., Melki, R., Brundin, P., and Angot, E. (2014). Alpha-synuclein transfers from neurons to oligodendrocytes. Glia 62, 387–398. doi: 10.1002/glia.22611
Rostami, J., Holmqvist, S., Lindstrom, V., Sigvardson, J., Westermark, G. T., Ingelsson, M., et al. (2017). Human astrocytes transfer aggregated alpha-synuclein via tunneling nanotubes. J. Neurosci. 37, 11835–11853. doi: 10.1523/JNEUROSCI.0983-17.2017
Rotermund, C., Truckenmuller, F. M., Schell, H., and Kahle, P. J. (2014). Diet-induced obesity accelerates the onset of terminal phenotypes in alpha-synuclein transgenic mice. J. Neurochem. 131, 848–858. doi: 10.1111/jnc.12813
Ruiz-Riquelme, A., Lau, H. H. C., Stuart, E., Goczi, A. N., Wang, Z., Schmitt-Ulms, G., et al. (2018). Prion-like propagation of beta-amyloid aggregates in the absence of APP overexpression. Acta Neuropathol. Commun. 6:26. doi: 10.1186/s40478-018-0529-x
Rusconi, R., Ulusoy, A., Aboutalebi, H., and Di Monte, D. A. (2018). Long-lasting pathological consequences of overexpression-induced alpha-synuclein spreading in the rat brain. Aging Cell 17:e12727. doi: 10.1111/acel.12727
Rustom, A., Saffrich, R., Markovic, I., Walther, P., and Gerdes, H. H. (2004). Nanotubular highways for intercellular organelle transport. Science 303, 1007–1010. doi: 10.1126/science.1093133
Sacino, A. N., Brooks, M., McGarvey, N. H., McKinney, A. B., Thomas, M. A., Levites, Y., et al. (2013). Induction of CNS alpha-synuclein pathology by fibrillar and non-amyloidogenic recombinant alpha-synuclein. Acta Neuropathol. Commun. 1:38. doi: 10.1186/2051-5960-1-38
Sacino, A. N., Brooks, M., Thomas, M. A., McKinney, A. B., Lee, S., Regenhardt, R. W., et al. (2014b). Intramuscular injection of alpha-synuclein induces CNS alpha-synuclein pathology and a rapid-onset motor phenotype in transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 111, 10732–10737. doi: 10.1073/pnas.1321785111
Sacino, A. N., Brooks, M., Thomas, M. A., McKinney, A. B., McGarvey, N. H., Rutherford, N. J., et al. (2014a). Amyloidogenic alpha-synuclein seeds do not invariably induce rapid, widespread pathology in mice. Acta Neuropathol. 127, 645–665. doi: 10.1007/s00401-014-1268-0
Sampson, T. R., Debelius, J. W., Thron, T., Janssen, S., Shastri, G. G., Ilhan, Z. E., et al. (2016). Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson's disease. Cell 167, 1469–1480.e1412. doi: 10.1016/j.cell.2016.11.018
Sanders, D. W., Kaufman, S. K., DeVos, S. L., Sharma, A. M., Mirbaha, H., Li, A., et al. (2014). Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron 82, 1271–1288. doi: 10.1016/j.neuron.2014.04.047
Sarnataro, D. (2018). Attempt to untangle the prion-like misfolding mechanism for neurodegenerative diseases. Int. J. Mol. Sci. 19:3081. doi: 10.3390/ijms19103081
Schell, H., Boden, C., Chagas, A. M., and Kahle, P. J. (2012). Impaired c-Fos and polo-like kinase 2 induction in the limbic system of fear-conditioned alpha-synuclein transgenic mice. PLoS ONE 7:e50245. doi: 10.1371/journal.pone.0050245
Schofield, E. C., Caine, D., Kril, J. J., Cordato, N. J., and Halliday, G. M. (2005). Staging disease severity in movement disorder tauopathies: brain atrophy separates progressive supranuclear palsy from corticobasal degeneration. Mov. Disord. 20, 34–39. doi: 10.1002/mds.20286
Schwarz, A. J., Shcherbinin, S., Slieker, L. J., Risacher, S. L., Charil, A., Irizarry, M. C., et al. (2018). Topographic staging of tau positron emission tomography images. Alzheimers Dement. 10, 221–231. doi: 10.1016/j.dadm.2018.01.006
Sebastian-Serrano, A., de Diego-Garcia, L., and Diaz-Hernandez, M. (2018). The neurotoxic role of extracellular tau protein. Int. J. Mol. Sci. 19:998. doi: 10.3390/ijms19040998
Sergeant, N., Bretteville, A., Hamdane, M., Caillet-Boudin, M. L., Grognet, P., Bombois, S., et al. (2008). Biochemistry of Tau in Alzheimer's disease and related neurological disorders. Expert Rev. Proteomics 5, 207–224. doi: 10.1586/14789450.5.2.207
Sergeant, N., Delacourte, A., and Buee, L. (2005). Tau protein as a differential biomarker of tauopathies. Biochim. Biophys. Acta 1739, 179–197. doi: 10.1016/j.bbadis.2004.06.020
Shimozawa, A., Ono, M., Takahara, D., Tarutani, A., Imura, S., Masuda-Suzukake, M., et al. (2017). Propagation of pathological alpha-synuclein in marmoset brain. Acta Neuropathol. Commun. 5:12. doi: 10.1186/s40478-017-0413-0
Singleton, A. B., Farrer, M., Johnson, J., Singleton, A., Hague, S., Kachergus, J., et al. (2003). alpha-Synuclein locus triplication causes Parkinson's disease. Science 302:841. doi: 10.1126/science.1090278
Smolek, T., Jadhav, S., Brezovakova, V., Cubinkova, V., Valachova, B., Novak, P., et al. (2019). First-in-rat study of human Alzheimer's disease tau propagation. Mol. Neurobiol. 56, 621–631. doi: 10.1007/s12035-018-1102-0
Sokolow, S., Henkins, K. M., Bilousova, T., Gonzalez, B., Vinters, H. V., Miller, C. A., et al. (2015). Pre-synaptic C-terminal truncated tau is released from cortical synapses in Alzheimer's disease. J. Neurochem. 133, 368–379. doi: 10.1111/jnc.12991
Sotiropoulos, I., Galas, M. C., Silva, J. M., Skoulakis, E., Wegmann, S., Maina, M. B., et al. (2017). Atypical, non-standard functions of the microtubule associated Tau protein. Acta Neuropathol. Commun. 5:91. doi: 10.1186/s40478-017-0489-6
Spillantini, M. G., Schmidt, M. L., Lee, V. M., Trojanowski, J. Q., Jakes, R., and Goedert, M. (1997). Alpha-synuclein in Lewy bodies. Nature 388, 839–840. doi: 10.1038/42166
St Martin, J. L., Klucken, J., Outeiro, T. F., Nguyen, P., Keller-McGandy, C., Cantuti-Castelvetri, I., et al. (2007). Dopaminergic neuron loss and up-regulation of chaperone protein mRNA induced by targeted over-expression of alpha-synuclein in mouse substantia nigra. J. Neurochem. 100, 1449–1457. doi: 10.1111/j.1471-4159.2006.04310.x
Stancu, I. C., Vasconcelos, B., Ris, L., Wang, P., Villers, A., Peeraer, E., et al. (2015). Templated misfolding of Tau by prion-like seeding along neuronal connections impairs neuronal network function and associated behavioral outcomes in Tau transgenic mice. Acta Neuropathol. 129, 875–894. doi: 10.1007/s00401-015-1413-4
Stanford, P. M., Shepherd, C. E., Halliday, G. M., Brooks, W. S., Schofield, P. W., Brodaty, H., et al. (2003). Mutations in the tau gene that cause an increase in three repeat tau and frontotemporal dementia. Brain 126, 814–826. doi: 10.1093/brain/awg090
Stockl, M. T., Zijlstra, N., and Subramaniam, V. (2013). alpha-Synuclein oligomers: an amyloid pore? Insights into mechanisms of alpha-synuclein oligomer-lipid interactions. Mol. Neurobiol. 47, 613–621. doi: 10.1007/s12035-012-8331-4
Sun, X., Wang, Y., Zhang, J., Tu, J., Wang, X. J., Su, X. D., et al. (2012). Tunneling-nanotube direction determination in neurons and astrocytes. Cell Death Dis. 3:e438. doi: 10.1038/cddis.2012.177
Sung, J. Y., Kim, J., Paik, S. R., Park, J. H., Ahn, Y. S., and Chung, K. C. (2001). Induction of neuronal cell death by Rab5A-dependent endocytosis of alpha-synuclein. J. Biol. Chem. 276, 27441–27448. doi: 10.1074/jbc.M101318200
Swanson, E., Breckenridge, L., McMahon, L., Som, S., McConnell, I., and Bloom, G. S. (2017). Extracellular tau oligomers induce invasion of endogenous tau into the somatodendritic compartment and axonal transport dysfunction. J. Alzheimers. Dis. 58, 803–820. doi: 10.3233/JAD-170168
Takahashi, M., Miyata, H., Kametani, F., Nonaka, T., Akiyama, H., Hisanaga, S., et al. (2015). Extracellular association of APP and tau fibrils induces intracellular aggregate formation of tau. Acta Neuropathol. 129, 895–907. doi: 10.1007/s00401-015-1415-2
Taneva, S. G., Lee, J. M., and Cornell, R. B. (2012). The amphipathic helix of an enzyme that regulates phosphatidylcholine synthesis remodels membranes into highly curved nanotubules. Biochim. Biophys. Acta 1818, 1173–1186. doi: 10.1016/j.bbamem.2012.01.006
Tardivel, M., Begard, S., Bousset, L., Dujardin, S., Coens, A., Melki, R., et al. (2016). Tunneling nanotube (TNT)-mediated neuron-to neuron transfer of pathological Tau protein assemblies. Acta Neuropathol. Commun. 4:117. doi: 10.1186/s40478-016-0386-4
Taschenberger, G., Garrido, M., Tereshchenko, Y., Bahr, M., Zweckstetter, M., and Kugler, S. (2012). Aggregation of alphaSynuclein promotes progressive in vivo neurotoxicity in adult rat dopaminergic neurons. Acta Neuropathol. 123, 671–683. doi: 10.1007/s00401-011-0926-8
Tofaris, G. K., Garcia Reitbock, P., Humby, T., Lambourne, S. L., O'Connell, M., Ghetti, B., et al. (2006). Pathological changes in dopaminergic nerve cells of the substantia nigra and olfactory bulb in mice transgenic for truncated human alpha-synuclein(1-120): implications for Lewy body disorders. J. Neurosci. 26, 3942–3950. doi: 10.1523/JNEUROSCI.4965-05.2006
Tosatto, L., Andrighetti, A. O., Plotegher, N., Antonini, V., Tessari, I., Ricci, L., et al. (2012). Alpha-synuclein pore forming activity upon membrane association. Biochim. Biophys. Acta 1818, 2876–2883. doi: 10.1016/j.bbamem.2012.07.007
Tozzi, A., de Iure, A., Bagetta, V., Tantucci, M., Durante, V., Quiroga-Varela, A., et al. (2016). Alpha-synuclein produces early behavioral alterations via striatal cholinergic synaptic dysfunction by interacting with GluN2D N-methyl-D-aspartate receptor subunit. Biol. Psychiatry 79, 402–414. doi: 10.1016/j.biopsych.2015.08.013
Trexler, A. J., and Rhoades, E. (2012). N-terminal acetylation is critical for forming alpha-helical oligomer of alpha-synuclein. Protein Sci. 21, 601–605. doi: 10.1002/pro.2056
Tsigelny, I. F., Sharikov, Y., Wrasidlo, W., Gonzalez, T., Desplats, P. A., Crews, L., et al. (2012). Role of alpha-synuclein penetration into the membrane in the mechanisms of oligomer pore formation. FEBS J. 279, 1000–1013. doi: 10.1111/j.1742-4658.2012.08489.x
Tuerde, D., Kimura, T., Miyasaka, T., Furusawa, K., Shimozawa, A., Hasegawa, M., et al. (2018). Isoform-independent and -dependent phosphorylation of microtubule-associated protein tau in mouse brain during postnatal development. J. Biol. Chem. 293, 1781–1793. doi: 10.1074/jbc.M117.798918
Tysnes, O. B., and Storstein, A. (2017). Epidemiology of Parkinson's disease. J. Neural. Transm. 124, 901–905. doi: 10.1007/s00702-017-1686-y
Ulusoy, A., Febbraro, F., Jensen, P. H., Kirik, D., and Romero-Ramos, M. (2010). Co-expression of C-terminal truncated alpha-synuclein enhances full-length alpha-synuclein-induced pathology. Eur. J. Neurosci. 32, 409–422. doi: 10.1111/j.1460-9568.2010.07284.x
Ulusoy, A., Musgrove, R. E., Rusconi, R., Klinkenberg, M., Helwig, M., Schneider, A., et al. (2015). Neuron-to-neuron alpha-synuclein propagation in vivo is independent of neuronal injury. Acta Neuropathol. Commun. 3:13. doi: 10.1186/s40478-015-0198-y
Ulusoy, A., Phillips, R. J., Helwig, M., Klinkenberg, M., Powley, T. L., and Di Monte, D. A. (2017). Brain-to-stomach transfer of alpha-synuclein via vagal preganglionic projections. Acta Neuropathol. 133, 381–393. doi: 10.1007/s00401-016-1661-y
Ulusoy, A., Rusconi, R., Perez-Revuelta, B. I., Musgrove, R. E., Helwig, M., Winzen-Reichert, B., et al. (2013). Caudo-rostral brain spreading of alpha-synuclein through vagal connections. EMBO Mol. Med. 5, 1119–1127. doi: 10.1002/emmm.201302475
Utton, M. A., Gibb, G. M., Burdett, I. D., Anderton, B. H., and Vandecandelaere, A. (2001). Functional differences of tau isoforms containing 3 or 4 C-terminal repeat regions and the influence of oxidative stress. J. Biol. Chem. 276, 34288–34297. doi: 10.1074/jbc.M011384200
Uversky, V. N. (2003). A protein-chameleon: conformational plasticity of alpha-synuclein, a disordered protein involved in neurodegenerative disorders. J. Biomol. Struct. Dyn. 21, 211–234. doi: 10.1080/07391102.2003.10506918
Uversky, V. N. (2011a). Intrinsically disordered proteins may escape unwanted interactions via functional misfolding. Biochim. Biophys. Acta 1814, 693–712. doi: 10.1016/j.bbapap.2011.03.010
Uversky, V. N. (2011b). Intrinsically disordered proteins from A to Z. Int. J. Biochem. Cell Biol. 43, 1090–1103. doi: 10.1016/j.biocel.2011.04.001
Uversky, V. N., and Fink, A. L. (2004). Conformational constraints for amyloid fibrillation: the importance of being unfolded. Biochim. Biophys. Acta 1698, 131–153. doi: 10.1016/j.bbapap.2003.12.008
Van der Perren, A., Toelen, J., Carlon, M., Van den Haute, C., Coun, F., Heeman, B., et al. (2011). Efficient and stable transduction of dopaminergic neurons in rat substantia nigra by rAAV 2/1, 2/2, 2/5, 2/6.2, 2/7, 2/8 and 2/9. Gene Ther. 18, 517–527. doi: 10.1038/gt.2010.179
van Swieten, J. C., Bronner, I. F., Azmani, A., Severijnen, L. A., Kamphorst, W., Ravid, R., et al. (2007). The DeltaK280 mutation in MAP tau favors exon 10 skipping in vivo. J. Neuropathol. Exp. Neurol. 66, 17–25. doi: 10.1097/nen.0b013e31802c39a4
Visanji, N. P., Wislet-Gendebien, S., Oschipok, L. W., Zhang, G., Aubert, I., Fraser, P. E., et al. (2011). Effect of Ser-129 phosphorylation on interaction of alpha-synuclein with synaptic and cellular membranes. J. Biol. Chem. 286, 35863–35873. doi: 10.1074/jbc.M111.253450
Vitale, F., Giliberto, L., Ruiz, S., Steslow, K., Marambaud, P., and d'Abramo, C. (2018). Anti-tau conformational scFv MC1 antibody efficiently reduces pathological tau species in adult JNPL3 mice. Acta Neuropathol. Commun. 6:82. doi: 10.1186/s40478-018-0585-2
Volles, M. J., and Lansbury, P. T. Jr. (2002). Vesicle permeabilization by protofibrillar alpha-synuclein is sensitive to Parkinson's disease-linked mutations and occurs by a pore-like mechanism. Biochemistry 41, 4595–4602. doi: 10.1021/bi0121353
Walker, L. C., Schelle, J., and Jucker, M. (2016). The prion-like properties of amyloid-beta assemblies: implications for Alzheimer's disease. Cold Spring Harb. Perspect. Med. 6:a024398. doi: 10.1101/cshperspect.a024398
Wang, Y., Balaji, V., Kaniyappan, S., Kruger, L., Irsen, S., Tepper, K., et al. (2017). The release and trans-synaptic transmission of Tau via exosomes. Mol. Neurodegener. 12:5. doi: 10.1186/s13024-016-0143-y
Wang, Y., and Mandelkow, E. (2016). Tau in physiology and pathology. Nat. Rev. Neurosci. 17, 5–21. doi: 10.1038/nrn.2015.1
Watts, J. C., Giles, K., Oehler, A., Middleton, L., Dexter, D. T., Gentleman, S. M., et al. (2013). Transmission of multiple system atrophy prions to transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 110, 19555–19560. doi: 10.1073/pnas.1318268110
Weingarten, M. D., Lockwood, A. H., Hwo, S. Y., and Kirschner, M. W. (1975). A protein factor essential for microtubule assembly. Proc. Natl. Acad. Sci. U.S.A. 72, 1858–1862. doi: 10.1073/pnas.72.5.1858
Wischik, C. M., Novak, M., Edwards, P. C., Klug, A., Tichelaar, W., and Crowther, R. A. (1988). Structural characterization of the core of the paired helical filament of Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 85, 4884–4888. doi: 10.1073/pnas.85.13.4884
Wood, J. G., Mirra, S. S., Pollock, N. J., and Binder, L. I. (1986). Neurofibrillary tangles of Alzheimer disease share antigenic determinants with the axonal microtubule-associated protein tau (tau). Proc. Natl. Acad. Sci. U.S.A. 83, 4040–4043. doi: 10.1073/pnas.83.11.4040
Wu, J. W., Herman, M., Liu, L., Simoes, S., Acker, C. M., Figueroa, H., et al. (2013). Small misfolded Tau species are internalized via bulk endocytosis and anterogradely and retrogradely transported in neurons. J. Biol. Chem. 288, 1856–1870. doi: 10.1074/jbc.M112.394528
Wu, J. W., Hussaini, S. A., Bastille, I. M., Rodriguez, G. A., Mrejeru, A., Rilett, K., et al. (2016). Neuronal activity enhances tau propagation and tau pathology in vivo. Nat. Neurosci. 19, 1085–1092. doi: 10.1038/nn.4328
Wu, K. P., and Baum, J. (2010). Detection of transient interchain interactions in the intrinsically disordered protein alpha-synuclein by NMR paramagnetic relaxation enhancement. J. Am. Chem. Soc. 132, 5546–5547. doi: 10.1021/ja9105495
Yamada, K., and Iwatsubo, T. (2018). Extracellular alpha-synuclein levels are regulated by neuronal activity. Mol. Neurodegener. 13:9. doi: 10.1186/s13024-018-0241-0
Yamada, M., Iwatsubo, T., Mizuno, Y., and Mochizuki, H. (2004). Overexpression of alpha-synuclein in rat substantia nigra results in loss of dopaminergic neurons, phosphorylation of alpha-synuclein and activation of caspase-9: resemblance to pathogenetic changes in Parkinson's disease. J. Neurochem. 91, 451–461. doi: 10.1111/j.1471-4159.2004.02728.x
Yang, W., Wang, G., Wang, C. E., Guo, X., Yin, P., Gao, J., et al. (2015). Mutant alpha-synuclein causes age-dependent neuropathology in monkey brain. J. Neurosci. 35, 8345–8358. doi: 10.1523/JNEUROSCI.0772-15.2015
Yang, Y., Qin, M., Bao, P., Xu, W., and Xu, J. (2017). Secretory carrier membrane protein 5 is an autophagy inhibitor that promotes the secretion of alpha-synuclein via exosome. PLoS ONE 12:e0180892. doi: 10.1371/journal.pone.0180892
Yekhlef, F., Ballan, G., Macia, F., Delmer, O., Sourgen, C., and Tison, F. (2003). Routine MRI for the differential diagnosis of Parkinson's disease, MSA, PSP, and CBD. J. Neural. Transm. 110, 151–169. doi: 10.1007/s00702-002-0785-5
Yuan, H., Du, L., Ge, P., Wang, X., and Xia, Q. (2018). Association of microtubule-associated protein tau gene polymorphisms with the risk of sporadic Alzheimer's disease: a meta-analysis. Int. J. Neurosci. 128, 577–585. doi: 10.1080/00207454.2017.1400972
Zabrocki, P., Bastiaens, I., Delay, C., Bammens, T., Ghillebert, R., Pellens, K., et al. (2008). Phosphorylation, lipid raft interaction and traffic of alpha-synuclein in a yeast model for Parkinson. Biochim. Biophys. Acta 1783, 1767–1780. doi: 10.1016/j.bbamcr.2008.06.010
Zaccai, J., Brayne, C., McKeith, I., Matthews, F., and Ince, P. G. (2008). Patterns and stages of alpha-synucleinopathy: relevance in a population-based cohort. Neurology 70, 1042–1048. doi: 10.1212/01.wnl.0000306697.48738.b6
Zarranz, J. J., Alegre, J., Gomez-Esteban, J. C., Lezcano, E., Ros, R., Ampuero, I., et al. (2004). The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann. Neurol. 55, 164–173. doi: 10.1002/ana.10795
Keywords: alpha-synuclein, Parkinson's disease, tau, Alzheimer's disease, spreading
Citation: Vasili E, Dominguez-Meijide A and Outeiro TF (2019) Spreading of α-Synuclein and Tau: A Systematic Comparison of the Mechanisms Involved. Front. Mol. Neurosci. 12:107. doi: 10.3389/fnmol.2019.00107
Received: 13 January 2019; Accepted: 09 April 2019;
Published: 25 April 2019.
Edited by:
Taher Darreh-Shori, Karolinska Institute (KI), SwedenReviewed by:
Rakez Kayed, The University of Texas Medical Branch at Galveston, United StatesCharles Robert Harrington, University of Aberdeen, United Kingdom;
Norbert Zilka, Institute of Neuroimmunology (SAS), Slovakia
Copyright © 2019 Vasili, Dominguez-Meijide and Outeiro. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tiago Fleming Outeiro, dGlhZ28ub3V0ZWlyb0BtZWQudW5pLWdvZXR0aW5nZW4uZGU=