 Gloria Lee
Gloria Lee Yi Zhou
Yi Zhou- Department of Biomedical Sciences, Florida State University College of Medicine, Tallahassee, FL, United States
The N-methyl-d-aspartate receptor (NMDAR) hypofunction hypothesis has been proposed to help understand the etiology and pathophysiology of schizophrenia. This hypothesis was based on early observations that NMDAR antagonists could induce a full range of symptoms of schizophrenia in normal human subjects. Accumulating evidence in humans and animal studies points to NMDAR hypofunctionality as a convergence point for various symptoms of schizophrenia. Here we review animal models of NMDAR hypofunction generated by pharmacological and genetic approaches, and how they relate to the pathophysiology of schizophrenia. In addition, we discuss the limitations of animal models of NMDAR hypofunction and their potential utility for therapeutic applications.
Introduction
Schizophrenia is a debilitating psychiatric disease that affects ~1% of the world population and places a major socio-economic burden (Blot et al., 2013). Patients exhibit positive and negative symptoms, as well as cognitive impairments, which typically emerge at early adolescence and worsen over time (Krystal et al., 1994; Morgan and Curran, 2006; Javitt, 2007; Howes et al., 2015). Both clinical and basic research suggest that schizophrenia is a neurodevelopmental disorder involving a variety of susceptibility genes, environmental risk factors, and epigenetic alterations (Cardno et al., 1999; Cannon et al., 2002; Shi et al., 2008; Walsh et al., 2008; Jaaro-Peled et al., 2009; Singh and O'Reilly, 2009), but it is unclear how these factors may contribute to the development of schizophrenic symptoms (Lewis and Gonzalez-Burgos, 2008; Faludi and Mirnics, 2011). Moreover, limited understanding on the pathophysiological mechanisms of schizophrenic brain has hindered the development of effective treatment for this disease. Current therapeutics is limited to antipsychotics (APDs) that mainly reduce positive symptoms but are ineffective for treating persistent negative or cognitive symptoms, which are often present before the onset of positive symptoms and lead to long-term functional impairments in schizophrenia patients (Howes et al., 2012; Citrome, 2014).
The glutamate hypothesis first emerged in the 1980s, and the N-methyl-D-aspartate receptor (NMDAR) hypofunction model was proposed afterwards upon observing that NMDAR antagonists could recapitulate a full range of positive, negative, and cognitive symptoms of schizophrenia in normal human subjects (Anis et al., 1983; Krystal et al., 1994). Consistent with this initial observation, findings from clinical, pharmacologic, and genetic studies suggested that NMDAR hypofunction may be one of the pathophysiological mechanism for schizophrenia (Goff and Coyle, 2001; Moghaddam, 2003; Jones et al., 2011; Javitt et al., 2012; Lin et al., 2012). NMDARs are ionotropic glutamate receptors that are comprised of the two obligatory NR1 subunits and two NR2 and/or NR3 subunits (Laurie and Seeburg, 1994). They function as a “coincidence detector” of pre- and post-synaptic activity and have crucial roles in glutamatergic neurotransmission, local rhythmic activity, and synaptic plasticity (Collingridge et al., 1988; Olney and Farber, 1995; Jensen and Lisman, 1996; Fellin et al., 2009). Thus, NMDARs are known to modulate cognition, memory, and higher-order brain functions (Moghaddam et al., 1997; Adams and Moghaddam, 1998; Palmer et al., 2008; Collingridge et al., 2013). In the last several decades, animal models of NMDAR hypofunction have been widely utilized to study the neurobiology of schizophrenia, as well as to test drugs for treating symptoms of schizophrenia. Here we review current literatures on animal models of NMDAR hypofunction using pharmacological and genetic approaches to induce NMDAR hypofunctionality.
Pharmacological Approaches to Induce NMDAR Hypofunction
Phencyclidine
Phencyclidine (PCP) is an abused drug that acts as a non-competitive blocker of NMDARs at lower doses. PCP administration was found to produce hallucinogenic activity in healthy human subjects (Itil et al., 1967). This had led to further investigation of the pathophysiological mechanisms for PCP-induced behavioral changes, particularly through inhibition of NMDARs (Carlsson and Carlsson, 1990; Johnson and Jones, 1990; Javitt and Zukin, 1991; Olney and Farber, 1995). To date, a wide range of animal studies have been conducted with different PCP administration regimen (acute, subchronic, or chronic), dosage, and co-administration with other drugs. The effects of PCP in animals at the molecular, electrophysiological, and behavioral levels were assessed by a variety of approaches.
In rodents, behaviors associated with positive symptoms of schizophrenia are assayed as novelty-induced hyperlocomotion in the open field test. Negative symptom-like behavioral alterations correlate with social withdrawal and anhedonia, which are commonly evaluated by social interaction, sucrose preference and forced swim tests (FST) (Sams-Dodd, 1996). Cognitive impairments are assessed using a variety of behavioral assays including radial arm maze, Morris water maze, Y-maze, T-maze, novel object recognition test, avoidance task learning test, attentional set-shifting task, modified hole board task, object and object-in-context recognition memory task, five-choice serial-reaction time test, and contextual fear conditioning. Moreover, the endophenotype corresponding to sensorimotor gating deficits in schizophrenics is detected as impaired prepulse inhibition (PPI) of acoustic startle response (McKibben et al., 2010). Acute administration of PCP induces a full spectrum of behavioral changes associated with positive, negative and cognitive symptoms of schizophrenia. Positive symptoms-related behaviors exhibited in acute PCP animal models include increased stereotypic behavior and ataxia, but there are conflicting reports regarding changes in locomotor activity. Negative symptoms-related behaviors are reflected as reduced social interaction, while cognitive deficits range from impaired latent learning, deficits in attention and cognitive flexibility to decreased sensorimotor gating (Sturgeon et al., 1979; Nabeshima et al., 1986; Mansbach and Geyer, 1989; Sams-Dodd, 1995, 1996; Martinez et al., 1999; Noda et al., 2001; Abdul-Monim et al., 2003; Egerton et al., 2005). These behavioral changes are accompanied by increased brain metabolism in areas of cortex, basal ganglia, and thalamus, decreased parvalbumin (PV) mRNA expression in the hippocampus and dorsal reticular nucleus of the thalamus, and altered zinc finger protein 225 (zif268) mRNA expression in the infralimbic cortex (Martinez et al., 1999; Egerton et al., 2005). Another study showed that acute PCP administration during early postnatal time period results in loss of PV-containing neurons from the primary somatosensory, motor, and retrosplenial cortices (Wang et al., 2008).
Subchronic (2–14 days) and chronic (15 days or longer) administration of PCP also generated a variety of schizophrenia-associated behavioral phenotypes (Noda et al., 1995; Sams-Dodd, 1995, 1996; Jentsch et al., 1997b, 1998; Stefani and Moghaddam, 2002; Balla et al., 2003; Abdul-Monim et al., 2006, 2007; Grayson et al., 2007; Egerton et al., 2008; Jenkins et al., 2008; Brigman et al., 2009; McKibben et al., 2010). Functional alterations induced by subchronic and chronic administration of PCP include reduced numbers of PV+ neurons in the hippocampus and frontal cortex. However, there are inconsistent reports on the PCP-induced changes in dopamine (DA) systems, as some studies observed potentiation of amphetamine-induced DA release and decreased basal DA metabolism in the prefrontal cortex (PFC) (Jentsch et al., 1997a; Balla et al., 2003; Cochran et al., 2003; Abdul-Monim et al., 2007; Jenkins et al., 2008; McKibben et al., 2010), whereas others reported reduced mesoprefrontal DA utilization (Jentsch et al., 1997a,b). In schizophrenia, dysregulation of DA systems is characterized by cortical hypodopaminergia and subcortical hyperdopaminergia (Slifstein et al., 2015). In that respect, there seems to be a discrepancy between the pharmacological rodent model and the disease.
Compared with acute exposure to PCP, subchronic and chronic exposure to PCP induces a more overt and sustained schizophrenia-like state in both rodents and in humans. As it provides a wider time frame to study long-lasting changes related to symptoms of schizophrenia, subchronic, and chronic PCP treated animal models are more commonly used to study the NMDAR hypofunctionality (Rainey and Crowder, 1975; Allen and Young, 1978; Cosgrove and Newell, 1991). In particular, these models have been applied to study the effects of NMDAR blockade on negative and cognitive symptoms-related behaviors, as well as to investigate the potential neurochemical and neuroanatomical basis underlying these behavioral changes (Jentsch and Roth, 1999; Lewis and Levitt, 2002; Table 1).
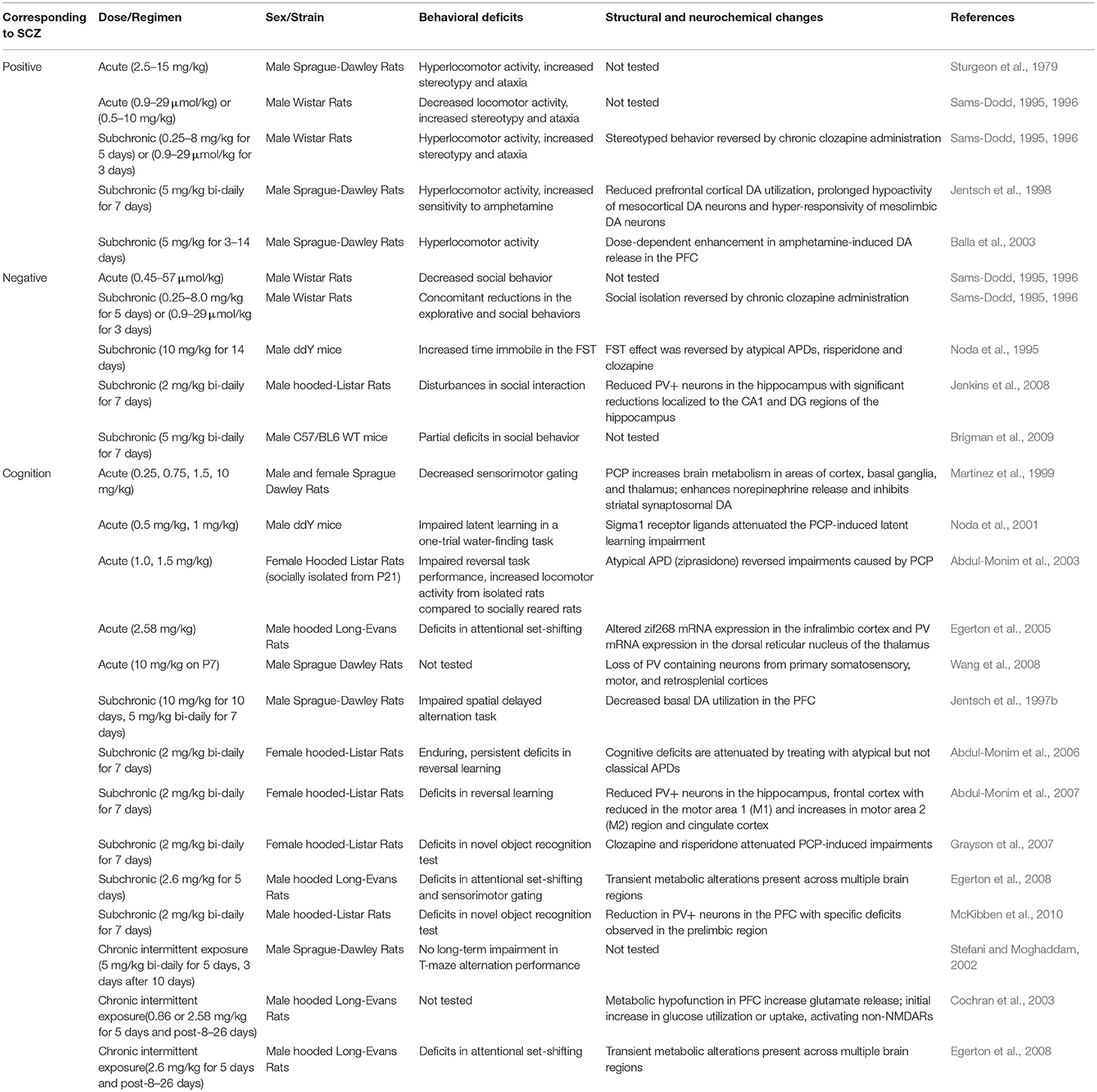
Table 1. Comparison of animal models of phencyclidine.
Ketamine
Ketamine was first synthesized as a PCP derivative back in the 1960s in an effort to minimize the side effects and neurotoxicity of PCP (Maddox et al., 1965). As a noncompetitive NMDAR antagonist, ketamine acts by specifically binding to a site near the channel pore (Roth et al., 2013). Ketamine was found to be safe and effective even with repeated administration with minimal side effects and was soon used as a clinical anesthetic (Domino et al., 1965; Corssen and Domino, 1966; Li and Vlisides, 2016). Ketamine administration has often been used in animal studies as a preclinical model to test the effects of APDs and novel compounds since atypical APDs are found to be effective in blocking ketamine's behavioral effects in both humans and rodents (Jentsch and Roth, 1999; Krystal et al., 1999, 2005; Becker et al., 2003; Lees et al., 2004; Gilmour et al., 2009; Neill et al., 2010). However, apart from the blockade of NMDARs, ketamine can act on a wide-range of targets in various cellular processes (Li and Vlisides, 2016).
Acute ketamine administration in animals results in cognitive deficits including reduced sensorimotor gating, spatial learning and memory impairments, along with changes in theta and gamma band activity (Verma and Moghaddam, 1996; de Bruin et al., 1999; Ehrlichman et al., 2009; Kittelberger et al., 2012; Szlachta et al., 2017; Coronel-Oliveros and Pacheco-Calderon, 2018). In contrast to cortical hypodopaminergic state observed in schizophrenia, these cognitive deficits are accompanied by increased DA release in the PFC (Verma and Moghaddam, 1996). Other studies also reported that cognitive deficits are associated with increased frontal cortical blood flow (Ingvar and Franzen, 1974; Vollenweider et al., 1997). Moreover, acute ketamine administration induces hyperlocomotor activity and stereotypic behaviors, which might be attributed to increased DA and serotonin turnover in the striatum and cortex (Irifune et al., 1991; Chatterjee et al., 2011, 2012; Coronel-Oliveros and Pacheco-Calderon, 2018). However, there are inconsistent reports as to the effect of acute ketamine administration on negative symptoms-related behaviors based on social interaction tests (Silvestre et al., 1997; Chatterjee et al., 2011; Coronel-Oliveros and Pacheco-Calderon, 2018). In fact, ketamine is known to have anti-depressant effects at low doses, which may oppose the induction of negative symptoms-related behaviors in animals (Zanos and Gould, 2018).
Subchronic ketamine administration, however, can induce negative symptoms-related behaviors, in addition to positive symptoms-related behaviors. These include increased time spent immobile during the forced swim test (FST), hyperlocomotor activity, and stereotypy (Becker et al., 2003; Chatterjee et al., 2011, 2012). Hyperlocomotor activity exhibited after a subchronic dose of ketamine is attributed to increased DA and serotonin levels in the cortex and striatum, which is thought to result from alternations in gene expression of DA and serotonin receptors (Chatterjee et al., 2011, 2012). On the other hand, negative symptom-related behavior in both acute and subchronic ketamine administration is associated with decreased levels of glycine in the striatum, hippocampus, and cortex (Chatterjee et al., 2012). This is consistent with previous findings from schizophrenia patients that linked reduced levels of glycine to negative symptoms of schizophrenia and increased levels of glycine for the treatment of negative symptoms (Javitt, 2010).
Subchronic ketamine administration also induce cognitive deficits, such as reversal learning and long-term spatial memory impairments (Featherstone et al., 2012; Szlachta et al., 2017). In addition, chronic ketamine administration results in decreased PV interneuron density in the hippocampus (Keilhoff et al., 2004; Kittelberger et al., 2012). Fast-spiking PV interneurons are important for encoding and storage of information required for working memory, and dysfunction in fast-spiking PV interneurons are known to cause cognitive deficits, potentially through disrupting theta and gamma oscillations (Bartos et al., 2007). Indeed, subchronic ketamine administration induce cognitive deficits that are accompanied by alterations in theta and gamma oscillation from the hippocampus and PFC (Featherstone et al., 2012). Moreover, audioradiographic imaging studies have reported dysconnectivity between PFC and hippocampus in both acute and subchronic ketamine animal models (Dawson et al., 2013).
Overall, ketamine can induce behavioral alterations associated with schizophrenia symptoms but with less excitotoxicity than PCP. When comparing treatment regimens, subchronic ketamine treatment models are more widely used, since it can induce persistent negative and cognitive symptoms-related behaviors with hallmark features of dysfunction in neuronal circuitry (Lahti et al., 2001; Morgan et al., 2009). However, behavioral deficits and cortical dysfunctions cannot be attributed to changes only in the glutamatergic system as it may involve changes in other neurotransmitter systems such as DA and serotonin (Grace, 2012; Table 2).
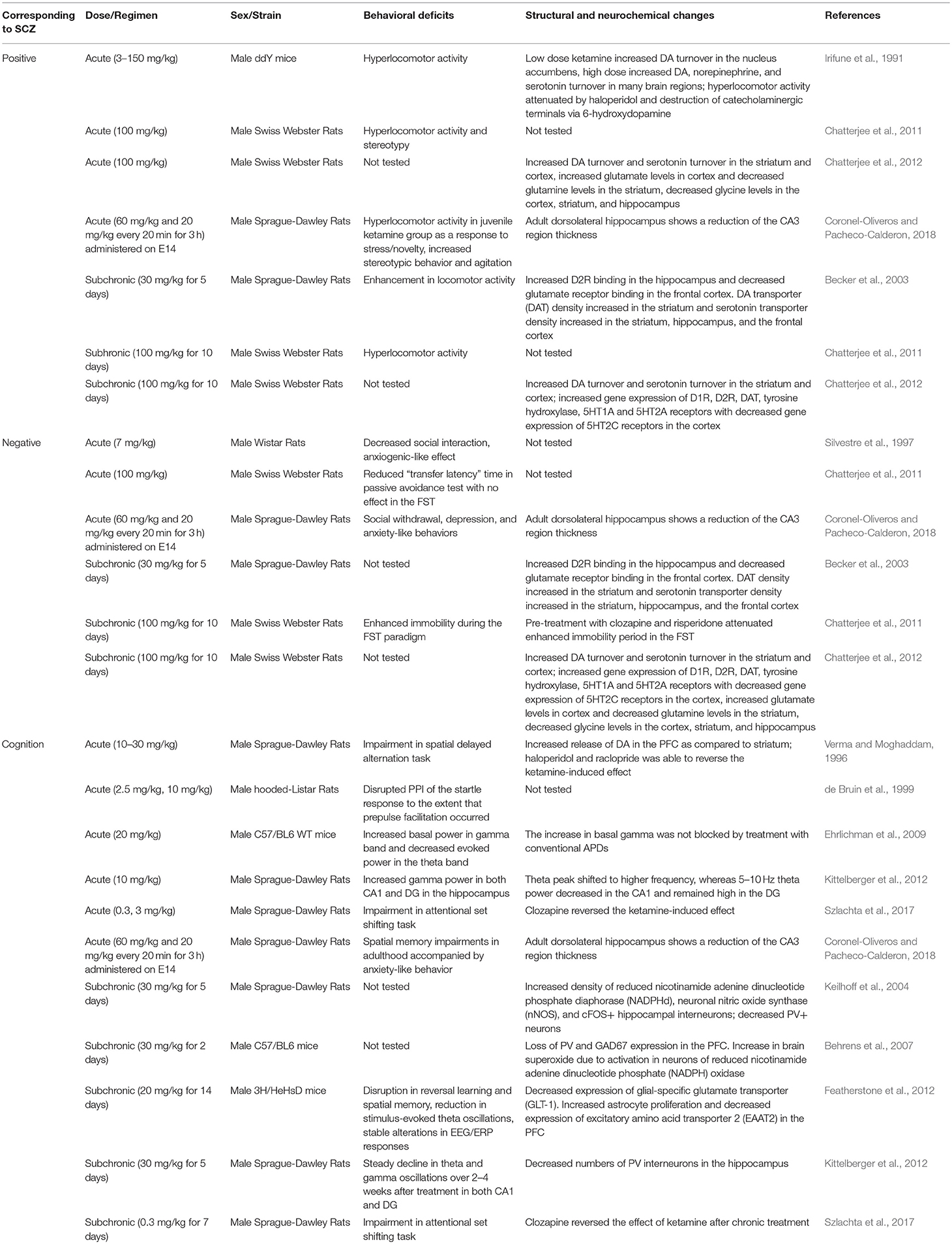
Table 2. Comparison of animal models of ketamine.
MK-801
Also known as dizocilpine, MK-801 is a non-competitive NMDAR antagonist that blocks the channel in a use- and voltage-dependent manner (Foster and Fagg, 1987; Huettner and Bean, 1988). Like ketamine, MK-801 seems to preferentially act on GABAergic interneurons. This selective action of MK-801 on interneurons reduces inhibitory influence on excitatory pyramidal neurons, leading to hyperexcitation in the PFC neuronal circuit (Yonezawa et al., 1998; Homayoun and Moghaddam, 2007). However, more recent studies showed that local MK-801 infusion into the PFC does not directly produce disinhibition (Gratton et al., 1987; Suzuki et al., 2002; Jodo et al., 2005), suggesting that hyper-glutamatergia in the PFC may require MK-801's effects in other brain regions such as the hippocampus (Jodo, 2013; Nakazawa et al., 2017). Moreover, MK-801 has substantially longer action of NMDAR blockade in rodents and higher specificity for NMDARs than ketamine (Miyamoto et al., 2000; Pinault, 2008; Hakami et al., 2009).
Perhaps due to its prolonged action, high potency and specificity for NMDARs, MK-801 can produce a full range of schizophrenia-related behavioral phenotypes in both acute and chronic treatment models. In rodents, acute MK-801 administration induces hyperlocomotor activity, decreased social interaction, and impairments in cognitive flexibility, latent learning, long-term spatial memory, working memory, and sensorimotor gating (Yamada et al., 1996; Bast et al., 2000; Noda et al., 2001; Harris et al., 2003; Rung et al., 2005; Abekawa et al., 2007; Manahan-Vaughan et al., 2008; Zou et al., 2008; Wiescholleck and Manahan-Vaughan, 2012). Cognitive deficits induced by acute MK-801 administration are associated with impaired long term potentiation (LTP) (Manahan-Vaughan et al., 2008; Wiescholleck and Manahan-Vaughan, 2012). Acute MK-801 treatment also results in decreased PV mRNA expression in the PFC, hippocampus, bed nucleus of the stria terminalis (BNST) amygdala, orbitofrontal and entorhinal cortex, with no changes in glutamate decarboxylase 67 kDa (GAD67) mRNA expression (Abekawa et al., 2007; Romon et al., 2011). Acute MK-801 administration is accompanied by increased gamma power in the hippocampus and decreased beta band power in the hippocampus (Kittelberger et al., 2012; Sullivan et al., 2015). In addition, acute ketamine treatment leads to increased synapse-associated protein 90/postsynaptic density protein 95 (SAP90/PSD95) and protein kinase C gamma (PKCγ) mRNA expression from cortical regions, decreased NR2C protein expression from the entorhinal cortex, and decreased NR2B protein expression from the parietal cortex (Linden et al., 2001).
Chronic MK-801 treatment also results in dysfunctional GABAergic interneurons as indicated by reduced PV interneuron density in the hippocampus (Braun et al., 2007). Similarly to acute treatment, chronic and subchronic MK-801 treatment leads to cognitive deficits (Latysheva and Rayevsky, 2003; Stefani and Moghaddam, 2005; Rujescu et al., 2006; Li et al., 2011; Xiu et al., 2014; Liu et al., 2017; Unal et al., 2018). In addition, chronic MK-801 treatment leads to sensorimotor gating and social interaction deficits, although there are controversial results on locomotor activity (Latysheva and Rayevsky, 2003; Eyjolfsson et al., 2006; Xiu et al., 2014; Unal et al., 2018). Another study showed that chronic MK-801 induce increased anxiety-like behavior and working memory deficits, which was accompanied by degenerative changes of myelin sheaths, decreased white matter and corpus callosum volume (Xiu et al., 2014). Early postnatal chronic MK-801 treatment model reported reduced locomotor activity, decreased rearing behavior, exploratory behavior, increased anxiety-like behavior as well as learning impairments (Latysheva and Rayevsky, 2003; Table 3). Results from MK-801 administration in awake rats suggest that NMDAR inhibition causes cortical excitation by disinhibition of pyramidal neurons in the PFC (Homayoun and Moghaddam, 2007). In addition, studies consistently report that chronic MK-801 administration leads to increased dopaminergic and serotonergic activity in the frontal cortex, nucleus accumbens and striatum (Loscher et al., 1991). As discussed earlier, an increased dopaminergic activity in the frontal cortex by chronic MK-801 administration may not be consistent with the cortical hypodopaminergic state in schizophrenia patients, which further points to the limitations in the use of pharmacological rodent models to recapitulate the dysregulation of DA systems.
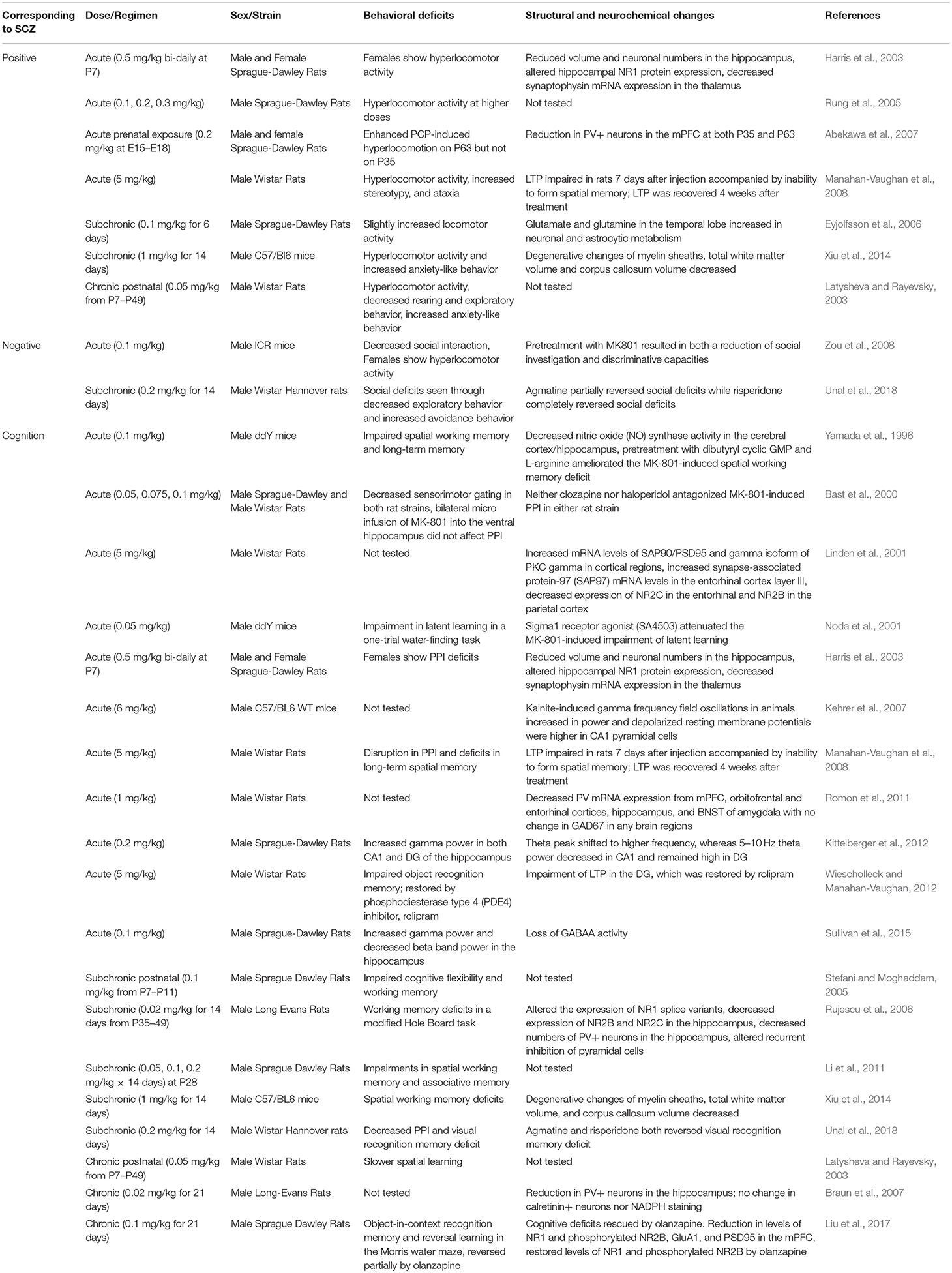
Table 3. Comparison of animal models of MK-801.
Genetic Approaches to Induce NMDAR Hypofunction
Knockout or Knockdown of NR1 Subunits
Based on genetic analyses, polymorphisms exist in both coding and promoter regions of glutamate receptor ionotropic NMDA type subunits (GRIN), impacting both NMDAR transcript levels and/or functions. Single nucleotide or dinucleotide-repeated polymorphisms of the NMDAR subunit genes, such as NR1 (GRIN1), NR2A (GRIN2A), and NR2B (GRIN2B), increase susceptibility to schizophrenia (Ohtsuki et al., 2001; Rice et al., 2001; Miyatake et al., 2002; Itokawa et al., 2003; Iwayama-Shigeno et al., 2005; Qin et al., 2005; Martucci et al., 2006; Tang et al., 2006; Zhao et al., 2006). Moreover, postmortem and genetic studies from schizophrenia patients support that abnormalities in glycine modulatory sites on the NMDARs may contribute to the pathophysiology of schizophrenia (Coyle, 2004).
Amongst various NMDAR subunits, NR1 is essential for formation and synaptic expression of NMDARs (Laurie and Seeburg, 1994). A complete loss of NR1 subunit in mice [NR1 knockout (KO)] is neonatally lethal, suggesting that NMDARs play a crucial role in early development (Forrest et al., 1994). To overcome neonatal lethality, a line of NR1 knockdown (KD) mice was generated by ectopic transgenic expression of one of the NR1 splice variants (NR1-1a) in the NR1 KO mice. This line of NR1 KD mice exhibits altered dendritic differentiation, branching, and somatosensory pattern, accompanied by increased axonal arborizations with faster and prematurely developed projection neurons of the corpus callosum (Iwasato et al., 1997; Lee et al., 2005). However, the average lifespan of this NR1 KD mice was dependent on the level of transgene expression. This limits the usage of these mice for further behavioral, molecular, and structural studies in adult mice. Another NR1 KD mice were created by inserting a neomycin cassette into an intronic region of the GRIN1 gene, leading to significantly lower levels of NR1 expression (Mohn et al., 1999). These mice are viable and exhibit a full range of behavioral phenotypes associated with schizophrenia, such as hyperlocomotor activity, stereotypy, self-injury, decreased anxiety-related behavior, reduced nest building, impaired social and sexual interactions, abnormal evoked response potentials (ERPs), cognitive inflexibility, abnormal selective attention, with spatial cognitive and sensorimotor gating deficits (Mohn et al., 1999; Duncan et al., 2004; Moy et al., 2006; Bickel et al., 2007; Dzirasa et al., 2009; Halene et al., 2009). These NR1 KD mice also exhibit reduced synapse-number in an age-dependent manner, decreased NMDA currents, reduced 2-deoxyglucose (DG) uptake in areas of the neocortex, increased amplitudes of auditory and visual ERPs, attenuated cortical and hippocampal theta-gamma phase coupling, increased dendritic length, and synapse-specific reductions in 14-3-3ε and disrupted in schizophrenia 1 (DISC1) protein expression (Duncan et al., 2002; Dzirasa et al., 2009; Halene et al., 2009; Ramsey et al., 2011). Others have also developed hypomorphic NR1 subunit mice with single or double point mutations in NMDAR glycine binding sites, GRIN1D481 and GRIN1K483. Hypomorphic NR1 mice with single point mutation in D481 (GRIN1D481N) are viable and exhibit increased startle reactivity, deficits in spatial recognition, spatial reference learning and memory, reduced sociability, anxiety, and sensitivity to NMDA-induced seizures, with impaired hippocampal LTP (Kew et al., 2000; Labrie et al., 2008). In contrast, GRIN1K483Q mice are perinatally lethal (Kew et al., 2000). Subsequently, Ballard et al. developed the compound heterozygote mice with point mutations in both glycine binding sites. These mice (GRIN1D481N/K483Q) are viable and display hyperlocomotor activity, stereotypy, disrupted nesting behavior, spatial learning, and sensorimotor gating deficits. These behavioral changes are accompanied by striatal dopaminergic and serotonergic hyperfunction. Interestingly, deficits in hippocampal LTP are rescued by D-serine administration (Ballard et al., 2002).
To understand how NMDAR hypofunction affects behaviors at cellular and circuit levels, various animal models with brain region- and/or cell-type specific deletion of NR1 have been created. Forebrain excitatory neuron-specific NR1 KO mice exhibit impairments in spatial memory, encoding and flexible expression of non-spatial memory with slower acquisition of trace fear conditioning. These mice also exhibit degraded co-activation of CA1 place cells during exploration and lack NMDAR-mediated synaptic currents and LTP in CA1 synapses (McHugh et al., 1996; Tsien et al., 1996; Huerta et al., 2000; Rondi-Reig et al., 2001). Dentate-gyrus (DG) granule cells-specific NR1 KO mice display deficits in the process of pattern separation, which is accompanied by reduced firing rate from cornu ammonis 3 (CA3) pyramidal cells during context-specific modulation. These mice also exhibit deficits in spatial working memory accompanied by impaired LTP in both medial and lateral perforant path inputs to the DG (McHugh et al., 2007; Niewoehner et al., 2007). CA1-specific NR1 KD rats were created by injecting NR1 antisense RNA into the CA1 region of the hippocampus. These rats exhibit impairments in avoidance task learning and memory formation (Cheli et al., 2002, 2006). Similarly, CA3 pyramidal cells-specific NR1 KO mice exhibit deficits in associative memory recall accompanied by impaired NMDAR-dependent LTP at the commissural/associational (C/A) pathway (Nakazawa et al., 2002). Another CA3-specific NR1 KO mice, created by an adeno-associated virus (AAV)-induced deletion of the NR1 gene, exhibit increased impulsive behavior, learning impairments, and decreased social approach behavior (Rajji et al., 2006; Finlay et al., 2015).
Mice with deletion of NR1 from cortical and hippocampal GABAergic interneurons during early postnatal development exhibit hyperlocomotor activity, anhedonia-like and anxiety-like behaviors, mating and nest-building deficits, as well as social memory, spatial working memory, and prepulse inhibition deficits. These changes are accompanied by increased firing of cortical excitatory neurons with reduced neuronal synchrony. Overall, these findings support that NMDAR hypofunction in corticolimbic GABAergic interneurons during early postnatal development can lead to the development of schizophrenia-related behavioral phenotypes (Belforte et al., 2010). Furthermore, PV-specific NR1 KO mice were generated by targeted deletion of NR1 subunits from a subset of GABAergic interneurons. These mice exhibit deficits in spatial working, short- and long-term recognition memory, which are correlated with profound changes in neural activity related to cognition including increased gamma oscillation power and decreased theta oscillation power of local field potentials (LFP) in the hippocampus (Korotkova et al., 2010). Moreover, other groups reported that the PV-specific NR1 KO mice display blunted MK-801-induced hyperlocomotor activity, suggesting that NMDARs in PV interneurons may be the site of MK-801 action (Belforte et al., 2010; Carlen et al., 2012). However, a more recent study reported that these mice are not protected against behavioral effects of MK-801, as MK-801 administration leads to increased stereotypy and pronounced catalepsy, which confound the locomotor readout (Bygrave et al., 2016).
NR1 deletion in forebrain pyramidal neurons also results in behavioral changes associated with schizophrenia. The forebrain pyramidal cells-specific NR1 KO mice display hyperlocomotor activity and decreased self-care, as well as social and cognitive impairments (Tatard-Leitman et al., 2015). In addition, these mice exhibit decreased expression of dopamine D2 receptor (D2R) and G-protein-regulated inward-rectifier potassium channel 2 (GIRK2) in the forebrain, increased baseline gamma power and pyramidal cell excitability (Tatard-Leitman et al., 2015). However, mPFC and sensory cortex pyramidal cells-specific NR1 KO mice only exhibit PPI and short-term memory impairments, suggesting that deletion of NR1 subunit in pyramidal neurons of broader forebrain regions may be required to induce a full range of symptoms of schizophrenia (Kehrer et al., 2008). Taken together, these studies demonstrate that NR1-mediated deficits in either pyramidal or GABAergic neurons could cause an imbalance of excitation and inhibition in the cortical neural circuit, leading to development of behavioral phenotypes related to schizophrenia (Table 4).
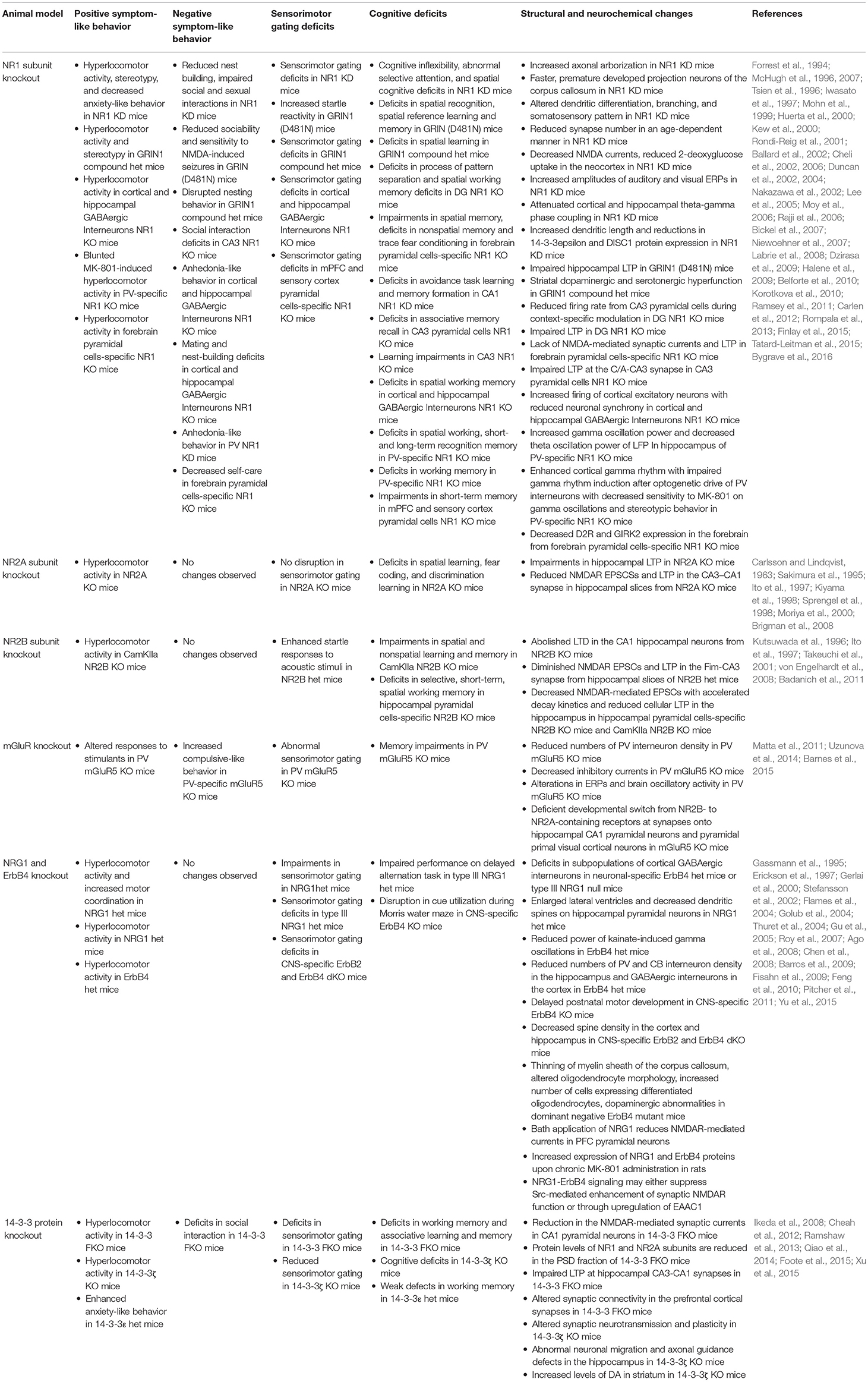
Table 4. Genetic animal models of NMDAR hypofunctionality.
Knockout of NR2A or NR2B Subunits
Amongst four NR2 subunits (A–D), the NR2A and NR2B subunits are known to predominantly function in the forebrain. They impart different characteristics on functional NMDARs, with NR2A-containing NMDARs having more rapid kinetics than NR2B-containing NMDARs (Kutsuwada et al., 1992; Monyer et al., 1994; Loftis and Janowsky, 2003). A switch of NR2B to NR2A subunit expression in the forebrain and sensory systems is linked with the timing of critical period for sensory plasticity, making NR2B and NR2A subunits particularly known for its involvement in postnatal brain development (Monyer et al., 1994; Sheng et al., 1994; Yashiro and Philpot, 2008). At birth, NR2B is the predominant subunit, whereas NR2A subunit expression begins around postnatal day 3 (P3), after which there is a gradual increase expression of NR2A subunits (Monyer et al., 1994; Sheng et al., 1994; Zhong et al., 1995). Furthermore, NR2A and NR2B subunits are known to interact differentially with binding partners in the postsynaptic density (PSD) and may potentially activate different downstream signaling pathways according to changes in NMDAR subunit composition during development (Massey et al., 2004). Thus, various genetic animal models with the loss of NR2A or NR2B subunits have been studied to understand how expression and function of these subunits may contribute to NMDAR hypofunctionality.
As suggested by its early expression in development, homozygous NR2B KO mice are perinatally lethal. These mice were examined shortly after birth in hippocampal slices. The loss of NR2B abolishes synaptic NMDA responses and long term depression (LTD) in the CA1 hippocampal neurons, suggesting that NR2B expression has an essential role in synaptic plasticity (Kutsuwada et al., 1996). Since homozygous NR2B KO mice died perinatally, the heterozygous NR2B mutant mice were generated to study the effect of NR2B deletion in adulthood. One study found diminished NMDAR excitatory postsynaptic currents (EPSCs) and LTP in the fimbrial (Fim)-CA3 synapse when recorded from hippocampal slices of the heterozygous NR2B mutant mice (Ito et al., 1997). A separate study reported that these mice exhibit enhanced startle responses to acoustic stimuli (Takeuchi et al., 2001).
Mouse models with deletion of NR2B subunits in certain brain regions and/or cell types were further studied to understand the role of NR2B subunits in cognitive functions. A forebrain pyramidal cells-specific NR2B KO mice were created by genetic deletion of NR2B beginning at early postnatal development using the calcium2+/calmodulin-dependent protein kinase II-alpha (CamKII-α) promoter. These mice display hyperlocomotor activity and exaggerated depressant effect (Badanich et al., 2011). Similarly, another study done on these forebrain pyramidal cells-specific NR2B KO mice reports impairment in spatial and non-spatial learning and memory, whereas hippocampal pyramidal cells-specific NR2B KO mice only display selective, spatial working memory deficits. Nevertheless, both forebrain and hippocampal pyramidal cells-specific NR2B KO mice exhibit decreased NMDAR-mediated EPSCs with accelerated decay kinetics and reduced cellular LTP in the hippocampus (von Engelhardt et al., 2008).
In contrast to NR2B KO mice, NR2A KO mice are viable. They exhibit hyperlocomotor activity and cognitive dysfunctions without any sensorimotor gating deficits (Sprengel et al., 1998; Brigman et al., 2008). The cognitive deficits exhibited in NR2A KO mice include spatial learning, fear coding, and discrimination learning impairments accompanied by impaired hippocampal LTP. When recorded from hippocampal slices generated from the NR2A KO mice, there was reduced NMDAR-mediated EPSCs and LTP in the CA3-CA1 synapse (Ito et al., 1997). Overall, the changes displayed in both genetic mutants of NR2A and NR2B subunits highlight the crucial role that both subunits play in synaptic plasticity and cognitive functions (Sakimura et al., 1995; Kiyama et al., 1998; Moriya et al., 2000). Due to overlapping roles of NR2A and NR2B subunits in learning and memory, NR2A and NR2B mutant mice would serve as great models to study the pathophysiology of cognitive symptoms of schizophrenia (Table 4).
Overexpression or Re-expression of NMDAR Subunits
Multiple studies have also examined behavioral and molecular consequences upon overexpressing different NMDAR subunit(s). In one study, overexpression of the NR1 subunit into the hippocampus of wild type mice via AAV-mediated delivery led to increased fear memory and neurogenesis as well as delayed onset of severe seizures (Kalev-Zylinska et al., 2009). Other studies reported that overexpression of NR2B subunits in the forebrain of a wild type mice improve learning and memory and enhance NMDAR-dependent synaptic potentiation (Tang et al., 1999, 2001). In addition, transgenic mice with forebrain pyramidal cells-specific overexpression of NR2B postnatally exhibit enhanced social recognition memory for different strains and animal species (Jacobs and Tsien, 2012). Another group found that overexpression of NR2B subunit in aged wild type mice enhances long-term spatial memory. On the other hand, transgenic mice with forebrain-specific NR2A overexpression display long-term memory deficits, suggesting that subtle shifts in subunit compositions can have major effects on native receptor function and highlights how different subunit compositions can produce receptors with different functional properties (Madden, 2002; Cui et al., 2013).
Behavioral deficits in genetic mutants of NMDAR subunit(s) could also be rescued by restoring NMDAR subunit expression levels. In the NR1-KD mice, the hypomorphic insertion mutation could be excised in the Cre recombinase-dependent manner to restore NR1 in a temporal-regulated fashion. This allowed to investigate behavioral phenotypic rescue at different stages of development upon global restoration of NMDARs (Mohn et al., 1999; Mielnik et al., 2017). The mice with inducible NR1 rescue during adolescence or in adulthood achieved similar levels of functional recovery in some of cognitive deficits and negative symptoms-related behavioral changes, with cortically-mediated behaviors completely or nearly rescued. However, subcortically-mediated behaviors, such as hyperlocomotor activity and anxiety-related behaviors were only partially rescued. This suggested that higher-order brain functions are potentially more amenable to treatment in adulthood and unencumbered by critical period (Mielnik et al., 2017). Overexpressing NR2B in aged wild type mice was able to rescue age-associated impairments in hippocampal-dependent spatial memory. This was accompanied by enhanced LTP and increased NMDAR-mediated EPSPs from hippocampal slices generated from these aged mice with increased NR2B subunit expression, suggesting that increasing NR2B expression in aged animals can also enhance memory and synaptic transmission (Brim et al., 2013). These studies further point to how re-expression or overexpression of NMDAR subunits rescue behavior deficits associated to symptoms of schizophrenia, suggesting that enhancing levels of NMDAR subunits may ameliorate NMDAR hypofunctionality. However, limitations in fully restoring NMDAR subunits or rescuing behavioral phenotypes associated with subcortically-mediated behaviors still remain to be a challenge and may require further studies.
Moreover, the NMDAR levels can be influenced by activity or experience. Multiple studies showed that both NR1 and NR2A subunit protein expressions increase in the hippocampus of 1, 2, and 3-month old rats following habituation to a new environment via open field test and following novel object recognition task (Baez et al., 2013; Cercato et al., 2016). Shanmugasundaram et al. reported that 6 h after training rats in a radial maze along 10 consecutive days, there is an increase in NR1 and NR2B protein levels in synaptosomal fractions from the hippocampus and an increase in synaptic NR1 and NR2A levels in the PFC (Shanmugasundaram et al., 2015). In conclusion, NMDAR levels are influenced by genetic overexpression, re-expression of NMDAR subunit(s) and are also experience- and activity-dependent.
Metabotropic Glutamate Receptors (mGluRs)
Glutamatergic neurotransmission is mediated by both metabotropic and ionotropic glutamate receptors. While ionotropic glutamate receptors such as NMDA and quisqualate/a-amino-3-hydroxy-5-methyl-5-isoxazolepropionic acid (AMPA) receptors are responsible for fast excitatory transmission, metabotropic receptors have a modulatory role (Kew and Kemp, 2005). Metabotropic glutamate receptors (mGluRs) are further subdivided into three groups, with Group I metabotropic glutamate receptors (mGluR1 and mGluR5) mainly localized postsynaptically and positively linked to phospholipase C (PLC), which is known to potentiate glutamate function at NMDARs. In contrast, Group II (mGluR2 and mGluR3) and III (mGluR4, mGluR6, mGluR7, and mGluR8) metabotropic glutamate receptors are primarily presynaptic and are negatively linked to adenylyl cyclase (AC), which is known for its role in modulating neurotransmitter release (Javitt, 2004). Moreover, Group I mGluRs induce the enhancement of NMDAR currents and are involved in the direct phosphorylation of the NMDARs (Cartmell et al., 2000; Pisani et al., 2001). In particular, mGluR5s at the PSDs are physically linked to NMDARs and are known to enhance NMDAR function, this makes mGluRs a leading target for novel therapeutics to treat cognitive symptoms of schizophrenia (Ehlers, 2002; Yang et al., 2004; Gray et al., 2009).
Animal model studies have reported that the excitatory glutamatergic neurotransmission through ionotropic and metabotropic glutamate receptors is necessary for the correct postnatal development of the GABAergic network, with mGluR5s having a fundamental role in the development of PV interneurons (Uzunova et al., 2014; Barnes et al., 2015). PV-specific mGluR5 KO mice display memory impairments, increased compulsive-like behaviors, abnormal sensorimotor gating, and altered responsiveness to stimulants. These behavioral changes are accompanied by reduced PV interneuron density, decreased inhibitory synaptic currents, as well as abnormal ERPs and brain oscillatory activity (Barnes et al., 2015) Another study showed that mGluR5 KO mice exhibit deficient developmental switch from NR2B- to NR2A-containing receptors at synapses onto hippocampal CA1 pyramidal neurons and pyramidal primary visual cortical neurons (Matta et al., 2011), suggesting that mGluR5 plays an important role in regulating age-dependent expression of NMDAR subunits.
In addition, pharmacological modulations of different mGluR subtypes may provide alternative mechanisms to normalize aberrant neurotransmission induced by NMDAR hypofunction (Moghaddam et al., 1997; Marino and Conn, 2002). A licensed drug originally discovered for the prevention of relapse in alcohol dependence, acamprosate, interferes with mGluR5-dependent glutamate release. Preclinical studies with acamprosate suggest that it normalizes glutamate release and NMDAR functions without altering the normal glutamatergic neurotransmission (De Witte et al., 2005). Other approaches have also been made to normalize glutamatergic neurotransmission by using glutamate release inhibitors, such as selective mGluR2/3 agonists (LY2140023), which alleviates positive and negative symptoms in schizophrenia patients during phase II clinical trials, but failed in phase III clinical trials (Patil et al., 2007; Li et al., 2015). Thus, far, mGluR-targeting compounds have yet to be clinically approved for schizophrenia patients (Moghaddam and Adams, 1998; Kinon et al., 2011; Hopkins, 2013). Further studies in different animal models may facilitate this process by establishing functional link between mGluRs and NMDAR hypofunctionality (Table 4).
Neuregulin 1 (NRG1) and ErbB4 Receptor
The neuregulins (NRGs) are a family of growth and differentiation factors encoded by four genes (NRG1-4). The NRGs bind to the ErbB family of tyrosine kinase transmembrane receptors (ErbB1-4). Amongst these receptors, ErbB4 is likely to be the major mediator of NRG1 functions in the brain (Mei and Xiong, 2008). Both NRG1 and ErbB4 receptor have been identified as candidate risk genes for schizophrenia through genetic studies (Stefansson et al., 2002, 2003; Yang et al., 2003; Corvin et al., 2004; Zhao et al., 2004; Owen et al., 2005; Hahn et al., 2006). Accumulating evidence supports that NRG1 and ErbB4 play crucial roles in neurodevelopment and in the modulation of NMDAR signaling (Ozaki et al., 1997; Rieff et al., 1999; Anton et al., 2004). NRG1 is expressed in the PFC, hippocampus, cerebellum, and substantia nigra, where it regulates the expression and function of NMDA, γ-aminobutyric acid (GABA), and acetylcholine receptors in both humans and in rodents (Kerber et al., 2003; Corfas et al., 2004; Law et al., 2004; O'Tuathaigh et al., 2007). ErbB4 receptor is involved in neurogenesis, synaptic plasticity, neuronal migration, synapse formation and the regulation of NMDAR-mediated neurotransmission (Rieff et al., 1999; Anton et al., 2004; Flames et al., 2004; Ghashghaei et al., 2006; Li et al., 2007).
In animal model studies, the NRG1 homozygous null mutant mice are found to be embryonically lethal (Erickson et al., 1997). This led to the studies of viable, NRG1 heterozygous null mutant mice. These mice exhibit hyperlocomotor activity and spatial learning impairments (Gerlai et al., 2000). Another NRG1 hypomorphic mice were created by deleting the transmembrane domain of the NRG1 protein. These heterozygous mice exhibit hyperlocomotor activity and impaired sensorimotor gating, which was accompanied by decreased MK-801 binding from the forebrain, suggesting fewer functional NMDARs (Stefansson et al., 2002). Subsequently, studies using mutant ErbB4 or NRG1 mouse lines revealed that the NRG1-ErbB signaling is crucial for tangential migration of cortical GABAergic interneurons, as the number of GABAergic interneurons in the embryonic cortex is decreased in either the neuronal specific NRG1 KO mice or a line of ErbB4 null mice (Flames et al., 2004). Another NRG1 heterozygous mice were generated by targeted disruption of type III NRG1 (Chen et al., 2008), which is one of many NRG1 isoforms highly expressed throughout embryonic and postnatal brain development and are implicated in schizophrenia (Meyer et al., 1997; Anton et al., 2004; Longart et al., 2004). These NRG1 heterozygous mice exhibit impaired performance on delayed alternation task and sensorimotor gating deficits. They also display enlarged lateral ventricles and decreased dendritic spine density on hippocampal pyramidal neurons (Chen et al., 2008).
The ErbB4 KO mice are also embryonically lethal (Gassmann et al., 1995). Thus, the heterozygous ErbB4 mutant mice were most widely used to study animal models of ErbB4 receptor. These mice mainly exhibit hyperlocomotor activity and reduced power of kainate-induced gamma oscillations, accompanied by reduced numbers of PV interneuron density in the hippocampus as well as reduced calbindin (CB) and GABAergic interneurons in the cortex (Stefansson et al., 2002; Flames et al., 2004; Fisahn et al., 2009). Other animal models with ErbB4 deletion in brain region and/or cell-type-specific manner have also been studied. Central nervous system (CNS)-specific ErbB4 KO mice exhibit delayed postnatal motor development, and disrupted cue utilization during Morris water maze (Golub et al., 2004). CNS-specific double KO (dKO) mice lacking both ErbB2 and ErbB4 at early embryonic stages display sensorimotor gating deficits, which is accompanied by decreased spine density in the cortex and hippocampus (Barros et al., 2009). Interestingly, another CNS-specific ErbB4 homozygous null mice did not exhibit any motor function impairments or any other behavioral deficits (Thuret et al., 2004). Moreover, expression of dominant negative ErbB4 in oligodendrocytes and myelination Schwann cells from E15 result in thinning of the myelin sheath of the corpus callosum, altered oligodendrocyte morphology, increased number of cells expressing differentiated oligodendrocytes as well as dopaminergic abnormalities, which is predicted to result from defective myelin (Roy et al., 2007; Table 4).
It is worth noting that most studies seem to suggest a link between increased NRG1-ErbB signaling and decreased NMDAR function in the PFC. Bath application of NRG1 reduces NMDAR-mediated currents in PFC pyramidal neurons (Gu et al., 2005). A postmortem tissue-stimulation study also reported that NRG1 stimulation suppresses NMDAR activation in the human PFC (Hahn et al., 2006). Conversely, blockade of NMDARs via chronic MK-801 administration in rats increases the expression of NRG1 and ErbB4 proteins (Feng et al., 2010). Moreover, others have found that NRG1-ErbB4 signaling may cause NMDAR hypofunction by either suppressing Src-mediated enhancement of synaptic NMDAR function or through upregulation of excitatory amino-acid carrier 1 (EAAC1) (Pitcher et al., 2011; Yu et al., 2015). Given that most of the animal models reviewed here were based on deficiency of the NRG1-ErbB4 signaling, caution must be exercised in interpreting these results in the context of NMDAR hypofunctionality.
14-3-3 Proteins
14-3-3 refers to a family of brain-enriched proteins (β, ε, η, γ, σ, θ, and ζ isoforms) encoded by seven genetic loci (YWHAB, YWHAE, YWHAH, YWHAG, YWHAS, YWHAQ, and YWHAZ) (Altar et al., 2009). 14-3-3 proteins exist as homo- or heterodimers and bind to target proteins via specific phosphoserine/phosphothreonine-containing motifs. By interacting with its target proteins, 14-3-3 modulates a wide range of cellular processes (Muslin et al., 1996; Berg et al., 2003). In the nervous system, 14-3-3 proteins are enriched at synapses and regulate synaptic transmission and plasticity (Martin et al., 1994; Broadie et al., 1997). Based on previous studies, 14-3-3 proteins play a crucial role in surface expression of NR2C subunit in cerebellar granule cells (Chen and Roche, 2009), and 14-3-3 family members are found to associate and colocalize with NMDARs (Taya et al., 2007).
We have generated the 14-3-3 functional KO (FKO) mice by transgenic expression of a dimeric fourteen-three-three peptide inhibitor (difopein), which antagonizes the binding of 14-3-3 proteins to their endogenous partners in an isoform-independent manner to disrupt 14-3-3 functions in the brain (Wang et al., 1999; Masters and Fu, 2001; Cao et al., 2010; Foote et al., 2015). Based on our behavioral analyses, 14-3-3 FKO mice exhibit a full range of endophenotypes associated with core symptoms of schizophrenia. These behavioral changes include hyperlocomotor activity, social interaction impairments, deficits in sensorimotor gating, working memory, and associative learning and memory. In 14-3-3 FKO mice, we also identified a significant reduction in the NMDAR-mediated synaptic currents in CA1 pyramidal neurons. The protein levels of NMDA receptors, particularly NR1 and NR2A subunits, are selectively reduced in the PSD fraction of 14-3-3 FKO mice. These observations thus provide in vivo evidence linking 14-3-3 dysfunction to NMDAR hypofunction in forebrain neurons (Qiao et al., 2014; Foote et al., 2015). In a recent study, we reported that AAV-mediated expression of the 14-3-3 inhibitor specifically within the hippocampus alone is sufficient to induce several behavioral deficits including hyperactivity, impaired associative learning and memory, and reduced sensorimotor gating. Conversely, selectively restoring the function of 14-3-3 in the forebrain of the 14-3-3 FKO mice attenuate hyperlocomotor activity of the 14-3-3 FKO mice. In addition, we show that postsynaptic NMDA receptor levels are regulated by acute 14-3-3 manipulations (Graham et al., 2019). Taken together, findings from these animal model studies directly link 14-3-3 inhibition and NMDAR hypofunction in specific forebrain regions to certain schizophrenia-associated behavioral deficits.
Previous genetic and postmortem studies have identified 14-3-3ζ (YWHAZ) and 14-3-3ε (YWHAE) as candidate risk genes for schizophrenia (Middleton et al., 2005; Wong et al., 2005; Ikeda et al., 2008). This led to animal model studies to determine whether the loss of 14-3-3ζ or 14-3-3ε isoform leads to behavioral changes related to symptoms of schizophrenia. The 14-3-3ζ KO mice display hyperlocomotor activity, sensorimotor gating and cognitive deficits. These mice also display alterations in synaptic neurotransmission and plasticity, abnormal neuronal migration and axonal guidance defects in the hippocampus, and increased levels of DA in the striatum (Cheah et al., 2012; Ramshaw et al., 2013; Xu et al., 2015). As 14-3-3ε KO mice are found to be neonatally lethal, 14-3-3ε heterozygous mice are studied for behavioral deficits related to schizophrenia. 14-3-3ε heterozygous mice display weak defects in working memory and enhanced anxiety-like behavior (Ikeda et al., 2008; Toyo-oka et al., 2014). Taken together, these results suggest a link between these 14-3-3 isoforms to schizophrenia. Further studies are required to understand whether NMDAR hypofunctionality may be mediating behavioral deficits associated with schizophrenia exhibited in 14-3-3 isoform-specific animal models (Table 4).
Conclusion
Here we reviewed various animal models of NMDAR hypofunctionality created through pharmacological and genetic approaches. Animal models of schizophrenia have also been generated by using dopaminergic agonists, such as amphetamine and apomorphine, which primarily induce behavioral changes limited to hyperlocomotor activity, stereotypy, and sensorimotor gating deficits (Kokkinidis and Anisman, 1980; Sharp et al., 1987; Swerdlow et al., 1994; Swerdlow and Geyer, 1998; Marcotte et al., 2001; Jones et al., 2011). Similarly, direct infusion of GABAA receptor antagonist picrotoxin into the mPFC only causes sensorimotor gating deficits in rats (Japha and Koch, 1999; Marcotte et al., 2001). In comparison, as reviewed in this article, NMDAR antagonists induce a full range of behaviors corresponding to symptoms of schizophrenia, with robust changes in negative and cognitive symptoms-related behaviors. Moreover, some behavioral deficits induced by NMDAR antagonists are reversed by administration of APDs, including haloperidol and clozapine (Irifune et al., 1991; Noda et al., 1995; Verma and Moghaddam, 1996; Sams-Dodd, 1999; Abdul-Monim et al., 2006; Szlachta et al., 2017). Thus, administration of NMDAR antagonists provides a relatively valid animal model for studying symptomologies and pathophysiology of schizophrenia.
However, there are limitations in the use of NMDAR antagonists to model NMDAR hypofunctionality. First, we must ask whether pharmacologically induced alterations in the brain are a true representation of dysfunctional neural circuitry in schizophrenic patients. Olney et al. argued that since NMDAR antagonists cause NMDAR hypofunctionality throughout the brain, these animal models cannot target NMDARs in a neural circuit specific manner, thus may fail to precisely recapitulate schizophrenia pathophysiology (Olney et al., 1999). Second, schizophrenia is considered a neurodevelopmental disorder with deficits occurring during early brain development, yet a majority of animal studies have been focused on NMDAR antagonist-induced changes in adulthood (Harrison and Weinberger, 2005; Rapoport et al., 2005; Fatemi and Folsom, 2009; Powell, 2010). Further studies should be extended to address schizophrenia as a neurodevelopmental disorder by administering NMDAR antagonists at either prenatal or postnatal time period and examine their impacts on behavior and neural circuitry during development. Third, it is debatable whether some of the drug-induced behavioral changes in rodents are direct or even true correspondence of symptoms exhibited by schizophrenia patients. For example, hyperlocomotor activity measured in the open field test may have low relevance to psychosis in human patients (Powell and Miyakawa, 2006; Moore, 2010). This should contribute to limitations in interpretation of results obtained from these animal models with currently used behavioral paradigms (Moore, 2010). Fourth, behavioral and neurophysiological phenotypes exhibited in animal models of NMDAR antagonists are not specifically corresponding to symptoms of schizophrenia. For example, subchronic ketamine treatment in rodents leads to increased aggression, which is not a schizophrenia-related behavioral phenotype (Becker et al., 2003). Other behavioral phenotypes in pharmacological models, such as social interaction deficits, hyper responsiveness to sensory stimuli as well as cognitive deficits, overlap with symptoms associated with autism spectrum disorder (ASD) (Crawley, 1999, 2012). Finally, NMDAR antagonists cause neurotoxicity, evidenced by neuronal vacuolization, neuronal necrosis, and other cytotoxic changes. This must be taken into consideration when interpreting results from those animal studies, especially for chronic models of NMDAR antagonist (Olney et al., 1989, 1991; Nakao et al., 2003). Neurotoxicity should also be considered by the researcher when deciding on the dosage, regimen, and the type of NMDAR antagonist proposed to be used depending on the aim of the study.
Compared to pharmacological models, genetic animal models have several advantages for studying NMDAR hypofunctionality in relation to schizophrenia. First, schizophrenia is a disease of common symptomatology with etiological heterogeneity caused by various genetic and environmental factors (Takahashi, 2013). Human genome wide association studies (GWAS) have identified NMDAR genes or genes encoding proteins that regulate NMDARs to be candidate risk genes for schizophrenia (Fromer et al., 2014; Purcell et al., 2014; Schizophrenia Working Group of the Psychiatric Genomics Consortium, 2014; Harrison, 2015). There is also evidence of reduced expression of some NMDAR-subunits in schizophrenia patients (Balu, 2016). Thus, genetic animal models of NMDAR hypofunction may have certain construct validity for schizophrenia. However, we must emphasize that NMDAR hypofunction itself does not guarantee the animal model of schizophrenia, since NMDAR hypofunction leads to different neurological and psychiatric conditions, which do not necessarily correspond to the symptoms of schizophrenia. Second, de novo mutations in schizophrenia patients are overrepresented in glutamatergic postsynaptic proteins comprising of NMDAR complexes, making genetic animal models particularly useful in dissecting the signaling pathways and molecular mechanisms leading to NMDAR hypofunction (Forrest et al., 1994; Fromer et al., 2014). As reviewed here, some of the genetic animal studies, such as NRG1/ErbB4 and 14-3-3 animal models, provided evidence to support NMDAR hypofunctionality as a potential convergence point for symptoms of schizophrenia (Snyder and Gao, 2013). Third, various genetic models have been created to either directly or indirectly downregulate NMDAR functions in a spatial and temporal controlled manner. Some of these models specifically examine the role of NMDAR functions in different cell types and/or brain regions, while others have studied the impact of NMDAR hypofunction at different neurodevelopment stages. These studies may help reveal “how” genetic and environmental factors (signaling pathways) lead to NMDAR hypofunctionality and behavioral deficits, as well as “where” (in which neural circuits) and ‘when' these changes occur during neurodevelopment. Such information will be crucial for our understanding of the disease progress. In the future, some of the animal models may provide us useful platforms for identifying novel therapeutics for schizophrenia patients.
Author Contributions
GL was responsible for drafting, writing, reviewing, and editing this manuscript. YZ was responsible for writing, reviewing, and editing the manuscript with suggestive ideas on formatting and outlining this manuscript.
Funding
This work was supported by a research grant from NIMH (MH115188A).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abdul-Monim, Z., Neill, J. C., and Reynolds, G. P. (2007). Sub-chronic psychotomimetic phencyclidine induces deficits in reversal learning and alterations in parvalbumin-immunoreactive expression in the rat. J. Psychopharmacol. 21, 198–205. doi: 10.1177/0269881107067097
Abdul-Monim, Z., Reynolds, G. P., and Neill, J. C. (2003). The atypical antipsychotic ziprasidone, but not haloperidol, improves phencyclidine-induced cognitive deficits in a reversal learning task in the rat. J. Psychopharmacol. 17, 57–65. doi: 10.1177/0269881103017001700
Abdul-Monim, Z., Reynolds, G. P., and Neill, J. C. (2006). The effect of atypical and classical antipsychotics on sub-chronic PCP-induced cognitive deficits in a reversal-learning paradigm. Behav. Brain Res. 169, 263–273. doi: 10.1016/j.bbr.2006.01.019
Abekawa, T., Ito, K., Nakagawa, S., and Koyama, T. (2007). Prenatal exposure to an NMDA receptor antagonist, MK-801 reduces density of parvalbumin-immunoreactive GABAergic neurons in the medial prefrontal cortex and enhances phencyclidine-induced hyperlocomotion but not behavioral sensitization to methamphetamine in postpubertal rats. Psychopharmacology 192, 303–316. doi: 10.1007/s00213-007-0729-8
Adams, B., and Moghaddam, B. (1998). Corticolimbic dopamine neurotransmission is temporally dissociated from the cognitive and locomotor effects of phencyclidine. J. Neurosci. 18, 5545–5554. doi: 10.1523/JNEUROSCI.18-14-05545.1998
Ago, Y., Arikawa, S., Yata, M., Yano, K., Abe, M., Takuma, K., et al. (2008). Antidepressant-like effects of the glucocorticoid receptor antagonist RU-43044 are associated with changes in prefrontal dopamine in mouse models of depression. Neuropharmacology 55, 1355–1363. doi: 10.1016/j.neuropharm.2008.08.026
Allen, R. M., and Young, S. J. (1978). Phencyclidine-induced psychosis. Am. J. Psychiatry 135, 1081–1084. doi: 10.1176/ajp.135.9.1081
Altar, C. A., Vawter, M. P., and Ginsberg, S. D. (2009). Target identification for CNS diseases by transcriptional profiling. Neuropsychopharmacology 34, 18–54. doi: 10.1038/npp.2008.172
Anis, N. A., Berry, S. C., Burton, N. R., and Lodge, D. (1983). The dissociative anaesthetics, ketamine and phencyclidine, selectively reduce excitation of central mammalian neurones by N-methyl-aspartate. Br. J. Pharmacol. 79, 565–575. doi: 10.1111/j.1476-5381.1983.tb11031.x
Anton, E. S., Ghashghaei, H. T., Weber, J. L., McCann, C., Fischer, T. M., Cheung, I. D., et al. (2004). Receptor tyrosine kinase ErbB4 modulates neuroblast migration and placement in the adult forebrain. Nat. Neurosci. 7, 1319–1328. doi: 10.1038/nn1345
Badanich, K. A., Doremus-Fitzwater, T. L., Mulholland, P. J., Randall, P. K., Delpire, E., and Becker, H. C. (2011). NR2B-deficient mice are more sensitive to the locomotor stimulant and depressant effects of ethanol. Genes Brain Behav. 10, 805–816. doi: 10.1111/j.1601-183X.2011.00720.x
Baez, M. V., Oberholzer, M. V., Cercato, M. C., Snitcofsky, M., Aguirre, A. I., and Jerusalinsky, D. A. (2013). NMDA receptor subunits in the adult rat hippocampus undergo similar changes after 5 minutes in an open field and after LTP induction. PLoS ONE 8:e55244. doi: 10.1371/journal.pone.0055244
Balla, A., Sershen, H., Serra, M., Koneru, R., and Javitt, D. C. (2003). Subchronic continuous phencyclidine administration potentiates amphetamine-induced frontal cortex dopamine release. Neuropsychopharmacology 28, 34–44. doi: 10.1038/sj.npp.1300019
Ballard, T. M., Pauly-Evers, M., Higgins, G. A., Ouagazzal, A. M., Mutel, V., Borroni, E., et al. (2002). Severe impairment of NMDA receptor function in mice carrying targeted point mutations in the glycine binding site results in drug-resistant nonhabituating hyperactivity. J. Neurosci. 22, 6713–6723. doi: 10.1523/JNEUROSCI.22-15-06713.2002
Balu, D. T. (2016). The NMDA receptor and schizophrenia: from pathophysiology to treatment. Adv. Pharmacol. 76, 351–382. doi: 10.1016/bs.apha.2016.01.006
Barnes, S. A., Pinto-Duarte, A., Kappe, A., Zembrzycki, A., Metzler, A., Mukamel, E. A., et al. (2015). Disruption of mGluR5 in parvalbumin-positive interneurons induces core features of neurodevelopmental disorders. Mol. Psychiatry 20, 1161–1172. doi: 10.1038/mp.2015.113
Barros, C. S., Calabrese, B., Chamero, P., Roberts, A. J., Korzus, E., Lloyd, K., et al. (2009). Impaired maturation of dendritic spines without disorganization of cortical cell layers in mice lacking NRG1/ErbB signaling in the central nervous system. Proc. Natl. Acad. Sci. U.S.A. 106, 4507–4512. doi: 10.1073/pnas.0900355106
Bartos, M., Vida, I., and Jonas, P. (2007). Synaptic mechanisms of synchronized gamma oscillations in inhibitory interneuron networks. Nat. Rev. Neurosci. 8, 45–56. doi: 10.1038/nrn2044
Bast, T., Zhang, W., Feldon, J., and White, I. M. (2000). Effects of MK801 and neuroleptics on prepulse inhibition: re-examination in two strains of rats. Pharmacol. Biochem. Behav. 67, 647–658. doi: 10.1016/S0091-3057(00)00409-3
Becker, A., Peters, B., Schroeder, H., Mann, T., Huether, G., and Grecksch, G. (2003). Ketamine-induced changes in rat behaviour: a possible animal model of schizophrenia. Prog. Neuropsychopharmacol. Biol. Psychiatry 27, 687–700. doi: 10.1016/S0278-5846(03)00080-0
Behrens, M. M., Ali, S. S., Dao, D. N., Lucero, J., Shekhtman, G., Quick, K. L., et al. (2007). Ketamine-induced loss of phenotype of fast-spiking interneurons is mediated by NADPH-oxidase. Science 318, 1645–1647. doi: 10.1126/science.1148045
Belforte, J. E., Zsiros, V., Sklar, E. R., Jiang, Z., Yu, G., Li, Y., et al. (2010). Postnatal NMDA receptor ablation in corticolimbic interneurons confers schizophrenia-like phenotypes. Nat. Neurosci. 13, 76–83. doi: 10.1038/nn.2447
Berg, D., Holzmann, C., and Riess, O. (2003). 14-3-3 proteins in the nervous system. Nat. Rev. Neurosci. 4, 752–762. doi: 10.1038/nrn1197
Bickel, S., Lipp, H. P., and Umbricht, D. (2007). Impaired attentional modulation of auditory evoked potentials in N-methyl-D-aspartate NR1 hypomorphic mice. Genes Brain Behav. 6, 558–568. doi: 10.1111/j.1601-183X.2006.00283.x
Blot, K., Bai, J., and Otani, S. (2013). The effect of non-competitive NMDA receptor antagonist MK-801 on neuronal activity in rodent prefrontal cortex: an animal model for cognitive symptoms of schizophrenia. J. Physiol. 107, 448–451. doi: 10.1016/j.jphysparis.2013.04.003
Braun, I., Genius, J., Grunze, H., Bender, A., Moller, H. J., and Rujescu, D. (2007). Alterations of hippocampal and prefrontal GABAergic interneurons in an animal model of psychosis induced by NMDA receptor antagonism. Schizophr. Res. 97, 254–263. doi: 10.1016/j.schres.2007.05.005
Brigman, J. L., Feyder, M., Saksida, L. M., Bussey, T. J., Mishina, M., and Holmes, A. (2008). Impaired discrimination learning in mice lacking the NMDA receptor NR2A subunit. Learn. Mem. 15, 50–54. doi: 10.1101/lm.777308
Brigman, J. L., Ihne, J., Saksida, L. M., Bussey, T. J., and Holmes, A. (2009). Effects of Subchronic Phencyclidine (PCP) treatment on social behaviors, and operant discrimination and reversal learning in C57BL/6J Mice. Front. Behav. Neurosci. 3:2. doi: 10.3389/neuro.08.002.2009
Brim, B. L., Haskell, R., Awedikian, R., Ellinwood, N. M., Jin, L., Kumar, A., et al. (2013). Memory in aged mice is rescued by enhanced expression of the GluN2B subunit of the NMDA receptor. Behav. Brain Res. 238, 211–226. doi: 10.1016/j.bbr.2012.10.026
Broadie, K., Rushton, E., Skoulakis, E. M., and Davis, R. L. (1997). Leonardo, a Drosophila 14-3-3 protein involved in learning, regulates presynaptic function. Neuron 19, 391–402. doi: 10.1016/S0896-6273(00)80948-4
Bygrave, A. M., Masiulis, S., Nicholson, E., Berkemann, M., Barkus, C., Sprengel, R., et al. (2016). Knockout of NMDA-receptors from parvalbumin interneurons sensitizes to schizophrenia-related deficits induced by MK-801. Transl. Psychiatry 6:e778. doi: 10.1038/tp.2016.44
Cannon, M., Jones, P. B., and Murray, R. M. (2002). Obstetric complications and schizophrenia: historical and meta-analytic review. Am. J. Psychiatry 159, 1080–1092. doi: 10.1176/appi.ajp.159.7.1080
Cao, W., Yang, X., Zhou, J., Teng, Z., Cao, L., Zhang, X., et al. (2010). Targeting 14-3-3 protein, difopein induces apoptosis of human glioma cells and suppresses tumor growth in mice. Apoptosis 15, 230–241. doi: 10.1007/s10495-009-0437-4
Cardno, A. G., Marshall, E. J., Coid, B., Macdonald, A. M., Ribchester, T. R., Davies, N. J., et al. (1999). Heritability estimates for psychotic disorders: the Maudsley twin psychosis series. Arch. Gen. Psychiatry 56, 162–168. doi: 10.1001/archpsyc.56.2.162
Carlen, M., Meletis, K., Siegle, J. H., Cardin, J. A., Futai, K., Vierling-Claassen, D., et al. (2012). A critical role for NMDA receptors in parvalbumin interneurons for gamma rhythm induction and behavior. Mol. Psychiatry 17, 537–548. doi: 10.1038/mp.2011.31
Carlsson, A., and Lindqvist, M. (1963). Effect of Chlorpromazine or Haloperidol on Formation of 3methoxytyramine and Normetanephrine in Mouse Brain. Acta Pharmacol. Toxicol. 20, 140–144. doi: 10.1111/j.1600-0773.1963.tb01730.x
Carlsson, M., and Carlsson, A. (1990). Interactions between glutamatergic and monoaminergic systems within the basal ganglia–implications for schizophrenia and Parkinson's disease. Trends Neurosci. 13, 272–276. doi: 10.1016/0166-2236(90)90108-M
Cartmell, J., Salhoff, C. R., Perry, K. W., Monn, J. A., and Schoepp, D. D. (2000). Dopamine and 5-HT turnover are increased by the mGlu2/3 receptor agonist LY379268 in rat medial prefrontal cortex, nucleus accumbens and striatum. Brain Res. 887, 378–384. doi: 10.1016/S0006-8993(00)03067-5
Cercato, M. C., Vazquez, C. A., Kornisiuk, E., Aguirre, A. I., Colettis, N., Snitcofsky, M., et al. (2016). GluN1 and GluN2A NMDA Receptor Subunits Increase in the Hippocampus during Memory Consolidation in the Rat. Front. Behav. Neurosci. 10:242. doi: 10.3389/fnbeh.2016.00242
Chatterjee, M., Ganguly, S., Srivastava, M., and Palit, G. (2011). Effect of ‘chronic' versus ‘acute' ketamine administration and its ‘withdrawal' effect on behavioural alterations in mice: implications for experimental psychosis. Behav. Brain Res. 216, 247–254. doi: 10.1016/j.bbr.2010.08.001
Chatterjee, M., Verma, R., Ganguly, S., and Palit, G. (2012). Neurochemical and molecular characterization of ketamine-induced experimental psychosis model in mice. Neuropharmacology 63, 1161–1171. doi: 10.1016/j.neuropharm.2012.05.041
Cheah, P. S., Ramshaw, H. S., Thomas, P. Q., Toyo-Oka, K., Xu, X., Martin, S., et al. (2012). Neurodevelopmental and neuropsychiatric behaviour defects arise from 14-3-3zeta deficiency. Mol. Psychiatry 17, 451–466. doi: 10.1038/mp.2011.158
Cheli, V., Adrover, M., Blanco, C., Ferrari, C., Cornea, A., Pitossi, F., et al. (2006). Knocking-down the NMDAR1 subunit in a limited amount of neurons in the rat hippocampus impairs learning. J. Neurochem. 97(Suppl. 1), 68–73. doi: 10.1111/j.1471-4159.2005.03592.x
Cheli, V. T., Adrover, M. F., Blanco, C., Rial Verde, E., Guyot-Revol, V., Vidal, R., et al. (2002). Gene transfer of NMDAR1 subunit sequences to the rat CNS using herpes simplex virus vectors interfered with habituation. Cell. Mol. Neurobiol. 22, 303–314. doi: 10.1023/A:1020720001865
Chen, B. S., and Roche, K. W. (2009). Growth factor-dependent trafficking of cerebellar NMDA receptors via protein kinase B/Akt phosphorylation of NR2C. Neuron 62, 471–478. doi: 10.1016/j.neuron.2009.04.015
Chen, Y. J., Johnson, M. A., Lieberman, M. D., Goodchild, R. E., Schobel, S., Lewandowski, N., et al. (2008). Type III neuregulin-1 is required for normal sensorimotor gating, memory-related behaviors, and corticostriatal circuit components. J. Neurosci. 28, 6872–6883. doi: 10.1523/JNEUROSCI.1815-08.2008
Citrome, L. (2014). Unmet needs in the treatment of schizophrenia: new targets to help different symptom domains. J. Clin. Psychiatry 75 (Suppl. 1), 21–26. doi: 10.4088/JCP.13049su1c.04
Cochran, S. M., Kennedy, M., McKerchar, C. E., Steward, L. J., Pratt, J. A., and Morris, B. J. (2003). Induction of metabolic hypofunction and neurochemical deficits after chronic intermittent exposure to phencyclidine: differential modulation by antipsychotic drugs. Neuropsychopharmacology 28, 265–275. doi: 10.1038/sj.npp.1300031
Collingridge, G. L., Herron, C. E., and Lester, R. A. (1988). Synaptic activation of N-methyl-D-aspartate receptors in the Schaffer collateral-commissural pathway of rat hippocampus. J. Physiol. 399, 283–300. doi: 10.1113/jphysiol.1988.sp017080
Collingridge, G. L., Volianskis, A., Bannister, N., France, G., Hanna, L., Mercier, M., et al. (2013). The NMDA receptor as a target for cognitive enhancement. Neuropharmacology 64, 13–26. doi: 10.1016/j.neuropharm.2012.06.051
Corfas, G., Roy, K., and Buxbaum, J. D. (2004). Neuregulin 1-erbB signaling and the molecular/cellular basis of schizophrenia. Nat. Neurosci. 7, 575–580. doi: 10.1038/nn1258
Coronel-Oliveros, C. M., and Pacheco-Calderon, R. (2018). Prenatal exposure to ketamine in rats: implications on animal models of schizophrenia. Dev. Psychobiol. 60, 30–42. doi: 10.1002/dev.21586
Corssen, G., and Domino, E. F. (1966). Dissociative anesthesia: further pharmacologic studies and first clinical experience with the phencyclidine derivative CI-581. Anesth. Analg. 45, 29–40. doi: 10.1213/00000539-196601000-00007
Corvin, A. P., Morris, D. W., McGhee, K., Schwaiger, S., Scully, P., Quinn, J., et al. (2004). Confirmation and refinement of an 'at-risk' haplotype for schizophrenia suggests the EST cluster, Hs.97362, as a potential susceptibility gene at the Neuregulin-1 locus. Mol. Psychiatry 9, 208–213. doi: 10.1038/sj.mp.4001412
Cosgrove, J., and Newell, T. G. (1991). Recovery of neuropsychological functions during reduction in use of phencyclidine. J. Clin. Psychol. 47, 159–69.
Coyle, J. T. (2004). The GABA-glutamate connection in schizophrenia: which is the proximate cause? Biochem. Pharmacol. 68, 1507–1514. doi: 10.1016/j.bcp.2004.07.034
Crawley, J. N. (1999). Behavioral phenotyping of transgenic and knockout mice: experimental design and evaluation of general health, sensory functions, motor abilities, and specific behavioral tests. Brain Res. 835, 18–26. doi: 10.1016/S0006-8993(98)01258-X
Crawley, J. N. (2012). Translational animal models of autism and neurodevelopmental disorders. Dialogues Clin. Neurosci. 14, 293–305.
Cui, Z., Feng, R., Jacobs, S., Duan, Y., Wang, H., Cao, X., et al. (2013). Increased NR2A:NR2B ratio compresses long-term depression range and constrains long-term memory. Sci. Rep. 3:1036. doi: 10.1038/srep01036
Dawson, N., Morris, B. J., and Pratt, J. A. (2013). Subanaesthetic ketamine treatment alters prefrontal cortex connectivity with thalamus and ascending subcortical systems. Schizophr. Bull. 39, 366–377. doi: 10.1093/schbul/sbr144
de Bruin, N. M., Ellenbroek, B. A., Cools, A. R., Coenen, A. M., and van Luijtelaar, L. (1999). Differential effects of ketamine on gating of auditory evoked potentials and prepulse inhibition in rats. Psychopharmacology 142, 9–17. doi: 10.1007/s002130050856
De Witte, P., Littleton, J., Parot, P., and Koob, G. (2005). Neuroprotective and abstinence-promoting effects of acamprosate: elucidating the mechanism of action. CNS Drugs 19, 517–537. doi: 10.2165/00023210-200519060-00004
Domino, E. F., Chodoff, P., and Corssen, G. (1965). Pharmacologic effects of Ci-581, a new dissociative anesthetic, in Man. Clin. Pharmacol. Ther. 6, 279–291. doi: 10.1002/cpt196563279
Duncan, G., Miyamoto, S., Gu, H., Lieberman, J., Koller, B., and Snouwaert, J. (2002). Alterations in regional brain metabolism in genetic and pharmacological models of reduced NMDA receptor function. Brain Res. 951, 166–176. doi: 10.1016/S0006-8993(02)03156-6
Duncan, G. E., Moy, S. S., Perez, A., Eddy, D. M., Zinzow, W. M., Lieberman, J. A., et al. (2004). Deficits in sensorimotor gating and tests of social behavior in a genetic model of reduced NMDA receptor function. Behav. Brain Res. 153, 507–519. doi: 10.1016/j.bbr.2004.01.008
Dzirasa, K., Ramsey, A. J., Takahashi, D. Y., Stapleton, J., Potes, J. M., Williams, J. K., et al. (2009). Hyperdopaminergia and NMDA receptor hypofunction disrupt neural phase signaling. J. Neurosci. 29, 8215–8224. doi: 10.1523/JNEUROSCI.1773-09.2009
Egerton, A., Reid, L., McGregor, S., Cochran, S. M., Morris, B. J., and Pratt, J. A. (2008). Subchronic and chronic PCP treatment produces temporally distinct deficits in attentional set shifting and prepulse inhibition in rats. Psychopharmacology 198, 37–49. doi: 10.1007/s00213-008-1071-5
Egerton, A., Reid, L., McKerchar, C. E., Morris, B. J., and Pratt, J. A. (2005). Impairment in perceptual attentional set-shifting following PCP administration: a rodent model of set-shifting deficits in schizophrenia. Psychopharmacology 179, 77–84. doi: 10.1007/s00213-004-2109-y
Ehlers, M. D. (2002). Molecular morphogens for dendritic spines. Trends Neurosci. 25, 64–67. doi: 10.1016/S0166-2236(02)02061-1
Ehrlichman, R. S., Gandal, M. J., Maxwell, C. R., Lazarewicz, M. T., Finkel, L. H., Contreras, D., et al. (2009). N-methyl-d-aspartic acid receptor antagonist-induced frequency oscillations in mice recreate pattern of electrophysiological deficits in schizophrenia. Neuroscience 158, 705–712. doi: 10.1016/j.neuroscience.2008.10.031
Erickson, S. L., O'Shea, K. S., Ghaboosi, N., Loverro, L., Frantz, G., Bauer, M., et al. (1997). ErbB3 is required for normal cerebellar and cardiac development: a comparison with ErbB2-and heregulin-deficient mice. Development 124, 4999–5011.
Eyjolfsson, E. M., Brenner, E., Kondziella, D., and Sonnewald, U. (2006). Repeated injection of MK801: an animal model of schizophrenia? Neurochem. Int. 48, 541–546. doi: 10.1016/j.neuint.2005.11.019
Faludi, G., and Mirnics, K. (2011). Synaptic changes in the brain of subjects with schizophrenia. Int. J. Dev. Neurosci. 29, 305–309. doi: 10.1016/j.ijdevneu.2011.02.013
Fatemi, S. H., and Folsom, T. D. (2009). The neurodevelopmental hypothesis of schizophrenia, revisited. Schizophr. Bull. 35, 528–548. doi: 10.1093/schbul/sbn187
Featherstone, R. E., Liang, Y., Saunders, J. A., Tatard-Leitman, V. M., Ehrlichman, R. S., and Siegel, S. J. (2012). Subchronic ketamine treatment leads to permanent changes in EEG, cognition and the astrocytic glutamate transporter EAAT2 in mice. Neurobiol. Dis. 47, 338–346. doi: 10.1016/j.nbd.2012.05.003
Fellin, T., Halassa, M. M., Terunuma, M., Succol, F., Takano, H., Frank, M., et al. (2009). Endogenous nonneuronal modulators of synaptic transmission control cortical slow oscillations in vivo. Proc. Natl. Acad. Sci. U.S.A. 106, 15037–15042. doi: 10.1073/pnas.0906419106
Feng, Y., Wang, X. D., Guo, C. M., Yang, Y., Li, J. T., Su, Y. A., et al. (2010). Expressions of neuregulin 1beta and ErbB4 in prefrontal cortex and hippocampus of a rat schizophrenia model induced by chronic MK-801 administration. J. Biomed. Biotechnol. 2010:859516. doi: 10.1155/2010/859516
Finlay, J. M., Dunham, G. A., Isherwood, A. M., Newton, C. J., Nguyen, T. V., Reppar, P. C., et al. (2015). Effects of prefrontal cortex and hippocampal NMDA NR1-subunit deletion on complex cognitive and social behaviors. Brain Res. 1600, 70–83. doi: 10.1016/j.brainres.2014.10.037
Fisahn, A., Neddens, J., Yan, L., and Buonanno, A. (2009). Neuregulin-1 modulates hippocampal gamma oscillations: implications for schizophrenia. Cereb. Cortex 19, 612–618. doi: 10.1093/cercor/bhn107
Flames, N., Long, J. E., Garratt, A. N., Fischer, T. M., Gassmann, M., Birchmeier, C., et al. (2004). Short- and long-range attraction of cortical GABAergic interneurons by neuregulin-1. Neuron 44, 251–261. doi: 10.1016/j.neuron.2004.09.028
Foote, M., Qiao, H., Graham, K., Wu, Y., and Zhou, Y. (2015). Inhibition of 14-3-3 Proteins Leads to schizophrenia-related behavioral phenotypes and synaptic defects in mice. Biol. Psychiatry 78, 386–395. doi: 10.1016/j.biopsych.2015.02.015
Forrest, D., Yuzaki, M., Soares, H. D., Ng, L., Luk, D. C., Sheng, M., et al. (1994). Targeted disruption of NMDA receptor 1 gene abolishes NMDA response and results in neonatal death. Neuron 13, 325–338. doi: 10.1016/0896-6273(94)90350-6
Foster, A. C., and Fagg, G. E. (1987). Neurobiology. Taking apart NMDA receptors. Nature 329, 395–396. doi: 10.1038/329395a0
Fromer, M., Pocklington, A. J., Kavanagh, D. H., Williams, H. J., Dwyer, S., Gormley, P., et al. (2014). De novo mutations in schizophrenia implicate synaptic networks. Nature 506, 179–184. doi: 10.1038/nature12929
Gassmann, M., Casagranda, F., Orioli, D., Simon, H., Lai, C., Klein, R., et al. (1995). Aberrant neural and cardiac development in mice lacking the ErbB4 neuregulin receptor. Nature 378, 390–394. doi: 10.1038/378390a0
Gerlai, R., Pisacane, P., and Erickson, S. (2000). Heregulin, but not ErbB2 or ErbB3, heterozygous mutant mice exhibit hyperactivity in multiple behavioral tasks. Behav. Brain Res. 109, 219–227. doi: 10.1016/S0166-4328(99)00175-8
Ghashghaei, H. T., Weber, J., Pevny, L., Schmid, R., Schwab, M. H., Lloyd, K. C., et al. (2006). The role of neuregulin-ErbB4 interactions on the proliferation and organization of cells in the subventricular zone. Proc. Natl. Acad. Sci. U.S.A. 103, 1930–1935. doi: 10.1073/pnas.0510410103
Gilmour, G., Pioli, E. Y., Dix, S. L., Smith, J. W., Conway, M. W., Jones, W. T., et al. (2009). Diverse and often opposite behavioural effects of NMDA receptor antagonists in rats: implications for NMDA antagonist modelling of schizophrenia. Psychopharmacology 205, 203–216. doi: 10.1007/s00213-009-1530-7
Goff, D. C., and Coyle, J. T. (2001). The emerging role of glutamate in the pathophysiology and treatment of schizophrenia. Am. J. Psychiatry 158, 1367–1377. doi: 10.1176/appi.ajp.158.9.1367
Golub, M. S., Germann, S. L., and Lloyd, K. C. (2004). Behavioral characteristics of a nervous system-specific erbB4 knock-out mouse. Behav. Brain Res. 153, 159–170. doi: 10.1016/j.bbr.2003.11.010
Grace, A. A. (2012). Dopamine system dysregulation by the hippocampus: implications for the pathophysiology and treatment of schizophrenia. Neuropharmacology 62, 1342–1348. doi: 10.1016/j.neuropharm.2011.05.011
Graham, K., Jiajing, Z., Haifa, Q., Yuying, W., and Yi, Z. (2019). Region-specific inhibition of 14-3-3 proteins induces psychomotor behaviors in mice. npj Schizophrenia 5:1. doi: 10.1038/s41537-018-0069-1
Gratton, A., Hoffer, B. J., and Freedman, R. (1987). Electrophysiological effects of phencyclidine in the medial prefrontal cortex of the rat. Neuropharmacology 26, 1275–1283. doi: 10.1016/0028-3908(87)90087-6
Gray, L. M., van den Buuse, S. E., Dean, B., and Hannan, A. J. (2009). Clozapine reverses schizophrenia-related behaviours in the metabotropic glutamate receptor 5 knockout mouse: association with N-methyl-D-aspartic acid receptor up-regulation. Int. J. Neuropsychopharmacol. 12, 45–60. doi: 10.1017/S1461145708009085
Grayson, B., Idris, N. F., and Neill, J. C. (2007). Atypical antipsychotics attenuate a sub-chronic PCP-induced cognitive deficit in the novel object recognition task in the rat. Behav. Brain Res. 184, 31–38. doi: 10.1016/j.bbr.2007.06.012
Gu, Z., Jiang, Q., Fu, A. K., Ip, N. Y., and Yan, Z. (2005). Regulation of NMDA receptors by neuregulin signaling in prefrontal cortex. J. Neurosci. 25, 4974–4984. doi: 10.1523/JNEUROSCI.1086-05.2005
Hahn, C. G., Wang, H. Y., Cho, D. S., Talbot, K., Gur, R. E., Berrettini, W. H., et al. (2006). Altered neuregulin 1-erbB4 signaling contributes to NMDA receptor hypofunction in schizophrenia. Nat. Med. 12, 824–828. doi: 10.1038/nm1418
Hakami, T., Jones, N. C., Tolmacheva, E. A., Gaudias, J., Chaumont, J., Salzberg, M., et al. (2009). NMDA receptor hypofunction leads to generalized and persistent aberrant gamma oscillations independent of hyperlocomotion and the state of consciousness. PLoS ONE 4:e6755. doi: 10.1371/journal.pone.0006755
Halene, T. B., Ehrlichman, R. S., Liang, Y., Christian, E. P., Jonak, G. J., Gur, T. L., et al. (2009). Assessment of NMDA receptor NR1 subunit hypofunction in mice as a model for schizophrenia. Genes Brain Behav. 8, 661–675. doi: 10.1111/j.1601-183X.2009.00504.x
Harris, L. W., Sharp, T., Gartlon, J., Jones, D. N., and Harrison, P. J. (2003). Long-term behavioural, molecular and morphological effects of neonatal NMDA receptor antagonism. Eur. J. Neurosci. 18, 1706–1710. doi: 10.1046/j.1460-9568.2003.02902.x
Harrison, P. J. (2015). Recent genetic findings in schizophrenia and their therapeutic relevance. J. Psychopharmacol. 29, 85–96. doi: 10.1177/0269881114553647
Harrison, P. J., and Weinberger, D. R. (2005). Schizophrenia genes, gene expression, and neuropathology: on the matter of their convergence. Mol. Psychiatry 10, 40–68. doi: 10.1038/sj.mp.4001686
Homayoun, H., and Moghaddam, B. (2007). NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. J. Neurosci. 27, 11496–11500. doi: 10.1523/JNEUROSCI.2213-07.2007
Hopkins, C. R. (2013). Is there a path forward for mGlu(2) positive allosteric modulators for the treatment of schizophrenia? ACS Chem. Neurosci. 4, 211–213. doi: 10.1021/cn400023y
Howes, O., McCutcheon, R., and Stone, J. (2015). Glutamate and dopamine in schizophrenia: an update for the 21st century. J. Psychopharmacol. 29, 97–115. doi: 10.1177/0269881114563634
Howes, O. D., Vergunst, F., Gee, S., McGuire, P., Kapur, S., and Taylor, D. (2012). Adherence to treatment guidelines in clinical practice: study of antipsychotic treatment prior to clozapine initiation. Br. J. Psychiatry 201, 481–485. doi: 10.1192/bjp.bp.111.105833
Huerta, P. T., Sun, L. D., Wilson, M. A., and Tonegawa, S. (2000). Formation of temporal memory requires NMDA receptors within CA1 pyramidal neurons. Neuron 25, 473–480. doi: 10.1016/S0896-6273(00)80909-5
Huettner, J. E., and Bean, B. P. (1988). Block of N-methyl-D-aspartate-activated current by the anticonvulsant MK-801: selective binding to open channels. Proc. Natl. Acad. Sci. U.S.A. 85, 1307–1311. doi: 10.1073/pnas.85.4.1307
Ikeda, M., Hikita, T., Taya, S., Uraguchi-Asaki, J. K., Toyo-oka, Wynshaw-Boris, A., Ujike, H., et al. (2008). Identification of YWHAE, a gene encoding 14-3-3epsilon, as a possible susceptibility gene for schizophrenia. Hum. Mol. Genet. 17, 3212–3222. doi: 10.1093/hmg/ddn217
Ingvar, D. H., and Franzen, G. (1974). Abnormalities of cerebral blood flow distribution in patients with chronic schizophrenia. Acta Psychiatr. Scand. 50, 425–462. doi: 10.1111/j.1600-0447.1974.tb09707.x
Irifune, M., Shimizu, T., and Nomoto, M. (1991). Ketamine-induced hyperlocomotion associated with alteration of presynaptic components of dopamine neurons in the nucleus accumbens of mice. Pharmacol. Biochem. Behav. 40, 399–407. doi: 10.1016/0091-3057(91)90571-I
Itil, T., Keskiner, A., Kiremitci, N., and Holden, J. M. (1967). Effect of phencyclidine in chronic schizophrenics. Can. Psychiatr. Assoc. J. 12, 209–212. doi: 10.1177/070674376701200217
Ito, I., Futai, K., Katagiri, H., Watanabe, M., Sakimura, K., Mishina, M., et al. (1997). Synapse-selective impairment of NMDA receptor functions in mice lacking NMDA receptor epsilon 1 or epsilon 2 subunit. J Physiol. 500(Pt. 2), 401–408. doi: 10.1113/jphysiol.1997.sp022030
Itokawa, M., Yamada, K., Yoshitsugu, K., Toyota, T., Suga, T., Ohba, H., et al. (2003). A microsatellite repeat in the promoter of the N-methyl-D-aspartate receptor 2A subunit (GRIN2A) gene suppresses transcriptional activity and correlates with chronic outcome in schizophrenia. Pharmacogenetics 13, 271–278. doi: 10.1097/00008571-200305000-00006
Iwasato, T., Erzurumlu, R. S., Huerta, P. T., Chen, D. F., Sasaoka, T., Ulupinar, E., et al. (1997). NMDA receptor-dependent refinement of somatotopic maps. Neuron 19, 1201–1210. doi: 10.1016/S0896-6273(00)80412-2
Iwayama-Shigeno, Y., Yamada, K., Itokawa, M., Toyota, T., Meerabux, J. M., Minabe, Y., et al. (2005). Extended analyses support the association of a functional (GT)n polymorphism in the GRIN2A promoter with Japanese schizophrenia. Neurosci. Lett. 378, 102–105. doi: 10.1016/j.neulet.2004.12.013
Jaaro-Peled, H., Hayashi-Takagi, A., Seshadri, S., Kamiya, A., Brandon, N. J., and Sawa, A. (2009). Neurodevelopmental mechanisms of schizophrenia: understanding disturbed postnatal brain maturation through neuregulin-1-ErbB4 and DISC1. Trends Neurosci. 32, 485–495. doi: 10.1016/j.tins.2009.05.007
Jacobs, S. A., and Tsien, J. Z. (2012). genetic overexpression of NR2B subunit enhances social recognition memory for different strains and species. PLoS ONE 7:e36387. doi: 10.1371/journal.pone.0036387
Japha, K., and Koch, M. (1999). Picrotoxin in the medial prefrontal cortex impairs sensorimotor gating in rats: reversal by haloperidol. Psychopharmacology 144, 347–354. doi: 10.1007/s002130051017
Javitt, D. C. (2004). Glutamate as a therapeutic target in psychiatric disorders. Mol. Psychiatry 9, 984–997. doi: 10.1038/sj.mp.4001551
Javitt, D. C. (2007). Glutamate and schizophrenia: phencyclidine, N-methyl-D-aspartate receptors, and dopamine-glutamate interactions. Int. Rev. Neurobiol. 78, 69–108. doi: 10.1016/S0074-7742(06)78003-5
Javitt, D. C. (2010). Glutamatergic theories of schizophrenia. Isr. J. Psychiatry Relat. Sci. 47, 4–16.
Javitt, D. C., and Zukin, S. R. (1991). Recent advances in the phencyclidine model of schizophrenia. Am. J. Psychiatry 148, 1301–1308. doi: 10.1176/ajp.148.10.1301
Javitt, D. C., Zukin, S. R., Heresco-Levy, U., and Umbricht, D. (2012). Has an angel shown the way? Etiological and therapeutic implications of the PCP/NMDA model of schizophrenia. Schizophr Bull. 38, 958–966. doi: 10.1093/schbul/sbs069
Jenkins, T. A., Harte, M. K., McKibben, C. E., Elliott, J. J., and Reynolds, G. P. (2008). Disturbances in social interaction occur along with pathophysiological deficits following sub-chronic phencyclidine administration in the rat. Behav. Brain Res. 194, 230–235. doi: 10.1016/j.bbr.2008.07.020
Jensen, O., and Lisman, J. E. (1996). Theta/gamma networks with slow NMDA channels learn sequences and encode episodic memory: role of NMDA channels in recall. Learn. Mem. 3, 264–278. doi: 10.1101/lm.3.2-3.264
Jentsch, J. D., Redmond, D. E. Jr., Elsworth, J. D., Taylor, J. R., Youngren, K. D., and Roth, R. H. (1997a). Enduring cognitive deficits and cortical dopamine dysfunction in monkeys after long-term administration of phencyclidine. Science 277, 953–955. doi: 10.1126/science.277.5328.953
Jentsch, J. D., and Roth, R. H. (1999). The neuropsychopharmacology of phencyclidine: from NMDA receptor hypofunction to the dopamine hypothesis of schizophrenia. Neuropsychopharmacology 20, 201–225. doi: 10.1016/S0893-133X(98)00060-8
Jentsch, J. D., Taylor, J. R., and Roth, R. H. (1998). Subchronic phencyclidine administration increases mesolimbic dopaminergic system responsivity and augments stress- and psychostimulant-induced hyperlocomotion. Neuropsychopharmacology 19, 105–113. doi: 10.1016/S0893-133X(98)00004-9
Jentsch, J. D., Tran, A., Le, D., Youngren, K. D., and Roth, R. H. (1997b). Subchronic phencyclidine administration reduces mesoprefrontal dopamine utilization and impairs prefrontal cortical-dependent cognition in the rat. Neuropsychopharmacology 17, 92–99. doi: 10.1016/S0893-133X(97)00034-1
Jodo, E. (2013). The role of the hippocampo-prefrontal cortex system in phencyclidine-induced psychosis: a model for schizophrenia. J. Physiol. Paris 107, 434–440. doi: 10.1016/j.jphysparis.2013.06.002
Jodo, E., Suzuki, Y., Katayama, T., Hoshino, K. Y., Takeuchi, S., Niwa, S., et al. (2005). Activation of medial prefrontal cortex by phencyclidine is mediated via a hippocampo-prefrontal pathway. Cereb. Cortex 15, 663–669. doi: 10.1093/cercor/bhh168
Johnson, K. M., and Jones, S. M. (1990). Neuropharmacology of phencyclidine: basic mechanisms and therapeutic potential. Annu. Rev. Pharmacol. Toxicol. 30, 707–750. doi: 10.1146/annurev.pa.30.040190.003423
Jones, C. A., Watson, D. J., and Fone, K. C. (2011). Animal models of schizophrenia. Br. J. Pharmacol. 164, 1162–1194. doi: 10.1111/j.1476-5381.2011.01386.x
Kalev-Zylinska, M. L., Symes, W., Young, D., and During, M. J. (2009). Knockdown and overexpression of NR1 modulates NMDA receptor function. Mol. Cell. Neurosci. 41, 383–396. doi: 10.1016/j.mcn.2009.04.003
Kehrer, C., Dugladze, T., Maziashvili, N., Wojtowicz, A., Schmitz, D., Heinemann, U., et al. (2007). Increased inhibitory input to CA1 pyramidal cells alters hippocampal gamma frequency oscillations in the MK-801 model of acute psychosis. Neurobiol. Dis. 25, 545–552. doi: 10.1016/j.nbd.2006.10.015
Kehrer, C., Maziashvili, N., Dugladze, T., and Gloveli, T. (2008). Altered excitatory-inhibitory balance in the NMDA-Hypofunction model of schizophrenia. Front. Mol. Neurosci. 1:6. doi: 10.3389/neuro.02.006.2008
Keilhoff, G., Becker, A., Grecksch, G., Wolf, G., and Bernstein, H. G. (2004). Repeated application of ketamine to rats induces changes in the hippocampal expression of parvalbumin, neuronal nitric oxide synthase and cFOS similar to those found in human schizophrenia. Neuroscience 126, 591–598. doi: 10.1016/j.neuroscience.2004.03.039
Kerber, G., Streif, R., Schwaiger, F. W., Kreutzberg, G. W., and Hager, G. (2003). Neuregulin-1 isoforms are differentially expressed in the intact and regenerating adult rat nervous system. J. Mol. Neurosci. 21, 149–165. doi: 10.1385/JMN:21:2:149
Kew, J. N., and Kemp, J. A. (2005). Ionotropic and metabotropic glutamate receptor structure and pharmacology. Psychopharmacology 179, 4–29. doi: 10.1007/s00213-005-2200-z
Kew, J. N., Koester, A., Moreau, J. L., Jenck, F., Ouagazzal, A. M., Mutel, V., et al. (2000). Functional consequences of reduction in NMDA receptor glycine affinity in mice carrying targeted point mutations in the glycine binding site. J. Neurosci. 20, 4037–4049. doi: 10.1523/JNEUROSCI.20-11-04037.2000
Kinon, B. J., Zhang, L., Millen, B. A., Osuntokun, O. O., Williams, J. E., Kollack-Walker, S., et al. (2011). A multicenter, inpatient, phase 2, double-blind, placebo-controlled dose-ranging study of LY2140023 monohydrate in patients with DSM-IV schizophrenia. J. Clin. Psychopharmacol. 31, 349–355. doi: 10.1097/JCP.0b013e318218dcd5
Kittelberger, K., Hur, E. E., Sazegar, S., Keshavan, V., and Kocsis, B. (2012). Comparison of the effects of acute and chronic administration of ketamine on hippocampal oscillations: relevance for the NMDA receptor hypofunction model of schizophrenia. Brain Struct. Funct. 217, 395–409. doi: 10.1007/s00429-011-0351-8
Kiyama, Y., Manabe, T., Sakimura, K., Kawakami, F., Mori, H., and Mishina, M. (1998). Increased thresholds for long-term potentiation and contextual learning in mice lacking the NMDA-type glutamate receptor epsilon1 subunit. J. Neurosci. 18, 6704–6712. doi: 10.1523/JNEUROSCI.18-17-06704.1998
Kokkinidis, L., and Anisman, H. (1980). Amphetamine models of paranoid schizophrenia: an overview and elaboration of animal experimentation. Psychol. Bull. 88, 551–579. doi: 10.1037/0033-2909.88.3.551
Korotkova, T., Fuchs, E. C., Ponomarenko, A., von Engelhardt, J., and Monyer, H. (2010). NMDA receptor ablation on parvalbumin-positive interneurons impairs hippocampal synchrony, spatial representations, and working memory. Neuron 68, 557–569. doi: 10.1016/j.neuron.2010.09.017
Krystal, J. H., Abi-Saab, W., Perry, E., D'Souza, D. C., Liu, N., Gueorguieva, R., et al. (2005). Preliminary evidence of attenuation of the disruptive effects of the NMDA glutamate receptor antagonist, ketamine, on working memory by pretreatment with the group II metabotropic glutamate receptor agonist, LY354740, in healthy human subjects. Psychopharmacology 179, 303–309. doi: 10.1007/s00213-004-1982-8
Krystal, J. H., D'Souza, D. C., Karper, L. P., Bennett, A., Abi-Dargham, A., Abi-Saab, D., et al. (1999). Interactive effects of subanesthetic ketamine and haloperidol in healthy humans. Psychopharmacology 145, 193–204. doi: 10.1007/s002130051049
Krystal, J. H., Karper, L. P., Seibyl, J. P., Freeman, G. K., Delaney, R., Bremner, J. D., et al. (1994). Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch. Gen. Psychiatry 51, 199–214. doi: 10.1001/archpsyc.1994.03950030035004
Kutsuwada, T., Kashiwabuchi, N., Mori, H., Sakimura, K., Kushiya, E., Araki, K., et al. (1992). Molecular diversity of the NMDA receptor channel. Nature 358, 36–41. doi: 10.1038/358036a0
Kutsuwada, T., Sakimura, K., Manabe, T., Takayama, C., Katakura, N., Kushiya, E., et al. (1996). Impairment of suckling response, trigeminal neuronal pattern formation, and hippocampal LTD in NMDA receptor epsilon 2 subunit mutant mice. Neuron 16, 333–344. doi: 10.1016/S0896-6273(00)80051-3
Labrie, V., Lipina, T., and Roder, J. C. (2008). Mice with reduced NMDA receptor glycine affinity model some of the negative and cognitive symptoms of schizophrenia. Psychopharmacology 200, 217–230. doi: 10.1007/s00213-008-1196-6
Lahti, A. C., Weiler, M. A., Tamara Michaelidis, B. A., Parwani, A., and Tamminga, C. A. (2001). Effects of ketamine in normal and schizophrenic volunteers. Neuropsychopharmacology 25, 455–467. doi: 10.1016/S0893-133X(01)00243-3
Latysheva, N. V., and Rayevsky, K. S. (2003). Chronic neonatal N-methyl-D-aspartate receptor blockade induces learning deficits and transient hypoactivity in young rats. Prog. Neuropsychopharmacol. Biol. Psychiatry 27, 787–794. doi: 10.1016/S0278-5846(03)00110-6
Laurie, D. J., and Seeburg, P. H. (1994). Ligand affinities at recombinant N-methyl-D-aspartate receptors depend on subunit composition. Eur. J. Pharmacol. 268, 335–345. doi: 10.1016/0922-4106(94)90058-2
Law, A. J., Shannon Weickert, C., Hyde, T. M., Kleinman, J. E., and Harrison, P. J. (2004). Neuregulin-1 (NRG-1) mRNA and protein in the adult human brain. Neuroscience 127, 125–136. doi: 10.1016/j.neuroscience.2004.04.026
Lee, L. J., Lo, F. S., and Erzurumlu, R. S. (2005). NMDA receptor-dependent regulation of axonal and dendritic branching. J. Neurosci. 25, 2304–2311. doi: 10.1523/JNEUROSCI.4902-04.2005
Lees, J., Hallak, J. E., Deakin, J. F., and Dursun, S. M. (2004). Gender differences and the effects of ketamine in healthy volunteers. J. Psychopharmacol. 18, 337–339. doi: 10.1177/026988110401800302
Lewis, D. A., and Gonzalez-Burgos, G. (2008). Neuroplasticity of neocortical circuits in schizophrenia. Neuropsychopharmacology 33, 141–165. doi: 10.1038/sj.npp.1301563
Lewis, D. A., and Levitt, P. (2002). Schizophrenia as a disorder of neurodevelopment. Annu. Rev. Neurosci. 25, 409–432. doi: 10.1146/annurev.neuro.25.112701.142754
Li, B., Woo, R. S., Mei, L., and Malinow, R. (2007). The neuregulin-1 receptor erbB4 controls glutamatergic synapse maturation and plasticity. Neuron 54, 583–597. doi: 10.1016/j.neuron.2007.03.028
Li, J. T., Su, Y. A., Guo, C. M., Feng, Y., Yang, Y., Huang, R. H., et al. (2011). Persisting cognitive deficits induced by low-dose, subchronic treatment with MK-801 in adolescent rats. Eur. J. Pharmacol. 652, 65–72. doi: 10.1016/j.ejphar.2010.10.074
Li, L., and Vlisides, P. E. (2016). Ketamine: 50 Years of Modulating the Mind. Front. Hum. Neurosci. 10:612. doi: 10.3389/fnhum.2016.00612
Li, M. L., Hu, X. Q., Li, F., and Gao, W. J. (2015). Perspectives on the mGluR2/3 agonists as a therapeutic target for schizophrenia: still promising or a dead end? Prog. Neuropsychopharmacol. Biol. Psychiatry 60, 66–76. doi: 10.1016/j.pnpbp.2015.02.012
Lin, C. H., Lane, H. Y., and Tsai, G. E. (2012). Glutamate signaling in the pathophysiology and therapy of schizophrenia. Pharmacol. Biochem. Behav. 100, 665–677. doi: 10.1016/j.pbb.2011.03.023
Linden, A. M., Vasanen, J., Storvik, M., Lakso, M., Korpi, E. R., Wong, G., et al. (2001). Uncompetitive antagonists of the N-methyl-D-aspartate (NMDA) receptors alter the mRNA expression of proteins associated with the NMDA receptor complex. Pharmacol. Toxicol. 88, 98–105. doi: 10.1034/j.1600-0773.2001.d01-89.x
Liu, X., Li, J., Guo, C., Wang, H., Sun, Y., Wang, H., et al. (2017). Olanzapine Reverses MK-801-Induced Cognitive Deficits and Region-Specific Alterations of NMDA Receptor Subunits. Front. Behav. Neurosci. 11:260. doi: 10.3389/fnbeh.2017.00260
Loftis, J. M., and Janowsky, A. (2003). The N-methyl-D-aspartate receptor subunit NR2B: localization, functional properties, regulation, and clinical implications. Pharmacol. Ther. 97, 55–85. doi: 10.1016/S0163-7258(02)00302-9
Longart, M., Liu, Y., Karavanova, I., and Buonanno, A. (2004). Neuregulin-2 is developmentally regulated and targeted to dendrites of central neurons. J. Comp. Neurol. 472, 156–172. doi: 10.1002/cne.20016
Loscher, W., Annies, R., and Honack, D. (1991). The N-methyl-D-aspartate receptor antagonist MK-801 induces increases in dopamine and serotonin metabolism in several brain regions of rats. Neurosci. Lett. 128, 191–194. doi: 10.1016/0304-3940(91)90258-U
Madden, D. R. (2002). The structure and function of glutamate receptor ion channels. Nat. Rev. Neurosci. 3, 91–101. doi: 10.1038/nrn725
Maddox, V. H., Godefroi, E. F., and Parcell, R. F. (1965). The synthesis of phencyclidine and other 1-Arylcyclohexylamines. J. Med. Chem. 8, 230–235. doi: 10.1021/jm00326a019
Manahan-Vaughan, D., von Haebler, D., Winter, C., Juckel, G., and Heinemann, U. (2008). A single application of MK801 causes symptoms of acute psychosis, deficits in spatial memory, and impairment of synaptic plasticity in rats. Hippocampus 18, 125–134. doi: 10.1002/hipo.20367
Mansbach, R. S., and Geyer, M. A. (1989). Effects of phencyclidine and phencyclidine biologs on sensorimotor gating in the rat. Neuropsychopharmacology 2, 299–308. doi: 10.1016/0893-133X(89)90035-3
Marcotte, E. R., Pearson, D. M., and Srivastava, L. K. (2001). Animal models of schizophrenia: a critical review. J. Psychiatry Neurosci. 26, 395–410.
Marino, M. J., and Conn, P. J. (2002). Direct and indirect modulation of the N-methyl D-aspartate receptor. Curr. Drug Targets CNS Neurol. Disord. 1, 1–16. doi: 10.2174/1568007023339544
Martin, H., Rostas, J., Patel, Y., and Aitken, A. (1994). Subcellular localisation of 14-3-3 isoforms in rat brain using specific antibodies. J. Neurochem. 63, 2259–2265. doi: 10.1046/j.1471-4159.1994.63062259.x
Martinez, Z. A., Ellison, G. D., Geyer, M. A., and Swerdlow, N. R. (1999). Effects of sustained phencyclidine exposure on sensorimotor gating of startle in rats. Neuropsychopharmacology 21, 28–39. doi: 10.1016/S0893-133X(98)00137-7
Martucci, L., Wong, A. H., De Luca, V., Likhodi, O., Wong, G. W., King, N., et al. (2006). N-methyl-D-aspartate receptor NR2B subunit gene GRIN2B in schizophrenia and bipolar disorder: polymorphisms and mRNA levels. Schizophr. Res. 84, 214–221. doi: 10.1016/j.schres.2006.02.001
Massey, P. V., Johnson, B. E., Moult, P. R., Auberson, Y. P., Brown, M. W., Molnar, E., et al. (2004). Differential roles of NR2A and NR2B-containing NMDA receptors in cortical long-term potentiation and long-term depression. J. Neurosci. 24, 7821–7828. doi: 10.1523/JNEUROSCI.1697-04.2004
Masters, S. C., and Fu, H. (2001). 14-3-3 proteins mediate an essential anti-apoptotic signal. J. Biol. Chem. 276, 45193–45200. doi: 10.1074/jbc.M105971200
Matta, J. A., Ashby, M. C., Sanz-Clemente, A., Roche, K. W., and Isaac, J. T. (2011). mGluR5 and NMDA receptors drive the experience- and activity-dependent NMDA receptor NR2B to NR2A subunit switch. Neuron 70, 339–351. doi: 10.1016/j.neuron.2011.02.045
McHugh, T. J., Blum, K. I., Tsien, J. Z., Tonegawa, S., and Wilson, M. A. (1996). Impaired hippocampal representation of space in CA1-specific NMDAR1 knockout mice. Cell 87, 1339–1349. doi: 10.1016/S0092-8674(00)81828-0
McHugh, T. J., Jones, M. W., Quinn, J. J., Balthasar, N., Coppari, R., Elmquist, J. K., et al. (2007). Dentate gyrus NMDA receptors mediate rapid pattern separation in the hippocampal network. Science 317, 94–99. doi: 10.1126/science.1140263
McKibben, C. E., Jenkins, T. A., Adams, H. N., Harte, M. K., and Reynolds, G. P. (2010). Effect of pretreatment with risperidone on phencyclidine-induced disruptions in object recognition memory and prefrontal cortex parvalbumin immunoreactivity in the rat. Behav. Brain Res. 208, 132–136. doi: 10.1016/j.bbr.2009.11.018
Mei, L., and Xiong, W. C. (2008). Neuregulin 1 in neural development, synaptic plasticity and schizophrenia. Nat. Rev. Neurosci. 9, 437–452. doi: 10.1038/nrn2392
Meyer, D., Yamaai, T., Garratt, A., Riethmacher-Sonnenberg, E., Kane, D., Theill, L. E., et al. (1997). Isoform-specific expression and function of neuregulin. Development 124, 3575–3586.
Middleton, F. A., Peng, L., Lewis, D. A., Levitt, P., and Mirnics, K. (2005). Altered expression of 14-3-3 genes in the prefrontal cortex of subjects with schizophrenia. Neuropsychopharmacology 30, 974–983. doi: 10.1038/sj.npp.1300674
Mielnik, C. A., Yuxiao, C., Mary Binko, A., Rehnuma, I., Marija, M., Wendy, H., et al. (2017). Consequences Of NMDA receptor deficiency can be rescued in the adult brain. bioRxiv [Preprint]. doi: 10.1101/140343
Miyamoto, S., Leipzig, J. N., Lieberman, J. A., and Duncan, G. E. (2000). Effects of ketamine, MK-801, and amphetamine on regional brain 2-deoxyglucose uptake in freely moving mice. Neuropsychopharmacology 22, 400–412. doi: 10.1016/S0893-133X(99)00127-X
Miyatake, R., Furukawa, A., and Suwaki, H. (2002). Identification of a novel variant of the human NR2B gene promoter region and its possible association with schizophrenia. Mol. Psychiatry 7, 1101–1106. doi: 10.1038/sj.mp.4001152
Moghaddam, B. (2003). Bringing order to the glutamate chaos in schizophrenia. Neuron 40, 881–884. doi: 10.1016/S0896-6273(03)00757-8
Moghaddam, B., Adams, B., Verma, A., and Daly, D. (1997). Activation of glutamatergic neurotransmission by ketamine: a novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. J. Neurosci. 17, 2921–2927. doi: 10.1523/JNEUROSCI.17-08-02921.1997
Moghaddam, B., and Adams, B. W. (1998). Reversal of phencyclidine effects by a group II metabotropic glutamate receptor agonist in rats. Science 281, 1349–1352. doi: 10.1126/science.281.5381.1349
Mohn, A. R., Gainetdinov, R. R., Caron, M. G., and Koller, B. H. (1999). Mice with reduced NMDA receptor expression display behaviors related to schizophrenia. Cell 98, 427–436. doi: 10.1016/S0092-8674(00)81972-8
Monyer, H., Burnashev, N., Laurie, D. J., Sakmann, B., and Seeburg, P. H. (1994). Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron 12, 529–540. doi: 10.1016/0896-6273(94)90210-0
Moore, H. (2010). The role of rodent models in the discovery of new treatments for schizophrenia: updating our strategy. Schizophr. Bull. 36, 1066–1072. doi: 10.1093/schbul/sbq106
Morgan, C. J., and Curran, H. V. (2006). Acute and chronic effects of ketamine upon human memory: a review. Psychopharmacology 188, 408–424. doi: 10.1007/s00213-006-0572-3
Morgan, C. J., Muetzelfeldt, L., and Curran, H. V. (2009). Ketamine use, cognition and psychological wellbeing: a comparison of frequent, infrequent and ex-users with polydrug and non-using controls. Addiction 104, 77–87. doi: 10.1111/j.1360-0443.2008.02394.x
Moriya, T., Kouzu, Y., Shibata, S., Kadotani, H., Fukunaga, K., Miyamoto, E., et al. (2000). Close linkage between calcium/calmodulin kinase II alpha/beta and NMDA-2A receptors in the lateral amygdala and significance for retrieval of auditory fear conditioning. Eur. J. Neurosci. 12, 3307–3314. doi: 10.1046/j.1460-9568.2000.00203.x
Moy, S. S., Perez, A., Koller, B. H., and Duncan, G. E. (2006). Amphetamine-induced disruption of prepulse inhibition in mice with reduced NMDA receptor function. Brain Res. 1089, 186–194. doi: 10.1016/j.brainres.2006.03.073
Muslin, A. J., Tanner, J. W., Allen, P. M., and Shaw, A. S. (1996). Interaction of 14-3-3 with signaling proteins is mediated by the recognition of phosphoserine. Cell 84, 889–897. doi: 10.1016/S0092-8674(00)81067-3
Nabeshima, T., Kozawa, T., Furukawa, H., and Kameyama, T. (1986). Phencyclidine-induced retrograde amnesia in mice. Psychopharmacology 89, 334–337. doi: 10.1007/BF00174370
Nakao, S., Nagata, A., Miyamoto, E., Masuzawa, M., Murayama, T., and Shingu, K. (2003). Inhibitory effect of propofol on ketamine-induced c-Fos expression in the rat posterior cingulate and retrosplenial cortices is mediated by GABAA receptor activation. Acta Anaesthesiol. Scand. 47, 284–290. doi: 10.1034/j.1399-6576.2003.00040.x
Nakazawa, K., Jeevakumar, V., and Nakao, K. (2017). Spatial and temporal boundaries of NMDA receptor hypofunction leading to schizophrenia. NPJ Schizophr 3:7. doi: 10.1038/s41537-016-0003-3
Nakazawa, K., Quirk, M. C., Chitwood, R. A., Watanabe, M., Yeckel, M. F., Sun, L. D., et al. (2002). Requirement for hippocampal CA3 NMDA receptors in associative memory recall. Science 297, 211–218. doi: 10.1126/science.1071795
Neill, J. C., Barnes, S., Cook, S., Grayson, B., Idris, N. F., McLean, S. L., et al. (2010). Animal models of cognitive dysfunction and negative symptoms of schizophrenia: focus on NMDA receptor antagonism. Pharmacol. Ther. 128, 419–432. doi: 10.1016/j.pharmthera.2010.07.004
Niewoehner, B., Single, F. N., Hvalby, O., Jensen, V. S., Meyer zum Alten, B., Seeburg, P. H., et al. (2007). Impaired spatial working memory but spared spatial reference memory following functional loss of NMDA receptors in the dentate gyrus. Eur. J. Neurosci. 25, 837–846. doi: 10.1111/j.1460-9568.2007.05312.x
Noda, A., Noda, Y., Kamei, H., Ichihara, K., Mamiya, T., Nagai, T., et al. (2001). Phencyclidine impairs latent learning in mice: interaction between glutamatergic systems and sigma(1) receptors. Neuropsychopharmacology 24, 451–460. doi: 10.1016/S0893-133X(00)00192-5
Noda, Y., Yamada, K., Furukawa, H., and Nabeshima, T. (1995). Enhancement of immobility in a forced swimming test by subacute or repeated treatment with phencyclidine: a new model of schizophrenia. Br. J. Pharmacol. 116, 2531–2537. doi: 10.1111/j.1476-5381.1995.tb15106.x
Ohtsuki, T., Sakurai, K., Dou, H., Toru, M., Yamakawa-Kobayashi, K., and Arinami, T. (2001). Mutation analysis of the NMDAR2B (GRIN2B) gene in schizophrenia. Mol. Psychiatry 6, 211–216. doi: 10.1038/sj.mp.4000808
Olney, J. W., and Farber, N. B. (1995). Glutamate receptor dysfunction and schizophrenia. Arch. Gen. Psychiatry 52, 998–1007. doi: 10.1001/archpsyc.1995.03950240016004
Olney, J. W., Labruyere, J., and Price, M. T. (1989). Pathological changes induced in cerebrocortical neurons by phencyclidine and related drugs. Science 244, 1360–1362. doi: 10.1126/science.2660263
Olney, J. W., Labruyere, J., Wang, G., Wozniak, D. F., Price, M. T., and Sesma, M. A. (1991). NMDA antagonist neurotoxicity: mechanism and prevention. Science 254, 1515–1518. doi: 10.1126/science.1835799
Olney, J. W., Newcomer, J. W., and Farber, N. B. (1999). NMDA receptor hypofunction model of schizophrenia. J. Psychiatr. Res. 33, 523–533. doi: 10.1016/S0022-3956(99)00029-1
O'Tuathaigh, C. M., Babovic, D., O'Meara, G., Clifford, J. J., Croke, D. T., et al. (2007). Susceptibility genes for schizophrenia: characterisation of mutant mouse models at the level of phenotypic behaviour. Neurosci. Biobehav. Rev. 31, 60–78. doi: 10.1016/j.neubiorev.2006.04.002
Owen, M. J., Craddock, N., and O'Donovan, M. C. (2005). Schizophrenia: genes at last? Trends Genet. 21, 518–525. doi: 10.1016/j.tig.2005.06.011
Ozaki, M., Sasner, M., Yano, R., Lu, H. S., and Buonanno, A. (1997). Neuregulin-beta induces expression of an NMDA-receptor subunit. Nature 390, 691–694. doi: 10.1038/37795
Palmer, C., Ellis, K. A., O'Neill, B. V., Croft, R. J., Leung, S., Oliver, C., et al. (2008). The cognitive effects of modulating the glycine site of the NMDA receptor with high-dose glycine in healthy controls. Hum. Psychopharmacol. 23, 151–159. doi: 10.1002/hup.904
Patil, S. T., Zhang, L., Martenyi, F., Lowe, S. L., Jackson, K. A., Andreev, B. V., et al. (2007). Activation of mGlu2/3 receptors as a new approach to treat schizophrenia: a randomized Phase 2 clinical trial. Nat. Med. 13, 1102–1107. doi: 10.1038/nm1632
Pinault, D. (2008). N-methyl d-aspartate receptor antagonists ketamine and MK-801 induce wake-related aberrant gamma oscillations in the rat neocortex. Biol. Psychiatry 63, 730–735. doi: 10.1016/j.biopsych.2007.10.006
Pisani, A., Gubellini, P., Bonsi, P., Conquet, F., Picconi, B., Centonze, D., et al. (2001). Metabotropic glutamate receptor 5 mediates the potentiation of N-methyl-D-aspartate responses in medium spiny striatal neurons. Neuroscience 106, 579–587. doi: 10.1016/S0306-4522(01)00297-4
Pitcher, G. M., Kalia, L. V., Ng, D., Goodfellow, N. M., Yee, K. T., Lambe, E. K., et al. (2011). Schizophrenia susceptibility pathway neuregulin 1-ErbB4 suppresses Src upregulation of NMDA receptors. Nat. Med. 17, 470–478. doi: 10.1038/nm.2315
Powell, C. M., and Miyakawa, T. (2006). Schizophrenia-relevant behavioral testing in rodent models: a uniquely human disorder? Biol. Psychiatry 59, 1198–1207. doi: 10.1016/j.biopsych.2006.05.008
Powell, S. B. (2010). Models of neurodevelopmental abnormalities in schizophrenia. Curr. Top. Behav. Neurosci. 4, 435–481. doi: 10.1007/7854_2010_57
Purcell, S. M., Moran, J. L., Fromer, M., Ruderfer, D., Solovieff, N., Roussos, P., et al. (2014). A polygenic burden of rare disruptive mutations in schizophrenia. Nature 506, 185–190. doi: 10.1038/nature12975
Qiao, H., Foote, M., Graham, K., Wu, Y., and Zhou, Y. (2014). 14-3-3 proteins are required for hippocampal long-term potentiation and associative learning and memory. J. Neurosci. 34, 4801–4808. doi: 10.1523/JNEUROSCI.4393-13.2014
Qin, S., Zhao, X., Pan, Y., Liu, J., Feng, G., Fu, J., et al. (2005). An association study of the N-methyl-D-aspartate receptor NR1 subunit gene (GRIN1) and NR2B subunit gene (GRIN2B) in schizophrenia with universal DNA microarray. Eur. J. Hum. Genet. 13, 807–814. doi: 10.1038/sj.ejhg.5201418
Rainey, J. M. Jr., and Crowder, M. K. (1975). Prolonged psychosis attributed to phencyclidine: report of three cases. Am. J. Psychiatry 132, 1076–1078. doi: 10.1176/ajp.132.10.1076
Rajji, T., Chapman, D., Eichenbaum, H., and Greene, R. (2006). The role of CA3 hippocampal NMDA receptors in paired associate learning. J. Neurosci. 26, 908–915. doi: 10.1523/JNEUROSCI.4194-05.2006
Ramsey, A. J., Milenkovic, M., Oliveira, A. F., Escobedo-Lozoya, Y., Seshadri, S., Salahpour, A., et al. (2011). Impaired NMDA receptor transmission alters striatal synapses and DISC1 protein in an age-dependent manner. Proc. Natl. Acad. Sci. U.S.A. 108, 5795–5800. doi: 10.1073/pnas.1012621108
Ramshaw, H., Xu, X., Jaehne, E. J., McCarthy, P., Greenberg, Z., Saleh, E., Schwarz, Q., et al. (2013). Locomotor hyperactivity in 14-3-3zeta KO mice is associated with dopamine transporter dysfunction. Transl. Psychiatry 3:e327. doi: 10.1038/tp.2013.99
Rapoport, J. L., Addington, A. M., Frangou, S., and Psych, M. R. (2005). The neurodevelopmental model of schizophrenia: update 2005. Mol. Psychiatry 10, 434–449. doi: 10.1038/sj.mp.4001642
Rice, S. R., Niu, N., Berman, D. B., Heston, L. L., and Sobell, J. L. (2001). Identification of single nucleotide polymorphisms (SNPs) and other sequence changes and estimation of nucleotide diversity in coding and flanking regions of the NMDAR1 receptor gene in schizophrenic patients. Mol. Psychiatry 6, 274–284. doi: 10.1038/sj.mp.4000838
Rieff, H. I., Raetzman, L. T., Sapp, D. W., Yeh, H. H., Siegel, R. E., and Corfas, G. (1999). Neuregulin induces GABA(A) receptor subunit expression and neurite outgrowth in cerebellar granule cells. J. Neurosci. 19, 10757–10766. doi: 10.1523/JNEUROSCI.19-24-10757.1999
Romon, T., Mengod, G., and Adell, A. (2011). Expression of parvalbumin and glutamic acid decarboxylase-67 after acute administration of MK-801. Implications for the NMDA hypofunction model of schizophrenia. Psychopharmacology 217, 231–238. doi: 10.1007/s00213-011-2268-6
Rompala, G. R., Zsiros, V., Zhang, S., Kolata, S. M., and Nakazawa, K. (2013). Contribution of NMDA receptor hypofunction in prefrontal and cortical excitatory neurons to schizophrenia-like phenotypes. PLoS ONE 8:e61278. doi: 10.1371/journal.pone.0061278
Rondi-Reig, L., Libbey, M., Eichenbaum, H., and Tonegawa, S. (2001). CA1-specific N-methyl-D-aspartate receptor knockout mice are deficient in solving a nonspatial transverse patterning task. Proc. Natl. Acad. Sci. U.S.A. 98, 3543–3548. doi: 10.1073/pnas.041620798
Roth, B. L., Gibbons, S., Arunotayanun, W., Huang, X. P., Setola, V., Treble, R., et al. (2013). The ketamine analogue methoxetamine and 3- and 4-methoxy analogues of phencyclidine are high affinity and selective ligands for the glutamate NMDA receptor. PLoS ONE 8:e59334. doi: 10.1371/journal.pone.0059334
Roy, K., Murtie, J. C., El-Khodor, B. F., Edgar, N., Sardi, S. P., Hooks, B. M., et al. (2007). Loss of erbB signaling in oligodendrocytes alters myelin and dopaminergic function, a potential mechanism for neuropsychiatric disorders. Proc. Natl. Acad. Sci. U.S.A. 104, 8131–8136. doi: 10.1073/pnas.0702157104
Rujescu, D., Bender, A., Keck, M., Hartmann, A. M., Ohl, F., Raeder, H., et al. (2006). A pharmacological model for psychosis based on N-methyl-D-aspartate receptor hypofunction: molecular, cellular, functional and behavioral abnormalities. Biol. Psychiatry 59, 721–729. doi: 10.1016/j.biopsych.2005.08.029
Rung, J. P., Carlsson, A., Ryden Markinhuhta, K., and Carlsson, M. L. (2005). (+)-MK-801 induced social withdrawal in rats; a model for negative symptoms of schizophrenia. Prog. Neuropsychopharmacol. Biol. Psychiatry 29, 827–832. doi: 10.1016/j.pnpbp.2005.03.004
Sakimura, K., Kutsuwada, T., Ito, I., Manabe, T., Takayama, C., Kushiya, E., et al. (1995). Reduced hippocampal LTP and spatial learning in mice lacking NMDA receptor epsilon 1 subunit. Nature 373, 151–155. doi: 10.1038/373151a0
Sams-Dodd, F. (1995). Distinct effects of d-amphetamine and phencyclidine on the social behaviour of rats. Behav. Pharmacol. 6, 55–65. doi: 10.1097/00008877-199501000-00009
Sams-Dodd, F. (1996). Phencyclidine-induced stereotyped behaviour and social isolation in rats: a possible animal model of schizophrenia. Behav. Pharmacol. 7, 3–23. doi: 10.1097/00008877-199601000-00001
Sams-Dodd, F. (1999). Phencyclidine in the social interaction test: an animal model of schizophrenia with face and predictive validity. Rev. Neurosci. 10, 59–90. doi: 10.1515/REVNEURO.1999.10.1.59
Schizophrenia Working Group of the Psychiatric Genomics Consortium (2014). Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427. doi: 10.1038/nature13595
Shanmugasundaram, B., Sase, A., Miklosi, A. G., Sialana, F. J., Subramaniyan, S., Aher, Y. D., et al. (2015). Frontal cortex and hippocampus neurotransmitter receptor complex level parallels spatial memory performance in the radial arm maze. Behav. Brain Res. 289, 157–168. doi: 10.1016/j.bbr.2015.04.043
Sharp, T., Zetterstrom, T., Ljungberg, T., and Ungerstedt, U. (1987). A direct comparison of amphetamine-induced behaviours and regional brain dopamine release in the rat using intracerebral dialysis. Brain Res. 401, 322–330. doi: 10.1016/0006-8993(87)91416-8
Sheng, M., Cummings, J., Roldan, L. A., Jan, Y. N., and Jan, L. Y. (1994). Changing subunit composition of heteromeric NMDA receptors during development of rat cortex. Nature 368, 144–147. doi: 10.1038/368144a0
Shi, J., Gershon, E. S., and Liu, C. (2008). Genetic associations with schizophrenia: meta-analyses of 12 candidate genes. Schizophr. Res. 104, 96–107. doi: 10.1016/j.schres.2008.06.016
Silvestre, J. S., Nadal, R., Pallares, M., and Ferre, N. (1997). Acute effects of ketamine in the holeboard, the elevated-plus maze, and the social interaction test in Wistar rats. Depress Anxiety 5, 29–33.
Singh, S. M., and O'Reilly, R. (2009). (Epi)genomics and neurodevelopment in schizophrenia: monozygotic twins discordant for schizophrenia augment the search for disease-related (epi)genomic alterations. Genome 52, 8–19. doi: 10.1139/G08-095
Slifstein, M., van de Giessen, E., Van Snellenberg, J., Thompson, J. L., Narendran, R., Gil, R., et al. (2015). Deficits in prefrontal cortical and extrastriatal dopamine release in schizophrenia: a positron emission tomographic functional magnetic resonance imaging study. JAMA Psychiatry 72, 316–324. doi: 10.1001/jamapsychiatry.2014.2414
Snyder, M. A., and Gao, W. J. (2013). NMDA hypofunction as a convergence point for progression and symptoms of schizophrenia. Front. Cell. Neurosci. 7:31. doi: 10.3389/fncel.2013.00031
Sprengel, R., Suchanek, B., Amico, C., Brusa, R., Burnashev, N., Rozov, A., et al. (1998). Importance of the intracellular domain of NR2 subunits for NMDA receptor function in vivo. Cell 92, 279–289. doi: 10.1016/S0092-8674(00)80921-6
Stefani, M. R., and Moghaddam, B. (2002). Effects of repeated treatment with amphetamine or phencyclidine on working memory in the rat. Behav. Brain Res. 134, 267–274. doi: 10.1016/S0166-4328(02)00040-2
Stefani, M. R., and Moghaddam, B. (2005). Systemic and prefrontal cortical NMDA receptor blockade differentially affect discrimination learning and set-shift ability in rats. Behav. Neurosci. 119, 420–428. doi: 10.1037/0735-7044.119.2.420
Stefansson, H., Sarginson, J., Kong, A., Yates, P., Steinthorsdottir, V., Gudfinnsson, E., et al. (2003). Association of neuregulin 1 with schizophrenia confirmed in a Scottish population. Am. J. Hum. Genet. 72, 83–87. doi: 10.1086/345442
Stefansson, H., Sigurdsson, E., Steinthorsdottir, V., Bjornsdottir, S., Sigmundsson, T., Ghosh, S., et al. (2002). Neuregulin 1 and susceptibility to schizophrenia. Am. J. Hum. Genet. 71, 877–892. doi: 10.1086/342734
Sturgeon, R. D., Fessler, R. G., and Meltzer, H. Y. (1979). Behavioral rating scales for assessing phencyclidine-induced locomotor activity, stereotyped behavior and ataxia in rats. Eur. J. Pharmacol. 59, 169–179. doi: 10.1016/0014-2999(79)90279-6
Sullivan, E. M., Timi, P., Hong, L. E., and O'Donnell, P. (2015). Reverse translation of clinical electrophysiological biomarkers in behaving rodents under acute and chronic NMDA receptor antagonism. Neuropsychopharmacology 40, 719–727. doi: 10.1038/npp.2014.228
Suzuki, Y., Jodo, E., Takeuchi, S., Niwa, S., and Kayama, Y. (2002). Acute administration of phencyclidine induces tonic activation of medial prefrontal cortex neurons in freely moving rats. Neuroscience 114, 769–779. doi: 10.1016/S0306-4522(02)00298-1
Swerdlow, N. R., Braff, D. L., Taaid, N., and Geyer, M. A. (1994). Assessing the validity of an animal model of deficient sensorimotor gating in schizophrenic patients. Arch. Gen. Psychiatry 51, 139–154. doi: 10.1001/archpsyc.1994.03950020063007
Swerdlow, N. R., and Geyer, M. A. (1998). Using an animal model of deficient sensorimotor gating to study the pathophysiology and new treatments of schizophrenia. Schizophr. Bull. 24, 285–301. doi: 10.1093/oxfordjournals.schbul.a033326
Szlachta, M., Pabian, P., Kusmider, M., Solich, J., Kolasa, M., Zurawek, D., et al. (2017). Effect of clozapine on ketamine-induced deficits in attentional set shift task in mice. Psychopharmacology 234, 2103–2112. doi: 10.1007/s00213-017-4613-x
Takahashi, S. (2013). Heterogeneity of schizophrenia: genetic and symptomatic factors. Am. J. Med. Genet. B Neuropsychiatr. Genet. 162B, 648–652. doi: 10.1002/ajmg.b.32161
Takeuchi, T., Kiyama, Y., Nakamura, K., Tsujita, M., Matsuda, I., Mori, H., et al. (2001). Roles of the glutamate receptor epsilon2 and delta2 subunits in the potentiation and prepulse inhibition of the acoustic startle reflex. Eur. J. Neurosci. 14, 153–160. doi: 10.1046/j.0953-816x.2001.01620.x
Tang, J., Chen, X., Xu, X., Wu, R., Zhao, J., Hu, Z., et al. (2006). Significant linkage and association between a functional (GT)n polymorphism in promoter of the N-methyl-D-aspartate receptor subunit gene (GRIN2A) and schizophrenia. Neurosci. Lett. 409, 80–82. doi: 10.1016/j.neulet.2006.09.022
Tang, Y. P., Shimizu, E., Dube, G. R., Rampon, C., Kerchner, G. A., Zhuo, M., et al. (1999). Genetic enhancement of learning and memory in mice. Nature 401, 63–69. doi: 10.1038/43432
Tang, Y. P., Wang, H., Feng, R., Kyin, M., and Tsien, J. Z. (2001). Differential effects of enrichment on learning and memory function in NR2B transgenic mice. Neuropharmacology 41, 779–790. doi: 10.1016/S0028-3908(01)00122-8
Tatard-Leitman, V. M., Jutzeler, C. R., Suh, J., Saunders, J. A., Billingslea, E. N., Morita, S., et al. (2015). Pyramidal cell selective ablation of N-methyl-D-aspartate receptor 1 causes increase in cellular and network excitability. Biol. Psychiatry 77, 556–568. doi: 10.1016/j.biopsych.2014.06.026
Taya, S., Shinoda, T., Tsuboi, D., Asaki, J., Nagai, K., Hikita, T., et al. (2007). DISC1 regulates the transport of the NUDEL/LIS1/14-3-3epsilon complex through kinesin-1. J. Neurosci. 27, 15–26. doi: 10.1523/JNEUROSCI.3826-06.2006
Thuret, S., Alavian, K. N., Gassmann, M., Lloyd, C. K., Smits, S. M., Smidt, M. P., et al. (2004). The neuregulin receptor, ErbB4, is not required for normal development and adult maintenance of the substantia nigra pars compacta. J. Neurochem. 91, 1302–1311. doi: 10.1111/j.1471-4159.2004.02809.x
Toyo-oka, K., Wachi, T., Hunt, R. F., Baraban, S. C., Taya, S., Ramshaw, H., et al. (2014). 14-3-3epsilon and zeta regulate neurogenesis and differentiation of neuronal progenitor cells in the developing brain. J. Neurosci. 34, 12168–12181. doi: 10.1523/JNEUROSCI.2513-13.2014
Tsien, J. Z., Huerta, P. T., and Tonegawa, S. (1996). The essential role of hippocampal CA1 NMDA receptor-dependent synaptic plasticity in spatial memory. Cell 87, 1327–1338. doi: 10.1016/S0092-8674(00)81827-9
Unal, G., Ates, A., and Aricioglu, F. (2018). Agmatine-attenuated cognitive and social deficits in subchronic MK-801 model of schizophrenia in rats. Psychiatry Clin. Psychopharmacol. 28, 245–253. doi: 10.1080/24750573.2018.1426696
Uzunova, G., Hollander, E., and Shepherd, J. (2014). The role of ionotropic glutamate receptors in childhood neurodevelopmental disorders: autism spectrum disorders and fragile x syndrome. Curr. Neuropharmacol. 12, 71–98. doi: 10.2174/1570159X113116660046
Verma, A., and Moghaddam, B. (1996). NMDA receptor antagonists impair prefrontal cortex function as assessed via spatial delayed alternation performance in rats: modulation by dopamine. J. Neurosci. 16, 373–379. doi: 10.1523/JNEUROSCI.16-01-00373.1996
Vollenweider, F. X., Leenders, K. L., Scharfetter, C., Antonini, A., Maguire, P., Missimer, J., et al. (1997). Metabolic hyperfrontality and psychopathology in the ketamine model of psychosis using positron emission tomography (PET) and [18F]fluorodeoxyglucose (FDG). Eur. Neuropsychopharmacol. 7, 9–24. doi: 10.1016/S0924-977X(96)00039-9
von Engelhardt, J., Doganci, B., Jensen, V., Hvalby, O., Gongrich, C., Taylor, A., et al. (2008). Contribution of hippocampal and extra-hippocampal NR2B-containing NMDA receptors to performance on spatial learning tasks. Neuron 60, 846–860. doi: 10.1016/j.neuron.2008.09.039
Walsh, T., McClellan, J. M., McCarthy, S. E., Addington, A. M., Pierce, S. B., Cooper, G. M., et al. (2008). Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science 320, 539–543. doi: 10.1126/science.1155174
Wang, B., Yang, H., Liu, Y. C., Jelinek, T., Zhang, L., Ruoslahti, E., et al. (1999). Isolation of high-affinity peptide antagonists of 14-3-3 proteins by phage display. Biochemistry 38, 12499–12504. doi: 10.1021/bi991353h
Wang, C. Z., Yang, S. F., Xia, Y., and Johnson, K. M. (2008). Postnatal phencyclidine administration selectively reduces adult cortical parvalbumin-containing interneurons. Neuropsychopharmacology 33, 2442–2455. doi: 10.1038/sj.npp.1301647
Wiescholleck, V., and Manahan-Vaughan, D. (2012). PDE4 inhibition enhances hippocampal synaptic plasticity in vivo and rescues MK801-induced impairment of long-term potentiation and object recognition memory in an animal model of psychosis. Transl. Psychiatry 2:e89. doi: 10.1038/tp.2012.17
Wong, A. H., Likhodi, O., Trakalo, J., Yusuf, M., Sinha, A., Pato, C. N., et al. (2005). Genetic and post-mortem mRNA analysis of the 14-3-3 genes that encode phosphoserine/threonine-binding regulatory proteins in schizophrenia and bipolar disorder. Schizophr. Res. 78, 137–146. doi: 10.1016/j.schres.2005.06.009
Xiu, Y., Kong, X. R., Zhang, L., Qiu, X., Chao, F. L., Peng, C., et al. (2014). White matter injuries induced by MK-801 in a mouse model of schizophrenia based on NMDA antagonism. Anat. Rec. 297, 1498–1507. doi: 10.1002/ar.22942
Xu, X., Jaehne, E. J., Greenberg, Z., McCarthy, P., Saleh, E., Parish, C. L., et al. (2015). 14-3-3zeta deficient mice in the BALB/c background display behavioural and anatomical defects associated with neurodevelopmental disorders. Sci. Rep. 5:12434. doi: 10.1038/srep12434
Yamada, K., Noda, Y., Hasegawa, T., Komori, Y., Nikai, T., Sugihara, H., et al. (1996). The role of nitric oxide in dizocilpine-induced impairment of spontaneous alternation behavior in mice. J. Pharmacol. Exp. Ther. 276, 460–466. doi: 10.1016/0306-4522(96)00161-3
Yang, J. Z., Si, T. M., Ruan, Y., Ling, Y. S., Han, Y. H., Wang, X. L., et al. (2003). Association study of neuregulin 1 gene with schizophrenia. Mol. Psychiatry 8, 706–709. doi: 10.1038/sj.mp.4001377
Yang, L., Mao, L., Tang, Q., Samdani, S., Liu, Z., and Wang, J. Q. (2004). A novel Ca2+-independent signaling pathway to extracellular signal-regulated protein kinase by coactivation of NMDA receptors and metabotropic glutamate receptor 5 in neurons. J. Neurosci. 24, 10846–10857. doi: 10.1523/JNEUROSCI.2496-04.2004
Yashiro, K., and Philpot, B. D. (2008). Regulation of NMDA receptor subunit expression and its implications for LTD, LTP, and metaplasticity. Neuropharmacology 55, 1081–1094. doi: 10.1016/j.neuropharm.2008.07.046
Yonezawa, Y., Kuroki, T., Kawahara, T., Tashiro, N., and Uchimura, H. (1998). Involvement of gamma-aminobutyric acid neurotransmission in phencyclidine-induced dopamine release in the medial prefrontal cortex. Eur. J. Pharmacol. 341, 45–56. doi: 10.1016/S0014-2999(97)01435-0
Yu, H. N., Park, W. K., Nam, K. H., Song, D. Y., Kim, H. S., Baik, T. K., et al. (2015). Neuregulin 1 controls glutamate uptake by up-regulating excitatory amino acid carrier 1 (EAAC1). J. Biol. Chem. 290, 20233–20244. doi: 10.1074/jbc.M114.591867
Zanos, P., and Gould, T. D. (2018). Mechanisms of ketamine action as an antidepressant. Mol. Psychiatry 23, 801–811. doi: 10.1038/mp.2017.255
Zhao, X., Li, H., Shi, Y., Tang, R., Chen, W., Liu, J., et al. (2006). Significant association between the genetic variations in the 5' end of the N-methyl-D-aspartate receptor subunit gene GRIN1 and schizophrenia. Biol. Psychiatry 59, 747–753. doi: 10.1016/j.biopsych.2005.10.023
Zhao, X., Shi, Y., Tang, J., Tang, R., Yu, L., Gu, N., et al. (2004). A case control and family based association study of the neuregulin1 gene and schizophrenia. J. Med. Genet. 41, 31–34. doi: 10.1136/jmg.2003.014977
Zhong, J., Carrozza, D. P., Williams, K., Pritchett, D. B., and Molinoff, P. B. (1995). Expression of mRNAs encoding subunits of the NMDA receptor in developing rat brain. J. Neurochem. 64, 531–539. doi: 10.1046/j.1471-4159.1995.64020531.x
Keywords: NMDAR, NMDAR hypofunction, NMDAR antagonists, knockout mice, schizophrenia, animal models, 14-3-3 proteins
Citation: Lee G and Zhou Y (2019) NMDAR Hypofunction Animal Models of Schizophrenia. Front. Mol. Neurosci. 12:185. doi: 10.3389/fnmol.2019.00185
Received: 15 May 2019; Accepted: 17 July 2019;
Published: 31 July 2019.
Edited by:
Xiaona Du, Hebei Medical University, ChinaReviewed by:
Dennis Kätzel, University of Ulm, GermanyJae-Ick Kim, Ulsan National Institute of Science and Technology, South Korea
Copyright © 2019 Lee and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yi Zhou, WWkuemhvdUBtZWQuZnN1LmVkdQ==