 Nurit Yanay
Nurit Yanay Malcolm Rabie
Malcolm Rabie Yoram Nevo
Yoram Nevo- 1Felsenstein Medical Research Center (FMRC), Tel-Aviv University, Tel-Aviv, Israel
- 2Institute of Neurology, Schneider Children’s Medical Center, Tel-Aviv University, Tel-Aviv, Israel
Muscle stem cells (MuSCs), known as satellite cells (SCs) have an incredible ability to regenerate, which enables the maintenance and growth of muscle tissue. In response to damaging stimuli, SCs are activated, proliferate, differentiate, and fuse to repair or generate a new muscle fiber. However, dystrophic muscles are characterized by poor muscle regeneration along with chronic inflammation and fibrosis. Indications for SC involvement in muscular dystrophy pathologies are accumulating, but their contribution to muscle pathophysiology is not precisely understood. In congenital muscular dystrophy type 1A (LAMA2-CMD), mutations in Lama2 gene cause either complete or partial absence in laminin-211 protein. Laminin-211 functions as a link between muscle extracellular matrix (ECM) and two adhesion systems in the sarcolemma; one is the well-known dystrophin–glycoprotein complex (DGC), and the second is the integrin complex. Because of its protein interactions and location, laminin-211 has a crucial role in muscle function and survival by maintaining sarcolemma integrity. In addition, laminin-211 is expressed in SCs and suggested to have a role in SC proliferation and differentiation. Downstream to the primary defect in laminin-211, several secondary genes and pathways accelerate disease mechanism, while at the same time there are unsuccessful attempts to regenerate as compensation for the dystrophic process. Lately, next-generation sequencing platforms have advanced our knowledge about the secondary events occurring in various diseases, elucidate the pathophysiology, and characterize new essential targets for development of new treatment strategies. This review will mainly focus on SC contribution to impaired regeneration in muscular dystrophies and specifically new findings suggesting SC involvement in LAMA2-CMD pathology.
Introduction
Skeletal muscle tissue is the most abundant tissue in the body. It is dynamic and in its healthy state adaptable to changes, such as exercise or injury. Skeletal muscle has the ability to fully regenerate in response to injury and to increase cell number or size accordingly. Muscle adaptive behavior is maintained by muscle stem cells (MuSCs), also known as satellite cells (SCs), located in a niche in close proximity to the muscle fiber. In response to either intrinsic or microenvironment (extrinsic) signaling, SCs are activated, proliferate, and fuse in a very controlled manner to repair or create new muscle fiber.
Muscular dystrophies are characterized by progressive skeletal muscle weakness and atrophy. The clinical deterioration is caused by substitution of muscle by fibrotic and fatty nonfunctional tissues. With new advanced diagnostic methods, accumulating data are emerging regarding decreased SC function and number, leading to impaired regeneration as a contributory mechanism to the pathology in muscular dystrophy. In this review, we mainly focus on impaired regeneration and SC involvement in the pathology of muscular dystrophies and the new findings in LAMA2-CMD.
Skeletal Muscle Regeneration
Adult healthy skeletal muscles have an excellent regeneration capacity to undergo constant repair and create new muscle fibers owing to MuSCs, also known as SCs. SCs are located in a niche between the myofiber sarcolemma and basement membrane, near the vasculature, and thus can act very rapidly, migrating and proliferating upon muscle injury (Mauro, 1961), and are the primary source of muscle regenerative cells (Ciciliot and Schiaffino, 2010; Almeida et al., 2016). Many other mononuclear cells, such as bone marrow stem cells, mesenchymal stem cells, and pericytes accompanying the muscle microenvironment, also have a role in muscle homeostasis and repair (Ferrari et al., 1998; Corti et al., 2002; Lee et al., 2005; Muskiewicz et al., 2005; Dellavalle et al., 2007; Liu et al., 2007; Negroni et al., 2009; de la Garza-Rodea et al., 2011). Following muscle injury, necrosis of damaged myofibers is followed by inflammatory responses, including recruitment of neutrophils and macrophages, which secrete inflammatory cytokines activating the quiescent SC mononuclear population in order to regenerate muscle (Otis et al., 2014). Muscle regeneration is a controlled finely tuned process, very similar to muscle formation during embryonic development (Allbrook, 1981), and can be subdivided into three main stages: proliferation, differentiation, and fusion (Randolph and Pavlath, 2015; Chal and Pourquié, 2017; Yanay et al., 2019), as can be seen in Figure 1A.
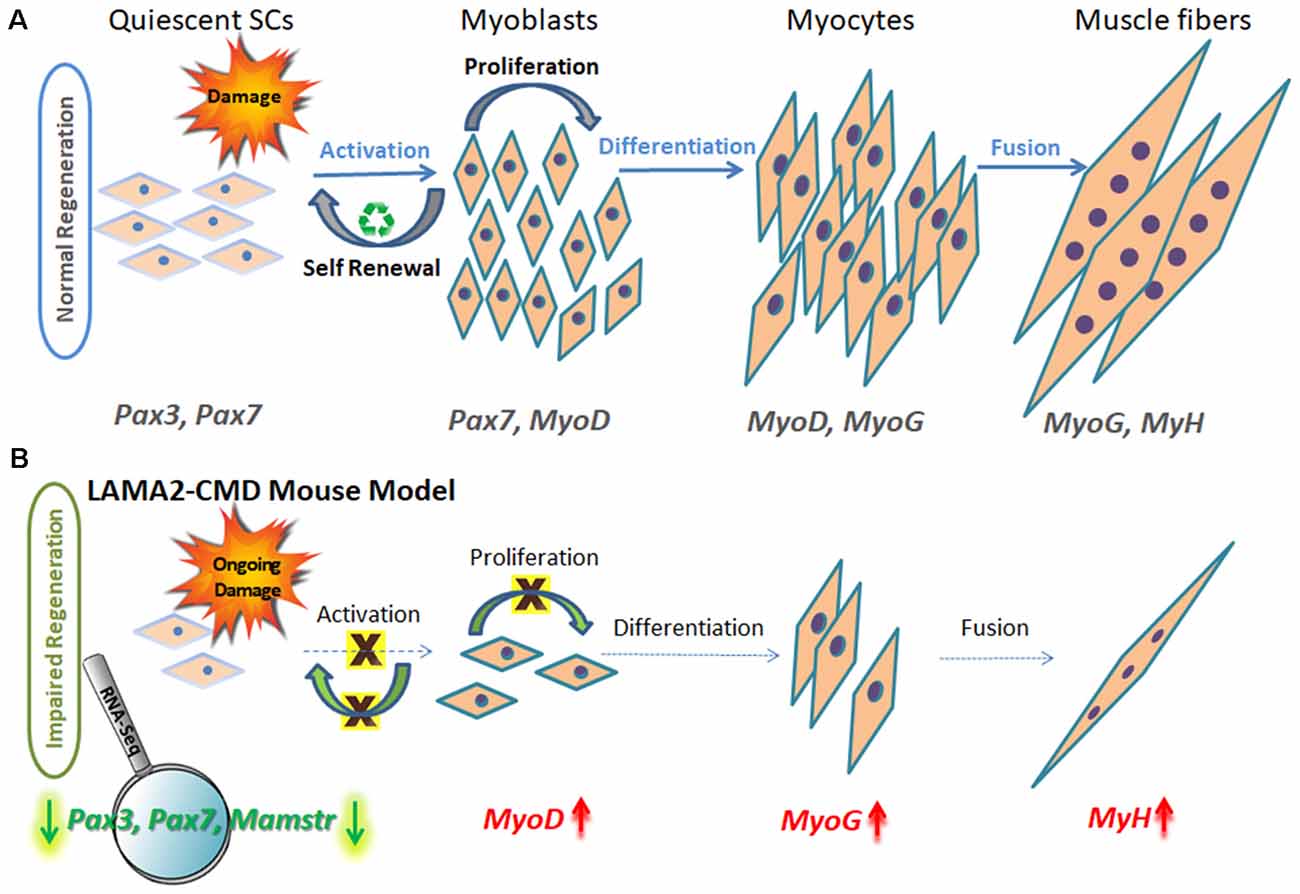
Figure 1. Schematic model of muscle regeneration process in normal and LAMA2-CMD mouse model. (A) In healthy muscle, satellite cells (SCs) are in a quiescence state as a default, located in a specialized niche, between the sarcolemma and basal lamina. Quiescent SCs express Pax7, and a subpopulation expresses also Pax3 transcription factors; however, MyoD is not expressed at this stage. Upon injury, SCs are activated and undergo proliferation. SCs can perform asymmetric or symmetric divisions. Asymmetric division allows self-renewal and maintenance of SC pool, and symmetric division allows myogenesis and generating myoblasts. Following proliferation stage, myoblasts express MyoD transcription factor and exit the cell cycle to promote differentiation. Upon differentiation, myoblasts differentiate into elongated myocytes expressing myogenin (MyoG). Myocytes can fuse forming myotubes, which become myofibers, the contractile unit of muscle, and express developmental myosin-heavy chain marker (MyH). (B) In dystrophic muscle, SCs have limited ability to compensate for muscle damage. In LAMA2-CMD dy2J/dy2J mouse model, RNA-Seq data indicated little or even absent SC population, due to significant downregulation of Pax7 and Pax3 genes in quadriceps muscle of 8-week-old mice (Yanay et al., 2019). Absence of those transcription factors disable proper proliferation and self-renewal; thus, the entire regeneration process is impaired. MyoD and MyoG upregulation is postulated to be an attempt to compensate for the unbalanced process and the reduced repair of the damage.
Each stage of muscle regeneration can be classified by a variety of molecular markers. The most familiar marker of all muscle SC state is paired box transcription factor 7 (Pax7), being essential for SC function during postnatal growth both in regeneration of skeletal muscle and maintaining a renewable SC pool (Seale et al., 2000; Olguin and Olwin, 2004; Zammit et al., 2004; Lepper et al., 2009; von Maltzahn et al., 2013). Subsets of SCs express premyoblast marker paired box transcription factor 3 (Pax3), important in the initial steps of muscle formation (Relaix et al., 2005, 2006); however, Pax3’s role in adult human muscle still remains to be established. The population of Pax3 and Pax7 double-positive stem cells expression is observed in the preliminary stage throughout embryonic and fetal development, as well as later in adult skeletal muscle SCs. Pax3 and Pax7 stimulate gene expression promoting proliferation and asymmetric divisions (known as self-renewal) and repress genes driving differentiation (Ben-Yair and Kalcheim, 2005; Kassar-Duchossoy et al., 2005; Relaix et al., 2005, 2006).
Upon SC activation, proliferating myoblasts coexpress Pax7 and MyoD transcription factors. From this stage, the differentiation process begins and is characterized by MyoD and MyoG expression.
Proliferating myoblasts either down-regulate Pax7 to differentiate or down-regulate MyoD to undergo self-renewal process, which maintains the SC pool. Maintaining the balance between myoblast proliferation and differentiation is crucial for the appropriate muscle regeneration process (Yablonka-Reuveni, 2011).
Under dystrophic conditions, muscle is persistently injured and degenerates (Otis et al., 2014) in a way that muscle necrosis surpasses regenerative capacity, and muscle repair cannot effectively compensate for damage. The muscle undergoes replacement by fibrotic tissue and fat leading to severe loss of muscle mass and function. Recently, there are cumulative indications of SC involvement in different muscular dystrophies in which SCs apparently fail to repair muscle damage efficiently (Logan et al., 2011; Ross et al., 2012; Urciuolo et al., 2013; Di Gioia et al., 2017).
The Contribution of Satellite Cell Regeneration Failure to Muscular Dystrophy Pathophysiology
Intrinsic and extrinsic defects may occur at each stage of this complex multipart muscle regeneration process. Several theories for the limited regenerative capacity of SCs have been proposed by Randolph and Pavlath (2015) and the 240th ENMC workshop on “The involvement of skeletal MuSCs in the pathology of muscular dystrophies” (Morgan et al., 2019). These theories can be divided into impairment of intrinsic and extrinsic factors:
Intrinsic Impairment in SCs
1. Reduction in SC pool. Defects in self-renewal, SC exhaustion, or senescence eventually lead to reduction in SC number. Self-renewal defects causing ineffective generation of SCs were demonstrated in sarcoglycanopathy LGMD2C and 2F patients (Higuchi et al., 1999), Col6A1 knockout mice (Urciuolo et al., 2013; Gattazzo et al., 2014a), Sepn1−/− murine model, and in selenoprotein-related myopathy (SEPN1-RM) patient muscle biopsies (Sacco et al., 2010; Castets et al., 2011). SC exhaustion caused by constant activation from ongoing cycles of degeneration and regeneration was demonstrated in mdx mouse model of Duchenne muscular dystrophy (DMD) and myoblasts isolated from aging DMD patients (Mouly et al., 2005; Sacco et al., 2010; Jiang et al., 2014). Lately self-renewal defect in SCs, in addition to exhaustion, was also observed in mdx mice indicating asymmetric division anomalies (Dumont et al., 2015). Senescence (premature aging) was demonstrated in both in vitro and in vivo models of LGMD2H mouse SCs (Kudryashova et al., 2012; Mokhonova et al., 2015) and myoblasts of human myotonic dystrophy type 1 (DM1) and 2 (DM2; Bigot et al., 2009; Beffy et al., 2010; Renna et al., 2014).
2. Defects in myoblast proliferation. Decreased proliferation of SCs has been observed in oculopharyngeal muscular dystrophy (OPMD) patients’ myoblasts (Périé et al., 2006), Emery–Dreifuss muscular dystrophy (EDMD) patient’s muscle tissue sections and cultured myoblasts (Meinke et al., 2015), muscle biopsies of a DM1 patient (Thornell et al., 2009), and in myoblasts of murine model of LGMD2O (Miyagoe-Suzuki et al., 2009). In patients and mouse model of Pompe disease, insufficient SC activation was noted, although their function and number seemed normal (Schaaf et al., 2018). Additionally, impaired SC transition from proliferation to differentiation has been demonstrated in biopsies from LGMD2A patients (Rosales et al., 2013).
3. Defect or delay in the differentiation stage was demonstrated for facioscapulohumeral muscular dystrophy (FSHD) in DUX4c-expressing C2C12 myoblasts (Bosnakovski et al., 2008). Defects in myoblast differentiation were also demonstrated in EDMD primary muscle and patient-derived myoblast cultures (Frock et al., 2006) and in C2C12 cells expressing mutated lamin A, representing the cellular model of AD EDMD (Favreau et al., 2004).
4. Defect or delay in the fusion stage was reported in FRG1 mouse model for FSHD primary myoblasts (Feeney et al., 2015) and limb-girdle muscular dystrophy type 2L (LGMD2L) Ano5−/− knockout mouse model (Whitlock et al., 2018).
Extrinsic Impairment in SC
1. Hostile microenvironment of dystrophic muscle may not be permissive for continued SC regeneration (Boldrin et al., 2012). Altered composition of the extracellular matrix (ECM), chronic inflammation (Wanschitz et al., 2013), defective autophagy (Tang and Rando, 2014), and fibrosis lead to a defect in SC niche and to cell senescence, as demonstrated in mouse models of dystroglycanopathy (Ross et al., 2012).
2. Alterations in signaling pathways as a result of the dystrophy may also underlie failure of SC regeneration. This was demonstrated in a limb-girdle muscular dystrophy patient with missense mutation in POGLUT1 (protein O-glucosyltransferase 1), in whom a decrease in Notch signaling was associated with muscle degeneration and loss of SCs (Servián-Morilla et al., 2016).
Intrinsic as well as extrinsic defects can be caused by the disease’s primary mutation.
In case that the mutation is in protein expressed within SCs, the primary mutation itself can impair SC function as demonstrated in DMD and LGMD2H mouse models (Kudryashova et al., 2012; Dumont et al., 2015). If not critical for SC function, the primary mutation may change structures of the ECM and basal lamina, altering SC niche, which may lead to microenviromental defects and ineffective SC activation (Gattazzo et al., 2014b).
Regeneration defects in more than one stage have been reported in several dystrophy types. However, the origin of these defects was elucidated in later studies, when new and powerful technologies enabled more accurate detection and analysis of the various components of the pathophysiology, beyond the primary mutation.
Until recently, the dystrophin gene, DMD, was considered to be expressed in myofibers and not SCs. Accordingly, the main contribution to DMD pathology was assumed to be limited to myofiber membrane fragility due to absence of dystrophin, leading to continuous necrosis with progressive fibrosis and muscle wasting (Morgan and Zammit, 2010). Hence, current therapies mainly focus on preventing dystrophy by targeting the myofiber. However, in a recent study, using RNA-sequencing (RNA-Seq) and microarray techniques, dystrophin expression was detected in a subset of SCs and postulated to have an important role in their polarity and asymmetric divisions. Therefore, a primary intrinsic SC dysfunction contributing to DMD pathogenesis has been suggested (Dumont et al., 2015). Evidences for active and unbalanced proliferation in DMD with no change in SC number compared to healthy muscle are available (Boldrin et al., 2009, 2015). However, this process of proliferation is impaired due to abnormal and uncontrolled SC divisions, impaired self-renewal, and eventual decline in the SC pool with time.
In accordance with these results, a recent study at our laboratory supports an intrinsic SC defect in muscle of the mdx mouse model of DMD. This study examined 8-week-old mdx mouse whole muscle transcriptome using RNA-Seq and found unsynchronized upregulation of all major regeneration transcription factors: Pax7, Myf5, MyoD, MyoG, and Mamster compared to WT muscle, indicating unbalanced active regeneration and SC uncontrolled activation, which may increase the proportion of abnormal cell divisions (Yanay et al., 2019).
Congenital Muscular Dystrophy Type 1A
Congenital muscular dystrophy type 1A (LAMA2-CMD), also known as merosin-deficient congenital muscular dystrophy type 1A (MDC1A), is a devastating incurable disease, caused by mutations in Lama2 gene encoding the α-subunit of laminin-211. Mutations in laminin-211, a key anchor basement membrane protein, disrupt the link between cytoskeleton, basement membrane, and ECM, rendering the muscle fiber sarcolemma fragile.
Laminin 211 interacts with α-dystroglycan, a member of the dystrophin–glycoprotein complex (DGC; Spence et al., 2002). This complex links ECM to the actin cytoskeleton of the myofiber, thereby protecting skeletal muscle membrane against contraction-induced damage (Petrof et al., 1993).
Independently, laminin-211 interacts with β1 integrin, a part of the α7β1 integrin complex that gathers a large number of proteins and, like the DGC, functions as a structural link between ECM and actin cytoskeleton, hence similarly playing a significant role in protecting skeletal muscle against contraction-induced injury. Recent studies suggest that the DGC and integrin complex have compensatory abilities in maintaining sarcolemma integrity, as demonstrated by much more severe muscle pathology in double-mutant knockout mice for dystrophin and α7β1 integrin than mice lacking either dystrophin or α7β1 individually (Allikian et al., 2004; Guo et al., 2006; Rooney et al., 2006).
In LAMA2-CMD, either partial or complete absence of laminin-211 expression leads to increased muscle vulnerability to injury with ensuing severe clinical features in children, as well as in LAMA2-CMD mouse models.
Several useful mouse models of LAMA2-CMD are available, demonstrating correlation between laminin-211 expression and disease severity, thus representing heterogeneity in their clinical presentation. Very severe forms of muscular dystrophy are represented by dyW/dyW and dy3k/dy3k models. These models have a similar severe dystrophic phenotype, with life spans of only a few weeks of age, commensurate with almost absent or completely absent laminin-211 expression in dyW/dyW and dy3k/dy3k models, respectively. Moderate severity with reduced life span of 6 months of age represents the dy/dy mouse, which has reduced laminin-211 expression and primary mutation still unknown. Mild muscular dystrophy represents the dy2J/dy2J mouse model, with spontaneous mutation in the LN domain resulting in partial deficiency, hind limb paralysis by 3–4 weeks of age, and decreased life span compared to wild type (Xu et al., 1994; Sunada et al., 1995; Miyagoe et al., 1997; Gawlik and Durbeej, 2011; Durbeej, 2015).
Impaired Regeneration in LAMA2-CMD
Previous studies have provided some clues that the proliferation stage in the regeneration process is impaired in LAMA2-CMD.
One clue for impaired regeneration in LAMA2-CMD, suggested by Kuang et al. (1999) is that a major contributor to muscle disease is abortive regeneration in the dyW mouse model. They reported immature myofibers and excessive mononuclear cell death in this model.
Next, in 2005, Girgenrath et al. (2005) showed in the same dyW model that mononucleated cells, and in particular muscle SC population, were decreased compared to WT and mdx mice, and therefore an altered proliferation stage was assumed. Thus, they hypothesize that SC poor proliferation may be one of the mechanisms underlying the lack of successful regeneration in LAMA2-CMD muscle (Girgenrath et al., 2005).
Only a few studies exploring LAMA2-CMD transcriptome and proteome have been published (Taniguchi et al., 2006; van Lunteren et al., 2006; Moreira Soares Oliveira et al., 2018). As for human data, muscle transcriptome was studied in a single LAMA2-CMD patient at the age of 8 months, using microarray technique with costume chip representing a limited number of 5,600 genes expressed in muscle. Most of the upregulated genes in this patients’ muscle were ECM components, which according to the author reflect active fibrosis and poor muscle regeneration (Taniguchi et al., 2006).
In mouse models, a single study demonstrating gene expression profiling in dy/dy using microarray technique found a limited number of genes differentially expressed in the diaphragm (van Lunteren et al., 2006). Genes with altered expression in the diaphragm belonged to cell motility, development, immune response, cellular adhesion, and collagen synthesis. In addition, a study by de Oliveira et al. (2014) described proteomic analysis in the dy3k/dy3k mouse model, showing approximately 100 differentially expressed proteins compared to WT, mainly involving metabolic processes, calcium binding, or ECM protein expression (fibrosis) in diaphragm and gastrocnemius muscles (de Oliveira et al., 2014).
Correlation between clinical phenotype and gene expression was mainly suggested for fibrosis in studies of LAMA2-CMD; thus, therapies so far have largely focused on amelioration or prevention of fibrosis (Nevo et al., 2010; Elbaz et al., 2012; Yu et al., 2013; Accorsi et al., 2016).
In our recent article, next-generation sequencing via RNA-Seq technique was applied in the dy2J/dy2J mouse (Yanay et al., 2019). Using this method, we detected a large number of novel significantly differentially expressed genes in quadriceps muscle of 8-week-old dy2J/dy2J mice.
The most significant finding was downregulation of three key myogenic stem cell factor genes: Pax3, Pax7, and Mamstr (Figure 1B). This demonstrated an abnormal regeneration process, which mainly points to impaired SC self-renewal in the dy2J/dy2J mouse model compared to WT.
These results are in agreement with ours and others histological results and previous clinical observation demonstrating poor proliferation and high degree of fibrosis in LAMA2-CMD patients and mouse models (Girgenrath et al., 2005; Yanay et al., 2019).
Earlier studies demonstrate that with absence of Pax3/Pax7 expression, cells undergo apoptosis or adopt alternative nonmuscle lineages (Soleimani et al., 2012), possibly explaining the mechanism underlining very early tissue replacement by fibrosis and fat in LAMA2-CMD models. Transient activation of Pax3 expression in cultures of primary myoblasts results in enhanced proliferation in these cells (Conboy and Rando, 2002; Kuang et al., 2006).
Not only the environment is important for SC functionality, but SCs may also have an impact on their own environment, as loss of SCs may also increase muscle fibrosis. Thus, the presence of normal SCs is required to maintain functional niches that support regeneration (Morrison and Spradling, 2008).
SC involvement in additional myopathies pathologies was reported for XL myotubular myopathy, SEPN1-related myopathies, and as a primary cause for EMARDD (early-onset myopathy with areflexia, respiratory distress, and dysphagia; Castets et al., 2011; Logan et al., 2011; Boyden et al., 2012; Lawlor et al., 2012; Di Gioia et al., 2017). Mutated genes expressed in myotubes as well as in SCs may alter myofiber maturation during embryogenesis, and early growth is postulated to be the cause for the early onset of pathology and weakness in these disorders. Similarly to these congenital myopathies, there are also indications for laminin-211 expression in LAMA2-CMD SCs (Schuler and Sorokin, 1995; Vachon et al., 1996; Morgan and Zammit, 2010). In addition to its roles in prevention of myofiber injury, laminin-211 has therefore been suggested to have a role in myoblast proliferation and SC differentiation and regulation (Girgenrath et al., 2005; Morgan and Zammit, 2010). SC dysfunction in LAMA2-CMD due to an intrinsic defect or extrinsic/microenvironment defects, or a combination of the two, should be further studied.
Understanding SC contribution to LAMA2-CMD muscle pathology may suggest new therapeutic strategies, as current therapies mainly target myofiber damage rather than the SC regeneration process.
Signaling Pathways Underlying Muscle Regeneration
Multiple signaling pathways are involved in skeletal muscle regeneration, and each of them is tightly controlled and regulated to enable efficient muscle repair. Notch, Wnt, Janus kinase/signal transducers and activators of transcription (JAK/STAT), Mitogen-activated protein kinase (MAPK), Nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), and Transforming growth factor beta (TGF-β) signaling pathways, and others have been intensely studied, and their contribution to developmental and adult muscle regeneration confirmed (Brack et al., 2007; Bjornson et al., 2012; Parker et al., 2012; von Maltzahn et al., 2012a; Tierney et al., 2014). Each signaling pathway is critical for accurate regeneration, but above all, their synchronization and balanced cross-talk are most important for proper regeneration (Figure 2). For instance, in adult skeletal muscle, SCs express high levels of Notch to remain in a quiescent state and prevent Pax7 induced differentiation (Conboy and Rando, 2012; Wen et al., 2012; Fujimaki et al., 2018). On the other hand, with upregulation of the canonical Wnt signaling pathway, SC differentiation is activated (von Maltzahn et al., 2012a). Thus, a precise timing to switch from Notch to Wnt signaling is required for proper SC differentiation (Brack et al., 2007). While the canonical Wnt signaling drives differentiation of SCs, mainly through the ligand Wnt3a, noncanonical Wnt signaling is responsible for mediating self-renewal and migration of SCs, and also growth of muscle fibers through Wnt7a ligand (von Maltzahn et al., 2012a).
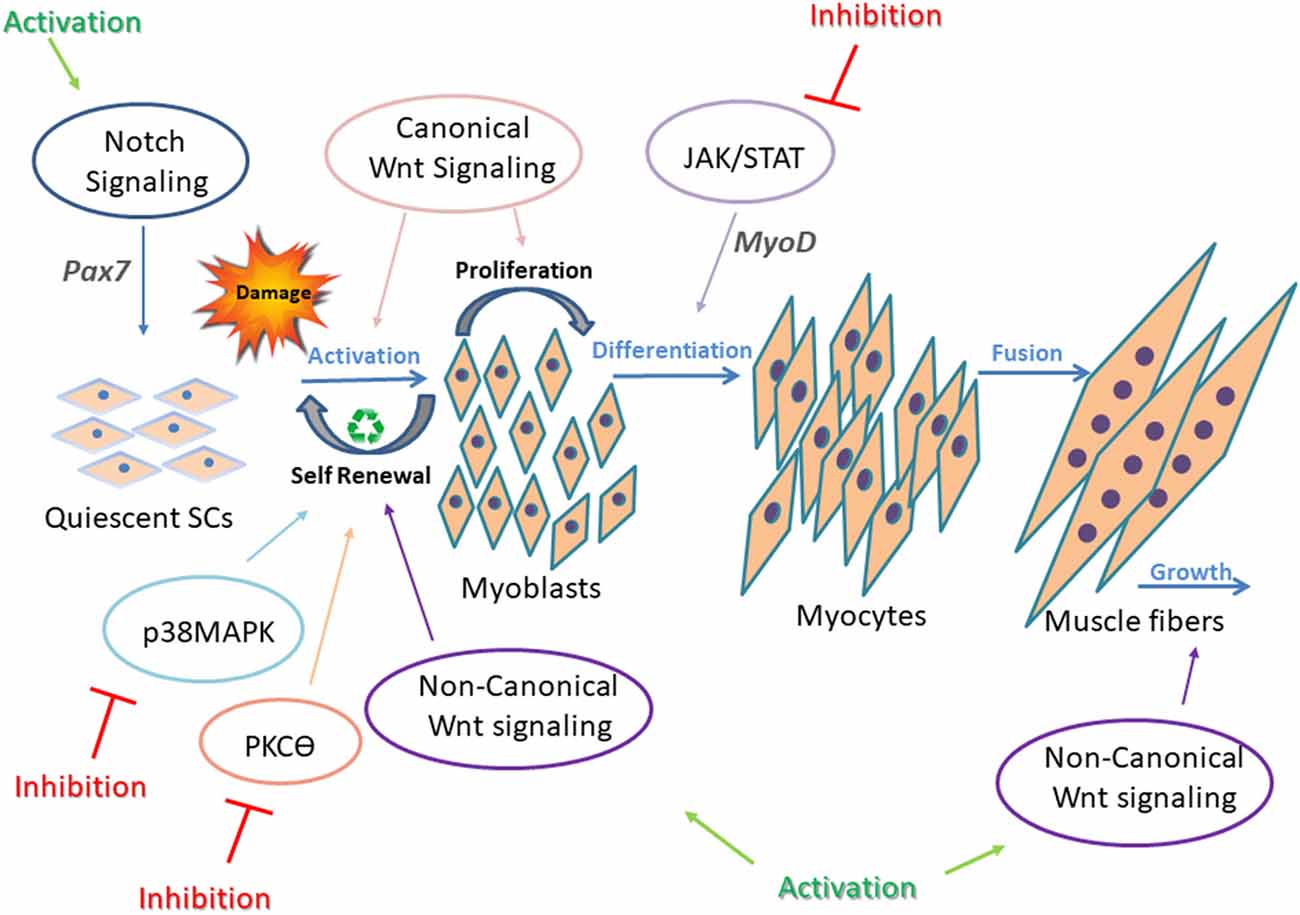
Figure 2. Underlying mechanisms in impaired regeneration during myogenesis. Main regeneration signaling pathways in muscular dystrophy and the corresponding targets for intervention are presented. In adult healthy skeletal muscle, all signaling pathways are tightly controlled and highly synchronized for proper SC differentiation. This is well demonstrated by the interplay between Notch and Wnt signaling. Upregulation of Notch maintains SCs in a quiescent state and promotes self-renewal of activated SCs through upregulation of Pax7. Canonical Wnt signaling antagonizes the effects of Notch signaling, thus allowing the progression through differentiation. Also, canonical and non-canonical Wnt signaling oppose each other, as canonical Wnt signaling regulates the differentiation of muscle SCs, and noncanonical Wnt signals (through ligand Wnt7a) mediate self-renewal (symmetric divisions), migration of SCs, and growth of muscle fibers. p38MAPK pathway regulates toward asymmetric division and SC self-renewal. In dystrophic muscle, signaling is altered and unsynchronized, resulting in defective regeneration. Continuous intervention to activate or inhibit abnormal signaling in SCs failed to improve muscle regeneration. Thus, a dynamic/cyclic regulation of signaling is necessary to balance regeneration of SCs in order to improve long-term regenerative defects of dystrophic muscles (Jiang et al., 2014; Tierney et al., 2014). Cyclic administration of Wnt7a, Notch activators, and JAK/STAT, MAPK, and PKCθ inhibitors have shown beneficial effects on the regeneration process in dystrophic muscles.
Examples for noncanonical Wnt signaling pathways include PCP (planar cell polarity), Wnt/Ca2+, and PI3K/AKT/mTOR signaling cascades. Interplay between these factors upon Wnt signaling results in activation of small GTPases, Rac, and Rho, leading to cytoskeletal remodeling, are essential for myoblast fusion.
Additional signaling pathways involved in SC regeneration include p38MAPK signaling, which is highly regulated to permit asymmetric division and SC self-renewal (Troy et al., 2012).
Also, JAK/STAT signaling pathway activates myogenic differentiation by regulating expression of specific genes such as MyoD (Wang et al., 2008).
Impaired Satellite Cell Regeneration Signaling Pathways in Muscular Dystrophy
Unsynchronized SC signaling pathways resulting in defective muscle regeneration has been described in several muscular dystrophies.
Decrease number of SCs in dystrophic muscle has been associated with a reduced Notch signaling pathway (Jiang et al., 2014; Servián-Morilla et al., 2016).
Activation of Notch signaling was shown to improve self-renewal capacity of SCs in mdx mice (Jiang et al., 2014) and ameliorate DMD phenotype in Golden Retriever muscular dystrophy dogs (GRMD; Vieira et al., 2015).
Recently, Fiore et al. (2020) suggested that lack or pharmacological inhibition of protein kinase C theta (PKCΘ), which modulates several signaling pathways in muscle, leads to increased Notch signaling and improved muscle repair and SC self-renewal ability in mdx mice. Muscles from limb-girdle muscular dystrophy patients show decreased Notch signaling and a dramatic reduction in SC pool (Servián-Morilla et al., 2016). Significant rescue of the myogenesis was demonstrated in these patients by increasing Notch signaling (Servián-Morilla et al., 2016).
Canonical Wnt signaling dysregulation has been reported in multiple muscle pathologies, such as DMD (Trensz et al., 2010), FSHD (Block et al., 2013), and OPMD (Abu-Baker et al., 2013).
Muscle lacking secreted factor, Wnt7a, a Wnt signaling ligand, exhibited a marked decrease in SC number following regeneration, whereas Wnt7a overexpression enhanced muscle regeneration and increased SC numbers. Wnt7a also induced myotube hypertrophy and a shift in fiber type toward slow-twitch in human primary myotubes (von Maltzahn et al., 2012b). Intramuscular treatment with Wnt7a increased activated SC number, myofiber size, and muscle force of mdx mice.
Inhibition of dysregulated p38MAPK signaling pathway in mdx mice improved SC self-renewal and mice phenotype (Smythe and Forwood, 2012; Wissing et al., 2014).
In addition, pharmacological inhibition of JAK/STAT signaling (by Calbiochem) increased numbers of SCs, enhanced muscle repair, and enhanced functional performance in aged healthy mice (Price et al., 2014).
Both canonical Wnt and TGF-β2 signaling are chronically elevated in mdx mouse muscle tissue (Biressi et al., 2014).
NF-κB and TGF-β signaling were overexpressed and involved in the pathophysiology of the dy2J/dy2J mouse (Elbaz et al., 2012, 2015). Also, it is well known that chronic activation of NF-κB signaling contributes to DMD pathology, promotes necrosis and inflammation, and inhibits muscle regeneration (Proto et al., 2015). NF-κB inhibition improves mdx dystrophic muscle regeneration by directly promoting lineage progression of muscle progenitor cells and by increasing progenitor cell survival (Proto et al., 2015).
Moreover, using canonical pathway analysis by Ingenuity Pathway Analysis, RhoA, NF-κB, epithelial-mesenchymal transition (EMT), TGF-β, and PKCθ were found to be altered in the LAMA2-CMD mouse model examined by RNA-Seq method (Yanay et al., 2019).
Potential Therapies to Enhance Regeneration
As previously mentioned, a fine-tuned balance between intrinsic signaling pathways and extrinsic factors is required to correctly control SC function. We therefore subdivide potential therapies to enhance regeneration in muscular dystrophy into those that affect intrinsic factors, extrinsic factors, or both (Figure 3).
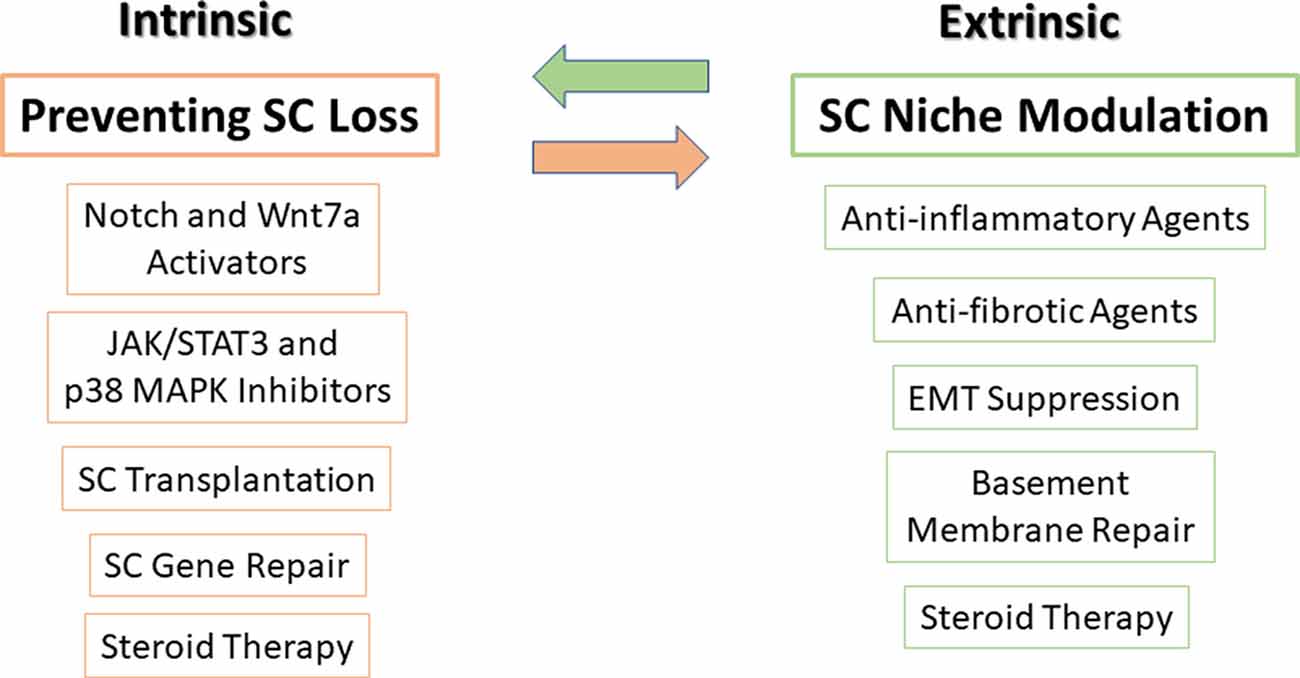
Figure 3. Potential therapies to target SCs and their niche. Very tightly controlled interactions exist between SCs and their microenvironment niche, thus, in order to optimize treatment, targeting both SCs (intrinsic factors) along with their microenvironment niche (extrinsic factors) should be carried out simultaneously. On the left side, SC transplantation, modulation of major signaling pathways in SCs, mediation of gene delivery and repair, and steroid treatment can prevent SC loss and improve their function. On the right side, using anti-inflammatory and antifibrotic agents, suppression of epithelial-mesenchymal transition (EMT), basement membrane protein repair, and steroid treatment can restore SC niche. Steroid treatment is presented in both sides as it affects both SCs and satellite niche.
As can be seen in Figure 3, pharmacological inhibitor or activator compounds balance and synchronize cell signaling of different intrinsic factors and may prevent SC loss and improve SC function.
Preliminary evidence for the use of SC transplantation as a potential therapeutic treatment was provided from the results of a phase II clinical trial in OPMD patients (www.clinicaltrials.gov NCT00773227; Périé et al., 2014). In that study, local autologous myoblast transplantation demonstrated significant improvement in patients’ swallowing abilities with no adverse side effects. Because this procedure was accepted as generally safe with good tolerance, it has been extended to a larger cohort of OPMD patients. However, many obstacles to SC transplantation still exist, such as SCs’ limited number, delivery methods, cell source (auto or allogeneic) as reviewed by Almeida et al. (2016), and need to be addressed before considering SC therapy for LAMA2-CMD patients or other muscular dystrophies with more widespread muscle involvement.
SC impaired genes can be targeted using viral vectors. Restoration of dystrophin expression using adeno-associated virus was shown to restore dystrophin in myotubes and restore dystrophin at low levels in SCs in the mdx mouse model (Tabebordbar et al., 2016). In addition, using lentiviral vector delivery of microdystrophin to neonatal SCs of mdx mice resulted in preservation for 2 years of dystrophin expression in myofibers, demonstrating stable transduction of SCs (Kimura et al., 2009).
Challenges in muscle formation may also be due to extrinsic factors in the microenvironment of SCs.
Host SC niche modulation should also be considered in order to produce efficient SC transplantation. Excess fibrosis and altered ECM composition alters SC functionality, resulting in impaired regeneration (Ross et al., 2012). A functional niche will provide an adjusted microenvironment for donor cells to be received by the hosts’ tissue (Boldrin et al., 2012). Thus, preservation of the defective components of the SC niche is critical in order to optimize stem cell therapies.
Contribution of fibrosis and also chronic inflammation to the pathogenesis of LAMA2-CMD are well documented (Girgenrath et al., 2005; Taniguchi et al., 2006; Elbaz et al., 2012, 2015; Yanay et al., 2019). Improved regeneration can be achieved using anti-inflammatory and antifibrotic agents as demonstrated over the years (Dadush et al., 2010; Nevo et al., 2011; Elbaz et al., 2012; Accorsi et al., 2016). Immunosuppressant cyclosporin A treatment maintained SC number in Col6a1−/− mice following multiple bouts of induced injury (Gattazzo et al., 2014a).
Our recent RNA-Seq results support the significant role not only, as expected, of fibrosis, but also of the immune system contribution to LAMA2-CMD pathogenesis. Our findings also proposed that epithelial–mesenchymal transition (EMT) contributes to the dystrophic tissue fibrosis (Yanay et al., 2019). Thus, development of therapeutic interventions, either anti-inflammatory therapies at disease onset or suppression of EMT, may decrease fibrosis in muscular dystrophy in the mouse model.
Repair and preservation of the basement membrane could also support SC niche and functionality and restore muscle regeneration. Laminin-411 was found to be overexpressed in LAMA2-CMD as an attempt to compensate for the abnormal laminin-211 protein (Patton et al., 1997; Ringelmann et al., 1999). Laminin-411 upregulation was also confirmed in our RNA-Seq of dy2J/dy2J. However, laminin-411 lacks binding domains to laminin-211 receptors. In the study by Reinhard et al. (2017), laminin-411 was used as a scaffold protein to link mini-agrin and αLNNd (a chimeric protein that contains laminin-211 binding parts to a basement membrane component nidogen-1) in order to stabilize the muscle basement membrane. Transgenic expression of mini-agrin composed of laminin-211 binding sites to α-dystroglycan and αLNNd, together with an increased laminin-411 level, restored basement membrane stability, muscle function and size, and animal survival in the dyW/dyW mouse model.
Steroid therapy is the gold standard treatment in DMD, and its effect is associated with elevated regeneration and reduced numbers of inflammatory cells (Sklar and Brown, 1991; Hussein et al., 2006).
Prednisolone treatment was found to upregulate the Mamstr gene in dystrophin-deficient mouse muscle compared to untreated controls (Chadwick et al., 2016) and upregulate Myh and dystrophin in human DMD primary culture (Sklar and Brown, 1991). In a clinical report by Hussein et al. (2010), 6 months’ prednisone therapy was associated with ultrastructural changes in dystrophic muscle and increased numbers of SCs, together with a decreased number of immune system cells (dendritic cells) and fibroblasts in DMD and BMD patients’ biopsies.
Summary
Recent findings point toward SC involvement in muscular dystrophy pathology.
The pathophysiology of LAMA2-CMD includes impaired SC regeneration in addition to muscle cell degeneration due to a fragile cell membrane, as a result of reduced laminin-211.
In support of this, we characterized in a recent study using RNA-Seq technique, a novel molecular signature of the contribution of specific key genes: Pax3, Pax7, and Mamstr, and signaling pathways, to the impaired muscle regeneration process in the dy2J/dy2J mouse model. These results suggest that the muscle phenotype in LAMA2-CMD may be ameliorated by therapies focused on recovering SC number and function.
In addition, preservation of the host SCs’ niche is also required in order to provide an optimal microenvironment to improve SC function or SCs/stem cell transplantation efficacy.
The lack of Pax3 may point to very early, possibly prenatal dysfunction of SCs; thus, early treatment may be considered, before massive fibrosis accumulation and as a consequence establishment of hostile microenvironments, which presents further difficulty to SC-induced regeneration.
Confirmation of this mouse model data with additional LAMA2-CMD human data is required to advance the development of additional new therapies in LAMA2-CMD.
Author Contributions
NY and YN wrote the manuscript with support of MR.
Funding
The authors declare that this study received funding from National Network of Excellence in neuroscience (NNE), Teva Pharmaceutical Industries Limited (Grant Nos. 3031 and 1059). The funder was not involved in the study design, collection, analysis, interpretation of data, the writing of this article or the decision to submit it for publication.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abu-Baker, A., Laganiere, J., Gaudet, R., Rochefort, D., Brais, B., Neri, C., et al. (2013). Lithium chloride attenuates cell death in oculopharyngeal muscular dystrophy by perturbing Wnt/β-catenin pathway. Cell Death Dis. 4:e821. doi: 10.1038/cddis.2013.342
Accorsi, A., Kumar, A., Rhee, Y., Miller, A., and Girgenrath, M. (2016). IGF-1/GH axis enhances losartan treatment in Lama2-related muscular dystrophy. Hum. Mol. Genet. 25, 4624–4634. doi: 10.1093/hmg/ddw291
Allbrook, D. (1981). Skeletal muscle regeneration. Muscle Nerve 4, 234–245. doi: 10.1002/mus.880040311
Allikian, M. J., Hack, A. A., Mewborn, S., Mayer, U., and McNally, E. M. (2004). Genetic compensation for sarcoglycan loss by integrin α7β1 in muscle. J. Cell Sci. 117, 3821–3830. doi: 10.1242/jcs.01234
Almeida, C. F., Fernandes, S. A., Ribeiro Junior, A. F., Keith Okamoto, O., and Vainzof, M. (2016). Muscle satellite cells: exploring the basic biology to rule them. Stem Cells Int. 2016:1078686. doi: 10.1155/2016/1078686
Beffy, P., Del Carratore, R., Masini, M., Furling, D., Puymirat, J., Masiello, P., et al. (2010). Altered signal transduction pathways and induction of autophagy in human myotonic dystrophy type 1 myoblasts. Int. J. Biochem. Cell Biol. 42, 1973–1983. doi: 10.1016/j.biocel.2010.08.010
Ben-Yair, R., and Kalcheim, C. (2005). Lineage analysis of the avian dermomyotome sheet reveals the existence of single cells with both dermal and muscle progenitor fates. Development 132, 689–701. doi: 10.1242/dev.01617
Bigot, A., Klein, A. F., Gasnier, E., Jacquemin, V., Ravassard, P., Butler-Browne, G., et al. (2009). Large CTG repeats trigger p16-dependent premature senescence in myotonic dystrophy type 1 muscle precursor cells. Am. J. Pathol. 174, 1435–1442. doi: 10.2353/ajpath.2009.080560
Biressi, S., Miyabara, E. H., Gopinath, S. D., Carlig, P. M., and Rando, T. A. (2014). A Wnt-TGFβ2 axis induces a fibrogenic program in muscle stem cells from dystrophic mice. Sci. Transl. Med. 6:267ra176. doi: 10.1126/scitranslmed.3008411
Bjornson, C. R., Cheung, T. H., Liu, L., Tripathi, P. V., Steeper, K. M., and Rando, T. A. (2012). Notch signaling is necessary to maintain quiescence in adult muscle stem cells. Stem Cells 30, 232–242. doi: 10.1002/stem.773
Block, G. J., Narayanan, D., Amell, A. M., Petek, L. M., Davidson, K. C., Bird, T. D., et al. (2013). Wnt/β-catenin signaling suppresses DUX4 expression and prevents apoptosis of FSHD muscle cells. Hum. Mol. Genet. 22, 4661–4672. doi: 10.1093/hmg/ddt314
Boldrin, L., Neal, A., Zammit, P. S., Muntoni, F., and Morgan, J. E. (2012). Donor satellite cell engraftment is significantly augmented when the host niche is preserved and endogenous satellite cells are incapacitated. Stem Cells 30, 1971–1984. doi: 10.1002/stem.1158
Boldrin, L., Zammit, P. S., and Morgan, J. E. (2015). Satellite cells from dystrophic muscle retain regenerative capacity. Stem Cell Res. 14, 20–29. doi: 10.1016/j.scr.2014.10.007
Boldrin, L., Zammit, P. S., Muntoni, F., and Morgan, J. E. (2009). Mature adult dystrophic mouse muscle environment does not impede efficient engrafted satellite cell regeneration and self-renewal. Stem Cells 27, 2478–2487. doi: 10.1002/stem.162
Bosnakovski, D., Lamb, S., Simsek, T., Xu, Z., Belayew, A., Perlingeiro, R., et al. (2008). DUX4c, an FSHD candidate gene, interferes with myogenic regulators and abolishes myoblast differentiation. Exp. Neurol. 214, 87–96. doi: 10.1016/j.expneurol.2008.07.022
Boyden, S. E., Mahoney, L. J., Kawahara, G., Myers, J. A., Mitsuhashi, S., Estrella, E. A., et al. (2012). Mutations in the satellite cell gene MEGF10 cause a recessive congenital myopathy with minicores. Neurogenetics 13, 115–124. doi: 10.1007/s10048-012-0315-z
Brack, A. S., Conboy, M. J., Roy, S., Lee, M., Kuo, C. J., Keller, C., et al. (2007). Increased Wnt signaling during aging alters muscle stem cell fate and increases fibrosis. Science 317, 807–810. doi: 10.1126/science.1144090
Castets, P., Bertrand, A. T., Beuvin, M., Ferry, A., Le Grand, F., Castets, M., et al. (2011). Satellite cell loss and impaired muscle regeneration in selenoprotein N deficiency. Hum. Mol. Genet. 20, 694–704. doi: 10.1093/hmg/ddq515
Chadwick, J. A., Bhattacharya, S., Lowe, J., Weisleder, N., and Rafael-Fortney, J. A. (2016). Renin-angiotensin-aldosterone system inhibitors improve membrane stability and change gene-expression profiles in dystrophic skeletal muscles. Am. J. Physiol. Cell Physiol. 312, C155–C168. doi: 10.1152/ajpcell.00269.2016
Chal, J., and Pourquié, O. (2017). Making muscle: skeletal myogenesis in vivo and in vitro. Development 144, 2104–2122. doi: 10.1242/dev.151035
Ciciliot, S., and Schiaffino, S. (2010). Regeneration of mammalian skeletal muscle. Basic mechanisms and clinical implications. Curr. Pharm. Des. 16, 906–914. doi: 10.2174/138161210790883453
Conboy, I. M., and Rando, T. A. (2002). The regulation of Notch signaling controls satellite cell activation and cell fate determination in postnatal myogenesis. Dev. Cell 3, 397–409. doi: 10.1016/s1534-5807(02)00254-x
Conboy, I. M., and Rando, T. A. (2012). Heterochronic parabiosis for the study of the effects of aging on stem cells and their niches. Cell Cycle 11, 2260–2267. doi: 10.4161/cc.20437
Corti, S., Strazzer, S., Del Bo, R., Salani, S., Bossolasco, P., Fortunato, F., et al. (2002). A subpopulation of murine bone marrow cells fully differentiates along the myogenic pathway and participates in muscle repair in the mdx dystrophic mouse. Exp. Cell Res. 277, 74–85. doi: 10.1006/excr.2002.5543
Dadush, O., Aga-Mizrachi, S., Ettinger, K., Tabakman, R., Elbaz, M., Fellig, Y., et al. (2010). Improved muscle strength and mobility in the dy2J/dy2J mouse with merosin deficient congenital muscular dystrophy treated with Glatiramer acetate. Neuromuscul. Disord. 20, 267–272. doi: 10.1016/j.nmd.2010.02.002
de la Garza-Rodea, A. S., van der Velde-van Dijke, I., Boersma, H., Gonçalves, M. A., van Bekkum, D. W., de Vries, A. A., et al. (2011). Myogenic properties of human mesenchymal stem cells derived from three different sources. Cell Transplant. 21, 153–173. doi: 10.3727/096368911x580554
de Oliveira, B. M., Matsumura, C. Y., Fontes-Oliveira, C. C., Gawlik, K. I., Acosta, H., Wernhoff, P., et al. (2014). Quantitative proteomic analysis reveals metabolic alterations, calcium dysregulation, and increased expression of extracellular matrix proteins in laminin α2 chain-deficient muscle. Mol. Cell. Proteomics 13, 3001–3013. doi: 10.1074/mcp.m113.032276
Dellavalle, A., Sampaolesi, M., Tonlorenzi, R., Tagliafico, E., Sacchetti, B., Perani, L., et al. (2007). Pericytes of human skeletal muscle are myogenic precursors distinct from satellite cells. Nat. Cell Biol. 9, 255–267. doi: 10.1038/ncb1542
Di Gioia, S. A., Connors, S., Matsunami, N., Cannavino, J., Rose, M. F., Gilette, N. M., et al. (2017). A defect in myoblast fusion underlies Carey-Fineman-Ziter syndrome. Nat. Commun. 8:16077. doi: 10.1038/ncomms16077
Dumont, N. A., Wang, Y. X., von Maltzahn, J., Pasut, A., Bentzinger, C. F., Brun, C. E., et al. (2015). Dystrophin expression in muscle stem cells regulates their polarity and asymmetric division. Nat. Med. 21, 1455–1463. doi: 10.1038/nm.3990
Durbeej, M. (2015). Laminin-α2 chain-deficient congenital muscular dystrophy: pathophysiology and development of treatment. Curr. Top. Membr. 76, 31–60. doi: 10.1016/bs.ctm.2015.05.002
Elbaz, M., Yanay, N., Aga-Mizrachi, S., Brunschwig, Z., Kassis, I., Ettinger, K., et al. (2012). Losartan, a therapeutic candidate in congenital muscular dystrophy: studies in the dy2J/dy2J mouse. Ann. Neurol. 71, 699–708. doi: 10.1002/ana.22694
Elbaz, M., Yanay, N., Laban, S., Rabie, M., Mitrani-Rosenbaum, S., and Nevo, Y. (2015). Life or death by NFκB, Losartan promotes survival in dy2J/dy2J mouse of MDC1A. Cell Death Dis. 6:e1690. doi: 10.1038/cddis.2015.60
Favreau, C., Higuet, D., Courvalin, J. C., and Buendia, B. (2004). Expression of a mutant lamin A that causes Emery-Dreifuss muscular dystrophy inhibits in vitro differentiation of C2C12 myoblasts. Mol. Cell. Biol. 24, 1481–1492. doi: 10.1128/mcb.24.4.1481-1492.2004
Feeney, S. J., McGrath, M. J., Sriratana, A., Gehrig, S. M., Lynch, G. S., D’Arcy, C. E., et al. (2015). FHL1 reduces dystrophy in transgenic mice overexpressing FSHD muscular dystrophy region gene 1 (FRG1). PLoS One 10:e0117665. doi: 10.1371/journal.pone.0117665
Ferrari, G., Cusella-De Angelis, G., Coletta, M., Paolucci, E., Stornaiuolo, A., Cossu, G., et al. (1998). Muscle regeneration by bone marrow-derived myogenic progenitors. Science 279, 1528–1530. doi: 10.1126/science.279.5356.1528
Fiore, P. F., Benedetti, A., Sandonà, M., Madaro, L., De Bardi, M., Saccone, V., et al. (2020). Lack of PKCtheta promotes regenerative ability of muscle stem cells in chronic muscle injury. Int. J. Mol. Sci. 21:E932. doi: 10.3390/ijms21030932
Frock, R. L., Kudlow, B. A., Evans, A. M., Jameson, S. A., Hauschka, S. D., and Kennedy, B. K. (2006). Lamin A/C and emerin are critical for skeletal muscle satellite cell differentiation. Genes Dev. 20, 486–500. doi: 10.1101/gad.1364906
Fujimaki, S., Seko, D., Kitajima, Y., Yoshioka, K., Tsuchiya, Y., Masuda, S., et al. (2018). Notch1 and Notch2 coordinately regulate stem cell function in the quiescent and activated states of muscle satellite cells. Stem Cells 36, 278–285. doi: 10.1002/stem.2743
Gattazzo, F., Molon, S., Morbidoni, V., Braghetta, P., Blaauw, B., Urciuolo, A., et al. (2014a). Cyclosporin A promotes in vivo myogenic response in collagen VI-deficient myopathic mice. Front. Aging Neurosci. 6:244. doi: 10.3389/fnagi.2014.00244
Gattazzo, F., Urciuolo, A., and Bonaldo, P. (2014b). Extracellular matrix: a dynamic microenvironment for stem cell niche. Biochim. Biophys. Acta 1840, 2506–2519. doi: 10.1016/j.bbagen.2014.01.010
Gawlik, K. I., and Durbeej, M. (2011). Skeletal muscle laminin and MDC1A: pathogenesis and treatment strategies. Skelet Muscle 1:9. doi: 10.1186/2044-5040-1-9
Girgenrath, M., Kostek, C. A., and Miller, J. B. (2005). Diseased muscles that lack dystrophin or laminin-α2 have altered compositions and proliferation of mononuclear cell populations. BMC Neurol. 5:7. doi: 10.1186/1471-2377-5-7
Guo, C., Willem, M., Werner, A., Raivich, G., Emerson, M., Neyses, L., et al. (2006). Absence of α 7 integrin in dystrophin-deficient mice causes a myopathy similar to Duchenne muscular dystrophy. Hum. Mol. Genet. 15, 989–998. doi: 10.1093/hmg/ddl018
Higuchi, I., Kawai, H., Kawajiri, M., Fukunaga, H., Horikiri, T., Niiyama, T., et al. (1999). Statistically significant differences in the number of CD24 positive muscle fibers and satellite cells between sarcoglycanopathy and age-matched Becker muscular dystrophy patients. Intern. Med. 38, 412–415. doi: 10.2169/internalmedicine.38.412
Hussein, M. R., Abu-Dief, E. E., Kamel, N. F., and Mostafa, M. G. (2010). Steroid therapy is associated with decreased numbers of dendritic cells and fibroblasts and increased numbers of satellite cells, in the dystrophic skeletal muscle. J. Clin. Pathol. 63, 805–813. doi: 10.1136/jcp.2010.078204
Hussein, M. R., Hamed, S. A., Mostafa, M. G., Abu-Dief, E. E., Kamel, N. F., and Kandil, M. R. (2006). The effects of glucocorticoid therapy on the inflammatory and dendritic cells in muscular dystrophies. Int. J. Exp. Pathol. 87, 451–461. doi: 10.1111/j.1365-2613.2006.00470.x
Jiang, C., Wen, Y., Kuroda, K., Hannon, K., Rudnicki, M. A., and Kuang, S. (2014). Notch signaling deficiency underlies age-dependent depletion of satellite cells in muscular dystrophy. Dis. Model. Mech. 7, 997–1004. doi: 10.1242/dmm.015917
Kassar-Duchossoy, L., Giacone, E., Gayraud-Morel, B., Jory, A., Gomès, D., and Tajbakhsh, S. (2005). Pax3/Pax7 mark a novel population of primitive myogenic cells during development. Genes Dev. 19, 1426–1431. doi: 10.1101/gad.345505
Kimura, E., Li, S., Gregorevic, P., Fall, B. M., and Chamberlain, J. S. (2009). Dystrophin delivery to muscles of mdx mice using lentiviral vectors leads to myogenic progenitor targeting and stable gene expression. Mol. Ther. 18, 206–213. doi: 10.1038/mt.2009.253
Kuang, S., Chargé, S. B., Seale, P., Huh, M., and Rudnicki, M. A. (2006). Distinct roles for Pax7 and Pax3 in adult regenerative myogenesis. J. Cell Biol. 172, 103–113. doi: 10.1083/jcb.200508001
Kuang, W., Xu, H., Vilquin, J. T., and Engvall, E. (1999). Activation of the lama2 gene in muscle regeneration: abortive regeneration in laminin α2-deficiency. Lab. Invest. 79, 1601–1613.
Kudryashova, E., Kramerova, I., and Spencer, M. J. (2012). Satellite cell senescence underlies myopathy in a mouse model of limb-girdle muscular dystrophy 2H. J. Clin. Invest. 122, 1764–1776. doi: 10.1172/jci59581
Lawlor, M. W., Alexander, M. S., Viola, M. G., Meng, H., Joubert, R., Gupta, V., et al. (2012). Myotubularin-deficient myoblasts display increased apoptosis, delayed proliferation and poor cell engraftment. Am. J. Pathol. 181, 961–968. doi: 10.1016/j.ajpath.2012.05.016
Lee, J. H., Kosinski, P. A., and Kemp, D. M. (2005). Contribution of human bone marrow stem cells to individual skeletal myotubes followed by myogenic gene activation. Exp. Cell Res. 307, 174–182. doi: 10.1016/j.yexcr.2005.03.008
Lepper, C., Conway, S. J., and Fan, C. M. (2009). Adult satellite cells and embryonic muscle progenitors have distinct genetic requirements. Nature 460, 627–631. doi: 10.1038/nature08209
Liu, Y., Yan, X., Sun, Z., Chen, B., Han, Q., Li, J., et al. (2007). Flk-1+ adipose-derived mesenchymal stem cells differentiate into skeletal muscle satellite cells and ameliorate muscular dystrophy in mdx mice. Stem Cells Dev. 16, 695–706. doi: 10.1089/scd.2006.0118
Logan, C. V., Lucke, B., Pottinger, C., Abdelhamed, Z. A., Parry, D. A., Szymanska, K., et al. (2011). Mutations in MEGF10, a regulator of satellite cell myogenesis, cause early onset myopathy, areflexia, respiratory distress and dysphagia (EMARDD). Nat. Genet. 43, 1189–1192. doi: 10.1038/ng.995
Mauro, A. (1961). Satellite cell of skeletal muscle fibers. J. Biophys. Biochem. Cytol. 9, 493–495. doi: 10.1083/jcb.9.2.493
Meinke, P., Schneiderat, P., Srsen, V., Korfali, N., Lê Thành, P., Cowan, G. J., et al. (2015). Abnormal proliferation and spontaneous differentiation of myoblasts from a symptomatic female carrier of X-linked Emery-Dreifuss muscular dystrophy. Neuromuscul. Disord. 25, 127–136. doi: 10.1016/j.nmd.2014.09.012
Miyagoe, Y., Hanaoka, K., Nonaka, I., Hayasaka, M., Nabeshima, Y., Arahata, K., et al. (1997). Laminin α2 chain-null mutant mice by targeted disruption of the Lama2 gene: a new model of merosin (laminin 2)-deficient congenital muscular dystrophy. FEBS Lett. 415, 33–39. doi: 10.1016/s0014-5793(97)01007-7
Miyagoe-Suzuki, Y., Masubuchi, N., Miyamoto, K., Wada, M. R., Yuasa, S., Saito, F., et al. (2009). Reduced proliferative activity of primary POMGnT1-null myoblasts in vitro. Mech. Dev. 126, 107–116. doi: 10.1016/j.mod.2008.12.001
Mokhonova, E. I., Avliyakulov, N. K., Kramerova, I., Kudryashova, E., Haykinson, M. J., and Spencer, M. J. (2015). The E3 ubiquitin ligase TRIM32 regulates myoblast proliferation by controlling turnover of NDRG2. Hum. Mol. Genet. 24, 2873–2883. doi: 10.1093/hmg/ddv049
Moreira Soares Oliveira, B., Gawlik, K. I., Durbeej, M., and Holmberg, J. (2018). Exploratory profiling of urine MicroRNAs in the dy2J/dy2J mouse model of LAMA2-CMD: relation to disease progression. PLoS Curr. 10:ecurrents.md.d0c203c018bc024f2f4c9791ecb05f88. doi: 10.1371/currents.md.d0c203c018bc024f2f4c9791ecb05f88
Morgan, J., Butler-Browne, G., Muntoni, F., and Patel, K. (2019). 240th ENMC workshop: the involvement of skeletal muscle stem cells in the pathology of muscular dystrophies 25–27 January 2019, Hoofddorp, The Netherlands. Neuromuscul. Disord. 29, 704–715. doi: 10.1016/j.nmd.2019.07.003
Morgan, J. E., and Zammit, P. S. (2010). Direct effects of the pathogenic mutation on satellite cell function in muscular dystrophy. Exp. Cell Res. 316, 3100–3108. doi: 10.1016/j.yexcr.2010.05.014
Morrison, S. J., and Spradling, A. C. (2008). Stem cells and niches: mechanisms that promote stem cell maintenance throughout life. Cell 132, 598–611. doi: 10.1016/j.cell.2008.01.038
Mouly, V., Aamiri, A., Bigot, A., Cooper, R. N., Di Donna, S., Furling, D., et al. (2005). The mitotic clock in skeletal muscle regeneration, disease and cell mediated gene therapy. Acta Physiol. Scand. 184, 3–15. doi: 10.1111/j.1365-201x.2005.01417.x
Muskiewicz, K. R., Frank, N. Y., Flint, A. F., and Gussoni, E. (2005). Myogenic potential of muscle side and main population cells after intravenous injection into sub-lethally irradiated mdx mice. J. Histochem. Cytochem. 53, 861–873. doi: 10.1369/jhc.4a6573.2005
Negroni, E., Riederer, I., Chaouch, S., Belicchi, M., Razini, P., Di Santo, J., et al. (2009). In vivo myogenic potential of human CD133+ muscle-derived stem cells: a quantitative study. Mol. Ther. 17, 1771–1778. doi: 10.1038/mt.2009.167
Nevo, Y., Aga-Mizrachi, S., Elmakayes, E., Yanay, N., Ettinger, K., Elbaz, M., et al. (2011). The Ras antagonist, farnesylthiosalicylic acid (FTS), decreases fibrosis and improves muscle strength in dy/dy mouse model of muscular dystrophy. PLoS One 6:e18049. doi: 10.1371/journal.pone.0018049
Nevo, Y., Halevy, O., Genin, O., Moshe, I., Turgeman, T., Harel, M., et al. (2010). Fibrosis inhibition and muscle histopathology improvement in laminin-α2-deficient mice. Muscle Nerve 42, 218–229. doi: 10.1002/mus.21706
Olguin, H. C., and Olwin, B. B. (2004). Pax-7 up-regulation inhibits myogenesis and cell cycle progression in satellite cells: a potential mechanism for self-renewal. Dev. Biol. 275, 375–388. doi: 10.1016/j.ydbio.2004.08.015
Otis, J. S., Niccoli, S., Hawdon, N., Sarvas, J. L., Frye, M. A., Chicco, A. J., et al. (2014). Pro-inflammatory mediation of myoblast proliferation. PLoS One 9:e92363. doi: 10.1371/journal.pone.0092363
Parker, M. H., von Maltzahn, J., Bakkar, N., Al-Joubori, B., Ishibashi, J., Guttridge, D., et al. (2012). MyoD-dependent regulation of NF-kappaB activity couples cell-cycle withdrawal to myogenic differentiation. Skelet. Muscle 2:6. doi: 10.1186/2044-5040-2-6
Patton, B. L., Miner, J. H., Chiu, A. Y., and Sanes, J. R. (1997). Distribution and function of laminins in the neuromuscular system of developing, adult, and mutant mice. J. Cell Biol. 139, 1507–1521. doi: 10.1083/jcb.139.6.1507
Périé, S., Mamchaoui, K., Mouly, V., Blot, S., Bouazza, B., Thornell, L. E., et al. (2006). Premature proliferative arrest of cricopharyngeal myoblasts in oculo-pharyngeal muscular dystrophy: therapeutic perspectives of autologous myoblast transplantation. Neuromuscul. Disord. 16, 770–781. doi: 10.1016/j.nmd.2006.07.022
Périé, S., Trollet, C., Mouly, V., Vanneaux, V., Mamchaoui, K., Bouazza, B., et al. (2014). Autologous myoblast transplantation for oculopharyngeal muscular dystrophy: a phase I/IIa clinical study. Mol. Ther. 22, 219–225. doi: 10.1038/mt.2013.155
Petrof, B. J., Shrager, J. B., Stedman, H. H., Kelly, A. M., and Sweeney, H. L. (1993). Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc. Natl. Acad. Sci. U S A 90, 3710–3714. doi: 10.1073/pnas.90.8.3710
Price, F. D., von Maltzahn, J., Bentzinger, C. F., Dumont, N. A., Yin, H., Chang, N. C., et al. (2014). Inhibition of JAK-STAT signaling stimulates adult satellite cell function. Nat. Med. 20, 1174–1181. doi: 10.1038/nm.3655
Proto, J. D., Tang, Y., Lu, A., Chen, W. C., Stahl, E., Poddar, M., et al. (2015). NF-kappaB inhibition reveals a novel role for HGF during skeletal muscle repair. Cell Death Dis. 6:e1730. doi: 10.1038/cddis.2015.66
Randolph, M. E., and Pavlath, G. K. (2015). A muscle stem cell for every muscle: variability of satellite cell biology among different muscle groups. Front. Aging Neurosci. 7:190. doi: 10.3389/fnagi.2015.00190
Reinhard, J. R., Lin, S., McKee, K. K., Meinen, S., Crosson, S. C., Sury, M., et al. (2017). Linker proteins restore basement membrane and correct LAMA2-related muscular dystrophy in mice. Sci. Transl. Med. 9:eaal4649. doi: 10.1126/scitranslmed.aal4649
Relaix, F., Montarras, D., Zaffran, S., Gayraud-Morel, B., Rocancourt, D., Tajbakhsh, S., et al. (2006). Pax3 and Pax7 have distinct and overlapping functions in adult muscle progenitor cells. J. Cell Biol. 172, 91–102. doi: 10.1083/jcb.200508044
Relaix, F., Rocancourt, D., Mansouri, A., and Buckingham, M. (2005). A Pax3/Pax7-dependent population of skeletal muscle progenitor cells. Nature 435, 948–953. doi: 10.1038/nature03594
Renna, L. V., Cardani, R., Botta, A., Rossi, G., Fossati, B., Costa, E., et al. (2014). Premature senescence in primary muscle cultures of myotonic dystrophy type 2 is not associated with p16 induction. Eur. J. Histochem. 58:2444. doi: 10.4081/ejh.2014.2444
Ringelmann, B., Roder, C., Hallmann, R., Maley, M., Davies, M., Grounds, M., et al. (1999). Expression of laminin α1, α2, α4, and α5 chains, fibronectin, and tenascin-C in skeletal muscle of dystrophic 129ReJ dy/dy mice. Exp. Cell Res. 246, 165–182. doi: 10.1006/excr.1998.4244
Rooney, J. E., Welser, J. V., Dechert, M. A., Flintoff-Dye, N. L., Kaufman, S. J., and Burkin, D. J. (2006). Severe muscular dystrophy in mice that lack dystrophin and α7 integrin. J. Cell Sci. 119, 2185–2195. doi: 10.1242/jcs.02952
Rosales, X. Q., Malik, V., Sneh, A., Chen, L., Lewis, S., Kota, J., et al. (2013). Impaired regeneration in LGMD2A supported by increased PAX7-positive satellite cell content and muscle-specific microrna dysregulation. Muscle Nerve 47, 731–739. doi: 10.1002/mus.23669
Ross, J., Benn, A., Jonuschies, J., Boldrin, L., Muntoni, F., Hewitt, J. E., et al. (2012). Defects in glycosylation impair satellite stem cell function and niche composition in the muscles of the dystrophic Large(myd) mouse. Stem Cells 30, 2330–2341. doi: 10.1002/stem.1197
Sacco, A., Mourkioti, F., Tran, R., Choi, J., Llewellyn, M., Kraft, P., et al. (2010). Short telomeres and stem cell exhaustion model Duchenne muscular dystrophy in mdx/mTR mice. Cell 143, 1059–1071. doi: 10.1016/j.cell.2010.11.039
Schaaf, G. J., van Gestel, T. J. M., In ’t Groen, S. L. M., de Jong, B., Boomaars, B., Tarallo, A., et al. (2018). Satellite cells maintain regenerative capacity but fail to repair disease-associated muscle damage in mice with Pompe disease. Acta Neuropathol. Commun. 6:119. doi: 10.1186/s40478-018-0620-3
Schuler, F., and Sorokin, L. M. (1995). Expression of laminin isoforms in mouse myogenic cells in vitro and in vivo. J. Cell Sci. 108, 3795–3805.
Seale, P., Sabourin, L. A., Girgis-Gabardo, A., Mansouri, A., Gruss, P., and Rudnicki, M. A. (2000). Pax7 is required for the specification of myogenic satellite cells. Cell 102, 777–786. doi: 10.1016/s0092-8674(00)00066-0
Servián-Morilla, E., Takeuchi, H., Lee, T. V., Clarimon, J., Mavillard, F., Area-Gómez, E., et al. (2016). A POGLUT1 mutation causes a muscular dystrophy with reduced Notch signaling and satellite cell loss. EMBO Mol. Med. 8, 1289–1309. doi: 10.15252/emmm.201505815
Sklar, R. M., and Brown, R. H. Jr. (1991). Methylprednisolone increases dystrophin levels by inhibiting myotube death during myogenesis of normal human muscle in vitro. J. Neurol. Sci. 101, 73–81. doi: 10.1016/0022-510x(91)90019-4
Smythe, G. M., and Forwood, J. K. (2012). Altered mitogen-activated protein kinase signaling in dystrophic (mdx) muscle. Muscle Nerve 46, 374–383. doi: 10.1002/mus.23312
Soleimani, V. D., Punch, V. G., Kawabe, Y., Jones, A. E., Palidwor, G. A., Porter, C. J., et al. (2012). Transcriptional dominance of Pax7 in adult myogenesis is due to high-affinity recognition of homeodomain motifs. Dev. Cell 22, 1208–1220. doi: 10.1016/j.devcel.2012.03.014
Spence, H. J., Chen, Y. J., and Winder, S. J. (2002). Muscular dystrophies, the cytoskeleton and cell adhesion. Bioessays 24, 542–552. doi: 10.1002/bies.10098
Sunada, Y., Bernier, S. M., Utani, A., Yamada, Y., and Campbell, K. P. (1995). Identification of a novel mutant transcript of laminin α 2 chain gene responsible for muscular dystrophy and dysmyelination in dy2J mice. Hum. Mol. Genet. 4, 1055–1061. doi: 10.1093/hmg/4.6.1055
Tabebordbar, M., Zhu, K., Cheng, J. K. W., Chew, W. L., Widrick, J. J., Yan, W. X., et al. (2016). in vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science 351, 407–411. doi: 10.1126/science.aad5177
Tang, A. H., and Rando, T. A. (2014). Induction of autophagy supports the bioenergetic demands of quiescent muscle stem cell activation. EMBO J. 33, 2782–2797. doi: 10.15252/embj.201488278
Taniguchi, M., Kurahashi, H., Noguchi, S., Sese, J., Okinaga, T., Tsukahara, T., et al. (2006). Expression profiling of muscles from Fukuyama-type congenital muscular dystrophy and laminin-α 2 deficient congenital muscular dystrophy; is congenital muscular dystrophy a primary fibrotic disease? Biochem. Biophys. Res. Commun. 342, 489–502. doi: 10.1016/j.nmd.2006.05.134
Thornell, L. E., Lindstom, M., Renault, V., Klein, A., Mouly, V., Ansved, T., et al. (2009). Satellite cell dysfunction contributes to the progressive muscle atrophy in myotonic dystrophy type 1. Neuropathol. Appl. Neurobiol. 35, 603–613. doi: 10.1111/j.1365-2990.2009.01014.x
Tierney, M. T., Aydogdu, T., Sala, D., Malecova, B., Gatto, S., Puri, P. L., et al. (2014). STAT3 signaling controls satellite cell expansion and skeletal muscle repair. Nat. Med. 20, 1182–1186. doi: 10.1038/nm.3656
Trensz, F., Haroun, S., Cloutier, A., Richter, M. V., and Grenier, G. (2010). A muscle resident cell population promotes fibrosis in hindlimb skeletal muscles of mdx mice through the Wnt canonical pathway. Am. J. Physiol. Cell Physiol. 299, C939–C947. doi: 10.1152/ajpcell.00253.2010
Troy, A., Cadwallader, A. B., Fedorov, Y., Tyner, K., Tanaka, K. K., and Olwin, B. B. (2012). Coordination of satellite cell activation and self-renewal by Par-complex-dependent asymmetric activation of p38α/β MAPK. Cell Stem Cell 11, 541–553. doi: 10.1016/j.stem.2012.05.025
Urciuolo, A., Quarta, M., Morbidoni, V., Gattazzo, F., Molon, S., Grumati, P., et al. (2013). Collagen VI regulates satellite cell self-renewal and muscle regeneration. Nat. Commun. 4:1964. doi: 10.1038/ncomms2964
Vachon, P. H., Loechel, F., Xu, H., Wewer, U. M., and Engvall, E. (1996). Merosin and laminin in myogenesis; specific requirement for merosin in myotube stability and survival. J. Cell Biol. 134, 1483–1497. doi: 10.1083/jcb.134.6.1483
van Lunteren, E., Moyer, M., and Leahy, P. (2006). Gene expression profiling of diaphragm muscle in α2-laminin (merosin)-deficient dy/dy dystrophic mice. Physiol. Genomics 25, 85–95. doi: 10.1152/physiolgenomics.00226.2005
Vieira, N. M., Elvers, I., Alexander, M. S., Moreira, Y. B., Eran, A., Gomes, J. P., et al. (2015). Jagged 1 rescues the duchenne muscular dystrophy phenotype. Cell 163, 1204–1213. doi: 10.1016/j.cell.2015.10.049
von Maltzahn, J., Jones, A. E., Parks, R. J., and Rudnicki, M. A. (2013). Pax7 is critical for the normal function of satellite cells in adult skeletal muscle. Proc. Natl. Acad. Sci. U S A 110, 16474–16479. doi: 10.1073/pnas.1307680110
von Maltzahn, J., Chang, N. C., Bentzinger, C. F., and Rudnicki, M. A. (2012a). Wnt signaling in myogenesis. Trends Cell Biol. 22, 602–609. doi: 10.1016/j.tcb.2012.07.008
von Maltzahn, J., Renaud, J. M., Parise, G., and Rudnicki, M. A. (2012b). Wnt7a treatment ameliorates muscular dystrophy. Proc. Natl. Acad. Sci. U S A 109, 20614–20619. doi: 10.1073/pnas.1215765109
Wang, K., Wang, C., Xiao, F., Wang, H., and Wu, Z. (2008). JAK2/STAT2/STAT3 are required for myogenic differentiation. J. Biol. Chem. 283, 34029–34036. doi: 10.1074/jbc.M803012200
Wanschitz, J. V., Dubourg, O., Lacene, E., Fischer, M. B., Hoftberger, R., Budka, H., et al. (2013). Expression of myogenic regulatory factors and myo-endothelial remodeling in sporadic inclusion body myositis. Neuromuscul. Disord. 23, 75–83. doi: 10.1016/j.nmd.2012.09.003
Wen, Y., Bi, P., Liu, W., Asakura, A., Keller, C., and Kuang, S. (2012). Constitutive Notch activation upregulates Pax7 and promotes the self-renewal of skeletal muscle satellite cells. Mol. Cell. Biol. 32, 2300–2311. doi: 10.1128/mcb.06753-11
Whitlock, J. M., Yu, K., Cui, Y. Y., and Hartzell, H. C. (2018). Anoctamin 5/TMEM16E facilitates muscle precursor cell fusion. J. Gen. Physiol. 150, 1498–1509. doi: 10.1085/jgp.201812097
Wissing, E. R., Boyer, J. G., Kwong, J. Q., Sargent, M. A., Karch, J., McNally, E. M., et al. (2014). P38α MAPK underlies muscular dystrophy and myofiber death through a Bax-dependent mechanism. Hum. Mol. Genet. 23, 5452–5463. doi: 10.1093/hmg/ddu270
Xu, H., Wu, X. R., Wewer, U. M., and Engvall, E. (1994). Murine muscular dystrophy caused by a mutation in the laminin α 2 (Lama2) gene. Nat. Genet. 8, 297–302. doi: 10.1038/ng1194-297
Yablonka-Reuveni, Z. (2011). The skeletal muscle satellite cell: still young and fascinating at 50. J. Histochem. Cytochem. 59, 1041–1059. doi: 10.1369/0022155411426780
Yanay, N., Elbaz, M., Konikov-Rozenman, J., Elgavish, S., Nevo, Y., Fellig, Y., et al. (2019). Pax7, Pax3 and mamstr genes are involved in skeletal muscle impaired regeneration of dy2J/dy2J mouse model of Lama2-CMD. Hum. Mol. Genet. 28, 3369–3390. doi: 10.1093/hmg/ddz180
Yu, Q., Sali, A., Van der Meulen, J., Creeden, B. K., Gordish-Dressman, H., Rutkowski, A., et al. (2013). Omigapil treatment decreases fibrosis and improves respiratory rate in dy2J mouse model of congenital muscular dystrophy. PLoS One 8:e65468. doi: 10.1371/journal.pone.0065468
Keywords: LAMA2-CMD, laminin-211, next-generation sequencing, satellite cells, dy2J/dy2J mouse model, muscular dystrophy
Citation: Yanay N, Rabie M and Nevo Y (2020) Impaired Regeneration in Dystrophic Muscle—New Target for Therapy. Front. Mol. Neurosci. 13:69. doi: 10.3389/fnmol.2020.00069
Received: 08 December 2019; Accepted: 08 April 2020;
Published: 25 May 2020.
Edited by:
Markus A. Ruegg, Universität Basel, SwitzerlandReviewed by:
Ashok Kumar, University of Houston, United StatesFrancesca Cencetti, University of Florence, Italy
Copyright © 2020 Yanay, Rabie and Nevo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yoram Nevo, eW9yYW1uZUBjbGFsaXQub3JnLmls