 Vanessa Castelli1
Vanessa Castelli1 Elisabetta Benedetti1Andrea Antonosante1Mariano Catanesi1Giuseppina Pitari1
Elisabetta Benedetti1Andrea Antonosante1Mariano Catanesi1Giuseppina Pitari1 Rodolfo Ippoliti1
Rodolfo Ippoliti1 Annamaria Cimini1,2*Michele d’Angelo1*
Annamaria Cimini1,2*Michele d’Angelo1*- 1Department of Life, Health and Environmental Sciences, University of L’Aquila, Abruzzo, Italy
- 2Sbarro Institute for Cancer Research and Molecular Medicine, Department of Biology, Temple University, Philadelphia, PA, United States
Brain cells normally respond adaptively to oxidative stress or bioenergetic challenges, resulting from ongoing activity in neuronal circuits. During aging and in neurodegenerative disorders, these mechanisms are compromised. In fact, neurons show unique age-related changes in functions and metabolism, resulting in greater susceptibility to insults and disease. Aging affects the nervous system as well as other organs. More precisely, as the nervous system ages, neuron metabolism may change, inducing glucose hypometabolism, impaired transport of critical substrates underlying metabolism, alterations in calcium signaling, and mitochondrial dysfunction. Moreover, in neuronal aging, an accumulation of impaired and aggregated proteins in the cytoplasm and in mitochondria is observed, as the result of oxidative stress: reduced antioxidant defenses and/or increase of reactive oxygen species (ROS). These changes lead to greater vulnerability of neurons in various regions of the brain and increased susceptibility to several diseases. Specifically, the first part of the review article will focus on the major neuronal cells’ rearrangements during aging in response to changes in metabolism and oxidative stress, while the second part will cover the neurodegenerative disease areas in detail.
Introduction
Aging affects the nervous system as well as other organs. In fact, neuronal cells are subjected to damaged protein accumulation, increased oxidative stress, perturbed energy homeostasis and mutations in nucleic acids. These alterations occur in normal aging and are exacerbated in vulnerable neuronal populations in neurodegenerative diseases.
During aging, the brain gradually declines leading to memory, learning, motor coordination and attention impairment (Alexander et al., 2012; Dykiert et al., 2012; Levin et al., 2014). Elderly people show difficulties in the understanding of rapid speech and complex syntaxes, due to cognitive impairment, but also due to hearing loss (Alexander et al., 2012). Brain decline parallels the aging of other organs, with a progressive deterioration after 50 years (Mendonca et al., 2017). At the 60-year threshold, most people become increasingly inclined to develop neurodegenerative diseases, such as Alzheimer’s disease (AD) and Parkinson’s disease (PD; Mattson, 2004; Kalia and Lang, 2015; Scheltens et al., 2016; Aarsland et al., 2017).
Interestingly, it has been reported that the human brain shrinks in an age-related manner, mostly in the temporal and frontal lobes (Peters, 2006). In fact, the development of brain atrophy during aging may anticipate the risk of developing cognitive impairment and dementia (Jack et al., 2005).
Aging
Oxidative Imbalance
Brain is susceptible to oxidative stress because of the elevated content of lipids—especially localized in the neuronal membrane—and the oxidative metabolism occurring in neurons. Tight balance between oxidative stress and the antioxidant system is necessary to preserve the structural integrity and the optimal brain functions (Birben et al., 2012; Castelli et al., 2018). Oxidative damage is involved in aging and age-associated cognitive impairment. During aging, neurons tend to accumulate impaired and aggregated proteins, and damaged mitochondria, as a consequence of oxidative stress. Increase of reactive oxygen species (ROS) production and/or decrease in antioxidant scavengers are the major players in this process (Halliwell, 2001; Figure 1).
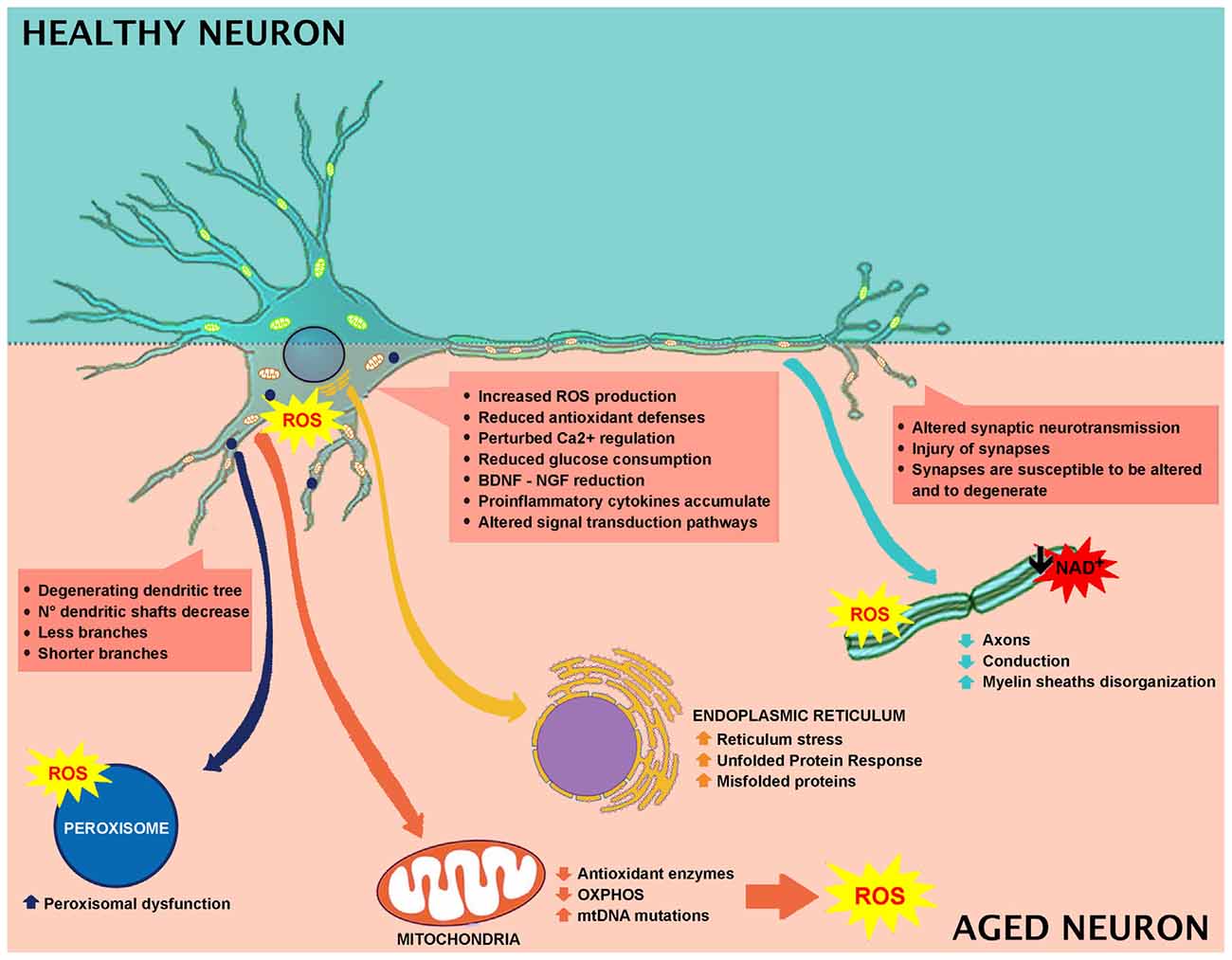
Figure 1. Effect of aging in neuronal cell and involved mechanisms.
Cognitive impairment (Berr et al., 2000; Serrano and Klann, 2004; Kim et al., 2015) and oxidative brain injuries are mainly due to lipid peroxidation products (Calabrese et al., 2004; Richwine et al., 2005; Zhu et al., 2006), protein oxidation (Abd El Mohsen et al., 2005; Rodrigues Siqueira et al., 2005; Poon et al., 2006), and oxidative alterations in nuclear and mitochondrial DNA (mtDNA; Hamilton et al., 2001). Elevated levels of protein carbonylation have been reported in different parts of the brain, among which includes the hippocampus (Abd El Mohsen et al., 2005; Rodrigues Siqueira et al., 2005). The principal neuronal cells ROS source originates from mitochondrial respiration and leads to increased intracellular calcium levels (Halliwell, 2001; Figure 1).
The age-related memory impairment is associated with decreased levels of antioxidants in the brain and plasma (Perkins et al., 1999; Berr et al., 2000; Rinaldi et al., 2003). In physiological conditions, two principal defense mechanisms required to counteract ROS-mediated damage are activated in the brain: the antioxidant enzymes and non-enzymatic antioxidants (Kohen et al., 2000; Kohen and Nyska, 2002). Among the ROS scavenger enzymes, superoxide dismutase (SOD), glutathione reductase, glyoxalase, glutathione peroxidase, and catalase (CAT; Griendling et al., 2000) are comprised. It has been widely demonstrated in various animal models that the intracellular glutathione concentration and the ratio of glutathione/glutathione disulfide (GSH/GSSG ratio) declines in an age-related manner (Sasaki et al., 2001; Sandhu and Kaur, 2002; Rebrin et al., 2003; Wang et al., 2003; Suh et al., 2004, 2005). This event has been described in all of the mammalian brain regions analyzed, including the hippocampus (Calabrese et al., 2004; Balu et al., 2005; Donahue et al., 2006; Zhu et al., 2006). In this condition, Nrf2 (nuclear factor erythroid 2-related factor) transcription factor switches on. The Nrf2 pathway is activated to counteract the accumulation of ROS and reactive nitrogen species since its activation increases the expression of several endogenous antioxidants. Aged animals show a decrease in Nrf2 activation compared to younger subjects (Safdar et al., 2010; Gounder et al., 2012). In agreement, in aged brains, as well as in other tissues, Nrf2 expression is decreased (Duan et al., 2009; Ungvari et al., 2011), paralleled by an increase in inflammatory genes in these organs (Ungvari et al., 2011). For this reason, Nrf2 has been suggested as a potential therapeutic target in diseases with neuroinflammatory and oxidative properties (Salim, 2017).
The mechanisms by which ROS leads to cerebral tissue damage is not clear. ROS may induce the activation of different molecular signaling pathways, including neuroinflammation and neuronal death (Gu et al., 2011; Figure 1), with the involvement of glutamate toxicity, aspartate receptor signaling and glucocorticoid receptor signaling (Albrecht et al., 2010; Nguyen et al., 2011). Biochemically, neurons have different susceptibilities to oxidative stress. For example, amygdala, hippocampus, and cerebellar granule cells are more vulnerable to oxidative stress (Wang and Michaelis, 2010), and accordingly are the first to degenerate. Interestingly, the increased vulnerability of the amygdala and the hippocampus to oxidative stress, together with the lack of antioxidant defenses, alter the biochemical integrity of the brain. Additionally, oxidative stress also affects glutamatergic receptors, responsible for the long-term potentiation and synaptic transmission (Haxaire et al., 2012; Lee et al., 2012; Rai et al., 2013). Altogether, these events underlay oxidative stress-induced cognitive decline.
Organelle Dynamics
The most typical responses to brain perturbations involve distinct organelles, including the endoplasmic reticulum, which allows the mediation of the Unfolded Protein Response (UPR), the mitochondria, which plays a leading role in regulating apoptosis, the nucleus, in which DNA lesions can trigger stress, and peroxisomes, which have a significant role in the response to oxidative stress (Figure 1).
Alterations in energy or nutrient balance, and changes in calcium signaling or redox status modify endoplasmic reticulum homeostasis, triggering reticulum stress, increased misfolded proteins, and upregulation of UPR. In fact, during aging, there is an imbalance between UPR protective adaptive response and pro-apoptotic signaling in favor of apoptosis (Paz Gavilán et al., 2006; Hussain and Ramaiah, 2007; Naidoo et al., 2008) with the consequent alteration of the capability to maintain proper protein folding and ER homeostasis (Figure 1).
Mitochondrial structure does not change during aging (Ledda et al., 2000; Martinelli et al., 2006). These organelles are mainly localized in neuronal axons and dendrites and are responsible for ATP generation, necessary for maintaining electrochemical neurotransmission and for cell repair (Mattson et al., 2008). One of the most usual consequences of the natural aging process is the reduced level of Ca2+-binding proteins.
Beyond their pivotal role in cellular energy metabolism production, mitochondria are also involved in cellular Ca2+ homeostasis and nuclear gene transcription regulation (Hou et al., 2012; Yun and Finkel, 2014; Raefsky and Mattson, 2017). Calcium signaling plays a fundamental role in neuronal function, regulation and the structural adaptive mechanism (Cohen et al., 2015). In particular, Ca2+ contributes to synaptic activity and is involved in the transmission of the depolarizing signal. During aging the ability of neuronal cells to maintain a proper energy level can be compromised, thus impacting on Ca2+ homeostasis and determining weakened control Ca2+ dynamics. As a consequence, an aberrant cytoplasmatic Ca2+ level occurs, paralleled by perturbed cytoskeletal dynamics and abnormal gene expression (Thibault et al., 2001; Toescu et al., 2004; Gant et al., 2006; Porte et al., 2008). It is clear that altered Ca2+ signaling is involved in impaired cognition. The restoration of Ca2+ homeostasis in aged rats leads, in fact, to an improvement of cognitive performances (Fukushima et al., 2008; Gant et al., 2015). Moreover, hippocampal neurons are more susceptible to Ca2+-excitotoxicity (Camandola and Mattson, 2011). Ca2+-dependent calpains activation is involved in neuronal injury and cell death, due to PARP1-mediated apoptosis (Mattson, 2000; Nixon, 2003; Fatokun et al., 2014).
Furthermore, the generation of membrane permeability transition pores is crucial in the apoptosis process (Mattson, 2000). Changes in brain energy metabolism can cause weakening of neuronal functions and alterations leading to neuronal death (Mattson et al., 2008; Grimm and Eckert, 2017; Figure 1).
As indicated above, in the aged brain, a failure in the normal antioxidant defense mechanisms occurs, which renders the brain more vulnerable to the lethal consequences of oxidative stress (Finkel and Holbrook, 2000). It has been reported that mitochondrial-free radicals are responsible for mtDNA damage. Post-mortem brains of elderly showed elevated levels of 8-hydroxy-2-deoxyguanosine in both nuclear DNA (nDNA) and in mtDNA. Moreover, some researchers indicated greater damage in mtDNA than in nDNA in aged rodents (Barja and Herrero, 2000; Barja, 2004; Yang et al., 2008).
During aging, mtDNA oxidative-induced mutations accumulate in post-mitotic tissues, including the brain (Chomyn and Attardi, 2003; Kraytsberg et al., 2003). A growing body of evidence indicates mtDNA mutations as the crucial mechanism leading to aging (Kujoth et al., 2005; Santos et al., 2013; Aon et al., 2016; Scheibye-Knudsen, 2016; DeBalsi et al., 2017; Kauppila et al., 2017; Figure 1). In a recent review, it has been highlighted that accumulation of mutations can induce an alteration in mtDNA replication, leading to electron transport chain activity loss and altered mitochondrial respiration (DeBalsi et al., 2017). In aging, organelle efficiency fails, leading to pathological status (Chauhan et al., 2014). Mitochondrial dynamics are protective in maintaining mitochondria integrity. In fact, during aging, synaptosomal mitochondria shift to a pro-fusion state occurs (Stauch et al., 2014). In the skeletal muscle, the lack of fusion, due to Mfn1 and Mfn2 ablation, induces accumulation of deletions and point mutations in the mitochondrial genome, together with muscle atrophy and mitochondrial alterations (Chen et al., 2010). Moreover, the absence of fusion, resulting from Drp1 ablation in mice adult forebrain, causes mitochondrial dysfunction and altered synapsis transmission, concomitant with decreased ATP production and oxygen consumption (Oettinghaus et al., 2016). The loss of Drp1 affects memory function and synapsis activity, evidencing the crucial role of mitochondrial fusion/fission activity in brain function. Drp1 deletion in post-mitotic Purkinje cells leads to mitochondrial swelling, oxidative damage, accumulation of autophagy markers, leading to neurodegeneration in the cerebellum (Kageyama et al., 2012).
The antioxidant treatment (MitoO and acetylcysteine), in Drp1 KO cells, reduced cell death and mitochondrial swelling (Kageyama et al., 2012). On this premise, efficient mitochondrial dynamics appear to be crucial for maintaining a healthy organelle population for potential therapeutic treatments.
Peroxisomes are essential organelles in eukaryotes and are involved in numerous metabolic pathways and redox homeostasis and contribute to essential brain metabolic processes. Their functional relevance in humans is emphasized by peroxisomal disorders (Trompier et al., 2014). Peroxisomes are implicated in cell aging since they are affected by altered ROS and RNS production (Giordano and Terlecky, 2012; Figure 1). During aging, cells are metabolically less active, and catalase is translocated from peroxisomes to the cytosol. It has been demonstrated that the restoration of peroxisomal catalase levels limits cellular senescence, and therefore, it could be used as a therapeutic target in aging (Giordano and Terlecky, 2012). Furthermore, aging depends on the peroxisome life cycle. After division, both young and old peroxisomes are present, with the latter being damaged by products of peroxisomal metabolism (Beach et al., 2012). The old peroxisomes are thus removed by autophagy to avoid compound accumulation and to maintain organelle homeostasis (Aksam et al., 2007).
Neuronal Metabolism
During aging, neuronal metabolism is impaired. The human brain has the highest energy requests with respect to any organ system, using more than 20% of the body’s energy, despite comprising only 2% of total body mass (Lin and Rothman, 2014). In vitro and in vivo studies demonstrated that the brain consumes a large amount of glucose for the maintenance of pre-synaptic and post-synaptic ion gradients for glutamate neurotransmission, and to preserve the resting potential of neurons (Attwell and Iadecola, 2002). Moreover, neurotransmitter signaling needs constant phospholipid remodeling and trans-membrane lipid asymmetries, which represent around 26% of the net energy uptake of the brain (Purdon et al., 2002). In the awake and unstimulated brain, the use of basal energy is already high, and when a stimulus occurs, the energy consumption is even higher.
In the brain, the metabolic fate of glucose varies depending on cell type and on the specific expression of metabolic enzymes. Neurons are mainly oxidative, while astrocytes are generally glycolytic (Ivanov et al., 2014). Neuronal glucose metabolism is multifaceted. It comprises the mechanisms controlling brain glucose consumption, such as the insulin signaling pathways. Brain glucose is transported across the endothelium into astrocytes through GLUT1 (glucose transporter) and transferred into neurons, mostly via GLUT3 and GLUT4, participating in the glycolytic pathway. In aged rat brains, decreased glucose uptake and a reduction in neuronal GLUTs have been reported (Yin et al., 2016). During aging, alterations in mitochondria energy-transducing ability, as well as glucose availability occur, together with an impairment in neuronal glucose uptake, increased oxidant production and decreased electron transport chain activity (Yin et al., 2016). A significant role is exerted by insulin and IGF-1 signaling, since they are involved in the regulation and maintenance of cognitive function and brain metabolism (de la Monte and Wands, 2005). Brain aging is closely related to decreased IGF-1 signaling, involving inactivation of the PI3K/Akt pathway (Jiang et al., 2013). Thus, during aging, it is more likely that brain glucose uptake, as well as systemic control of glucose, will fail (Reger et al., 2006). Several preclinical and clinical studies highlight a link between low brain glucose uptake and mild cognitive impairment (Cunnane et al., 2011). In aged brain, glucose hypometabolism and mitochondrial alteration are observed, which are initial indicators of age-related impairment (Small et al., 2000; Mosconi et al., 2008). Moreover, decreased glucose consumption has been described in different brain areas in human subjects with the use of the positron emission tomography analyses of fluorodeoxyglucose uptake (Zuendorf et al., 2003). Rats showed an age-dependent decline in glucose consumption in the hippocampus and cortex, which is linked to impaired cognitive performance (Gage et al., 1984). Synaptic spines are the site of neurotransmission and need a high rate of ATP for maintaining neurotransmitter transporter activities and Na+ and Ca+ pumps, and for restoring gradients after synapsis activity (Attwell and Laughlin, 2001; Alle et al., 2009; Harris et al., 2012; Rangaraju et al., 2014). During aging, neurons are not able to generate ATP, thus synapses are susceptible to alterations and degeneration (Harris et al., 2012). Numerous factors concur to age-dependent brain hypometabolism. Some clinical studies demonstrated that cerebral blood flow and age are negatively connected (Schultz et al., 1999; Fabiani et al., 2014). During aging, blood-brain barrier (BBB) impairment and brain hypoperfusion lead to decreased import of nutrients, and/or removal of toxins and parenchymal accumulation of blood-derived proteins and immune cells (Zlokovic, 2011; Rosenberg, 2012). Aged mice showed a reduction in white matter, ultrastructural mitochondria modifications and a weak correlation between mitochondria and endoplasmic reticulum (Stahon et al., 2016). Aged brains showed decreased NADH, total NAD and NAD+ levels (Bai et al., 2011; Pittelli et al., 2011; Zhu et al., 2015). GLUT1 levels are reduced in an age-dependent manner, in compromised cerebral blood flow, cerebral capillary mass, and glucose uptake, concomitant with augmented BBB leakage (Winkler et al., 2015). These rearrangements precede dendritic spine loss in CA1 hippocampal neurons and are linked to behavioral decline (Winkler et al., 2015).
Morphological Rearrangement
Wide loss of nerve cells is not present in normal aging, and the regions affected by a neuronal loss are restricted (no more than 10%). The principal age-related neuronal structural alterations involve dendrites reduction in length and number, with a loss of various dendritic spines (Figure 1). Hippocampal DG-CA3 system is involved in regenerative ability, structural plasticity and in the regulation of neurotrophic factors like brain-derived neurotrophic factor (BDNF; Wang and Michaelis, 2010). Oxidative injury of DG-CA could impair remodeling ability, decrease cell proliferation, modify structural plasticity, and alter neurogenesis, collectively impairing normal synaptic neurotransmission. Moreover, dendritic shrinking and amygdala hyperactivity, that could increase synaptic instabilities by altering the hippocampus-amygdala connectivity, have been observed (Kreibich and Blendy, 2004; Brown et al., 2005; Radley et al., 2006; Wood et al., 2010). Synaptic injury is paralleled by axons reduction and disorganized myelin sheaths undergoing segmental demyelination followed by remyelination. These changes contribute to cognitive decline and behavioral impairment that often occurs during normal aging (Peters et al., 2000; Sandell and Peters, 2001, 2003; Peters and Sethares, 2002; Bowley et al., 2010). Myelin is important for insulating axons and guarantees rapid propagation of action potentials, thus alterations in myelin structure induce weak conduction velocity along axons. Additionally, the remyelination process generates shorter myelin internodes (Peters et al., 2000; Sandell and Peters, 2001, 2003; Peters and Sethares, 2002; Bowley et al., 2010). The decrease in conduction velocity along axons, due to changes in myelin sheaths and internodes, renders reaction times longer and interferes with neuronal synchrony, which is implicated in cognitive performance (including attention and memory), as well as sensory and motor functions (Jermakowicz and Casagrande, 2007).
One of the first changes studied in neuronal cells was the accumulation of lipofuscin granules (Moreno-García et al., 2018). Lipofuscin granules, which range between 1 and 3 μm in diameter, appear as brownish particles in neuronal cytoplasm and are probably indigestible residues of lysosomes materials. Since, for many years, lipofuscin was the only one change recognized, its accumulation was an indicative marker of the nervous system aging. Several studies reported that pigmentation of lipofuscin accumulation varies in different regions of the nervous system and that it is accumulated at different rates depending on the brain region. Aggregation of lipofuscin does not strongly affect neuronal metabolism and functional activities (Pannese, 2011).
All the studies on non-human species reported that, during aging, the neuronal loss is restricted to some central nervous system (CNS) areas; however, in human it is still difficult to estimate the percentage of loss but is restricted to specific areas (Merrill et al., 2001; Mohammed and Santer, 2001; Keuker et al., 2004).
It has been further reported that aged animals and humans’ axons may present degenerated mitochondria, glycogen inclusions and filaments accumulations (Geoffroy et al., 2016; Salvadores et al., 2017). These modifications trigger axonal impairments. In fact, in aged animals and humans, degenerated or degenerating axons, as well as the reduction in axons number, have been observed (Sandell and Peters, 2001, 2003; Marner et al., 2003; Cepurna et al., 2005; Bowley et al., 2010; Peters et al., 2010). In brain aging and neurodegeneration, the levels of Aβ-peptide and pro-inflammatory cytokines accumulate at an early stage during the pathogenic process (Figures 1, 2). These accumulations trigger the alteration of signal transduction pathways crucial for neuronal health. Neurotrophins signaling is crucial in memory, learning, synaptic function and plasticity, and neuronal cell survival (Smith, 1996; Huang and Reichardt, 2001).
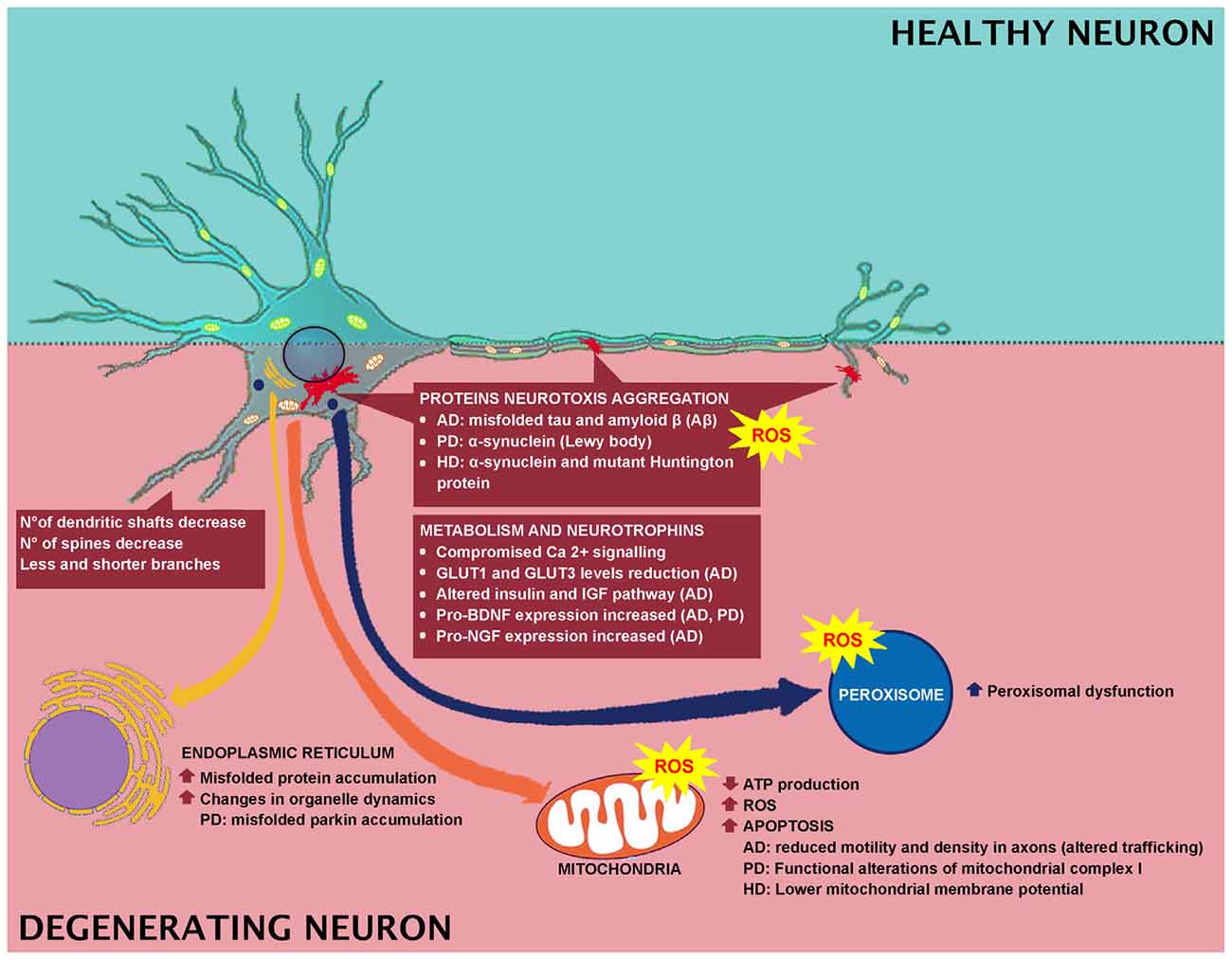
Figure 2. Effect of degeneration in neuronal cell and involved mechanisms.
In fact, a preclinical study in aged animals demonstrated that chronic BDNF deficiency induced an age-dependent impairment, thus suggesting that BDNF positively affects brain performance and neuronal survival (Petzold et al., 2015). It has been indicated that during aging, TrkB receptors decrease and that BDNF helps some pituitary function as an intracellular messenger (Rage et al., 2007). BDNF is protective and pro-survival (Castelli et al., 2018), is involved in energy homeostasis control (Xu and Xie, 2016), and is strongly reduced during aging.
Another neurotrophin, Glial cell line-derived neurotrophic factor (GDNF), is altered during aging. GDNF increases in an age-dependent manner in the frontal cortex but not in the hippocampus, suggesting that it exerts its trophic action locally (Matsunaga et al., 2006). Moreover, GDNF is involved in glutamate release and altered glutamate transporters expression, inducing excitotoxicity and triggering neurodegeneration (Farrand et al., 2015).
The glycoprotein nerve growth factor (NGF) is involved in cognitive functions and is strongly decreased during aging. These shreds of evidences suggested that this neurotrophin influences age-dependent cognitive impairment, the maintenance and survival of cholinergic neurons and memory (Yang et al., 2014).
In preclinical studies in transgenic animals, increased BDNF, NGF, and NT3 levels, paralleled by a reduction in number of hippocampal positive amyloid plaques, were reported after insulin-like growth factor 2 (IGF2) treatments (Mellott et al., 2014).
Neurodegenerative Disease
Oxidative Imbalance
Neurodegenerative disorders, commonly associated with muscular and cognitive deficits, have pathological hallmarks, including brain atrophy, plaques, neurofibrillary tangles, and aggregates (Kipps et al., 2005; Obeso et al., 2008; Gandhi and Abramov, 2012). In AD, PD and Huntington disease (HD), the main feature is the neurotoxic aggregation of specific proteins in the brain (Figure 2). In specific, AD is characterized by the accumulation of misfolded tau and amyloid β (Aβ) proteins, while in PD and Huntington’s diseases, α-synuclein (α-syn) and mutant Huntington protein (mHtt) accumulate, respectively. Researchers have indicated a link between oxidative stress and development of neuronal plaque, α-synuclein, and mHtt (Li et al., 2013), and a link between the Aβ protein formation and ROS has been reported as well (Behl et al., 1997; Abramov and Duchen, 2005; Shelat et al., 2008). In PD, oxidative stress leads to α-synuclein aggregation in dopaminergic neurons, which in turn induces intracellular ROS formation (Xiang et al., 2013). While in HD, in vitro studies indicated that free radicals are implicated in misfolding and accumulation of mHtt-induced neurotoxicity (Pitts et al., 2012). In AD brain, antioxidant enzyme activity is strongly reduced (Ansari and Scheff, 2010) and Aβ-mediated ROS production induces lipid peroxidation, causing reduced membrane permeability and triggering excitotoxicity mechanisms by increased calcium influx (Figure 2). These events lead to altered neurotransmission and cognitive impairment. In fact, ROS has been related to Aβ-induced damage in LTP, resulting in learning and memory impairment, due to altered neuronal transmission (Dumont et al., 2009; Ma et al., 2011; Ma and Klann, 2012; Parajuli et al., 2013). In addition, oxidative stress could be linked to clearance of Aβ. It has been reported that Aβ oxidizes LRP1, thus inducing to the accumulation of the neurotoxic peptide Aβ in the brain. For instance, LRP1 is a multifaceted protein, which controls the efflux of Aβ from the brain to the blood, across the BBB, and LRP1 activity is decreased in AD (Jeynes and Provias, 2008). Therefore, Aβ disrupts this clearance through oxidizing LRP1. LRP1 oxidation is confirmed by the presence of HNE-LRP1 protein adducts in the AD hippocampus (Owen et al., 2010). The altered Aβ clearance triggers Aβ accumulation in the brain, a key factor in AD pathogenesis (Cheignon et al., 2018). Another target for oxidative stress in AD is represented by protein Tau. Indeed, HNE induces alteration in the protein Tau conformation, thus supporting the involvement of the oxidative stress, particularly induced by Aβ, in the AD pathogenesis, by inducing neurofibrillary tangle formation (Liu et al., 2005; Cheignon et al., 2018).
Organelle Dynamics
In a transgenic model of neurodegenerative diseases, misfolded protein accumulation induces changes in the organelle dynamics, and in the reticulum (Reddy et al., 1999; Rao et al., 2002). UPR impairment has also been described in PD. In fact, with parkin function loss, misfolded parkin substrates accumulate in the endoplasmic reticulum of the dopaminergic neurons of the substantia nigra, inducing ER stress and neuronal death (Imai et al., 2001). This event occurs in autosomal recessive juvenile parkinsonism in particular, where mutations in the Parkin gene trigger reticulum stress (Imai et al., 2001). Studies indicated that targeting UPR by inactivation or inhibition represents a potential therapy for neurodegenerative diseases (Brown and Naidoo, 2012).
Mitochondria exert pivotal functions in most neurodegenerative diseases. When mitochondrial dynamics and activities are impaired, low ATP production, high levels of ROS, and apoptosis occur (Suárez-Rivero et al., 2016). Interestingly, mitochondria extracted from lymphoblasts of Huntington’s disease patients show lower mitochondrial membrane potential with respect to the control group (Panov et al., 2002). This finding was also confirmed by mitochondria extracted from the brains of transgenic mice expressing mutant huntingtin protein. The physiological alteration, mitochondrial-mediated oxidative stress and calcium perturbations of the mitochondria induced the onset of behavioral and pathological abnormalities (Mattson et al., 2008). As shown in Figure 2, Ca2+ signaling is compromised in neurodegenerative disorders. Interestingly, neurons expressing elevated levels of Ca2+–binding proteins are preserved by AD, whereas neurons expressing these proteins at lower levels are subjected to wide impairment. One of the causes of the high vulnerability of AD neurons is the decreased Ca2+buffering ability of the neuronal cytosol. Neurons of AD aged patients showed activation of Ca2+-dependent proteases (calpain family). Calpain is activated as a response to the increased levels of Ca2+ in the cytosol and cleaves various proteins essential for the regular neuronal activity, triggering, and as a consequence, neuronal impairment and apoptosis. PD pathogenesis is attributable to Ca2+ dysregulation (Surmeier et al., 2012), therefore, the handling of mitochondrial Ca2+ buffering capacity and the pharmacological modulation of L-type channel activity can represent therapeutic strategies for reducing PD progression (Calì et al., 2011, 2014). Functional alterations of mitochondrial complex I are responsible for PD onset (Hu and Wang, 2016). Sporadic PD patients showed reduced complex I in various brain regions, neural and extra-neural tissues (Parker et al., 2008). Complex I deficiency is linked to increased ROS generation (Swerdlow et al., 1996). Moreover, mutations in α-synuclein are typical of PD pathology (Guardia-Laguarta et al., 2014). PD can also be caused by mutations in Parkin and PTEN-induced kinase 1 (PINK1). The Parkin gene is highly expressed in brain tissues, including the substantia nigra (Kitada et al., 1998). PINK1 is a mitochondrially-located molecule and has a positive role. A mutation in its kinase domain renders cells susceptible to oxidative stress (Valente et al., 2004).
In AD neuritis, the concomitant depletion of mitochondria or fusion and fission regulators, as well as deficits in axonal transportation and axonopathy, indicate that mitochondrial transport represents one of the causes of impairment (Massano and Bhatia, 2012). Mitochondria Aβ neurons or neurons expressing Amyloid precursor protein exhibit reduced motility and density in axons (Gao et al., 2008; Massano and Bhatia, 2012). Comparably, tau, particularly in its mutant form, alters mitochondrial trafficking in the neuronal cell. This altered trafficking can be reduced by the inhibition of mitochondrial fragmentation (Kausar et al., 2018), suggesting that the impairment of mitochondrial transport is strictly related to mitochondrial fragmentation. This correlation should also be considered in AD, in order to evaluate, in future investigations, if restoring mitochondrial trafficking could prevent Aβ or tau-induced mitochondrial and neuronal impairment. However, it should be explored as to whether alterations of mitochondria in neurodegenerative diseases constitute a primary or a secondary event or are just part of a larger multifactorial pathogenic process.
Numerous evidence indicated that peroxisomes have a crucial role in aging and altered peroxisome functions support the onset of age-related pathologies. In fact, in an in vitro study, a connection between peroxisomes and AD has been reported. Primary rat hippocampal neurons were treated with Wy-14.463, a peroxisome proliferator agonist, and it was observed that peroxisomal proliferation protected neurons against Aβ peptide-induced cell death (Santos et al., 2005; Cimini et al., 2009). Moreover, an induction in ABCD3 and ACOX1 expression has been reported in a transgenic AD mouse model that may represent an efficient fatty acid β-oxidation necessary to counteract mitochondrial dysfunctions (Fanelli et al., 2013). Finally, peroxisomal dysfunction induced by thioridazine triggers increased APP and β-secretase expression (Shi et al., 2012). All these pieces of evidence support the relationship between neurodegeneration and peroxisomal dysfunction (Figure 2).
Neuronal Metabolism
In AD patients, alteration in BBB integrity occurs (Zipser et al., 2007; Zlokovic, 2011). Moreover, the disease-dependent alterations in BBB are faster with respect to normal subjects (Montagne et al., 2015; van de Haar et al., 2016). In addition, an alteration in nutrient and metabolic enzymes has been reported in AD. For instance, GLUT1 and GLUT3 levels are reduced in the AD patients’ brains (Simpson et al., 1994; Harr et al., 1995) and correlate with the reduction of brain glucose consumption and consequent cognitive impairment (Landau et al., 2010; Figure 2).
People with mild cognitive impairment or AD showed a strongly reduced glucose consumption compared to normal aging (Kato et al., 2016). The hypothesis that AD may be considered a type of diabetes mellitus that specifically affects the brain has been recently discussed. In fact, recently, the scientific literature has focused on the fact that AD may be triggered by insulin resistance and insulin deficiency. It has been reported that type 2 diabetes mellitus induces insulin resistance, cognitive impairment, and oxidative stress, mimicking AD neurodegeneration. Moreover, the altered insulin and IGF pathway, as indicated above, represents early and progressive alterations that can trigger the main molecular, histopathological and biochemical lesions of AD patients (Figure 2). Furthermore, it has been reviewed that, as a consequence of the diabetes induction through intracerebral inoculation of streptozotocin, an alkylating antineoplastic agent, animals showed closer features of AD. In addition, common pathophysiological alterations and signaling, such as inflammatory, oxidative stress and PI3K-GSK3β, between AD and diabetes have been indicated. In light of these overlaps, AD has been referred to as type 3 diabetes, since it mimics a kind of diabetes that affects the brain since the AD molecular and cellular characteristics are similar to those observed in type 1 and type 2 diabetes (de la Monte and Wands, 2008; Kandimalla et al., 2017).
Morphological Rearrangement
In AD there is a substantial loss of neurons but this is not the main factor inducing cognitive impairment (Figure 2). Researchers have explained these phenomena identifying changes in dendrites and axons. For instance, in diseased neurons, the dendritic tree undergoes faster decline, in fact, a decrease in the number of dendritic shafts occurs and the few remaining show fewer and shorter branches, as well as a reduced number of spines (Dickstein et al., 2007; Wang et al., 2018). However, not all the spines are affected in the same manner, since only the thin spines, which are plastic, strongly motile and are involved in learning, are lost (Dumitriu et al., 2010). BDNF is also involved in neurodegeneration (Figure 2) and its strong decrease indicates more vulnerable neuronal populations. This highlights the necessity to detect a potential therapy that targets BDNF signaling (Tapia-Arancibia et al., 2008), since in AD and PD, there is also a concomitant increase in pro-BDNF expression (Budni et al., 2015).
On the other hand, it has been reported that GDNF also has a neuroprotective effect on substantia nigra neurons in PD patients (Gill et al., 2003). In contrast to NGF, an increase in pro-NGF is linked to AD and to mild cognitive impairment. In light of this, NGF, as well as proNGF, could be used as AD targets.
Conclusion
Aging is a stochastic process dependent on the predictable and random features leading to the accumulation of unrepaired cellular damage, weakened cellular repair, an imbalance in antioxidant defenses and altered metabolism. Healthy aging depends on a combination of lifestyle, individual genetic factors, and external environmental factors. Several reports indicated that glucose hypometabolism, impaired transport of critical substrates, alterations in calcium signaling, mitochondrial dysfunction, and oxidative stress, are mechanisms of aging that render neurons susceptible to degeneration. All of these events trigger morphological rearrangements in neurons, leading to malfunction, compromised transmission, loss of memory and performance (Figure 1).
On this basis, age-related neurodegenerative disorders show a picture far worse with respect to normal aging (Figure 2), characterized by an increase of misfolded proteins, increase of oxidative stress and reduced scavenging capability resulting in massive neuronal loss; mainly in a specific brain area, and leading to heavy alterations in performance, memory and a change in personality.
These findings point towards a multifactorial portrait where different agents, many interdependent, play a significant role during brain aging, and where exacerbation of these factors may expose to the onset of age-related neurodegenerative disease.
Author Contributions
VC: oxidative stress and general organization of manuscript. EB and MC: neurotrophins. AA and GP: mitochondria. Md’A: energetic metabolism and Figures 1, 2. RI and AC: supervising and editing the manuscript. GP: declare that substantially contributes to the review article in object.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Aarsland, D., Creese, B., Politis, M., Chaudhuri, K. R., Ffytche, D. H., Weintraub, D., et al. (2017). Cognitive decline in Parkinson disease. Nat. Rev. Neurol. 13, 217–231. doi: 10.1038/nrneurol.2017.27
Abd El Mohsen, M. M., Iravani, M. M., Spencer, J. P. E., Rose, S., Fahim, A. T., Motawi, T. M. K., et al. (2005). Age-associated changes in protein oxidation and proteasome activities in rat brain: modulation by antioxidants. Biochem. Biophys. Res. Commun. 336, 386–391. doi: 10.1016/j.bbrc.2005.07.201
Abramov, A. Y., and Duchen, M. R. (2005). The role of an astrocytic NADPH oxidase in the neurotoxicity of amyloid β peptides. Philos. Trans. R. Soc. Lond. B Biol. Sci. 360, 2309–2314. doi: 10.1098/rstb.2005.1766
Aksam, E. B., Koek, A., Kiel, J. A. K. W., Jourdan, S., Veenhuis, M., and van der Klei, I. J. (2007). A peroxisomal lon protease and peroxisome degradation by autophagy play key roles in vitality of Hansenula polymorpha cells. Autophagy 3, 96–105. doi: 10.4161/auto.3534
Albrecht, P., Lewerenz, J., Dittmer, S., Noack, R., Maher, P., and Methner, A. (2010). Mechanisms of oxidative glutamate toxicity: the glutamate/cystine antiporter system xc as a neuroprotective drug target. CNS Neurol. Disord. Drug Targets 9, 373–382. doi: 10.2174/187152710791292567
Alexander, G. E., Ryan, L., Bowers, D., Foster, T. C., Bizon, J. L., Geldmacher, D. S., et al. (2012). Characterizing cognitive aging in humans with links to animal models. Front. Aging Neurosci. 4:21. doi: 10.3389/fnagi.2012.00021
Alle, H., Roth, A., and Geiger, J. R. P. (2009). Energy-efficient action potentials in hippocampal mossy fibers. Science 325, 1405–1408. doi: 10.1126/science.1174331
Ansari, M. A., and Scheff, S. W. (2010). Oxidative stress in the progression of Alzheimer disease in the frontal cortex. J. Neuropathol. Exp. Neurol. 69, 155–167. doi: 10.1097/NEN.0b013e3181cb5af4
Aon, M. A., Cortassa, S., Juhaszova, M., and Sollott, S. J. (2016). Mitochondrial health, the epigenome and healthspan. Clin. Sci. 130, 1285–1305. doi: 10.1042/CS20160002
Attwell, D., and Iadecola, C. (2002). The neural basis of functional brain imaging signals. Trends Neurosci. 25, 621–625. doi: 10.1016/s0166-2236(02)02264-6
Attwell, D., and Laughlin, S. B. (2001). An energy budget for signaling in the grey matter of the brain. J. Cereb. Blood Flow Metab. 21, 1133–1145. doi: 10.1097/00004647-200110000-00001
Bai, P., Cantó, C., Oudart, H., Brunyánszki, A., Cen, Y., Thomas, C., et al. (2011). PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab. 13, 461–468. doi: 10.1016/j.cmet.2011.03.004
Balu, M., Sangeetha, P., Murali, G., and Panneerselvam, C. (2005). Age-related oxidative protein damages in central nervous system of rats: modulatory role of grape seed extract. Int. J. Dev. Neurosci. 23, 501–507. doi: 10.1016/j.ijdevneu.2005.06.001
Barja, G. (2004). Free radicals and aging. Trends Neurosci. 27, 595–600. doi: 10.1016/j.tins.2004.07.005
Barja, G., and Herrero, A. (2000). Oxidative damage to mitochondrial DNA is inversely related to maximum life span in the heart and brain of mammals. FASEB J. 14, 312–318. doi: 10.1096/fasebj.14.2.312
Beach, A., Burstein, M. T., Richard, V. R., Leonov, A., Levy, S., and Titorenko, V. I. (2012). Integration of peroxisomes into an endomembrane system that governs cellular aging. Front. Physiol. 3:283. doi: 10.3389/fphys.2012.00283
Behl, C., Trapp, T., Skutella, T., and Holsboer, F. (1997). Protection against oxidative stress-induced neuronal cell death—a novel role for RU486. Eur. J. Neurosci. 9, 912–920. doi: 10.1111/j.1460-9568.1997.tb01442.x
Berr, C., Balansard, B., Arnaud, J., Roussel, A. M., and Alpérovitch, A. (2000). Cognitive decline is associated with systemic oxidative stress: the EVA study. Etude du Vieillissement Artériel. J. Am. Geriatr. Soc. 48, 1285–1291. doi: 10.1111/j.1532-5415.2000.tb02603.x
Birben, E., Sahiner, U. M., Sackesen, C., Erzurum, S., and Kalayci, O. (2012). Oxidative stress and antioxidant defense. World Allergy Organ. J. 5, 9–19. doi: 10.1097/WOX.0b013e3182439613
Bowley, M. P., Cabral, H., Rosene, D. L., and Peters, A. (2010). Age changes in myelinated nerve fibers of the cingulate bundle and corpus callosum in the rhesus monkey. J. Comp. Neurol. 518, 3046–3064. doi: 10.1002/cne.22379
Brown, S. M., Henning, S., and Wellman, C. L. (2005). Mild, short-term stress alters dendritic morphology in rat medial prefrontal cortex. Cereb. Cortex 15, 1714–1722. doi: 10.1093/cercor/bhi048
Brown, M. K., and Naidoo, N. (2012). The endoplasmic reticulum stress response in aging and age-related diseases. Front. Physiol. 3:263. doi: 10.3389/fphys.2012.00263
Budni, J., Bellettini-Santos, T., Mina, F., Garcez, M. L., and Zugno, A. I. (2015). The involvement of BDNF, NGF and GDNF in aging and Alzheimer’s disease. Aging Dis. 6, 331–341. doi: 10.14336/AD.2015.0825
Calabrese, V., Scapagnini, G., Ravagna, A., Colombrita, C., Spadaro, F., Butterfield, D. A., et al. (2004). Increased expression of heat shock proteins in rat brain during aging: relationship with mitochondrial function and glutathione redox state. Mech. Ageing Dev. 125, 325–335. doi: 10.1016/j.mad.2004.01.003
Calì, T., Ottolini, D., and Brini, M. (2011). Mitochondria, calcium and endoplasmic reticulum stress in Parkinson’s disease. Biofactors 37, 228–240. doi: 10.1002/biof.159
Calì, T., Ottolini, D., and Brini, M. (2014). Calcium signaling in Parkinson’s disease. Cell Tissue Res. 357, 439–454. doi: 10.1007/s00441-014-1866-0
Camandola, S., and Mattson, M. P. (2011). Aberrant subcellular neuronal calcium regulation in aging and Alzheimer’s disease. Biochim. Biophys. Acta 1813, 965–973. doi: 10.1016/j.bbamcr.2010.10.005
Castelli, V., Grassi, D., Bocale, R., d’Angelo, M., Antonosante, A., Cimini, A., et al. (2018). Diet and brain health: which role for polyphenols? Curr. Pharm. Des. 24, 227–238. doi: 10.2174/1381612824666171213100449
Cepurna, W. O., Kayton, R. J., Johnson, E. C., and Morrison, J. C. (2005). Age related optic nerve axonal loss in adult brown norway rats. Exp. Eye Res. 80, 877–884. doi: 10.1016/j.exer.2004.12.021
Chauhan, A., Vera, J., and Wolkenhauer, O. (2014). The systems biology of mitochondrial fission and fusion and implications for disease and aging. Biogerontology 15, 1–12. doi: 10.1007/s10522-013-9474-z
Cheignon, C., Tomas, M., Bonnefont-Rousselot, D., Faller, P., Hureau, C., and Collin, F. (2018). Oxidative stress and the amyloid β peptide in Alzheimer’s disease. Redox Biol. 14, 450–464. doi: 10.1016/j.redox.2017.10.014
Chen, H., Vermulst, M., Wang, Y. E., Chomyn, A., Prolla, T. A., McCaffery, J. M., et al. (2010). Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell 141, 280–289. doi: 10.1016/j.cell.2010.02.026
Chomyn, A., and Attardi, G. (2003). MtDNA mutations in aging and apoptosis. Biochem. Biophys. Res. Commun. 304, 519–529. doi: 10.1016/s0006-291x(03)00625-9
Cimini, A., Moreno, S., D’Amelio, M., Cristiano, L., D’Angelo, B., Falone, S., et al. (2009). Early biochemical and morphological modifications in the brain of a transgenic mouse model of Alzheimer’s disease: a role for peroxisomes. J. Alzheimers Dis. 18, 935–952. doi: 10.3233/JAD-2009-1199
Cohen, S. M., Li, B., Tsien, R. W., and Ma, H. (2015). Evolutionary and functional perspectives on signaling from neuronal surface to nucleus. Biochem. Biophys. Res. Commun. 460, 88–99. doi: 10.1016/j.bbrc.2015.02.146
Cunnane, S., Nugent, S., Roy, M., Courchesne-Loyer, A., Croteau, E., Tremblay, S., et al. (2011). Brain fuel metabolism, aging and Alzheimer’s disease. Nutrition 27, 3–20. doi: 10.1016/j.nut.2010.07.021
DeBalsi, K. L., Hoff, K. E., and Copeland, W. C. (2017). Role of the mitochondrial DNA replication machinery in mitochondrial DNA mutagenesis, aging and age-related diseases. Ageing Res. Rev. 33, 89–104. doi: 10.1016/j.arr.2016.04.006
de la Monte, S. M., and Wands, J. R. (2005). Review of insulin and insulin-like growth factor expression, signaling and malfunction in the central nervous system: relevance to Alzheimer’s disease. J. Alzheimers Dis. 7, 45–61. doi: 10.3233/jad-2005-7106
de la Monte, S. M., and Wands, J. R. (2008). Alzheimer’s disease is type 3 diabetes-evidence reviewed. J. Diabetes Sci. Technol. 2, 1101–1113. doi: 10.1177/193229680800200619
Dickstein, D. L., Kabaso, D., Rocher, A. B., Luebke, J. I., Wearne, S. L., and Hof, P. R. (2007). Changes in the structural complexity of the aged brain. Aging Cell 6, 275–284. doi: 10.1111/j.1474-9726.2007.00289.x
Donahue, A. N., Aschner, M., Lash, L. H., Syversen, T., and Sonntag, W. E. (2006). Growth hormone administration to aged animals reduces disulfide glutathione levels in hippocampus. Mech. Ageing Dev. 127, 57–63. doi: 10.1016/j.mad.2005.09.003
Duan, W., Zhang, R., Guo, Y., Jiang, Y., Huang, Y., Jiang, H., et al. (2009). Nrf2 activity is lost in the spinal cord and its astrocytes of aged mice. In Vitro Cell. Dev. Biol. Anim. 45, 388–397. doi: 10.1007/s11626-009-9194-5
Dumitriu, D., Hao, J., Hara, Y., Kaufmann, J., Janssen, W. G. M., Lou, W., et al. (2010). Selective changes in thin spine density and morphology in monkey prefrontal cortex correlate with aging-related cognitive impairment. J. Neurosci. 30, 7507–7515. doi: 10.1523/JNEUROSCI.6410-09.2010
Dumont, M., Wille, E., Stack, C., Calingasan, N. Y., Beal, M. F., and Lin, M. T. (2009). Reduction of oxidative stress, amyloid deposition and memory deficit by manganese superoxide dismutase overexpression in a transgenic mouse model of Alzheimer’s disease. FASEB J. 23, 2459–2466. doi: 10.1096/fj.09-132928
Dykiert, D., Der, G., Starr, J. M., and Deary, I. J. (2012). Age differences in intra-individual variability in simple and choice reaction time: systematic review and meta-analysis. PLoS One 7:e45759. doi: 10.1371/journal.pone.0045759
Fabiani, M., Low, K. A., Tan, C.-H., Zimmerman, B., Fletcher, M. A., Schneider-Garces, N., et al. (2014). Taking the pulse of aging: mapping pulse pressure and elasticity in cerebral arteries with optical methods. Psychophysiology 51, 1072–1088. doi: 10.1111/psyp.12288
Fanelli, F., Sepe, S., D’Amelio, M., Bernardi, C., Cristiano, L., Cimini, A., et al. (2013). Age-dependent roles of peroxisomes in the hippocampus of a transgenic mouse model of Alzheimer’s disease. Mol. Neurodegener. 8:8. doi: 10.1186/1750-1326-8-8
Farrand, A. Q., Gregory, R. A., Scofield, M. D., Helke, K. L., and Boger, H. A. (2015). Effects of aging on glutamate neurotransmission in the substantia nigra of Gdnf heterozygous mice. Neurobiol. Aging 36, 1569–1576. doi: 10.1016/j.neurobiolaging.2014.11.017
Fatokun, A. A., Dawson, V. L., and Dawson, T. M. (2014). Parthanatos: mitochondrial-linked mechanisms and therapeutic opportunities. Br. J. Pharmacol. 171, 2000–2016. doi: 10.1111/bph.12416
Finkel, T., and Holbrook, N. J. (2000). Oxidants, oxidative stress and the biology of ageing. Nature 408, 239–247. doi: 10.1038/35041687
Fukushima, H., Maeda, R., Suzuki, R., Suzuki, A., Nomoto, M., Toyoda, H., et al. (2008). Upregulation of calcium/calmodulin-dependent protein kinase IV improves memory formation and rescues memory loss with aging. J. Neurosci. 28, 9910–9919. doi: 10.1523/JNEUROSCI.2625-08.2008
Gage, F. H., Kelly, P. A., and Björklund, A. (1984). Regional changes in brain glucose metabolism reflect cognitive impairments in aged rats. J. Neurosci. 4, 2856–2865. doi: 10.1523/jneurosci.04-11-02856.1984
Gandhi, S., and Abramov, A. Y. (2012). Mechanism of oxidative stress in neurodegeneration. Oxid. Med. Cell. Longev. 2012:428010. doi: 10.1155/2012/428010
Gant, J. C., Chen, K.-C., Kadish, I., Blalock, E. M., Thibault, O., Porter, N. M., et al. (2015). Reversal of aging-related neuronal Ca2+ dysregulation and cognitive impairment by delivery of a transgene encoding FK506-binding protein 12.6/1b to the hippocampus. J. Neurosci. 35, 10878–10887. doi: 10.1523/JNEUROSCI.1248-15.2015
Gant, J. C., Sama, M. M., Landfield, P. W., and Thibault, O. (2006). Early and simultaneous emergence of multiple hippocampal biomarkers of aging is mediated by Ca2+-induced Ca2+ release. J. Neurosci. 26, 3482–3490. doi: 10.1523/JNEUROSCI.4171-05.2006
Gao, L., Laude, K., and Cai, H. (2008). Mitochondrial pathophysiology, reactive oxygen species and cardiovascular diseases. Vet. Clin. North Am. Small Anim. Pract. 38, 137–155. doi: 10.1016/j.cvsm.2007.10.004
Geoffroy, C. G., Hilton, B. J., Tetzlaff, W., and Zheng, B. (2016). Evidence for an age-dependent decline in axon regeneration in the adult Mammalian central nervous system. Cell Rep. 15, 238–246. doi: 10.1016/j.celrep.2016.03.028
Gill, S. S., Patel, N. K., Hotton, G. R., O’Sullivan, K., McCarter, R., Bunnage, M., et al. (2003). Direct brain infusion of glial cell line-derived neurotrophic factor in Parkinson disease. Nat. Med. 9, 589–595. doi: 10.1038/nm850
Giordano, C. R., and Terlecky, S. R. (2012). Peroxisomes, cell senescence and rates of aging. Biochim. Biophys. Acta 1822, 1358–1362. doi: 10.1016/j.bbadis.2012.03.013
Gounder, S. S., Kannan, S., Devadoss, D., Miller, C. J., Whitehead, K. J., Whitehead, K. S., et al. (2012). Impaired transcriptional activity of Nrf2 in age-related myocardial oxidative stress is reversible by moderate exercise training. PLoS One 7:e45697. doi: 10.1371/journal.pone.0045697
Griendling, K. K., Sorescu, D., Lassègue, B., and Ushio-Fukai, M. (2000). Modulation of protein kinase activity and gene expression by reactive oxygen species and their role in vascular physiology and pathophysiology. Arterioscler. Thromb. Vasc. Biol. 20, 2175–2183. doi: 10.1161/01.atv.20.10.2175
Grimm, A., and Eckert, A. (2017). Brain aging and neurodegeneration: from a mitochondrial point of view. J. Neurochem. 143, 418–431. doi: 10.1111/jnc.14037
Gu, Y., Dee, C. M., and Shen, J. (2011). Interaction of free radicals, matrix metalloproteinases and caveolin-1 impacts blood-brain barrier permeability. Front. Biosci. 3, 1216–1231. doi: 10.2741/s222
Guardia-Laguarta, C., Area-Gomez, E., Rüb, C., Liu, Y., Magrané, J., Becker, D., et al. (2014). α-synuclein is localized to mitochondria-associated ER membranes. J. Neurosci. 34, 249–259. doi: 10.1523/JNEUROSCI.2507-13.2014
Halliwell, B. (2001). Role of free radicals in the neurodegenerative diseases: therapeutic implications for antioxidant treatment. Drugs Aging 18, 685–716. doi: 10.2165/00002512-200118090-00004
Hamilton, M. L., Van Remmen, H., Drake, J. A., Yang, H., Guo, Z. M., Kewitt, K., et al. (2001). Does oxidative damage to DNA increase with age? Proc. Natl. Acad. Sci. U S A 98, 10469–10474. doi: 10.1073/pnas.171202698
Harr, S. D., Simonian, N. A., and Hyman, B. T. (1995). Functional alterations in Alzheimer’s disease: decreased glucose transporter 3 immunoreactivity in the perforant pathway terminal zone. J. Neuropathol. Exp. Neurol. 54, 38–41. doi: 10.1097/00005072-199501000-00005
Harris, J. J., Jolivet, R., and Attwell, D. (2012). Synaptic energy use and supply. Neuron 75, 762–777. doi: 10.1016/j.neuron.2012.08.019
Haxaire, C., Turpin, F. R., Potier, B., Kervern, M., Sinet, P.-M., Barbanel, G., et al. (2012). Reversal of age-related oxidative stress prevents hippocampal synaptic plasticity deficits by protecting D-serine-dependent NMDA receptor activation. Aging Cell 11, 336–344. doi: 10.1111/j.1474-9726.2012.00792.x
Hou, Y., Ouyang, X., Wan, R., Cheng, H., Mattson, M. P., and Cheng, A. (2012). Mitochondrial superoxide production negatively regulates neural progenitor proliferation and cerebral cortical development. Stem Cells 30, 2535–2547. doi: 10.1002/stem.1213
Hu, Q., and Wang, G. (2016). Mitochondrial dysfunction in Parkinson’s disease. Transl. Neurodegener. 5:14. doi: 10.1186/s40035-016-0060-6
Huang, E. J., and Reichardt, L. F. (2001). Neurotrophins: roles in neuronal development and function. Annu. Rev. Neurosci. 24, 677–736. doi: 10.1146/annurev.neuro.24.1.677
Hussain, S. G., and Ramaiah, K. V. A. (2007). Reduced eIF2α phosphorylation and increased proapoptotic proteins in aging. Biochem. Biophys. Res. Commun. 355, 365–370. doi: 10.1016/j.bbrc.2007.01.156
Imai, Y., Soda, M., Inoue, H., Hattori, N., Mizuno, Y., and Takahashi, R. (2001). An unfolded putative transmembrane polypeptide, which can lead to endoplasmic reticulum stress, is a substrate of Parkin. Cell 105, 891–902. doi: 10.1016/s0092-8674(01)00407-x
Ivanov, A. I., Malkov, A. E., Waseem, T., Mukhtarov, M., Buldakova, S., Gubkina, O., et al. (2014). Glycolysis and oxidative phosphorylation in neurons and astrocytes during network activity in hippocampal slices. J. Cereb. Blood Flow Metab. 34, 397–407. doi: 10.1038/jcbfm.2013.222
Jack, C. R., Shiung, M. M., Weigand, S. D., O’Brien, P. C., Gunter, J. L., Boeve, B. F., et al. (2005). Brain atrophy rates predict subsequent clinical conversion in normal elderly and amnestic MCI. Neurology 65, 1227–1231. doi: 10.1212/01.wnl.0000180958.22678.91
Jermakowicz, W. J., and Casagrande, V. A. (2007). Neural networks a century after Cajal. Brain Res. Rev. 55, 264–284. doi: 10.1016/j.brainresrev.2007.06.003
Jeynes, B., and Provias, J. (2008). Evidence for altered LRP/RAGE expression in Alzheimer lesion pathogenesis. Curr. Alzheimer Res. 5, 432–437. doi: 10.2174/156720508785908937
Jiang, X., Zhou, J., Giobbie-Hurder, A., Wargo, J., and Hodi, F. S. (2013). The activation of MAPK in melanoma cells resistant to BRAF inhibition promotes PD-L1 expression that is reversible by MEK and PI3K inhibition. Clin. Cancer Res. 19, 598–609. doi: 10.1158/1078-0432.CCR-12-2731
Kageyama, Y., Zhang, Z., and Roda, R. (2012). Mitochondrial division ensures the survival of postmitotic neurons by suppressing oxidative damage. J. Cell Biol. 197, 535–551. doi: 10.1083/jcb.201110034
Kalia, L. V., and Lang, A. E. (2015). Parkinson’s disease. Lancet 386, 896–912. doi: 10.1016/S0140-6736(14)61393-3
Kandimalla, R., Thirumala, V., and Reddy, P. H. (2017). Is Alzheimer’s disease a type 3 diabetes? A critical appraisal. Biochim. Biophys. Acta Mol. Basis Dis. 1863, 1078–1089. doi: 10.1016/j.bbadis.2016.08.018
Kato, T., Inui, Y., Nakamura, A., and Ito, K. (2016). Brain fluorodeoxyglucose (FDG) PET in dementia. Ageing Res. Rev. 30, 73–84. doi: 10.1016/j.arr.2016.02.003
Kauppila, T. E. S., Kauppila, J. H. K., and Larsson, N.-G. (2017). Mammalian mitochondria and aging: an update. Cell Metab. 25, 57–71. doi: 10.1016/j.cmet.2016.09.017
Kausar, S., Wang, F., and Cui, H. (2018). The role of mitochondria in reactive oxygen species generation and its implications for neurodegenerative diseases. Cells 7:12. doi: 10.3390/cells7120274
Keuker, J. I. H., de Biurrun, G., Luiten, P. G. M., and Fuchs, E. (2004). Preservation of hippocampal neuron numbers and hippocampal subfield volumes in behaviorally characterized aged tree shrews. J. Comp. Neurol. 468, 509–517. doi: 10.1002/cne.10996
Kim, G. H., Kim, J. E., Rhie, S. J., and Yoon, S. (2015). The role of oxidative stress in neurodegenerative diseases. Exp. Neurobiol. 24, 325–340. doi: 10.5607/en.2015.24.4.325
Kipps, C. M., Duggins, A. J., Mahant, N., Gomes, L., Ashburner, J., and McCusker, E. A. (2005). Progression of structural neuropathology in preclinical Huntington’s disease: a tensor based morphometry study. J. Neurol. Neurosurg. Psychiatry 76, 650–655. doi: 10.1136/jnnp.2004.047993
Kitada, T., Asakawa, S., Hattori, N., Matsumine, H., Yamamura, Y., Minoshima, S., et al. (1998). Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392, 605–608. doi: 10.1038/33416
Kohen, R., and Nyska, A. (2002). Oxidation of biological systems: oxidative stress phenomena, antioxidants, redox reactions and methods for their quantification. Toxicol. Pathol. 30, 620–650. doi: 10.1080/01926230290166724
Kohen, R., Vellaichamy, E., Hrbac, J., Gati, I., and Tirosh, O. (2000). Quantification of the overall reactive oxygen species scavenging capacity of biological fluids and tissues. Free Radic. Biol. Med. 28, 871–879. doi: 10.1016/s0891-5849(00)00191-x
Kraytsberg, Y., Nekhaeva, E., Bodyak, N. B., and Khrapko, K. (2003). Mutation and intracellular clonal expansion of mitochondrial genomes: two synergistic components of the aging process? Mech. Ageing Dev. 124, 49–53. doi: 10.1016/s0047-6374(02)00169-0
Kreibich, A. S., and Blendy, J. A. (2004). cAMP response element-binding protein is required for stress but not cocaine-induced reinstatement. J. Neurosci. 24, 6686–6692. doi: 10.1523/JNEUROSCI.1706-04.2004
Kujoth, G. C., Hiona, A., Pugh, T. D., Someya, S., Panzer, K., Wohlgemuth, S. E., et al. (2005). Mitochondrial DNA mutations, oxidative stress and apoptosis in mammalian aging. Science 309, 481–484. doi: 10.1126/science.1112125
Landau, S. M., Harvey, D., Madison, C. M., Reiman, E. M., Foster, N. L., Aisen, P. S., et al. (2010). Comparing predictors of conversion and decline in mild cognitive impairment. Neurology 75, 230–238. doi: 10.1212/WNL.0b013e3181e8e8b8
Ledda, M., Barni, L., Altieri, L., and Pannese, E. (2000). Decrease in the nucleo-cytoplasmic volume ratio of rabbit spinal ganglion neurons with age. Neurosci. Lett. 286, 171–174. doi: 10.1016/s0304-3940(00)01116-2
Lee, D. Z., Chung, J. M., Chung, K., and Kang, M.-G. (2012). Reactive oxygen species (ROS) modulate AMPA receptor phosphorylation and cell-surface localization in concert with pain-related behavior. Pain 153, 1905–1915. doi: 10.1016/j.pain.2012.06.001
Levin, O., Fujiyama, H., Boisgontier, M. P., Swinnen, S. P., and Summers, J. J. (2014). Aging and motor inhibition: a converging perspective provided by brain stimulation and imaging approaches. Neurosci. Biobehav. Rev. 43, 100–117. doi: 10.1016/j.neubiorev.2014.04.001
Li, Y.-J., Xu, M., Gao, Z.-H., Wang, Y.-Q., Yue, Z., Zhang, Y.-X., et al. (2013). Alterations of serum levels of BDNF-related miRNAs in patients with depression. PLoS One 8:e63648. doi: 10.1371/journal.pone.0063648
Lin, A.-L., and Rothman, D. L. (2014). What have novel imaging techniques revealed about metabolism in the aging brain? Future Neurol. 9, 341–354. doi: 10.2217/fnl.14.13
Liu, Q., Smith, M. A., Avilá, J., DeBernardis, J., Kansal, M., Takeda, A., et al. (2005). Alzheimer-specific epitopes of tau represent lipid peroxidation-induced conformations. Free Radic. Biol. Med. 38, 746–754. doi: 10.1016/j.freeradbiomed.2004.11.005
Ma, T., Hoeffer, C. A., Wong, H., Massaad, C. A., Zhou, P., Iadecola, C., et al. (2011). Amyloid β-induced impairments in hippocampal synaptic plasticity are rescued by decreasing mitochondrial superoxide. J. Neurosci. 31, 5589–5595. doi: 10.1523/JNEUROSCI.6566-10.2011
Ma, T., and Klann, E. (2012). Amyloid β: linking synaptic plasticity failure to memory disruption in Alzheimer’s disease. J. Neurochem. 120, 140–148. doi: 10.1111/j.1471-4159.2011.07506.x
Marner, L., Nyengaard, J. R., Tang, Y., and Pakkenberg, B. (2003). Marked loss of myelinated nerve fibers in the human brain with age. J. Comp. Neurol. 462, 144–152. doi: 10.1002/cne.10714
Martinelli, C., Sartori, P., Ledda, M., and Pannese, E. (2006). A study of mitochondria in spinal ganglion neurons during life: quantitative changes from youth to extremely advanced age. Tissue Cell 38, 93–98. doi: 10.1016/j.tice.2005.12.002
Massano, J., and Bhatia, K. P. (2012). Clinical approach to 2012 Parkinson’s disease: features, diagnosis and principles of management. Cold Spring Harb. Perspect. Med. 2:a008870. doi: 10.1101/cshperspect.a008870
Matsunaga, W., Isobe, K., and Shirokawa, T. (2006). Involvement of neurotrophic factors in aging of noradrenergic innervations in hippocampus and frontal cortex. Neurosci. Res. 54, 313–318. doi: 10.1016/j.neures.2005.12.013
Mattson, M. P. (2000). Apoptosis in neurodegenerative disorders. Nat. Rev. Mol. Cell Biol. 1, 120–129. doi: 10.1038/35040009
Mattson, M. P. (2004). Pathways towards and away from Alzheimer’s disease. Nature 430, 631–639. doi: 10.1038/nature02621
Mattson, M. P., Gleichmann, M., and Cheng, A. (2008). Mitochondria in neuroplasticity and neurological disorders. Neuron 60, 748–766. doi: 10.1016/j.neuron.2008.10.010
Mellott, T. J., Pender, S. M., Burke, R. M., Langley, E. A., and Blusztajn, J. K. (2014). IGF2 ameliorates amyloidosis, increases cholinergic marker expression and raises BMP9 and neurotrophin levels in the hippocampus of the APPswePS1dE9 Alzheimer’s disease model mice. PLoS One 9:e94287. doi: 10.1371/journal.pone.0094287
Mendonca, G. V., Pezarat-Correia, P., Vaz, J. R., Silva, L., and Heffernan, K. S. (2017). Impact of aging on endurance and neuromuscular physical performance: the role of vascular senescence. Sports Med. 47, 583–598. doi: 10.1007/s40279-016-0596-8
Merrill, D. A., Chiba, A. A., and Tuszynski, M. H. (2001). Conservation of neuronal number and size in the entorhinal cortex of behaviorally characterized aged rats. J. Comp. Neurol. 438, 445–456. doi: 10.1002/cne.1327
Mohammed, H. A., and Santer, R. M. (2001). Total neuronal numbers of rat lumbosacral primary afferent neurons do not change with age. Neurosci. Lett. 304, 149–152. doi: 10.1016/s0304-3940(01)01781-5
Montagne, A., Barnes, S. R., Sweeney, M. D., Halliday, M. R., Sagare, A. P., Zhao, Z., et al. (2015). Blood-brain barrier breakdown in the aging human hippocampus. Neuron 85, 296–302. doi: 10.1016/j.neuron.2014.12.032
Moreno-García, A., Kun, A., Calero, O., Medina, M., and Calero, M. (2018). An overview of the role of lipofuscin in age-related neurodegeneration. Front. Neurosci. 12:464. doi: 10.3389/fnins.2018.00464
Mosconi, L., De Santi, S., Li, J., Tsui, W. H., Li, Y., Boppana, M., et al. (2008). Hippocampal hypometabolism predicts cognitive decline from normal aging. Neurobiol. Aging 29, 676–692. doi: 10.1016/j.neurobiolaging.2006.12.008
Naidoo, N., Ferber, M., Master, M., Zhu, Y., and Pack, A. I. (2008). Aging impairs the unfolded protein response to sleep deprivation and leads to proapoptotic signaling. J. Neurosci. 28, 6539–6548. doi: 10.1523/JNEUROSCI.5685-07.2008
Nguyen, D., Alavi, M. V., Kim, K.-Y., Kang, T., Scott, R. T., Noh, Y. H., et al. (2011). A new vicious cycle involving glutamate excitotoxicity, oxidative stress and mitochondrial dynamics. Cell Death Dis. 2:e240. doi: 10.1038/cddis.2011.117
Nixon, R. A. (2003). The calpains in aging and aging-related diseases. Ageing Res. Rev. 2, 407–418. doi: 10.1016/s1568-1637(03)00029-1
Obeso, J. A., Rodríguez-Oroz, M. C., Benitez-Temino, B., Blesa, F. J., Guridi, J., Marin, C., et al. (2008). Functional organization of the basal ganglia: therapeutic implications for Parkinson’s disease. Mov. Disord. 23, S548–S559. doi: 10.1002/mds.22062
Oettinghaus, B., Schulz, J. M., Restelli, L. M., Licci, M., Savoia, C., Schmidt, A., et al. (2016). Synaptic dysfunction, memory deficits and hippocampal atrophy due to ablation of mitochondrial fission in adult forebrain neurons. Cell Death Differ. 23, 18–28. doi: 10.1038/cdd.2015.39
Owen, J. B., Sultana, R., Aluise, C. D., Erickson, M. A., Price, T. O., and Bu, G. (2010). Oxidative modification to LDL receptor-related protein 1 in hippocampus from subjects with Alzheimer disease: implications for Aβ accumulation in AD brain. Free Radic. Biol. Med. 49, 1798–1803. doi: 10.1016/j.freeradbiomed.2010.09.013
Pannese, E. (2011). Morphological changes in nerve cells during normal aging. Brain Struct. Funct. 216, 85–89. doi: 10.1007/s00429-011-0308-y
Panov, A. V., Gutekunst, C. A., Leavitt, B. R., Hayden, M. R., Burke, J. R., Strittmatter, W. J., et al. (2002). Early mitochondrial calcium defects in Huntington’s disease are a direct effect of polyglutamines. Nat. Neurosci. 5, 731–736. doi: 10.1038/nn884
Parajuli, B., Sonobe, Y., Horiuchi, H., Takeuchi, H., Mizuno, T., and Suzumura, A. (2013). Oligomeric amyloid β induces IL-1β processing via production of ROS: implication in Alzheimer’s disease. Cell Death Dis. 4:e975. doi: 10.1038/cddis.2013.503
Parker, W. D., Parks, J. K., and Swerdlow, R. H. (2008). Complex I deficiency in Parkinson’s disease frontal cortex. Brain Res. 1189, 215–218. doi: 10.1016/j.brainres.2007.10.061
Paz Gavilán, M., Vela, J., Castaño, A., Ramos, B., del Río, J. C., Vitorica, J., et al. (2006). Cellular environment facilitates protein accumulation in aged rat hippocampus. Neurobiol. Aging 27, 973–982. doi: 10.1016/j.neurobiolaging.2005.05.010
Perkins, A. J., Hendrie, H. C., Callahan, C. M., Gao, S., Unverzagt, F. W., Xu, Y., et al. (1999). Association of antioxidants with memory in a multiethnic elderly sample using the Third National Health and Nutrition Examination Survey. Am. J. Epidemiol. 150, 37–44. doi: 10.1093/oxfordjournals.aje.a009915
Peters, J. M., Lee, S. S., Li, W., Ward, J. M., Gavrilova, O., Everett, C., et al. (2000). Growth, adipose, brain, and skin alterations resulting from targeted disruption of the mouse peroxisome proliferator-activated receptor β (δ). Mol. Cell. Biol. 20, 5119–5128. doi: 10.1128/mcb.20.14.5119-5128.2000
Peters, A., and Sethares, C. (2002). Aging and the myelinated fibers in prefrontal cortex and corpus callosum of the monkey. J. Comp. Neurol. 442, 277–291. doi: 10.1002/cne.10099
Peters, A., Sethares, C., and Moss, M. B. (2010). How the primate fornix is affected by age. J. Comp. Neurol. 518, 3962–3980. doi: 10.1002/cne.22434
Petzold, A., Psotta, L., Brigadski, T., Endres, T., and Lessmann, V. (2015). Chronic BDNF deficiency leads to an age-dependent impairment in spatial learning. Neurobiol. Learn. Mem. 120, 52–60. doi: 10.1016/j.nlm.2015.02.009
Pittelli, M., Felici, R., Pitozzi, V., Giovannelli, L., Bigagli, E., Cialdai, F., et al. (2011). Pharmacological effects of exogenous NAD on mitochondrial bioenergetics, DNA repair, and apoptosis. Mol. Pharmacol. 80, 1136–1146. doi: 10.1124/mol.111.073916
Pitts, A., Dailey, K., Newington, J. T., Chien, A., Arseneault, R., Cann, T., et al. (2012). Dithiol-based compounds maintain expression of antioxidant protein peroxiredoxin 1 that counteracts toxicity of mutant huntingtin. J. Biol. Chem. 287, 22717–22729. doi: 10.1074/jbc.m111.334565
Poon, H. F., Calabrese, V., Calvani, M., and Butterfield, D. A. (2006). Proteomics analyses of specific protein oxidation and protein expression in aged rat brain and its modulation by L-acetylcarnitine: insights into the mechanisms of action of this proposed therapeutic agent for CNS disorders associated with oxidative stress. Antioxid. Redox Signal. 8, 381–394. doi: 10.1089/ars.2006.8.381
Porte, Y., Buhot, M.-C., and Mons, N. (2008). Alteration of CREB phosphorylation and spatial memory deficits in aged 129T2/Sv mice. Neurobiol. Aging 29, 1533–1546. doi: 10.1016/j.neurobiolaging.2007.03.023
Purdon, A. D., Rosenberger, T. A., Shetty, H. U., and Rapoport, S. I. (2002). Energy consumption by phospholipid metabolism in mammalian brain. Neurochem. Res. 27, 1641–1647. doi: 10.1023/A:1021635027211
Radley, J. J., Rocher, A. B., Miller, M., Janssen, W. G. M., Liston, C., Hof, P. R., et al. (2006). Repeated stress induces dendritic spine loss in the rat medial prefrontal cortex. Cereb. Cortex 16, 313–320. doi: 10.1093/cercor/bhi104
Raefsky, S. M., and Mattson, M. P. (2017). Adaptive responses of neuronal mitochondria to bioenergetic challenges: roles in neuroplasticity and disease resistance. Free Radic. Biol. Med. 102, 203–216. doi: 10.1016/j.freeradbiomed.2016.11.045
Rage, F., Silhol, M., Binamé, F., Arancibia, S., and Tapia-Arancibia, L. (2007). Effect of aging on the expression of BDNF and TrkB isoforms in rat pituitary. Neurobiol. Aging 28, 1088–1098. doi: 10.1016/j.neurobiolaging.2006.05.013
Rai, S., Kamat, P. K., Nath, C., and Shukla, R. (2013). A study on neuroinflammation and NMDA receptor function in STZ (ICV) induced memory impaired rats. J. Neuroimmunol. 254, 1–9. doi: 10.1016/j.jneuroim.2012.08.008
Rangaraju, V., Calloway, N., and Ryan, T. A. (2014). Activity-driven local ATP synthesis is required for synaptic function. Cell 156, 825–835. doi: 10.1016/j.cell.2013.12.042
Rao, R. V., Peel, A., Logvinova, A., del Rio, G., Hermel, E., Yokota, T., et al. (2002). Coupling endoplasmic reticulum stress to the cell death program: role of the ER chaperone GRP78. FEBS Lett. 514, 122–128. doi: 10.1016/s0014-5793(02)02289-5
Rebrin, I., Kamzalov, S., and Sohal, R. S. (2003). Effects of age and caloric restriction on glutathione redox state in mice. Free Radic. Biol. Med. 35, 626–635. doi: 10.1016/s0891-5849(03)00388-5
Reddy, P. H., Williams, M., and Tagle, D. A. (1999). Recent advances in understanding the pathogenesis of Huntington’s disease. Trends Neurosci. 22, 248–255. doi: 10.1016/S0166-2236(99)01415-0
Reger, M. A., Watson, G. S., Frey, W. H. II., Baker, L. D., Cholerton, B., Keeling, M. L., et al. (2006). Effects of intranasal insulin on cognition in memory-impaired older adults: modulation by APOE genotype. Neurobiol. Aging 27, 451–458. doi: 10.1016/j.neurobiolaging.2005.03.016
Richwine, A. F., Godbout, J. P., Berg, B. M., Chen, J., Escobar, J., Millard, D. K., et al. (2005). Improved psychomotor performance in aged mice fed diet high in antioxidants is associated with reduced ex vivo brain interleukin-6 production. Brain Behav. Immun. 19, 512–520. doi: 10.1016/j.bbi.2004.12.005
Rinaldi, P., Polidori, M. C., Metastasio, A., Mariani, E., Mattioli, P., Cherubini, A., et al. (2003). Plasma antioxidants are similarly depleted in mild cognitive impairment and in Alzheimer’s disease. Neurobiol. Aging 24, 915–919. doi: 10.1016/s0197-4580(03)00031-9
Rodrigues Siqueira, I., Fochesatto, C., da Silva Torres, I. L., Dalmaz, C., and Alexandre Netto, C. (2005). Aging affects oxidative state in hippocampus, hypothalamus and adrenal glands of Wistar rats. Life Sci. 78, 271–278. doi: 10.1016/j.lfs.2005.04.044
Rosenberg, G. A. (2012). Neurological diseases in relation to the blood-brain barrier. J. Cereb. Blood Flow Metab. 32, 1139–1151. doi: 10.1038/jcbfm.2011.197
Safdar, A., deBeer, J., and Tarnopolsky, M. A. (2010). Dysfunctional Nrf2-Keap1 redox signaling in skeletal muscle of the sedentary old. Free Radic. Biol. Med. 49, 1487–1493. doi: 10.1016/j.freeradbiomed.2010.08.010
Salim, S. (2017). Oxidative stress and the central nervous system. J. Pharmacol. Exp. Ther. 360, 201–205. doi: 10.1124/jpet.116.237503
Salvadores, N., Sanhueza, M., Manque, P., and Court, F. A. (2017). Axonal degeneration during aging and its functional role in neurodegenerative disorders. Front. Neurosci. 11:451. doi: 10.3389/fnins.2017.00451
Sandell, J. H., and Peters, A. (2001). Effects of age on nerve fibers in the rhesus monkey optic nerve. J. Comp. Neurol. 429, 541–553. doi: 10.1002/1096-9861(20010122)429:4<541::aid-cne3>3.0.co;2-5
Sandell, J. H., and Peters, A. (2003). Disrupted myelin and axon loss in the anterior commissure of the aged rhesus monkey. J. Comp. Neurol. 466, 14–30. doi: 10.1002/cne.10859
Sandhu, S. K., and Kaur, G. (2002). Alterations in oxidative stress scavenger system in aging rat brain and lymphocytes. Biogerontology 3, 161–173. doi: 10.1023/A:1015643107449
Santos, R. X., Correia, S. C., Zhu, X., Smith, M. A., Moreira, P. I., Castellani, R. J., et al. (2013). Mitochondrial DNA oxidative damage and repair in aging and Alzheimer’s disease. Antioxid. Redox Signal. 18, 2444–2457. doi: 10.1089/ars.2012.5039
Santos, M. J., Quintanilla, R. A., Toro, A., Grandy, R., Dinamarca, M. C., Godoy, J. A., et al. (2005). Peroxisomal proliferation protects from β-amyloid neurodegeneration. J. Biol. Chem. 280, 41057–41068. doi: 10.1074/jbc.m505160200
Sasaki, T., Senda, M., Kim, S., Kojima, S., and Kubodera, A. (2001). Age-related changes of glutathione content, glucose transport and metabolism and mitochondrial electron transfer function in mouse brain. Nucl. Med. Biol. 28, 25–31. doi: 10.1016/s0969-8051(00)00180-3
Scheibye-Knudsen, M. (2016). Neurodegeneration in accelerated aging. Dan. Med. J. 63:B5308. doi: 10.1093/med/9780190497996.003.0024
Scheltens, P., Blennow, K., Breteler, M. M. B., de Strooper, B., Frisoni, G. B., Salloway, S., et al. (2016). Alzheimer’s disease. Lancet 388, 505–517. doi: 10.1016/S0140-6736(15)01124-1
Schultz, S. K., O’Leary, D. S., Boles Ponto, L. L., Watkins, G. L., Hichwa, R. D., and Andreasen, N. C. (1999). Age-related changes in regional cerebral blood flow among young to mid-life adults. Neuroreport 10, 2493–2496. doi: 10.1097/00001756-199908200-00011
Serrano, F., and Klann, E. (2004). Reactive oxygen species and synaptic plasticity in the aging hippocampus. Ageing Res. Rev. 3, 431–443. doi: 10.1016/j.arr.2004.05.002
Shelat, P. B., Chalimoniuk, M., Wang, J.-H., Strosznajder, J. B., Lee, J. C., Sun, A. Y., et al. (2008). Amyloid β peptide and NMDA induce ROS from NADPH oxidase and AA release from cytosolic phospholipase A2 in cortical neurons. J. Neurochem. 106, 45–55. doi: 10.1111/j.1471-4159.2008.05347.x
Shi, R., Zhang, Y., Shi, Y., Shi, S., and Jiang, L. (2012). Inhibition of peroxisomal β-oxidation by thioridazine increases the amount of VLCFAs and Aβ generation in the rat brain. Neurosci. Lett. 528, 6–10. doi: 10.1016/j.neulet.2012.08.086
Simpson, I. A., Vannucci, S. J., and Maher, F. (1994). Glucose transporters in mammalian brain. Biochem. Soc. Trans. 22, 671–675. doi: 10.1042/bst0220671
Small, G. W., Ercoli, L. M., Silverman, D. H., Huang, S. C., Komo, S., Bookheimer, S. Y., et al. (2000). Cerebral metabolic and cognitive decline in persons at genetic risk for Alzheimer’s disease. Proc. Natl. Acad. Sci. U S A 97, 6037–6042. doi: 10.1073/pnas.090106797
Smith, M. A. (1996). Hippocampal vulnerability to stress and aging: possible role of neurotrophic factors. Behav. Brain Res. 78, 25–36. doi: 10.1016/0166-4328(95)00220-0
Stahon, K. E., Bastian, C., Griffith, S., Kidd, G. J., Brunet, S., and Baltan, S. (2016). Age-related changes in axonal and mitochondrial ultrastructure and function in white matter. J. Neurosci. 36, 9990–10001. doi: 10.1523/JNEUROSCI.1316-16.2016
Stauch, K. L., Purnell, P. R., and Fox, H. S. (2014). Aging synaptic mitochondria exhibit dynamic proteomic changes while maintaining bioenergetic function. Aging 6, 320–334. doi: 10.18632/aging.100657
Suárez-Rivero, J. M., Villanueva-Paz, M., de la Cruz-Ojeda, P., de la Mata, M., Cotán, D., Oropesa-Ávila, M., et al. (2016). Mitochondrial dynamics in mitochondrial diseases. Diseases 5:E1. doi: 10.3390/diseases5010001
Suh, J. H., Moreau, R., Heath, S.-H. D., and Hagen, T. M. (2005). Dietary supplementation with (R)-α-lipoic acid reverses the age-related accumulation of iron and depletion of antioxidants in the rat cerebral cortex. Redox Rep. 10, 52–60. doi: 10.1179/135100005x21624
Suh, J. H., Wang, H., Liu, R.-M., Liu, J., and Hagen, T. M. (2004). (R)-α-lipoic acid reverses the age-related loss in GSH redox status in post-mitotic tissues: evidence for increased cysteine requirement for GSH synthesis. Arch. Biochem. Biophys. 423, 126–135. doi: 10.1016/j.abb.2003.12.020
Surmeier, D. J., Guzman, J. N., Sanchez, J., and Schumacker, P. T. (2012). Physiological phenotype and vulnerability in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2:a009290. doi: 10.1101/cshperspect.a009290
Swerdlow, R. H., Parks, J. K., Miller, S. W., Davis, R. E., Tuttle, J. B., Trimmer, P. A., et al. (1996). Origin and functional consequences of the complex I defect in Parkinson’s disease. Ann. Neurol. 40, 663–671. doi: 10.1002/ana.410400417
Tapia-Arancibia, L., Aliaga, E., Silhol, M., and Arancibia, S. (2008). New insights into brain BDNF function in normal aging and Alzheimer disease. Brain Res. Rev. 59, 201–220. doi: 10.1016/j.brainresrev.2008.07.007
Thibault, O., Hadley, R., and Landfield, P. W. (2001). Elevated postsynaptic [Ca2+]i and L-type calcium channel activity in aged hippocampal neurons: relationship to impaired synaptic plasticity. J. Neurosci. 21, 9744–9756. doi: 10.1523/JNEUROSCI.21-24-09744.2001
Toescu, E. C., Verkhratsky, A., and Landfield, P. W. (2004). Ca2+ regulation and gene expression in normal brain aging. Trends Neurosci. 27, 614–620. doi: 10.1016/j.tins.2004.07.010
Trompier, D., Vejux, A., Zarrouk, A., Gondcaille, C., Geillon, F., Nury, T., et al. (2014). Brain peroxisomes. Biochimie 98, 102–110. doi: 10.1016/j.biochi.2013.09.009
Ungvari, Z., Bailey-Downs, L., Gautam, T., Sosnowska, D., Wang, M., Monticone, R. E., et al. (2011). Age-associated vascular oxidative stress, Nrf2 dysfunction, and NF-κB activation in the nonhuman primate Macaca mulatta. J. Gerontol. A Biol. Sci. Med. Sci. 66, 866–875. doi: 10.1093/gerona/glr092
Valente, E. M., Abou-Sleiman, P. M., Caputo, V., Muqit, M. M., Harvey, K., Gispert, S., et al. (2004). Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 304, 1158–1160. doi: 10.1126/science.1096284
van de Haar, H. J., Burgmans, S., Jansen, J. F. A., van Osch, M. J. P., van Buchem, M. A., Muller, M., et al. (2016). Blood-brain barrier leakage in patients with early alzheimer disease. Radiology 281, 527–535. doi: 10.1148/radiol.2016152244
Wang, H., Liu, H., and Liu, R. M. (2003). Gender difference in glutathione metabolism during aging in mice. Exp. Gerontol. 38, 507–517. doi: 10.1016/s0531-5565(03)00036-6
Wang, X., and Michaelis, E. K. (2010). Selective neuronal vulnerability to oxidative stress in the brain. Front. Aging Neurosci. 2:12. doi: 10.3389/fnagi.2010.00012
Wang, S., Zhu, J., and Xu, T. (2018). 17β-estradiol (E2) promotes growth and stability of new dendritic spines via estrogen receptor β pathway in intact mouse cortex. Brain Res. Bull. 137, 241–248. doi: 10.1016/j.brainresbull.2017.12.011
Winkler, E. A., Nishida, Y., Sagare, A. P., Rege, S. V., Bell, R. D., Perlmutter, D., et al. (2015). GLUT1 reductions exacerbate Alzheimer’s disease vasculo-neuronal dysfunction and degeneration. Nat. Neurosci. 18, 521–530. doi: 10.1038/nn.3966
Wood, S. K., Walker, H. E., Valentino, R. J., and Bhatnagar, S. (2010). Individual differences in reactivity to social stress predict susceptibility and resilience to a depressive phenotype: role of corticotropin-releasing factor. Endocrinology 151, 1795–1805. doi: 10.1210/en.2009-1026
Xiang, W., Schlachetzki, J. C. M., Helling, S., Bussmann, J. C., Berlinghof, M., Schäffer, T. E., et al. (2013). Oxidative stress-induced posttranslational modifications of α-synuclein: specific modification of α-synuclein by 4-hydroxy-2-nonenal increases dopaminergic toxicity. Mol. Cell. Neurosci. 54, 71–83. doi: 10.1016/j.mcn.2013.01.004
Xu, B., and Xie, X. (2016). Neurotrophic factor control of satiety and body weight. Nat. Rev. Neurosci. 17, 282–292. doi: 10.1038/nrn.2016.24
Yang, J., Brown, M. S., Liang, G., Grishin, N. V., and Goldstein, J. L. (2008). Identification of the acyltransferase that octanoylates ghrelin, an appetite-stimulating peptide hormone. Cell 132, 387–396. doi: 10.1016/j.cell.2008.01.017
Yang, C., Liu, Y., Ni, X., Li, N., Zhang, B., and Fang, X. (2014). Enhancement of the nonamyloidogenic pathway by exogenous NGF in an Alzheimer transgenic mouse model. Neuropeptides 48, 233–238. doi: 10.1016/j.npep.2014.04.005
Yin, F., Sancheti, H., Patil, I., and Cadenas, E. (2016). Energy metabolism and inflammation in brain aging and Alzheimer’s disease. Free Radic. Biol. Med. 100, 108–122. doi: 10.1016/j.freeradbiomed.2016.04.200
Yun, J., and Finkel, T. (2014). Mitohormesis. Cell Metab. 19, 757–766. doi: 10.1016/j.cmet.2014.01.011
Zhu, Y., Carvey, P. M., and Ling, Z. (2006). Age-related changes in glutathione and glutathione-related enzymes in rat brain. Brain Res. 1090, 35–44. doi: 10.1016/j.brainres.2006.03.063
Zhu, X.-H., Lu, M., Lee, B.-Y., Ugurbil, K., and Chen, W. (2015). In vivo NAD assay reveals the intracellular NAD contents and redox state in healthy human brain and their age dependences. Proc. Natl. Acad. Sci. U S A 112, 2876–2881. doi: 10.1073/pnas.1417921112
Zipser, B. D., Johanson, C. E., Gonzalez, L., Berzin, T. M., Tavares, R., Hulette, C. M., et al. (2007). Microvascular injury and blood-brain barrier leakage in Alzheimer’s disease. Neurobiol. Aging 28, 977–986. doi: 10.1016/j.neurobiolaging.2006.05.016
Zlokovic, B. V. (2011). Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat. Rev. Neurosci. 12, 723–738. doi: 10.1038/nrn3114
Keywords: aging, neurodegeneration, energetic metabolism, mitochondrial dysfunction, oxidative stress
Citation: Castelli V, Benedetti E, Antonosante A, Catanesi M, Pitari G, Ippoliti R, Cimini A and d’Angelo M (2019) Neuronal Cells Rearrangement During Aging and Neurodegenerative Disease: Metabolism, Oxidative Stress and Organelles Dynamic. Front. Mol. Neurosci. 12:132. doi: 10.3389/fnmol.2019.00132
Received: 23 January 2019; Accepted: 07 May 2019;
Published: 28 May 2019.
Edited by:
Pier Giorgio Mastroberardino, Erasmus University Rotterdam, NetherlandsReviewed by:
Anna Fracassi, Roma Tre University, ItalyCésar Payán-Gómez, Universidad del Rosario, Colombia
Copyright © 2019 Castelli, Benedetti, Antonosante, Catanesi, Pitari, Ippoliti, Cimini and d’Angelo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Annamaria Cimini, YW5uYW1hcmlhLmNpbWluaUB1bml2YXEuaXQ=; Michele d’Angelo, ZGFuZ2Vsby1taWNoZWxlQGhvdG1haWwuY29t