 Wei Lei
Wei Lei David I. Duron1
David I. Duron1 Carrie Stine
Carrie Stine Brian S. J. Blagg
Brian S. J. Blagg John M. Streicher
John M. Streicher- 1Department of Pharmacology, College of Medicine, University of Arizona, Tucson, AZ, United States
- 2Department of Pharmaceutical and Administrative Sciences, School of Pharmacy, Presbyterian College, Clinton, SC, United States
- 3Department of Chemistry & Biochemistry, College of Science, University of Notre Dame, Notre Dame, IN, United States
Opioid activation of the mu opioid receptor (MOR) promotes signaling cascades that evoke both analgesic responses to pain and side effects like addiction and dependence. Manipulation of these cascades, such as by biased agonism, has great promise to improve opioid therapy. However, the signaling cascades of the MOR are in general poorly understood, providing few targets for drug development. In our earlier work, we identified Heat shock protein 90 (Hsp90) as a novel and crucial regulator of opioid anti-nociception in the brain by promoting ERK MAPK activation. In this study, we sought to identify the molecular isoforms and co-chaperones by which Hsp90 carried out this role, which could provide specific targets for future clinical intervention. We used novel selective small molecule inhibitors as well as CRISPR/Cas9 gene editing constructs delivered by the intracerebroventricular (icv) route to the brains of adult CD-1 mice to target Hsp90 isoforms (Hsp90α/β, Grp94) and co-chaperones (p23, Cdc37, Aha1). We found that inhibition of the isoform Hsp90α fully blocked morphine anti-nociception in a model of post-surgical paw incision pain, while blocking ERK and JNK MAPK activation, suggesting Hsp90α as the main regulator of opioid response in the brain. We further found that inhibition of the co-chaperones p23 and Cdc37 blocked morphine anti-nociception, suggesting that these co-chaperones assist Hsp90α in promoting opioid anti-nociception. Lastly, we used cycloheximide treatment in the brain to demonstrate that rapid protein translation within 30 min of opioid treatment is required for Hsp90 regulation of opioid response. Together these studies provide insight into the molecular mechanisms by which Hsp90 promotes opioid anti-nociception. These findings thus both improve our basic science knowledge of MOR signal transduction and could provide future targets for clinical intervention to improve opioid therapy.
Introduction
The mu opioid receptor (MOR) evokes complex signal transduction cascades upon activation by opioid ligands like morphine. For decades now it has been understood that the MOR represses cAMP production by inhibiting adenylyl cyclase via the GαI subunit, and activates G protein-coupled inwardly-rectifying potassium channels via the Gβ/γ subunit, which summate on neuronal hyperpolarization and subsequent inhibition of nociceptive inputs (Al-Hasani and Bruchas, 2011). However, it is clear that signaling regulators beyond this simple cascade have a strong impact on opioid anti-nociception and side effects, including other G proteins, ERK MAPK (Macey et al., 2009), Src (Zhang et al., 2017), CaMKII (Li et al., 2016), RSK2 (Darcq et al., 2012), and others. These signaling regulators could provide important targets for opioid drug development; for instance βarrestin2 was shown to reduce opioid anti-nociception while promoting side effects like tolerance and dependence, leading to the development of βarrestin2 biased agonists with reduced side effects (Bohn et al., 1999; Raehal et al., 2005; Dewire et al., 2013; Manglik et al., 2016; Schmid et al., 2017). However, in general, the mechanisms by which these signaling regulators impact opioid physiology are not known, and very few targets like βarrestin2 have been validated for drug development (Al-Hasani and Bruchas, 2011; Olson et al., 2017). This gap illustrates the need for investigation into the signalosome of the MOR and the mechanisms by which these regulators impact opioid physiology.
To this end, in our earlier work, we identified the central signaling regulator Heat shock protein 90 (Hsp90) as a novel and crucial regulator of opioid signaling in the brain, that promoted opioid anti-nociception by promoting ERK MAPK activation (Lei et al., 2017). Hsp90 is a major regulator of protein folding via chaperone activity in concert with other Hsps like Hsp70 (Li and Buchner, 2013). However, Hsp90 also has a major role in signal transduction by regulating signaling molecule localization, complex/scaffold formation, and acute signaling activation (Streicher, 2019). Despite the importance of Hsp90 in regulating signaling, only two previous studies directly linked Hsp90 to opioid signaling. An in vitro study found that Hsp90 inhibition decreased cAMP superactivation, a marker for opioid dependence (Koshimizu et al., 2010); supporting these findings, an in vivo mouse study found that injection of Hsp90 inhibitor reduced the somatic signs of morphine withdrawal (Abul-Husn et al., 2011). Our study was thus the first to directly link Hsp90 regulation of MOR signaling to opioid anti-nociception.
Our study did show that Hsp90 inhibition very strongly decreased morphine anti-nociception in models of acute and chronic pain, and identified a signaling mechanism via ERK MAPK (Lei et al., 2017). However, this study only took the first small step in identifying the role of Hsp90 in regulating opioid signaling. We used the ATP-pocket inhibitor 17-AAG, which is non-selective between the four Hsp90 isoforms (Hsp90α/β, Grp94, TRAP1). These isoforms differ in their subcellular localization and protein targets, with Hsp90α/β localized to the cytoplasm, Grp94 to the endoplasmic reticulum, and TRAP1 to the mitochondria (Liu et al., 2015; Kim et al., 2016; Mishra et al., 2017). We also did not identify any of the crucial co-chaperones, which mediate and target the specific activity of Hsp90 in different cells and tissues (Li and Buchner, 2013). Co-chaperones have specific roles, like Cdc37 having a key role in signaling kinase targeting, suggesting their possible involvement in MOR signaling (Hinz et al., 2007). Identifying the isoforms and co-chaperones involved in Hsp90 regulation of opioid signaling will thus reveal key details of the molecular mechanism by which Hsp90 promotes anti-nociception. Identifying these refined molecular targets could also provide more selective targets for clinical intervention, which has been done in an analogous way for Hsp70 (Assimon et al., 2013, 2015).
In this study, we thus sought to identify specific Hsp90 isoforms and co-chaperones responsible for the promotion of opioid anti-nociception by Hsp90. We utilized novel selective inhibitors and in vivo CRISPR/Cas9 gene editing in the brains of adult CD-1 mice to test Hsp90 isoforms (Hsp90α/β, Grp94) and co-chaperones (p23, Cdc37, Aha1). Through these studies, we found that the isoform Hsp90α and the co-chaperones p23 and Cdc37 strongly promoted MOR signaling and opioid anti-nociception in the brain. These findings expand our knowledge of the specific molecular mechanisms by which Hsp90 regulates opioid anti-nociception, and could provide more selective targets for clinical intervention.
Materials and Methods
Drugs
KUNA115 (Mishra et al., under review), KUNB106 (Mishra et al., in press), KUNG65 (compound 30 in Crowley et al., 2017), KU-32 (compound A4 in Ansar et al., 2007), and KU177 (compound 12c in Zhao et al., 2011) were synthesized by the Blagg laboratory using the cited protocols. The identity of the ligands was confirmed by high resolution mass spectrometry and nuclear magnetic resonance, while the purity of the compounds was confirmed to >95% by high performance liquid chromatography. Gedunin (#33-871-0), Celastrol (#32-031-0), 17-AAG (#AAJ66960MC), and Cycloheximide (#AC357420010) were obtained from Fisher Scientific. DAMGO (#1171) was obtained from Tocris/R&D. Morphine sulfate pentahydrate was obtained from the NIDA Drug Supply Program. All compounds except for DAMGO and morphine were prepared as DMSO stock solutions and diluted into a vehicle solution prior to injection. DAMGO was prepared in a stock solution of sterile USP water and morphine in sterile USP saline; morphine was prepared fresh prior to each experiment. Matched vehicle controls were included for each drug injection. The vehicles used were: 2% DMSO and 98% sterile USP water for KUNA115, KUNB106, KUNG65, KU177, Cycloheximide; 1% DMSO and 99% sterile USP water for KU-32 and 17-AAG; 10% DMSO, 10% Tween80, and 80% sterile USP water for gedunin and celastrol; sterile USP water for DAMGO; and sterile USP saline for morphine. Drug powders were stored at −20°C under desiccation or as recommended by the manufacturer, and stock solutions were stored at −20°C.
CRISPR/Cas9 DNA Constructs
CRISPR gene editing constructs were obtained from Genecopoeia as all-in-one DNA vectors containing universal promoters driving expression of the gRNA and Cas9 gene, along with a neomycin resistance gene for mammalian cell selection and an mCherry gene for visualization (pCRISPR-CG vector). The constructs were pre-designed by Genecopoeia to target each mouse gene. They included a universal negative control vector that expresses all the same elements with a non-targeting gRNA (#CCPCTR01-CG01-B), and constructs to target Hsp90α (#MCP229411-CG01), p23 (#MCP232080-CG12), Cdc37 (#MCP231406-CG12), and PEBP1 (#MCP231756-CG01) as a further negative control.
Each DNA vector was amplified for use using standard bacterial transformation, and an endotoxin-free maxi-prep kit to reduce inflammation upon injection. Each vector was also validated by restriction digest. The Hsp90α vector was validated in an in vitro experiment. Mouse 66.1 breast cancer cells were cultured as described in Edwards et al. (2018). The cells were electroporated with 10 μg of DNA per cuvette, then selected with 500 μg/ml of G418 until the cells recovered and began growing again. At this point, the cells were harvested and analyzed by Western blot as described below. The in vivo delivery and validation of all vectors are also described below.
Animals
Male and female CD-1 (a.k.a. ICR) mice from 4 to 8 weeks of age were used for all experiments and were obtained from Charles River Laboratories. The mice were recovered for at least 5 days after shipping prior to use and housed no more than five per cage. All mice were housed in the University of Arizona’s AAALAC-accredited vivarium with temperature and humidity control, 12 h light/dark cycles, and standard chow and water available ad libitum. All experiments performed were approved by the University of Arizona’s IACUC, and all experiments were in accordance with the NIH Care and Use of Laboratory Animals handbook.
Paw Incision Model
Mice were randomly assigned to experimental groups in age-matched cohorts, and the experimenter was blinded to treatment group identity by the delivery of coded drug vials. We utilized a post-surgical paw incision pain model, with the surgery performed as described in our earlier work (Lei et al., 2017). Drugs or CRISPR DNA constructs were delivered by the intracerebroventricular (icv) route in a 5 μl volume, also performed as described in Lei et al. (2017). For drug treatments, the paw incision surgery was performed, and while the mice were still under anesthesia, an icv injection of inhibitor drug or vehicle was performed. The mice then recovered from both the surgery and injection for 1 or 24 h prior to opioid injection and pain measurement. For CRISPR experiments, 4 μg of DNA was complexed with Turbofect in vivo transfection reagent (#FERR0541 from Fisher Scientific) according to the manufacturer’s instructions and injected icv daily from days 1 to 3. The mice then recovered, with the paw incision surgery performed on day 9 and opioid injection and pain measurement performed on day 10. Our in vivo CRISPR protocol is based on the protocol reported in Sandweiss et al. (2017).
Mechanical pain/allodynia on the incised paw was measured using Von Frey filaments with the up-down method, as performed in our earlier work and the literature (Chaplan et al., 1994; Lei et al., 2017; Edwards et al., 2018). Pre- and post-CRISPR and pre- and post-surgical baselines were measured to determine any impact of the treatment on baseline responses prior to the injection of morphine. Mechanical allodynia was measured in a 2–3 h time course after the injection of morphine.
Signaling Protein Analysis by Western Blot
To analyze brain signaling changes, mice were injected icv with KUNA115 as above for 24 h, followed by icv injection of DAMGO for 10 min. The mice were sacrificed by rapid cervical dislocation, and the periaqueductal gray (PAG) region was rapidly dissected on an ice-cooled metal block and snap-frozen in liquid nitrogen. G418-selected populations of 66.1 cells transfected with CRISPR constructs described above were also harvested for protein analysis. The cells were washed with ice-cold dPBS and the cells recovered by adding lysis buffer and scraping with a plastic cell spatula. The methods for protein extraction from both brain regions and cell lysates, the composition of our lysis buffer, and the protocol for performing the Western blot are all reported in our earlier work (Lei et al., 2017).
We used the following antibodies for our Western analysis: phospho-Akt (#50-191-224, Fisher Scientific); total-Akt (#50-190-279, Fisher Scientific); phospho-ERK (#50-191-932, Fisher Scientific); total-ERK (#50-191-129, Fisher Scientific); phospho-JNK (#9255, Cell Signaling); total-JNK (#9252, Cell Signaling); Hsp70 (#4872, Cell Signaling); STAT3 (#9139, Cell Signaling); GAPDH (#PIMA515738, Fisher Scientific); Hsp90α (#MA110892, Fisher Scientific); and mCherry (#NBP196752SS, Fisher Scientific). The antibodies were generally used at 1:1,000 in 5% BSA in TBST rocking overnight at 4°C. We used goat anti-rabbit or goat anti-mouse IRDye 680 or 800 secondary antibodies from LiCor Biosciences at 1:5,000–10,000 in 5% non-fat dry milk in TBST at room temperature for 1 h. The resulting data was imaged using an Odyssey Fc imager from LiCor Biosciences. The data were quantitated using Scion Image, derived from NIH ImageJ. Phospho-protein signal was normalized to total protein signal from the same sample (e.g., pERK normalized to tERK), while total protein signal was normalized to the housekeeping gene GAPDH (e.g., STAT3 normalized to GAPDH). These normalized data were further normalized to the vehicle-treated control animals within each experiment.
Immunohistochemistry
CRISPR-mediated knockdown in the brain of the target proteins Hsp90α, p23, and Cdc37 was validated post hoc in treated mice as above using immunohistochemistry. The mice were first perfused with 4% paraformaldehyde in saline, and the brains removed and frozen as a block in OTC medium. The brains were sectioned using a cryotome with 20 μm sections and mounted on Leica Xtra slides (#NC0215141, Fisher Scientific); the frozen sections were dried at room temperature for 15 min and stored at −20°C until use.
For Hsp90α: sections warmed to room temperature, then washed in TBS (20 mM Tris, 150 mM NaCl, pH 7.2) for 10 min. Blocked for 2 h in a humidified chamber at room temperature (10% goat serum, 0.3% Triton-X100 in TBS). Incubated with primary antibody (#380-003, Synaptic Systems) at 1:50 in 5% goat serum, 0.3% Triton-X100 in TBS overnight at 4°C. Washed 3 × 10 min in TBS, followed by anti-rabbit Alexa 488 secondary antibody (#A11034, Fisher Scientific) at 1:200 in 5% goat serum, 0.3% Triton-X100 in TBS for 1 h at room temperature. The slides were then washed 3 × 10 min in TBS, dried at room temperature for 10 min, then mounted.
For p23 and Cdc37: sections warmed to room temperature, then washed in phosphate buffered saline (PBS) for 10 min. Heat-induced antigen retrieval performed for 20 min at 95°C in sodium citrate buffer (10 mM sodium citrate, 0.05% Tween20, pH 6.0). The slides were cooled to room temperature for 20 min, then washed in PBS with 0.1% Tween20 (PBST) twice, and PBS once. The sections were then blocked for 30 min in a humidified chamber at room temperature [3% fetal bovine serum (FBS) in PBS]. After blocking, the sections were incubated with primary antibody overnight at 4°C (p23-1:1,000 of #MA3414 from Fisher Scientific; Cdc37-1:100 of #MA3029 from Fisher Scientific; both in 3% FBS in PBS). The slides were then washed 3 × 10 min in PBST, followed by secondary antibody at room temperature (p23-1:200 of anti-mouse Alexa594, #A11032 from Fisher Scientific, in 3% FBS in PBS for 1 h. Cdc37-1:200 of anti-mouse Alexa488, #A11031 from Fisher Scientific, in 3% FBS in PBST for 30 min). The slides were then washed 3 × 10 min in PBST and dried at room temperature for 10 min before mounting.
All sections were imaged using a standard fluorescent microscope using the appropriate filters for Alexa488 (blue/green) and Alexa594 (green/red). The knockdown of all targets was broadly apparent across the brain. The pontine reticular nucleus (PRN) was chosen as the site of imaging and quantitation. Images were taken from 4 to 6 adjacent sections from each animal. The fluorescence intensity divided by the area of the image was calculated for each section, and the 4–6 sections per animal averaged to produce a single mean value counted as N = 1.
Data Analysis
All data is reported as the mean ± SEM. All statistical analysis was performed using GraphPad Prism 8.0. All behavioral data is reported as raw threshold values without normalization; Western blot and immunohistochemistry data normalized as described in those sections above. Statistical comparisons for behavioral data and ERK/JNK/Akt Western data performed using a two-way ANOVA with Fisher’s Least Significant Difference post hoc test. Comparisons of immunohistochemistry data and Hsp70/STAT3 Western data performed using an Unpaired 2-Tailed t-test. In all cases significance was set as a p-value of <0.05. For the dose/response experiment using KUNA115, the area under the curve (AUC) was calculated for each dose and treatment using Prism 8.0, and graphed by log dose and treatment group. Linear regression analysis was performed, and the parameters of those fitted lines used to calculate the potency (A50) as previously described (Lei et al., 2017). The sample sizes and technical replicates for each experiment are described in the Figure Legends.
Results
Isoform-Selective Inhibitor Screen Identifies Hsp90α
To identify the active Hsp90 isoform in regulating opioid anti-nociception in the brain, we performed a screen of isoform-selective small molecule inhibitors. KUNA115 is selective for Hsp90α (Mishra et al., under review), KUNB106 for Hsp90β (Mishra et al., in press), and KUNG65 for Grp94 (Crowley et al., 2017). All inhibitors were delivered at a screening dose of 0.1 nmol by the icv route for 24 h, a model predicated on our earlier studies with the non-selective Hsp90 inhibitor 17-AAG (Lei et al., 2017). Over a full morphine dose range of 1–10 mg/kg, we found that KUNA115 strongly blocked morphine anti-nociception in paw incision pain, suggesting the involvement of the isoform Hsp90α (Figure 1A). Dose/response analysis for this experiment revealed an A50 potency value of morphine of 8.95 mg/kg for Vehicle-treated mice, in line with literature values, validating the experiment (Figure 1B). The dose/response curve for KUNA115 meanwhile was so flat it did not give a feasible value (calculated A50 of 88, 139, 382 mg/kg; Figure 1B). These results show that an Hsp90α-selective inhibitor strongly blocked morphine anti-nociception in this pain model in line with our previous results using a non-selective inhibitor (Lei et al., 2017). We also found a similar result with female mice, suggesting no sex differences with this target and model (Figure 1C).
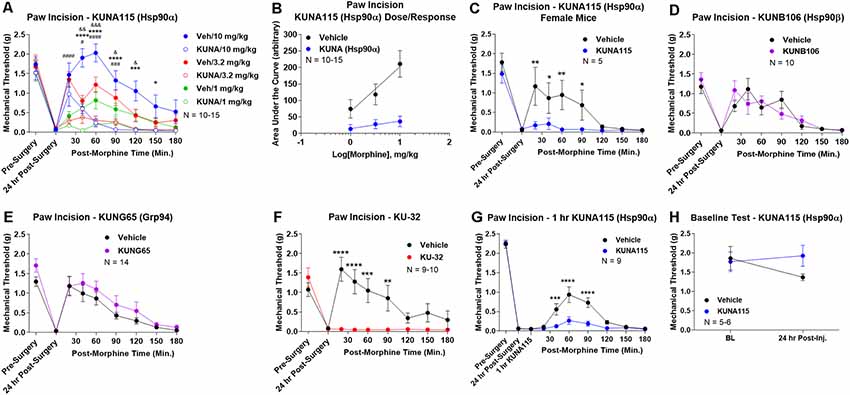
Figure 1. Heat shock protein 90 (Hsp90) isoform-selective inhibitor screen identifies Hsp90α in promoting opioid anti-nociception in the brain. CD-1 mice had the paw incision surgery performed, with concurrent injection of vehicle or 0.1 nmol of inhibitor intracerebroventricular (icv). Mice recovered 24 h, followed by sc injection of morphine. Mechanical thresholds measured before and after surgery, and in a time course after morphine injection. Pre- and post-surgery baselines did not differ for any group (p > 0.05). All data reported as the mean ± SEM, with sample sizes of mice/group noted in each graph. All statistics performed by two-way ANOVA with Fisher’s Least Significant Difference post hoc test. (A) Male mice tested with KUNA115 (Hsp90α selective) and a 1–10 mg/kg morphine dose range. *,***,****p < 0.05, 0.001, 0.0001 between Veh and KUNA groups at same time point at 10 mg/kg; #,###,####p < 0.05, 0.001, 0.0001 between Veh and KUNA groups at same time point at 3.2 mg/kg; &,&&,&&&p < 0.05, 0.01, 0.001 between Veh and KUNA groups at same time point at 1 mg/kg. Performed in 2–3 technical replicates. (B) Dose/response curves constructed from the data in (A) and analyzed as described in “Materials and Methods” section. A50: Vehicle = 8.95 mg/kg; KUNA115 = 88, 139, 382 mg/kg (too flat for accurate calculation). (C) KUNA115 experiment performed in female mice with 3.2 mg/kg morphine. Performed in one technical replicate. For remaining graphs: *,**,***,****p < 0.05, 0.01, 0.001, 0.0001 vs. same time point inhibitor treatment group. (D) Male mice tested with KUNB106 (Hsp90β selective) with 3.2 mg/kg morphine. Performed in two technical replicates. (E) Male mice tested with KUNG65 (Grp94 selective) with 3.2 mg/kg morphine. Performed in three technical replicates. (F) Male mice tested with KU-32 (non-selective) with 3.2 mg/kg morphine. Performed in two technical replicates. (G) Male and female mice had paw incision performed, with 23 h recovery. KUNA115 or Vehicle then injected as above, with 1 h treatment prior to 3.2 mg/kg morphine. Performed in two technical replicates. (H) Male and female mice had KUNA115 or Vehicle injected as above with a 24 h recovery. Pre- and post-injection baselines measured without any surgery, pain state, or opioids present. Performed in two technical replicates by different experimenters.
When we tested the other isoform-selective inhibitors, we found no differences for KUNB106 (Hsp90β; Figure 1D) and KUNG65 (Grp94; Figure 1E). These results do suggest that Hsp90α alone is active in the brain for opioid signaling regulation. As a further control, we tested the impact of an alternate site inhibitor KU-32, which binds to the C-terminal region of Hsp90 unlike the ATP pocket targeted 17-AAG but is similarly non-selective between isoforms (Ansar et al., 2007). The results with KU-32 are the same as for KUNA115 above and 17-AAG (Lei et al., 2017), further validating the results and suggesting a bona fide role for Hsp90 in regulating opioid anti-nociception (Figure 1F).
We also performed additional experiments to further define the impact of KUNA115/Hsp90α on opioid anti-nociception. All experiments above were carried out with a 24 h recovery, leaving the time course of KUNA115 unknown. We thus performed a paw incision experiment as above with only a 1 h KUNA115 treatment; this resulted in a strong loss of morphine anti-nociception, similar to the 24 h results above (Figure 1G). This finding suggests that KUNA115 has a relatively rapid onset that is sustained for 24 h or more. We also controlled for potential impacts of KUNA115 on mechanical thresholds without pain or opioids present. We found that a 24 h KUNA115 treatment as above had no impact on baseline mechanical thresholds, suggesting the results above are due to a specific impact on the opioid system (Figure 1H). This conclusion is further supported by our earlier work in which we found no impact of Hsp90 inhibitor treatment on motor performance in the Rotarod test (Lei et al., 2017).
Hsp90α Regulates Opioid Signal Transduction
We next sought to measure the impact of Hsp90α-selective inhibition on opioid signaling. We combined KUNA115 treatment with DAMGO stimulation in the brain, which is a highly selective, potent, and efficacious MOR agonist. We analyzed ERK and JNK MAPK, Akt, Hsp70, and STAT3 by Western blot in the PAG region of the brain (Figure 2A). This region was chosen based on our earlier studies using non-selective inhibitors (Lei et al., 2017); the PAG is also a key region in the pain modulatory circuitry (Heinricher et al., 2009). We found that both ERK and JNK MAPK phosphorylation was stimulated by DAMGO in Vehicle-treated mice, however, stimulation over baseline was lost with KUNA115 treatment (Figure 2B). With JNK MAPK, KUNA115 significantly raised the unstimulated baseline, as we saw for ERK MAPK with 17-AAG treatment (Lei et al., 2017). With the kinase Akt, KUNA115 treatment tended to increase both unstimulated and DAMGO-stimulated phospho-Akt levels, so that the KUNA115/DAMGO group was significantly elevated over Vehicle/Vehicle baseline (Figure 2B). Lastly, KUNA115 treatment had no impact on Hsp70 protein levels, unlike 17-AAG treatment (Lei et al., 2017), while it significantly decreased total protein levels of the signaling regulator STAT3 (Figure 2B). Together these results demonstrate that Hsp90α has a specific role in regulating opioid signal transduction in the brain suggesting potential involvement in opioid anti-nociception, similar to what we showed for 17-AAG treatment and ERK MAPK (Lei et al., 2017).
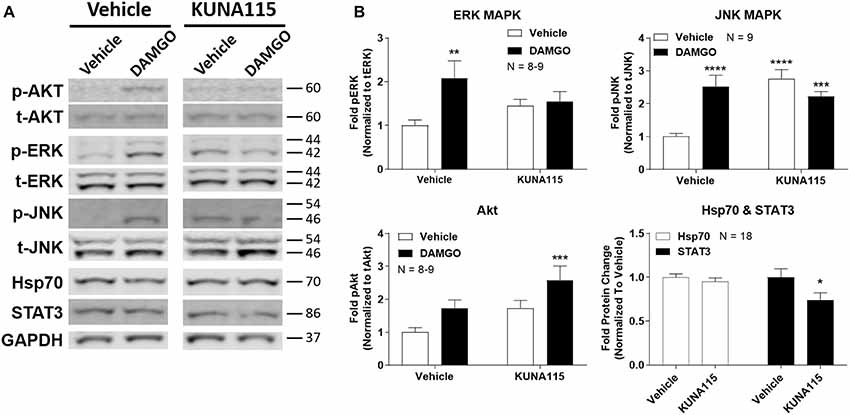
Figure 2. Hsp90α regulates opioid signal transduction in the brain. Male CD-1 mice had vehicle or 0.1 nmol KUNA115 injected icv, 24 h, followed by vehicle or 0.1 nmol DAMGO icv, 10 min. Periaqueductal gray region analyzed by Western blot. All data reported as the mean ± SEM, with sample sizes of mice/group noted in each graph; all experiments performed in four technical replicates. For ERK, JNK and Akt, data analyzed by two-way ANOVA with Fisher’s Least Significant Difference post hoc test; **,***,****p < 0.01, 0.001, 0.0001 vs. Vehicle:Vehicle group. For Hsp70 and STAT3 data analyzed by Unpaired 2-Tailed t-test; *p < 0.05 vs. same target Vehicle group. (A) Representative sample blots shown for each target, with MW indicated for each protein. Each pair of images for one target (e.g., p-Akt) were from the same blot, but discontinuous, so they are separated to denote this fact. (B) All Western data quantitated by target. ERK, JNK, and Akt are phosphorylated protein signal normalized to total protein. Hsp70 and STAT3 are normalized to GAPDH. KUNA115 treatment caused a loss of ERK and JNK stimulation over baseline by DAMGO, and a loss of STAT3 protein expression.
CRISPR Knockdown of Hsp90α in Adult Mouse Brain
To confirm the role of Hsp90α in regulating opioid anti-nociception, we used CRISPR/Cas9 DNA constructs to knockdown Hsp90α protein expression broadly across the brain in adult mice (similar approach to Sandweiss et al., 2017). We first validated our CRISPR construct in vitro using mouse 66.1 cells. Transfection of a negative control CRISPR vector or a vector targeting the protein PEBP had no impact on Hsp90α protein levels, while our vector targeting Hsp90α reduced protein levels by ~90% (Figure 3A). We could also detect expression of the mCherry protein in all cells with a CRISPR vector, verifying successful transfection of all constructs (Figure 3A).
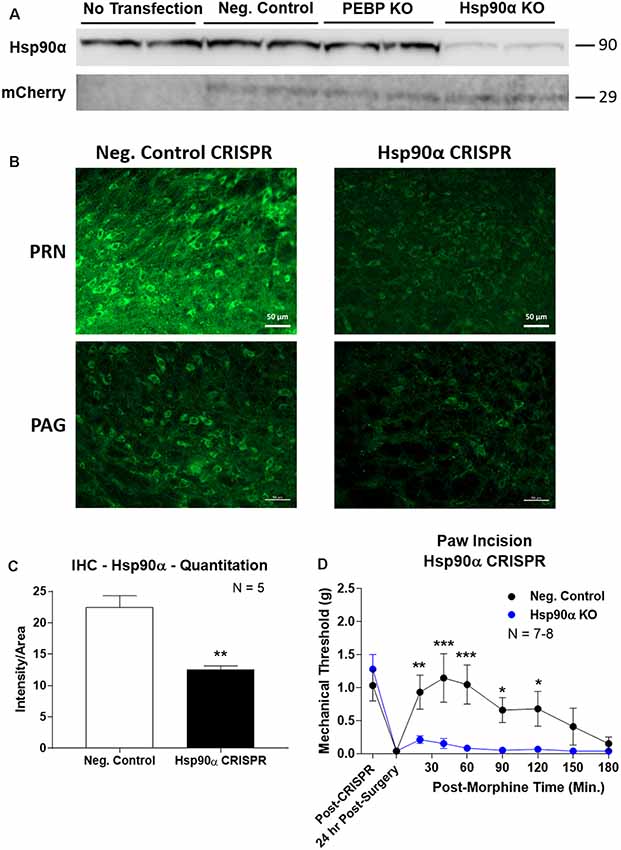
Figure 3. CRISPR/Cas9 gene editing of Hsp90α in adult mouse brain. CRISPR constructs for all targets prepared and delivered as described in “Materials and Methods” section. All quantitative data reported as mean ± SEM. (A) Hsp90α CRISPR construct validated in 66.1 cells. Western blot shown with replicate wells of cells in each lane, with MW indicated for each target. Hsp90α protein levels reduced by ~90% only in the presence of Hsp90α-targeted CRISPR construct. Other constructs (Negative Control, PEBP) have no effect. mCherry protein levels are present in all transfected cells, demonstrating successful transfection of CRISPR DNA. (B) Hsp90α or negative control CRISPR delivered to CD-1 male mouse brains and analyzed for protein knockdown on day 10. Representative images shown from pontine reticular nucleus (PRN) and periaqueductal gray (PAG). Hsp90α (green signal) is present in cell bodies and dendritic trees, and the signal is reduced by CRISPR treatment. (C) Quantitation of all data from (B) performed as described in “Materials and Methods” section. Sample size of mice/group noted in graph. **p < 0.01 vs. Negative Control group by Unpaired 2-Tailed t-test. Mice treated in one technical replicate, with the resulting tissue stained and analyzed in more than one technical replicate. CRISPR treatment reduced Hsp90α signal by 43.9%. (D) CRISPR-treated CD-1 male mice had paw incision surgery performed on day 9, with injection of 3.2 mg/kg morphine sc on day 10. Sample size of mice/group noted in graph, performed in two technical replicates. *,**,***p < 0.05, 0.01, 0.001 vs. same time point Hsp90α group by two-way ANOVA with Fisher’s Least Significant Difference post hoc test.
We next treated mice with the Hsp90α or negative control CRISPR vectors as described in the “Materials and Methods” section, and validated successful target knockdown by immunohistochemistry. We found broad knockdown across the entire brain and selected the PRN and PAG for analysis. We could detect a strong signal in both cell bodies and apparent dendritic fields that was strongly reduced by Hsp90α CRISPR treatment (Figure 3B). Quantitation of fluorescent signal revealed a significant reduction of 43.9% (Figure 3C). Thus validated, we next tested the impact of Hsp90α knockdown on morphine anti-nociception (Figure 3D). We found that Hsp90α CRISPR treatment fully blocked anti-nociception in paw incision pain (Figure 3D), very similarly to KUNA115 (Figure 1) and 17-AAG (Lei et al., 2017), confirming the role of Hsp90α in regulating opioid anti-nociception in the brain.
Hsp90 Co-chaperones p23 and Cdc37 Regulate Opioid Anti-nociception in the Brain
We first used the co-chaperone-selective inhibitors gedunin (p23, Brandt et al., 2008), celastrol (Cdc37, Zhang et al., 2008), and KU177 (Aha1, Zhao et al., 2011) in paw incision pain as for the isoform inhibitors above. Both gedunin (p23; Figure 4A) and celastrol (Cdc37; Figure 4B) strongly reduced morphine anti-nociception in paw incision pain, very similar to the Hsp90 inhibitors above. However, KU177 (Aha1) had only a slight impact on opioid anti-nociception, suggesting it may not have a significant role (Figure 4C).
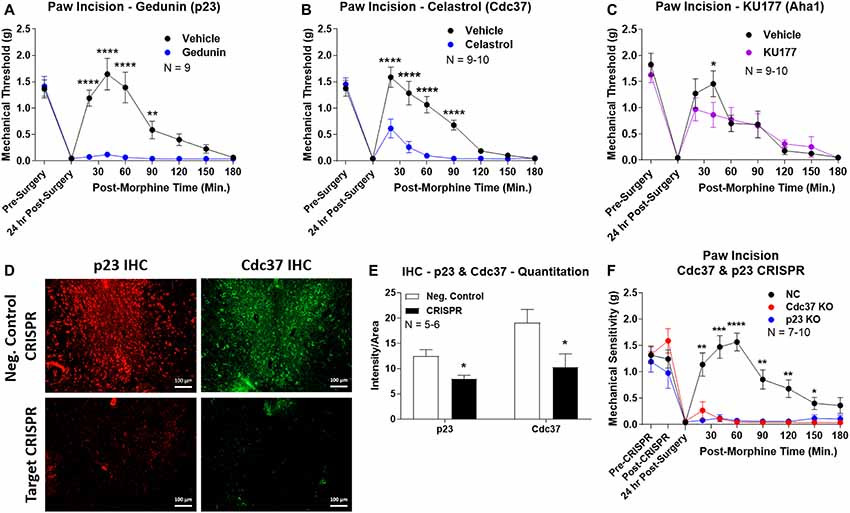
Figure 4. Identification of the co-chaperones p23 and Cdc37 as promoters of opioid anti-nociception in the brain. Male CD-1 mice used for all experiments, data reported as the mean ± SEM, with sample sizes of mice/group noted in each graph. (A–C) Ten nanomoles of Gedunin (p23, A), 10 nmol of Celastrol (Cdc37, B), or 0.1 nmol of KU177 (Aha1, C) or Vehicle control injected icv concurrently with paw incision surgery with a 24 h recovery, followed by 3.2 mg/kg morphine sc. Experiments performed with two technical replicates for each drug. *,**,****p < 0.05, 0.01, 0.0001 vs. same time point inhibitor treatment group by two-way ANOVA with Fisher’s Least Significant Difference post hoc test. (D) p23, Cdc37, or Negative Control CRISPR-treated mice with icv delivery of constructs analyzed by IHC for target knockdown on day 10. Representative images shown from the PRN. Both targets (p23 – red, Cdc37 – green) have a similar staining pattern to Hsp90α, and both are clearly reduced by CRISPR treatment. (E) Data from (D) for all mice quantitated as described in the “Materials and Methods” section. All mice treated in one technical replicate, with staining and analysis of the resulting tissue performed in more than one technical replicate. *p < 0.05 vs. same target Negative Control group by Unpaired 2-Tailed t-test. CRISPR treatment reduced p23 signal by 36.3% and Cdc37 by 46.0%. (F) CRISPR-treated mice had paw incision surgery performed on day 9, with injection of 3.2 mg/kg morphine sc on day 10. Performed in two technical replicates. *,**,***,****p < 0.05, 0.01, 0.001, 0.0001 vs. both same time point p23/Cdc37 CRISPR groups by two-way ANOVA with Fisher’s Least Significant Difference post hoc test.
We next moved forward with CRISPR/Cas9 gene editing to confirm that p23 and Cdc37 regulate opioid anti-nociception as we did for Hsp90α above. IHC analysis showed a similar broad knockdown across the brain with CRISPR treatment, particularly apparent in the PRN (Figure 4D). Quantitation revealed significant decreases of 36.3% for p23 and 46.0% for Cdc37 (Figure 4E). We next tested the impact of targeted CRISPR treatment for these proteins in paw incision pain, and found that both p23 and Cdc37 CRISPR knockdown fully blocked morphine anti-nociception, very similar to Hsp90α inhibition above or 17-AAG treatment (Lei et al., 2017; Figure 4F). These results confirm that both p23 and Cdc37 regulate opioid anti-nociception in the brain.
Rapid Translation Required for Hsp90 Regulation of Opioid Anti-nociception
Lastly, we sought to identify part of the molecular biology mechanism by which Hsp90 regulates opioid anti-nociception. We combined 24 h Hsp90 inhibition as above and in our previous work (Lei et al., 2017) with treatment of the translation inhibitor cycloheximide in the brain 30 min prior to morphine treatment. We found that cycloheximide had no impact on morphine anti-nociception in vehicle-treated mice; however, cycloheximide fully restored morphine anti-nociception back to vehicle-treated levels in mice treated with the pan-Hsp90 inhibitor 17-AAG (Figure 5A). We found the same results with KUNA115, confirming that Hsp90α regulates translation during morphine anti-nociception (Figure 5B). These results strongly suggest that rapid translation within 30 min of morphine treatment is required for Hsp90 inhibition to impact opioid signaling and anti-nociception.
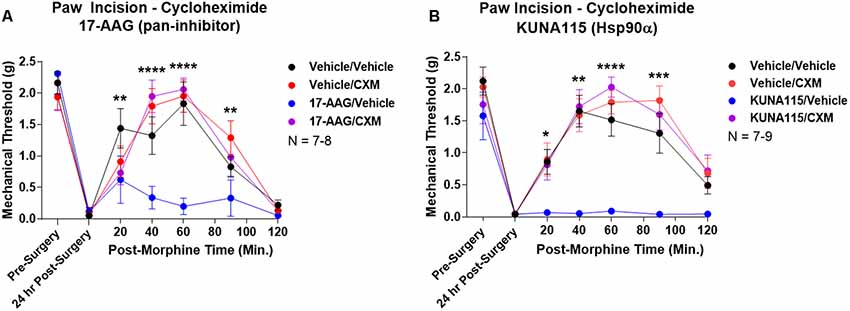
Figure 5. Rapid protein translation is required for Hsp90 inhibition to impact opioid anti-nociception. Male and female CD-1 mice treated with 0.5 nmol 17-AAG (A) or 0.1 nmol KUNA115 (B) or vehicle icv, 24 h, followed by 85 nmol cycloheximide or vehicle icv, 30 min, followed by 10 mg/kg morphine sc. No difference between approximately equal male and female groups (p > 0.05) so they were combined for this analysis. Data reported as the mean ± SEM with the sample size of mice/group noted in the graph. Performed in three technical replicates by different experimenters. *,**,***,****p < 0.05, 0.01, 0.001, 0.0001 for the 17-AAG/Vehicle group vs. any of the other three groups at the same time point by two-way ANOVA with Fisher’s Least Significant Difference post hoc test. There were no differences between the Vehicle/Vehicle, Vehicle/CXM, or 17-AAG/CXM groups.
Discussion
In this study, we have identified the isoform Hsp90α as a key player in promoting opioid anti-nociception and signaling in the brain using both selective small molecule inhibitors (Figures 1, 2) and CRISPR/Cas9 gene editing in the brains of adult mice (Figure 3). Using these techniques, we further identified the co-chaperones p23 and Cdc37 as key promoters of opioid anti-nociception (Figure 4). We also found that rapid protein translation is part of the molecular mechanism by which Hsp90 regulates opioid anti-nociception (Figure 5). When combined with the results of our previous study using a non-selective Hsp90 inhibitor (Lei et al., 2017), our findings suggest that Hsp90α in concert with p23 and Cdc37 promote ERK MAPK activation by the MOR in the brain, and that inhibiting these proteins reverses these roles summating in loss of opioid ERK activation and anti-nociception. This model is diagrammed in Figure 6.
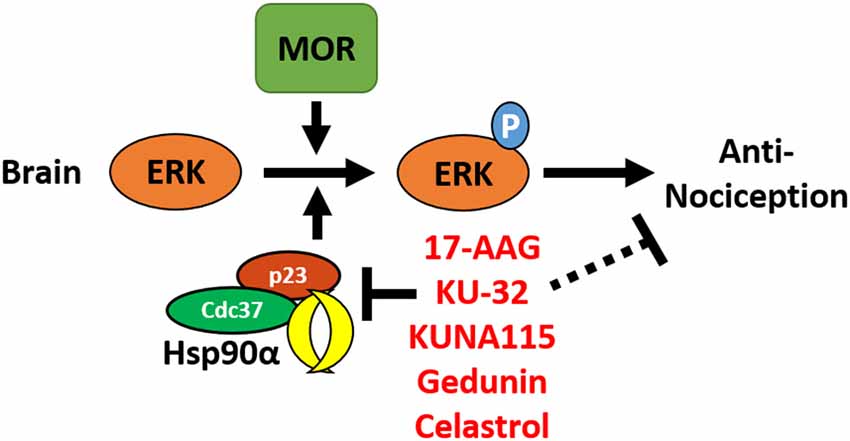
Figure 6. Model for the regulation of mu opioid receptor (MOR) signaling by Hsp90 in the brain. This summary model combines results from this manuscript with our earlier work using non-selective Hsp90 inhibitors in the brain (Lei et al., 2017). Together this data suggests that Hsp90α, p23, and Cdc37 act in concert in the brain to promote the phosphorylation of ERK MAPK by the MOR in response to opioid drugs, thus promoting opioid anti-nociception. Treatment with non-selective or selective inhibitors blocks this role, thus blocking ERK MAPK phosphorylation and blocking anti-nociception in response to opioids.
Understanding the specific molecules involved in promoting opioid anti-nociception may provide future targets for refined clinical intervention. First generation non-selective Hsp90 inhibitors like 17-AAG failed clinical trials due to liver toxicity (Sidera and Patsavoudi, 2014). Second generation and alternate site inhibitors like KU-32 have shown higher tolerability, and indeed KU-32 has been shown to be neuroprotective (Urban et al., 2010, 2012; Ma et al., 2015). However, non-selective targeting of Hsp90 has an inherently higher risk of unacceptable side effects, especially since Hsp90 is a ubiquitous protein with a very high expression level and numerous client proteins (Li and Buchner, 2013). The four Hsp90 isoforms have different cellular locations (cytoplasm for Hsp90α/β, endoplasmic reticulum for Grp94, mitochondria for TRAP1) and client proteins, meaning that targeting only one necessary isoform, such as Hsp90α identified above, should inherently decrease the risk of side effects (Echeverría et al., 2011). Similarly, identifying specific co-chaperones like p23 and Cdc37 identified above will also provide specific targets that should reduce potential side effects. This is particularly true with co-chaperones since there are many more of them and each co-chaperone is more selective by role and tissue expression, providing further selectivity (Li and Buchner, 2013). Along these lines, recent studies have identified ligands that interfere with specific Hsp90:co-chaperone interactions, such as celastrol used in our study here, that impact specific functions without the broad sledgehammer of inhibiting the entire Hsp90 protein (Zhang et al., 2008). The specific proteins identified through this study could thus be the first step in identifying improved therapies to modulate opioid treatment; an analogous approach has been used for Hsp70 (Assimon et al., 2013).
The identification of these specific molecules may also lend insight into the molecular mechanism by which Hsp90 regulates opioid signaling. As mentioned above, Hsp90α is cytoplasmic with its own unique complement of client proteins (Bergmayr et al., 2013; Liu et al., 2015). This specificity narrows down the field of potential mechanisms by which Hsp90α regulates opioid signaling, especially to the Hsp90α-specific signaling changes in ERK, JNK, and STAT3 that we have identified (Figure 2). Further differences include the lack of Hsp70 induction caused by KUNA115 treatment in Figure 2. Earlier studies have shown that non-selective inhibitors like 17-AAG impact both the heat shock response (Hsp70 induction) and protein folding activity of Hsp90, while the C-terminal inhibitor KU-32 only impacts the heat shock response (Ansar et al., 2007). All three compounds, 17-AAG, KU-32, and KUNA115 (Figure 1; Lei et al., 2017), impact opioid anti-nociception in the same way, providing a clue that the protein activity of Hsp90α responsible will be the one impacted by all three classes of compound (and which does not require Hsp70 upregulation).
Similarly, the co-chaperone Cdc37 that we have identified has been shown in numerous studies to be crucial for targeting kinases to Hsp90 in both canonical and non-canonical pathways that are crucial for kinase function (Hinz et al., 2007; Gould et al., 2009; Ota et al., 2010). Importantly, these studies have shown that Cdc37 does not simply assist in kinase folding and maturation, but also assists in complex formation that is required for acute activation. Considering the signaling kinases impacted, especially ERK MAPK which we have identified as a mechanism of Hsp90 regulation of anti-nociception (Figure 2; Lei et al., 2017), it makes sense that Cdc37 would be implicated in the Hsp90 mechanism of action. The co-chaperone p23 also has a canonical role in assisting Hsp90 in the protein folding cascade, and has also been linked to acute regulation of signal transduction, as by the A2A receptor (Bergmayr et al., 2013; Li and Buchner, 2013). The functional overlap between the specific molecules identified and the drugs used will allow us to identify likely mechanisms of signaling regulation in future studies, further informed by our results in Figure 5 suggesting that active protein translation is required for Hsp90 inhibition to impact opioid anti-nociception. Our results showing that Hsp90 inhibition is impactful within 1 h and that baseline mechanical response is not altered provides further mechanistic guidance (Figures 1G,H).
While positively identifying Hsp90α, p23, and Cdc37, our studies demonstrated no response to other isoform and co-chaperone inhibitors. This does suggest that Hsp90β, Grp94, and Aha1 are not involved in regulating opioid anti-nociception in the brain. However, this data must be interpreted with caution. We did not exhaustively test these molecules by CRISPR and other methods as we did for Hsp90α, p23, and Cdc37, leaving open the possibility that these other molecules could still be involved. Thus our data should be interpreted most strongly as identifying Hsp90α, p23, and Cdc37 without necessarily ruling out other players. Future studies can address this question more exhaustively, as well as test for the involvement of numerous other co-chaperones not tested in this study. These studies were also all performed broadly across the forebrain without testing the impact of the spinal cord or the periphery, or sub-regions and circuits within the brain. Future studies will also need to address whether these other regions have different mechanisms by which Hsp90 could regulate opioid response.
Data Availability Statement
All datasets generated for this study are included in the article.
Ethics Statement
The animal study was reviewed and approved by IACUC, University of Arizona.
Author Contributions
WL collaboratively developed the initial idea for the project, participated in study design, performed most experiments, and analyzed the data. DD performed some paw incision and cycloheximide experiments, and analyzed the data. CS performed one of the cycloheximide experiments and analyzed the data. SM synthesized, purified, and characterized the novel small molecule inhibitors described above. BB supervised SM in the course of the chemistry work, and collaboratively developed the initial idea for the project. JS supervised WL, DD, and CS in the course of their work, conceived the initial idea for the project, participated in study design, analyzed some of the data, and wrote the manuscript. All authors had editorial input into the manuscript.
Funding
We acknowledge the support of an Arizona Biomedical Research Commission New Investigator Award #ADHS18-198875 and institutional funds from the University of Arizona to JS. Institutional funds from Presbyterian College to WL also supported this study, along with National Institutes of Health (NIH) R01CA213566 to BB.
Conflict of Interest
JS has grants from the NIH and Arizona Biomedical Research Commission (ABRC), as well as research contracts with Depomed, Inc. and Divine Healing, LLC. JS also is a founder with an equity stake in Teleport Pharmaceuticals, LLC. None of these commercial involvements concern the topic of the research study at hand, or Hsp90 research more broadly. BB has grants from the NIH and is a founder with an equity stake in Grannus Therapeutics, a virtual startup for developing novel Hsp90 inhibitors.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to acknowledge the assistance of Mr. Justin LaVigne with the in vitro validation of CRISPR constructs.
References
Abul-Husn, N. S., Annangudi, S. P., Ma’ayan, A., Ramos-Ortolaza, D. L., Stockton, S. D.Jr., Gomes, I., et al. (2011). Chronic morphine alters the presynaptic protein profile: identification of novel molecular targets using proteomics and network analysis. PLoS One 6:e25535. doi: 10.1371/journal.pone.0025535
Al-Hasani, R., and Bruchas, M. R. (2011). Molecular mechanisms of opioid receptor-dependent signaling and behavior. Anesthesiology 115, 1363–1381. doi: 10.1097/aln.0b013e318238bba6
Ansar, S., Burlison, J. A., Hadden, M. K., Yu, X. M., Desino, K. E., Bean, J., et al. (2007). A non-toxic Hsp90 inhibitor protects neurons from Abeta-induced toxicity. Bioorg. Med. Chem. Lett. 17, 1984–1990. doi: 10.1016/j.bmcl.2007.01.017
Assimon, V. A., Gillies, A. T., Rauch, J. N., and Gestwicki, J. E. (2013). Hsp70 protein complexes as drug targets. Curr. Pharm. Des. 19, 404–417. doi: 10.2174/138161213804143699
Assimon, V. A., Southworth, D. R., and Gestwicki, J. E. (2015). Specific binding of tetratricopeptide repeat proteins to heat shock protein 70 (Hsp70) and heat shock protein 90 (Hsp90) is regulated by affinity and phosphorylation. Biochemistry 54, 7120–7131. doi: 10.1021/acs.biochem.5b00801
Bergmayr, C., Thurner, P., Keuerleber, S., Kudlacek, O., Nanoff, C., Freissmuth, M., et al. (2013). Recruitment of a cytoplasmic chaperone relay by the A2A adenosine receptor. J. Biol. Chem. 288, 28831–28844. doi: 10.1074/jbc.M113.464776
Bohn, L. M., Lefkowitz, R. J., Gainetdinov, R. R., Peppel, K., Caron, M. G., and Lin, F. T. (1999). Enhanced morphine analgesia in mice lacking beta-arrestin 2. Science 286, 2495–2498. doi: 10.1126/science.286.5449.2495
Brandt, G. E., Schmidt, M. D., Prisinzano, T. E., and Blagg, B. S. (2008). Gedunin, a novel hsp90 inhibitor: semisynthesis of derivatives and preliminary structure-activity relationships. J. Med. Chem. 51, 6495–6502. doi: 10.1021/jm8007486
Chaplan, S. R., Bach, F. W., Pogrel, J. W., Chung, J. M., and Yaksh, T. L. (1994). Quantitative assessment of tactile allodynia in the rat paw. J. Neurosci. Methods 53, 55–63. doi: 10.1016/0165-0270(94)90144-9
Crowley, V. M., Huard, D. J. E., Lieberman, R. L., and Blagg, B. S. J. (2017). Second generation Grp94-selective inhibitors provide opportunities for the inhibition of metastatic cancer. Chemistry 23, 15775–15782. doi: 10.1002/chem.201703398
Darcq, E., Befort, K., Koebel, P., Pannetier, S., Mahoney, M. K., Gaveriaux-Ruff, C., et al. (2012). RSK2 signaling in medial habenula contributes to acute morphine analgesia. Neuropsychopharmacology 37, 1288–1296. doi: 10.1038/npp.2011.316
Dewire, S. M., Yamashita, D. S., Rominger, D. H., Liu, G., Cowan, C. L., Graczyk, T. M., et al. (2013). A G protein-biased ligand at the mu-opioid receptor is potently analgesic with reduced gastrointestinal and respiratory dysfunction compared to morphine. J. Pharmacol. Exp. Ther. 344, 708–717. doi: 10.1124/jpet.112.201616
Echeverría, P. C., Bernthaler, A., Dupuis, P., Mayer, B., and Picard, D. (2011). An interaction network predicted from public data as a discovery tool: application to the Hsp90 molecular chaperone machine. PLoS One 6:e26044. doi: 10.1371/journal.pone.0026044
Edwards, K. A., Havelin, J. J., Mcintosh, M. I., Ciccone, H. A., Pangilinan, K., Imbert, I., et al. (2018). A kappa opioid receptor agonist blocks bone cancer pain without altering bone loss, tumor size, or cancer cell proliferation in a mouse model of cancer-induced bone pain. J. Pain 19, 612–625. doi: 10.1016/j.jpain.2018.01.002
Gould, C. M., Kannan, N., Taylor, S. S., and Newton, A. C. (2009). The chaperones Hsp90 and Cdc37 mediate the maturation and stabilization of protein kinase C through a conserved PXXP motif in the C-terminal tail. J. Biol. Chem. 284, 4921–4935. doi: 10.1074/jbc.m808436200
Heinricher, M. M., Tavares, I., Leith, J. L., and Lumb, B. M. (2009). Descending control of nociception: specificity, recruitment and plasticity. Brain Res. Rev. 60, 214–225. doi: 10.1016/j.brainresrev.2008.12.009
Hinz, M., Broemer, M., Arslan, S. C., Otto, A., Mueller, E. C., Dettmer, R., et al. (2007). Signal responsiveness of IkappaB kinases is determined by Cdc37-assisted transient interaction with Hsp90. J. Biol. Chem. 282, 32311–32319. doi: 10.1074/jbc.m705785200
Kim, H., Yang, J., Kim, M. J., Choi, S., Chung, J. R., Kim, J. M., et al. (2016). Tumor necrosis factor receptor-associated protein 1 (TRAP1) mutation and TRAP1 inhibitor gamitrinib-triphenylphosphonium (G-TPP) induce a forkhead box O (FOXO)-dependent cell protective signal from mitochondria. J. Biol. Chem. 291, 1841–1853. doi: 10.1074/jbc.m115.656934
Koshimizu, T. A., Tsuchiya, H., Tsuda, H., Fujiwara, Y., Shibata, K., Hirasawa, A., et al. (2010). Inhibition of heat shock protein 90 attenuates adenylate cyclase sensitization after chronic morphine treatment. Biochem. Biophys. Res. Commun. 392, 603–607. doi: 10.1016/j.bbrc.2010.01.089
Lei, W., Mullen, N., Mccarthy, S., Brann, C., Richard, P., Cormier, J., et al. (2017). Heat-shock protein 90 (Hsp90) promotes opioid-induced anti-nociception by an ERK mitogen-activated protein kinase (MAPK) mechanism in mouse brain. J. Biol. Chem. 292, 10414–10428. doi: 10.1074/jbc.m116.769489
Li, J., and Buchner, J. (2013). Structure, function and regulation of the hsp90 machinery. Biomed. J. 36, 106–117. doi: 10.4103/2319-4170.113230
Li, Z., Li, C., Yin, P., Wang, Z. J., and Luo, F. (2016). Inhibition of CaMKIIalpha in the central nucleus of amygdala attenuates fentanyl-induced hyperalgesia in rats. J. Pharmacol. Exp. Ther. 359, 82–89. doi: 10.1124/jpet.116.233817
Liu, W., Vielhauer, G. A., Holzbeierlein, J. M., Zhao, H., Ghosh, S., Brown, D., et al. (2015). KU675, a concomitant heat-shock protein inhibitor of Hsp90 and Hsc70 that manifests isoform selectivity for Hsp90alpha in prostate cancer cells. Mol. Pharmacol. 88, 121–130. doi: 10.1124/mol.114.097303
Ma, J., Pan, P., Anyika, M., Blagg, B. S., and Dobrowsky, R. T. (2015). Modulating molecular chaperones improves mitochondrial bioenergetics and decreases the inflammatory transcriptome in diabetic sensory neurons. ACS Chem. Neurosci. 6, 1637–1648. doi: 10.1021/acschemneuro.5b00165
Macey, T. A., Bobeck, E. N., Hegarty, D. M., Aicher, S. A., Ingram, S. L., and Morgan, M. M. (2009). Extracellular signal-regulated kinase 1/2 activation counteracts morphine tolerance in the periaqueductal gray of the rat. J. Pharmacol. Exp. Ther. 331, 412–418. doi: 10.1124/jpet.109.152157
Manglik, A., Lin, H., Aryal, D. K., Mccorvy, J. D., Dengler, D., Corder, G., et al. (2016). Structure-based discovery of opioid analgesics with reduced side effects. Nature 537, 185–190. doi: 10.1038/nature19112
Mishra, S. J., Ghosh, S., Stothert, A. R., Dickey, C. A., and Blagg, B. S. (2017). Transformation of the non-selective aminocyclohexanol-based Hsp90 inhibitor into a Grp94-seletive scaffold. ACS Chem. Biol. 12, 244–253. doi: 10.1021/acschembio.6b00747
Mishra, S. J., Kent, C. N., Beebe, K., Liu, W., Taylor, J. A., Neckers, L. M., et al. (in press). Hsp90beta-selective inhibitors exhibit nanomolar potency and overcome detriments associated with pan-Hsp90 inhibition. J. Med. Chem.
Olson, K. M., Lei, W., Keresztes, A., Lavigne, J., and Streicher, J. M. (2017). Novel molecular strategies and targets for opioid drug discovery for the treatment of chronic pain. Yale J. Biol. Med. 90, 97–110.
Ota, A., Zhang, J., Ping, P., Han, J., and Wang, Y. (2010). Specific regulation of noncanonical p38alpha activation by Hsp90-Cdc37 chaperone complex in cardiomyocyte. Circ. Res. 106, 1404–1412. doi: 10.1161/circresaha.109.213769
Raehal, K. M., Walker, J. K., and Bohn, L. M. (2005). Morphine side effects in beta-arrestin 2 knockout mice. J. Pharmacol. Exp. Ther 314, 1195–1201. doi: 10.1124/jpet.105.087254
Sandweiss, A. J., Mcintosh, M. I., Moutal, A., Davidson-Knapp, R., Hu, J., Giri, A. K., et al. (2017). Genetic and pharmacological antagonism of NK1 receptor prevents opiate abuse potential. Mol. Psychiatry 23, 1745–1755. doi: 10.1038/mp.2017.102
Schmid, C. L., Kennedy, N. M., Ross, N. C., Lovell, K. M., Yue, Z., Morgenweck, J., et al. (2017). Bias factor and therapeutic window correlate to predict safer opioid analgesics. Cell 171, 1165.e1113–1175.e1113. doi: 10.1016/j.cell.2017.10.035
Sidera, K., and Patsavoudi, E. (2014). HSP90 inhibitors: current development and potential in cancer therapy. Recent Pat. Anticancer. Drug Discov. 9, 1–20. doi: 10.2174/15748928113089990031
Streicher, J. M. (2019). The role of heat shock proteins in regulating receptor signal transduction. Mol. Pharm. 111, 91–103. doi: 10.1016/bs.vh.2019.05.010
Urban, M. J., Li, C., Yu, C., Lu, Y., Krise, J. M., Mcintosh, M. P., et al. (2010). Inhibiting heat-shock protein 90 reverses sensory hypoalgesia in diabetic mice. ASN Neuro 2:e00040. doi: 10.1042/an20100015
Urban, M. J., Pan, P., Farmer, K. L., Zhao, H., Blagg, B. S., and Dobrowsky, R. T. (2012). Modulating molecular chaperones improves sensory fiber recovery and mitochondrial function in diabetic peripheral neuropathy. Exp. Neurol. 235, 388–396. doi: 10.1016/j.expneurol.2012.03.005
Zhang, L., Kibaly, C., Wang, Y. J., Xu, C., Song, K. Y., Mcgarrah, P. W., et al. (2017). Src-dependent phosphorylation of mu-opioid receptor at Tyr(336) modulates opiate withdrawal. EMBO Mol. Med. 9, 1521–1536. doi: 10.15252/emmm.201607324
Zhang, T., Hamza, A., Cao, X., Wang, B., Yu, S., Zhan, C. G., et al. (2008). A novel Hsp90 inhibitor to disrupt Hsp90/Cdc37 complex against pancreatic cancer cells. Mol. Cancer Ther. 7, 162–170. doi: 10.1158/1535-7163.mct-07-0484
Keywords: heat shock protein 90 (Hsp90), p23, Cdc37, opioid, pain, anti-nociception, CRISPR, translation
Citation: Lei W, Duron DI, Stine C, Mishra S, Blagg BSJ and Streicher JM (2019) The Alpha Isoform of Heat Shock Protein 90 and the Co-chaperones p23 and Cdc37 Promote Opioid Anti-nociception in the Brain. Front. Mol. Neurosci. 12:294. doi: 10.3389/fnmol.2019.00294
Received: 28 June 2019; Accepted: 15 November 2019;
Published: 29 November 2019.
Edited by:
Felix Viana, Institute of Neurosciences of Alicante (IN), SpainReviewed by:
Wei Xiong, University of Science and Technology of China, ChinaBo Zhao, Indiana University, United States
Copyright © 2019 Lei, Duron, Stine, Mishra, Blagg and Streicher. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: John M. Streicher, anN0cmVpY2hlckBlbWFpbC5hcml6b25hLmVkdQ==