 Lavrentii G. Danilov1
Lavrentii G. Danilov1 Andrew G. Matveenko1Varvara E. Ryzhkova1Mikhail V. Belousov1,2Olga I. Poleshchuk1Daria V. Likholetova1Petr A. Sokolov3Nina A. Kasyanenko3
Andrew G. Matveenko1Varvara E. Ryzhkova1Mikhail V. Belousov1,2Olga I. Poleshchuk1Daria V. Likholetova1Petr A. Sokolov3Nina A. Kasyanenko3 Andrey V. Kajava4,5
Andrey V. Kajava4,5 Galina A. Zhouravleva1,6*
Galina A. Zhouravleva1,6* Stanislav A. Bondarev1,6*
Stanislav A. Bondarev1,6*- 1Department of Genetics and Biotechnology, St. Petersburg State University, St. Petersburg, Russia
- 2Laboratory for Proteomics of Supra-Organismal Systems, All-Russia Research Institute for Agricultural Microbiology (ARRIAM), St. Petersburg, Russia
- 3Department of Molecular Biophysics and Polymer Physics, St. Petersburg State University, St. Petersburg, Russia
- 4Centre de Recherche en Biologie cellulaire de Montpellier (CRBM), UMR 5237 CNRS, Université Montpellier, Montpellier, France
- 5Institut de Biologie Computationnelle (IBC), Universitè Montpellier, Montpellier, France
- 6Laboratory of Amyloid Biology, St. Petersburg State University, St. Petersburg, Russia
A number of [PSI+]-no-more (PNM) mutations, eliminating [PSI+] prion, were previously described in SUP35. In this study, we designed and analyzed a new PNM mutation based on the parallel in-register β-structure of Sup35 prion fibrils suggested by the known experimental data. In such an arrangement, substitution of non-charged residues by charged ones may destabilize the fibril structure. We introduced Q33K/A34K amino acid substitutions into the Sup35 protein, corresponding allele was called sup35-M0. The mutagenized residues were chosen based on ArchCandy in silico prediction of high inhibitory effect on the amyloidogenic potential of Sup35. The experiments confirmed that Sup35-M0 leads to the elimination of [PSI+] with high efficiency. Our data suggested that the elimination of the [PSI+] prion is associated with the decreased aggregation properties of the protein. The new mutation can induce the prion with very low efficiency and is able to propagate only weak [PSI+] prion variants. We also showed that Sup35-M0 protein co-aggregates with the wild-type Sup35 in vivo. Moreover, our data confirmed the utility of the strategy of substitution of non-charged residues by charged ones to design new mutations to inhibit a prion formation.
1. Introduction
Prions are self-propagating and transmissible protein isoforms that cause fatal neurodegenerative disease in humans or heritable traits in lower eukaryotes. The most known hallmark of almost all prions is a formation of amyloid aggregates (Liebman and Chernoff, 2012). These aggregates have a set of specific properties, such as resistance to detergents and proteases, interaction with dyes Thioflavin T and S, birefringence when stained with the Congo Red dye, and cross-β-structure (Baxa et al., 2006). The first discovered prion, PrPSc (“prion protein” scrapie), causes severe infectious neurodegenerative diseases in mammals (Prusiner, 2013). Discovery of prions in lower eukaryotes (Wickner, 1994) revealed that this phenomenon is widespread and based on common mechanisms. Thus, unicellular organisms such as yeast can be used for investigation of prionization and the data obtained can be extrapolated to mammalian prions (Liebman and Chernoff, 2012).
One of the best-studied prions to date is [PSI+], an isoform of Sup35 protein (Cox, 1965; Wickner, 1994; Wickner et al., 1995), which is a eukaryotic release factor 3 (Stansfield et al., 1995; Zhouravleva et al., 1995). This protein is divided into three domains (N, M, and C) (Kushnirov et al., 1988). The C-terminal part of Sup35 contains four GTP binding sites (Stansfield et al., 1995; Zhouravleva et al., 1995) and it is essential for the cell viability and termination of translation (Ter-Avanesyan et al., 1993). N-domain is required for [PSI+] maintenance (Ter-Avanesyan et al., 1994) and formation of stress-inducible condensates (Franzmann et al., 2018). N-domain consists of two parts — the Q/N rich segment (1–39 aa) and the oligopeptide repeats region (OR), containing one incomplete and five complete repeats (40–112 aa) (Kushnirov et al., 1988). The charged M-domain represents an unfolded linker that also affects [PSI+] prion maintenance (Liu et al., 2002; Helsen and Glover, 2012).
Cells bearing [PSI+] prion have a reduced amount of monomeric Sup35 protein, that increases the frequency of the read-through of premature stop codons (Liebman and Chernoff, 2012). Nonsense mutation ade1-14, which lead to the synthesis of a truncated non-functional Ade1 protein and to the inability of cells to synthesize adenine, is often used to test for the nonsense suppression caused by the prion. The accumulation of the adenine biosynthesis intermediate results in the red color of colonies growing on the 1/4 YEPD medium. The appearance of [PSI+] prion leads to the suppression of ade1-14 nonsense mutation and the formation of full-length Ade1. Phenotypically it can be detected by growth on media lacking adenine and the white colony color. This manifestation can vary depending on the structure of Sup35 aggregates (prion variant), templated upon prion propagation. The term “variant” is used hereafter for different prion variants, and “strain” — only for yeast strains. Cells bearing weak variants of the [PSI+] prion demonstrate weak growth on adenineless media, i.e., weak nonsense suppression, while the strong [PSI+] variants lead to almost complete masking of the ade1-14 mutant phenotype (Liebman and Chernoff, 2012).
Different approaches may be used for investigation of Sup35 aggregates in [PSI+] cells. They can be decorated by transiently overproduced Sup35NM-GFP and visualized with fluorescence microscopy (Osherovich et al., 2004). Sup35 aggregates can also be directly analyzed with biochemical approaches: differential centrifugation, SDD-AGE (Kryndushkin et al., 2003) or modifications of SDS-PAGE (Kushnirov et al., 2006).
Oligopeptide repeats in the Sup35 N-domain significantly affect [PSI+] prion maintenance. At least two first ORs are essential for the prion propagation (Liu and Lindquist, 1999; Osherovich et al., 2004; Shkundina et al., 2006). At the same time OR expansion leads to increased fragmentation of Sup35 aggregates, while a decrease in the number of repeats has an opposite effect (Langlois et al., 2016). Previously, using the T-REKS algorithm (Jorda and Kajava, 2009), we identified an additional OR in the Sup35 N-domain, located from 28 to 40 amino acid residues (Bondarev et al., 2013). The mutation within this OR, named sup35-M0, was designed based on the model of a superpleated-β-structure, proposed for Sup35 aggregates (Kajava et al., 2004). According to this model, charged amino acid residues located inside the fibril, can destabilize this structure, due to the electrostatic repulsion. In this work, we investigated the effect of this mutation on the prion propagation and properties of Sup35 aggregates.
2. Materials and Methods
2.1. Strains, Media, and Growth Condition
Saccharomyces cerevisiae strain 7A-D832 [psi–] and its isogenic [PSI+] derivative 10-7A-D832 (Bondarev et al., 2013) were used in this study unless otherwise specified. Both strains contain the sup35::TRP1 knockout on the chromosome, compensated by plasmid(s) bearing the SUP35 gene. For the experiments with protein transformation, the [psi–] [pin–] strain 2-OT56 (Matveenko et al., 2016) was used.
Yeast strain 12-D1682, used for the induction of new [PSI+] variants, was constructed as follows. Strain GT671 was transformed by pRSU2 plasmid, carrying the URA3 marker. Transformant with the Ura+Leu– phenotype was selected and designated as U-GT671. Yeast strain GT159 (Chernoff et al., 1999) was transformed by the pRSU1 plasmid (Volkov et al., 2002), carrying LEU2 marker, and mated with the U-GT671 strain. Diploids were selected on SC-Ura-Leu media. Then random ascospore isolates were obtained, and MATa Ura–Leu+ segregant was selected and named 12-D1682 (Table 1). This strain was transformed with a pRS316CUP-NM-GFP plasmid for overproduction of Sup35NM-GFP. The prion induction was performed as described below and seven prion variants were isolated. Clones that lost pRS316CUP-NM-GFP were selected after several passages on YEPD.

Table 1. Strains of S. cerevisiae used in this study.
Yeast cultures were maintained on the YEPD (yeast extract/peptone/dextrose medium) or synthetic complete minimal medium (SC) (Kaiser et al., 1994). Solid media were prepared with the addition of agar (2%). SC media with 5'-FOA (1 mg/ml) was used for counter-selection of plasmids bearing URA3 (Kaiser et al., 1994). Nonsense suppression in [PSI+] cells was detected by the ability to grow on the SC medium lacking adenine (SC-Ade) or by the colony color on 1/4 YEPD (Eaglestone et al., 2000). Yeast cells were grown at 30°C.
2.2. Plasmids
Plasmids bearing the new sup35 mutation were constructed by site-directed mutagenesis. We amplified the vector using highly processive DNA polymerase (AccuPrime Pfx, Invitrogen) (the primer sequences are available upon request). The vectors pRSU1 (Volkov et al., 2002), pRSU2 (Volkov et al., 2002), pRS316CUP-NM-GFP (Serio et al., 1999), pRS315CUP-NM-GFP and pET-20b-SUP35NM-His6 (Allen et al., 2005) were used as templates. Next, the PCR mixture was treated with DpnI (Thermo Scientific) to remove the template DNA. Then, this solution was used for transformation of E. coli competent cells. All mutations were verified by sequencing. To construct the pRS315CUP-NM-GFP plasmid, we ligated the region with the CUP1 promoter, Sup35NM and GFP from pRS316CUP-NM-GFP (Serio et al., 1999) into the polylinker site of the pRS315 plasmid (Sikorski and Hieter, 1989). The region of interest in pRS316CUP-NM-GFP and the polylinker site were digested by XhoI and SacI enzymes. Sticky-end ligation was performed with T4 DNA-ligase according to Thermo Scientific protocol. pRS315CG was obtained analogously from pRS316CG (Serio et al., 1999) and pRS315. pR16CUP-NM-yTagRFP-T plasmid was obtained by insertion of the XhoI-XhoI fragment from pCUP-NM-His6 (Kiktev et al., 2015) in place of the XhoI-SalI fragment of pR16CUP-SFP1C-yTagRFP-T which in turn resulted from the substitution of the PstI-PstI fragment in pR16CUP-SFP1-Cerulean (Matveenko et al., 2016) for the PstI-PstI fragment from pIM35 (Malcova et al., 2016). TagRFP-T is a TagRFP derivative containing one additional substitution (Shaner et al., 2008). All the plasmids are listed in Table 2.

Table 2. Plasmids used in this study.
2.3. Genetic and Microbiological Procedures
Standard microbiological approaches were used for all manipulations with yeast and bacterial colonies (Sambrook and Fritsch, 1989). Yeast protein transformation was performed as described previously (Tanaka and Weissman, 2006). Direct plasmid shuffle (from wild-type to mutant allele) was performed as follows: the [PSI+] sup35::TRP1 strain with the SUP35 gene on a URA3 plasmid was transformed with LEU2 plasmids bearing the wild-type or mutant SUP35 alleles. Transformants, selected on SC medium lacking uracil and leucine (SC-Ura-Leu), were tested for suppression of the ade1-14 mutation to determine the presence of [PSI+]. These transformants were replica plated on media with 5′-FOA for counter-selection of the plasmid with SUP35, and then on the SC-Leu and SC-Ura media to prove the loss of the plasmid. The suppressor phenotype of the obtained strains was analyzed on SC-Ade or 1/4 YEPD. Reverse plasmid shuffle (from mutant allele to wild-type) was performed as follows: the strains after direct shuffle were transformed with plasmids bearing the wild-type allele. Transformants were selected on SC-Ura-Leu medium and then streaked out on YEPD media to allow spontaneous plasmid loss. Colonies were replica plated on SC-Leu or SC-Ura medium to identify clones which contain only wild-type SUP35 allele. After selection, the cells were tested for suppression of the ade1-14 mutation to determine the presence of [PSI+].
The [PSI+] prion loss and transmission were scored according to the previously described procedure (Afanasieva et al., 2011) with minor modifications. The [PSI+] strain (10-7A-D832) was transformed with a LEU2 plasmid bearing the SUP35 or sup35-M0. To estimate [PSI+] curing, caused by the presence of mutated sup35 allele, three transformants for each allele were replica plated three times on a medium lacking uracil, then resuspended in water and plated on 1/4 YEPD medium to obtain single colonies and to reveal the nonsense suppressor phenotype. Then these clones were replica plated on media lacking uracil or leucine. The frequency of prion loss was estimated as a fraction of Ura+Leu– [PSI+] colonies. To determine the efficiency of [PSI+] transmission from Sup35 to Sup35-M0, 50 transformants for each combination of SUP35 and sup35-M0 alleles were replica plated three times on medium lacking leucine containing uracil to enable the cells to lose plasmid containing wild-type SUP35. To estimate the efficiency of [PSI+] transmission, the fraction of Ade+ colonies was scored amongst Ura–Leu+ isolates.
2.4. [PSI+] Induction
Plasmids bearing SUP35NM-GFP or GFP under control of CUP1 promoter were used for the prion induction. Strains [psi–][PIN+] with corresponding plasmids were grown in selective media at 30°C to logarithmic phase. For the induction of CUP1 promoter, CuSO4 was added into the media to the final concentration of 100 μM for the 7A-D832 strain or 50 μM for 12-D1682. Before induction and after 24 h, the aliquots of cultures were plated on 1/4 YEPD to count the number of white clones and evaluate the frequency of their appearance. To compare the amounts of Sup35NM for different constructions, other aliquots were taken at the same time points. Cell lysates were obtained with alkaline lysis (Zhang et al., 2011) and subsequently analyzed with SDS-PAGE (Sambrook and Fritsch, 1989). The same cells were used for fluorescence microscopy.
2.5. Decoration of Sup35 Aggregates in vivo
Two combinations of isogenic strains were used in this experiment: P-74-D694 and 74-D694, or 10-7A-D832 and 7A-D832. The first pair (P-74-D694 and 74-D694) was transformed with plasmids for production of Sup35NM fused with different fluorescent proteins (Sup35NM-TagRFP-T and Sup35NM-GFP with substitutions) and corresponding control constructs (TagRFP-T and GFP). For the TagRFP-T production, cells with plasmid pIM35 were grown overnight in the liquid media lacking methionine. For overproduction of the other constructs with fluorescent proteins, CuSO4 was added to a final concentration of 50 μM. The second pair of strains (10-7A-D832 and 7A-D832) was transformed with plasmids for production of Sup35NM-GFP, Sup35NM-M0-GFP or GFP. Overproduction of these proteins was induced by addition of CuSO4 to a final concentration of 100 μM. In all cases, the induction time was 4 h.
2.6. Propagon Counts
The transformants of 10-7A-D832 with pRS316CUP-NM-GFP and pRS316CUP-NM-M0-GFP were used for the propagon counts. The cells were grown in liquid SC-Ura medium with additional adenine to the early logarithmic phase (OD600 = 0.2). Then CuSO4 was added to a final concentration of 25 μM. Cells were plated on YEPD supplemented with 3 mM GuHCl to obtain single colonies before the addition of CuSO4 and after one cell culture division (estimated by OD600). The number of propagons in cells was determined using a previously described colony-based method (Cox et al., 2003).
2.7. Fluorescence Microscopy
Cells were gently pelleted (2000-3000 rpm) and resuspended in 50% glycerol. Fluorescence was analyzed using a Zeiss AxioScope.A1 wide-field fluorescence microscope. Images were taken with a QIClick-F-CLR-12 (QImaging) camera using QCAPTURE PRO 7 software.
2.8. Protein Analysis
The amount of Sup35 in different strains was quantified using Western Blotting with rabbit polyclonal anti-Sup35 antibodies (Chabelskaya et al., 2004). Monoclonal anti-tubulin antibodies (T6074, Sigma) were used for tubulin detection. Densitometry measurements were performed in ImageJ software (Schneider et al., 2012). SDS-PAGE with additional boiling (Kushnirov et al., 2006) was performed to detect Sup35NM-GFP and Sup35 in the aggregated and soluble fractions. For the analysis of Sup35 amyloid aggregates, SDD-AGE was used (Kryndushkin et al., 2003).
2.9. Protein Purification From Escherichia coli and Fibril Preparation
For Sup35NM purification, pET-20b-SUP35NM-His6 (Allen et al., 2005) plasmid or its derivative for Sup35NM-M0 overproduction were used. For protein purification, E. coli strain BL21(DE3) was used (Studier and Moffattf, 1986). Overproduction of recombinant proteins was carried out in 2TYa media with 1 mM IPTG. Cultures were grown at 37°C for 6 h. Proteins were purified in denaturing conditions (in the presence of 8 M urea) according to previously published protocols (Glover et al., 1997; Serio et al., 1999). The purification was performed with a two-step procedure with Ni-NTA agarose (Invitrogen) and Q-sepharose (GE Healthcare) columns. Proteins were concentrated with a centrifuge concentrator with molecular weight cutoff of 30 kDa (Millipore).
The obtained Sup35NM proteins were diluted at least 100-fold into fibril assembly buffer (5 mM potassium phosphate pH 7.5, 150 mM NaCl) to a final protein concentration of 0.5 mg/ml. In these conditions, Sup35NM spontaneously forms aggregates. Samples were incubated at 26°C with slow overhead rotation (rotator Bio RS-24, Biosan). To monitor amyloid fibril formation, aliquots were removed every 12 h up to 24 h of incubation. The rate of aggregated protein was estimated by SDS-PAGE with boiled and unboiled samples.
2.10. TEM and AFM
For fibrils visualization, Jeol JEM-2100 transmission electron microscope and Bruker Nanoscope V atomic force microscope were used. The negative staining with a 1% aqueous solution of uranyl acetate was used for TEM measurements. Samples were prepared by applying 5 μl of the Sup35NM fibril solution with concentration 0.5 mg/ml on a substrate, followed by washing with distilled water and drying. The fibrils were immobilized on freshly-cleaved mica surface for AFM analysis and formvar coated copper grids for TEM measurements (Sokolov et al., 2018).
2.11. Statistical Analysis
To compare the protein amounts the Mann-Whitney U-test was used (Mann and Whitney, 1947). The Fisher's exact test (Fisher, 1935) was used to compare the proportion of cells with a particular phenotype. All statistical tests were performed in R (R Core Team, 2018).
3. Results
3.1. Design of a New sup35 Mutation
In the previous work, we constructed five mutant sup35 alleles, each of them leading to substitutions of two consecutive polar residues to charged ones (lysines) in the middle of one of the oligopeptide repeats (OR1 - OR5). Such mutations are incompatible with Sup35p aggregates with superpleated β-structure spanning the ORs with the respective mutations. These mutations were named sup35KK, and each was designated according to the number of ORs (from sup35-M1 to sup35-M5) (Bondarev et al., 2013). We introduced mutations in all previously known ORs of Sup35 (Kushnirov et al., 1988). However, using T-REKS program, we identified additional repeat upstream of the known ORs (28–40 aa) (Bondarev et al., 2013). To complete the set of sup35KK alleles, we substituted two residues in the middle of newly identified OR0 (Q33K/A34K) to lysines and designated this mutation sup35-M0. The potential effect of this mutation on the Sup35 aggregation was evaluated with ArchCandy program (Ahmed et al., 2015). Previously it was shown that this tool accurately predicts the impact of amino acid substitutions on aggregation properties of a protein (Ahmed et al., 2015; Bondarev et al., 2015; Roche et al., 2017). According to the analysis, sup35-M0 mutation could significantly decrease the amyloidogenic potential of Sup35 and have the highest effect on this parameter compared to known PNM2 mutation (Doel et al., 1994) and other sup35KK, which were shown to eliminate the [PSI+] prion (Figure 1) (Bondarev et al., 2013).
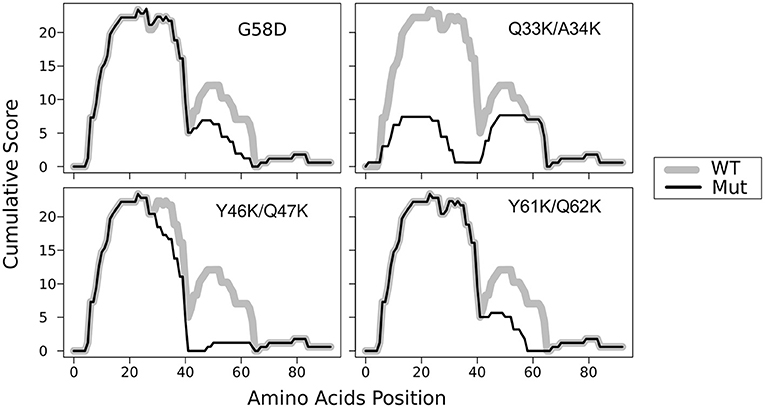
Figure 1. Substitutions Q33K/A34K within N-domain of Sup35 decrease the amyloidogenic potential of the protein. The ArchCandy program (Ahmed et al., 2015) was used to predict amyloidogenic properties. Cumulative scores (sum of β-arch scores counted for each amino acid residue) are presented on the plot. WT — wild-type protein; G58D, Q33K/A34K, Y46K/Q47K, and Q61K/Q62K substitutions that correspond to mutations PNM2, sup35-M0, -M1, and -M2, respectively.
3.2. The sup35-M0 Mutation Efficiently Eliminates the [PSI+] Prion
To analyze the effect of the mutation, we used previously described isogenic [PSI+] and [psi–] strains with SUP35 deletion compensated by a copy of this gene on a URA3 plasmid (Bondarev et al., 2013). In this system, we can change the alleles of SUP35 by plasmid shuffling. The presence of the nonsense mutation ade1-14 in these strains allows monitoring the prion propagation by the cell phenotype. As [PSI+] strains are able to suppress ade1-14 mutation, we test for the prion loss by detecting the decrease in growth on medium lacking adenine accompanied by increased accumulation of the red pigment on 1/4 YEPD. To check the effect of sup35-M0, we transformed isogenic [PSI+] and [psi–] strains with a plasmid bearing sup35-M0 or SUP35 (control). All independent [PSI+] transformants bearing the mutant allele demonstrated a significant decrease in nonsense suppression phenotype on 1/4 YEPD (Figure 2A) or SC media without adenine (data not shown). The complete elimination of prion phenotype was observed after loss of the wild-type SUP35 allele (Figure 2A). Yeast cells did not restore nonsense suppressor phenotype after replacement of a mutant allele by the wild-type (Figure 2A) suggesting that sup35-M0 mutation leads to the [PSI+] prion loss.
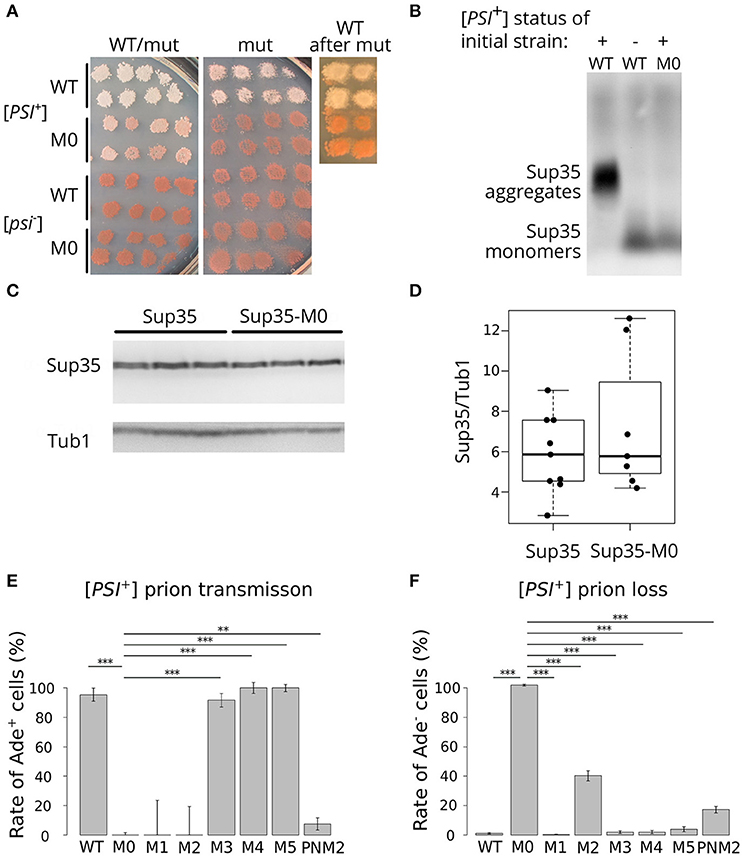
Figure 2. sup35-M0 efficiently and irreversibly eliminates [PSI+] prion. (A) The phenotype of strains with different combinations of SUP35 and sup35-M0 alleles in [PSI+] and [psi–] strains on 1/4 YEPD is shown (images were taken after 4 days of incubation). Transformants bearing two plasmids with two wild-type alleles or combination of sup35-M0 and SUP35 are presented on the panel “WT/mut” (at least 16 transformants were analyzed). The phenotype of cells after the plasmid loss is shown on panel “mut”. Finally, sup35-M0 (or SUP35 as a control) were replaced with SUP35 by the reverse plasmid shuffling, phenotype of obtained strains is presented on panel “WT after mut.” (B) The sup35-M0 allele leads to the elimination of Sup35 aggregates according to SDD-AGE results. Antibodies against Sup35 were used for Western Blotting. (C) Result of Western Blot hybridization after SDS-PAGE analysis of protein lysates from the [psi–] strain with mutant or wild-type allele of SUP35 with anti-Sup35 and anti-Tub1 antibodies. (D) The densitometry analysis of Sup35 protein level (ten replicates) revealed no difference in Sup35 protein level in strains with sup35-M0 compared to SUP35. (E) [PSI+] transmission from the wild-type to the indicated sup35 allele. Fraction of cells that retained the prion after loss of the wild-type allele is shown on graph. (F) [PSI+] loss induced by transient expression of the sup35KK alleles and PNM2 mutation. Fraction of cells that have lost the prion after the loss of sup35KK allele is shown. **p-value < 0.01 and ***p-value < 0.001 according to Fisher's exact test.
Elimination of the prion should be accompanied by the elimination of Sup35 aggregates from the cells. We checked for the disappearance of aggregates in the transformants, which lost the wild-type SUP35, with SDD-AGE (Kryndushkin et al., 2003) and did not find Sup35 aggregates in cells bearing only sup35-M0 (Figure 2B). This fact confirmed our assumption that the mutation eliminates the prion. We also compared the relative amount of the Sup35 protein for strains with the wild-type and the mutant SUP35 allele and found no difference (Figures 2C,D), suggesting that the prion loss was not caused by a decreased amount of Sup35.
To compare the effects of the new mutation on the prion replication, we estimated the prion loss and transmission in the presence of sup35-M0 allele according to special protocols for each parameter [(Afanasieva et al., 2011), see Materials and Methods section for details]. We did not observe cases of prion transmission to the sup35-M0 allele (Figure 2E). At the same time, the rate of [PSI+] loss was 97.12 ± 0.55% that significantly exceeds the same parameter for other sup35KK mutations with a maximum value of ~40% in case of substitutions within the second OR (Figure 2F) (Bondarev et al., 2013). Based on this data we concluded that new mutation very efficiently eliminates the [PSI+] prion.
3.3. The Sup35NM-M0 Protein Forms Infectious Amyloid Aggregates
The highly efficient loss of the prion caused by sup35-M0 suggested that Sup35 with amino acid changes Q33K/A34K might be unable to form aggregates and induce [PSI+] formation. To check this hypothesis, we constructed plasmids for purification of the Sup35NM-M0 protein from E. coli cells. We chose only the N-terminal part of the protein because it is sufficient for aggregation (Glover et al., 1997). Wild-type Sup35NM protein was used as a control. After 24 h of incubation in nondenaturing conditions, both proteins formed SDS-resistant aggregates, detected by comparing the amount of the proteins in boiled and unboiled samples analyzed with SDS-PAGE (Figure 3A). Using atomic force microscopy (AFM) and transmission electron microscopy (TEM) we investigated the morphology of Sup35NM and Sup35-M0 aggregates and found no detectable difference between them (Figure 3B). To test infectious properties of the obtained fibrils, we used the protein transformation technique (Tanaka and Weissman, 2006) and demonstrated that aggregates of both proteins were infectious and led to [PSI+] appearance (Figure 3C). In all cases, the observed nonsense suppressor phenotype was prion-mediated because it was lost after cell growth on the media with GuHCl (data not shown), which is known to cause the loss of [PSI+] (Tuite et al., 1981). At the same time, cells transformed with monomeric protein did not acquire prion phenotype. Thus, charged residues within OR0 in Sup35 do not significantly change its ability to form infectious aggregates in vitro.
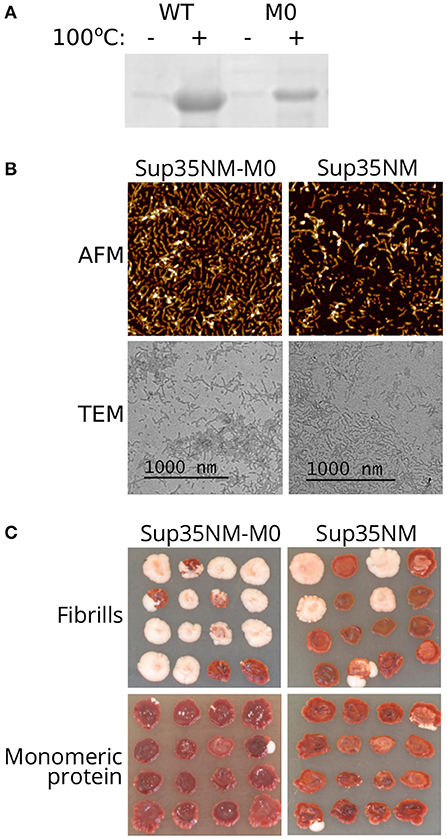
Figure 3. Sup35NM-M0 forms infectious amyloid aggregates. (A) Result of Coomassie staining of the gel after SDS-PAGE analysis of Sup35 fibrils formed in vitro. Sup35-M0 forms SDS-resistant aggregates similar to wild-type Sup35. (B) AFM and TEM images of fibrils formed by Sup35NM or Sup35NM-M0. (C) Phenotype of [psi–][pin–] strain (2-OT56) transformed with fibrillar or monomeric proteins on 1/4 YEPD (16 independent transformants are shown for each case, images were taken after 5 days of incubation). The appearance of [PSI+] phenotype (white color) after protein transformation suggested infectious properties of the aggregates.
3.4. The sup35-M0 Allele Can Induce and Propagate the [PSI+] Prion but With Low Efficiency
Next, we analyzed the effect of sup35-M0 allele on the prion induction in vivo. The presence of another prion [PIN+] is required for [PSI+] de novo formation in yeast cells For this experiment [psi–][PIN+] strains (derivatives of 7A-D832 or 12-D1682 bearing SUP35 or sup35-M0) were transformed with plasmids for Sup35NM-GFP (positive control), Sup35NM-M0-GFP or GFP (negative control) overproduction. The presence of sup35-M0 in cells significantly decreased the frequency of [PSI+] formation in different yeast strains. Furthermore, the overproduction of Sup35NM-M0-GFP induced [PSI+] with very low efficiency (Figures 4A,B). This result allowed us to conclude that investigated substitutions significantly decrease the aggregation propensity of the protein, as was predicted by the ArchCandy program (Figure 1). We also compared patterns of Sup35NM-GFP and Sup35NM-M0-GFP fluorescence upon their overproduction. In all cases, we found that Sup35 aggregates formed during [PSI+] induction: rings, ribbons, or dots (Figure 4C), which are detected on the different stages of the prion life cycle (Tyedmers, 2012).
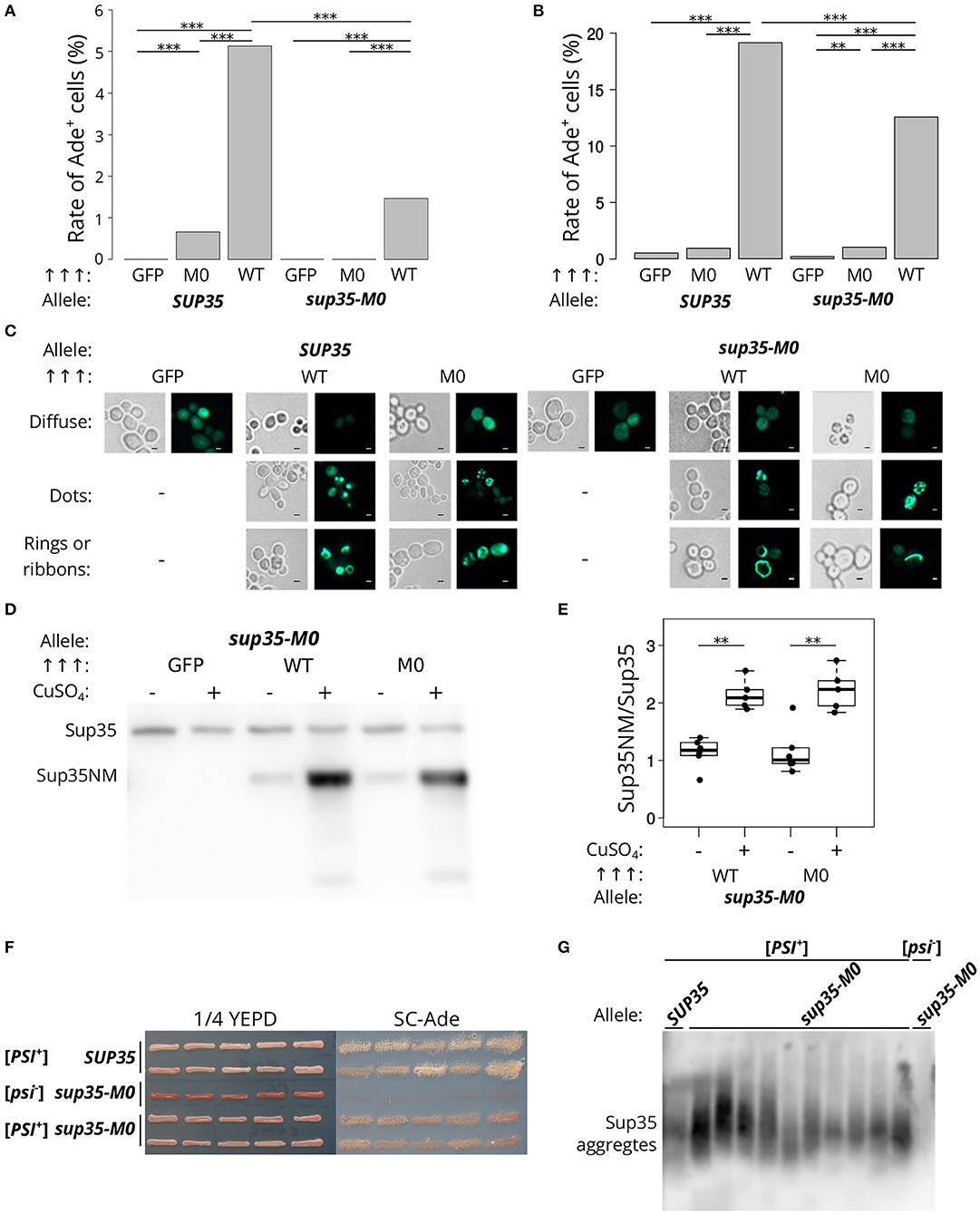
Figure 4. sup35-M0 can induce and propagate [PSI+] prion but with low efficiency. (A) Frequencies of [PSI+] induction in the [psi–][PIN+] 7A-D832 (A) and 12-D1682 (B) cells upon overexpression of SUP35NM-GFP (WT) or sup35NM-M0-GFP (M0) in the presence of wild-type or mutant allele of SUP35. Overproduction of GFP hereafter was used as a negative control. All constructions were under control of CUP1 promoter, CuSO4 was used for the 24 h induction. All experiments were repeated six times. Our results demonstrated that the mutation has a dramatically lower potential to induce [PSI+] prion than SUP35 (***p-value < 0.001 according to Fisher's exact test). The “Allele” designates allele of full-length SUP35 present in cells. (C) The cells tested on panel A were analyzed with the fluorescence microscopy (scale bar equals 5 μm). Various types of prion aggregates (dots, rings, and ribbons) were detected in the presence of both alleles (SUP35 and sup35-M0). (D) Results of Western Blot hybridization after SDS-PAGE analysis of protein lysates of strains used for [PSI+] induction. (E) Densitometry analysis of the Western Blotting. The level of N-terminal domain of Sup35 fused to GFP was normalized to the full-length Sup35-M0 which is unchanged in cells with wild-type and mutant sup35 allele according to the results presented on the Figure 2 (**p-value < 0.01 according to Mann-Whitney U-test). (F) The nonsense suppressor phenotype of several [PSI+] variants induced in presence of SUP35 or sup35-M0 in the 12-D1682 strain. Ten independent isolates are shown for each case. Cells were grown for 4 days on 1/4 YEPD and 5 days on SC-Ade. (G) The results of SDD-AGE analysis of protein lysates of typical [PSI+] variants induced in the presence of sup35-M0, antibodies against Sup35 were used for Western Blotting.
The low frequency of the prion induction upon sup35NM-M0 overexpression may also be explained by the effect of the mutation on the protein stability. However, the levels of corresponding proteins upon their overproduction are the same (Figures 4D,E), which contradicts this hypothesis.
To check that the cells with nonsense suppressor phenotype and bearing sup35-M0 were [PSI+] we isolated several corresponding clones of 12-D1682. The suppressor phenotype of these clones was preserved after several passages (Figure 4F) and in the absence of the plasmid, used for [PSI+] induction, but eliminated after the growth on GuHCl containing media (data not shown). We also found aggregates of Sup35 in all analyzed strains with prion variants (Figure 4G). These results prove the ability of sup35-M0 to maintain the [PSI+] prion. Nevertheless, it should be mentioned that all [PSI+] variants formed in presence of sup35-M0 had weak suppressor phenotype (Figure 4F).
3.5. The Sup35-M0 Protein Can Incorporate Into Fibrils of the Wild-Type Protein in vivo
We analyzed the ability of the protein with Q33K/A34K substitutions to incorporate into pre-existing Sup35 aggregates in vivo. Transient overproduction of Sup35NM fused with fluorescent protein leads to the decoration of existing Sup35 aggregates and formation of detectable fluorescent foci in cells (Osherovich et al., 2004). [PSI+][PIN+] and [psi–][PIN+] yeast strains (P-74-D694 and 74-D694, respectively) were transformed with the plasmids for overproduction of Sup35NM fused to a red fluorescent protein, TagRFP-T, in combination with either Sup35NM-GFP, or Sup35NM-M0-GFP, and analyzed with fluorescence microscopy. This experiment showed that the aggregates of Sup35NM and Sup35NM-M0 decorate [PSI+] aggregates (Figure 5A). One possible explanation of these results is that independently formed Sup35-M0 fibrils might co-localize with the wild-type fibrils, however, colocalization of the fibrils forming de novo in [psi–] strains seems less likely than co-aggregation of the wild-type and mutant proteins. Then we rechecked the ability of Sup35-M0 to embed into aggregates of the prion variant used in experiments with plasmid shuffle. The isogenic [PSI+][PIN+] and [psi–][PIN+] strains (10-7A-D832 and 7A-D832, respectively) were analyzed for the aggregate formation of Sup35NM-GFP or Sup35NM-M0-GFP. We detected fluorescent foci for both proteins only in [PSI+] strain (Figure 5B), which suggested the inclusion of the proteins into pre-existing aggregates, rather than de novo aggregation of the overproduced proteins. Then the incorporation of Sup35NM-M0 into amyloid aggregates was analyzed with SDS-PAGE with modifications, which allowed to evaluate the distribution of the protein with substitutions between fractions of detergent-resistant aggregates and monomers (Figure 5C). The results clearly demonstrated that Sup35NM-M0 was converted into amyloid-like conformation in the investigated [PSI+] strain. Taken together these data demonstrated the ability of the Sup35-M0 to incorporate into various Sup35 aggregates in vivo.
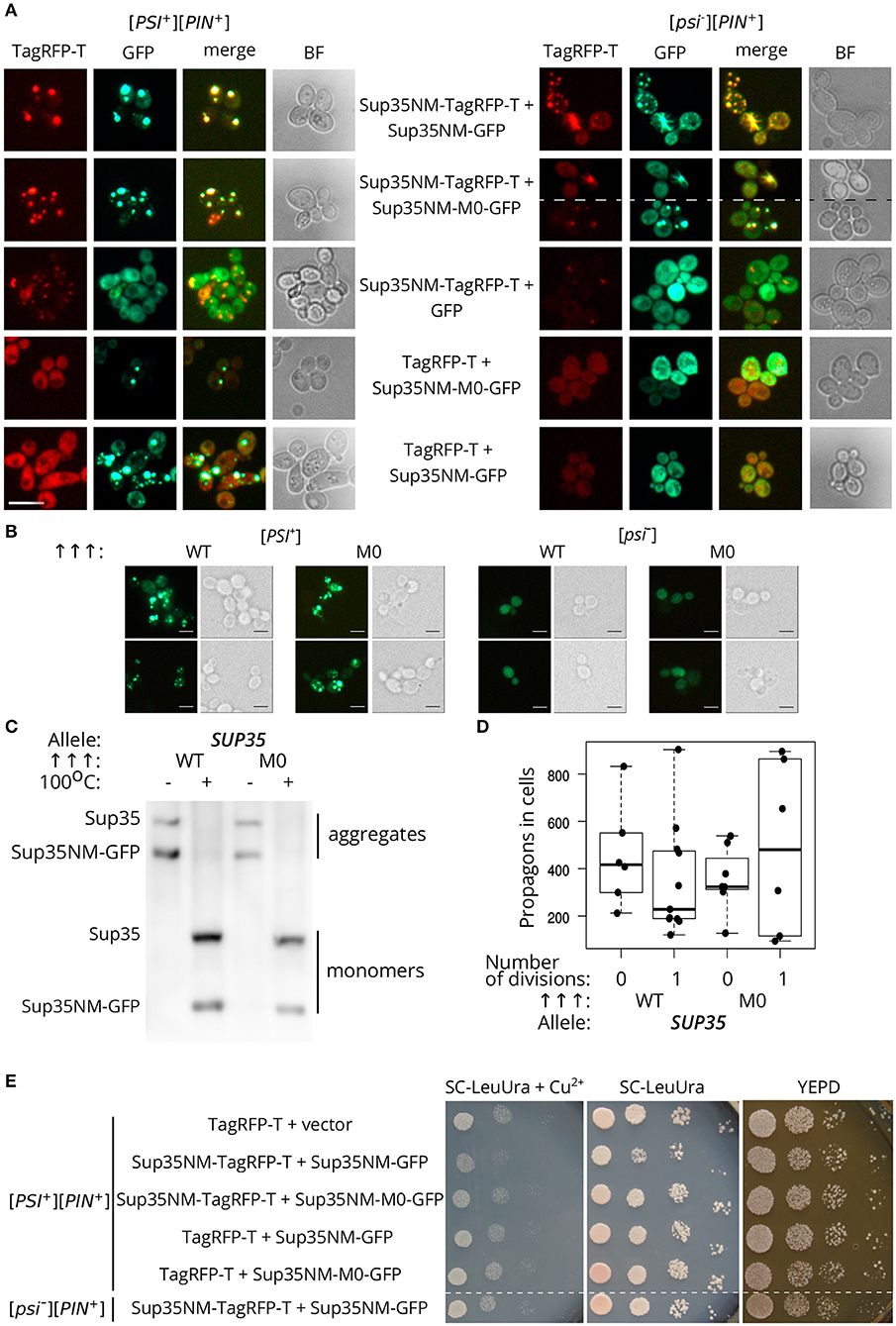
Figure 5. The Sup35-M0 protein can incorporate into fibrils of wild-type protein in vivo. (A) [psi–][PIN+] (74-D694) and [PSI+][PIN+] (P-74-D694) yeast strains were transformed with the plasmids for overproduction of Sup35NM-yTagRFP-T, in combination with either Sup35NM-GFP or Sup35NM-M0-GFP. We observed that the aggregates of Sup35NM and Sup35NM-M0 colocalize in [PSI+], as well as in [psi–] cells (scale bar equals 10 μm). (B) The transformants of [psi–][PIN+] (7A-D832) and [PSI+][PIN+] (10-7A-D832) with overproduced Sup35NM-GFP (WT), or Sup35NM-M0-GFP (M0) were analyzed with fluorescence microscopy (scale bar equals 5 μm). We detected foci of both proteins only in [PSI+], but not in [psi–], strain, which indicates inclusion of the proteins into existing aggregates. (C) The result of SDS-PAGE with boiled gel for strains from the panel B was shown. The “Allele” designates allele of full-length SUP35 present in cells. The Sup35NM-GFP (WT) and Sup35NM-M0-GFP (M0) proteins can incorporate into the existing prion aggregates upon transient overproduction in [PSI+] strain (both are detected in a fraction of aggregates). (D) Production of Sup35NM-M0-GFP does not affect the number of propagons. The cells from the panel B were used to calculate number of propagons before and after mild overproduction of Sup35NM-GFP or Sup35NM-M0-GFP; 25 μM CuSO4 was used for the induction. (E) Cells with overproduction of Sup35NM-TagRFP-T together with Sup35-NM-M0-GFP or Sup35NM-GFP were analyzed for prion toxicity. Cells were plated in 10-fold serial dilutions and grown for 2 days on SC-LeuUra + Cu2+ or YEPD and 4 days on SC-UraLeu. TagRFP-T production was used as a control; vector — pRS315.
Incorporation of Sup35-M0 into the existing prion aggregates may have different effects. Previously we proposed that the analogous mutation in the second OR (sup35-M2) leads to formation of non-heritable fold and as a result to the prion loss (Bondarev et al., 2013). The main mechanism responsible for the prion transmission is a fragmentation of the prion aggregates by chaperones (Liebman and Chernoff, 2012). An impairment of this process should lead to the decrease in number of prion “seeds,” called propagons, and affects the transmission of the prion upon cell division. We analyzed the effect of Sup35NM-M0-GFP production on the propagon number after one generation and found no significant differences compared to Sup35NM-GFP (Figure 5D). Thus, we considered that sup35-M0 has negligible effect on the aggregate fragmentation.
Finally, using phenotypic assay, we investigated the influence of the Sup35NM-M0 incorporation into wild-type Sup35 aggregates on the [PSI+] prion properties. One of the effects of increased Sup35 aggregation in [PSI+] cells is a reduction in cell viability as overproduction of Sup35NM in [PSI+] strains may lead to increased prion-dependent lethality (Derkatch, 1998; Vishveshwara et al., 2009). We checked whether Sup35NM-M0 retained the toxic properties of the wild-type protein. In contrast to Sup35NM-GFP, overexpression of Sup35NM-M0-GFP did not lead to a decrease in cell viability (Figure 5E). Overall, our data imply that the mutant Sup35 is able to co-aggregate with the wild-type protein, but their coaggregation may destabilize prion propagation of the native protein.
3.6. The Effect of sup35-M0 Mutation Is Variant-Unspecific
The effect of sup35 mutations on [PSI+] usually depends on the prion variant (Derkatch et al., 1999; King, 2001). We checked whether the effects of sup35-M0 are variant-specific. We obtained seven new [PSI+] prion variants in the strain with a single copy of SUP35 on the plasmid (see Materials and Methods section for details). These strains had different strengths of the nonsense suppressor phenotype on medium lacking adenine, and differed in size of Sup35 aggregates (Figures 6A,B). The replacement of SUP35 by sup35-M0 in these strains led to the loss of nonsense suppressor phenotype (Figure 6C) and Sup35 aggregates (verified for two investigated strains with SDD-AGE, data not shown). This allowed us to conclude that the sup35-M0 allele eliminates [PSI+] independently of the prion variant.

Figure 6. The sup35-M0 mutation destabilizes different [PSI+] variants. (A) The suppressor phenotype of the obtained [PSI+] variants (designated by numbers 1-7). [PSI+] designates the 10-7A-D832 strain. Cells were plated in 10-fold serial dilutions and grown for 3 days on YEPD and 5 days on SC-Ade. (B) The comparison of Sup35 aggregate size in strains from the panel A. [psi–] is the 12-D1682 strain. (C) The suppressor phenotype of the same strains after the replacement of SUP35 by sup35-M0. Cells were grown for 3 days on YEPD or 1/4 YEPD and 7 days on SC-Ade.
4. Discussion
The N-terminal domain of Sup35 is traditionally subdivided onto QN-rich (1–39 aa) and oligopeptide repeats regions (40–112 aa) (Kushnirov et al., 1988). The minimal region essential for [PSI+] propagation was assumed to comprise the first 57 residues, i.e., all QN region and two first ORs (Osherovich et al., 2004; Shkundina et al., 2006). In this work, we described the effects of substitutions (Q33K/A34K) within previously uncharacterized oligopeptide repeat of Sup35 on the [PSI+] prion propagation. We designated this new mutation as sup35-M0 and showed that it is able to eliminate [PSI+]. This fact is in good agreement with position of the mutation, as well as the fact, that the majority of known PNM mutations is located within the region which was shown to be important for [PSI+] maintenance (DePace et al., 1998; King, 2001).
The sup35KK mutations, as it was shown previously, may have different effects on [PSI+] propagation (Bondarev et al., 2013). Taken together with current results, we can conclude that proteins with corresponding substitutions within OR0–OR2 eliminate the prion, but can incorporate into pre-existing prion aggregates, induce [PSI+] appearance and form amyloid aggregates in vitro (Bondarev et al., 2013 and paper in prep). Despite these common features only sup35-M2 and the sup35-M0 eliminate the prion even in presence of the wild-type allele (Figure 2F). This allows us to speculate that the role of first OR in the prion propagation is different from OR0 or OR2.
The hallmark of sup35-M0 is the higher efficiency of [PSI+] elimination (97.12 ± 0.55%, Figure 2F) compared to the other previously characterized PNM mutations. For example, the prion loss in the presence of PNM2 or sup35-M2 mutations reachs 20 and 40%, respectively (Bondarev et al., 2013). The shuffle of SUP35 from S. cerevisiae to the homologs from other yeast species (S. paradoxus, S. bayanus, S. mikatae, S. kudriavzevii) may also lead to the [PSI+] elimination but with lower efficiency than sup35-M0 (Afanasieva et al., 2011). Furthermore, the prion elimination by the sup35-M0 mutation can destabilize different [PSI+] variants (Figure 6C), while the effects of all previously described PNM mutations were variant-specific (Derkatch et al., 1999; King, 2001). Another specific feature of sup35-M0 is a very low ability to induce and propagate the prion. Our results suggest that this mutation can maintain only limited number of weak prion variants (Figure 4F).
The strong effect of sup35-M0 was not linked to the stability of the protein as the relative amounts of wild-type and mutant proteins did not differ (Figures 2C,D). Also, the elimination of the prion in the presence of sup35-M0 could not be explained by the complete inability of the protein to propagate [PSI+]. Sup35NM-M0 can form infectious aggregates in vitro (Figure 3C), overproduction of Sup35NM-M0-GFP leads to the prion induction in vivo, and, finally, sup35-M0 can maintain the prion, though, with low efficiency (Figures 4A,B). The low prion induction rate upon overproduction of Sup35NM-M0-GFP and in the presence of sup35-M0 may be explained by the reduced ability of the soluble protein to aggregate with itself, as was demonstrated for Sup35-M1 and Sup35-M2 (Khan et al., 2018).
It is noteworthy that our experimental results once again illustrate the accuracy of ArchCandy prediction (Ahmed et al., 2015; Bondarev et al., 2015; Roche et al., 2017). We found that sup35-M0 significantly decreases the frequency of the prion induction de novo and has lower prionogenic potential in vivo (Figure 4A). These data are in a good agreement with the bioinformatics predictions of ArchCandy, according to which lysines in 33–34 aa positions significantly decrease the amyloidogenic potential of Sup35 protein (Figure 1).
Prion loss caused by a certain SUP35 allele may occur due to different mechanisms. In case of the interspecies barrier, three mechanisms were proposed: the inability of the heterologous protein to incorporate into prion aggregates, the block of aggregation by the protein and formation of the non-heritable fold of aggregates (Afanasieva et al., 2011). Detailed investigation of [PSI+] elimination caused by PNM2 revealed two potential processes that may explain nonheritable properties of aggregates: increased fragmentation leading to solubilization of aggregates and impairment of prion transmission to the daughter cell (DiSalvo et al., 2011; Verges et al., 2011; Pei et al., 2017). We found that Sup35NM-M0 can incorporate into pre-existing Sup35 aggregates in vivo (Figures 5B,C), but this does not affect the number of propagons (Figure 5D) and thus the fragmentation of aggregates. It seems that both mechanisms could not explain the effect of sup35-M0. We suppose that the prion loss caused by sup35-M0 is rather linked with the decreased aggregation propensity of the protein, followed by solubilization of aggregates by cellular chaperones. This hypothesis is in good agreement with very low [PSI+] induction rate in the presence of sup35-M0 (Figure 4A) and prediction of the ArchCandy (Figure 1). However, this disagrees with the high efficiency of co-aggregation of Sup35NM-M0 with Sup35 (Figure 5), but we suggest that differences in aggregation rate in this experiment may be hidden due to the overproduction of the protein.
In summary, here we described a new mutation in SUP35, which can efficiently eliminate [PSI+] factor in a variant-independent manner. The sup35-M0 possess very low amyloidogenic potential and can protect cells from the spontaneous appearance of the prion. We suggest that the investigated mutation may be widely used for fast and non-specific elimination of the [PSI+] prion or for design of yeast strains which almost never undergo transition to the [PSI+] state. Moreover, our discovery may serve as a proof of concept for the design of a prion-eliminating mutations using specific bioinformatic tools. In mammals at least two analogous mutations, eliminating the PrP prion in presence of the wild type allele, were described. Both of them lead to substitutions of polar residue to the charged one (Q167R or Q218K) Thus, our study supports the design of analogous mutations that could block propagation of mammalian prion and amyloid proteins and thus may be useful for amyloidosis therapy.
Data Availability Statement
The datasets generated for this study are available on request to the corresponding author.
Author Contributions
LD, AM, PS, and SB designed the experiments. LD, VR, AM, MB, OP, DL, PS, and SB performed the experiments. LD, VR, AM, PS, and SB prepared figures. LD, AM, and SB wrote original draft. LD, AM, MB, DL, PS, NK, AK, GZ, and SB performed review and editing the manuscript.
Funding
The authors acknowledge St. Petersburg State University for the research grant (id: 28229898, SB, the ArchCandy analysis). This work was also supported by RFBR grants (19-04-00173, GZ, the analysis of sup35-M0 effects on [PSI+] propagation; 17-54-150002, GZ, and PRC CNRS grant PRC1524, AK, the in vitro experiments; 18-34-00536, AM, the analysis of colocalization of Sup35NM-M0 with the wild-type protein in vivo) and RSF grant (18-14-00050, GZ, the analysis of sup35-M0 effect on the translation termination).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to Yury O. Chernoff and Ivana Malcova for plasmids and strains. We thank Ksenia V. Sukhanova, Anton B. Matiiv, Yury A. Barbitoff and Ekaterina Davydova for critical reading of the manuscript and suggestions. The work was supported by the RRC MCT SPbSU and we thank Maxim Vorobyov, Nikolai Kostin and Alexei Masharskiy from this facility for help with experiments.
References
Afanasieva, E. G., Kushnirov, V. V., Tuite, M. F., and Ter-Avanesyan, M. D. (2011). Molecular basis for transmission barrier and interference between closely related prion proteins in yeast. J. Biol. Chem. 286, 15773–15780. doi: 10.1074/jbc.M110.183889
Ahmed, A. B., Znassi, N., Château, M.-T., and Kajava, A. V. (2015). A structure-based approach to predict predisposition to amyloidosis. Alzheimers Dement. 11, 681–690. doi: 10.1016/j.jalz.2014.06.007
Allen, K. D., Wegrzyn, R. D., Chernova, T. A., Müller, S., Newnam, G. P., Winslett, P. A., et al. (2005). Hsp70 chaperones as modulators of prion life cycle: novel effects of Ssa and Ssb on the Saccharomyces cerevisiae prion [PSI+]. Genetics 169, 1227–1242. doi: 10.1534/genetics.104.037168
Baxa, U., Cassese, T., Kajava, A. V., and Steven, A. C. (2006). Structure, function, and amyloidogenesis of fungal prions: filament polymorphism and prion variants. Adv. Protein. Chem. 73, 125–180. doi: 10.1016/S0065-3233(06)73005-4
Bondarev, S. A., Shchepachev, V. V., Kajava, A. V., and Zhouravleva, G. A. (2013). Effect of charged residues in the N-domain of Sup35 protein on prion [PSI+] stability and propagation. J. Biol. Chem. 288, 28503–28513. doi: 10.1074/jbc.M113.471805
Bondarev, S. A., Zhouravleva, G. A., Belousov, M. V., and Kajava, A. V. (2015). Structure-based view on [PSI+] prion properties. Prion 9, 190–199. doi: 10.1080/19336896.2015.1044186
Chabelskaya, S., Kiktev, D., Inge-Vechtomov, S., Philippe, M., and Zhouravleva, G. (2004). Nonsense mutations in the essential gene SUP35 of Saccharomyces cerevisiae are non-lethal. Mol. Genet. Genomics 272, 297–307. doi: 10.1007/s00438-004-1053-1
Chernoff, Y. O., Newnam, G. P., Kumar, J., Allen, K., and Zink, A. D. (1999). Evidence for a protein mutator in yeast: role of the Hsp70-related chaperone ssb in formation, stability, and toxicity of the [PSI+] prion. Mol. Cell. Biol. 19, 8103–8112. doi: 10.1128/MCB.19.12.8103
Cox, B., Ness, F., and Tuite, M. (2003). Analysis of the generation and segregation of propagons: entities that propagate the [PSI+] prion in yeast. Genetics 165, 23–33.
DePace, A. H., Santoso, A., Hillner, P., and Weissman, J. S. (1998). A critical role for amino-terminal glutamine/asparagine repeats in the formation and propagation of a yeast prion. Cell 93, 1241–1252.
Derkatch, I. L., Bradley, M. E., and Liebman, S. W. (1998). Overexpression of the SUP45 gene encoding a Sup35p-binding protein inhibits the induction of the de novo appearance of the [PSI+] prion. Proc. Natl. Acad. Sci. U.S.A. 95, 2400–2405. doi: 10.1073/pnas.95.5.2400
Derkatch, I. L., Bradley, M. E., Zhou, P., Chernoff, Y. O., and Liebman, S. W. (1997). Genetic and environmental factors affecting the de novo appearance of the [PSI+] prion in Saccharomyces cerevisiae. Genetics 147, 507–519.
Derkatch, I. L., Bradley, M. E., Zhou, P., and Liebman, S. W. (1999). The PNM2 mutation in the prion protein domain of SUP35 has distinct effects on different variants of the [PSI+] prion in yeast. Curr. Genet. 35, 59–67.
DiSalvo, S., Derdowski, A., Pezza, J. A., and Serio, T. R. (2011). Dominant prion mutants induce curing through pathways that promote chaperone-mediated disaggregation. Nat. Struct. Mol. Biol. 18, 486–492. doi: 10.1038/nsmb.2031
Doel, S. M., McCready, S. J., Nierras, C. R., and Cox, B. S. (1994). The dominant PNM2- mutation which eliminates the [PSI+] factor of Saccharomyces cerevisiae is the result of a missense mutation in the SUP35 gene. Genetics 137, 659–670.
Drozdova, P. B., Tarasov, O. V., Matveenko, A. G., Radchenko, E. A., Sopova, J. V., Polev, D. E., et al. (2016). Genome sequencing and comparative analysis of Saccharomyces cerevisiae strains of the Peterhof genetic collection. PLoS ONE 11:e0154722. doi: 10.1371/journal.pone.0154722
Eaglestone, S. S., Ruddock, L. W., Cox, B. S., Tuite, M. F., and Lindquist, S. L. (2000). Guanidine hydrochloride blocks a critical step in the propagation of the prion-like determinant [PSI+] of Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A. 97:240–244. doi: 10.1073/pnas.97.1.240
Franzmann, T. M., Jahnel, M., Pozniakovsky, A., Mahamid, J., Holehouse, A. S., Nüske, E., et al. (2018). Phase separation of a yeast prion protein promotes cellular fitness. Science 359:eaao5654. doi: 10.1126/science.aao5654
Glover, J. R., Kowal, A. S., Schirmer, E. C., Patino, M. M., Liu, J. J., and Lindquist, S. (1997). Self-seeded fibers formed by Sup35, the protein determinant of [PSI+], a heritable prion-like factor of S. cerevisiae. Cell 89, 811–819.
Helsen, C. W., and Glover, J. R. (2012). A new perspective on hsp104-mediated propagation and curing of the yeast prion [PSI+]. Prion 6, 234–239. doi: 10.4161/pri.19913
Jorda, J., and Kajava, A. V. (2009). T-REKS: identification of tandem REpeats in sequences with a K-meanS based algorithm. Bioinformatics 25, 2632–2638. doi: 10.1093/bioinformatics/btp482
Kaiser, C., Michaelis, S., and Mitchell, A. (1994). Methods in Yeast Genetics. New York, NY: Cold Spring Harbour laboratory press.
Kajava, A. V., Baxa, U., Wickner, R. B., and Steven, A. C. (2004). A model for Ure2p prion filaments and other amyloids: the parallel superpleated β-structure. Proc. Natl. Acad. Sci. U.S.A. 101, 7885–7890. doi: 10.1073/pnas.0402427101
Khan, T., Kandola, T., Wu, J., Venkatesan, S., Ketter, E., Lange, J. J., et al. (2018). Quantifying nucleation in vivo reveals the physical basis of prion-like phase behavior. Mol. Cell. 71, 155–168. doi: 10.1016/j.molcel.2018.06.016
Kiktev, D. A., Melomed, M. M., Lu, C. D., Newnam, G. P., and Chernoff, Y. O. (2015). Feedback control of prion formation and propagation by the ribosome-associated chaperone complex. Mol. Microbiol. 96, 621–632. doi: 10.1111/mmi.12960
King, C. Y. (2001). Supporting the structural basis of prion strains: induction and identification of [PSI+] variants. J. Mol. Biol. 307, 1247–1260. doi: 10.1006/jmbi.2001.4542
Kryndushkin, D. S., Alexandrov, I. M., Ter-Avanesyan, M. D., and Kushnirov, V. V. (2003). Yeast [PSI+] prion aggregates are formed by small Sup35 polymers fragmented by Hsp104. J. Biol. Chem. 278, 49636–49643. doi: 10.1074/jbc.M307996200
Kushnirov, V. V., Alexandrov, I. M., Mitkevich, O. V., Shkundina, I. S., and Ter-Avanesyan, M. D. (2006). Purification and analysis of prion and amyloid aggregates. Methods 39, 50–55. doi: 10.1016/j.ymeth.2006.04.007
Kushnirov, V. V., Ter-Avanesyan, M. D., Telckov, M. V., Surguchov, A. P., Smirnov, V. N., and Inge-Vechtomov, S. G. (1988). Nucleotide sequence of the SUP2 (SUP35) gene of Saccharomyces cerevisiae. Gene 66, 45–54.
Langlois, C. R., Pei, F., Sindi, S. S., and Serio, T. R. (2016). Distinct prion domain sequences ensure efficient amyloid propagation by promoting chaperone binding or processing in vivo. PLoS Genet. 12:e1006417. doi: 10.1371/journal.pgen.1006417
Lee, C. I., Yang, Q., Perrier, V., and Baskakov, I. V. (2007). The dominant-negative effect of the Q218K variant of the prion protein does not require protein X. Protein Sci. 16, 2166–2173. doi: 10.1110/ps.072954607
Liebman, S. W., and Chernoff, Y. O. (2012). Prions in yeast. Genetics 191, 1041–1072. doi: 10.1534/genetics.111.137760
Liu, J. J., and Lindquist, S. (1999). Oligopeptide-repeat expansions modulate 'protein-only' inheritance in yeast. Nature 400, 573–576.
Liu, J. J., Sondheimer, N., and Lindquist, S. L. (2002). Changes in the middle region of Sup35 profoundly alter the nature of epigenetic inheritance for the yeast prion [PSI+]. Proc. Natl. Acad. Sci. U.S.A. 99(Suppl. 4), 16446–16453. doi: 10.1073/pnas.252652099
Malcova, I., Farkasovsky, M., Senohrabkova, L., Vasicova, P., and Hasek, J. (2016). New integrative modules for multicolor-protein labeling and live-cell imaging in Saccharomyces cerevisiae. FEMS Yeast Res. 16:fow027. doi: 10.1093/femsyr/fow027
Mann, H. B., and Whitney, D. R. (1947). On a test of whether one of two random variables is stochastically larger than the other. Ann. Math. Statis. 18, 50–60.
Matveenko, A. G., Drozdova, P. B., Belousov, M. V., Moskalenko, S. E., Bondarev, S. A., Barbitoff, Y. A., et al. (2016). SFP1-mediated prion-dependent lethality is caused by increased Sup35 aggregation and alleviated by Sis1. Genes Cells. 21, 1290–1308. doi: 10.1111/gtc.12444
Osherovich, L. Z., Cox, B. S., Tuite, M. F., and Weissman, J. S. (2004). Dissection and design of yeast prions. PLoS Biol. 2, 442–451. doi: 10.1371/journal.pbio.0020086
Pei, F., DiSalvo, S., Sindi, S. S., and Serio, T. R. (2017). A dominant-negative mutant inhibits multiple prion variants through a common mechanism. PLoS Genet. 13:e1007085. doi: 10.1371/journal.pgen.1007085
Perrier, V., Kaneko, K., Safar, J., Vergara, J., Tremblay, P., DeArmond, S. J., et al. (2002). Dominant-negative inhibition of prion replication in transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 99, 13079–13084. doi: 10.1073/pnas.182425299
Prusiner, S. B. (2013). Biology and genetics of prions causing neurodegeneration. Ann. Rev. Genet. 47, 601–623. doi: 10.1146/annurev-genet-110711-155524
R Core Team (2018). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Roche, D. B., Villain, E., and Kajava, A. V. (2017). Usage of a dataset of NMR resolved protein structures to test aggregation vs. solubility prediction algorithms. Protein Sci. 26, 1864–1869. doi: 10.1002/pro.3225
Sambrook, J., and Fritsch, E., M. T. (1989). Molecular Cloning: A Laboratory Manual, 2nd Edn. NY: Cold Spring Harbor Laboratory Press; Cold Spring Harbour.
Schneider, C. A., Rasband, W. S., and Eliceiri, K. W. (2012). NIH image to ImageJ : 25 years of image analysis. Nat. Methods 9, 671–675. doi: 10.1038/nmeth.2089
Serio, T. R., Cashikar, A. G., Moslehi, J. J., Kowal, A. S., and Lindquist, S. L. (1999). Yeast prion [PSI+] and its determinant, Sup35p. Method. Enzymol. 309, 649–673.
Shaner, N. C., Lin, M. Z., McKeown, M. R., Steinbach, P. A., Hazelwood, K. L., Davidson, M. W., and Tsien, R. Y. (2008). Improving the photostability of bright monomeric orange and red fluorescent proteins. Nat. Methods 5, 545–551. doi: 10.1038/nmeth.1209
Shkundina, I. S., Kushnirov, V. V., Tuite, M. F., and Ter-Avanesyan, M. D. (2006). The role of the N-terminal oligopeptide repeats of the yeast Sup35 prion protein in propagation and transmission of prion variants. Genetics 172, 827–835. doi: 10.1534/genetics.105.048660
Sikorski, R. S., and Hieter, P. (1989). A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122, 19–27.
Sokolov, P. A., Bondarev, S. A., Belousov, M. V., Zhouravleva, G. A., and Kasyanenko, N. A. (2018). Sup35NMp morphology evaluation on Au, Si, formvar and mica surfaces using AFM, SEM and TEM. J. Struct. Biol. 201, 5–14. doi: 10.1016/j.jsb.2017.10.006
Stansfield, I., Jones, K. M., Kushnirov, V. V., Dagkesamanskayal, A. R., Poznyakovski, A., Paushkin, S. V., et al. (1995). The products of the SUP45 (eRF1) and SUP35 genes interact to mediate translation termination in a Saccharomyces cerevisiae. EMBO J. 14, 4365–4373.
Studier, F. W., and Moffattf, B. A. (1986). Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J. Mol. Biol. 189, 113–130.
Tanaka, M., and Weissman, J. S. (2006). An efficient protein transformation protocol for introducing prions into yeast. Method. Enzymol. 412, 185–200. doi: 10.1016/S0076-6879(06)12012-1
Ter-Avanesyan, M. D., Dagkesamanskaya, A. R., Kushnirov, V. V., and Smirnov, V. N. (1994). The SUP35 omnipotent suppressor gene is involved in the maintenance of the non-mendelian determinant [PSI+] in the yeast Saccharomyces cerevisiae. Genetics 137, 671–676.
Ter-Avanesyan, M. D., Kushnirov, V. V., Dagkesamanskaya, A. R., Didichenko, S. A., Chernoff, Y. O., Inge-Vechtomov, S. G., et al. (1993). Deletion analysis of the SUP35 gene of the yeast Saccharomyces cerevisiae reveals two non-overlapping functional regions in the encoded protein. Mol. Microbiol. 7, 683–692.
Tuite, M. F., Mundy, C. R., and Cox, B. S. (1981). Agents that cause a high frequency of genetic change from [PSI+] to [psi–] in Saccharomyces cerevisiae. Genetics 98, 691–711.
Tyedmers, J. (2012). Patterns of [PSI+] aggregation allow insights into cellular organization of yeast prion aggregates. Prion 6, 191–200. doi: 10.4161/pri.18986
Verges, K. J., Smith, M. H., Toyama, B. H., and Weissman, J. S. (2011). Strain conformation, primary structure and the propagation of the yeast prion [PSI+]. Nat. Struct. Mol. Biol. 18, 493–499. doi: 10.1038/nsmb.2030
Vishveshwara, N., Bradley, M. E., and Liebman, S. W. (2009). Sequestration of essential proteins causes prion associated toxicity in yeast. Mol. Microbiol. 73, 1101–1114. doi: 10.1111/j.1365-2958.2009.06836.x
Volkov, K. V., Aksenova, A. Y., Soom, M. J., Osipov, K. V., Svitin, A. V., Kurischko, C., et al. (2002). Novel non-mendelian determinant involved in the control of translation accuracy in Saccharomyces cerevisiae. Genetics 160, 25–36.
Wickner, R. (1994). [URE3] as an altered URE2 protein: evidence for a prion analog in Saccharomyces cerevisiae. Science 264, 566–569.
Wickner, R. B., Masison, D. C., and Edskes, H. K. (1995). [PSI+] and [URE3] as yeast prions. Yeast 11, 1671–1685.
Zhang, T., Lei, J., Yang, H., Xu, K., Wang, R., and Zhang, Z. (2011). An improved method for whole protein extraction from yeast Saccharomyces cerevisiae. Yeast 28, 795–798. doi: 10.1002/yea.1905
Zhouravleva, G., Frolova, L., Le Goff, X., Le Guellec, R., Inge-Vechtomov, S., Kisselev, L., et al. (1995). Termination of translation in eukaryotes is governed by two interacting polypeptide chain release factors, eRF1 and eRF3. EMBO J. 14, 4065–4072.
Keywords: [PSI+], amyloid, ArchCandy, prion, Saccharomyces cerevisiae, SUP35 mutation, superpleated-β-structure
Citation: Danilov LG, Matveenko AG, Ryzhkova VE, Belousov MV, Poleshchuk OI, Likholetova DV, Sokolov PA, Kasyanenko NA, Kajava AV, Zhouravleva GA and Bondarev SA (2019) Design of a New [PSI+]-No-More Mutation in SUP35 With Strong Inhibitory Effect on the [PSI+] Prion Propagation. Front. Mol. Neurosci. 12:274. doi: 10.3389/fnmol.2019.00274
Received: 30 August 2019; Accepted: 28 October 2019;
Published: 19 November 2019.
Edited by:
Maria Rosário Almeida, University of Porto, PortugalReviewed by:
Heinrich J. G. Matthies, University of Alabama at Birmingham, United StatesIlia V. Baskakov, University of Maryland, Baltimore, United States
Copyright © 2019 Danilov, Matveenko, Ryzhkova, Belousov, Poleshchuk, Likholetova, Sokolov, Kasyanenko, Kajava, Zhouravleva and Bondarev. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Galina A. Zhouravleva, emhvdXJhdmxldmFAcmFtYmxlci5ydQ==; Zy56aHVyYXZsZXZhQHNwYnUucnU=; Stanislav A. Bondarev, c3RhbmlzbGF2c3BiZ3VAZ21haWwuY29t; cy5ib25kYXJldkBzcGJ1LnJ1