 Raman Kumar1†
Raman Kumar1† Elizabeth Palmer2,3†Alison E. Gardner1Renee Carroll1Siddharth Banka4,5Ola Abdelhadi5Dian Donnai4,5
Elizabeth Palmer2,3†Alison E. Gardner1Renee Carroll1Siddharth Banka4,5Ola Abdelhadi5Dian Donnai4,5 Ype Elgersma6,7Cynthia J. Curry8Alice Gardham9Mohnish Suri10Rishikesh Malla11
Ype Elgersma6,7Cynthia J. Curry8Alice Gardham9Mohnish Suri10Rishikesh Malla11 Lauren Ilana Brady12
Lauren Ilana Brady12 Mark Tarnopolsky12Dimitar N. Azmanov13,14Vanessa Atkinson13,14Michael Black13,14
Mark Tarnopolsky12Dimitar N. Azmanov13,14Vanessa Atkinson13,14Michael Black13,14 Gareth Baynam15Lauren Dreyer16,17Robin Z. Hayeems18Christian R. Marshall19Gregory Costain20Marja W. Wessels21Julia Baptista22
Gareth Baynam15Lauren Dreyer16,17Robin Z. Hayeems18Christian R. Marshall19Gregory Costain20Marja W. Wessels21Julia Baptista22 James Drummond23Melanie Leffler2
James Drummond23Melanie Leffler2 Michael Field2
Michael Field2 Jozef Gecz1,24*
Jozef Gecz1,24*- 1Adelaide Medical School and the Robinson Research Institute, The University of Adelaide, Adelaide, SA, Australia
- 2Genetics of Learning Disability Service, Hunter Genetics, Waratah, NSW, Australia
- 3School of Women’s and Children’s Health, University of New South Wales, Randwick, NSW, Australia
- 4Faculty of Biology, Medicine and Health, Division of Evolution and Genomic Sciences, School of Biological Sciences, University of Manchester, Manchester, United Kingdom
- 5Manchester Centre for Genomic Medicine, St. Mary’s Hospital, Manchester University NHS Foundation Trust, Health Innovation Manchester, Manchester, United Kingdom
- 6Department of Neuroscience, Erasmus MC University Medical Center, Rotterdam, Netherlands
- 7ENCORE Expertise Centre for Neurodevelopmental Disorders, Erasmus MC University Medical Center, Rotterdam, Netherlands
- 8Genetic Medicine, Department of Pediatrics, University of California, San Francisco, San Francisco, CA, United States
- 9North West Thames Regional Genetics Service, Northwick Park Hospital, Harrow, United Kingdom
- 10Nottingham Clinical Genetics Service, Nottingham University Hospitals NHS Trust, and the 100,000 Genomes Project and the Genomics England Research Consortium, Nottingham, United Kingdom
- 11Division of Pediatric Neurology, Medical University of South Carolina, Charleston, SC, United States
- 12Department of Pediatrics, McMaster University Medical Centre, Hamilton, ON, Canada
- 13Department of Diagnostic Genomics, PathWest, Nedlands, WA, Australia
- 14Division of Pathology and Laboratory Medicine, Medical School, University of Western Australia, Crawley, WA, Australia
- 15Faculty of Health and Medical Sciences, University of Western Australia Medical School, Perth, WA, Australia
- 16Genetic Services of Western Australia, Undiagnosed Diseases Program, Department of Health, Government of Western Australia, Perth, WA, Australia
- 17Linear Clinical Research, Perth, WA, Australia
- 18Child Health Evaluative Sciences, Research Institute, The Hospital for Sick Children, and Institute of Health Policy Management and Evaluation, University of Toronto, Toronto, ON, Canada
- 19Genome Diagnostics, Department of Paediatric Laboratory Medicine, The Hospital for Sick Children, and Laboratory Medicine and Pathobiology, University of Toronto, Toronto, ON, Canada
- 20Department of Paediatrics, Division of Clinical and Metabolic Genetics, The Hospital for Sick Children, Toronto, ON, Canada
- 21Department of Clinical Genetics, Erasmus MC University Medical Center, Rotterdam, Netherlands
- 22Royal Devon and Exeter NHS Foundation Trust, Exeter, United Kingdom
- 23Neuroradiology, Royal North Shore Hospital, Sydney, NSW, Australia
- 24Childhood Disability Prevention, South Australian Health and Medical Research Institute, Adelaide, SA, Australia
Multiple TREX mRNA export complex subunits (e.g., THOC1, THOC2, THOC5, THOC6, THOC7) have now been implicated in neurodevelopmental disorders (NDDs), neurodegeneration and cancer. We previously implicated missense and splicing-defective THOC2 variants in NDDs and a broad range of other clinical features. Here we report 10 individuals from nine families with rare missense THOC2 variants including the first case of a recurrent variant (p.Arg77Cys), and an additional individual with an intragenic THOC2 microdeletion (Del-Ex37-38). Ex vivo missense variant testing and patient-derived cell line data from current and published studies show 9 of the 14 missense THOC2 variants result in reduced protein stability. The splicing-defective and deletion variants result in a loss of small regions of the C-terminal THOC2 RNA binding domain (RBD). Interestingly, reduced stability of THOC2 variant proteins has a flow-on effect on the stability of the multi-protein TREX complex; specifically on the other NDD-associated THOC subunits. Our current, expanded cohort refines the core phenotype of THOC2 NDDs to language disorder and/or ID, with a variable severity, and disorders of growth. A subset of affected individuals’ has severe-profound ID, persistent hypotonia and respiratory abnormalities. Further investigations to elucidate the pathophysiological basis for this severe phenotype are warranted.
Introduction
Neurodevelopmental disorders (NDDs) caused by genetic, epigenetic and environmental factors are clinically heterogeneous conditions affecting the central nervous system in children and include intellectual disability (ID), epilepsy, autism and cerebral palsy. NDDs affect more than 3% of children worldwide. Genetic causes are attributed to variants at over 1,000 loci and include single nucleotide changes (single nucleotide variants, SNV; coding and non-coding variants) and larger losses, gains or rearrangements (structural variants; Tărlungeanu and Novarino, 2018).
Many molecular causes of NDDs disrupt diverse cellular pathways, particularly affecting neuronal development, proliferation and/or migration of cells to cause learning and behavioral disabilities (Srivastava and Schwartz, 2014). One such pathway—that has been the focus of our ongoing investigations—is of the highly conserved TREX (TRanscription-EXport) mRNA export pathway. Multiple TREX complex subunits (e.g., THOC1, THOC2, THOC5, THOC6, and THOC7) have been implicated in NDDs and human disease (Kumar et al., 2015, 2018; Heath et al., 2016; Amos et al., 2017; Mattioli et al., 2019). TREX-mediated mRNA export from the nucleus to the cytoplasm is a complex and highly conserved pathway in all eukaryotes. The TREX complex is composed of a THO sub-complex of a set of six stoichiometric and stable subunits (THOC1-3, 5–7) and accessory proteins (UAP56, UIF, Aly, CIP29, PDIP3, ZC11A, SRRT, Chtop; Heath et al., 2016). THOC subunits are ubiquitously expressed, including in the human brain (Uhlén et al., 2015). The TREX complex has critical roles in developmental processes such as pluripotency maintenance and hematopoiesis as well as in gene regulation, 3′ mRNA processing, stress responses, mitotic progression and genome stability in mammalian cells (Mancini et al., 2010; Yamazaki et al., 2010). We have previously reported genetic and molecular evidence implicating a large number of missense and splicing-defective variants in THOC2–which codes for the largest subunit of the TREX mRNA export complex—in NDDs (Mental retardation, X-linked 12/35 MIM 300957; Kumar et al., 2015, 2018). Here we present genetic and molecular evidence on a set of novel THOC2 variants, and using aggregate data, refine the core clinical phenotype of THOC2 NDDs.
Materials and Methods
Clinical Studies
Through direct contact with clinicians, facilitated by the genotype-phenotype database DECIPHER and the Human Disease Gene Web series, where we moderate a THOC2-related disorder site1, 10 individuals from 9 families were identified with rare (absent from gnomAD 2.1) missense variants or an intragenic microdeletion. Eight of these variants were novel and one recurrent (p.Arg77Cys) that was a maternally inherited in individual 8 as previously reported by us (detailed in Supplementary Data; Kumar et al., 2018). In our previous report, we designated p.Arg77Cys as a variant of uncertain clinical significance in the absence of functional studies at the time (Kumar et al., 2018). All families consented to publication of de-identified clinical information, neuroimaging and, for seven families, clinical photographs, in line with local ethics board regulations. The variants have been submitted to ClinVar2; accession numbers SCV001132790-SCV001132797.
Molecular Studies
RNA extraction and RT-qPCR (primers listed in Supplementary Tables S1, S2) were performed as reported previously (Kumar et al., 2015). We used THOC2 Del-Ex37-38 (lymphoblastoid cell lines, LCLs and skin fibroblasts) and p.Asn666Asp (skin fibroblasts) cells from the affected individuals and their carrier heterozygous mothers. However, we used THOC2 p.Arg77Cys and p.Tyr881Cys variant LCLs of only probands. THOC2 Del-Ex37-38 (LCLs and skin fibroblasts) cDNAs (generated by reverse transcribing the total RNAs with Superscript IV reverse transcriptase; Life Technologies, VIC, Australia) were amplified using KAPA HiFi PCR Kit with GC buffer (Kapa Biosystems, IN, USA) and hTHOC2-4326F/P276 and hTHOC3-3′UTR-R2/P392 primers (Supplementary Table S1) at 95°C for 3 min, 35 cycles of 98°C-10 s, 59°C-10 s, 72°C-80 s, incubation at 72°C for 10 min, gel purified (Qiagen MinElute Gel Extraction kit; Qiagen, Victoria, Australia) and Sanger sequenced using the same primers. Genomic deleted region in THOC2 Del-Ex-37-38 carrier mother and affected son was identified by PCR amplification of the target regions from their blood gDNAs using LongAmp Hot Start Taq 2× Master Mix (Promega, Alexandria, NSW, Australia) and hTHOC2-4460-F/P390 and hTHOC2-gDNA-R1/P415 primers (Supplementary Table S1) at 94°C for 30 s, 35 cycles of 94°C-15 s, 64°C-15 s, 65°C-8 min 30 s, incubation at 65°C for 10 min. Appropriate PCR products were gel-purified (Qiagen MinElute Gel Extraction Kit) and Sanger sequenced using hTHOC2-gDNA-F7/P421 and hTHOC2-gDNA-R7/P422 primers (Supplementary Table S1).
Cellular Studies
We performed THOC2 immunofluorescence staining in Del-Ex37-38 and p.Asn666Asp skin fibroblasts using two anti-THOC2 antibodies; anti-THOC2-I to region between amino acids 1,400–1,450 (Bethyl Laboratories A303-629A, Montgomery, TX, USA) and anti-THOC2-II to a region between amino acids 1543–1593 (Bethyl Laboratories A303-630A) of the THOC2 protein. Both the antibodies were used for detecting the THOC2 protein in Del-Ex37-38 affected individuals and his carrier heterozygous mother fibroblasts but only anti-THOC2-I for detecting the THOC2 p.Asn666Asp in the affected individuals and his carrier heterozygous mother fibroblasts.
Western Blotting
The Epstein–Barr virus (EBV)-immortalized B-cell lines (LCLs) established from peripheral blood lymphocytes of affected individuals and controls were maintained in RPMI1640 supplemented with 10% fetal bovine serum, 2 mM L-glutamine and 1% Penicillin-Streptomycin at 37°C with 5% CO2. Affected individual-derived fibroblasts were cultured in Dulbecco’s modified Eagle’s medium (Sigma) containing 10% fetal bovine serum (Life Technologies), 2 mM L-glutamine, and 1% Penicillin-Streptomycin at 37°C with 5% CO2. Total fibroblast proteins were extracted in 50 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.1% Triton-X-100, 1 mM EDTA, 50 mM NaF, 1× Protease inhibitor/no EDTA cocktail and 0.1 mM Na3VO4 and LCLs in 50 mM Tris-HCl pH 7.5, 5 mM EDTA, 50 mM KCl, 0.1% NP-40 and 1× Protease inhibitor/no EDTA cocktail (Roche protease inhibitor cocktail; Sigma–Aldrich, Castle Hill, NSW, Australia) by low frequency sonication for 12 s and centrifugation at 13,000× g. Proteins were assayed using Pierce BCA Protein Assay Kit (Thermo Scientific) according to manufacturer’s protocol. Eight to Ten microgram of protein was resolved on NuPAGE 3–8% Tris-Acetate gel (THOC2) or 8% SDA-PAGE (Laemmli, 1970), transferred to nitrocellulose membranes and western blotted with the following antibodies: anti-THOC1 (Bethyl Laboratories A302-839A), anti-THOC2 (Bethyl Laboratories A303-630A and A303-629A), anti-THOC3 (HPA044009; Sigma–Aldrich, Castle Hill, NSW, Australia), anti-THOC5 (Bethyl Laboratories A302-120A), anti-β-Tubulin (Ab6046; Abcam, Melbourne, VIC, Australia) and polyclonal goat anti-rabbit IgG/HRP (Dako; P0448) as secondary antibody. Signal was detected by Clarity Western ECL Substrate (BIO-RAD 170-5061) and captured using Gel documentation System (BIO-RAD, Gladesville, NSW, Australia).
In silico Pathogenicity Prediction
We used CADD v1.3 (Kircher et al., 2014), SIFT (Sim et al., 2012), Provean (Choi and Chan, 2015), GERP++ (Davydov et al., 2010), PhyloP (Pollard et al., 2010), gnomAD frequency (v2.1.1; Karczewski et al., 2019), Polyphen2 (Adzhubei et al., 2010), MutPred2 (Pejaver et al., 2017), VEST3 (Carter et al., 2013), and Mutation Assessor Score and Pred (Reva et al., 2011) tools for in silico prediction of the pathogenicity of different variants (Table 1).
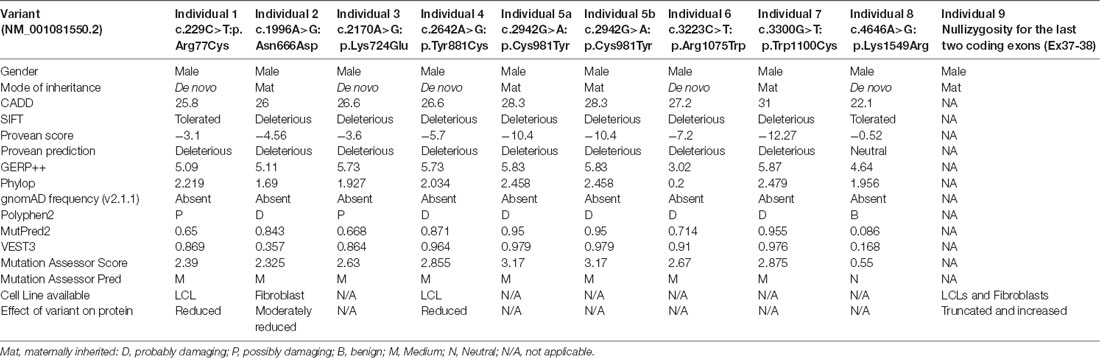
Table 1. Pathogenicity predictions and descriptions for the THOC2 variants.
Results
Identification of THOC2 Variants
We previously implicated missense and splicing-defective THOC2 variants in NDDs with a broad range of clinical features (Kumar et al., 2015, 2018). Here we report 10 previously unreported affected individuals with THOC2 variants, including the first case of a recurrent THOC2 variant (c.229C>T; p.Arg77Cys). We also identify a novel intragenic THOC2 microdeletion (Del-Ex37-Ex38) [NM_001081550.2 c.4678–572_4782 + 1215; p.(1559fs16)] (Tables 1, 2, Figures 1, 2). These variants were identified from either whole exome (WES) or whole-genome sequencing (WGS) of the affected individuals and confirmed by Sanger sequencing of the PCR amplified variant-carrying region from gDNAs of the parents and the affected individuals (detailed in Supplementary Data). The missense THOC2 variants affect amino acids that are highly conserved (Supplementary Figure S1), are absent in the gnomAD database and are predicted to be pathogenic based on a number of in silico analyses tools (Table 1). We have also performed cellular and molecular investigations on variants p.Arg77Cys, p.Tyr881Cys, p.Asn666Asp and Del-Ex37-38, for which we had access to affected individual-derived cells (Table 1; also see below).
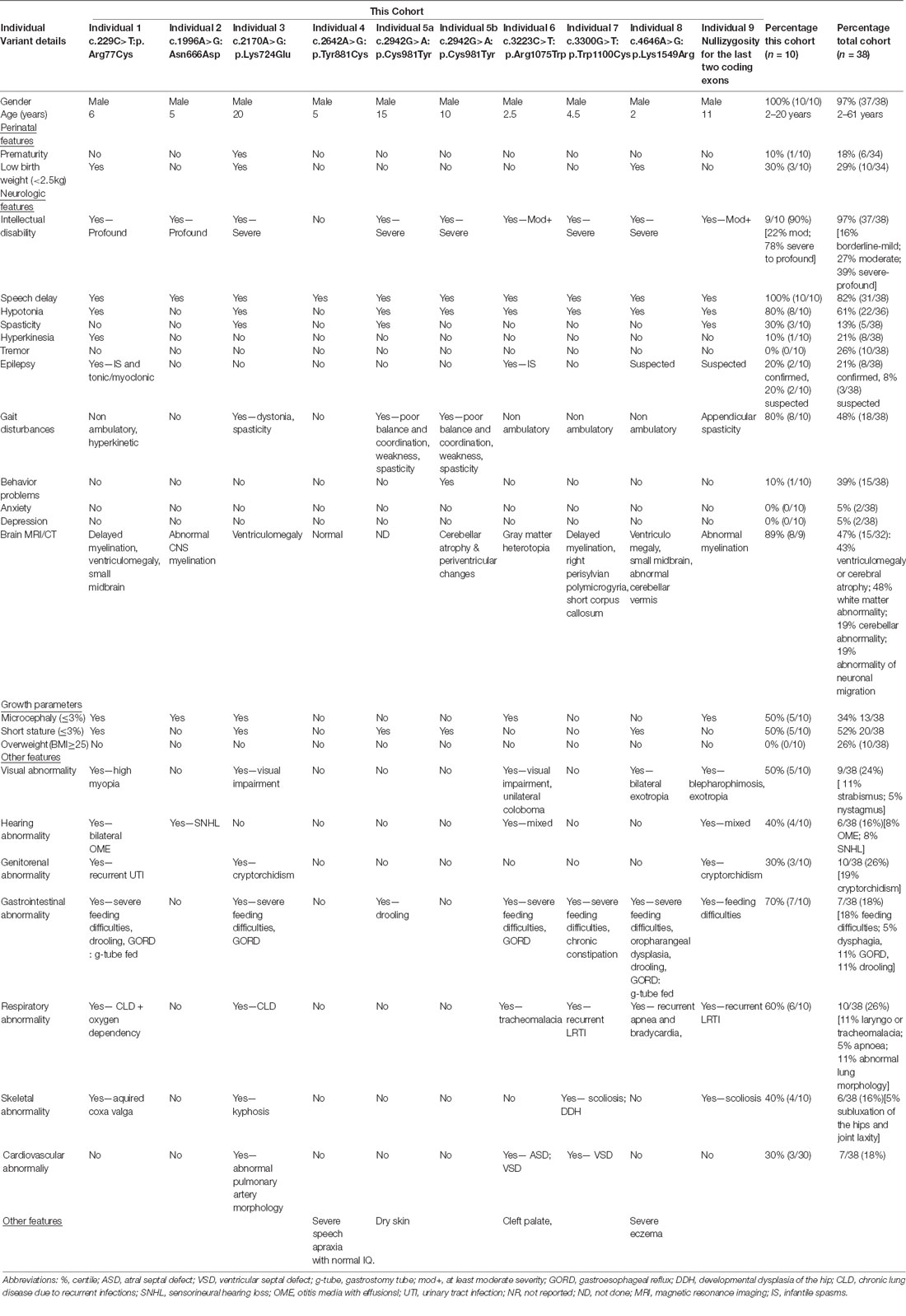
Table 2. Summary of clinical data of THOC2 variants.
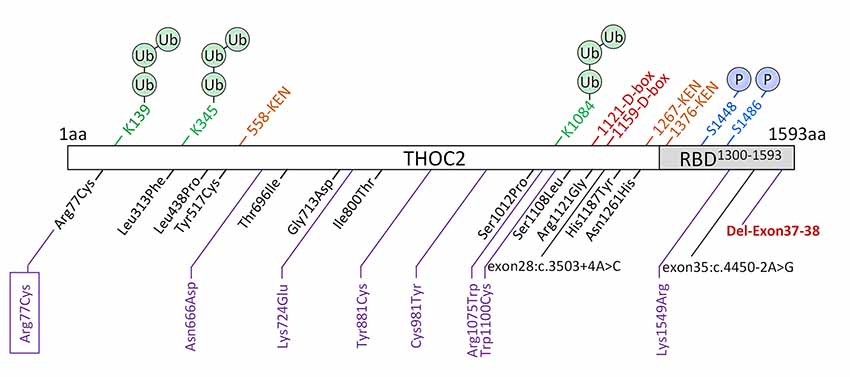
Figure 1. New THOC2 variants (purple). These include the first Arg77Cys recurrent (boxed) and Del-Exon-37-38 variants (red bold). Previously published THOC2 variants (black) and structural features are also shown (Kumar et al., 2015, 2018).
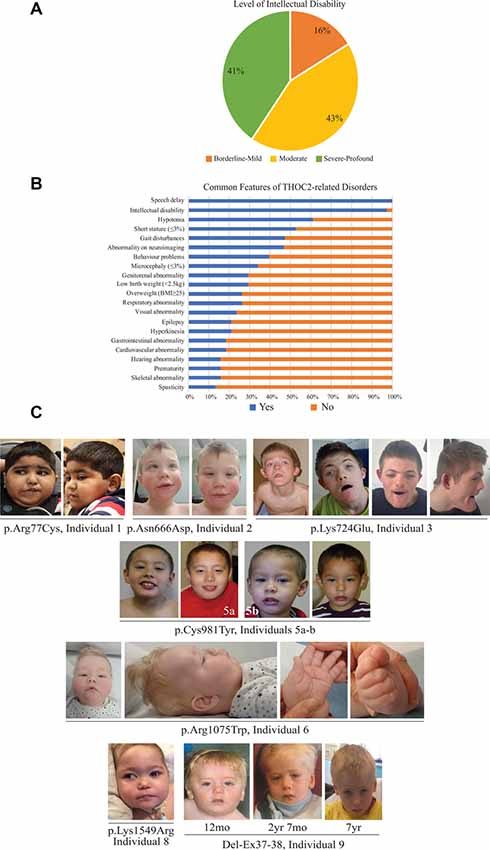
Figure 2. Clinical presentations of the affected individuals identified so far. (A) Percentage of individuals with different levels of ID. (B) Percentage of individuals with common features. (C) Photographs of the affected individuals in the current cohort.
Clinical Presentations
The clinical phenotype of affected individuals with THOC2 variants in this cohort is presented in Table 2 and summarized in Figures 2A,B. The available photographs are shown in Figure 2C. Additional information relating the clinical features is included in case reports in the Supplementary Data and Supplementary Table S2, using an HPO (Human Phenotype Ontology) term framework (Köhler et al., 2019). The main phenotypic features were compared to the individuals reported previously by our group (Table 2, Kumar et al., 2015, 2018). This allowed us to make some general observations about the clinical phenotype of the n = 38 individuals with THOC2 variants (this cohort; Kumar et al., 2015, 2018). First, there is no clear genotypic-phenotypic correlation between the severity or spectrum of phenotypic differences and either the type of genetic variant (missense, splicing-defective or microdeletion) or the part of the protein affected by the variant (Table 2). Second, compared to the first published cohort, which consisted of hemizygous affected males in multi-generational families, the second and this cohort also include many males with de novo THOC2 variants or THOC2 variants in siblings which, on further segregation, were shown to be de novo in the heterozygous mother. The initial cohort described males with typically mild to moderate ID. Of 20 individuals, 30% (6/20) had borderline-mild ID, 55% (11/20) moderate ID and only 15% (3/20) severe ID (Kumar et al., 2015). In contrast, our last (Kumar et al., 2018) and current cohort includes a higher proportion [75% this cohort; 62.5% (Kumar et al., 2018)] of males with severe to profound ID. In striking contrast to our overall findings is individual 4 with the p.Tyr881Cys variant. This male, in whom pathogenicity of p.Tyr881Cys variant is supported by evidence from functional studies (Figures 5B,C: detailed results below), is unique in our cohort as having no evidence of general cognitive dysfunction. On psychometric testing, he had a performance IQ of 104, within the normal range. He did have significant language disorder, with a formal diagnosis of speech apraxia. At the age of 5, he could effectively communicate by sign language. He has a history of mild gross motor delay. Therefore, this publication clarifies that the range of cognitive disability in THOC2-related disorder is very broad—ranging from a normal IQ to a profound ID (Figure 2A).
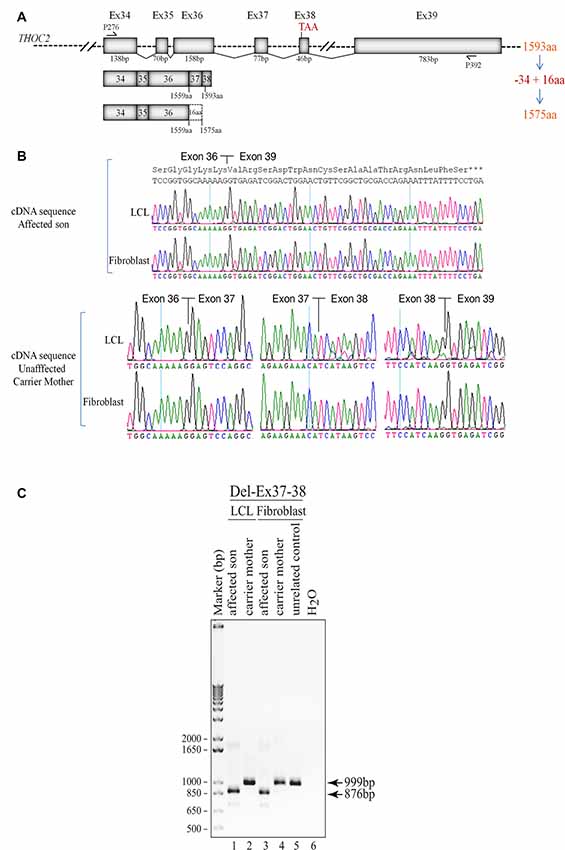
Figure 3. Del-Ex37-38 results in the loss of 34 C-terminal amino acids in the affected individual. (A) Part of the THOC2 gene showing Ex34 to Ex39 and positions of primers. Ex37-38 deleted mRNA translates a 1575 amino acid protein, lacking 34 C-terminal amino acids but adding 16 amino acids encoded from the 3′ UTR region of the deleted mRNAs. (B) Sanger sequencing chromatograms of DNA amplified from LCL and fibroblast cDNAs of the affected individual and his heterozygous unaffected carrier mother using primers P276/P392 located within Ex34 and Ex39. Wild type (1593 amino acid) and C-terminal deleted THOC2 protein (1575 amino acids) translated from Del-Ex37-38 mRNA is also shown. (C) Ex37-38 coding sequence is deleted in both fibroblast and LCL THOC2 mRNAs of the affected son but not in the unaffected carrier mother. Total fibroblast and LCL RNAs were reverse transcribed and PCR amplified using primers P276/P392. PCR products (999 bp from the carrier mother and 876 bp from the affected son) were gel purified and Sanger sequenced using P276 and P392 primers.
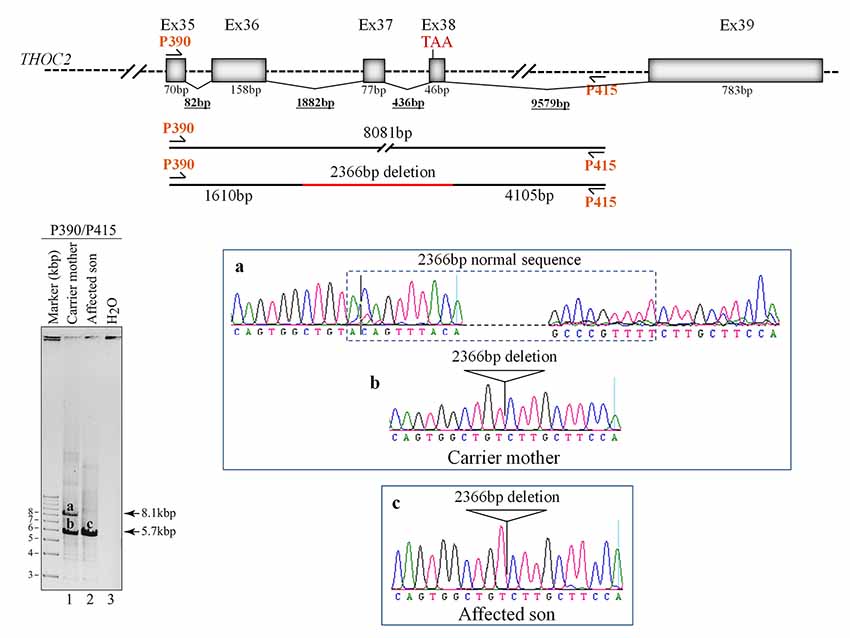
Figure 4. Del-Ex37-38 results from the deletion of approximately 2.4 kb genomic DNA (chrX:122743574-122745939) flanking the Ex37-38. Sequences around the deleted target region were amplified from the affected son and carrier mother’s blood gDNA using P390/P415 primers using LongAmp Hot Start Taq 2× Master Mix, resolved on agarose gel and bands a-c were eluted and Sanger sequenced. Positions of the P390/P415 primers and sequencing chromatograms (a–c) around the deleted gDNA regions are shown.
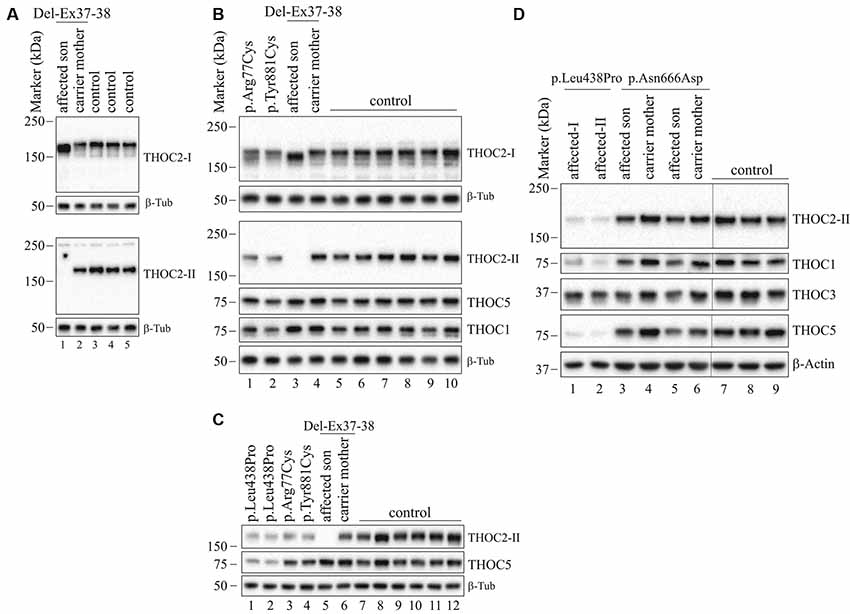
Figure 5. THOC2 variant protein analysis in patient-derived cell lines. (A) THOC2 protein in affected son with Del-Ex37-38, unaffected carrier mother and control fibroblasts. Total protein lysates were western blotted with an anti-THOC2-I antibody that binds to a region between amino acids 1,400–1,450 coded by Ex32-34 mRNA (upper panel) and anti-THOC2-II antibody that binds to a region between amino acids 1543–1593 coded by Ex37-38 mRNA (lower panel). Samples were probed for β-Tubulin as a loading control. (B) THOC2 protein in p.Arg77Cys, p.Tyr881Cys, affected son with Del-Ex37-38, unaffected carrier mother and control LCLs. Total protein lysates were western blotted with anti-THOC2-I, anti-THOC2-II, anti-THOC1, anti-THOC5 and anti-β-Tubulin (loading control) antibodies. Del-Ex37-38 affected fibroblasts (Lane 1 in panel A) and LCLs (Lane 3 in panel B) showing presence of a C-terminally deleted smaller but higher levels of the THOC2 protein when western blotted with anti-THOC2-I antibody (upper panels) that is absent in the affected fibroblast lysates probed with anti-THOC2-II antibody (lower panels). However, normal full-length THOC2 protein is present in the carrier mother and controls western blotted with both the anti-THOC2-I and anti-THOC2-II antibodies (Lane 2 in upper and lower panels in A and Lane 4 in upper and lower panels in B). The p.Arg77Cys and p.Tyr881Cys THOC2 levels are reduced. (C) THOC2 protein levels in p.Leu438Pro (reported to have reduced protein stability; Kumar et al., 2015), p.Arg77Cys, p.Tyr881Cys, Del-Ex37-38 affected son, his unaffected carrier mother and control LCLs. Total protein lysates were western blotted with anti-THOC2-II, anti-THOC5 and anti-β-Tubulin (loading control) antibodies. (D) The p.Asn666Asp levels are moderately reduced in the affected fibroblasts. p.Asn666Asp affected son, his unaffected carrier mother and control fibroblast total lysates were western blotted with the antibodies as shown. p.Leu438Pro fibroblasts previously reported having reduced THOC2 protein stability were included as controls (Kumar et al., 2015). Vertical lines on western blot images indicate the site of deleted sample lanes.
Third, data in Figure 2B and Table 2 suggest that THOC2-related disorder should be considered a multi-systemic disorder in a significant proportion of individuals. The most common extra-neurological feature is disorders of growth. Growth disorders are common across the whole expanded cohort (Figure 2B); a half (52%) have persistent short stature, a third (34%) are microcephalic and 29% have intrauterine growth restriction (IUGR). There was no relationship between IUGR in the offspring and carrier status in the mother. 26% of the expanded cohort have increased body weight, with obesity in older males particularly noted. Congenital anomalies of the cardiorespiratory, genitorenal and skeletal systems, severe feeding difficulties and gastroesophageal reflux, and visual and hearing impairments are present in a significant proportion (10–30%). Therefore, comprehensive evaluation including detailed systems review by a pediatrician, cardiac echocardiogram, hearing screening and ophthalmological review in all newly diagnosed patients is highly recommended. Photographs where available are shown in Figure 2C. As was the case in our previous publications, an easily recognizable facial gestalt is not apparent, although children with a more severe phenotype (Individuals 1, 6, 8 and 9) have downturned corners of their mouth and deep-set almond-shaped eyes: a facial appearance, which may reflect reduced tone.
Fourth, a more complex neuromuscular phenotype is emerging in a proportion of the cohort: that of severe to profound ID, persistent muscular hypotonia, recurrent aspiration and respiratory tract infections, excessive salivation and an increased risk of congenital anomalies, including laryngo/tracheomalacia, cardiac and palatal anomalies. This phenotype was present in the individual with p.Arg77Cys variant for which initial functional studies using exogenous THOC2 variant expression in HEK293T cells were not conclusive (Kumar et al., 2018). We classified this as a variant of uncertain significance, also in view of this patient’s phenotype, which was quite different from the majority of the cohort. We are now more confident that this variant is pathogenic. The variant is recurrent and de novo in individual 1 in this cohort and molecular studies from patient-derived cell lines available from this second family are consistent with reduced protein levels. Both individuals with the p.Arg77Cys variant has strikingly overlapping phenotypes (see Supplemental Data for detailed case reports). Individuals 6, 8 and 9 from the current cohort also have similarities to this phenotype. This more severe phenotype is also frequently associated with neuroradiological abnormalities and an increased chance of a seizure disorder. Seizure disorder, although not very common over the whole cohort (Figure 2B: 21%), can be problematic – with a severe developmental and epileptic encephalopathy picture, commonly including infantile spasms. Therefore, the clinicians should have a low threshold for investigating neurological symptoms or signs with an EEG and MRI brain.
Fifth, although for many individuals the phenotype is static, a more complex neurological phenotype also emerges with age in several males; in the expanded cohort 13% develop hypertonia and spasticity or contractures, and 53% have some abnormality of motor coordination, stereotypic or involuntary movements. In the current cohort, three individuals had received an additional diagnosis of cerebral palsy. Of the nine affected individuals included in this cohort, cases 1, 4, 5, 6, 7 have had multi-timepoint MRI neuro-imaging reviewed by a single Neuroradiology Attending Consultant with dedicated pediatric expertise (data not shown). Although no unifying neuroradiological abnormalities were delineated from this small cohort, one affected individual demonstrated interval cerebellar hemispheric volume loss after a 6-year period, and another demonstrated volume loss of the corpus callosum and mild reduction in myelination at an early post-natal 4-month scan. A further patient had reported features of nodular heterotopia although this imaging was not available for review. No other definitive migrational anomaly, midline defect or development abnormality was identified. The potential implications on white matter development and degeneration in this cohort require long term follow-up imaging.
Lastly, all heterozygous female carriers in this cohort had normal intelligence, as was the case for the entire cohort, with the exception of the one female reported to date with a de novo THOC2 variant with a developmental and epileptic encephalopathy (Kumar et al., 2018). When tested, asymptomatic female carriers are highly skewed for X inactivation (Kumar et al., 2015, 2018).
In summary, this expanded cohort has clarified that the core phenotype of THOC2 related disorder in hemizygous males is that of language disorder and ID, with a wide variability in severity, and that disorders of growth are common, and multi-systemic involvement is present in a significant proportion. A subpopulation of patients has a very severe phenotype with severe-profound ID, persistent hypotonia, respiratory abnormalities and a higher chance of other congenital anomalies. Some affected males show the progression of neurological symptoms and progressive neuroradiological features. Further research to explore the underlying pathophysiological basis for these more severe phenotypes is warranted, but likely reflect the involvement of THOC2 in multi-organ development and function.
Molecular Studies
We used primary skin fibroblasts and/or immortalized B-lymphocytes (LCLs) derived from affected individuals and where available, also their heterozygous carrier mothers.
Del-Ex37-38 THOC2 Variant
We found an affected male with deletion of THOC2 Ex37-38 (NM_001081550.2) as identified by massively parallel sequencing (see Figure 3). We have subsequently validated this as a 2.4 kb X-chromosome gDNA microdeletion (encompassing Ex37-38 sequence) which was also present in his carrier mother (Figure 4). In addition, we PCR amplified the Ex35-Ex39 coding region from cDNAs prepared by reverse transcribing the LCL and fibroblast mRNAs from the affected individual and his normal carrier mother (Figure 3C). Sanger sequencing of the amplified products showed deletion of Ex37-38, but not Ex39 coding sequences, in the affected individual (Figures 3B,C). The carrier mother’s cells showed mRNAs with Ex37-38 sequences consistent with her X-inactivation skewing (10:90; Figures 3A–C). We detected a slightly smaller THOC2 protein (due to Ex37-38-encoded C-terminal 34 amino acid deletion) in affected son’s LCLs and fibroblasts but normal full-length THOC2 protein in the unaffected carrier mother on western blots probed with anti-THOC2-I antibody that binds to a region between amino acids 1,400–1,450 coded by Ex32-33 mRNA sequences (compare lanes 1 and 2, upper panel, Figure 5A and lanes 3 and 4, Figure 5B). Notably, C-terminal THOC2 truncated protein levels were higher in both the affected LCLs and fibroblasts (Figures 5A,B). As expected, the anti-THOC2-II antibody that binds to a region between amino acids 1543–1593 coded by Ex37-38 mRNA sequences (lower panel) did not detect the THOC2 protein in affected son’s LCLs and fibroblasts that express a C-terminally-deleted THOC2 protein. This antibody however detected THOC2 protein in the carrier mother-derived cells as she expressed normal full-length THOC2 protein (compare lanes 1 and 2, lower panel, Figure 5A and lanes 3 and 4, Figure 5B). THOC1 and THOC5 levels were similar in fibroblasts of the affected son and carrier mother (Figure 5A). Likewise, anti-THOC2-I antibody detected nuclear THOC2 protein in both the affected son and carrier mother’s fibroblasts by immunofluorescence but anti-THOC2-II antibody, as expected, detected the THOC2 protein in the mother’s but not son’s fibroblasts (Figures 6A–C). Normal nuclear THOC2 localization was detected in fibroblasts of the affected son and carrier mother (Figure 6F). We observed no significant difference in THOC2 mRNA level in LCLs and fibroblasts of the affected son and carrier mother (Figure 7A).
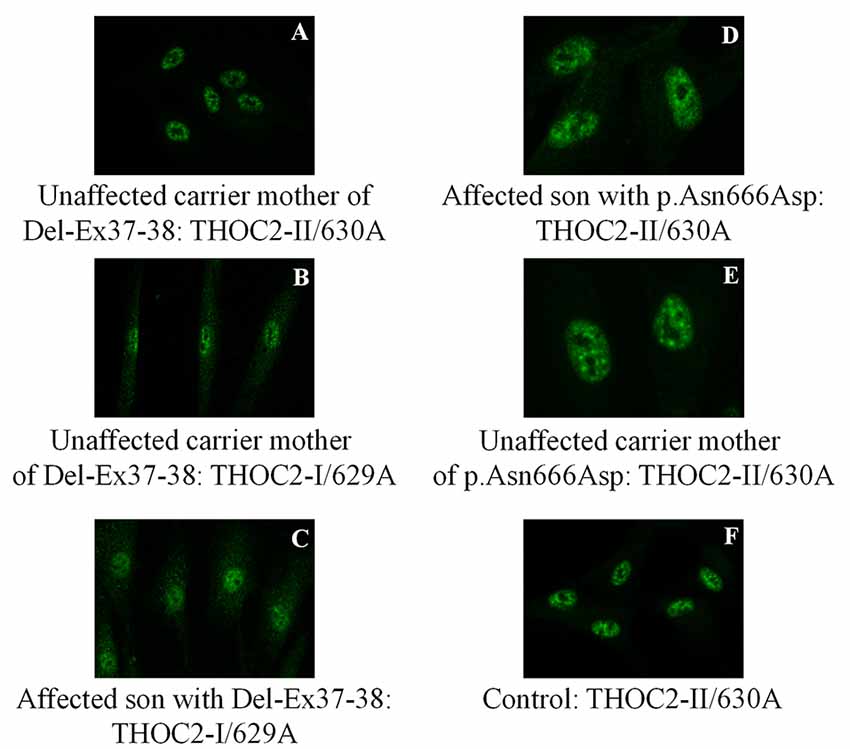
Figure 6. THOC2 localization is unaltered in variant fibroblasts. THOC2-II antibody detects THOC2 protein in fibroblasts of the Del-Ex37-38 unaffected carrier mother (A) but not affected son (not shown, as we detected no fluorescence signal). However, the THOC2-I antibody detects THOC2 protein in fibroblasts of unaffected carrier mother (B) and the affected son with Del-Ex37-38 (C). Localization p.Asn666Asp THOC2 protein in fibroblasts of an affected son (D), his unaffected carrier mother (E) and control (F).
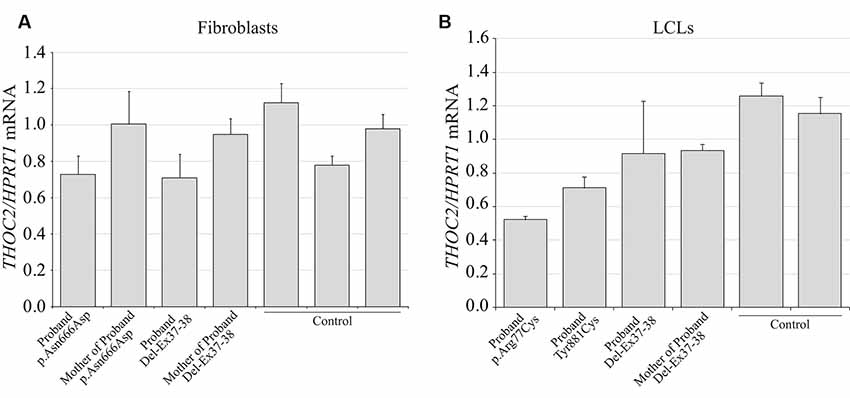
Figure 7. THOC2 mRNA expression in the variant cell lines. cDNA was generated by reverse transcribing the total RNA extracted from variant fibroblasts (A), LCLs (B) and appropriate controls using SuperScript IV reverse transcriptase and assayed for THOC2 expression (relative to HPRT1 housekeeping gene) with SYBR green master mix and primer pairs listed in Supplementary Table S1. Assays were performed two times independently and error bars show SDs.
THOC2 Missense Variants
LCLs were available from affected individuals’ hemizygous for the p.Arg77Cys and p.Tyr881Cys variants and fibroblasts from the affected individual and his carrier mother with the Trp.Asn666Asp variant (Figure 5). p.Arg77Cys and p.Tyr881Cys THOC2 variants resulted in reduced protein levels compared to control LCLs (Figures 5B,C). THOC1 and THOC5 levels were also moderately reduced in these LCLs (Figures 5B,C). Interestingly, a reduction in p.Arg77Cys and p.Tyr881Cys THOC2 variant protein levels were similar to unstable p.Leu438Pro variant THOC2 protein reported previously (Figure 5C; Kumar et al., 2015). Note that RT-qPCR assays in p.Arg77Cys and p.Tyr881Cys THOC2 variant LCLs also showed reduced mRNA expression compared to unrelated normal controls (Figure 7B). Western blotting on p.Asn666Asp fibroblasts also showed a small but reproducible reduction in THOC2 protein and related THO complex subunits THOC1, THOC3 and THOC5 compared to his unaffected carrier mother and unrelated normal controls (Figure 5D). p.Asn666Asp THOC2 reduction was much less pronounced than that of highly unstable p.Leu438Pro THOC2 as well as THOC1 and THOC5 proteins in affected fibroblasts (Figure 5D). p.Asn666Asp THOC2 localization was not affected (Figures 6D,E). Levels of mRNA expression in p.Asn666Asp affected LCLs were similar to that of the patient’s carrier mother and unrelated controls (Figure 7A).
Discussion
TREX-mediated mRNA export is a fundamental process that is essential for the export of mRNA from the cytoplasm to the nucleus to allow translation of the normal level of protein required for efficient growth and development. We and others have presented evidence showing that perturbed mRNA export leads to altered cellular function, thus causing a range of diseases (Di Gregorio et al., 2013; Kumar et al., 2015, 2018; Heath et al., 2016; Mattioli et al., 2019). During the last few years, we have focussed on the association of variation in the THOC2 gene and NDDs. THOC2 is intolerant to complete loss of function [probability of loss of function or pLI = 1; o/e = 0.02 (0.01–0.08): gnomAD v.2.1.1]. We first reported four THOC2 missense inherited variants in the affected individuals with variable degrees of ID and commonly observed features such as speech delay, elevated BMI, short stature, seizure disorders, gait disturbance, and tremors (Kumar et al., 2015). In 2018, we reported an additional six affected individuals from five unrelated families with two de novo and three maternally inherited pathogenic or likely pathogenic missense and splicing-defective THOC2 variants. The study also included a de novo variant in a female with epileptic encephalopathy. We observed a core ID phenotype and common behavioral features, infantile hypotonia, gait disturbance and growth impairment (Kumar et al., 2018). In the current cohort, we present molecular and clinical findings on ten additional THOC2 variant individuals, confirming a more severe THOC2-related phenotype associated with severe hypotonia, respiratory and other congenital anomalies, and summarize the phenotypic impact of all THOC2 variants to date in 38 individuals. This publication, therefore, may serve as the most up to date reference of this X-linked NDD condition.
Variants in TREX subunit proteins can alter their stability (Kumar et al., 2015, 2018; e.g., THOC2 ID variants), interaction (e.g., THOC6 syndromic ID variants; Mattioli et al., 2019) and/or localization (e.g., THOC6 syndromic ID variants; Mattioli et al., 2019). We observed reduced levels of p.Arg77Cys THOC2 (although the stability of this variant exogenously expressed in HEK293T cells was uninformative, see Figure 3 in Kumar et al., 2018) and p.Tyr881Cys THOC2 in the affected LCLs and p.Asn666Asp THOC2 affected fibroblasts (without THOC2 mislocalization). Variant THOC2 protein reduction also impacted the THOC1, THOC3 and THOC5 stability in the variant cells, which is consistent with a number of reports showing that instability of THO subunits—either by missense changes or siRNA-mediated knockdown—results in reduced levels of other THO subunits (Chi et al., 2013; Kumar et al., 2015, 2018). As the THOC2 mRNA levels in the affected LCLs or fibroblasts were comparable to the controls, the reduced THOC2 variant protein levels are most likely due to enhanced proteasome-mediated degradation of ubiquitinated THOC2 (Lopitz-Otsoa et al., 2012). Reduced or depleted THO subunit proteins have been shown to cause severe to mild nuclear mRNA retention (Chi et al., 2013) and hence can cause variable molecular and cellular consequences both in mammalian cells and lower organisms. For example, THOC2 depletion impacts chromosome alignment, mitotic progression, and genomic stability in human HeLa cells (Yamazaki et al., 2010). Thoc2 knockdown in Drosophila S2 cells inhibits cell proliferation and heat-shock mRNA export (Rehwinkel et al., 2004). Thoc2 depletion results in a significant increase in the length of neurites in cultured rat primary hippocampal neurons and C. elegans thoc2 knockout worms are slow-growing, sterile, have functional defects in specific sensory neurons and die prematurely (Di Gregorio et al., 2013). Danio rerio Thoc2 is essential for embryonic development (Amsterdam et al., 2004). Interestingly, a de novo translocation creating a PTK2-THOC2 fusion that reduced the expression of the two genes in a female patient was implicated in cognitive impairment and cerebellar hypoplasia (also reported in 4/19 of our cases; Di Gregorio et al., 2013). THOC2 or THOC5 depletion leads to dedifferentiation of vascular smooth muscle cells, characterized by increased migration and proliferation (Yuan et al., 2018). Thoc2 and Thoc5 depletion was shown to cause retention of self-renewal (Oct4) and pluripotent gene transcripts (Nanog, Sox2, Esrrb, and Klf4) as well as increased differentiation gene expression (Cdx2, Gata3 and Gata4) in mouse embryonic stem cells, resulting in a decrease in cell pluripotency and proliferation (Wang et al., 2013).
We observed accumulation of 34 C-terminal amino acid THOC2 truncated protein in Del-Ex37-38 LCLs and fibroblasts that is similar to the accumulation of 110 C-terminal amino acid deleted THOC2 protein in fibroblasts of the affected individual carrying the exon35:c.4450–2A>G splice-variant and potential loss of the 1,300–1,593 amino acid putative RNA binding domain (RBD) in a proportion of blood cells with the exon28:c.3503 + 4A>C defective splice-variant (Kumar et al., 2018). The truncated proteins in these individuals had normal nuclear localization. Together, the affected individuals (at least in Del-Ex37-38 and exon35:c.4450–2A>G splice-variant) not only have truncated THOC2 protein (where a part of the RBD is lost) but actually more of it, which may indicate presence of a degradation signal within this region and/or stabilization due to structural changes to the THOC2 protein that may or may not be impacting interactions with other TREX subunits. That C-terminally truncated THOC2 protein potentially has perturbed function is consistent with yeast Tho2 studies showing that Tho2Δ1408–1597 (small C-terminal RBD deletion) grows slower than the wild-type strain and ThoΔ1271–1597 (complete RBD deletion) strain barely grows at restrictive temperature (Peña et al., 2012).
THOC2 depletion leads to almost complete retention of mRNAs in the cell nucleus, suggesting that it is an essential mRNA export factor whose complete knockout can be potentially toxic to the cell (Chi et al., 2013). We, therefore, suggest that all the identified THOC2 variants result in partial loss-of-function that alters normal mRNA export in neuronal and likely other cells, thereby causing a broad range of clinical presentations in the affected individuals carrying the THOC2 variants. However, although THOC2 depletion causes bulk mRNA nuclear retention in HeLa cells (Chi et al., 2013), whether THOC2 variants affect—bulk or specific—mRNA export in neuronal cells has not been elucidated. TREX complex was originally thought to be a bulk mRNA export pathway but a number of reports have shown its role in specific mRNA export. For example, the Thoc1/2 and Thoc5 or Thoc6 are responsible for nuclear export of only a subset of mRNAs (e.g., heat shock mRNAs) in Drosophila and mammalian cells, respectively (Rehwinkel et al., 2004; Katahira et al., 2009; Guria et al., 2011). Thoc2 and Thoc5 selectively bind and regulate the export of mRNAs involved in the maintenance of pluripotency (e.g., Nanog, Sox2, Esrrb, and Klf4 mRNAs (Wang et al., 2013) and Thoc5 in hematopoiesis (Mancini et al., 2010).
The published data in conjunction with our molecular and cellular data, and broader clinical presentations in our cohort indicate that THOC2 reduction (and consequently other THO subunits) or potential alterations in TREX subunit interactions due to intragenic deletion of C-terminal RBD and/or structural interactions as a result of THOC2 missense changes result in perturbed mRNA export, causing cellular dysfunction and NDDs. This view is supported by the published evidence showing that slight perturbations in mRNA export can lead to NDDs (Beaulieu et al., 2013; Kumar et al., 2015, 2018; Mattioli et al., 2019), neurodegenerative disease (Woerner et al., 2016) and cancer (Domínguez-Sánchez et al., 2011). There are also examples showing how variants in the human mRNA export mediator GLE1 result in a severe fetal motor neuron disease (Nousiainen et al., 2008) and amyotrophic lateral sclerosis (ALS; Kaneb et al., 2015), and toxic CUG expansion in the 3′ untranslated region of the DM protein kinase mRNA can cause myotonic dystrophy type I by impairing RNA transport out of the nucleus (Brook et al., 1992).
Based on clinical and, where available, molecular data on 38 individuals, we have come to a realization that the THOC2-linked NDDs can present with highly variable phenotypes. Intriguingly, we also identify a case of severe expressive dysphasia without obvious intellectual impairment. This particular case and observation still awaits replication and as such further validation. In hindsight, it is perhaps not surprising that even subtle perturbation to a crucial cellular mechanism like TREX mRNA export can yield complex and often variable clinical presentations which can not only be driven by the THOC2 gene variation itself, but can also be modified by genome-wide polygenic risk (Niemi et al., 2018) or other yet to be identified factors.
Data Availability Statement
The datasets have been uploaded to ClinVar and are available here: https://www.ncbi.nlm.nih.gov/clinvar/ SCV001132790[clv_acc] under accession numbers SCV001132790, SCV001132791, SCV001132792, SCV001132793, SCV001132794, SCV001132795, SCV001132796 and SCV001132797.
Ethics Statement
The studies involving human participants were reviewed and approved by WCHN Human Research Ethics Committee and University of Adelaide, Adelaide. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author Contributions
RK, EP, MF, and JG designed the experiments and wrote the article. RK performed the molecular studies. AEG helped with pathogenicity predictions. RC with tissue culture. SB, OA, DD, YE, CC, AG, MS, RM, LB, MT, DNA, VA, MB, GB, LD, RH, CM, GC, MW, and JB contributed THOC2 variants, photos, clinical data, and in some cases, the patient-derived cells for molecular investigations. JD reviewed MRI neuro-images. EP and ML collated the clinical data. All authors read and made suggestions on the manuscript.
Funding
This work was supported by National Health and Medical Research Council (NHMRC) Program Grant APP1091593 (JG), NHMRC Project Grant APP1163240 (JG, RK), Australian Research Council Discovery Project 170103090 (JG, RK) and Senior Research Fellowship APP1041920 (JG). The Toronto group (RH, CM, GC) acknowledges funding support from the Norm Saunders Complex Care Initiative, SickKids Centre for Genetic Medicine, and the University of Toronto McLaughlin Centre. We thank the individuals and families for their contribution to this study. The c.3300G>T p.(Trp1100Cys) THOC2 variant was identified as part of the 100,000 Genomes Project that is managed by Genomics England Limited (a wholly-owned company of the Department of Health and Social Care) and funded by the National Institute for Health Research and NHS England. The Wellcome Trust, Cancer Research UK and the Medical Research Council have also funded research infrastructure. The 100,000 Genomes Project uses data provided by patients and collected by the National Health Service as part of their care and support.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
- ^ https://humandiseasegenes.nl/thoc2/
- ^ https://www.ncbi.nlm.nih.gov/clinvar/?term=“GOLD%20service%2C%20Hunter%20New%20England%20Health”[submitter]+AND+“THOC2”[gene]
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnmol.2020.00012/full#supplementary-material.
References
Adzhubei, I. A., Schmidt, S., Peshkin, L., Ramensky, V. E., Gerasimova, A., Bork, P., et al. (2010). A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249. doi: 10.1038/nmeth0410-248
Amos, J. S., Huang, L., Thevenon, J., Kariminedjad, A., Beaulieu, C. L., Masurel-Paulet, A., et al. (2017). Autosomal recessive mutations in THOC6 cause intellectual disability: syndrome delineation requiring forward and reverse phenotyping. Clin. Genet. 91, 92–99. doi: 10.1111/cge.12793
Amsterdam, A., Nissen, R. M., Sun, Z., Swindell, E. C., Farrington, S., and Hopkins, N. (2004). Identification of 315 genes essential for early zebrafish development. Proc. Natl. Acad. Sci. U S A 101, 12792–12797. doi: 10.1073/pnas.0403929101
Beaulieu, C. L., Huang, L., Innes, A. M., Akimenko, M. A., Puffenberger, E. G., Schwartz, C., et al. (2013). Intellectual disability associated with a homozygous missense mutation in THOC6. Orphanet J. Rare Dis. 8:62. doi: 10.1186/1750-1172-8-62
Brook, J. D., Mccurrach, M. E., Harley, H. G., Buckler, A. J., Church, D., Aburatani, H., et al. (1992). Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell 69:385. doi: 10.1016/0092-8674(92)90418-c
Carter, H., Douville, C., Stenson, P. D., Cooper, D. N., and Karchin, R. (2013). Identifying Mendelian disease genes with the variant effect scoring tool. BMC Genomics 14:S3. doi: 10.1186/1471-2164-14-s3-s3
Chi, B., Wang, Q., Wu, G., Tan, M., Wang, L., Shi, M., et al. (2013). Aly and THO are required for assembly of the human TREX complex and association of TREX components with the spliced mRNA. Nucleic Acids Res. 41, 1294–1306. doi: 10.1093/nar/gks1188
Choi, Y., and Chan, A. P. (2015). PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 31, 2745–2747. doi: 10.1093/bioinformatics/btv195
Davydov, E. V., Goode, D. L., Sirota, M., Cooper, G. M., Sidow, A., and Batzoglou, S. (2010). Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput. Biol. 6:e1001025. doi: 10.1371/journal.pcbi.1001025
Di Gregorio, E., Bianchi, F. T., Schiavi, A., Chiotto, A. M., Rolando, M., Verdun Di Cantogno, L., et al. (2013). A de novo X;8 translocation creates a PTK2-THOC2 gene fusion with THOC2 expression knockdown in a patient with psychomotor retardation and congenital cerebellar hypoplasia. J. Med. Genet. 50, 543–551. doi: 10.1136/jmedgenet-2013-101542
Domínguez-Sánchez, M. S., Saez, C., Japon, M. A., Aguilera, A., and Luna, R. (2011). Differential expression of THOC1 and ALY mRNP biogenesis/export factors in human cancers. BMC Cancer 11:77. doi: 10.1186/1471-2407-11-77
Guria, A., Tran, D. D., Ramachandran, S., Koch, A., El Bounkari, O., Dutta, P., et al. (2011). Identification of mRNAs that are spliced but not exported to the cytoplasm in the absence of THOC5 in mouse embryo fibroblasts. RNA 17, 1048–1056. doi: 10.1261/rna.2607011
Heath, C. G., Viphakone, N., and Wilson, S. A. (2016). The role of TREX in gene expression and disease. Biochem. J. 473, 2911–2935. doi: 10.1042/bcj20160010
Kaneb, H. M., Folkmann, A. W., Belzil, V. V., Jao, L. E., Leblond, C. S., Girard, S. L., et al. (2015). Deleterious mutations in the essential mRNA metabolism factor, hGle1, in amyotrophic lateral sclerosis. Hum. Mol. Genet. 24, 1363–1373. doi: 10.1093/hmg/ddu545
Karczewski, K. J., Francioli, L. C., Tiao, G., Cummings, B. B., Alföldi, J., Wang, Q., et al. (2019). Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. bioRxiv [Preprint]. doi: 10.1101/531210
Katahira, J., Inoue, H., Hurt, E., and Yoneda, Y. (2009). Adaptor Aly and co-adaptor Thoc5 function in the Tap-p15-mediated nuclear export of HSP70 mRNA. EMBO J. 28, 556–567. doi: 10.1038/emboj.2009.5
Kircher, M., Witten, D. M., Jain, P., O’Roak, B. J., Cooper, G. M., and Shendure, J. (2014). A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 46, 310–315. doi: 10.1038/ng.2892
Köhler, S., Carmody, L., Vasilevsky, N., Jacobsen, J. O. B., Danis, D., Gourdine, J. P., et al. (2019). Expansion of the human phenotype ontology (HPO) knowledge base and resources. Nucleic Acids Res. 47, D1018–D1027. doi: 10.1093/nar/gky1105
Kumar, R., Corbett, M. A., Van Bon, B. W., Woenig, J. A., Weir, L., Douglas, E., et al. (2015). THOC2 mutations implicate mRNA-export pathway in X-linked intellectual disability. Am. J. Hum. Genet. 97, 302–310. doi: 10.1016/j.ajhg.2015.05.021
Kumar, R., Gardner, A., Homan, C. C., Douglas, E., Mefford, H., Wieczorek, D., et al. (2018). Severe neurocognitive and growth disorders due to variation in THOC2, an essential component of nuclear mRNA export machinery. Hum. Mutat. 39, 1126–1138. doi: 10.1002/humu.23557
Laemmli, U. K. (1970). Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685. doi: 10.1038/227680a0
Lopitz-Otsoa, F., Rodriguez-Suarez, E., Aillet, F., Casado-Vela, J., Lang, V., Matthiesen, R., et al. (2012). Integrative analysis of the ubiquitin proteome isolated using tandem ubiquitin binding entities (TUBEs). J. Proteomics 75, 2998–3014. doi: 10.1016/j.jprot.2011.12.001
Mancini, A., Niemann-Seyde, S. C., Pankow, R., El Bounkari, O., Klebba-Farber, S., Koch, A., et al. (2010). THOC5/FMIP, an mRNA export TREX complex protein, is essential for hematopoietic primitive cell survival in vivo. BMC Biol. 8:1. doi: 10.1186/1741-7007-8-1
Mattioli, F., Isidor, B., Abdul-Rahman, O., Gunter, A., Huang, L., Kumar, R., et al. (2019). Clinical and functional characterization of recurrent missense variants implicated in THOC6-related intellectual disability. Hum. Mol. Genet. 28, 952–960. doi: 10.1093/hmg/ddy391
Niemi, M. E. K., Martin, H. C., Rice, D. L., Gallone, G., Gordon, S., Kelemen, M., et al. (2018). Common genetic variants contribute to risk of rare severe neurodevelopmental disorders. Nature 562, 268–271. doi: 10.1038/s41586-018-0566-4
Nousiainen, H. O., Kestilä, M., Pakkasjärvi, N., Honkala, H., Kuure, S., Tallila, J., et al. (2008). Mutations in mRNA export mediator GLE1 result in a fetal motoneuron disease. Nat. Genet. 40, 155–157. doi: 10.1038/ng.2007.65
Pejaver, V., Urresti, J., Lugo-Martinez, J., Pagel, K. A., Lin, G. N., Nam, H.-J., et al. (2017). MutPred2: inferring the molecular and phenotypic impact of amino acid variants. bioRxiv [Preprint]. doi: 10.1101/134981
Peña, A., Gewartowski, K., Mroczek, S., Cuellar, J., Szykowska, A., Prokop, A., et al. (2012). Architecture and nucleic acids recognition mechanism of the THO complex, an mRNP assembly factor. EMBO J. 31, 1605–1616. doi: 10.1038/emboj.2012.10
Pollard, K. S., Hubisz, M. J., Rosenbloom, K. R., and Siepel, A. (2010). Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 20, 110–121. doi: 10.1101/gr.097857.109
Rehwinkel, J., Herold, A., Gari, K., Kocher, T., Rode, M., Ciccarelli, F. L., et al. (2004). Genome-wide analysis of mRNAs regulated by the THO complex in Drosophila melanogaster. Nat. Struct. Mol. Biol. 11, 558–566. doi: 10.1038/nsmb759
Reva, B., Antipin, Y., and Sander, C. (2011). Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res. 39:e118. doi: 10.1093/nar/gkr407
Sim, N. L., Kumar, P., Hu, J., Henikoff, S., Schneider, G., and Ng, P. C. (2012). SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 40, W452–W457. doi: 10.1093/nar/gks539
Srivastava, A. K., and Schwartz, C. E. (2014). Intellectual disability and autism spectrum disorders: causal genes and molecular mechanisms. Neurosci. Biobehav. Rev. 46, 161–174. doi: 10.1016/j.neubiorev.2014.02.015
Tărlungeanu, D. C., and Novarino, G. (2018). Genomics in neurodevelopmental disorders: an avenue to personalized medicine. Exp. Mol. Med. 50:100. doi: 10.1038/s12276-018-0129-7
Uhlén, M., Fagerberg, L., Hallström, B. M., Lindskog, C., Oksvold, P., Mardinoglu, A., et al. (2015). Proteomics. Tissue-based map of the human proteome. Science 347:1260419. doi: 10.1126/science.1260419
Wang, L., Miao, Y.-L., Zheng, X., Lackford, B., Zhou, B., Han, L., et al. (2013). The THO complex regulates pluripotency gene mRNA export and controls embryonic stem cell self-renewal and somatic cell reprogramming. Cell Stem Cell 13, 676–690. doi: 10.1016/j.stem.2013.10.008
Woerner, A. C., Frottin, F., Hornburg, D., Feng, L. R., Meissner, F., Patra, M., et al. (2016). Cytoplasmic protein aggregates interfere with nucleocytoplasmic transport of protein and RNA. Science 351, 173–176. doi: 10.1126/science.aad2033
Yamazaki, T., Fujiwara, N., Yukinaga, H., Ebisuya, M., Shiki, T., Kurihara, T., et al. (2010). The closely related RNA helicases, UAP56 and URH49, preferentially form distinct mRNA export machineries and coordinately regulate mitotic progression. Mol. Biol. Cell 21, 2953–2965. doi: 10.1091/mbc.e09-10-0913
Keywords: mRNA export, THOC2, intellectual disability, neurodevelopmental disorders, microdeletion
Citation: Kumar R, Palmer E, Gardner AE, Carroll R, Banka S, Abdelhadi O, Donnai D, Elgersma Y, Curry CJ, Gardham A, Suri M, Malla R, Brady LI, Tarnopolsky M, Azmanov DN, Atkinson V, Black M, Baynam G, Dreyer L, Hayeems RZ, Marshall CR, Costain G, Wessels MW, Baptista J, Drummond J, Leffler M, Field M and Gecz J (2020) Expanding Clinical Presentations Due to Variations in THOC2 mRNA Nuclear Export Factor. Front. Mol. Neurosci. 13:12. doi: 10.3389/fnmol.2020.00012
Received: 18 October 2019; Accepted: 15 January 2020;
Published: 11 February 2020.
Edited by:
Robert J. Harvey, University of the Sunshine Coast, AustraliaReviewed by:
Jia Nee Foo, Nanyang Technological University, SingaporeFerdinando Di Cunto, University of Turin, Italy
Copyright © 2020 Kumar, Palmer, Gardner, Carroll, Banka, Abdelhadi, Donnai, Elgersma, Curry, Gardham, Suri, Malla, Brady, Tarnopolsky, Azmanov, Atkinson, Black, Baynam, Dreyer, Hayeems, Marshall, Costain, Wessels, Baptista, Drummond, Leffler, Field and Gecz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jozef Gecz, am96ZWYuZ2VjekBhZGVsYWlkZS5lZHUuYXU=
† These authors have contributed equally to this work