 Antonio Monaco
Antonio Monaco Alessandro Fraldi
Alessandro Fraldi- 1Telethon Institute of Genetics and Medicine, Pozzuoli, Italy
- 2Department of Translational Medicine, University of Naples “Federico II,” Naples, Italy
Many neurodegenerative conditions are characterized by the deposition of protein aggregates (mainly amyloid-like) in the central nervous system (CNS). In post-mitotic CNS cells protein aggregation causes cytotoxicity by interfering with various cellular functions. Mutations in different genes may directly cause protein aggregation. However, genetic factors together with aging may contribute to the onset of protein aggregation also by affecting cellular degradative functions, in particular the autophagy-lysosomal pathway (ALP). Increasing body of evidence show that ALP dysfunction and protein aggregation are functionally interconnected and induce each other during neurodegenerative processes. We will summarize the findings supporting these concepts by focusing on lysosomal storage diseases (LSDs), a class of metabolic inherited conditions characterized by global lysosomal dysfunction and often associated to a severe neurodegenerative course. We propose a model by which the inherited lysosomal defects initiate aggregate-prone protein deposition, which, in turns, worsen ALP degradation function, thus generating a vicious cycle, which boost neurodegenerative cascades.
Protein Aggregation in Neurodegeneration Diseases
A hallmark of many neurodegenerative diseases is the progressive formation of insoluble protein aggregates that, in most cases, are composed by amyloidogenic proteins (Chiti and Dobson, 2006). Indeed, under different stress conditions, several intrinsically disordered proteins (normally soluble) misfold and undergo structural changes and self-assembly that ultimately lead to their aggregation into insoluble deposits, referred to as amyloids (Dobson, 2003). Amyloid deposits are characterized by a fibrillar morphology and a cross-β structure, whereby intermolecular main-chain hydrogen bonding acts as one major stabilizing interaction (Chiti and Dobson, 2006). Although in some neurodegenerations (e.g., in the polyglutamine diseases; see below) aggregation per se could be not the cause of the observed neurotoxicity, generally, amyloid aggregation represents a therapeutic target for neurological conditions since it can cause cytotoxicity either by directly interfering with various cellular functions or because the aggregates sequester other proteins, which play essential cellular functions (Ciechanover and Kwon, 2015; Gallardo et al., 2016). Nevertheless, the mechanisms underlying neurotoxicity driven by amyloid deposition are not completely understood.
Amyloid deposits found in neurodegenerative diseases are often characterized by one main component; however, in some neurodegenerative conditions several amyloidogenic proteins may contribute to amyloid deposition (Table 1). Alzheimer’s disease (AD), the most common neurodegenerative disorder is characterized by deposition of amyloid plaques, whose main component is the amyloid-beta (Aβ) protein (Goedert and Spillantini, 2006). α-Synuclein accumulation and aggregation within Lewy bodies and neurites of the CNS in the form of amyloid fibrils plays a central role in the pathophysiology of Parkinson’s disease (PD) and in a subset of neurodegenerative conditions known as dementias with Lewy bodies (Spillantini et al., 1997). Polyglutamine (polyQ) expansions in unrelated proteins and consequent intracellular accumulation of the mutant protein in inclusion bodies is the underlying cause of a number of inherited rare neurodegenerative disorders, including Huntington’s disease (HD) (polyQ expansion in the huntingtin protein), spinal and bulbar muscular atrophy (SBMA) (polyQ expansion in the androgen receptor protein), and some forms of spinocerebellar ataxias (polyQ expansion in ataxin protein) (Perutz, 1999). Neurofibrillary tangles, which consists of fibrillar aggregates of hyperphosphorylated tau protein, are commonly seen in aging and AD brain and are correlated with decline of brain functions in these conditions (Goedert and Spillantini, 2006). Frontotemporal dementia (FTD), another neuropathy with protein aggregation has also been associated with toxic intracellular aggregates of hyperphosphorylated tau (Lee et al., 2001). Interestingly, some forms of FTD are negative for tau inclusions, while are positive for inclusions containing misfolded TAR DNA-binding protein 43 (TDP-43) (Kwong et al., 2007). TDP-43 inclusions are also found in the amyotrophic lateral sclerosis (ALS), the most common forms of motor neuron disease (Kwong et al., 2007). Aggregate containing the carboxy terminal fragment of APP (APP-βCTF) have been found in Down Syndrome, a neurodevelopmental disorder with pathological features common to the early onset forms of AD (Ying et al., 2019). Amyloid aggregates containing misfolded prion protein (PrP) cause the so-called prion diseases, a group of rare neurodegenerative conditions characterized by the capability of misfolded PrP to transmit their pathological shape onto normal variants of the same protein (Aguzzi and Heikenwalder, 2006). The accumulation of different unrelated misfolded proteins, including the neuronal intermediate filaments (NFs), is a hallmark of the Charcot–Marie–Tooth disease, the most common inherited neuromuscular disease (Theocharopoulou and Vlamos, 2015; Didonna and Opal, 2019). Aggregates containing NFs are frequently observed also in other motor neuron diseases. Lysosomal storage diseases (LSDs) are a group of metabolic diseases caused by inherited defects in lysosomal or non-lysosomal proteins leading to lysosomal storage and global dysfunction often associated with neurodegeneration (Schultz et al., 2011; Platt et al., 2012, 2018). In several LSDs the primary storage caused by the specific inherited lysosomal defect is associated to the deposition of amyloidogenic proteins. Accumulation of α-synuclein has been shown to trigger neurotoxicity through aggregation-dependent mechanisms in Gaucher disease, a severe neurological LSD belonging to the sphingolipidoses, a family of LSDs characterized by primary lipid storage (Mazzulli et al., 2011). α-Synuclein aggregation and neurofibrillary tangles have been observed also in other sphingolipidoses, such as the Niemann–Pick and the Krabbe diseases (Suzuki et al., 1995; Saito et al., 2004; Smith et al., 2014). Accumulation and amyloidogenic processing of an oversialylated APP in lysosomes, and extracellular release of Aβ peptides have been observed in a mouse model of sialidosis, an LSDs caused by the deficiency of the lysosomal sialidase NEU1 (Annunziata et al., 2013). Accumulation of APP-βCTF was found in GM1 gangliosidosis, an LSD characterized by primary lysosomal storage of GM1 ganglioside in neurons (Zha et al., 2004). Mucopolysaccharidoses (MPS) are a family of LSDs with primary storage of glycosaminoglycans (GAGs) due to the deficiency of lysosomal enzymes required for GAG stepwise degradation (Clarke, 2008). Amyloid aggregation has been observed in different types of MPSs, including the MPS type IIIA, one of the most common and severe form of neurodegenerative LSD (Ginsberg et al., 1999; Hamano et al., 2008; Ohmi et al., 2009; Martins et al., 2015; Beard et al., 2017; Sambri et al., 2017; Monaco et al., 2020). In particular, by studying a mouse model of MPS-IIIA, we have shown that brain deposition of α-synuclein together with other amyloidogenic proteins including tau, Aβ, and PrP trigger neurodegenerative processes by both loss-of-function (LOF) (Sambri et al., 2017) and gain of toxic function mechanisms (Monaco et al., 2020) (see next sections for further discussion).
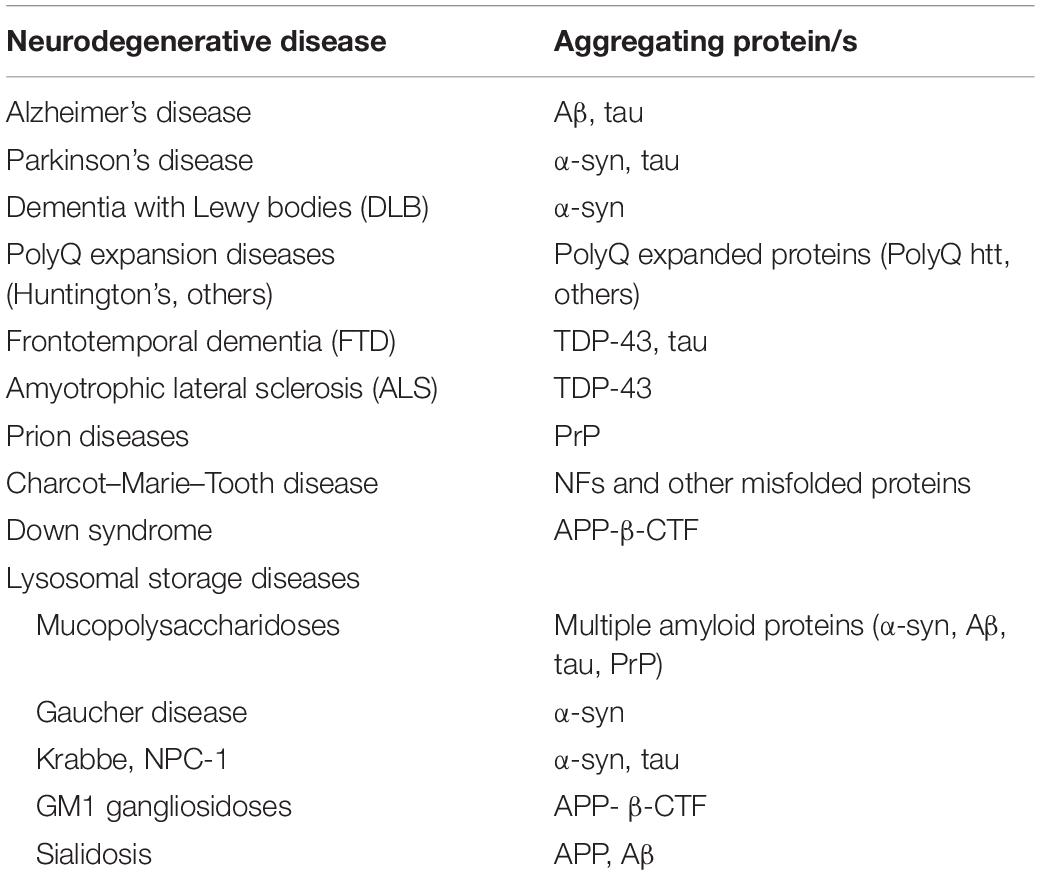
Table 1. Protein aggregation in neurodegenerative diseases.
Factors Determining Protein Aggregation
There are two main factors that cause protein aggregation in neurodegenerative diseases: Gain-of-function (GOF) dominant mutations in genes encoding aggregate-prone proteins and the decline of cellular degradation functions, in particular of the autophagy-lysosomal system (Figure 1).
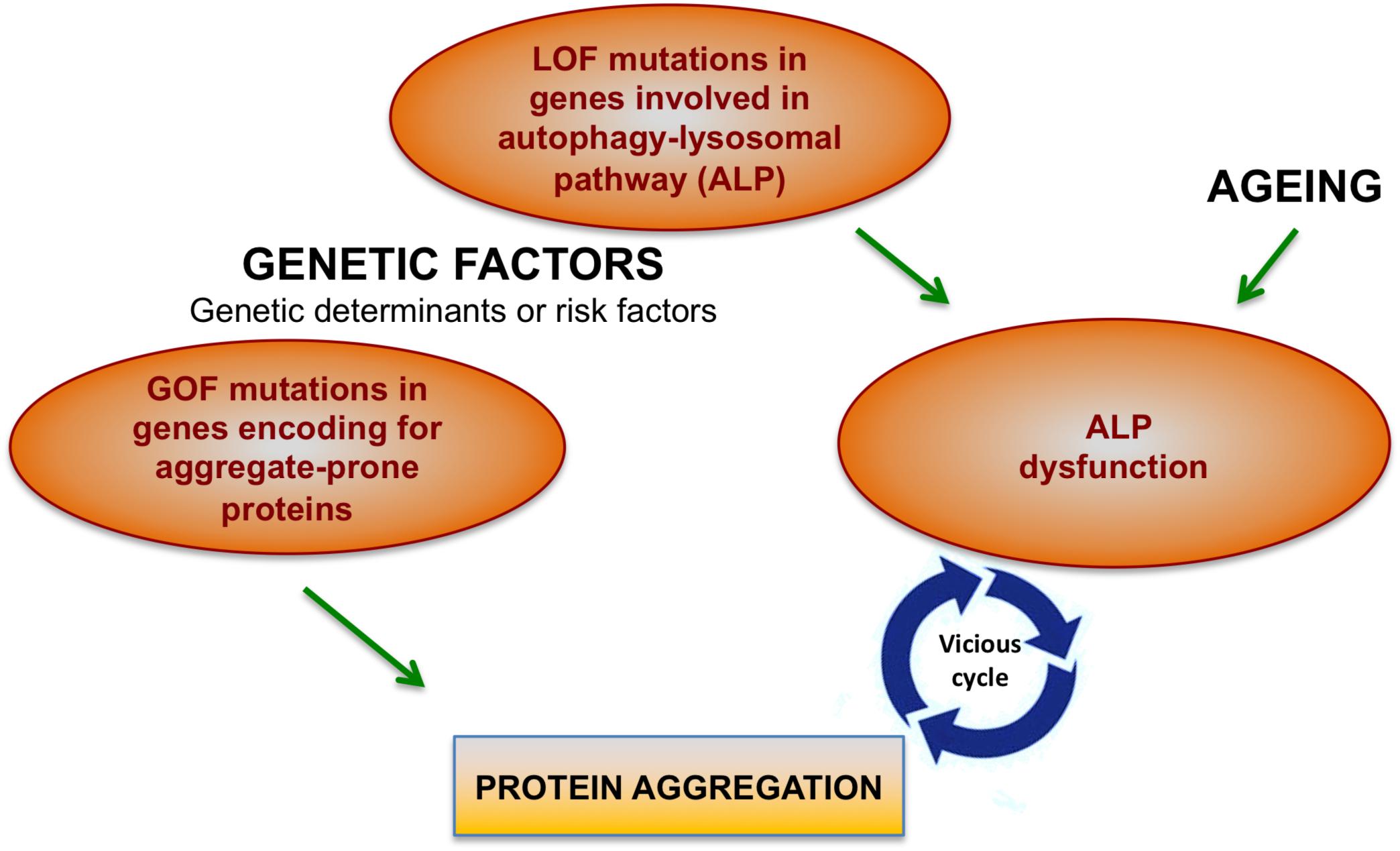
Figure 1. Factors contributing to protein aggregation in neurodegenerative conditions. Mutations in different genes may directly cause protein aggregation. However, genetic factors together with aging may contribute to the onset of protein aggregation also by affecting cellular degradative functions, in particular the autophagy-lysosomal pathway (ALP). Increasing body of evidence show that ALP dysfunction and protein aggregation are functionally and closely interconnected and induce each other during neurodegenerative processes.
GOF Mutations in Genes Encoding Aggregate-Prone Proteins
Protein aggregation may be directly caused by dominant GOF mutations in gene encoding aggregate-prone proteins or precursors of aggregate-prone proteins. GOF mutations in the gene encoding huntingtin lead to polyQ expansion and huntingtin aggregation, thus causing HD (Perutz, 1999). In the AD, ∼5% of the forms are Mendelian and are caused by mutations in the genes encoding the amyloid precursor protein (APP) and presenilin1/2 (PSEN1/2). These genes are directly involved in the “amyloidogenic cascade” by which the APP protein is sequentially cleaved and processed to generate aggregate-prone Aβ peptides (Hardy and Selkoe, 2002). While PSEN1/2 mutations increase the activity of γ-secretase that enhances Aβ peptides production, GOF mutations in APP gene increase the generation of Aβ peptides either by making APP a better substrate for its processing or by changing the biophysical properties of the Aβ peptide, thus rendering it more likely to aggregate. Among the rare Mendelian forms of PD a number of cases are associated with dominant GOF mutations in the gene encoding α-synuclein (SNCA), which lead to the abnormal aggregation of the protein (Bras et al., 2015). Some GOF mutations in the PRNP gene (encoding the PrP protein) produce altered misfolded-prone versions of PrP, thus causing prion disease (Beck et al., 2010). Genetic studies identified GOF mutations in the MAPT gene encoding tau protein in some familial cases of the FTD (Rainero et al., 2017). GOF mutations in the TARDBP gene (encoding TDP-43) are associated with FTD and ALS (Kwong et al., 2007). In Down syndrome the extra gene copy of APP gene (on chromosome 21) leads to increased production of APP-βCTF (Ying et al., 2019). Finally, several GOF mutations in unrelated genes have been found to cause misfolding and accumulation of the corresponding proteins in Charcot–Marie–Tooth disease (Braathen, 2012).
Decline of the Autophagy-Lysosomal Pathway
In many neurodegenerative conditions protein aggregation may occurs without specific GOF mutations in genes encoding aggregate-prone proteins. In these conditions protein aggregation is associated to the decline of cellular degradative functions, specifically of the autophagy-lysosomal pathway (ALP) (Figure 1). ALP is a major process for degrading intracellular macromolecules and generating energy or building blocks to make other macromolecules. ALP relies on the engulfment of cargos to be degraded (macromolecules or damaged organelles) in double-membrane vesicles (autophagosomes), which, therefore, fuse with endosomes/lysosomes to form autolysosomes, where autophagosome contents are degraded by lysosomal enzymes (Yu et al., 2018). ALP plays a key role in protein homeostasis and in the clearance of protein aggregates (processes that are particularly important in non-dividing neurons). Therefore, ALP dysfunction may determine/contribute to the toxic aggregation in neurodegenerative conditions (Nixon, 2013; Fraldi et al., 2016). Accordingly, mice KO for key ALP components exhibit neuronal accumulation of aggregate-prone proteins and neurodegeneration (Hara et al., 2006; Komatsu et al., 2006). Furthermore, in the case of Aβ aggregation, it has been reported that the functionality of endo-lysosomal recycling trafficking is critical for determining the amyloidogenic cascade (Rajendran and Annaert, 2012; Das et al., 2013) and, therefore, any detrimental effect on the endo-lysosomal transport results in alterations of Aβ production (Nixon, 2017).
Autophagy-lysosomal pathway decline may be caused by genetic factors (LOF – mutations inherited in a dominant or recessive fashion), environmental factors (mainly aging) which are known to impact on degradative capability of cells (Nixon et al., 2008) or by protein aggregation itself (see next paragraph). Therefore, genetic factors trigger protein aggregation in neurodegenerative diseases either directly (GOF mutations in gene encoding aggregate-prone proteins) or indirectly (LOF mutations in ALP genes). Importantly, genetic factors can represent either the genetic determinant or a risk factor that contributes to neurodegenerative conditions (Figure 1).
Lysosomal storage diseases are the paradigm of neurodegenerative diseases associated to ALP dysfunction caused by genetic factors (Fraldi et al., 2016). Indeed, in LSDs LOF mutations in lysosomal hydrolases or in proteins involved in lysosomal biology cause lysosomal storage and global dysfunction associated to the impairment of the autophagy flux (Lieberman et al., 2012; Platt et al., 2012). A number of AD patients carrying LOF mutations in PSEN1 show lysosomal and autophagic dysfunction (Lee et al., 2010). Lysosome dysfunction in these patients can be explained by two different mechanisms, one involving defects in the lysosomal acidification machinery and the other in lysosomal Ca+2 homeostasis (Coen et al., 2012; Lee et al., 2015). Some Mendelian forms of PD are caused by mutations in ALP genes. Mutations in ATP13A2 encoding a component of the lysosomal acidification machinery (ATPase type 13A2) are associated with lysosomal dysfunction and defective autophagosomes clearance in PD (Ramirez et al., 2006). PD caused by mutations in the LRRK2 gene showed lysosomal stress and accumulation of abnormal autophagosomes (reviewed in Jin and Klionsky, 2014). PD with mutations in VPS35 is associated to defects in the retrograde transport between endosomes and the trans-Golgi network (Zimprich et al., 2011). Mutations in PINK (PTEN-induced putative kinase) or PARKIN (PD protein) genes cause PD forms characterized by defective mitophagy (Geisler et al., 2010). Dysfunction of ALP has been associated with specific mutations also in ALS (Song et al., 2012) and in CMT disease (Lee et al., 2012; BasuRay et al., 2013). LOF mutations in the genes whose products are involved in endo-lysosomal function (such as CHMP2B, progranulin, and TMEM106B genes) have been identified as the causative factors in familiar forms of FTD (Rainero et al., 2017).
Mutations in ALP genes may also represent risk/predisposing factors for disease pathogenesis. Interestingly, in PD many ALP gene mutations represent risk factors when they are in heterozygosis, while cause a specific LSD when they are in homozygosis, thus providing a strong genetic evidence linking between PD and LSDs (Shachar et al., 2011; Robak et al., 2017). The most known example of this genetic link is provided by the GBA gene encoding for the glucocerebrosidase (GCase), a lysosomal enzyme involved in the degradation of glucosylceramide. When GBA is mutated in homozygosis causes the Gaucher’s diseases, while when it is mutated in heterozygosis represents a major risk factor for PD (Sidransky et al., 2009).
Protein Aggregation May Affect ALP Generating a Vicious Cycle in Neurodegenerative Diseases
As discussed in the previous section, ALP dysfunction may contribute to the toxic aggregation in neurodegenerative diseases. On the other hand, mounting evidence also show that protein aggregation itself may affect ALP, thus generating a vicious cycle, which boost protein aggregation and toxicity (Figure 1). Although mechanisms underlying these processes are still poorly understood, these indirect pathways may explain why ALP became dysfunctional in neurodegenerative conditions caused by GOF mutations in aggregate-prone proteins.
How Protein Aggregation Affect ALP
Different works have shown that the aggregated forms of α-synuclein can bind the lysosome, thus impairing the chaperone-mediated autophagy, a selective autophagy pathway for degradation of cytosolic proteins (Cuervo et al., 2004; Martinez-Vicente et al., 2008) or inducing lysosomal rupture (Freeman et al., 2013). Moreover, α-synuclein overexpression may compromise ALP by inhibiting autophagy initiation via Rab1a inhibition (Winslow et al., 2010). In addition, α-synuclein toxicity has been reported to be associated with a progressive decline in markers of lysosome function due to cytoplasmic retention of TFEB, a master transcription factor regulating lysosomal biogenesis and function (Settembre et al., 2011; Decressac et al., 2013). Similarly, the polyglutamine-expanded androgen receptor (polyQ-AR) associated to SBMA, interferes with TFEB transactivation, which accounts for autophagic flux defects present in SBMA motor neuron-like cells (Cortes et al., 2014). Abnormal toxic polyQ expansions of htt protein may affect the efficiency of autophagy by inhibiting cargo recognition by autophagosomes (Martinez-Vicente et al., 2010) and/or by inhibiting autophagosome biogenesis and transport (Wong and Holzbaur, 2014; Rui et al., 2015). In Down syndrome increased production of APP-βCTF has been shown to impair lysosomal acidification and function (Ying et al., 2019).
The Paradigm of Lysosomal Storage Diseases
The interplay between ALP dysfunction, protein aggregation, and neurodegenerative processes is well represented in LSDs (Figure 2). Here, we will provide some key examples of LSDs in which the mechanisms underlying this interconnection have been studied more in depth.
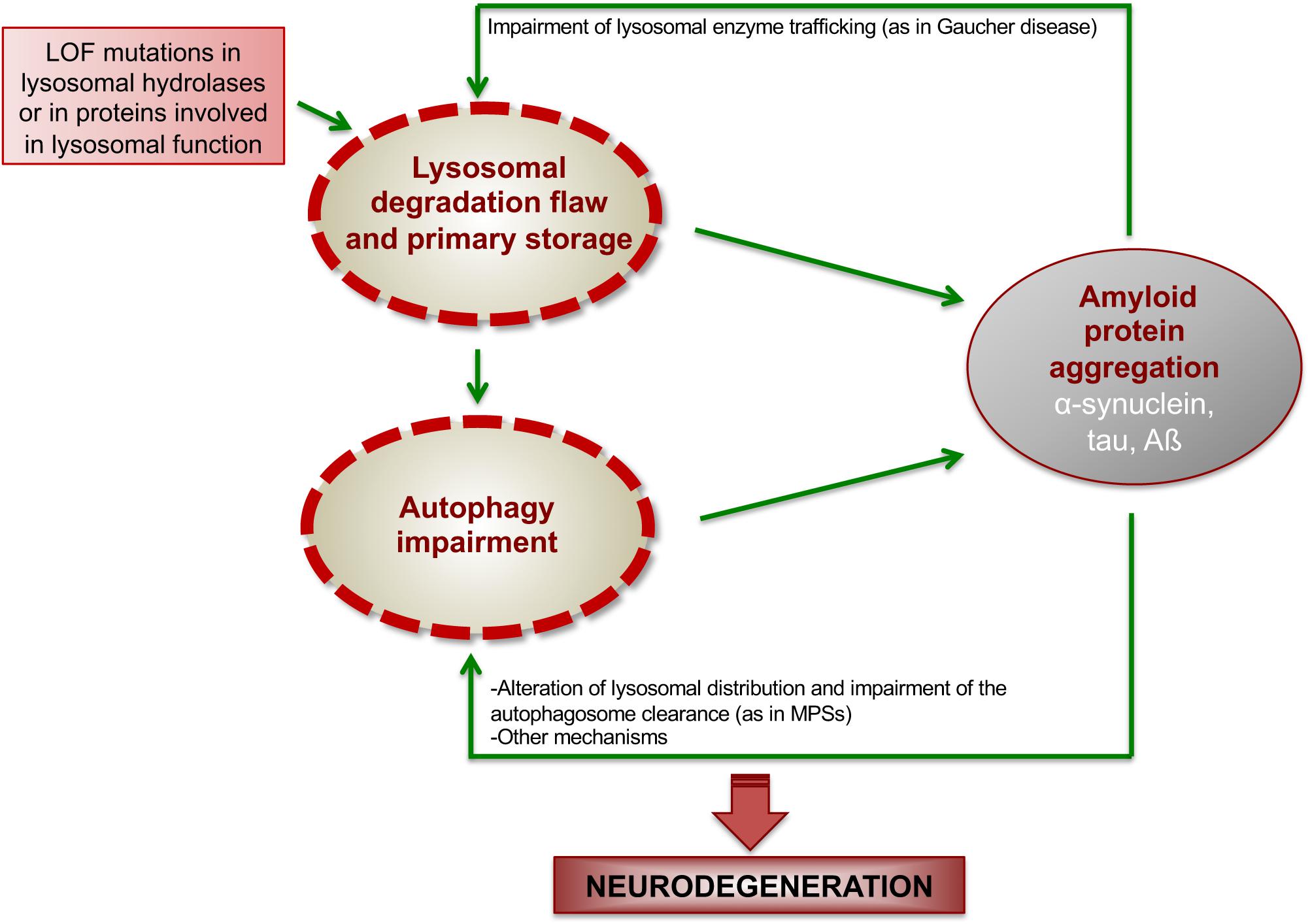
Figure 2. Proposed model showing how ALP dysfunction and protein aggregation generate a vicious cycle in LSDs. In LSDs, the inherited LOF of a specific lysosomal enzyme causes lysosomal degradation flaw and primary storage, which promotes the initial deposition of amyloidogenic proteins. Such amyloid deposition, in turns, worsens lysosomal degradation capability and impairs autophagy function, thus generating a vicious cycle, which boost neurodegenerative cascades.
In Gaucher disease lower levels of GCase in the lysosomes lead to the increased accumulation of glucosylceramide, which stabilizes soluble oligomeric α-synuclein intermediates that, in turn, are converted into amyloid fibrils (Mazzulli et al., 2011). The accumulation of α-synuclein inhibits the trafficking of newly synthesized GCase from ER to Golgi, thus reducing the amount of functional GCase in the lysosomes and further amplifying glucosylceramide accumulation. Therefore, the loss of GCase creates a positive feedback loop of reduced lysosomal function and α-synuclein accumulation that ultimately leading to neurodegeneration (Mazzulli et al., 2011).
As discussed above several MPSs show the presence of amyloidogenic protein aggregates in the brain. Nevertheless, the neuropathogenic relevance of these amyloid deposits in the context of MPSs and the underlying mechanisms remain largely unexplored. Recently, we have demonstrated that α-synuclein accumulates as neuronal insoluble aggregates in a mouse model of MPS-IIIA, and showed that this accumulation depletes synaptic α-synuclein, contributing to neurodegeneration by a LOF mechanism (Sambri et al., 2017). Further studies in MPS-IIIA mice have showed that α-synuclein progressively accumulates together with other amyloid proteins, including PrP, tau, and Aβ mostly into the lysosomes of neuronal cell bodies, thus exerting a gain of neurotoxic function by affecting ALP (Monaco et al., 2020). Indeed, inhibiting amyloid aggregation in MPS-IIIA mice by using CLR01, a “molecular tweezer” that acts as a broad-spectrum inhibitor of protein self-assembly (Attar and Bitan, 2014) reduced lysosomal enlargement and re-activates autophagy, thus ameliorating neurodegenerative signs (Monaco et al., 2020). Mechanistically, our preliminary data in MPS-IIIA mouse brain indicate that the build-up of multiple amyloid proteins into the lysosomes of neurons leads to lysosomal clustering in cell body and to the concomitant depletion of the axonal pool of lysosomes, which are critical for autophagosome encountering and clearance. As a consequence, LAMP1-negative autophagosomes massively accumulate in the cell periphery and axons. Therefore, our data suggest a model in which amyloid aggregation impairs the autophagic flux in neurons by disrupting normal lysosomal distribution and, thus preventing lysosomes to encounter and fuse with autophagosomes. Importantly, a similar neuropathogenic link between amyloid deposition and ALP is likely to occur also in other MPSs showing both amyloid deposition (see previous section) and autophagy impairment (Pierzynowska et al., 2019). Furthermore, in addition to the inhibition of lysosomal-mediated clearance of autophagosomes, other mechanisms (such as those discussed above) may contribute to amyloid-induced autophagy impairment in MPSs. Nevertheless, an open question is: if amyloid accumulation accounts for autophagy degradative dysfunction, what triggers initial deposition of amyloid proteins in the MPS brain? In the case of Gaucher disease it has been demonstrated that the primary storage of glucosylceramide stabilizes α-synuclein intermediates, promoting amyloid fibrils deposition (Mazzulli et al., 2011). It is likely that the primary storage of other sphingolipids may trigger amyloid aggregation in other forms of sphingolipidoses where, indeed, amyloid protein deposition has been observed (see the previous section). Similarly, GAGs could initiate and stabilize amyloid deposition in the case of MPSs. Supporting this hypothesis, it has been reported that GAGs provide a scaffold promoting amyloid aggregation (Iannuzzi et al., 2015; Liu et al., 2016).
In summary, findings in Gaucher disease and MPSs suggest a model in which primary lysosomal storage due to the inherited lysosomal deficiency triggers initial amyloid deposition, which, in turns, affect lysosomal functions, including autophagy degradation, thus generating a vicious loop between ALP and amyloid deposition, which boost neurodegeneration (Figure 2).
Conclusion
The interplay between protein aggregation and ALP dysfunction is crucial in driving neurodegenerative processes in a number of neurological conditions, among which LSDs represent the paradigm. In LSDs genetic factors directly cause the failure in lysosomal degradation function and the storage of undegraded materials into the lysosome. Although the underlying mechanisms are still unclear, it is likely that the primary storage due to the inherited lysosomal defect may promote the initial deposition of amyloidogenic proteins into the lysosomal compartment. Such compartmentalized deposition, in turns, worsens autophagy-lysosomal degradation capability of neurons through different mechanisms that may involve reduced trafficking of lysosomal enzymes to the lysosomes (as in the case of Gaucher diseases), impaired autophagosome clearance (as we demonstrated in MPSs) and, likely, others. These mechanisms generate a vicious loop that boost neurodegenerative processes in LSDs, thus allowing, on the other hand, the possibility to identify new attractive therapeutic targets to treat these severe neurological conditions.
Data Availability Statement
The datasets generated for this study are available on request to the corresponding author.
Ethics Statement
The animal studies were conducted in accordance with the guidelines of the Animal Care and Use Committee of TIGEM in Naples and authorized by the Italian Ministry of Health.
Author Contributions
AF conceived and wrote the manuscript. AM contributed to conceiving the manuscript and co-wrote the manuscript. All authors listed approved it for publication.
Funding
We acknowledge the Cure Sanfilippo Foundation (Grant to AF) and the Telethon Foundation (Grant to AF).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Aguzzi, A., and Heikenwalder, M. (2006). Pathogenesis of prion diseases: current status and future outlook. Nat. Rev. Microbiol. 4, 765–775. doi: 10.1038/nrmicro1492
Annunziata, I., Patterson, A., Helton, D., Hu, H., Moshiach, S., Gomero, E., et al. (2013). Lysosomal NEU1 deficiency affects amyloid precursor protein levels and amyloid-beta secretion via deregulated lysosomal exocytosis. Nat. Commun. 4:2734. doi: 10.1038/ncomms3734
Attar, A., and Bitan, G. (2014). Disrupting self-assembly and toxicity of amyloidogenic protein oligomers by “molecular tweezers” – from the test tube to animal models. Curr. Pharm. Des. 20, 2469–2483. doi: 10.2174/13816128113199990496
BasuRay, S., Mukherjee, S., Romero, E. G., Seaman, M. N., and Wandinger-Ness, A. (2013). Rab7 mutants associated with Charcot-Marie-Tooth disease cause delayed growth factor receptor transport and altered endosomal and nuclear signaling. J. Biol. Chem. 288, 1135–1149. doi: 10.1074/jbc.M112.417766
Beard, H., Hassiotis, S., Gai, W. P., Parkinson-Lawrence, E., Hopwood, J. J., and Hemsley, K. M. (2017). Axonal dystrophy in the brain of mice with Sanfilippo syndrome. Exp. Neurol. 295, 243–255. doi: 10.1016/j.expneurol.2017.06.010
Beck, J. A., Poulter, M., Campbell, T. A., Adamson, G., Uphill, J. B., Guerreiro, R., et al. (2010). PRNP allelic series from 19 years of prion protein gene sequencing at the MRC Prion Unit. Hum. Mutat. 31, E1551–E1563. doi: 10.1002/humu.21281
Braathen, G. J. (2012). Genetic epidemiology of Charcot-Marie-Tooth disease. Acta Neurol. Scand. Suppl. 126, iv–22. doi: 10.1111/ane.12013
Bras, J., Guerreiro, R., and Hardy, J. (2015). SnapShot: genetics of Parkinson’s disease. Cell 160, 570–570.e1. doi: 10.1016/j.cell.2015.01.019
Chiti, F., and Dobson, C. M. (2006). Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 75, 333–366. doi: 10.1146/annurev.biochem.75.101304.123901
Ciechanover, A., and Kwon, Y. T. (2015). Degradation of misfolded proteins in neurodegenerative diseases: therapeutic targets and strategies. Exp. Mol. Med. 47:e147. doi: 10.1038/emm.2014.117
Clarke, L. A. (2008). The mucopolysaccharidoses: a success of molecular medicine. Expert Rev. Mol. Med. 10:e1. doi: 10.1017/S1462399408000550
Coen, K., Flannagan, R. S., Baron, S., Carraro-Lacroix, L. R., Wang, D., Vermeire, W., et al. (2012). Lysosomal calcium homeostasis defects, not proton pump defects, cause endo-lysosomal dysfunction in PSEN-deficient cells. J. Cell Biol. 198, 23–35. doi: 10.1083/jcb.201201076
Cortes, C. J., Miranda, H. C., Frankowski, H., Batlevi, Y., Young, J. E., Le, A., et al. (2014). Polyglutamine-expanded androgen receptor interferes with TFEB to elicit autophagy defects in SBMA. Nat. Neurosci. 17, 1180–1189. doi: 10.1038/nn.3787
Cuervo, A. M., Stefanis, L., Fredenburg, R., Lansbury, P. T., and Sulzer, D. (2004). Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 305, 1292–1295. doi: 10.1126/science.1101738
Das, U., Scott, D. A., Ganguly, A., Koo, E. H., Tang, Y., and Roy, S. (2013). Activity-induced convergence of APP and BACE-1 in acidic microdomains via an endocytosis-dependent pathway. Neuron 79, 447–460. doi: 10.1016/j.neuron.2013.05.035
Decressac, M., Mattsson, B., Weikop, P., Lundblad, M., Jakobsson, J., and Bjorklund, A. (2013). TFEB-mediated autophagy rescues midbrain dopamine neurons from alpha-synuclein toxicity. Proc. Natl. Acad. Sci. U.S.A. 110, E1817–E1826. doi: 10.1073/pnas.1305623110
Didonna, A., and Opal, P. (2019). The role of neurofilament aggregation in neurodegeneration: lessons from rare inherited neurological disorders. Mol. Neurodegener. 14:19. doi: 10.1186/s13024-019-0318-4
Fraldi, A., Klein, A. D., Medina, D. L., and Settembre, C. (2016). Brain disorders due to lysosomal dysfunction. Annu. Rev. Neurosci. 39, 277–295. doi: 10.1146/annurev-neuro-070815-14031
Freeman, D., Cedillos, R., Choyke, S., Lukic, Z., McGuire, K., Marvin, S., et al. (2013). Alpha-synuclein induces lysosomal rupture and cathepsin dependent reactive oxygen species following endocytosis. PLoS One 8:e62143. doi: 10.1371/journal.pone.0062143
Gallardo, R., Ramakers, M., De Smet, F., Claes, F., Khodaparast, L., Khodaparast, L., et al. (2016). De novo design of a biologically active amyloid. Science 354:aah4949. doi: 10.1126/science.aah4949
Geisler, S., Holmstrom, K. M., Treis, A., Skujat, D., Weber, S. S., Fiesel, F. C., et al. (2010). The PINK1/Parkin-mediated mitophagy is compromised by PD-associated mutations. Autophagy 6, 871–878. doi: 10.4161/auto.6.7.13286
Ginsberg, S. D., Galvin, J. E., Lee, V. M., Rorke, L. B., Dickson, D. W., Wolfe, J. H., et al. (1999). Accumulation of intracellular amyloid-beta peptide (A beta 1-40) in mucopolysaccharidosis brains. J. Neuropathol. Exp. Neurol. 58, 815–824. doi: 10.1097/00005072-199908000-4
Goedert, M., and Spillantini, M. G. (2006). A century of Alzheimer’s disease. Science 314, 777–781. doi: 10.1126/science.1132814
Hamano, K., Hayashi, M., Shioda, K., Fukatsu, R., and Mizutani, S. (2008). Mechanisms of neurodegeneration in mucopolysaccharidoses II and IIIB: analysis of human brain tissue. Acta Neuropathol. 115, 547–559. doi: 10.1007/s00401-007-0325-3
Hara, T., Nakamura, K., Matsui, M., Yamamoto, A., Nakahara, Y., Suzuki-Migishima, R., et al. (2006). Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441, 885–889. doi: 10.1038/nature04724
Hardy, J., and Selkoe, D. J. (2002). The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297, 353–356. doi: 10.1126/science.1072994
Iannuzzi, C., Irace, G., and Sirangelo, I. (2015). The effect of glycosaminoglycans (GAGs) on amyloid aggregation and toxicity. Molecules 20, 2510–2528. doi: 10.3390/molecules20022510
Jin, M., and Klionsky, D. J. (2014). Regulation of autophagy: modulation of the size and number of autophagosomes. FEBS Lett. 588, 2457–2463. doi: 10.1016/j.febslet.2014.06.015
Komatsu, M., Waguri, S., Chiba, T., Murata, S., Iwata, J., Tanida, I., et al. (2006). Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441, 880–884. doi: 10.1038/nature04723
Kwong, L. K., Neumann, M., Sampathu, D. M., Lee, V. M., and Trojanowski, J. Q. (2007). TDP-43 proteinopathy: the neuropathology underlying major forms of sporadic and familial frontotemporal lobar degeneration and motor neuron disease. Acta Neuropathol. 114, 63–70. doi: 10.1007/s00401-007-0226-5
Lee, J. H., McBrayer, M. K., Wolfe, D. M., Haslett, L. J., Kumar, A., Sato, Y., et al. (2015). Presenilin 1 maintains lysosomal Ca(2+) homeostasis via TRPML1 by regulating vATPase-mediated lysosome acidification. Cell Rep. 12, 1430–1444. doi: 10.1016/j.celrep.2015.07.050
Lee, J. H., Yu, W. H., Kumar, A., Lee, S., Mohan, P. S., Peterhoff, C. M., et al. (2010). Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 141, 1146–1158. doi: 10.1016/j.cell.2010.05.008
Lee, S. M., Chin, L. S., and Li, L. (2012). Charcot-Marie-Tooth disease-linked protein SIMPLE functions with the ESCRT machinery in endosomal trafficking. J. Cell Biol. 199, 799–816. doi: 10.1083/jcb.201204137
Lee, V. M., Goedert, M., and Trojanowski, J. Q. (2001). Neurodegenerative tauopathies. Annu. Rev. Neurosci. 24, 1121–1159. doi: 10.1146/annurev.neuro.24.1.1121
Lieberman, A. P., Puertollano, R., Raben, N., Slaugenhaupt, S., Walkley, S. U., and Ballabio, A. (2012). Autophagy in lysosomal storage disorders. Autophagy 8, 719–730. doi: 10.4161/auto.19469
Liu, C. C., Zhao, N., Yamaguchi, Y., Cirrito, J. R., Kanekiyo, T., Holtzman, D. M., et al. (2016). Neuronal heparan sulfates promote amyloid pathology by modulating brain amyloid-beta clearance and aggregation in Alzheimer’s disease. Sci. Transl. Med. 8:332ra44. doi: 10.1126/scitranslmed.aad3650
Martinez-Vicente, M., Talloczy, Z., Kaushik, S., Massey, A. C., Mazzulli, J., Mosharov, E. V., et al. (2008). Dopamine-modified alpha-synuclein blocks chaperone-mediated autophagy. J. Clin. Invest. 118, 777–788. doi: 10.1172/JCI32806
Martinez-Vicente, M., Talloczy, Z., Wong, E., Tang, G., Koga, H., Kaushik, S., et al. (2010). Cargo recognition failure is responsible for inefficient autophagy in Huntington’s disease. Nat. Neurosci. 13, 567–576. doi: 10.1038/nn.2528
Martins, C., Hulkova, H., Dridi, L., Dormoy-Raclet, V., Grigoryeva, L., Choi, Y., et al. (2015). Neuroinflammation, mitochondrial defects and neurodegeneration in mucopolysaccharidosis III type C mouse model. Brain 138(Pt 2), 336–355. doi: 10.1093/brain/awu355
Mazzulli, J. R., Xu, Y. H., Sun, Y., Knight, A. L., McLean, P. J., Caldwell, G. A., et al. (2011). Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 146, 37–52. doi: 10.1016/j.cell.2011.06.001
Monaco, A., Maffia, V., Sorrentino, N., Sambri, I., Ezhova, Y., Giuliano, T., et al. (2020). The amyloid self-assembly inhibitor CLR01 relieves autophagy and ameliorates neuropathology in a severe lysosomal storage disease. Mol. Ther. doi: 10.1016/j.ymthe.2020.02.005 [Epub ahead of print].
Nixon, R. A. (2013). The role of autophagy in neurodegenerative disease. Nat. Med. 19, 983–997. doi: 10.1038/nm.3232
Nixon, R. A. (2017). Amyloid precursor protein and endosomal-lysosomal dysfunction in Alzheimer’s disease: inseparable partners in a multifactorial disease. FASEB J. 31, 2729–2743. doi: 10.1096/fj.201700359
Nixon, R. A., Yang, D. S., and Lee, J. H. (2008). Neurodegenerative lysosomal disorders: a continuum from development to late age. Autophagy 4, 590–599. doi: 10.4161/auto.6259
Ohmi, K., Kudo, L. C., Ryazantsev, S., Zhao, H. Z., Karsten, S. L., and Neufeld, E. F. (2009). Sanfilippo syndrome type B, a lysosomal storage disease, is also a tauopathy. Proc. Natl. Acad. Sci. U.S.A. 106, 8332–8337. doi: 10.1073/pnas.0903223106
Perutz, M. F. (1999). Glutamine repeats and neurodegenerative diseases: molecular aspects. Trends Biochem. Sci. 24, 58–63. doi: 10.1016/s0968-0004(98)01350-4
Pierzynowska, K., Gaffke, L., Podlacha, M., Brokowska, J., and Wegrzyn, G. (2019). Mucopolysaccharidosis and autophagy: controversies on the contribution of the process to the pathogenesis and possible therapeutic applications. Neuromol. Med. 22, 25–30. doi: 10.1007/s12017-019-08559-1
Platt, F. M., Boland, B., and van der Spoel, A. C. (2012). The cell biology of disease: lysosomal storage disorders: the cellular impact of lysosomal dysfunction. J. Cell Biol. 199, 723–734. doi: 10.1083/jcb.201208152
Platt, F. M., d’Azzo, A., Davidson, B. L., Neufeld, E. F., and Tifft, C. J. (2018). Lysosomal storage diseases. Nat. Rev. Dis. Primers 4:27. doi: 10.1038/s41572-018-0025-4
Rainero, I., Rubino, E., Michelerio, A., D’Agata, F., Gentile, S., and Pinessi, L. (2017). Recent advances in the molecular genetics of frontotemporal lobar degeneration. Funct. Neurol. 32, 7–16. doi: 10.11138/fneur/2017.32.1.007
Rajendran, L., and Annaert, W. (2012). Membrane trafficking pathways in Alzheimer’s disease. Traffic 13, 759–770. doi: 10.1111/j.1600-0854.2012.01332.x
Ramirez, A., Heimbach, A., Grundemann, J., Stiller, B., Hampshire, D., Cid, L. P., et al. (2006). Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat. Genet. 38, 1184–1191. doi: 10.1038/ng1884
Robak, L. A., Jansen, I. E., van Rooij, J., Uitterlinden, A. G., Kraaij, R., Jankovic, J., et al. (2017). Excessive burden of lysosomal storage disorder gene variants in Parkinson’s disease. Brain 140, 3191–3203. doi: 10.1093/brain/awx285
Rui, Y. N., Xu, Z., Patel, B., Chen, Z., Chen, D., Tito, A., et al. (2015). Huntingtin functions as a scaffold for selective macroautophagy. Nat. Cell Biol. 17, 262–275. doi: 10.1038/ncb3101
Saito, Y., Suzuki, K., Hulette, C. M., and Murayama, S. (2004). Aberrant phosphorylation of alpha-synuclein in human Niemann-Pick type C1 disease. J. Neuropathol. Exp. Neurol. 63, 323–328. doi: 10.1093/jnen/63.4.323
Sambri, I., D’Alessio, R., Ezhova, Y., Giuliano, T., Sorrentino, N. C., Cacace, V., et al. (2017). Lysosomal dysfunction disrupts presynaptic maintenance and restoration of presynaptic function prevents neurodegeneration in lysosomal storage diseases. EMBO Mol. Med. 9, 112–132. doi: 10.15252/emmm.201606965
Schultz, M. L., Tecedor, L., Chang, M., and Davidson, B. L. (2011). Clarifying lysosomal storage diseases. Trends Neurosci. 34, 401–410. doi: 10.1016/j.tins.2011.05.006
Settembre, C., Di Malta, C., Polito, V. A., Garcia Arencibia, M., Vetrini, F., Erdin, S., et al. (2011). TFEB links autophagy to lysosomal biogenesis. Science 332, 1429–1433. doi: 10.1126/science.1204592
Shachar, T., Lo Bianco, C., Recchia, A., Wiessner, C., Raas-Rothschild, A., and Futerman, A. H. (2011). Lysosomal storage disorders and Parkinson’s disease: Gaucher disease and beyond. Mov. Disord. 26, 1593–1604. doi: 10.1002/mds.23774
Sidransky, E., Nalls, M. A., Aasly, J. O., Aharon-Peretz, J., Annesi, G., Barbosa, E. R., et al. (2009). Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N. Engl. J. Med. 361, 1651–1661. doi: 10.1056/NEJMoa0901281
Smith, B. R., Santos, M. B., Marshall, M. S., Cantuti-Castelvetri, L., Lopez-Rosas, A., Li, G., et al. (2014). Neuronal inclusions of alpha-synuclein contribute to the pathogenesis of Krabbe disease. J. Pathol. 232, 509–521. doi: 10.1002/path.4328
Song, C. Y., Guo, J. F., Liu, Y., and Tang, B. S. (2012). Autophagy and its comprehensive impact on ALS. Int. J. Neurosci. 122, 695–703. doi: 10.3109/00207454.2012.714430
Spillantini, M. G., Schmidt, M. L., Lee, V. M., Trojanowski, J. Q., Jakes, R., and Goedert, M. (1997). Alpha-synuclein in Lewy bodies. Nature 388, 839–840. doi: 10.1038/42166
Suzuki, K., Parker, C. C., Pentchev, P. G., Katz, D., Ghetti, B., D’Agostino, A. N., et al. (1995). Neurofibrillary tangles in Niemann-Pick disease type C. Acta Neuropathol. 89, 227–238. doi: 10.1007/bf00309338
Theocharopoulou, G., and Vlamos, P. (2015). Modeling protein misfolding in charcot-marie-tooth disease. Adv. Exp. Med. Biol. 820, 91–102. doi: 10.1007/978-3-319-09012-2_7
Winslow, A. R., Chen, C. W., Corrochano, S., Acevedo-Arozena, A., Gordon, D. E., Peden, A. A., et al. (2010). alpha-Synuclein impairs macroautophagy: implications for Parkinson’s disease. J. Cell Biol. 190, 1023–1037. doi: 10.1083/jcb.201003122
Wong, Y. C., and Holzbaur, E. L. (2014). The regulation of autophagosome dynamics by huntingtin and HAP1 is disrupted by expression of mutant huntingtin, leading to defective cargo degradation. J. Neurosci. 34, 1293–1305. doi: 10.1523/JNEUROSCI.1870-13.2014
Ying, J., Sato, Y., Im, E., Berg, M., Bordi, M., Darji, S., et al. (2019). Lysosomal dysfunction in Down syndrome is APP-dependent and mediated by APP-betaCTF (C99). J. Neurosci. 39, 5255–5268. doi: 10.1523/JNEUROSCI.0578-19.2019
Yu, L., Chen, Y., and Tooze, S. A. (2018). Autophagy pathway: cellular and molecular mechanisms. Autophagy 14, 207–215. doi: 10.1080/15548627.2017.1378838
Zha, Q., Ruan, Y., Hartmann, T., Beyreuther, K., and Zhang, D. (2004). GM1 ganglioside regulates the proteolysis of amyloid precursor protein. Mol. Psychiatry 9, 946–952. doi: 10.1038/sj.mp.4001509
Keywords: lysosome, lysosomal storage disease, autophagy, amyloid aggregation, molecular therapy of neurodegenerative diseases
Citation: Monaco A and Fraldi A (2020) Protein Aggregation and Dysfunction of Autophagy-Lysosomal Pathway: A Vicious Cycle in Lysosomal Storage Diseases. Front. Mol. Neurosci. 13:37. doi: 10.3389/fnmol.2020.00037
Received: 16 December 2019; Accepted: 24 February 2020;
Published: 11 March 2020.
Edited by:
Sandra Macedo-Ribeiro, University of Porto, PortugalReviewed by:
Sabine Hilfiker, Spanish National Research Council, SpainClevio Nobrega, University of Algarve, Portugal
Copyright © 2020 Monaco and Fraldi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alessandro Fraldi, ZnJhbGRpQHRpZ2VtLml0