 Xinyue Liu1
Xinyue Liu1 Jing Zhao1Yingkai Zhang2Iban Ubarretxena-Belandia3,4
Jing Zhao1Yingkai Zhang2Iban Ubarretxena-Belandia3,4 Scott Forth1,5Raquel L. Lieberman6Chunyu Wang1,5,7*
Scott Forth1,5Raquel L. Lieberman6Chunyu Wang1,5,7*- 1Center for Biotechnology and Interdisciplinary Studies, Rensselaer Polytechnic Institute, Troy, NY, United States
- 2Department of Chemistry, New York University, New York, NY, United States
- 3Instituto Biofisika (UPV/EHU, CSIC), University of the Basque Country, Leioa, Spain
- 4Ikerbasque, Basque Foundation for Science, Bilbao, Spain
- 5Department of Biological Sciences, Rensselaer Polytechnic Institute, Troy, NY, United States
- 6School of Chemistry and Biochemistry, Georgia Institute of Technology, Atlanta, GA, United States
- 7Department of Chemistry and Chemical Biology, Rensselaer Polytechnic Institute, Troy, NY, United States
Intramembrane-cleaving proteases (I-CLiPs) catalyze the hydrolysis of peptide bonds within the transmembrane regions of membrane protein substrates, releasing bioactive fragments that play roles in many physiological and pathological processes. Based on their catalytic mechanism and nucleophile, I-CLiPs are classified into metallo, serine, aspartyl, and glutamyl proteases. Presenilin is the most prominent among I-CLiPs, as the catalytic subunit of γ-secretase (GS) complex responsible for cleaving the amyloid precursor protein (APP) and Notch, as well as many other membrane substrates. Recent cryo-electron microscopy (cryo-EM) structures of GS provide new details on how presenilin recognizes and cleaves APP and Notch. First, presenilin transmembrane helix (TM) 2 and 6 are dynamic. Second, upon binding to GS, the substrate TM helix is unwound from the C-terminus, resulting in an intermolecular β-sheet between the substrate and presenilin. The transition of the substrate C-terminus from α-helix to β-sheet is proposed to expose the scissile peptide bond in an extended conformation, leaving it susceptible to protease cleavage. Despite the astounding new insights in recent years, many crucial questions remain unanswered regarding the inner workings of γ-secretase, however. Key unanswered questions include how the enzyme recognizes and recruits substrates, how substrates are translocated from an initial docking site to the active site, how active site aspartates recruit and coordinate catalytic water, and the nature of the mechanisms of processive trimming of the substrate and product release. Answering these questions will have important implications for drug discovery aimed at selectively reducing the amyloid load in Alzheimer’s disease (AD) with minimal side effects.
Four Classes of Intramembrane-Cleaving Proteases
Intramembrane-cleaving proteases (I-CLiPs, also called IMPAS) carry out regulated intramembrane proteolysis (RIP). They hydrolyze peptide bonds buried inside the membrane lipid bilayer (Brown et al., 2000) and release bioactive fragments (Haze et al., 1999; Niwa et al., 1999; Lal and Caplan, 2011; Lichtenthaler et al., 2011). Numerous I-CLiP substrates have been discovered, including the sterol regulatory element-binding proteins (SREBPs; Brown and Goldstein, 1997), the membrane receptor Notch (Selkoe and Kopan, 2003), and the amyloid precursor protein (APP; Annaert and De Strooper, 1999). I-CLiPs therefore play crucial roles in a variety of biological processes, including embryonic development, immune responses, and normal function of the nervous system. In addition, I-CLiPs contribute to many diseases such as cancer and Alzheimer’s disease (AD; Winter-Vann and Casey, 2005; Lichtenthaler et al., 2011; Düsterhöft et al., 2017).
Based on their catalytic mechanisms, I-CLiPs are classified into four families: rhomboid serine proteases (Wu et al., 2006), S2P-metalloproteases (Feng et al., 2007), di-aspartyl proteases (Fluhrer et al., 2009), and glutamyl proteases (Manolaridis et al., 2013). Although six classes of soluble proteases are known, I-CLiPs using cysteine or threonine as catalytic residue have not yet been identified. In the 3D structures of I-CLiPs, the polar catalytic residues are located well below the membrane surface, shielded from hydrophobic membrane environment by surrounding transmembrane helices (TMs), whereas water molecules are readily accesible to the catalytic residues through a hydrophilic chamber or channel.
Serine I-CLiPs
Rhomboids constitute a large superfamily of serine I-CLiPs, which are involved in developmental signaling in Drosophila (Wasserman and Freeman, 1997), host invasion of protozoan parasites (Sibley, 2013), and human diseases such as cancer and neurodegeneration (Bergbold and Lemberg, 2013; Düsterhöft et al., 2017). Rhomboids have been intensely studied as model I-CLiP and also for their biological importance (see an excellent review by Strisovsky et al., 2009; Tichá et al., 2018). The rhomboid fold is composed of six TMs named TM1 to TM6 (Figure 1A). The catalytic dyad, serine (on TM4) and histidine (on TM6), is located at a V-shaped cavity accessible to the aqueous phase at a distance of 10–12 Å below the membrane surface (Wang et al., 2006; Wu et al., 2006; Ben-Shem et al., 2007; Figure 1A). During intramembrane proteolysis, the histidine activates the catalytic serine for a nucleophilic attack on substrates (Lemieux et al., 2007). Rhomboids recognize the helical TMs and a linear segment adjacent to the TMs of their substrates (Strisovsky et al., 2009). Structural and modeling studies proposed that the TMs of the substrates may bind the rhomboid at the interface of TM2 and TM5, where TM5 plays the role of the substrate gate (Baker et al., 2007; Xue and Ha, 2013; Zoll et al., 2014; Shokhen and Albeck, 2017). Binding studies reveal a role of allostery in catalysis. Dimerization of rhomboids is required for the formation of an exosite and subsequent allosteric substrate binding and activation (Arutyunova et al., 2014).
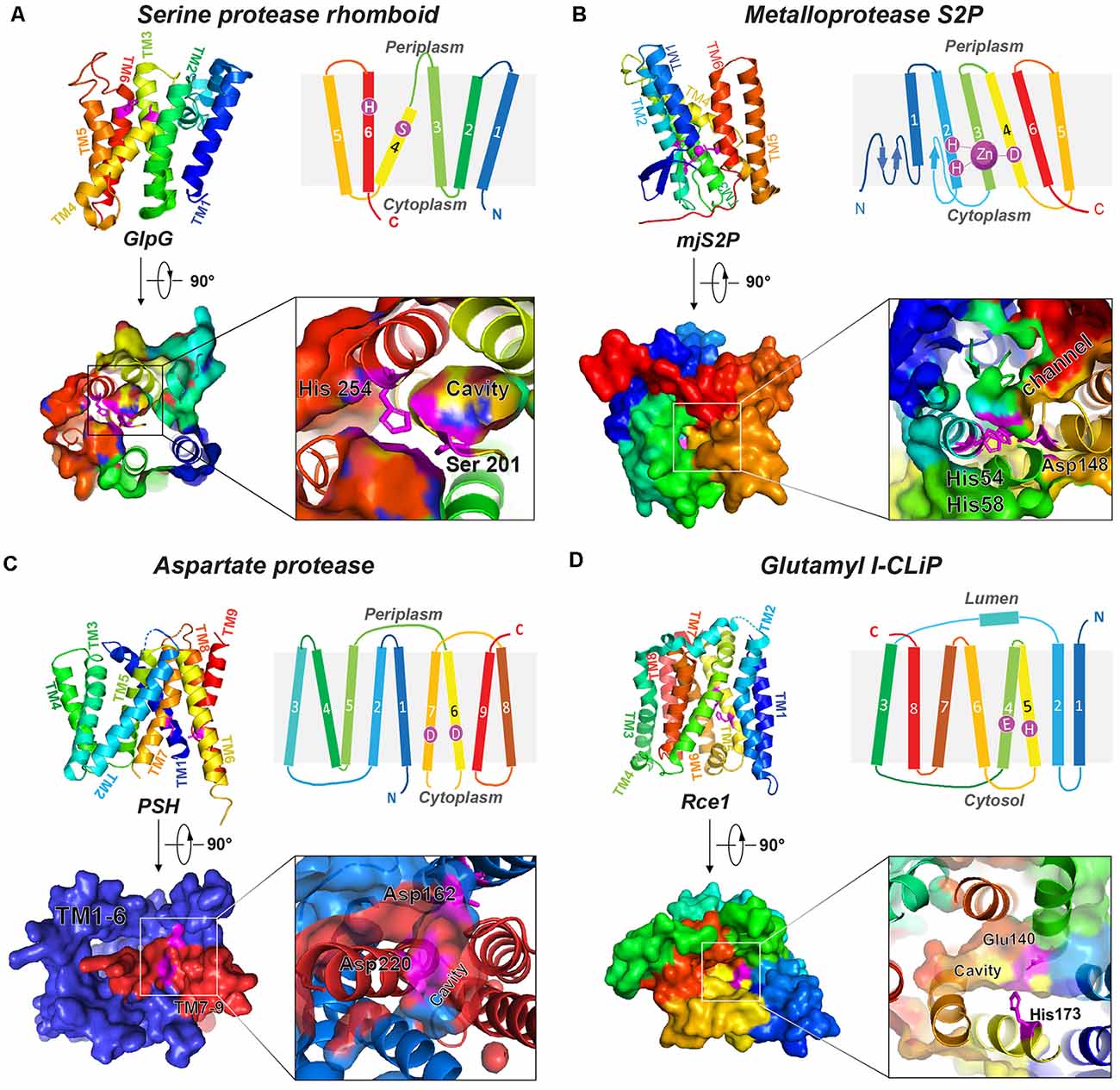
Figure 1. Representative structures of four I-CLiP families. Catalytic residues are labeled on the schematic structure, and the catalytic cavities are shown in the crystal structure from either the extracellular (or luminal) side or cytoplasmic side. (A) Serine protease rhomboid (GlpG, PDB: 2NRF). (B) Metalloprotease S2P (mjS2P PDB: 3B4R). (C) Aspartate protease MCMJR1, aka presenilin homolog (PSH, PDB: 4HYG). (D) Glutamyl I-CLiP (mmRce1 PDB: 4CAD).
Metalloproteases
Site-2 proteases (S2Ps) constitute another family of metalloproteases, which activate membrane-bound transcription factors through RIP. S2Ps have been well studied in the context of cholesterol metabolism, with a zinc ion at its active site (Sun et al., 2016). After site-1 protease (S1P) cleavage, S2P cleaves SREBPs. The N-terminus of SREBP is then released and enters the nucleus to activate genes for biosynthesis and uptake of cholesterol (Sakai et al., 1996; Brown and Goldstein, 1997). An X-ray structure of Methanocaldococcus jannaschii S2P (mjS2P; Figure 1B), an S2P ortholog, revealed six TMs and three β-strands. The zinc ion, ~14 Å below the membrane surface, is coordinated by two histidine residues in an HEXXH motif (“H” is histidine, “E” is glutamate, and “X” is any amino acid) in TM2 and an aspartate in TM4 (Feng et al., 2007). Two conformations were identified: an open state and a closed state (Figure 2A). In the closed conformation, water accesses zinc via a polar channel open to the cytoplasmic side. In the open conformation, the TM1 and TM6 are separated by 10–12 Å, forming a cleft for substrate entry and positioning the catalytic zinc towards the substrate (Figure 2B).
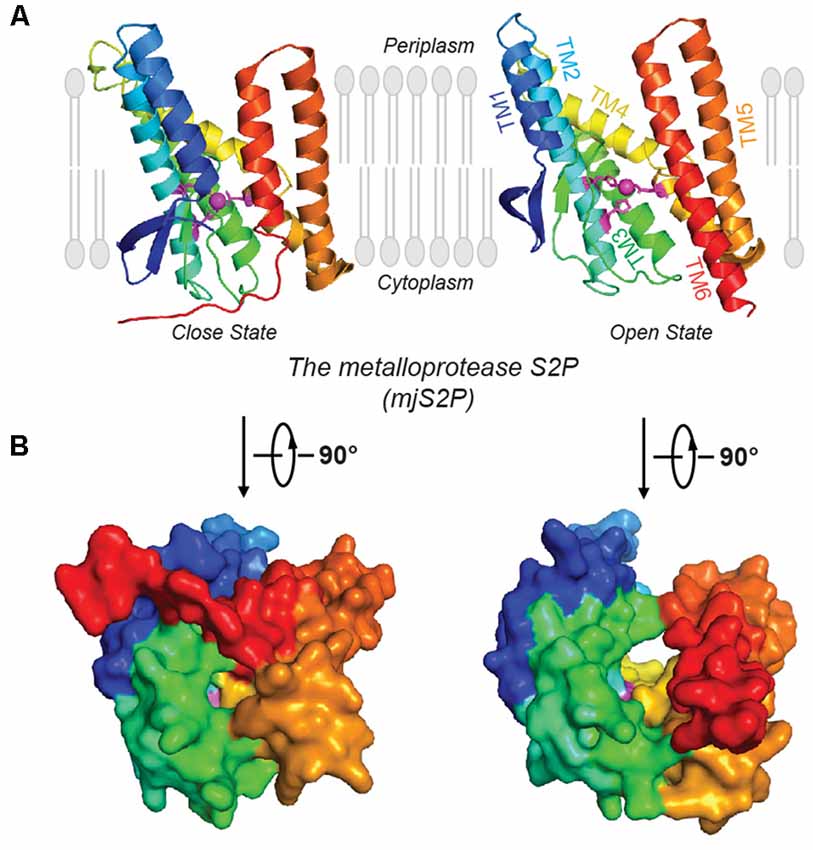
Figure 2. Open and closed conformations of mjS2P. (A) Crystal structures of the closed and open states of mjS2P, a metallo I-CLiP, and (B) cytoplasmic view of the catalytic cavity in the closed state and the cleft in the open state.
Di-Aspartyl Proteases
Di-aspartyl intramembrane proteases are characterized by a pair of catalytic aspartates. One of their catalytic aspartates is contained within the signature GXGD motif (“G” is glycine, “X” is any amino acid, and “D” is aspartate; Steiner et al., 2000; Fluhrer et al., 2009). Di-aspartyl intramembrane proteases are involved in many fundamental processes such as cell differentiation, development, immune surveillance, and virus maturation. This family has two key members: presenilin (PS) and signal peptide peptidase (SPP; Weihofen et al., 2002). PS is the catalytic subunit of γ-secretase (GS; Wolfe et al., 1999; Li et al., 2000), which cleaves Notch and APP transmembrane domain (TMD; Francis et al., 2002; Haass and Steiner, 2002), among over 90 substrates (Beel and Sanders, 2008). PS homologs (PSHs) can also cleave APP at the two major cleavage sites of PS (Torres-Arancivia et al., 2010; Naing et al., 2018a): the γ-site and the ε-site, generating Aβ42 and Aβ48, respectively (Naing et al., 2018a). A ~3.3-Å resolution crystal structure of an ortholog from Methanoculleus marisnigri (MCMJR1) showed nine TMs (Figure 1C) with TM1–TM6 equivalent to the amino-terminal fragment [N-terminal fragment (NTF)] and TM7–TM9 equivalent to the C-terminal fragment (CTF) of PS formed by autoproteolysis of GS (Li et al., 2013). TM1–TM6 tilt at angles of 15–35° away from the lipid membrane surface and form a horseshoe-shaped structure surrounding the CTF TMs. The active site aspartates (Asp 162 on TM6 and Asp 220 on TM7) are located in a cavity accessible from the cytoplasmic side, approximately 8 Å from the membrane surface. The structure of MCMJR1 characterized by small angle neutron scattering (SANS) is smaller than the crystal structure, indicating that the enzyme may be more compact in solution (Naing et al., 2018b).
Glutamyl Proteases
Ras converting enzyme 1 (Rce1) is a glutamate intramembrane protease (Manolaridis et al., 2013) found in the endoplasmic reticulum. Rce1 carries out posttranslational modifications of proteins with a C-terminus CAAX motif (“C” is cysteine, “A” is an aliphatic amino acid, and “X” is any amino acid residue; Figure 3; Boyartchuk et al., 1997). Substrates of Rce1 include Ras and prelamin A. Rce1 cleavage of these substrates is necessary for their function. The posttranslational modifications of CAAX proteins include cysteine isoprenylation, −AAX release, and methylation of the exposed C-terminal carboxyl of isoprenylcysteine (Figure 3; Schmidt et al., 1998). The Rce1 is the prenyl endopeptidase responsible for the release of the C-terminal −AAX peptide. These modifications are required for proper localization of the Ras protein (Michaelson et al., 2005) and can affect various signaling pathways during differentiation, proliferation, and oncogenesis (Winter-Vann and Casey, 2005; Christiansen et al., 2011). A crystal structure of the Rce1 ortholog from Methanococcus maripaludis (MmRce1) reveals eight TMs (Figure 1D; Manolaridis et al., 2013). TMs 4–7 form a conical cavity with an opening towards the cytosol, allowing solvent access and prenylated substrate accommodation. The catalytic dyad, a glutamate and a histidine, is located in the cavity approximately 10 Å away from the membrane surface.
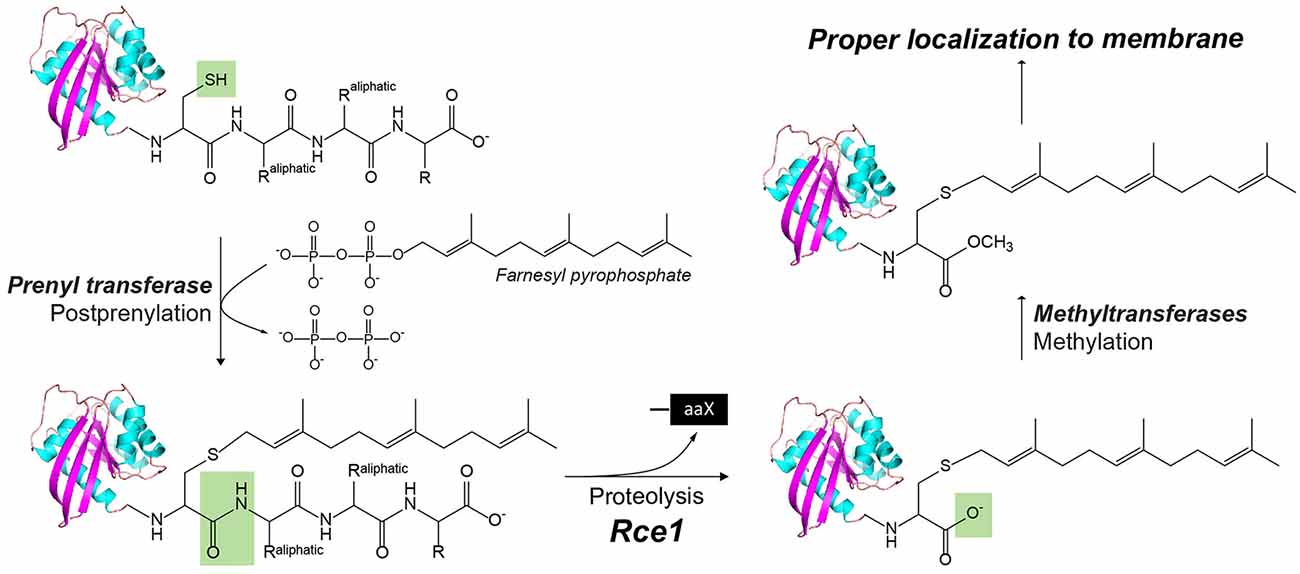
Figure 3. The posttranslational modification of proteins with a C-terminus CAAX motif by Rce1, a glutamyl IMP. In CaaX, “C” is cysteine, “A” is an aliphatic amino acid, and “X” is any amino acid. The posttranslational modifications of CAAX proteins include the cysteine isoprenylation, the −aaX release, and carboxyl methylation of the exposed isoprenylcysteine. The Rce1 is the prenyl endopeptidase for the release of the C-terminal −aaX peptide. These modifications are required for proper localization of the Ras to the membrane.
Finally, a hybrid I-CLiP, ZMPSTE24, is a zinc metalloprotease that matures lamin A, a nuclear scaffold protein, through recognizing a CAAX motif (Pendás et al., 2002). Mutations in ZMPSTE24 are associated with premature aging, such as in Hutchinson–Guilford progeria syndrome (HGPS; Navarro et al., 2014). ZMPSTE24 resides in the inner nuclear membrane and is also known as farnesylated-protein converting enzyme 1 (FACE-1), and Ste24 in yeast. After farnesylation of the C-terminal CAAX motif, prelamin A is cleaved by either Rce1 or ZMPSTE24, and then the C-terminal cysteine residue is carboxymethylated (Figure 3). ZMPSTE24 further cleaves a 15-residue CTF, resulting in mature lamin and its release from the nuclear membrane. In progeroid conditions caused by ZMPSTE24 mutation, farnesylated and methylated prelamin accumulates in the nuclear membrane. ZMPSTE24 contains an extraordinary intramembrane chamber, large enough to accommodate a ~10-kDa protein or ~450 water molecules (Pryor et al., 2013). The active site residues are facing the chamber, with an arrangement almost identical to bacterial thermolysin.
Gs in Health and Disease
GS is reported to cleave over 90 substrates (Beel and Sanders, 2008). Conversely, aberrant GS cleavage is associated with many diseases, including cancer, skin disorder, and neurodegenerative diseases (Shih and Wang, 2007; Kelleher and Shen, 2010). Here, we highlight the two most prominent GS substrates, APP and Notch, which are involved in AD and cancer, respectively.
GS and AD
Amyloid plaques are a hallmark of AD pathology, which are mainly composed of aggregated amyloid-β (Aβ) peptides. Aβ deposits have been proposed as the initial trigger in the decade-long progression towards neurodegeneration in AD (Tanzi and Bertram, 2005), which leads to tau pathology and eventually widespread neuroinflammation. Aβ peptides are produced from APP by the consecutive action of two proteases, β-secretase and GS. β-Secretase sheds the ectodomain of APP, generating C99 and the N-terminus of the subsequent Aβ species (Mullard, 2017). GS is the I-CLiP that cleaves within the TM of APP (APPTM), releasing different lengths of Aβ peptides into the extracellular matrix or endosome lumen (Qi-Takahara et al., 2005; Takami et al., 2009). Longer Aβ peptides (e.g., Aβ42 and Aβ43) are particularly prone to aggregation.
There are two APP processing pathways (Figure 4). In the non-amyloidogenic pathway, APP is first cleaved by α-secretase to generate C83, and further cleavage of GS can no longer generate Aβ. In the amyloidogenic pathway, APP is first cleaved by β-secretase to generate a membrane-bound CTF containing 99 amino acid residues (C99). C99 is then the substrate of GS to generate Aβ, the pathogenic peptide for AD (Lichtenthaler et al., 2011), while the APP intracellular domain (AICD) is liberated into the cytoplasm (Haass and Steiner, 2002). β-Secretase and GS both localize to the lipid rafts of cell or organellar membranes, and cholesterol plays an important role in the enzyme activity (Tun et al., 2002; Urano et al., 2005). The observation of different lengths of Aβ peptides suggests a successive C-terminal trimming mechanism of GS after the initial ε-cleavage (Qi-Takahara et al., 2005; Takami et al., 2009). In addition to Aβ40 and Aβ42, Aβ38, Aβ43, Aβ45, Aβ46, and Aβ48 are also identified. Starting from two initial ε-cleavage sites ε48 and ε49, Aβ40, Aβ43, and Aβ46 are generated from Aβ49 through successive shedding of tripeptides. Non-transitional state GS inhibitors (GSI), DAPT and Compound E, suppress intracellular Aβ40 production while increasing Aβ43 and in turn Aβ46 levels (Qi-Takahara et al., 2005). Aβ45, Aβ42, and Aβ38 are generated from Aβ48 (Figure 4). These two product lines have been established using LC–MS/MS (Takami et al., 2009). The stepwise cleavage sites are named ε48/ε49, ζ45/ζ46, ζ42/ζ43, and γ38/γ40 (Lichtenthaler et al., 2011; De Strooper and Chávez Gutiérrez, 2015; Langosch and Steiner, 2017).
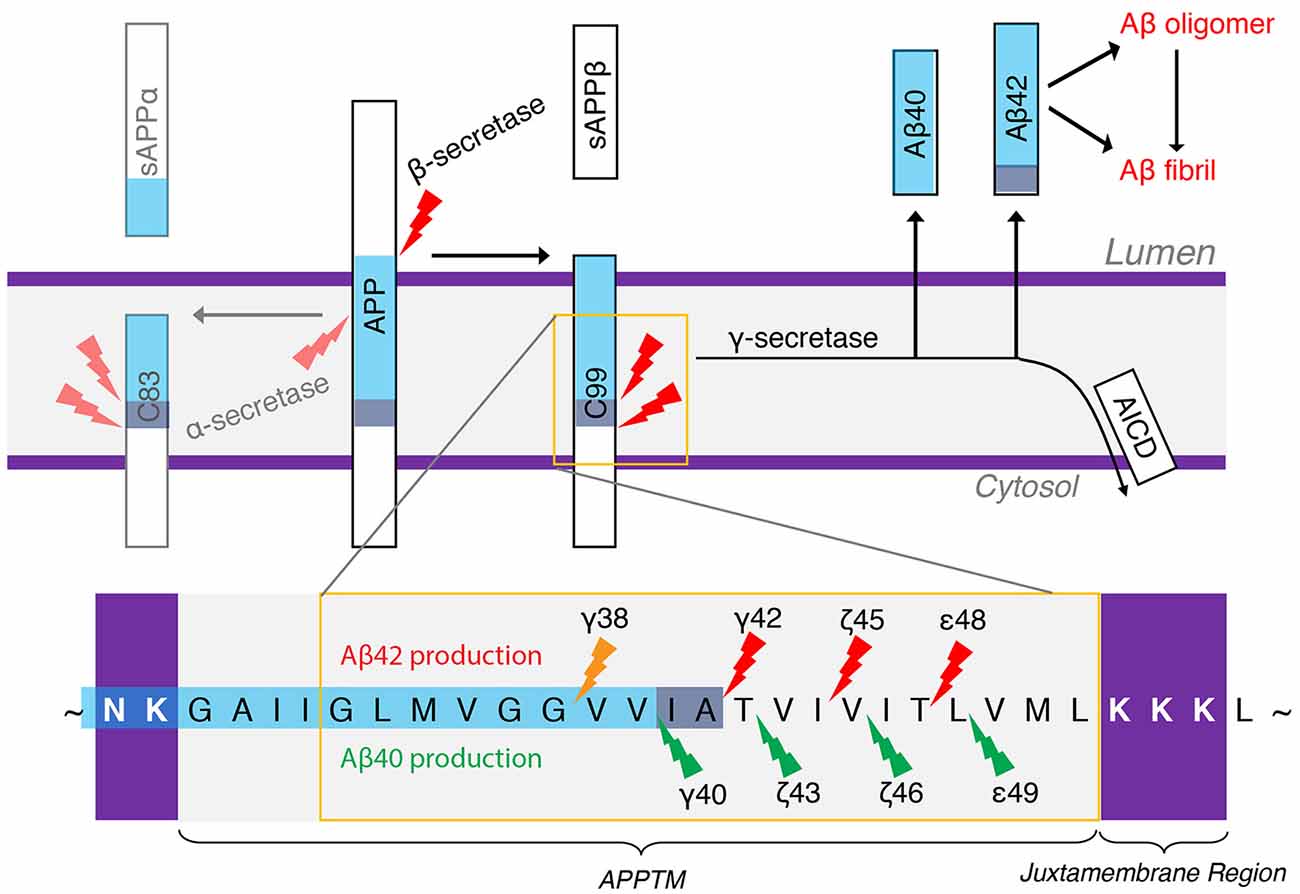
Figure 4. The generation of Aβ40 and Aβ42 from amyloid precursor protein (APP). α-Secretase and β-secretase are the sheddases generating C83 and C99 from APP, respectively. γ-secretase (GS) is the I-CLiP that carries out intramembrane proteolysis of C99 to generate Aβ, a pathogenic peptide in Alzheimer’s disease (AD).
Aβ peptides can aggregate into oligomers and fibrils. Longer Aβ forms, such as Aβ42 and Aβ43, are especially prone to aggregation and are therefore much more toxic (Makin, 2018). Mutations in APP on chromosome 21q (Levy et al., 1990; Goate et al., 1991; Tanzi and Bertram, 2005; Bertram et al., 2010) and in PS 1 and 2 genes (PSEN1 and PSEN2, respectively) on chromosomes 14 and 1 (Levy-Lahad et al., 1995; Rogaev et al., 1995; Sherrington et al., 1995) can cause early-onset familial Alzheimer’s disease (FAD), characterized by an increased Aβ42/Aβ40 ratio biochemically. The most common FAD mutations occur in PS, underlining the important biological role for GS. The successive cleavage of the APP substrates progressively destabilizes the GS–Aβn complex with the shortening of the Aβn. It has been shown that PSEN mutations will further destabilize the Aβn–GS complex, resulting in the release of longer Aβn (Szaruga et al., 2017) and raising the Aβ42/Aβ40 ratio.
As a GS substrate, the local conformation and dynamics of APPTM contribute to the observed cleavage sites. A right-handed APPTM helical dimer was characterized by nuclear magnetic resonance (NMR) in solution (Figure 5A). In the same study, FAD mutations V44M and V44A within APPTM were found to selectively expose the T48 site for fast solvent exchange. This may promote T48 for the initial ε-cleavage over L49 and consequently shift cleavage preference towards Aβ42 production (Chen et al., 2014).
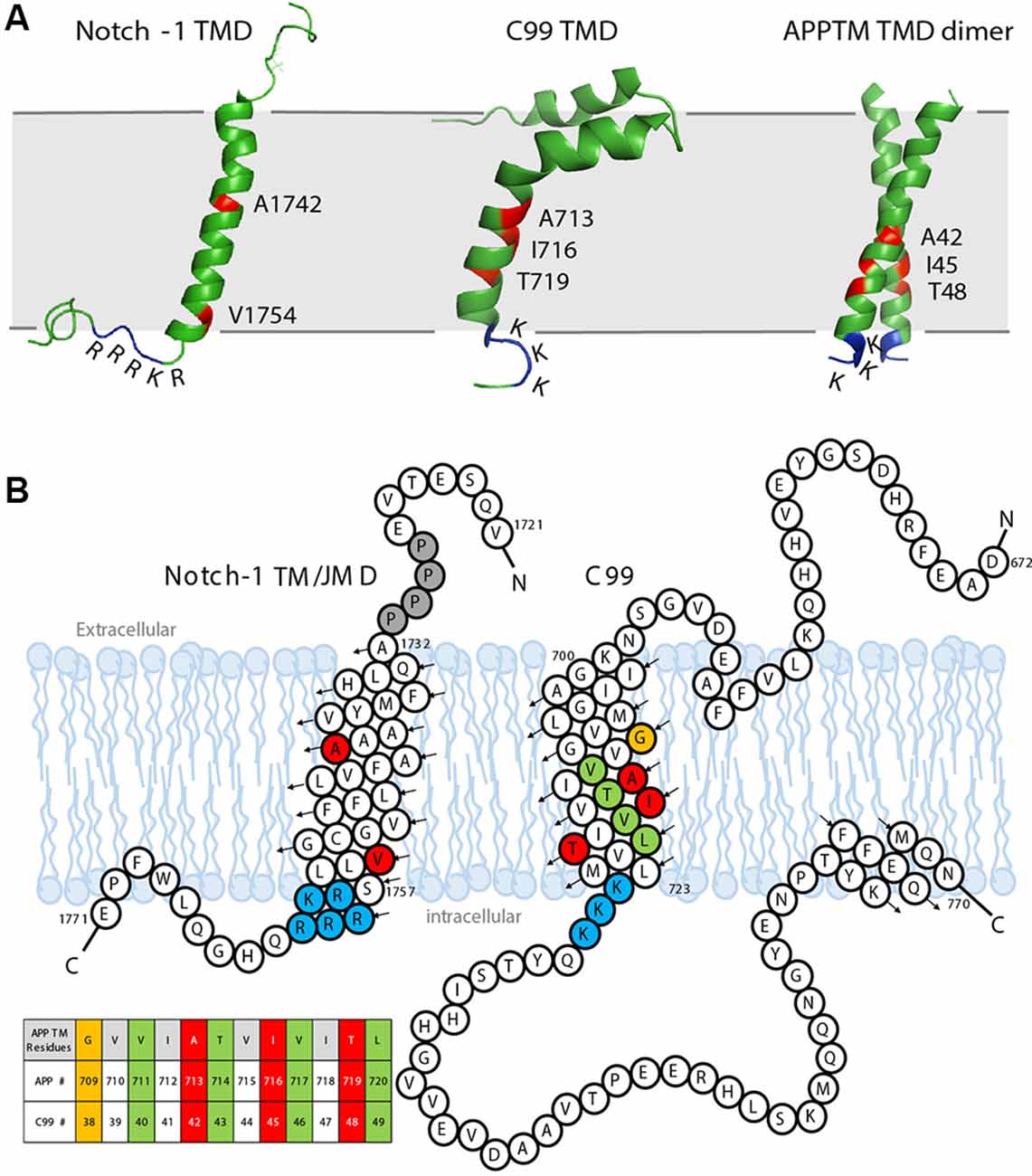
Figure 5. The sequence and solution structure of the transmembrane domain (TMD) of Notch-1 and APP. (A) The solution nuclear magnetic resonance (NMR) structure of Notch-1 TMD (PDB: 5KZO), C99 TMD (PDB: 2LP1), and APPTM TMD dimer (PDB: 2LZ3). The major cleavage S3 site on Notch-1 and Aβ42 cleavage sites on C99/APPTM are labeled red. The juxtamembrane domains lysine (K) and arginine (R) are indicated. (B) The sequence and topology of Notch-1 TMD (adapted with permission from Deatherage et al., 2017; copyright 2017 American Association for the Advancement of Science) and APP-C99 (adapted with permission from Beel et al., 2008; copyright 2008 American Chemical Society). Black arrows indicate the direction of the helices from N- to C-termini. Important residues are color coded: the positively charged juxtamembrane domain residues are labeled blue. In Notch-1, the major S3 cleavage site Val1754 and major S4 cleavage site A1742 are labeled red. In APP-C99, the Aβ40 cleavage sites are labeled green, and the Aβ42 cleavages sites are labeled red/orange. A table of APPTM residue numbering is provided in the context of both APP and C99.
GS and Notch Signaling
Notch signaling is involved in neurogenesis, synapse growth and plasticity, and neuronal death in vertebrates (Kopan and Ilagan, 2009). The Notch receptor is a single-span membrane protein like APP. For Notch-1, the TMD is from residues Ala1732 to Ser1757, terminated by a cluster of basic residues: 1758RKRRR1762, similar to the APP intracellular juxtamembrane region 724KKK726 (Deatherage et al., 2017; Figures 5A,B). In the Notch signaling pathway, Notch precursors are cleaved by a furin-like convertase at Site-1 (S1), generating the mature Notch receptor, a 2,500-residue membrane protein. The shedding of the Notch ectodomain following S1 cleavage is carried out by ADAM, a metalloprotease, which is referred to as Site-2 (S2) cleavage. After shedding, the Notch receptor undergoes cleavage by GS, which, like APP, is Processive (van Tetering and Vooijs, 2011). For Notch-1, the initial cleavage, which is called the Site-3 (S3) cleavage, mainly occurs at Val1754 (Figure 5B), releasing a large Notch intracellular domain (NICD; Deatherage et al., 2017). The NICD translocates to the nucleus, forming an activator complex (Kitagawa, 2015). The processive cleavage stops at Site-4 (S4), mainly at Ala1742, and an extracellular domain (ECD) peptide (Nβ) terminating at residue 1741 is released (Deatherage et al., 2017). PS1 mutations associated with FAD also cause a shift in the Nβ cleavage site, in a similar manner to Aβ (Okochi et al., 2006).
Targeting GS for AD Drug Discovery
A major theme in AD drug discovery is to reduce amyloid by inhibiting GS. To date, however, clinical trials of GSIs have failed due to severe side effects and worsening cognitive functions in patients. The so-called Notch-sparing APP-selective inhibitors, which preferentially inhibit APP cleavage over Notch by GS, did not show reduced toxicity (Crump et al., 2012; Tong et al., 2012). Another strategy in AD drug discovery is to develop GS modulators (GSM), which bias GS activity towards generating shorter, less toxic Aβ peptides (Bursavich et al., 2016). Given the complexity of the role of GS in biology beyond Notch and APP, it is imperative that the molecular details of GS interactions with substrates be understood to inform an effective strategy for discovering disease-modifying drugs in AD.
Structures of APO GS and its Subunits
There are four essential components of GS: PS (also abbreviated as PSEN), nicastrin (NCT), anterior pharynx-defective 1 (APH-1), and PS enhancer 2 (PEN-2; Kimberly et al., 2003; Figure 6A). The catalytic subunit, PS, consists of nine TMs with two catalytic aspartates, Asp257 and Asp385, located in TM6 and TM7, respectively (Wolfe et al., 1999; Li et al., 2013; Bai et al., 2015b). GS is matured and activated only after PS undergoes autoproteolysis, cleaving itself between TM6 and TM7 and dividing PS into an NTF and a CTF (Thinakaran et al., 1996; Knappenberger et al., 2004). NCT, which has a large, heavily glycosylated ECD and a single TM segment (Xie et al., 2014), is involved in the initial binding of substrate and likely inhibits the docking of substrates with long N-termini prior to the action of a sheddase. APH-1 contains seven TMs and is mainly responsible for the assembly, scaffolding, and stabilization of the GS complex (Brunkan et al., 2005). PEN-2, composed of three TMs is required for PS autoproteolysis and stabilizes PS NTF and CTF (Luo et al., 2003; Prokop et al., 2004). Although these four components are sufficient for performing cleavage, additional proteins are possibly involved in the modulation of the GS cleavage activity (Wakabayashi et al., 2009). For example, TMP21, a member of the p24 cargo protein family, is reported to be a component of PS complexes and regulates GS cleavage (Chen et al., 2006).
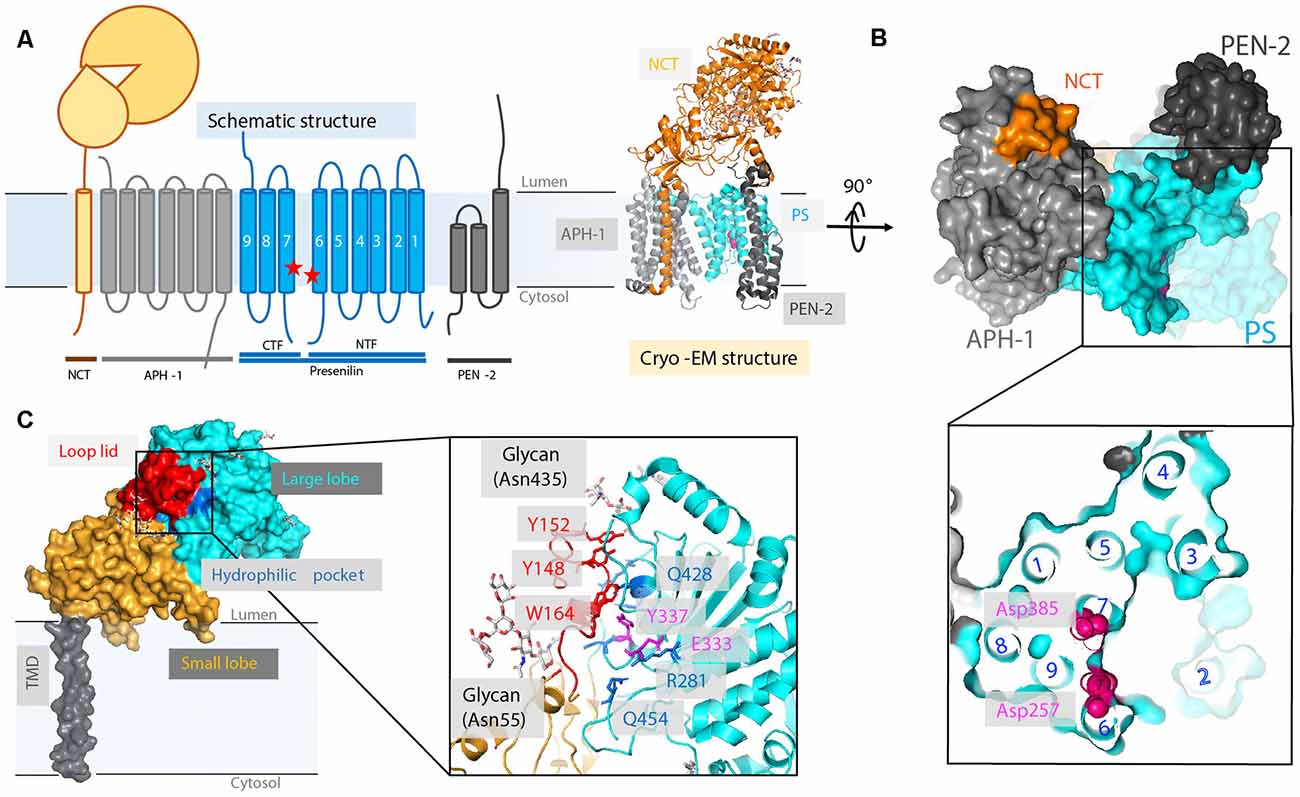
Figure 6. High-resolution cryo-electron microscopy (cryo-EM) structure of apo γ-secretase (GS; PDB: 5A63). (A) Schematics of GS complex. (B) The horseshoe shape arrangement of the GS TMs, with two catalytic aspartates located on the convex side of the transmembrane helix (TM) horseshoe: Asp257 on TM6 and Asp385 on TM7. The location of TM2 is drawn based on a bound-state GS structure (PDB: 5FN3). (C) The cryo-EM structure of a nicastrin subunit; a close-up view of the hydrophilic pocket is shown.
X-Ray Structure of a PSH From Methanoculleus marisnigri JR1 (MCMJR1)
A PS ortholog was discovered from Methanoculleus marisnigri JR1 (Torres-Arancivia et al., 2010), and its X-ray structure (Figure 1) was solved soon thereafter (Li et al., 2013). The two catalytic aspartate residues are ~9–10 Å apart. This distance is too far for the coordination of a catalytic water when compared to soluble aspartate proteases. In pepsin, the two catalytic aspartates are ~3 Å away from each other, and in HIV protease, the two catalytic aspartates are only 2.3 Å apart (Kovalevsky et al., 2007; Weber et al., 2013). Several explanations may account for the MCMJR1 structure being in an inactive conformation. Limited proteolysis was used during crystallization, which likely removed linker regions between TMs that in turn allow new motions to occur. Another possibility is that the apo state of the enzyme is an inactive conformation, and substrate binding triggers a conformational change to move the two aspartate residues closer together to carry out catalysis, as suggested by structures of GS (see below and Bai et al., 2015a).
X-Ray Structure of the NCT Homolog From Dictyostelium purpureum (DpNCT)
The structure of NCT was first solved for a eukaryotic homolog from Dictyostelium purpureum (DpNCT), which shares 40% sequence identity with human NCT (HsNCT). The 1.95-Å resolution crystal structure reveals a large ECD and a single TM helix (Xie et al., 2014). The ECD of DpNCT contains a large lobe and a small lobe, interacting with each other through numerous van der Waals contacts at the center of the interface and 11 hydrogen bonds at the periphery of the interface. A pocket in the large lobe is surrounded by hydrophilic side chains, which may be responsible for anchoring hydrophilic N-termini of the substrates such as APP and Notch. An extended loop from the small lobe forms a lid that hovers above the pocket, likely gating substrate entry. Conformational changes are needed for substrate recruitment (Li et al., 2014; Xie et al., 2014).
Cryo-Electron Microscopy Structure of GS
After intensive cryo-electron microscopy (cryo-EM) efforts (Lu et al., 2014; Sun et al., 2015), a 3.4-Å map of GS was obtained with excellent main-chain connectivity and discernable side-chain features (Bai et al., 2015b; Figures 6A,B). Among the 20 TMs identified, TM2 of PS1 shows the highest degree of flexibility. Except for TM2 and TM6, the other 18 TMs were observed with good side-chain density, including the seven TMs of APH-1, the other seven TMs of PS1, the three TMs of PEN-2, and the lone TM of NCT. Overall, the TMs form a horseshoe shape (Bai et al., 2015b; Sun et al., 2015), with PS1 and APH-1 at the center and PEN-2 and NCT at the tips of the horseshoe. The two catalytic residues (Asp257 and Asp385 of PS1) are on the convex side of the TM horseshoe (Figure 6B). The cryo-EM structure of PS solved here is largely superimposable with the PSH from MCMJR1. The ECD of NCT directly interacts with PEN-2. The TMs predominantly interact through van der Waals contacts among hydrophobic side chains.
The flexibility of PS1 TM2 and TM6 seen in the cryo-EM structure suggests a pathway for the substrate entrance and conformational changes during substrate docking and translocation. Masked classification of the apo-state GS cryo-EM dataset revealed three major classes of conformations (Figure 7; Bai et al., 2015a). In class 1, TM2 from PS1 is ordered, and there is unassigned density corresponding to a kinked α-helix, which may be a fortuitously co-purified cellular substrate or product. In class 2, the TM2 helix could be also observed but not well defined. In class 3, no substrate or TM2 could be observed. PEN-2 rotates away from PS1, together with PS1 TM3 and TM4, while PS1 TM5/TM6 move towards the extracellular/lumenal space and TM6 rotates towards TM7. In the cryo-EM structure of GS in complexes with the peptidomimetic inhibitor DAPT (Bai et al., 2015a), the conformation of PS1 is very similar to class 1. Both PS1 TM2 and the linkers between TM2 and TM1 and TM2 and TM3 become ordered in the presence of DAPT, as well as part of the long linker between TM6 and TM7. TM6 displays a kink near the active site, forming a hydrophobic binding pocket with TM2, TM3, TM5, and TM7 for DAPT, the same pocket that APP and Notch substrates occupy revealed by later cryo-EM structures (see “Interaction of GS With Substrates” section). Crucial structural features and interactions of PS1 are listed in Table 1.
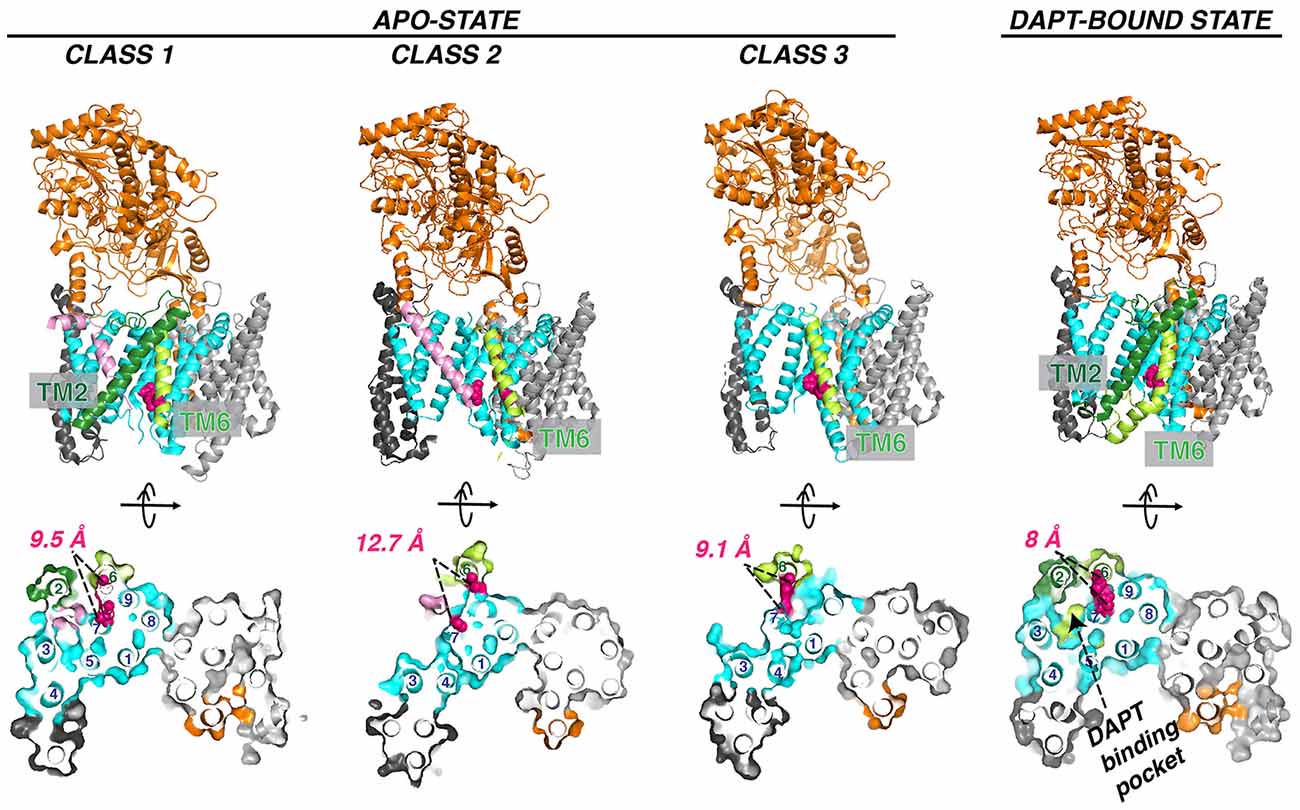
Figure 7. Three classes of apo GS conformation and DAPT-bound GS structure. The pink helix in class 1 (PDB: 5FN3) and class 2 (PDB: 5FN4) represents an unidentified substrate co-purified with GS. TM2 observed in class 1 and DAPT bound state (PDB: 5FN2) is in forest green. The two catalytic aspartate residues are colored red. Helix numbering of the PS1 subunit is labeled in the orthogonal view from the cytosolic side.
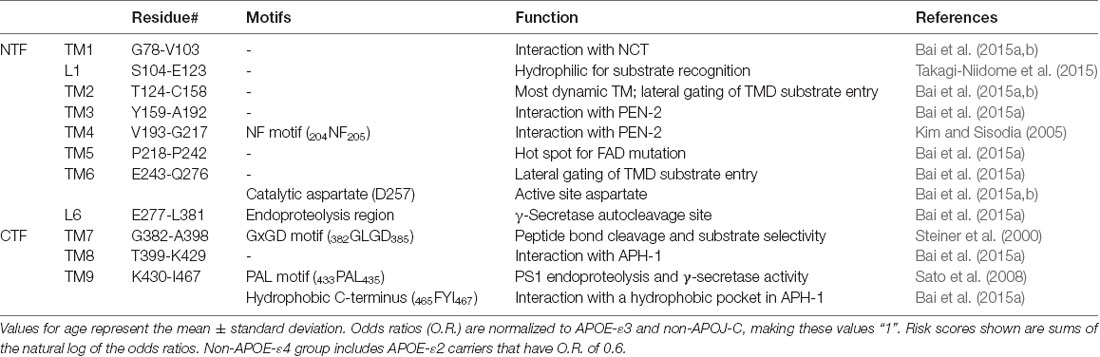
Table 1. Function and motifs in the transmembrane domains (TM) and loops (L) of PS1.
The cryo-EM structure of GS also reveals new details regarding HsNCT (Bai et al., 2015b; Figure 6C). First, the residues involved in GS substrate recognition, Glu333 and Tyr337, are located in a hydrophilic pocket. Charged arginine residues (Arg281, Arg285, Arg429, and Arg432) in this buried pocket may also mediate specific hydrogen bonding and salt bridges for substrate recruitment. Second, 11 glycosylation sites were identified on the large lobe. This heavy glycosylation likely contributes to substrate recruitment (Shah et al., 2005) and in ECD folding and stability. Two glycans on Asn55 and Asn435 from the large lobe flank the lid from the small lobe.
Interaction of GS With Substrates
Several interaction models have been put forth to explain the successive cleavage of APP substrate by GS [see “GS and AD” section]. First, a “piston model” was proposed in which APP–C99 remains in a helical conformation but shifts successively downward towards the active site of PS (Takagi et al., 2010). However, downward shifting of the substrate may make it harder for the product to be released as processive cleavage progresses. Second, a substrate “bending model” was put forward based on C99 TM backbone dynamics and the bend of a co-purified substrate observed in the class I cryo-EM structure of GS. In this model, C99 presents the scissile bond by bending the TM helix (Scharnagl et al., 2014; Langosch et al., 2015). Lastly, as elaborated in this section, growing evidence supports a substrate TM unwinding model to generate the scissile peptide bond in extended conformation, favoring the extended β-strand conformation that binds productively to the active site of proteases (Madala et al., 2010).
Docking Site Mapping by Nuclear Magnetic Resonance (NMR)
Solution NMR has been utilized to probe substrate docking of APPTM, using PS orthologs that are catalytically active towards the TM segment of APP (APPTM). Chemical shift perturbation (CSP) showed that juxtamembrane regions of APPTM mediate its docking to MCMJR1. The largest CSP occurred at residues K28 and K54 of APPTM (Figure 8), likely mediating electrostatic interactions with the MCMJR1 (Clemente et al., 2018). Binding of the substrate to MCMJR1 decreased the magnitude of amide proton chemical shifts δH at the C-terminal half of the substrate APPTM. Because amide δH has a strong positive correlation with hydrogen bond strength, the pattern of decreasing δH indicates that the docking to the enzyme weakens helical hydrogen bonds and unwinds the substrate TM helix around the initial ε-cleavage site. The APPTM V44M substitution linked to FAD caused more CSP and helical unwinding around the ε-cleavage site. MAMRE50, another archaeal ortholog of PSH, which cleaved APPTM at a higher rate, also caused more CSP and helical unwinding in APPTM than in MCMJR1. These data suggest that docking of the substrate TM helix and helix unwinding are coupled in intramembrane proteolysis by PS and its ortholog, and FAD mutations can modify enzyme–substrate interaction.
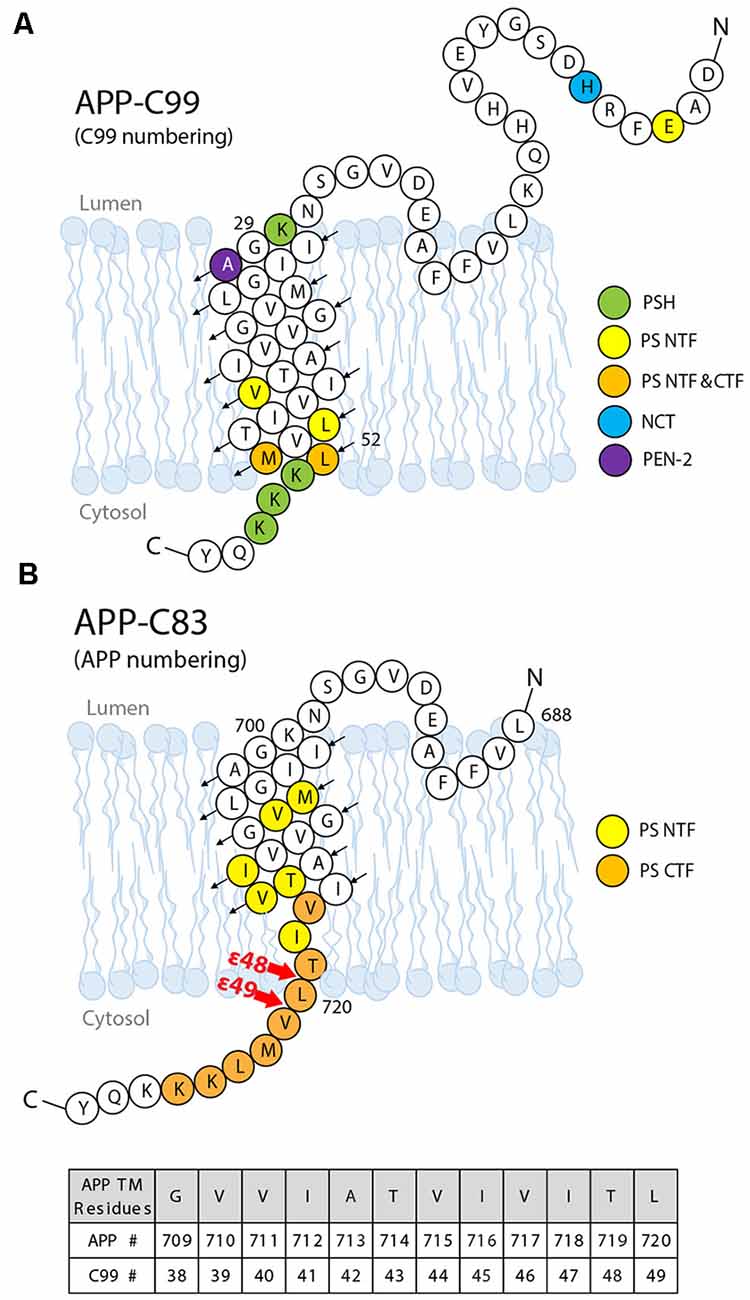
Figure 8. The substrate–enzyme interaction sites on the APP substrate. (A) Interaction sites identified by NMR titration (green) and photoaffinity cross-linking (other colors). (B) Interaction sites between the presenilin subunit and APP identified by cryo-EM. Upon I-CLiP binding, C-terminus unwinding occurs, and a β-strand is formed from L720 to K725.
Interaction Mapping by Photoaffinity Cross-Linking
A comprehensive mapping of the interaction between APP C99 and GS at residue resolution was accomplished by photoaffinity mapping (Fukumori and Steiner, 2016). Sixty-eight His-tagged C99 constructs containing photo-active amino acid para-benzoyl-L-phenylalanine (Bpa) substitution, from residues D1 to D68, were produced. After incubation with CHAPSO-solubilized GS and UV irradiation, the Bpa residue photo-cross-linked with nearby GS residues, within ~3 Å. Cross-linked substrates and GS components were isolated by Ni-NTA affinity pulldown followed by dissociation of GS for photoaffinity mapping.
Photoaffinity mapping showed that APP C99 residues Val44, Leu49, Met51, and Leu52 are cross-linked to PS1 NTF, representing major substrate–enzyme interaction sites. Cross-linking at an exosite was also observed. C99 Glu3 was cross-linked to PS1 NTF, most likely through interaction with the loop L1 between TM1 and TM2. His6 and Ala30 cross-linked with NCT and PEN-2, respectively. Ala30 is not close to PEN-2 in the cryo-EM structure of the GS–APP complex, indicating that major conformation changes occur during substrate–GS interaction. Met51 and Leu52 also cross-linked to PS1 CTF (Figure 8), as expected. To distinguish between interactions for substrate recruitment and for cleavage, “substrate-binding chase” experiments were carried out: first, C99 “binding” and cross-linking to GS were performed at 4°C to inhibit enzyme cleavage, followed by a 37°C cleavage “chase” experiment. When the substrate was cross-linked with PS1 NTF, it could be cleaved under 37°C and could also be inhibited by GSIs. However, when the substrate and NCT/PEN-2 are cross-linked, the substrate cannot be cleaved, indicating that exosite cross-linking blocked the substrate passage from the GS exosite to the active site. Furthermore, the cross-linking of PS1 NTF was suppressed by GSIs while cross-linking involving PEN-2 was increased with GSI’s presence. These data further confirmed the existence of a substrate docking site distinct from the active site. In summary, these studies show that the GS substrate binds to GS in two steps: first, the substrate binds to the exosite, likely formed by NCT, PEN-2, and NTF, and then the substrate translocates to the active site formed by PS1 NTF/CTF. Compared with the interaction sites identified in the cryo-EM structure of the GS–APP complex (Zhou et al., 2019; Figure 8B), this photoaffinity mapping showed additional interaction sites during substrate docking and translocation.
Biophysical Studies of Substrate TM Unwinding
Solid-state NMR revealed that the TM helix of C99 unravels downstream of the ε-sites (Sato et al., 2009). Under isotopic labeling, deep-ultraviolet resonance Raman (dUVRR) spectra of Gurken, a substrate for GlpG rhomboid and MCMJR1 (Torres-Arancivia et al., 2010), displays both α-helical and 310-helical geometry; 310-helical unwinding was observed during binding to the enzyme (Brown et al., 2018). When the 310-helical content was suppressed using a proline-to-alanine mutation, binding was not affected, but cleavage was inhibited. This result is consistent with the fact that the initial docking site is distinct from the active site proposed for GS (Fukumori and Steiner, 2016) and rhomboids (Arutyunova et al., 2014). As mentioned above, hydrogen bond weakening and helical unwinding in the APPTM C-terminus upon binding to MCMJR1 were also observed in solution NMR (Clemente et al., 2018).
Cryo-EM Structure of GS in Complex With Notch and APP
The unwinding of the substrate TM helix at the carboxyl terminus was confirmed in cryo-EM structures of human GS in complex with mouse Notch-100 (Yang et al., 2019) and APP-C83 fragment (Zhou et al., 2019). To stabilize the GS–substrate complexes, disulfide-cross-linked GS–APP/Notch complexes were generated with human GS containing an active site mutation (PS1-Q112C/D385A, PEN-2, APH-1aL, and NCT) and APP-C83 (V695C; Zhou et al., 2019) or Notch-100 (P1728C; Yang et al., 2019). In the highest-resolution (2.6–2.7 Å) complex structure, TM6 extends to having two helices (TM6 and TM6a; Figure 9); TM2 and the loop between TM6/TM7 of PS are more ordered compared to free GS (Bai et al., 2015b).
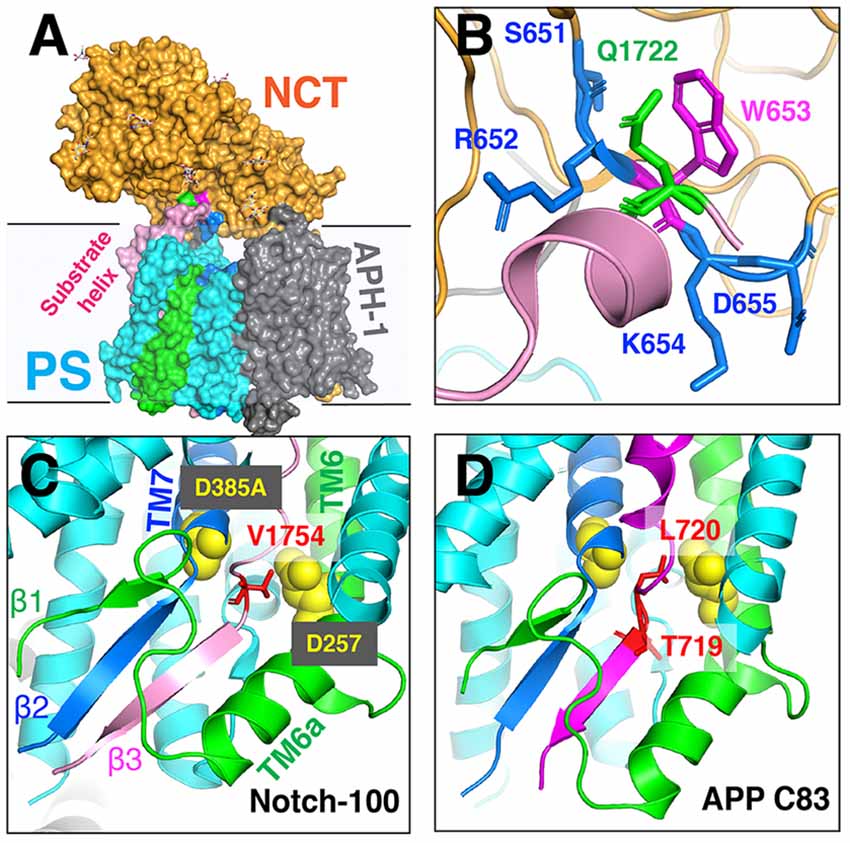
Figure 9. The cryo-EM structure of the GS–substrate complex with Notch-100 and APP-C83. Nicastrin (NCT) is colored orange and PS1 cyan. TM6 and TM7 from PS1 are colored green and blue, respectively. Substrates are in pink/magenta. (A) The overall complex structure. (B) A close-up view of the NCT hydrophilic pocket interacting with the Notch substrate. Q1722 is on Notch-100. 651SRWKD655 is on NCT. (C) The intermolecular β-sheet around Notch-100 C-terminal cleavage sites. TM6 extends to two helices (TM6/TM6a). The hybrid β-sheet consists of β1 from TM6, β2 from TM7, and β3 from the substrate. Two catalytic aspartates are at the S3 cleavage site. (D) A similar hybrid β-sheet between APP-C83 and PS TM6/TM7. The ε-cleavage sites are in extended conformation.
The structures reveal that the C-termini of both APP and Notch adopt a β-strand conformation, forming an intermolecular, antiparallel β-sheet with two induced β-strands from PS1 NTF (TM6) and CTF (TM7). In this β-strand mode, the cleavage sites on substrate TM are in a more extended conformation and become more exposed. The ε-cleavage sites (residues T719 and L720) in APPTM are fully extended (Figure 9D), as is the S3 cleavage sites (V1754) at the C-terminal part of Notch TM (Figure 9C).
Additional details of the participation of the NCT ECD in substrate recruitment (Xie et al., 2014) were revealed in the complex structures. In addition to the hydrophilic pocket reported in DpNCT (Xie et al., 2014), another hydrophilic pocket (Ser651, Arg652, Lys654, and Asp655) located at the small lobe near the membrane was identified (Figure 9A). A short helix of Notch-100 is inserted into the hydrophilic pocket (Yang et al., 2019). Kinetic data showed that the binding affinity between GS and Notch is driven by TMD interaction and that the affinity decreases with increasing ectodomain length and structure (Bolduc et al., 2016). Substrates with longer ectodomains could only be efficiently cleaved after disrupting the NCT fold. The sterical hindrance of NCT likely contributes to the selectivity of the GS substrate.
Open Questions and Future Directions
Despite the tremendous progress detailed above, our molecular picture of GS remains far from complete. We do not know the effect of the lipid composition of the lipid bilayer, hence how the cellular location of APP affects GS cleavage and how FAD mutations affect Aβ production and increase the Aβ42/Aβ40 ratio. We still do not have full clarity on how GS interacts with its substrate. In Figure 10, we outline the major steps of APP C99 interaction with GS, which ultimately results in the production of Aβ, a pathogenic peptide in AD. In each step, there are many important, unanswered questions:
1. C99 docks to the GS exosite, coupled with helical unwinding near the initial cleavage site (Clemente et al., 2018). However, we do not know the molecular identity of the exosite. Most likely the exosite is not too far from the active site and may be composed of both NCT, TM2, and loop 1 of PS. The exosite may be mapped by blocking substrate entry into the GS active site using an active site GSI. Disulfide gates may be engineered to probe the exosite and substrate translocation pathway, as was carried out with rhomboids (Baker et al., 2007).
2. From the exosite, C99 translocates to the enzyme active site, forming an intermolecular β-sheet with PS (Zhou et al., 2019). We do not know the pathway of substrate translocation, partly because we do not know the exact substrate docking site. TM2 and TM6 are the most dynamic TMs in PS1 and therefore are mostly likely involved in the lateral gating mechanism of substrate translocation. The detailed dynamics of substrate translocation can be elucidated by combining the power of molecular dynamics simulations and cutting-edge experimental structural determination methods for membrane proteins.
3. Initial ε-cleavage occurs at T48 or L49, releasing the AICD and forming Aβ48 or Aβ49, precursor peptides of the Aβ42 or Aβ42 production line, respectively. Here, the catalytic mechanism is not known, nor how the two active site aspartates coordinate a catalytic water molecule to facilitate hydrolysis. In all of the solved structures of GS and MCMJR1, the catalytic aspartates appear to be too far away from each other to coordinate a catalytic water. Thus, we have yet to capture the conformation of the GS active site in a catalytically competent state. Because of the stability of hybrid β-sheet at the C-terminus of C83, a large conformational change is needed for reducing this intermolecular interaction to facilitate the release of AICD. How this happens also remains an open question.
4. Following ε-cleavage, carboxypeptidase activity of GS trims Aβ48/Aβ49 processively (Figure 10), shedding tripeptides to produce Aβ42 and Aβ40. The mechanism of processive cleavage is not known. Based on biochemical evidence, Wolfe et al. proposed a tripeptide binding pocket in the GS active site for P1′P2′P3′ (Wolfe, 2020), which is not obvious in the GS–C83 complex. How the active site aspartates get to the next cleavage site on the substrate, as well as the driving force for this process, is not clear. It is straightforward to speculate that it involves concerted conformational changes and dynamics in both GS and the substrate. The catalytic aspartates in PS may move towards more N-terminal cleavage sites in APPTM while GS continues to unwind the substrate. The timing of AICD release and C-terminal trimming is not clear, for example, whether they are concurrent, sequential, or of random order. We suggest that MD simulations will be extremely helpful in providing clues for experimentalists in this area.
5. Finally, following processive cleavage by GS, the shorter and more hydrophilic Aβ fragment dissociates from the enzyme and exits the membrane. The mechanism of Aβ peptide or AICD release has been little studied. What are the kinetics and pathway of Aβ release? How does it involve NCT and other components of GS? During processive cleavage, Aβ fragments may either be released or undergo one more step of trimming (e.g., Aβ42 is released vs. Aβ42 is cut down to Aβ38). How is this bifurcation in the Aβ production pathway determined mechanistically? Both equilibrium (Szaruga et al., 2017) and kinetic stability of the Aβ/GS complex might be critical determinants in this situation. Answers to these questions have important implications for the design and discovery of new GSMs and selective GSIs.
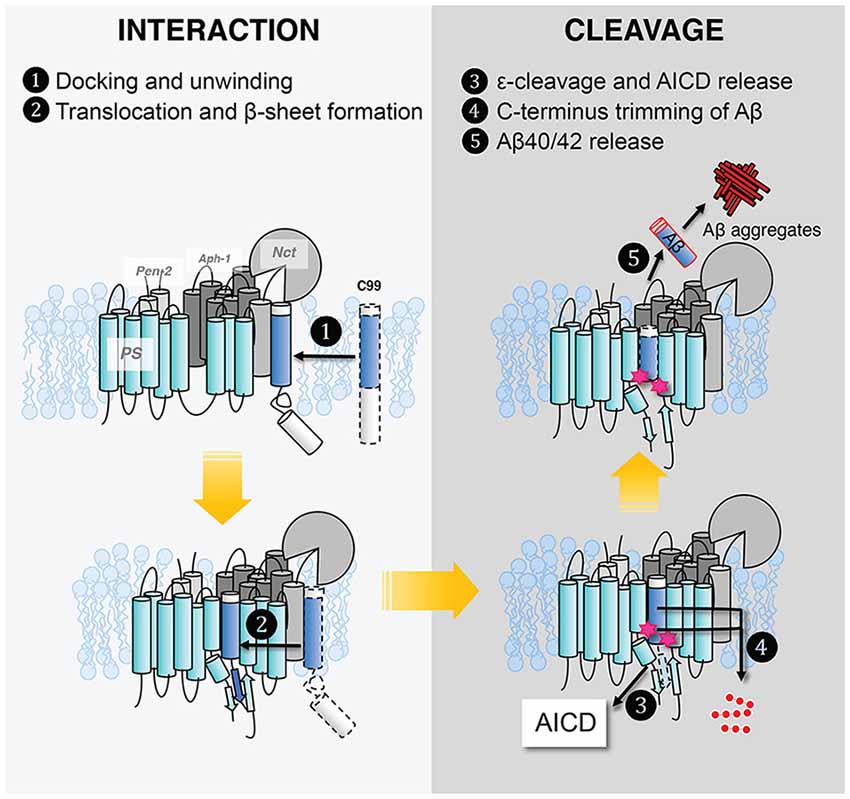
Figure 10. Five steps in γ-secretase–substrate interaction and cleavage to produce Aβ.
Given the recent structural insights, an intriguing question for AD drug discovery is whether selective GSIs can be designed or discovered. Yang et al. (2019) pointed out several distinct pockets in the GS–C83 complex that have different shapes and dimensions compared with the GS–Notch complex (Zhou et al., 2019), which may be targeted for rational drug design. However, it is important to note that GS is highly dynamic, and binding pockets can stretch and/or shrink. Thus, for selective GSI, we may still need to rely on docking coupled with long-time-course MD simulation, high-throughput (HT), or ultra-HT methods such as DNA encoded libraries which enable screening of tens of billions of compounds in a single test tube (Satz, 2018).
Conclusion
There has been tremendous progress in the structural and mechanistic investigation of the substrate–enzyme interaction in intramembrane proteolysis, especially in light of the recent cryo-EM structures of GS–C83 and GS–Notch complexes. In particular, cryo-EM revealed the formation of a hybrid, intermolecular β-sheet between GS and its substrates, which is consistent with numerous biochemical and biophysical studies. However, our knowledge of how GS interacts with its substrates, which is crucial for developing selective amyloid reduction agents, remains far from complete.
Author Contributions
XL and CW prepared the text and figures. JZ, YZ, IU-B, SF, and RL edited and revised the manuscript.
Funding
This work was supported by a grant NS109926 to CW from the National Institute of Neurological Disorders and Stroke (NINDS), and a grant to RL from NSF MCB-1817796.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
I-CLiPs, intramembrane-cleaving proteases; GS, γ-secretase; APP, amyloid precursor protein; cryo-EM, cryo-electron microscopy; SREBPs, sterol regulatory element-binding proteins; AD, Alzheimer’s disease; S2P, site-2-protease; S1P, site-1-protease; TM, transmembrane helix; IAP, intramembrane aspartate protease; PS, presenilin; SPP, signal peptide peptidase; TMD, transmembrane domain; PSH, presenilin homolog; NTF, amino-terminal fragment; CTF, carboxy-terminal fragment; SANS, small angle neutron scattering; Rce1, Ras and a-factor converting enzyme 1; MmRce1, Methanococcus maripaludis homolog of Rce1; ZMPSTE24, zinc metallopeptidase STE24; Aβ, amyloid-β peptide; APPTM, transmembrane domain of APP; C99, C-terminal fragment containing 99 amino acid residues; AICD, APP intracellular domain; FAD, familial Alzheimer’s disease; PSEN1 and PSEN2, presenilin 1 and 2 genes; NICD, Notch intracellular domain; NCT, nicastrin; APH-1, anterior pharynx-defective 1; PEN-2, presenilin enhancer 2; DpNCT, Dictyostelium purpureum homolog of nicastrin; HsNCT, human nicastrin; ECD, extracellular domain; CSP, chemical shift perturbation; GSIs, γ-secretase inhibitors; dUVRR, deep-ultraviolet resonance Raman spectra.
References
Annaert, W., and De Strooper, B. (1999). Presenilins: molecular switches between proteolysis and signal transduction. Trends Neurosci. 22, 439–443. doi: 10.1016/s0166-2236(99)01455-1
Arutyunova, E., Panwar, P., Skiba, P. M., Gale, N., Mak, M. W., and Lemieux, M. J. (2014). Allosteric regulation of rhomboid intramembrane proteolysis. EMBO J. 33, 1869–1881. doi: 10.15252/embj.201488149
Bai, X.-C, Rajendra, E., Yang, G., Shi, Y., and Scheres, S. H. W. (2015a). Sampling the conformational space of the catalytic subunit of human γ-secretase. Elife 4:e11182. doi: 10.7554/eLife.11182
Bai, X.-C., Yan, C., Yang, G., Lu, P., Ma, D., Sun, L., et al. (2015b). An atomic structure of human γ-secretase. Nature 525, 212–217. doi: 10.1038/nature14892
Baker, R. P., Young, K., Feng, L., Shi, Y., and Urban, S. (2007). Enzymatic analysis of a rhomboid intramembrane protease implicates transmembrane helix 5 as the lateral substrate gate. Proc. Natl. Acad. Sci. U S A 104, 8257–8262. doi: 10.1073/pnas.0700814104
Beel, A. J., Mobley, C. K., Kim, H. J., Tian, F., Hadziselimovic, A., Jap, B., et al. (2008). Structural studies of the transmembrane C-terminal domain of the amyloid precursor protein (APP): does APP function as a cholesterol sensor? Biochemistry 47, 9428–9446. doi: 10.1021/bi800993c
Beel, A. J., and Sanders, C. R. (2008). Substrate specificity of γ-secretase and other intramembrane proteases. Cell. Mol. Life Sci. 65, 1311–1334. doi: 10.1007/s00018-008-7462-2
Ben-Shem, A., Fass, D., and Bibi, E. (2007). Structural basis for intramembrane proteolysis by rhomboid serine proteases. Proc. Natl. Acad. Sci. U S A 104, 462–466. doi: 10.1073/pnas.0609773104
Bergbold, N., and Lemberg, M. K. (2013). Emerging role of rhomboid family proteins in mammalian biology and disease. Biochim. Biophys. Acta 1828, 2840–2848. doi: 10.1016/j.bbamem.2013.03.025
Bertram, L., Lill, C. M., and Tanzi, R. E. (2010). The genetics of Alzheimer disease: back to the future. Neuron 68, 270–281. doi: 10.1016/j.neuron.201010.013
Bolduc, D. M., Montagna, D. R., Gu, Y., Selkoe, D. J., and Wolfe, M. S. (2016). Nicastrin functions to sterically hinder γ-secretase-substrate interactions driven by substrate transmembrane domain. Proc. Natl. Acad. Sci. U S A 113, E509–E518. doi: 10.1073/pnas.1512952113
Boyartchuk, V. L., Ashby, M. N., and Rine, J. (1997). Modulation of Ras and a-factor function by carboxyl-terminal proteolysis. Science 275, 1796–1800. doi: 10.1126/science.275.5307.1796
Brown, M. C., Abdine, A., Chavez, J., Schaffner, A., Torres-Arancivia, C., Lada, B., et al. (2018). Unwinding of the substrate transmembrane helix in intramembrane proteolysis. Biophys. J. 114, 1579–1589. doi: 10.1016/j.bpj.2018.01.043
Brown, M. S., and Goldstein, J. L. (1997). The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell 89, 331–340. doi: 10.1016/s0092-8674(00)80213-5
Brown, M. S., Ye, J., Rawson, R. B., and Goldstein, J. L. (2000). Regulated intramembrane proteolysis: a control mechanism conserved from bacteria to humans.. Cell 100, 391–398. doi: 10.1016/s0092-8674(00)80675-3
Brunkan, A. L., Martinez, M., Walker, E. S., and Goate, A. M. (2005). Presenilin endoproteolysis is an intramolecular cleavage. Mol. Cell. Neurosci. 29, 65–73. doi: 10.1016/j.mcn.2004.12.012
Bursavich, M. G., Harrison, B. A., and Blain, J.-F. (2016). γ secretase modulators: new Alzheimer’s drugs on the horizon? J. Med. Chem. 59, 7389–7409. doi: 10.1021/acs.jmedchem.5b01960
Chen, W., Gamache, E., Rosenman, D. J., Xie, J., Lopez, M. M., Li, Y.-M., et al. (2014). Familial Alzheimer’s mutations within APPTM increase Aβ42 production by enhancing accessibility of ε-cleavage site. Nat. Commun. 5:3037. doi: 10.1038/ncomms4037
Chen, F., Hasegawa, H., Schmitt-Ulms, G., Kawarai, T., Bohm, C., Katayama, T., et al. (2006). TMP21 is a presenilin complex component that modulates γ-secretase but not ε-secretase activity. Nature 440, 1208–1212. doi: 10.1038/nature04667
Christiansen, J. R., Kolandaivelu, S., Bergo, M. O., and Ramamurthy, V. (2011). RAS-converting enzyme 1-mediated endoproteolysis is required for trafficking of rod phosphodiesterase 6 to photoreceptor outer segments. Proc. Natl. Acad. Sci. U S A 108, 8862–8866. doi: 10.1073/pnas.1103627108
Clemente, N., Abdine, A., Ubarretxena-Belandia, I., and Wang, C. (2018). Coupled transmembrane substrate docking and helical unwinding in intramembrane proteolysis of amyloid precursor protein. Sci. Rep. 8:12411. doi: 10.1038/s41598-018-30015-6
Crump, C. J., Castro, S. V., Wang, F., Pozdnyakov, N., Ballard, T. E., Sisodia, S. S., et al. (2012). BMS-708,163 targets presenilin and lacks notch-sparing activity. Biochemistry 51, 7209–7211. doi: 10.1021/bi301137h
De Strooper, B., and Chávez Gutiérrez, L. (2015). Learning by failing: ideas and concepts to tackle γ-secretases in Alzheimer’s disease and beyond. Annu. Rev. Pharmacol. Toxicol. 55, 419–437. doi: 10.1146/annurev-pharmtox-010814-124309
Deatherage, C. L., Lu, Z., Kroncke, B. M., Ma, S., Smith, J. A., Voehler, M. W., et al. (2017). Structural and biochemical differences between the Notch and the amyloid precursor protein transmembrane domains. Sci. Adv. 3:e1602794. doi: 10.1126/sciadv.1602794
Düsterhöft, S., Künzel, U., and Freeman, M. (2017). Rhomboid proteases in human disease: mechanisms and future prospects. Biochim. Biophys. Acta Mol. Cell Res. 1864, 2200–2209. doi: 10.1016/j.bbamcr.2017.04.016
Feng, L., Yan, H., Wu, Z., Yan, N., Wang, Z., Jeffrey, P. D., et al. (2007). Structure of a site-2 protease family intramembrane metalloprotease. Science 318, 1608–1612. doi: 10.1126/science.1150755
Fluhrer, R., Steiner, H., and Haass, C. (2009). Intramembrane Proteolysis by signal peptide peptidases: a comparative discussion of GXGD-type aspartyl proteases. J. Biol. Chem. 284, 13975–13979. doi: 10.1074/jbc.r800040200
Francis, R., McGrath, G., Zhang, J., Ruddy, D. A., Sym, M., Apfeld, J., et al. (2002). Aph-1 and pen-2 are required for notch pathway signaling, γ-secretase cleavage of βAPP and presenilin protein accumulation. Dev. Cell 3, 85–97. doi: 10.1016/s1534-5807(02)00189-2
Fukumori, A., and Steiner, H. (2016). Substrate recruitment of γ-secretase and mechanism of clinical presenilin mutations revealed by photoaffinity mapping. EMBO J. 35, 1628–1643. doi: 10.15252/embj.201694151
Goate, A., Chartier-Harlin, M.-C., Mullan, M., Brown, J., Crawford, F., Fidani, L., et al. (1991). Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 349, 704–706. doi: 10.1038/349704a0
Li, X., Dang, S., Yan, C., Gong, X., Wang, J., and Shi, Y. (2013). Structure of a presenilin family intramembrane aspartate protease. Nature 493, 56–61. doi: 10.1038/nature11801
Haass, C., and Steiner, H. (2002). Alzheimer disease γ-secretase: a complex story of GxGD-type presenilin proteases. Trends Cell Biol. 12, 556–562. doi: 10.1016/s0962-8924(02)02394-2
Haze, K., Yoshida, H., Yanagi, H., Yura, T., and Mori, K. (1999). Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell 10, 3787–3799. doi: 10.1091/mbc.10.11.3787
Kelleher, R. J., and Shen, J. (2010). γ-secretase and human disease. Science 330, 1055–1056. doi: 10.1126/science.1198668
Kim, S. H., and Sisodia, S. S. (2005). Evidence that the “NF” motif in transmembrane domain 4 of presenilin 1 is critical for binding with PEN-2. J. Biol. Chem. 280, 41953–41966. doi: 10.1074/jbc.M509070200
Kimberly, W. T., LaVoie, M. J., Ostaszewski, B. L., Ye, W., Wolfe, M. S., and Selkoe, D. J. (2003). γ-Secretase is a membrane protein complex comprised of presenilin, nicastrin, aph-1, and pen-2. Proc. Natl. Acad. Sci. U S A 100, 6382–6387. doi: 10.1073/pnas.1037392100
Kitagawa, M. (2015). Notch signalling in the nucleus: roles of Mastermind-like (MAML) transcriptional coactivators. J. Biochem. 159, 287–294. doi: 10.1093/jb/mvv123
Knappenberger, K. S., Tian, G., Ye, X., Sobotka-Briner, C., Ghanekar, S. V., Greenberg, B. D., et al. (2004). Mechanism of γ-secretase cleavage activation: is γ-secretase regulated through autoinhibition involving the presenilin-1 exon 9 loop? Biochemistry 43, 6208–6218. doi: 10.1016/j.mechmachtheory.2004
Kopan, R., and Ilagan, M. X. G. (2009). The canonical notch signaling pathway: unfolding the activation mechanism. Cell 137, 216–233. doi: 10.1016/j.cell.2009.03.045
Kovalevsky, A. Y., Chumanevich, A. A., Liu, F., Louis, J. M., and Weber, I. T. (2007). Caught in the Act: the 1.5 Å resolution crystal structures of the HIV-1 protease and the I54V mutant reveal a tetrahedral reaction intermediate. Biochemistry 46, 14854–14864. doi: 10.1021/bi700822g
Lal, M., and Caplan, M. (2011). Regulated intramembrane proteolysis: signaling pathways and biological functions. Physiology 26, 34–44. doi: 10.1152/physiol.00028.2010
Langosch, D., Scharnagl, C., Steiner, H., and Lemberg, M. K. (2015). Understanding intramembrane proteolysis: from protein dynamics to reaction kinetics. Trends Biochem. Sci. 40, 318–327. doi: 10.1016/j.tibs.2015.04.001
Langosch, D., and Steiner, H. (2017). Substrate processing in intramembrane proteolysis by γ-secretase—the role of protein dynamics. Biol. Chem. 398, 441–453. doi: 10.1515/hsz-2016-0269
Lemieux, M. J., Fischer, S. J., Cherney, M. M., Bateman, K. S., and James, M. N. G. (2007). The crystal structure of the rhomboid peptidase from Haemophilus influenzae provides insight into intramembrane proteolysis. Proc. Natl. Acad. Sci. U S A 104, 750–754. doi: 10.1073/pnas.0609981104
Levy, E., Carman, M., Fernandez-Madrid, I., Power, M., Lieberburg, I., van Duinen, S., et al. (1990). Mutation of the Alzheimer’s disease amyloid gene in hereditary cerebral hemorrhage, Dutch type. Science 248, 1124–1126. doi: 10.1126/science.2111584
Levy-Lahad, E., Wasco, W., Poorkaj, P., Romano, D., Oshima, J., Pettingell, W., et al. (1995). Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science 269, 973–977. doi: 10.1126/science.7638622
Li, Y., Lu, S. H.-J., Tsai, C.-J., Bohm, C., Qamar, S., Dodd, R. B., et al. (2014). Structural interactions between inhibitor and substrate docking sites give insight into mechanisms of human PS1 complexes. Structure 22, 125–135. doi: 10.1016/j.str.2013.09.018
Li, Y. M., Xu, M., Lai, M. T., Huang, Q., Castro, J. L., DiMuzlo-Mower, J., et al. (2000). Photoactivated γ-secretase inhibitors directed to the active site covalently label presenilin 1. Nature 405, 689–694. doi: 10.1038/35015085
Lichtenthaler, S. F., Haass, C., and Steiner, H. (2011). Regulated intramembrane proteolysis—lessons from amyloid precursor protein processing. J. Neurochem. 117, 779–796. doi: 10.1111/j.1471-4159.2011.07248.x
Lu, P., Bai, X., Ma, D., Xie, T., Yan, C., Sun, L., et al. (2014). Three-dimensional structure of human γ-secretase. Nature 512, 166–170. doi: 10.1038/nature13567
Luo, W., Wang, H., Li, H., Kim, B. S., Shah, S., Lee, H.-J., et al. (2003). PEN-2 and APH-1 coordinately regulate proteolytic processing of presenilin 1. J. Biol. Chem. 278, 7850–7854. doi: 10.1074/jbc.c200648200
Madala, P. K., Tyndall, J. D. A., Nall, T., and Fairlie, D. P. (2010). Update 1 of: proteases universally recognize β strands in their active sites. Chem. Rev. 110, PR1–PR31. doi: 10.1021/cr900368a
Makin, S. (2018). The amyloid hypothesis on trial. Nature 559, S4–S7. doi: 10.1038/d41586-018-05719-4
Manolaridis, I., Kulkarni, K., Dodd, R. B., Ogasawara, S., Zhang, Z., Bineva, G., et al. (2013). Mechanism of farnesylated CAAX protein processing by the intramembrane protease Rce1. Nature 504, 301–305. doi: 10.1038/nature12754
Michaelson, D., Ali, W., Chiu, V. K., Bergo, M., Silletti, J., Wright, L., et al. (2005). Postprenylation CAAX processing is required for proper localization of Ras but not Rho GTPases. Mol. Biol. Cell 16, 1606–1616. doi: 10.1091/mbc.e04-11-0960
Mullard, A. (2017). BACE inhibitor bust in Alzheimer trial. Nat. Rev. Drug Discov. 16:155. doi: 10.1038/nrd.2017.43
Naing, S.-H., Kalyoncu, S., Smalley, D. M., Kim, H., Tao, X., George, J. B., et al. (2018a). Both positional and chemical variables control in vitro proteolytic cleavage of a presenilin ortholog. J. Biol. Chem. 293, 4653–4663. doi: 10.1074/jbc.ra117.001436
Naing, S.-H., Oliver, R. C., Weiss, K. L., Urban, V. S., and Lieberman, R. L. (2018b). Solution structure of an intramembrane aspartyl protease via small angle neutron scattering. Biophys. J. 114, 602–608. doi: 10.1016/j.bpj.2017.12.017
Navarro, C. L., Esteves-Vieira, V., Courrier, S., Boyer, A., Duong Nguyen, T., Huong, L. T. T., et al. (2014). New ZMPSTE24 (FACE1) mutations in patients affected with restrictive dermopathy or related progeroid syndromes and mutation update. Eur. J. Hum. Genet. 22, 1002–1011. doi: 10.1038/ejhg.2013.258
Niwa, M., Sidrauski, C., Kaufman, R. J., and Walter, P. (1999). A role for presenilin-1 in nuclear accumulation of ire1 fragments and induction of the mammalian unfolded protein response. Cell 99, 691–702. doi: 10.1016/s0092-8674(00)81667-0
Okochi, M., Fukumori, A., Jiang, J., Itoh, N., Kimura, R., Steiner, H., et al. (2006). Secretion of the Notch-1 Aβ-like peptide during Notch signaling. J. Biol. Chem. 281, 7890–7898. doi: 10.1074/jbc.m513250200
Pendás, A. M., Zhou, Z., Cadiñanos, J., Freije, J. M. P., Wang, J., Hultenby, K., et al. (2002). Defective prelamin A processing and muscular and adipocyte alterations in Zmpste24 metalloproteinase-deficient mice. Nat. Genet. 31, 94–99. doi: 10.1038/ng871
Prokop, S., Shirotani, K., Edbauer, D., Haass, C., and Steiner, H. (2004). Requirement of PEN-2 for stabilization of the presenilin N-/C-terminal fragment heterodimer within the γ-secretase complex. J. Biol. Chem. 279, 23255–23261. doi: 10.1074/jbc.m401789200
Pryor, E. E., Horanyi, P. S., Clark, K. M., Fedoriw, N., Connelly, S. M., Koszelak-Rosenblum, M., et al. (2013). Structure of the integral membrane protein CAAX protease Ste24p. Science 339, 1600–1604. doi: 10.1126/science.1232048
Qi-Takahara, Y., Morishima-Kawashima, M., Tanimura, Y., Dolios, G., Hirotani, N., Horikoshi, Y., et al. (2005). Longer forms of amyloid β protein: implications for the mechanism of intramembrane cleavage by γ-secretase. J. Neurosci. 25, 436–445. doi: 10.1523/JNEUROSCI.1575-04.2005
Rogaev, E. I., Sherrington, R., Rogaeva, E. A., Levesque, G., Ikeda, M., Liang, Y., et al. (1995). Familial Alzheimer’s disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer’s disease type 3 gene. Nature 376, 775–778. doi: 10.1038/376775a0
Sakai, J., Duncan, E. A., Rawson, R. B., Hua, X., Brown, M. S., and Goldstein, J. L. (1996). Sterol-regulated release of SREBP-2 from cell membranes requires two sequential cleavages, one within a transmembrane segment. Cell 85, 1037–1046. doi: 10.1016/s0092-8674(00)81304-5
Takami, M., Nagashima, Y., Sano, Y., Ishihara, S., Morishima-Kawashima, M., Funamoto, S., et al. (2009). γ-secretase: successive tripeptide and tetrapeptide release from the transmembrane domain of -carboxyl terminal fragment. J. Neurosci. 29, 13042–13052. doi: 10.1523/JNEUROSCI.2362-09.2009
Sato, C., Takagi, S., Tomita, T., and Iwatsubo, T. (2008). The C-terminal PAL motif and transmembrane domain 9 of presenilin 1 are involved in the formation of the catalytic pore of the -secretase. J. Neurosci. 28, 6264–6271. doi: 10.1523/JNEUROSCI.1163-08.2008
Sato, T., Tang, T.-C., Reubins, G., Fei, J. Z., Fujimoto, T., Kienlen-Campard, P., et al. (2009). A helix-to-coil transition at the -cut site in the transmembrane dimer of the amyloid precursor protein is required for proteolysis. Proc. Natl. Acad. Sci. U S A 106, 1421–1426. doi: 10.1073/pnas.0812261106
Satz, A. L. (2018). What do you get from DNA-encoded libraries? ACS Med. Chem. Lett. 9, 408–410. doi: 10.1021/acsmedchemlett.8b00128
Scharnagl, C., Pester, O., Hornburg, P., Hornburg, D., Götz, A., and Langosch, D. (2014). Side-chain to main-chain hydrogen bonding controls the intrinsic backbone dynamics of the amyloid precursor protein transmembrane helix. Biophys. J. 106, 1318–1326. doi: 10.1016/j.bpj.2014.02.013
Schmidt, W. K., Tam, A., Fujimura-Kamada, K., and Michaelis, S. (1998). Endoplasmic reticulum membrane localization of Rce1p and Ste24p, yeast proteases involved in carboxyl-terminal CAAX protein processing and amino-terminal a-factor cleavage. Proc. Natl. Acad. Sci. U S A 95, 11175–11180. doi: 10.1073/pnas.95.19.11175
Selkoe, D., and Kopan, R. (2003). Notch And Presenilin: regulated intramembrane proteolysis links development and degeneration. Annu. Rev. Neurosci. 26, 565–597. doi: 10.1146/annurev.neuro.26.041002.131334
Shah, S., Lee, S. F., Tabuchi, K., Hao, Y. H., Yu, C., LaPlant, Q., et al. (2005). Nicastrin functions as a γ-secretase-substrate receptor. Cell 122, 435–447. doi: 10.1016/j.cell.2005.05.022
Sherrington, R., Rogaev, E. I., Liang, Y., Rogaeva, E. A., Levesque, G., Ikeda, M., et al. (1995). Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature 375, 754–760. doi: 10.1038/375754a0
Shih, I. M., and Wang, T. L. (2007). Notch signaling, γ-secretase inhibitors, and cancer therapy. Cancer Res. 67, 1879–1882. doi: 10.1158/0008-5472.CAN-06-3958
Shokhen, M., and Albeck, A. (2017). How does the exosite of rhomboid protease affect substrate processing and inhibition? Protein Sci. 26, 2355–2366. doi: 10.1002/pro.3294
Sibley, L. D. (2013). The roles of intramembrane proteases in protozoan parasites. Biochim. Biophys. Acta 1828, 2908–2915. doi: 10.1016/j.bbamem.2013.04.017
Steiner, H., Kostka, M., Romig, H., Basset, G., Pesold, B., Hardy, J., et al. (2000). Glycine 384 is required for presenilin-1 function and is conserved in bacterial polytopic aspartyl proteases. Nat. Cell Biol. 2, 848–851. doi: 10.1038/35041097
Strisovsky, K., Sharpe, H. J., and Freeman, M. (2009). Sequence-specific intramembrane proteolysis: identification of a recognition motif in rhomboid substrates. Mol. Cell 36, 1048–1059. doi: 10.1016/j.molcel.2009.11.006
Sun, L., Li, X., and Shi, Y. (2016). Structural biology of intramembrane proteases: mechanistic insights from rhomboid and S2P to γ-secretase. Curr. Opin. Struct. Biol. 37, 97–107. doi: 10.1016/j.sbi.2015.12.008
Sun, L., Zhao, L., Yang, G., Yan, C., Zhou, R., Zhou, X., et al. (2015). Structural basis of human γ-secretase assembly. Proc. Natl. Acad. Sci. U S A 112, 6003–6008. doi: 10.1073/pnas.1506242112
Szaruga, M., Munteanu, B., Lismont, S., Veugelen, S., Horré, K., Mercken, M., et al. (2017). Alzheimer’s-causing mutations shift Aβ length by destabilizing γ-secretase-Aβn interactions. Cell 170, 443.e14–456.e14. doi: 10.1016/j.cell.2017.07.004
Takagi, S., Tominaga, A., Sato, C., Tomita, T., and Iwatsubo, T. (2010). Participation of transmembrane domain 1 of presenilin 1 in the catalytic pore structure of the γ-secretase. J. Neurosci. 30, 15943–15950. doi: 10.1523/JNEUROSCI.3318-10.2010
Takagi-Niidome, S., Sasaki, T., Osawa, S., Sato, T., Morishima, K., Cai, T., et al. (2015). Cooperative roles of hydrophilic loop 1 and the c-terminus of presenilin 1 in the substrate-gating mechanism of -secretase. J. Neurosci. 35, 2646–2656. doi: 10.1523/JNEUROSCI.3164-14.2015
Tanzi, R. E., and Bertram, L. (2005). Twenty years of the Alzheimer’s disease amyloid hypothesis: a genetic perspective. Cell 120, 545–555. doi: 10.1016/j.cell.2005.02.008
Thinakaran, G., Borchelt, D. R., Lee, M. K., Slunt, H. H., Spitzer, L., Kim, G., et al. (1996). Endoproteolysis of presenilin 1 and accumulation of processed derivatives in vivo. Neuron 17, 181–190. doi: 10.1016/s0896-6273(00)80291-3
Tichá, A., Collis, B., and Strisovsky, K. (2018). The rhomboid superfamily: structural mechanisms and chemical biology opportunities. Trends Biochem. Sci. 43, 726–739. doi: 10.1016/j.tibs.2018.06.009
Tong, G., Wang, J. S., Sverdlov, O., Huang, S. P., Slemmon, R., Croop, R., et al. (2012). Multicenter, randomized, double-blind, placebo-controlled, single-ascending dose study of the oral γ-secretase inhibitor BMS-708163 (Avagacestat): tolerability profile, pharmacokinetic parameters, and pharmacodynamic markers. Clin. Ther. 34, 654–667. doi: 10.1016/j.clinthera.2012.01.022
Torres-Arancivia, C., Ross, C. M., Chavez, J., Assur, Z., Dolios, G., Mancia, F., et al. (2010). Identification of an archaeal presenilin-like intramembrane protease. PLoS One 5:e13072. doi: 10.1371/journal.pone.0013072
Tun, H., Marlow, L., Pinnix, I., Kinsey, R., and Sambamurti, K. (2002). Lipid rafts play an important role in Aβ biogenesis by regulating the β-secretase pathway. J. Mol. Neurosci. 19, 31–35. doi: 10.1007/s12031-002-0007-5
Urano, Y., Hayashi, I., Isoo, N., Reid, P. C., Shibasaki, Y., Noguchi, N., et al. (2005). Association of active γ-secretase complex with lipid rafts. J. Lipid Res. 46, 904–912. doi: 10.1194/jlr.M400333-JLR200
van Tetering, G., and Vooijs, M. (2011). Proteolytic cleavage of notch: “HIT and RUN”. Curr. Mol. Med. 11, 255–269. doi: 10.2174/156652411795677972
Wakabayashi, T., Craessaerts, K., Bammens, L., Bentahir, M., Borgions, F., Herdewijn, P., et al. (2009). Analysis of the γ-secretase interactome and validation of its association with tetraspanin-enriched microdomains. Nat. Cell Biol. 11, 1340–1346. doi: 10.1038/ncb1978
Wang, Y., Zhang, Y., and Ha, Y. (2006). Crystal structure of a rhomboid family intramembrane protease. Nature 444, 179–180. doi: 10.1038/nature05255
Wasserman, J. D., and Freeman, M. (1997). Control of EGF receptor activation in Drosophila. Trends Cell Biol. 7, 431–436. doi: 10.1016/s0962-8924(97)01143-4
Weber, I. T., Waltman, M. J., Mustyakimov, M., Blakeley, M. P., Keen, D. A., Ghosh, A. K., et al. (2013). Joint X-ray/neutron crystallographic study of HIV-1 protease with clinical inhibitor amprenavir: insights for drug design. J. Med. Chem. 56, 5631–5635. doi: 10.1021/jm400684f
Weihofen, A., Binns, K., Lemberg, M. K., Ashman, K., and Martoglio, B. (2002). Identification of signal peptide peptidase, a presenilin-type aspartic protease. Science 296, 2215–2218. doi: 10.1126/science.1070925
Winter-Vann, A. M., and Casey, P. J. (2005). Post-prenylation-processing enzymes as new targets in oncogenesis. Nat. Rev. Cancer 5, 405–412. doi: 10.1038/nrc1612
Wolfe, M. S. (2020). Substrate recognition and processing by γ-secretase. Biochim. Biophys. Acta Biomembr. 1862:183016. doi: 10.1016/j.bbamem.2019.07.004
Wolfe, M. S., Xia, W., Ostaszewski, B. L., Diehl, T. S., Kimberly, W. T., and Selkoe, D. J. (1999). Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and γ-secretase activity. Nature 398, 513–517. doi: 10.1038/19077
Wu, Z., Yan, N., Feng, L., Oberstein, A., Yan, H., Baker, R. P., et al. (2006). Structural analysis of a rhomboid family intramembrane protease reveals a gating mechanism for substrate entry. Nat. Struct. Mol. Biol. 13, 1084–1091. doi: 10.1038/nsmb1179
Xie, T., Yan, C., Zhou, R., Zhao, Y., Sun, L., Yang, G., et al. (2014). Crystal structure of the γ-secretase component nicastrin. Proc. Natl. Acad. Sci. U S A 111, 13349–13354. doi: 10.1073/pnas.1414837111
Xue, Y., and Ha, Y. (2013). Large lateral movement of transmembrane helix S5 is not required for substrate access to the active site of rhomboid intramembrane protease. J. Biol. Chem. 288, 16645–16654. doi: 10.1074/jbc.m112.438127
Yang, G., Zhou, R., Zhou, Q., Guo, X., Yan, C., Ke, M., et al. (2019). Structural basis of Notch recognition by human γ-secretase. Nature 565, 192–197. doi: 10.1038/s41586-018-0813-8
Zhou, R., Yang, G., Guo, X., Zhou, Q., Lei, J., and Shi, Y. (2019). Recognition of the amyloid precursor protein by human γ-secretase. Science 363:eaaw0930. doi: 10.1126/science.aaw0930
Keywords: I-CLiPs, γ-secretase, substrate, interaction, Alzheimer’s disease
Citation: Liu X, Zhao J, Zhang Y, Ubarretxena-Belandia I, Forth S, Lieberman RL and Wang C (2020) Substrate–Enzyme Interactions in Intramembrane Proteolysis: γ-Secretase as the Prototype. Front. Mol. Neurosci. 13:65. doi: 10.3389/fnmol.2020.00065
Received: 19 December 2019; Accepted: 03 April 2020;
Published: 19 May 2020.
Edited by:
Maria Rosário Almeida, University of Porto, PortugalReviewed by:
Homira Behbahani, Karolinska Institutet (KI), SwedenHong Qing, Beijing Institute of Technology, China
Copyright © 2020 Liu, Zhao, Zhang, Ubarretxena-Belandia, Forth, Lieberman and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chunyu Wang, d2FuZ2M1QHJwaS5lZHU=