 Paraskevi Krashia
Paraskevi Krashia Annalisa Nobili
Annalisa Nobili Marcello D'Amelio
Marcello D'Amelio- 1Laboratory of Molecular Neurosciences, Department of Experimental Neurosciences, IRCCS Santa Lucia Foundation, Rome, Italy
- 2Unit of Molecular Neurosciences, Department of Medicine, University Campus-Biomedico, Rome, Italy
The term “dementia” describes a syndrome caused by several illnesses in which there is progressive decline in multiple areas of brain function. The main form of dementia is Alzheimer's disease (AD), currently affecting 47 million people, 0.5% of the global population, according to the 2016 World Alzheimer Report (Banerjee, 2012; ADI, 2016). AD is an irreversible and progressive brain disorder characterized by the presence of extracellular amyloid β (Aβ) plaques and intracellular neurofibrillary (tau) tangles, brain atrophy, inflammation and progressive loss of memory and cognition (D'Amelio and Rossini, 2012; Ismail et al., 2016). Comorbidities like epileptic seizures (Noebels, 2011; Pandis and Scarmeas, 2012), neuropsychiatric symptoms, mainly apathy, depression, aggression, and circadian rhythm disturbances (Lyketsos et al., 2011; Masters et al., 2015; Alves et al., 2017; Musiek et al., 2018), impose an additional burden on patients and their families.
There are two forms of AD, early-onset and late-onset (Guerreiro et al., 2012). Early-onset AD represents < 5% of cases and occurs in people aged 30–60. Most patients suffer from the late-onset form, occurring in people over-60. Early-onset AD is strongly linked to autosomal dominant mutations in amyloid precursor protein (APP) and presenilin genes [PSEN1, PSEN2; (Blennow et al., 2006)] while the most common genetic variant for late-onset AD is apolipoprotein E (APOE), a three-allele polymorphism (ε2, ε3, and ε4) where ε4 is the high-risk allele (Corder et al., 1993). However, other than old age, the aetiological factors for late-onset AD remain to be determined, but increasing evidence points to the potential risk roles of vascular diseases and lifestyle factors such as smoking, high blood pressure, high-fat diet, obesity, diabetes, hypercholesterolemia, and cerebrovascular lesions. Conversely, active social engagement, physical exercise, balanced diet, and mentally-stimulating activity have been suggested as beneficial for delaying the clinical manifestation of AD (Kivipelto et al., 2001; Qiu et al., 2009; Mayeux and Stern, 2012; Knight et al., 2014; Winblad et al., 2016).
The broadly-accepted hypothesis of AD pathogenesis states that the generation and accumulation of Aβ peptides represent the cause of the progressive and massive neuronal loss that primarily affects the hippocampus and cortex. Hyperphosphorylated-tau deposition, contributing to neuronal dysfunction and cognitive symptoms, is generally regarded as a downstream event in the amyloid cascade (Hardy and Selkoe, 2002; Scheff et al., 2006; D'Amelio and Rossini, 2012). However, the amyloid cascade hypothesis seems most applicable in cases of early-onset AD, representing a primary cerebral amyloidosis. Whether individuals with late-onset AD also carry genetic variations that promote a primary amyloidosis remains to be shown. Nonetheless, attempts to clear Aβ from the brain led to failure of several long and promising clinical trials (Doody et al., 2013; Cummings et al., 2014; Gauthier et al., 2016; Vandenberghe et al., 2016; Anderson et al., 2017; Carroll, 2017; Coleman and Mastroeni, 2017; Honig et al., 2018). Additionally, amyloid imaging demonstrates that many normal patients can show Aβ plaque deposits, or that AD patients can show low Aβ plaque load (Edison et al., 2007; Li et al., 2008), while in the brain of elderly non-demented patients the distribution of Aβ plaques can be as extensive as that of dementia patients (Davis et al., 1999; Fagan et al., 2009; Price et al., 2009; Chételat et al., 2013). Together, these data suggest that Aβ deposition might be a phenomenon of aging or a consequence of an upstream event and not the causative reason for AD, with scientists now questioning the validity of the amyloid hypothesis (Hardy and Selkoe, 2002; Tanzi and Bertram, 2005; Chételat, 2013; Herrup, 2015).
An important evidence against the amyloid hypothesis is the recent discovery that prior to the accumulation of Aβ depositions, and much earlier than the observation of hippocampal cell death, there is evidence from both an early-AD genetic mouse model (Tg2576) as well as from late-onset patients that the dopamine neurons in the Ventral Tegmental Area (VTA) are compromised (Nobili et al., 2017; De Marco and Venneri, 2018; Serra et al., 2018). The VTA is the origin of tyrosine hydroxylase positive (TH+) axons forming the mesocorticolimbic dopaminergic pathway [Figure 1A; (Gasbarri et al., 1994; Björklund and Dunnett, 2007)], projecting primarily to the prefrontal cortex, hippocampus, nucleus accumbens (NAc), olfactory bulb, and amygdala. Degeneration of VTA dopamine neurons in Tg2576 mice is evident as early as 3 months of age and is accompanied by significant levels of local inflammation, while cell death worsens with age (Nobili et al., 2017; D'Amelio and Nisticò, 2018). The degeneration of dopamine neurons results in lower dopamine outflow in the hippocampus and NAc and correlates temporally with impairments in hippocampal neuronal excitability, synaptic plasticity, memory, and reward performance (Nobili et al., 2017; Cordella et al., 2018). Other brain regions containing TH+ neurons, in particular the Substantia Nigra pars compacta (SNpc) and Locus Coeruleus (LC), are intact in Tg2576 mice at least at early stages (Nobili et al., 2017), suggesting that the observed degeneration primarily affects the VTA. In line with data from Tg2576 mice, TH+ neuron loss has been associated to Aβ load in two other frequently used AD models, the 3 × Tg-AD and APPswe/PS1ΔE9 mice (Liu et al., 2008; Moreno-Castilla et al., 2016), highlighting the role of the VTA and LC in the disease progression. Consistently with the aforementioned observations, pharmacological manipulations aimed at increasing the dopaminergic transmission in the hippocampus and cortex can improve synaptic functions, cognitive impairment and memory deficits in AD patients and in experimental models, emphasizing the critical role of dopamine for proper brain function (Monteverde et al., 1990; Tsunekawa et al., 2008; Himeno et al., 2011; Jürgensen et al., 2011; Koch et al., 2011, 2014; Guzmán-Ramos et al., 2012; Pazini et al., 2013; Martorana and Koch, 2014; Nobili et al., 2017).
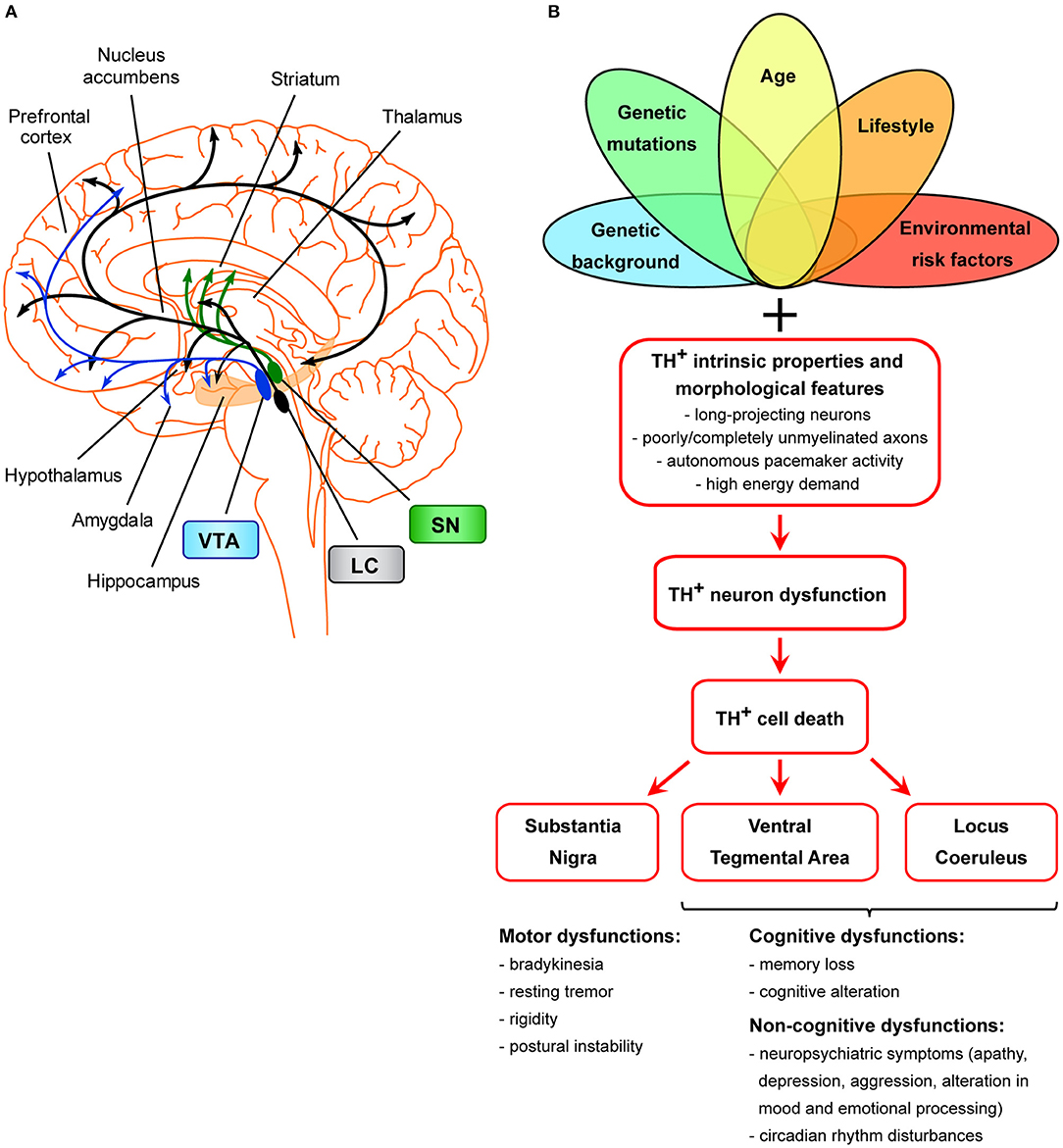
Figure 1. Scheme of the unifying hypothesis of dopamine neuron loss in neurodegenerative diseases. (A) Scheme of the dopaminergic projections arising from the VTA, SNpc, and LC. The dopaminergic neurons of the SNpc project mainly to the dorsal striatum (caudate and putamen), mediating the control of voluntary movements via the direct and indirect pathways (Calabresi et al., 2014). Degeneration of these neurons is the main cause of motor symptoms in PD (Surmeier, 2007). LC TH+ neurons innervate most of the cortex and subcortical regions and have been shown to be important for cognitive and non-cognitive functions, memory consolidation, attention, sleep-wake cycle, mood, and emotional processing (Palmer et al., 1987; Sara, 2009; Robertson et al., 2013; Kempadoo et al., 2016; Roy et al., 2016; Takeuchi et al., 2016; Borodovitsyna et al., 2017). The VTA is the origin of TH+ axons forming the mesocortical and the mesolimbic dopaminergic pathways, together called the mesocorticolimbic system (Gasbarri et al., 1994; Björklund and Dunnett, 2007). The mesocorticolimbic system projects primarily to the prefrontal cortex, hippocampus, NAc, olfactory bulb, amygdala, and anterior cingulate cortex. This system is implicated in motivated behaviors, reward and emotional processing, memory, and learning. Loss of VTA and LC TH+ neurons in AD is thought to be the basis for memory deficits, cognitive, and neuropsychiatric symptoms (Kelly et al., 2017; Nobili et al., 2017; D'Amelio et al., 2018a; Serra et al., 2018), while loss of VTA and LC TH+ neurons in PD is linked to the appearance of neuropsychiatric symptoms and dementia (Jellinger, 1991; German et al., 1992; Surmeier, 2007; Costa et al., 2012; Alberico et al., 2015; Caminiti et al., 2017). (B) Scheme of TH+ cell death in age-related neurodegeneration. Several factors (such as genetic background, genetic mutations, age, environmental or lifestyle risk factors), either alone or in combinations, might selectively dispose TH+ neurons of a specific area (SNpc, VTA, LC) to degenerate. Moreover, these neurons are more prone than others to cell death due to features that make them particularly sensitive: these cells are long-projecting neurons with poorly or completely unmyelinated axons that provide massive synaptic innervations in projecting areas (Braak et al., 2006), they have autonomous pacemaker activity due to which they have high requirements for energy and therefore also require efficient mitochondrial function. From a clinical point of view, the manifestation of clinical symptoms is strictly associated with the functions of SNpc-, VTA-, and/or LC-projecting areas.
Due to the strong involvement of LC TH+ neurons in prodromal AD subjects (Kelly et al., 2017), a clinical study made an effort to consolidate the events of TH+ neuron degeneration in AD by clarifying the temporal evolution of changes in the VTA and LC. The study recruited a large sample of patients with either amnestic mild cognitive impairment (MCI) or late-onset AD and healthy non-demented subjects and used resting-state functional MRI to assess the functional connectivity between the VTA or LC and other brain areas (Serra et al., 2018). The rational was that loss of neurons within a specific nucleus such as the VTA or LC would result in reduced connectivity between that nucleus and its projection areas. In line with the data from Tg2576 mice (Nobili et al., 2017), the study showed a significant disconnection between the VTA and the thalamus, medial-temporal structures and the parietal lobe in MCI patients, while these patients show no significant changes in LC connectivity. LC disconnection was only observed in late-AD patients and is distributed to the posterior parietal cortex (Serra et al., 2018). In MCI patients the VTA disconnection is predominant with hippocampal and parietal regions, while in AD patients it involves all nodes of the default-mode network (Gili et al., 2011; Bozzali et al., 2012; Serra et al., 2018). A second study used structural MRI to show that the volume of the VTA can be associated with the typical clinical markers of AD, namely the hippocampal size and memory index (D'Amelio et al., 2018b; De Marco and Venneri, 2018). The main finding was that a smaller VTA corresponds to worse indices of memory and hippocampal size. The authors did not find association between the SNpc and the size of the hippocampus in AD patients, excluding a generic reduction of the midbrain volume and confirming a prominent involvement of the VTA (De Marco and Venneri, 2018). These studies suggest that the VTA volume or connectivity might represent prodromal clinical markers, with the potential of predicting the conversion from healthy state to clinical AD.
From a clinical point of view, the VTA degeneration in AD could be particularly relevant for the manifestation of neuropsychiatric symptoms since mesocorticolimbic dopamine broadly regulates non-motor behavior like motivation, reward, pleasure, attention, emotional processing, and mood conditioning [Figure 1B; (Grace et al., 2007; Bromberg-Martin et al., 2010; Russo and Nestler, 2013)]. In fact, although AD is considered principally a cognitive and memory disorder, almost all patients develop neuropsychiatric symptoms and personality changes that could be explained by the reduced connectivity of the VTA with the default-mode network (Serra et al., 2018). These symptoms, particularly depression, aggression, personality changes and circadian rhythm disturbances, are frequently observed very early in AD (Lyketsos et al., 2011; Masters et al., 2015; Ismail et al., 2016; Alves et al., 2017; Musiek et al., 2018) and persist in parallel with the deterioration of cognitive function. Depression and apathy are most frequent in people with MCI and early AD, although the development of verbal and physical agitation is also quite evident. As the disease progresses, delusions, hallucinations, and aggression become more frequent, while apathy becomes the most persistent neuropsychiatric symptom throughout AD progression. Additionally, population studies show that circadian rhythm disturbances and the frequency of mood symptoms are much higher in people with AD or MCI compared to the general population (Lyketsos et al., 2002, 2011; Taragano et al., 2009). In fact, the MCI stage, today recognized as an intermediate state of clinical dementia (Petersen, 2009), is particularly important for early intervention because MCI patients progress to AD at a much higher rate if they show depression (Taragano et al., 2009). Direct comparison of patients with and without neuropsychiatric symptoms confirmed that patients with apathy, depression or anxiety have more severe functional disconnection between the VTA and the default-mode network compared to patients that lacked such symptoms (Serra et al., 2018). Surprisingly, besides AD, deficits in the mesolimbic dopamine system are considered the leading cause of neuropsychiatric symptoms and cognitive dysfunction occurring also in Parkinson's disease (PD). Indeed the VTA dopamine neuron loss has been associated to several non-motor symptoms which may precede parkinsonism by many years, reflecting the insurgence of dementia, anxiety and/or depression (Elgh et al., 2009; Gustafsson et al., 2015; Castrioto et al., 2016). In line with this, today it is generally recognized that, although neurodegenerative events affect more severely the nigrostriatal dopamine system in PD, the mesolimbic system is also compromised, even if to a lesser degree (Jellinger, 1991; Alberico et al., 2015; Caminiti et al., 2017). In fact, several analysis of PD brains showed about 50% loss of TH+ neurons in the A10 region corresponding to the VTA (Hirsch et al., 1988; German et al., 1989; Alberico et al., 2015).
Thus, it appears that the vulnerability of midbrain dopamine neurons is a common feature both in PD and AD, and the appearance of neuropsychiatric symptoms can be an important prognostic tool.
A Unifying Hypothesis of Dopamine Cell Death in Age-Related Neurodegeneration?
Although the Tg2576 mouse is a genetic model of AD (Hsiao et al., 1996) and early-onset AD is overall attributed to mutations in specific genes (Blennow et al., 2006), the experimental evidence that the first neurons to degenerate in Tg2576 mice are the dopaminergic neurons in the VTA (Nobili et al., 2017) suggests that these neurons are more susceptible than others to cell death. This indicates that several factors other than APP overexpression could contribute to their susceptibility. This observation is in line with the fact that alterations in the functional connectivity or volume of the VTA are observed in late-onset AD patients, overall suggesting that the susceptibility of dopamine neurons is a common theme in both early- and late-onset AD (D'Amelio et al., 2018a).
The increased vulnerability of VTA dopaminergic neurons in AD could be considered a symmetrical picture to the increased vulnerability of SNpc dopaminergic neurons in PD, or the degeneration of TH+ neurons in the LC in both AD and PD patients (German et al., 1992; Liu et al., 2008; Kelly et al., 2017; D'Amelio et al., 2018a). In fact, LC neurons might be considered as “atypical” dopaminergic cells that co-express dopamine with noradrenaline (Kempadoo et al., 2016; Takeuchi et al., 2016; Beas et al., 2018). These three TH+ regions (SNpc, VTA, LC) project to a widespread number of brain areas, each with a specific function (Figure 1B). Therefore, it is logical to assume that the different manifestation of clinical symptoms in AD, PD, or even PD with dementia, would be dependent on the brain areas that suffer mostly from the dopamine loss and on the degree of TH+ neuron degeneration.
What, then, causes TH+ cell death in age-related neurodegeneration? These neurons in SNpc, VTA and LC share common features that can increase their susceptibility. They are long-projecting neurons with poorly or completely unmyelinated axons that provide massive synaptic innervations in projecting areas (Braak et al., 2006). As such, efficient mechanisms of axonal transport and autophagy are essential for removal of damaged mitochondria and for regeneration of synaptic components. Additionally, these cells have autonomous pacemaker activity that might contribute to their vulnerability, due to the high requirements for energy and therefore efficient mitochondrial function.
These functional and structural features could lead to further compromise by the metabolism of dopamine, mitochondrial dysfunction, oxidative stress, impaired protein degradation, metal homeostasis, neurochemistry of metalloproteins and neuroinflammation (Schapira, 2001; Liss et al., 2005; Surmeier, 2007; Lim et al., 2016; Burbulla et al., 2017; Mor et al., 2017; Adlard and Bush, 2018). All these factors have been extensively studied in PD to explain the loss of SNpc neurons [reviewed in (Sulzer, 2007; Surmeier et al., 2011; Tambasco et al., 2018)] but their contribution to the vulnerability of VTA or LC dopamine neurons is yet to be defined. Thus, mitochondrial stress, deficits in calcium buffering or increased oxidation by dopamine by-products might all worsen the ability of dopamine neurons to defend themselves from internal or external risk factors.
Unlike the common features mentioned above, these neurons also have specific intrinsic characteristics that differentiate them from each other, such as different potassium and calcium conductances, calcium-binding proteins, or genetic profiling (Neuhoff et al., 2002; Liss et al., 2005; Lammel et al., 2008; Guzman et al., 2009; Khaliq and Bean, 2010; Panman et al., 2014; Poulin et al., 2014). These characteristics, either alone or in combinations also with genetic mutations, age, environmental, or lifestyle risk factors, can induce a selective vulnerability of different SNpc, VTA, and LC TH+ neuron subpopulations to cell death. Indeed, the enormous intrinsic variability in dopamine neuron subpopulations (Neuhoff et al., 2002; Lammel et al., 2008; Roeper, 2013; Krashia et al., 2017) might result in different pathophysiological responses to metabolic, genetic, environmental or lifestyle risk factors, and in this way might contribute to the differential vulnerability.
To summarize, in recent years increasing evidence has proven a strong association between deficits in dopaminergic signaling and several cognitive and neuropsychiatric alterations related to AD (Martorana and Koch, 2014; D'Amelio et al., 2018a). Novel knowledge on the vulnerability of VTA dopamine neurons in AD, explaining these deficits, is just starting to emerge. However, the unifying hypothesis of dopamine cell death in age-related neurodegeneration proposed here is highly appealing: lessons learned from PD studies can be essential for disentangling the enigma of dopamine neuron death in AD. We can conclude that, like PD, AD could be considered as a complex disease with multiple etiologies, external and internal, all converging at massive dopamine neuronal loss. This unifying hypothesis of dopamine cell death in age-related neurodegeneration is an interesting hypothesis that merits more thought.
Author Contributions
PK, AN, and MD equally contribute in writing the article.
Funding
MD was supported by an Alzheimer's Association's Research Grant (AARG-18-566270) and by the Italian Ministry of Health (Progetto Ricerca Finalizzata code RF-2018-12365527). AN was supported by a post-doctoral Fellowship by Collegio Ghislieri. PK was supported by a post-doctoral Fellowship by the Veronesi Foundation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
ADI (2016). World Alzheimer Report 2016—Improving Healthcare for People Living With Dementia: Coverage, Quality and Costs Now and in the Future.
Adlard, P. A., and Bush, A. I. (2018). Metals and Alzheimer's disease: how far have we come in the clinic? J. Alzheimers Dis. 62, 1369–1379. doi: 10.3233/JAD-170662
Alberico, S. L., Cassell, M. D., and Narayanan, N. S. (2015). The vulnerable ventral tegmental area in Parkinson's disease. Basal Ganglia 5, 51–55. doi: 10.1016/j.baga.2015.06.001
Alves, G. S., Carvalho, A. F., de Amorim de Carvalho, L., Sudo, F. K., Siqueira-Neto, J. I., Oertel-Knochel, V., et al. (2017). Neuroimaging findings related to behavioral disturbances in alzheimer's disease: a systematic review. Curr. Alzheimer Res. 14, 61–75. doi: 10.2174/1567205013666160603010203
Anderson, R. M., Hadjichrysanthou, C., Evans, S., and Wong, M. M. (2017). Why do so many clinical trials of therapies for Alzheimer's disease fail? Lancet 390, 2327–2329. doi: 10.1016/S0140-6736(17)32399-1
Banerjee, S. (2012). The macroeconomics of dementia–will the world economy get Alzheimer's disease? Arch. Med. Res. 43, 705–709. doi: 10.1016/j.arcmed.2012.10.006
Beas, B. S., Wright, B. J., Skirzewski, M., Leng, Y., Hyun, J. H., Koita, O., et al. (2018). The locus coeruleus drives disinhibition in the midline thalamus via a dopaminergic mechanism. Nat. Neurosci. 21, 963–973. doi: 10.1038/s41593-018-0167-4
Björklund, A., and Dunnett, S. B. (2007). Dopamine neuron systems in the brain: an update. Trends Neurosci. 30, 194–202. doi: 10.1016/j.tins.2007.03.006
Blennow, K., de Leon, M. J., and Zetterberg, H. (2006). Alzheimer's disease. Lancet 368, 387–403. doi: 10.1016/S0140-6736(06)69113-7
Borodovitsyna, O., Flamini, M., and Chandler, D. (2017). Noradrenergic modulation of cognition in health and disease. Neural Plast. 2017:6031478. doi: 10.1155/2017/6031478
Bozzali, M., Giulietti, G., Basile, B., Serra, L., Spanò, B., Perri, R., et al. (2012). Damage to the cingulum contributes to Alzheimer's disease pathophysiology by deafferentation mechanism. Hum. Brain Mapp. 33, 1295–1308. doi: 10.1002/hbm.21287
Braak, H., Bohl, J. R., Müller, C. M., Rüb, U., de Vos, R. A. I., and Tredici, K. D. (2006). Stanley fahn lecture 2005: The staging procedure for the inclusion body pathology associated with sporadic Parkinson's disease reconsidered. Mov. Disord. 21, 2042–2051. doi: 10.1002/mds.21065
Bromberg-Martin, E. S., Matsumoto, M., and Hikosaka, O. (2010). Dopamine in motivational control: rewarding, aversive, and alerting. Neuron 68, 815–834. doi: 10.1016/j.neuron.2010.11.022
Burbulla, L. F., Song, P., Mazzulli, J. R., Zampese, E., Wong, Y. C., Jeon, S., et al. (2017). Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson's disease. Science 357, 1255–1261. doi: 10.1126/science.aam9080
Calabresi, P., Picconi, B., Tozzi, A., Ghiglieri, V., and Di Filippo, M. (2014). Direct and indirect pathways of basal ganglia: a critical reappraisal. Nat. Neurosci. 17, 1022–1030. doi: 10.1038/nn.3743
Caminiti, S. P., Presotto, L., Baroncini, D., Garibotto, V., Moresco, R. M., Gianolli, L., et al. (2017). Axonal damage and loss of connectivity in nigrostriatal and mesolimbic dopamine pathways in early Parkinson's disease. Neuroimage Clin. 14, 734–740. doi: 10.1016/j.nicl.2017.03.011
Carroll, J. (2017). Another Alzheimer's Drug Flops in Pivotal Clinical Trial. AAAS. Science. Available online at: http://www.sciencemag.org/news/2017/02/another-alzheimers-drug-flops-pivotal-clinical-trial (accessed January 21, 2019).
Castrioto, A., Thobois, S., Carnicella, S., Maillet, A., and Krack, P. (2016). Emotional manifestations of PD: neurobiological basis. Mov. Disord. 31, 1103–1113. doi: 10.1002/mds.26587
Chételat, G. (2013). Alzheimer disease: Aβ-independent processes-rethinking preclinical AD. Nat. Rev. Neurol. 9, 123–124. doi: 10.1038/nrneurol.2013.21
Chételat, G., La Joie, R., Villain, N., Perrotin, A., de La Sayette, V., Eustache, F., et al. (2013). Amyloid imaging in cognitively normal individuals, at-risk populations and preclinical Alzheimer's disease. Neuroimage Clin. 2, 356–365. doi: 10.1016/j.nicl.2013.02.006
Coleman, P. D., and Mastroeni, D. (2017). A call for new approaches to Alzheimer's disease research. neurobiol. Aging 57, iii–iv. doi: 10.1016/j.neurobiolaging.2017.04.027
Cordella, A., Krashia, P., Nobili, A., Pignataro, A., La Barbera, L., Viscomi, M. T., et al. (2018). Dopamine loss alters the hippocampus-nucleus accumbens synaptic transmission in the Tg2576 mouse model of Alzheimer's disease. Neurobiol. Dis. 116, 142–154. doi: 10.1016/j.nbd.2018.05.006
Corder, E. H., Saunders, A. M., Strittmatter, W. J., Schmechel, D. E., Gaskell, P. C., Small, G. W., et al. (1993). Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science 261, 921–923.
Costa, C., Sgobio, C., Siliquini, S., Tozzi, A., Tantucci, M., Ghiglieri, V., et al. (2012). Mechanisms underlying the impairment of hippocampal long-term potentiation and memory in experimental Parkinson's disease. Brain 135, 1884–1899. doi: 10.1093/brain/aws101
Cummings, J. L., Morstorf, T., and Zhong, K. (2014). Alzheimer's disease drug-development pipeline: few candidates, frequent failures. Alzheimers Res. Ther. 6:37. doi: 10.1186/alzrt269
D'Amelio, M., and Nisticò, R. (2018). Unlocking the secrets of dopamine in Alzheimer's disease. Pharmacol. Res. 128:399. doi: 10.1016/j.phrs.2017.06.018
D'Amelio, M., Puglisi-Allegra, S., and Mercuri, N. (2018a). The role of dopaminergic midbrain in Alzheimer's disease: translating basic science into clinical practice. Pharmacol. Res. 130, 414–419. doi: 10.1016/j.phrs.2018.01.016
D'Amelio, M., and Rossini, P. M. (2012). Brain excitability and connectivity of neuronal assemblies in Alzheimer's disease: from animal models to human findings. Prog. Neurobiol. 99, 42–60. doi: 10.1016/j.pneurobio.2012.07.001
D'Amelio, M., Serra, L., and Bozzali, M. (2018b). Ventral tegmental area in prodromal Alzheimer's disease: bridging the gap between mice and humans. J. Alzheimers Dis. 63, 181–183. doi: 10.3233/JAD-180094
Davis, D. G., Schmitt, F. A., Wekstein, D. R., and Markesbery, W. R. (1999). Alzheimer neuropathologic alterations in aged cognitively normal subjects. J. Neuropathol. Exp. Neurol. 58, 376–388.
De Marco, M., and Venneri, A. (2018). Volume and connectivity of the ventral tegmental area are linked to neurocognitive signatures of Alzheimer's disease in humans. J. Alzheimers Dis. 63, 167–180. doi: 10.3233/JAD-171018
Doody, R. S., Raman, R., Farlow, M., Iwatsubo, T., Vellas, B., Joffe, S., et al. (2013). A phase 3 trial of semagacestat for treatment of Alzheimer's disease. N. Engl. J. Med. 369, 341–350. doi: 10.1056/NEJMoa1210951
Edison, P., Archer, H. A., Hinz, R., Hammers, A., Pavese, N., Tai, Y. F., et al. (2007). Amyloid, hypometabolism, and cognition in Alzheimer disease: an [11C]PIB and [18F]FDG PET study. Neurology 68, 501–508. doi: 10.1212/01.wnl.0000244749.20056.d4
Elgh, E., Domellöf, M., Linder, J., Edström, M., Stenlund, H., and Forsgren, L. (2009). Cognitive function in early Parkinson's disease: a population-based study. Eur. J. Neurol. 16, 1278–1284. doi: 10.1111/j.1468-1331.2009.02707.x
Fagan, A. M., Mintun, M. A., Shah, A. R., Aldea, P., Roe, C. M., Mach, R. H., et al. (2009). Cerebrospinal fluid tau and ptau(181) increase with cortical amyloid deposition in cognitively normal individuals: implications for future clinical trials of Alzheimer's disease. EMBO Mol. Med. 1, 371–380. doi: 10.1002/emmm.200900048
Gasbarri, A., Verney, C., Innocenzi, R., Campana, E., and Pacitti, C. (1994). Mesolimbic dopaminergic neurons innervating the hippocampal formation in the rat: a combined retrograde tracing and immunohistochemical study. Brain Res. 668, 71–79.
Gauthier, S., Albert, M., Fox, N., Goedert, M., Kivipelto, M., Mestre-Ferrandiz, J., et al. (2016). Why has therapy development for dementia failed in the last two decades? Alzheimers Dement. 12, 60–64. doi: 10.1016/j.jalz.2015.12.003
German, D. C., Manaye, K., Smith, W. K., Woodward, D. J., and Saper, C. B. (1989). Midbrain dopaminergic cell loss in Parkinson's disease: computer visualization. Ann. Neurol. 26, 507–514. doi: 10.1002/ana.410260403
German, D. C., Manaye, K. F., White, C. L., Woodward, D. J., McIntire, D. D., Smith, W. K., et al. (1992). Disease-specific patterns of locus coeruleus cell loss. Ann. Neurol. 32, 667–676. doi: 10.1002/ana.410320510
Gili, T., Cercignani, M., Serra, L., Perri, R., Giove, F., Maraviglia, B., et al. (2011). Regional brain atrophy and functional disconnection across Alzheimer's disease evolution. J. Neurol. Neurosurg. Psychiatry 82, 58–66. doi: 10.1136/jnnp.2009.199935
Grace, A. A., Floresco, S. B., Goto, Y., and Lodge, D. J. (2007). Regulation of firing of dopaminergic neurons and control of goal-directed behaviors. Trends Neurosci. 30, 220–227. doi: 10.1016/j.tins.2007.03.003
Guerreiro, R. J., Gustafson, D. R., and Hardy, J. (2012). The genetic architecture of Alzheimer's disease: beyond APP, PSENs and APOE. Neurobiol. Aging 33, 437–456. doi: 10.1016/j.neurobiolaging.2010.03.025
Gustafsson, H., Nordström, A., and Nordström, P. (2015). Depression and subsequent risk of Parkinson disease: a nationwide cohort study. Neurology 84, 2422–2429. doi: 10.1212/WNL.0000000000001684
Guzman, J. N., Sánchez-Padilla, J., Chan, C. S., and Surmeier, D. J. (2009). Robust pacemaking in substantia nigra dopaminergic neurons. J. Neurosci. 29, 11011–11019. doi: 10.1523/JNEUROSCI.2519-09.2009
Guzmán-Ramos, K., Moreno-Castilla, P., Castro-Cruz, M., McGaugh, J. L., Martínez-Coria, H., LaFerla, F. M., et al. (2012). Restoration of dopamine release deficits during object recognition memory acquisition attenuates cognitive impairment in a triple transgenic mice model of Alzheimer's disease. Learn. Mem. 19, 453–460. doi: 10.1101/lm.026070.112
Hardy, J., and Selkoe, D. J. (2002). The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297, 353–356. doi: 10.1126/science.1072994
Herrup, K. (2015). The case for rejecting the amyloid cascade hypothesis. Nat. Neurosci. 18, 794–799. doi: 10.1038/nn.4017
Himeno, E., Ohyagi, Y., Ma, L., Nakamura, N., Miyoshi, K., Sakae, N., et al. (2011). Apomorphine treatment in Alzheimer mice promoting amyloid-β degradation. Ann. Neurol. 69, 248–256. doi: 10.1002/ana.22319
Hirsch, E., Graybiel, A. M., and Agid, Y. A. (1988). Melanized dopaminergic neurons are differentially susceptible to degeneration in Parkinson's disease. Nature 334, 345–348. doi: 10.1038/334345a0
Honig, L. S., Vellas, B., Woodward, M., Boada, M., Bullock, R., Borrie, M., et al. (2018). Trial of solanezumab for mild dementia due to Alzheimer's disease. N. Engl. J. Med. 378, 321–330. doi: 10.1056/NEJMoa1705971
Hsiao, K., Chapman, P., Nilsen, S., Eckman, C., Harigaya, Y., Younkin, S., et al. (1996). Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 274, 99–102.
Ismail, Z., Smith, E. E., Geda, Y., Sultzer, D., Brodaty, H., Smith, G., et al. (2016). Neuropsychiatric symptoms as early manifestations of emergent dementia: Provisional diagnostic criteria for mild behavioral impairment. Alzheimers Dement. 12, 195–202. doi: 10.1016/j.jalz.2015.05.017
Jellinger, K. A. (1991). Pathology of Parkinson's disease. Changes other than the nigrostriatal pathway. Mol. Chem. Neuropathol. 14, 153–197.
Jürgensen, S., Antonio, L. L., Mussi, G. E. A., Brito-Moreira, J., Bomfim, T. R., De Felice, F. G., et al. (2011). Activation of D1/D5 dopamine receptors protects neurons from synapse dysfunction induced by amyloid-beta oligomers. J. Biol. Chem. 286, 3270–3276. doi: 10.1074/jbc.M110.177790
Kelly, S. C., He, B., Perez, S. E., Ginsberg, S. D., Mufson, E. J., and Counts, S. E. (2017). Locus coeruleus cellular and molecular pathology during the progression of Alzheimer's disease. Acta Neuropathol Commun. 5:8. doi: 10.1186/s40478-017-0411-2
Kempadoo, K. A., Mosharov, E. V., Choi, S. J., Sulzer, D., and Kandel, E. R. (2016). Dopamine release from the locus coeruleus to the dorsal hippocampus promotes spatial learning and memory. Proc. Natl. Acad. Sci. U.S.A. 113, 14835–14840. doi: 10.1073/pnas.1616515114
Khaliq, Z. M., and Bean, B. P. (2010). Pacemaking in dopaminergic ventral tegmental area neurons: depolarizing drive from background and voltage-dependent sodium conductances. J. Neurosci. 30, 7401–7413. doi: 10.1523/JNEUROSCI.0143-10.2010
Kivipelto, M., Helkala, E. L., Laakso, M. P., Hänninen, T., Hallikainen, M., Alhainen, K., et al. (2001). Midlife vascular risk factors and Alzheimer's disease in later life: longitudinal, population based study. BMJ 322, 1447–1451. doi: 10.1136/bmj.322.7300.1447
Knight, E. M., Martins, I. V. A., Gümüsgöz, S., Allan, S. M., and Lawrence, C. B. (2014). High-fat diet-induced memory impairment in triple-transgenic Alzheimer's disease (3xTgAD) mice is independent of changes in amyloid and tau pathology. Neurobiol. Aging 35, 1821–1832. doi: 10.1016/j.neurobiolaging.2014.02.010
Koch, G., Di Lorenzo, F., Bonnì, S., Giacobbe, V., Bozzali, M., Caltagirone, C., et al. (2014). Dopaminergic modulation of cortical plasticity in Alzheimer's disease patients. Neuropsychopharmacology 39, 2654–2661. doi: 10.1038/npp.2014.119
Koch, G., Esposito, Z., Codecà, C., Mori, F., Kusayanagi, H., Monteleone, F., et al. (2011). Altered dopamine modulation of LTD-like plasticity in Alzheimer's disease patients. Clin. Neurophysiol. 122, 703–707. doi: 10.1016/j.clinph.2010.10.033
Krashia, P., Martini, A., Nobili, A., Aversa, D., D'Amelio, M., Berretta, N., et al. (2017). On the properties of identified dopaminergic neurons in the mouse substantia nigra and ventral tegmental area. Eur. J. Neurosci. 45, 92–105. doi: 10.1111/ejn.13364
Lammel, S., Hetzel, A., Häckel, O., Jones, I., Liss, B., and Roeper, J. (2008). Unique properties of mesoprefrontal neurons within a dual mesocorticolimbic dopamine system. Neuron 57, 760–773. doi: 10.1016/j.neuron.2008.01.022
Li, Y., Rinne, J. O., Mosconi, L., Pirraglia, E., Rusinek, H., DeSanti, S., et al. (2008). Regional analysis of FDG and PIB-PET images in normal aging, mild cognitive impairment, and Alzheimer's disease. Eur. J. Nucl. Med. Mol. Imaging 35, 2169–2181. doi: 10.1007/s00259-008-0833-y
Lim, S., Chun, Y., Lee, J. S., and Lee, S.-J. (2016). Neuroinflammation in synucleinopathies. Brain Pathol. 26, 404–409. doi: 10.1111/bpa.12371
Liss, B., Haeckel, O., Wildmann, J., Miki, T., Seino, S., and Roeper, J. (2005). K-ATP channels promote the differential degeneration of dopaminergic midbrain neurons. Nat. Neurosci. 8, 1742–1751. doi: 10.1038/nn1570
Liu, Y., Yoo, M.-J., Savonenko, A., Stirling, W., Price, D. L., Borchelt, D. R., et al. (2008). Amyloid pathology is associated with progressive monoaminergic neurodegeneration in a transgenic mouse model of Alzheimer's disease. J. Neurosci. 28, 13805–13814. doi: 10.1523/JNEUROSCI.4218-08.2008
Lyketsos, C. G., Carrillo, M. C., Ryan, J. M., Khachaturian, A. S., Trzepacz, P., Amatniek, J., et al. (2011). Neuropsychiatric symptoms in Alzheimer's disease. Alzheimers Dement. 7, 532–539. doi: 10.1016/j.jalz.2011.05.2410
Lyketsos, C. G., Lopez, O., Jones, B., Fitzpatrick, A. L., Breitner, J., and DeKosky, S. (2002). Prevalence of neuropsychiatric symptoms in dementia and mild cognitive impairment: results from the cardiovascular health study. JAMA 288, 1475–1483. doi: 10.1001/jama.288.12.1475
Martorana, A., and Koch, G. (2014). Is dopamine involved in Alzheimer's disease? Front Aging Neurosci. 6:252. doi: 10.3389/fnagi.2014.00252
Masters, M. C., Morris, J. C., and Roe, C. M. (2015). “Noncognitive” symptoms of early Alzheimer disease: a longitudinal analysis. Neurology 84, 617–622. doi: 10.1212/WNL.0000000000001238
Mayeux, R., and Stern, Y. (2012). Epidemiology of Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2:a006239. doi: 10.1101/cshperspect.a006239
Monteverde, A., Gnemmi, P., Rossi, F., Monteverde, A., and Finali, G. C. (1990). Selegiline in the treatment of mild to moderate Alzheimer-type dementia. Clin. Ther. 12, 315–322.
Mor, D. E., Tsika, E., Mazzulli, J. R., Gould, N. S., Kim, H., Daniels, M. J., et al. (2017). Dopamine induces soluble α-synuclein oligomers and nigrostriatal degeneration. Nat. Neurosci. 20, 1560–1568. doi: 10.1038/nn.4641
Moreno-Castilla, P., Rodriguez-Duran, L. F., Guzman-Ramos, K., Barcenas-Femat, A., Escobar, M. L., and Bermudez-Rattoni, F. (2016). Dopaminergic neurotransmission dysfunction induced by amyloid-β transforms cortical long-term potentiation into long-term depression and produces memory impairment. Neurobiol. Aging 41, 187–199. doi: 10.1016/j.neurobiolaging.2016.02.021
Musiek, E. S., Bhimasani, M., Zangrilli, M. A., Morris, J. C., Holtzman, D. M., and Ju, Y.-E. S. (2018). Circadian rest-activity pattern changes in aging and preclinical Alzheimer Disease. JAMA Neurol. 75, 582–590. doi: 10.1001/jamaneurol.2017.4719
Neuhoff, H., Neu, A., Liss, B., and Roeper, J. (2002). I(h) channels contribute to the different functional properties of identified dopaminergic subpopulations in the midbrain. J. Neurosci. 22, 1290–1302. doi: 10.1523/JNEUROSCI.22-04-01290.2002
Nobili, A., Latagliata, E. C., Viscomi, M. T., Cavallucci, V., Cutuli, D., Giacovazzo, G., et al. (2017). Dopamine neuronal loss contributes to memory and reward dysfunction in a model of Alzheimer's disease. Nat. Commun. 8:14727. doi: 10.1038/ncomms14727
Noebels, J. (2011). A perfect storm: converging paths of epilepsy and Alzheimer's dementia intersect in the hippocampal formation. Epilepsia 1, 39–46. doi: 10.1111/j.1528-1167.2010.02909.x
Palmer, A. M., Wilcock, G. K., Esiri, M. M., Francis, P. T., and Bowen, D. M. (1987). Monoaminergic innervation of the frontal and temporal lobes in Alzheimer's disease. Brain Res. 401, 231–238.
Pandis, D., and Scarmeas, N. (2012). Seizures in Alzheimer disease: clinical and epidemiological data. Epilepsy Curr. 12, 184–187. doi: 10.5698/1535-7511-12.5.184
Panman, L., Papathanou, M., Laguna, A., Oosterveen, T., Volakakis, N., Acampora, D., et al. (2014). Sox6 and Otx2 control the specification of substantia nigra and ventral tegmental area dopamine neurons. Cell Rep. 8, 1018–1025. doi: 10.1016/j.celrep.2014.07.016
Pazini, A. M., Gomes, G. M., Villarinho, J. G., da Cunha, C., Pinheiro, F., Ferreira, A. P. O., et al. (2013). Selegiline reverses aβ25–35-induced cognitive deficit in male mice. Neurochem. Res. 38, 2287–2294. doi: 10.1007/s11064-013-1137-6
Petersen, R. C. (2009). Early diagnosis of Alzheimer's disease: is MCI too late? Curr. Alzheimer Res. 6, 324–330. doi: 10.2174/156720509788929237
Poulin, J.-F., Zou, J., Drouin-Ouellet, J., Kim, K.-Y. A., Cicchetti, F., and Awatramani, R. B. (2014). Defining midbrain dopaminergic neuron diversity by single-cell gene profiling. Cell Rep. 9, 930–943. doi: 10.1016/j.celrep.2014.10.008
Price, J. L., McKeel, D. W., Buckles, V. D., Roe, C. M., Xiong, C., Grundman, M., et al. (2009). Neuropathology of nondemented aging: presumptive evidence for preclinical Alzheimer disease. Neurobiol. Aging 30, 1026–1036. doi: 10.1016/j.neurobiolaging.2009.04.002
Qiu, C., Kivipelto, M., and von Strauss, E. (2009). Epidemiology of Alzheimer's disease: occurrence, determinants, and strategies toward intervention. Dialogues Clin. Neurosci. 11, 111–128.
Robertson, S. D., Plummer, N. W., de Marchena, J., and Jensen, P. (2013). Developmental origins of central norepinephrine neuron diversity. Nat. Neurosci. 16, 1016–1023. doi: 10.1038/nn.3458
Roeper, J. (2013). Dissecting the diversity of midbrain dopamine neurons. Trends Neurosci. 36, 336–342. doi: 10.1016/j.tins.2013.03.003
Roy, D. S., Arons, A., Mitchell, T. I., Pignatelli, M., Ryan, T. J., and Tonegawa, S. (2016). Memory retrieval by activating engram cells in mouse models of early Alzheimer's disease. Nature 531, 508–512. doi: 10.1038/nature17172
Russo, S. J., and Nestler, E. J. (2013). The brain reward circuitry in mood disorders. Nat. Rev. Neurosci. 14, 609–625. doi: 10.1038/nrn3381
Sara, S. J. (2009). The locus coeruleus and noradrenergic modulation of cognition. Nat. Rev. Neurosci. 10, 211–223. doi: 10.1038/nrn2573
Schapira, A. H. (2001). Causes of neuronal death in Parkinson's disease. Adv. Neurol. 86, 155–162. doi: 10.1007/s10495-008-0309-3
Scheff, S. W., Price, D. A., Schmitt, F. A., and Mufson, E. J. (2006). Hippocampal synaptic loss in early Alzheimer's disease and mild cognitive impairment. Neurobiol. Aging 27, 1372–1384. doi: 10.1016/j.neurobiolaging.2005.09.012
Serra, L., D'Amelio, M., Di Domenico, C., Dipasquale, O., Marra, C., Mercuri, N. B., et al. (2018). In vivo mapping of brainstem nuclei functional connectivity disruption in Alzheimer's disease. Neurobiol. Aging 72, 72–82. doi: 10.1016/j.neurobiolaging.2018.08.012
Sulzer, D. (2007). Multiple hit hypotheses for dopamine neuron loss in Parkinson's disease. Trends Neurosci. 30, 244–250. doi: 10.1016/j.tins.2007.03.009
Surmeier, D. J. (2007). Calcium, ageing, and neuronal vulnerability in Parkinson's disease. Lancet Neurol. 6, 933–938. doi: 10.1016/S1474-4422(07)70246-6
Surmeier, D. J., Guzman, J. N., Sanchez-Padilla, J., and Schumacker, P. T. (2011). The role of calcium and mitochondrial oxidant stress in the loss of substantia nigra pars compacta dopaminergic neurons in Parkinson's disease. Neuroscience 198, 221–231. doi: 10.1016/j.neuroscience.2011.08.045
Takeuchi, T., Duszkiewicz, A. J., Sonneborn, A., Spooner, P. A., Yamasaki, M., Watanabe, M., et al. (2016). Locus coeruleus and dopaminergic consolidation of everyday memory. Nature 537, 357–362. doi: 10.1038/nature19325
Tambasco, N., Romoli, M., and Calabresi, P. (2018). Selective basal ganglia vulnerability to energy deprivation: experimental and clinical evidences. Prog. Neurobiol. 169, 55–75. doi: 10.1016/j.pneurobio.2018.07.003
Tanzi, R. E., and Bertram, L. (2005). Twenty years of the Alzheimer's disease amyloid hypothesis: a genetic perspective. Cell 120, 545–555. doi: 10.1016/j.cell.2005.02.008
Taragano, F., Allegri, R., Krupitzki, H., Sarasola, D., Serrano, C., Loñ, L., et al. (2009). Mild behavioral impairment and risk of dementia. J. Clin. Psychiatry 70, 584–592. doi: 10.4088/JCP.08m04181
Tsunekawa, H., Noda, Y., Mouri, A., Yoneda, F., and Nabeshima, T. (2008). Synergistic effects of selegiline and donepezil on cognitive impairment induced by amyloid beta (25–35). Behav. Brain Res. 190, 224–232. doi: 10.1016/j.bbr.2008.03.002
Vandenberghe, R., Rinne, J. O., Boada, M., Katayama, S., Scheltens, P., Vellas, B., et al. (2016). Bapineuzumab for mild to moderate Alzheimer's disease in two global, randomized, phase 3 trials. Alzheimers Res. Ther. 8:18. doi: 10.1186/s13195-016-0189-7
Keywords: Alzheimer, dopamine neuron, Parkinson, VTA = ventral tegmental area, degeneration, neuropsychiatric symptoms, midbrain, Tg2576
Citation: Krashia P, Nobili A and D'Amelio M (2019) Unifying Hypothesis of Dopamine Neuron Loss in Neurodegenerative Diseases: Focusing on Alzheimer's Disease. Front. Mol. Neurosci. 12:123. doi: 10.3389/fnmol.2019.00123
Received: 02 February 2019; Accepted: 25 April 2019;
Published: 17 May 2019.
Edited by:
Rubén Quintana-Cabrera, University of Salamanca, SpainReviewed by:
Tito Cali', University of Padova, ItalyPaula Merino-Serrais, Cajal Institute (CSIC), Spain
Copyright © 2019 Krashia, Nobili and D'Amelio. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marcello D'Amelio, bS5kYW1lbGlvQHVuaWNhbXB1cy5pdA==
†These authors have contributed equally to this work