 Yousuke Tsuneoka
Yousuke Tsuneoka Hiromasa Funato
Hiromasa Funato- 1Department of Anatomy, Faculty of Medicine, Toho University, Tokyo, Japan
- 2International Institutes for Integrative Sleep Medicine (WPI-IIIS), University of Tsukuba, Ibaraki, Japan
The visualization of multiple gene expressions in well-preserved tissues is crucial for the elucidation of physiological and pathological processes. In situ hybridization chain reaction (HCR) is a method to visualize specific mRNAs in diverse organisms by applying a HCR that is an isothermal enzyme-free nucleotide polymerization method using hairpin DNAs. Although in situ HCR is a versatile method, this method is not widely used by researchers because of their higher cost than conventional in situ hybridization (ISH). Here, we redesigned hairpin DNAs so that their lengths were half the length of commonly used hairpin DNAs. We also optimized the conjugated fluorophores and linkers. Modified in situ HCR showed sufficient fluorescent signals to detect various mRNAs such as Penk, Oxtr, Vglut2, Drd1, Drd2, and Moxd1 in mouse neural tissues with a high signal-to-noise ratio. The sensitivity of modified in situ HCR in detecting the Oxtr mRNA was better than that of fluorescent ISH using tyramide signal amplification. Notably, the modified in situ HCR does not require proteinase K treatment so that it enables the preservation of morphological structures and antigenicity. The modified in situ HCR simultaneously detected the distributions of c-Fos immunoreactivity and Vglut2 mRNA, and detected multiple mRNAs with a high signal-noise ratio at subcellular resolution in mouse brains. These results suggest that the modified in situ HCR using short hairpin DNAs is cost-effective and useful for the visualization of multiple mRNAs and proteins.
Introduction
To elucidate physiological and pathological processes in living organisms, it is crucial to visualize gene expression at good spatial resolution in a well-preserved morphological context. In situ hybridization (ISH) is a commonly used technique for detecting specific mRNAs in cells, tissues or whole bodies (Jensen, 2014). ISH was originally developed with the use of a radioisotope-labeled antisense nucleotide (Krumlauf et al., 1987; Marcus et al., 2001), which was subsequently replaced by a digoxigenin-labeled probe that enabled alkaline phosphatase- or peroxidase-based chromogenic reactions (Funato et al., 2000; Moorman et al., 2001). The use of two or more chromogens or fluorophores in combination enables the visualization of more than one mRNA. To increase the sensitivity of the ISH method to detect less abundant mRNAs, a method called tyramide signal amplification has been developed (Zaidi et al., 2000). Recent progress in ISH includes locked nucleic acid probes that are commonly applied for the detection of small RNAs (Urbanek et al., 2015), and rolling cycle amplification that has been reported to detect a single mRNA in situ (Larsson et al., 2010).
Currently, enzyme-based amplification using digoxigenin-labeled probes is the most used detection technique. To visualize multiple mRNAs, however, the procedures from probe hybridization to the chromogenic reaction generally need to be conducted twice serially, which takes a substantial amount of time and requires great labor. Also, when detecting low abundance mRNAs, artificial signals are inevitably produced due to nonspecific probe hybridization and nonspecific chromogenic enzyme reactions (Jensen, 2014).
The hybridization chain reaction (HCR) is an isothermal enzyme-free polymerization method that uses two different hairpin nucleotides: H1 and H2 (Figure 1A; Dirks and Pierce, 2004). The hairpin molecules are composed of toehold, stem, and loop domains and are self-assembling and metastable in the absence of initiator nucleotides that have a specific sequence complementary to the toehold and stem domains of an H1 hairpin (Figure 1A). In the presence of an initiator nucleotide, the toehold and stem domains of an H1 hairpin hybridize with the initiator through strand displacement. The remaining single-strand part of the opened H1 hairpin that was originally the loop and stem domains, hybridizes with H2 hairpin and produces a single strand part that has a sequence identical to the initiator, which in turn hybridizes with an H1 hairpin. Therefore, once an H1 hairpin is hybridized with an initiator, the polymerization of H1 and H2 hairpins continues and forms long nicked double-helices (Figures 1A,D and Supplementary Figure S1; Dirks and Pierce, 2004).
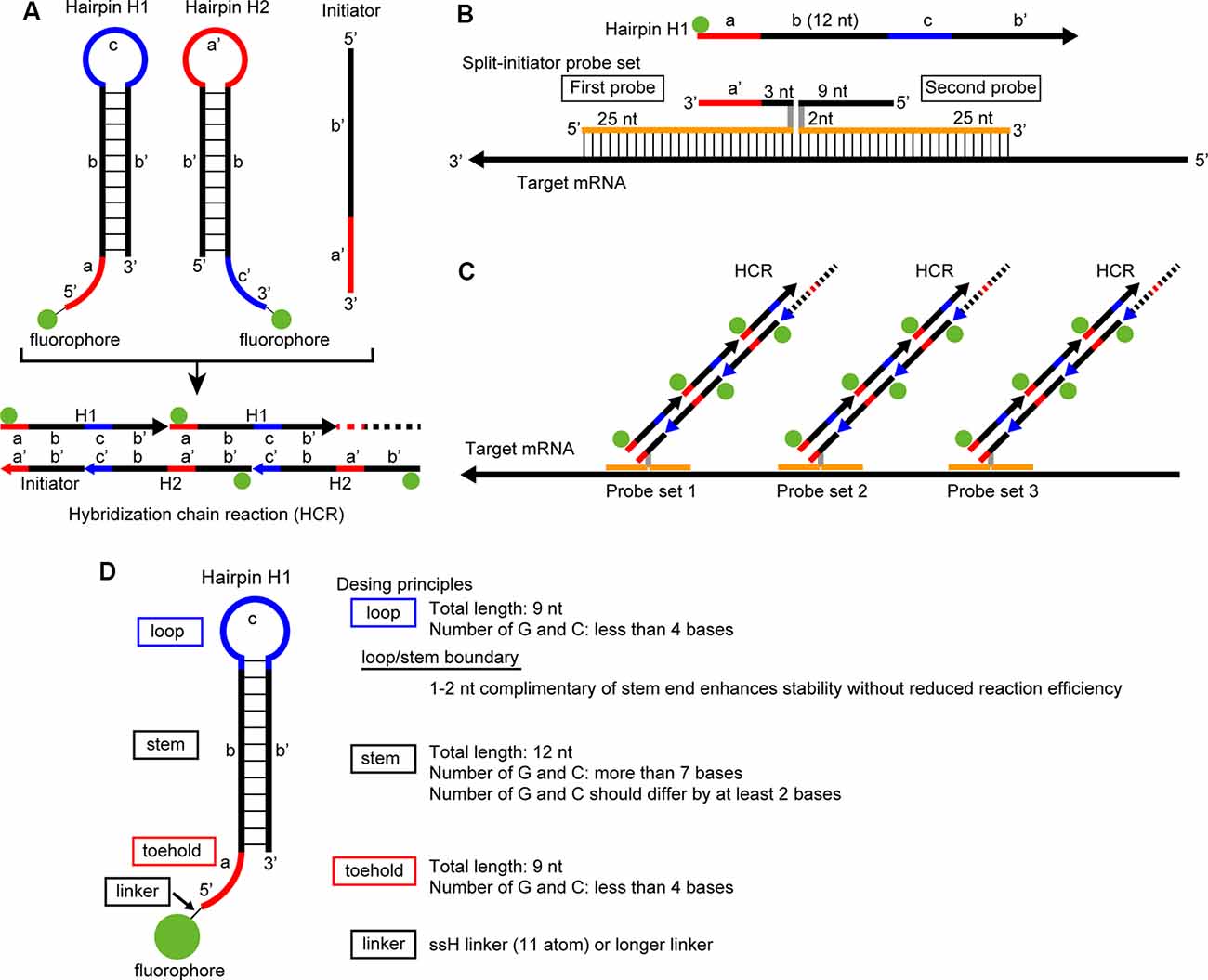
Figure 1. Principle of in situ hybridization chain reaction (HCR) and short hairpin design. (A) Each hairpin DNA (H1, H2) has toehold, stem and loop domains and is conjugated to a fluorophore. Whereas the sequence of the toehold domain of H1 (a) is complementary to that of the loop domain of H2 (a’), the sequence of the loop domain of H1 (c) is complementary to that of the toehold domain of H2 (c’). In the presence of an initiator that is composed of a’ and b’, the initiator hybridizes with the toehold and stem domains (a,b) of H1. Then, the remaining part of H1 (c,b’) hybridizes with the toehold and stem domains (c’,b) of H2. Thereafter, H1 and H2 continue to hybridizes with each other (lower). (B) Split-initiator probe set. One probe has sequence a’, first 3 nucleotide (nt) of sequence b’, 2 nt spacer sequence, and 25 nt sequence complementary to the target mRNA. Another probe has 25 nt sequence complementary to the target mRNA, 2 nt spacer sequence and the last 9 nt of sequence b’. (C) In situ HCR using split-initiator probes. Three sets of split-initiator probes hybridize with the target mRNA and lead to HCR. (D) Design principles of short hairpin DNA. A set of short hairpin DNAs and split-initiator probes with actual nucleotide sequences are shown in Supplementary Figure S1.
In situ HCR is a method that applies the HCR technique to the visualization of a specific nucleotide in cells, tissues, and whole-mount samples using an RNA probe that has a sequence enabling the hybridization to a target mRNA and a sequence to work as an initiator of HCR (Choi et al., 2010). Choi et al. (2010) successfully visualized the localization of target mRNA in zebrafish using HCR with fluorescently labeled RNA hairpins. In situ HCR has been further advanced by the use of a DNA hairpin (Choi et al., 2014; Yamaguchi et al., 2015), and the use of different fluorophore-labeled DNA hairpins for the detection of multiple mRNAs (Choi et al., 2016). Although in situ HCR frequently has been accompanied by false-positive signals and background signals due to nonspecific probe bindings, the “third-generation” in situ HCR method using a pair of split probes successfully reduced nonspecific signals (Choi et al., 2018). The advantages of in situ HCR using split probes are a high signal-to-noise ratio, a high sensitivity that enables single-molecule imaging, and an easy protocol for multiplex staining. In situ HCR using split-probes does not require stringent conditions for probe hybridization, and both prehybridization and hybridization are conducted under a mild condition of 37°C, which leads to decreased damage to tissues and well-preserved morphology.
Despite the technical advantages of in situ HCR, this method has not become standard procedure to detect mRNAs because in situ HCR that uses 72-nucleotide (nt)-long DNA hairpins costs more than conventional ISH methods. The cost of oligonucleotides increases in proportion to their length, and the yield of full-length oligos decreases as their lengths increase. Thus, shorter DNA hairpins are favorable as long as they progress HCR.
In this study, we optimized and shortened DNA hairpins and initiators for in situ HCR with split probes. We also simplified the in situ HCR protocol by removing proteinase K treatment. Our modified in situ HCR protocol is sensitive to a low abundance mRNA, is easier to handle and enables better antigenicity preservation for immunohistochemistry at a decreased cost.
Materials and Methods
Design and Synthesis of DNA Probes
All nonlabeled oligo DNAs were synthesized as standard desalted oligos (Integrated DNA Technologies). We modified and optimized the design of DNA probes using short hairpin DNAs from previous studies (Choi et al., 2014, 2018) as follows. Split-initiator DNA probes were designed to minimize off-target complementarity using a homology search by NCBI Blastn1, and they were designed to have 45–55% GC content in their mRNA binding sites. A pair of split-initiator probes were a 39-nt long DNA (25-nt long binding sites, 2-nt long spacer, and 12-nt long split-initiator sequence) and a 36-nt long DNA (25-nt long binding sites, 2-nt-long spacer, and 9-nt-long split-initiator sequence). The split-initiator sequence of the 39-nt-long DNA has a 9-nt-long toehold and a 3-nt-long sequence that is complementary to the first 3-nt of the stem domain of the hairpins. The split-initiator sequence of the 36-nt-long DNA probe has a 9-nt-long sequence that is complementary to the following 9-nt of the stem domain of the hairpins (Figure 1B and Supplementary Figure S1). Five or 10 sets of 36-nt and 39-nt DNA probes were designed for each target mRNA (Supplementary Table S2). All probe sets for each target mRNA were prepared, mixed, and stored in TE (10 mM Tris-HCl pH 8.0 and 1 mM EDTA). Each probe mixture was subsequently purified by denaturing polyacrylamide gel electrophoresis (PAGE) using 20% polyacrylamide gels (1:40 bis and linear acrylamide). After purification, the probes were diluted in TE to 2 μM.
Design and Synthesis of DNA Hairpin Amplifiers
To find the shortest sequences of DNA for hairpins that reliably trigger and continue HCR, we tested 36-44-nt long hairpins containing a 12-nt long stem sequence (Supplementary Table S1). The hairpin DNAs were designed using the multistate sequence design feature of NUPACK2, to produce target secondary structures shown in Figure 1A. After the NUPACK random design, the sequences were manually edited according to the previously reported criteria: less than 40% GC content in a toehold domain and greater than 60% GC content in a stem (Ang and Yung, 2016). The manually adjusted hairpin sequences were subsequently assessed by a NUPACK simulation to avoid undesirable secondary structures.
DNA hairpins labeled at the 5′ end with an amino linker, ssH, were synthesized (FASMAC). ssH-labeling of the 5′ end of DNAs enables subsequent coupling to a fluorophore (Komatsu et al., 2008). For comparison, C6-amino linker-labeled DNAs were also prepared (FASMAC). The fluorophores that were conjugated with succinimidyl esters were FAM (Sigma–Aldrich #21878), ATTO390, ATTO488, ATTO550, ATTO565 (ATTO-TEC), Alexa Fluor488, Alexa Fluor568, and Alexa Fluor647 (Thermo Fisher Scientific; Figures 2D–K). Following chloroform purification and ethanol precipitation with MgCl2, ssH-labeled DNAs were dissolved in 0.1 M borate buffer, pH 9.0, at 1 mM concentration of the DNA. One-third volume of fluorophore-conjugated succinimidyl ester (10 mg/ml in dimethylformamide) was mixed with the DNA solution and incubated for 4 h at room temperature to allow the coupling reaction of succinimidyl ester with the ssH-amino linker conjugated to the DNA. Fluorophore-conjugated DNAs were purified by denaturing PAGE using 20% polyacrylamide gels to remove incorporated fluorophores and incorrectly synthesized shorter oligonucleotides. The fluorescence of DNA bands corresponding to expected sizes was visualized by a hand-made LED illuminator (OptoSupply) and filter sets (LEE filters) to minimize DNA damage caused by UV light; then the bands were excised from the gel. The excised gel bands were crushed and soaked in TE at 4°C overnight, and the DNA recovered by ethanol precipitation as described above. The fluorophore-labeled DNAs were eluted in TE with 150 mM NaCl, and the concentration was adjusted to 3 μM based on the 260 nm absorbance. It is noted that the exact concentration of fluorophore-labeled DNAs cannot be determined because the fluorophores absorb 260 nm light to some extent, and the stem domain (double-strand) has a lower 260 nm absorbance per nucleotide than the toehold and loop domains (single strand).
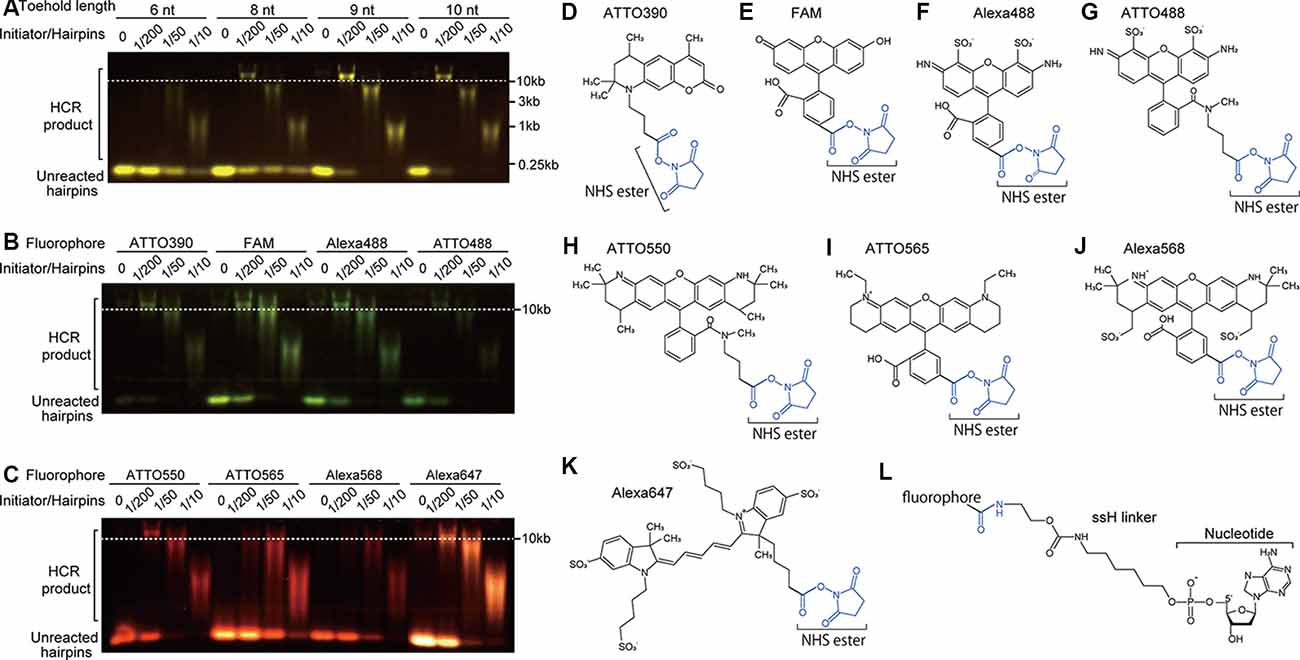
Figure 2. Effect of toehold length and fluorophores on HCR. (A) Effect of toehold length of hairpin DNAs on HCR efficiency. Short hairpin DNAs with a toehold length of 6, 8, 9, and 10 nt were examined. The ratios of initiator’s concentration to hairpin DNAs’ concentration were 0, 1/200, 1/50, and 1/10 with a constant concentration of 0.5 μM hairpin DNAs. HCR products were visualized after agarose gel electrophoresis. HCR product bands longer than 10 kb indicate efficient HCR. Unreacted hairpin DNAs were recognized as bands shorter than the position of 0.25 kb. Used hairpin DNAs were #S4 (6-nt toehold), #S8 (8-nt toehold), #S41 (9-nt toehold), and #S9 (10-nt toehold; Supplementary Table S2). (B,C) Effect of fluorophores on HCR efficiency. Hairpin DNAs with 9-nt toehold (#S41) conjugated with either ATTO390, FAM, Alexa488, ATTO488, ATTO550, ATTO565, ATTO568 or Alexa647 were used with different initiator/hairpin concentration ratios. Captured images taken with appropriate filter sets were overlaid based on the loading position. Dashed lines indicate a band size of 10 kb dsDNA. (D–K) Chemical structures of N-hydroxysuccinimide (NHS) ester (blue)-conjugated fluorophores. (L) Chemical structure of ssH linker that is conjugated with a fluorophore via an amido bond and with a nucleotide via a phosphoric acid.
Nonlabeled DNA hairpins were used only for in vitro study in microtubes, not for in situ HCR. Nonlabeled DNAs were purified by denaturing PAGE like the process used for the labeled DNAs, except for post staining of the polyacrylamide gel using GelGreen (Biotium).
HCR Verification of Hairpin DNAs in Microtubes
All hairpin DNAs were snap-cooled (heated to 95°C for 2 min and cooled to room temperature for 30 min) to form a hairpin structure before use. HCR was performed in 5× standard saline citrate (SSC) with 0.1% Tween 20 (5× SSCT). Each microtube was prepared by adding 2 μl of 3 μM hairpin DNAs, 6 μl of 0, 0.005, 0.02 or 0.1 μM initiator DNA, and an appropriate amount of 20× SSC with 10% Tween 20 to bring the reaction volume to 12 μl. Two hours after mixing the DNAs, the samples were supplemented with prestained loading dye (Bio-craft) and loaded into native 1% agarose gels. The gels were electrophoresed in sodium borate buffer (10 mM NaOH and 35 mM boric acid) at 135 V for 30 min and then were imaged. The intensity of each band was analyzed by ImageJ to quantify the amount of HCR products.
We determined that a pair of hairpins showed strong amplification when HCR products exceeded 10 kb with 1/200 of the initiator-to-short hairpin ratio. We also judged that initiator-independent HCR (leakage) was low when a very weak signal of the HCR product was observed after 2 h of HCR without initiator nucleotides. Also, HCR efficiency was evaluated based on the amount of unreacted hairpin (see Figure 2).
In situ Hybridization
All animal procedures were conducted following the Guidelines for Animal Experiments of Toho University and were approved by the Institutional Animal Care and Use Committee of Toho University (Approved protocol ID #19-51-405). Breeding pairs of C57BL/6J mice were obtained from Japan SLC and CLEA Japan. Mice were raised in our breeding colony under controlled conditions (12 h light/dark cycle; lights on at 8:00 A.M., 23 ± 2°C, 55 ± 5% humidity; and ad libitum access to water and food). Male mice (20–30 weeks old) were used except for c-Fos staining after maternal behavior. Female mice (20–30 weeks old) were sacrificed 2 h after pup presentation for c-Fos immunohistochemistry (Tsuneoka et al., 2013). At least three mice were used for each experiment to confirm reproducibility. Mice were anesthetized with sodium pentobarbital (50 mg/kg, i.p.), and then were transcardially perfused with 4% paraformaldehyde (PFA) in phosphate-buffered saline (PBS). The brains were postfixed in 4% PFA at 4°C overnight, which was followed by cryoprotection in 30% sucrose in PBS for 2 days, embedded in Surgipath (FSC22, Leica Biosystems), and were stored at −80°C until use. The brains were cryosectioned coronally at a thickness of 40 μm.
All the sections were stained by the free-floating method, which enables better preservation of tissue morphology and uniform staining of the sections. The handling and prehybridization procedures were the same as those in our published protocol (Tsuneoka et al., 2015, 2017b). Briefly, the sections were washed with PBS containing 0.1% Tween-20 (PBST), treated with or without proteinase K (Roche, 10 mg/ml in PBST) for 10 min and postfixed with 4% PFA in PBS for 10 min at 37°C. The sections were immersed in methanol containing 0.3% H2O2 for 10 min, followed by acetylation with 0.25% acetic anhydride in 0.1 M triethanolamine (pH 8.0) for 20 min. After washing, the sections were prehybridized for 10 min at 37°C in a hybridization buffer containing 30% formamide, 10% dextran sulfate, 5× SSC, 10 mM citric acid, 0.1% Tween 20, 50 μg/ml heparin, 1× Denhardt’s solution as described (Choi et al., 2018). The sections were moved to another hybridization solution containing a mixture of 1 nM split-initiator probes, and incubated overnight at 37°C. In the case of staining for multiple targets, the probes were added simultaneously. After hybridization, the sections were washed three times for 10 min in 5× SSCT with 30% formamide at 37°C, followed by three washes for 10 min in 5× SSCT without formamide at room temperature.
For in situ HCR (Figure 1C), 3 μM hairpin DNA solutions were separately snap-cooled before use. The sections were incubated in amplification buffer (10% dextran sulfate in 5× SSCT) with 60 nM hairpin DNA pairs for 45 min, 2 h or overnight at 25°C. In the case of multiple staining, the hairpin DNAs were added simultaneously. Then, the samples were washed with 5× SSCT and PBST three times at room temperature.
In the case of combined ISH and immunohistochemistry, the sections were blocked using 0.8% Block Ace/PBST (Dainihon-Seiyaku), which was followed by overnight incubation with rabbit anti-c-Fos antibody (1:2,500, sc-52, Santa Cruz) in 0.4% Block Ace/PBST at 4°C. After washing three times with PBST, sections were incubated with an Alexa488-conjugated donkey anti-rabbit goat antibody (1:500, 711-545-152, Jackson ImmunoResearch) with Hoechst 33342 (1 μg/ml) for an hour at room temperature. The sections were mounted on the slide glass and cover-slipped with mounting media containing antifade (1% n-propyl gallate and 10% Mowiol4–88 in PBS).
To compare the sensitivity between in situ HCR and enzyme-based ISH, we also performed chromogenic ISH and fluorescent ISH using tyramide signal amplification. The cDNA fragment of oxytocin receptor (Oxtr) mRNA (GenBank ID: NM_001081147,1869-3843) was amplified, inserted into the pGEM-T plasmid (A3600, Promega), which was used into DH5α E. coli. After confirmation that the DNA sequence was correct, template cDNA was produced using polymerase chain reaction with specific primers (5′-ATTTAGGTGACACTATAG-3′) and (5′-TAATACGACTCACTATAGGG-3′). The probe was transcribed by SP6 RNA polymerase (P1085; Promega) in the presence of digoxigenin-labeled UTP (Dig labeling mix; Roche Diagnostics, Switzerland), which was followed by precipitation with LiCl with ethanol. The riboprobe was digested by alkaline hydrolysis to reduce the average size to 500 bases. Although hydrolyzed riboprobes sometimes increase nonspecific hybridization, our ISH protocol using a stringent wash, RNase A treatment, and non-excessive hydrolysis suppresses the background.
The prehybridization procedure was identical to that of in situ HCR. After acetylation, sections were washed with PBST, which was followed by incubation at 57°C overnight in a hybridization mixture containing 1 μg/ml riboprobe, 50% deionized formamide, 5× SSC (pH 7.0), 5 mM EDTA (pH 8.0), 0.2 mg/ml yeast tRNA, 0.2% Tween-20, 0.2% sodium dodecyl sulfate, 10% dextran sulfate, and 0.1 mg/ml heparin. After hybridization, the sections were washed twice with 2× SSC containing 50% formamide at 57°C for 10 min, incubated with RNAse A solution (20 μg/ml) at 37°C for 30 min, rinsed twice with 2× SSC and 0.2× SSC at 37°C (10 min each), and incubated in an alkaline phosphatase-conjugated or peroxidase-conjugated anti-digoxigenin antiserum (1:5,000 and 1:10,000, respectively; Roche) for 2 h at room temperature. Then the alkaline phosphatase-labeled sections were washed with 100 mM Tris-HCl (pH 8.0) and 150 mM NaCl and were incubated with BCIP/NBT (Roche) in 100 mM Tris-HCl (pH 9.5), 150 mM NaCl, 1 mM MgCl2 and 10% polyvinyl alcohol for 3 days at room temperature. The sections were dehydrated by treatment with methanol, ethanol, and xylene, and were mounted with Marinol (Muto Pure Chemical). The peroxidase-labeled sections were washed and immersed in 0.1 M boric buffer (pH 8.5) containing 10 μM Alexa568-labeled tyramide, 10% dextran sulfate, 0.05 mg/ml iodophenol and 0.003% H2O2 for 30 min. After washing, the sections were mounted as they were in the in situ HCR experiments.
Histological Analysis
Fluorescent photomicrographs were obtained using a Nikon Eclipse Ni microscope equipped with the A1R confocal detection system under 20×, 40× and 100× objective lenses (Nikon Instruments Inc., Tokyo, Japan). The photomicrographs for the bright field observation were taken by a Nikon AZ-100 microscope equipped with a digital camera (Sony α7s). Images were analyzed using ImageJ software (version 1.50i, NIH, USA). Quantification of the fluorescent photographs was performed at the same threshold and adjustment of contrast.
Results
Reliable HCR Using 42-nt-long DNA Hairpins
To find the shortest sequences of DNA hairpins that reliably proceed with the HCR, we systematically examined hairpin DNAs with different lengths for their HCR efficiency based on the appearance of HCR products larger than 10 kb and the intensity of the unreacted hairpin DNA band. Because the commonly used DNA hairpins have toehold and loop domains of equal length and are 72-nt long containing 12-nt toehold, 24-nt stem and 12-nt loop domains (Choi et al., 2014, 2016, 2018), we first tested 7 pairs of nonlabeled 36-nt hairpin DNAs containing 6-nt toehold, 12-nt stem, and 6-nt loop domains. However, they all showed low HCR efficiency (#S1-S7 in Supplementary Table S1). Even in the presence of a high concentration of an initiator DNA, an HCR that was allowed to occur for 2 h left a majority of hairpin DNAs unreacted (Figure 2A). Thus, 36-nt hairpin DNAs were not suitable for HCR.
Next, we tested non-labeled hairpin DNAs which have 8-nt, 9-nt, or 10-nt long toehold and loop domains and 12-nt stem domains (8-nt: #S8, 9-nt: #S41, and 10-nt: #S9 in Supplementary Table S1). HCR with 8-nt long toehold and loop domains successfully produced ~10 kb products, but unreacted hairpin DNAs remained in all initiator concentrations. HCR with 9-nt and 10-nt long toehold and loop domains produced ~10 kb products, and only a small amount of hairpin DNA was unreacted (Figure 2A). There was no apparent difference in the amount of incorporated hairpin DNA between the 9-nt and 10-nt toehold/loop after a 2-h HCR, indicating that the 9-nt-long toehold and loop domains were sufficient for efficient HCR (Figure 2A, #S4, #S8–9, and #S41 in Supplementary Table S1), which is consistent with our NUPACK simulation (data not shown). Thus, we used 42-nt hairpins containing 9-nt toehold, 12-nt stem, and 9-nt loop domains thereafter.
The Sequence of the Stem and Transition Between the Stem and Loop Affected Hairpin Stability
Since insufficient stability of hairpin DNAs leads to initiator-independent HCR, we optimized the sequence of the stem domains of short hairpin pair based on the abundance of initiator-independent HCR. Through this optimization using short hairpins with different nucleotide contents, we noticed that short hairpin pair with a similar number of G and C on one strand of the stem domain tended to result in frequent initiator-independent HCR (Table 1). In contrast, when the number difference between G and C on one strand of the stem domain is 4 or 5, the incidence of initiator-independent HCR was low (Table 1). The difference between the numbers of G and C on one strand of the stem domain significantly correlated with the initiator-independent HCR as evaluated by band intensity (Spearman’s rank correlation test, rho = −0.40, S = 6941.8, p = 0.026).
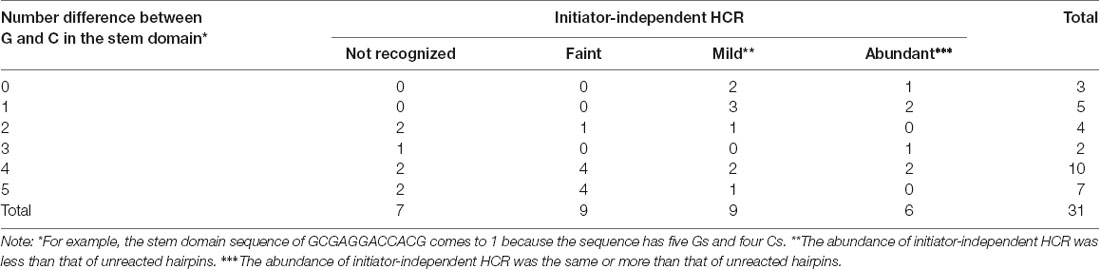
Table 1. Number of short hairpin pairs with different G and C number on one strand of the stem domain and their initiator-independent hybridization chain reaction (HCR).
Also, we noticed that the shift of the transition site between the stem and loop domains by one or two nucleotides sometimes decreased initiator-independent HCR (Figure 1D). For example, the shift by one nucleotide produces a hairpin containing 9-nt toehold, 13-nt stem, and 7-nt loop domains, in which the toehold sequence hybridized with the loop domain and a part of the stem domain. However, the shift of the transition site sometimes led to decreased HCR efficiency due to the higher stability of the hairpin. Thus, the 42-nt DNA hairpins used in this study have some differences in the length of the stem and loop domains. Because of the high HCR efficiency and a small amount of initiator-independent HCR, we used #S41 hairpin DNAs to examine eight fluorophores and two linkers thereafter.
Conjugated Fluorophores Affected HCR Efficiency
We examined which fluorophore to use affects HCR efficiency in microtubes. The conjugation of ATTO390, FAM, ATTO488, Alexa488, ATTO550, and Alexa647 (Figures 2D–K) to 42-nt DNA hairpins did not interfere with HCR, while that of ATTO565 and Alexa568 resulted in decreased HCR efficiency (Figures 2B,C). ATTO565- or Alexa568-conjugated hairpin DNAs produced a small amount of ~10 kb products with a 1:200 ratio of initiator and hairpin DNAs and left a large amount of hairpin DNAs unreacted (Figure 2C). When compared with the C6 linker, the ssH linker showed higher HCR efficiency. C6-linked ATTO550-conjugated hairpin DNA did not form ~10 kb long products with 1/200 amount of initiator DNA (Supplementary Figure S2), indicating that ssH linker is better at least for ATTO550.
In situ HCR Using Short Hairpin DNAs
Next, we performed in situ HCR for mouse brain using the fluorophore-labeled short hairpin DNAs described above and the reagents and buffers with reaction conditions reported in previous studies (Choi et al., 2014, 2018). Consistent with in vitro HCR, 42-nt hairpin DNAs provided stronger in situ HCR signals for Proenkephalin (Penk) mRNA than 36-nt hairpin DNA (Supplementary Figure S3). Thus, 42-nt hairpin DNAs were used for in situ HCR to detect mRNAs in this study. Penk mRNA-positive cells were abundantly detected in the striatum after HCR amplification initiated by five split-initiator probe sets for Penk mRNA (Figure 3). Extending the incubation time with Alexa647-conjugated hairpin pairs (#S41 in Supplementary Table S1) from 45 min to overnight enhanced the fluorescence signal for Penk mRNA. The incubation of split-initiator probes with one of the hairpin pairs (H1) or the incubation of hairpin pairs (H1, H2) without split-initiator probes did not generate any signals, indicating the in situ HCR specific to Penk mRNA (Figure 3). In the absence of initiator probes, background signals slightly increased as the incubation continued.
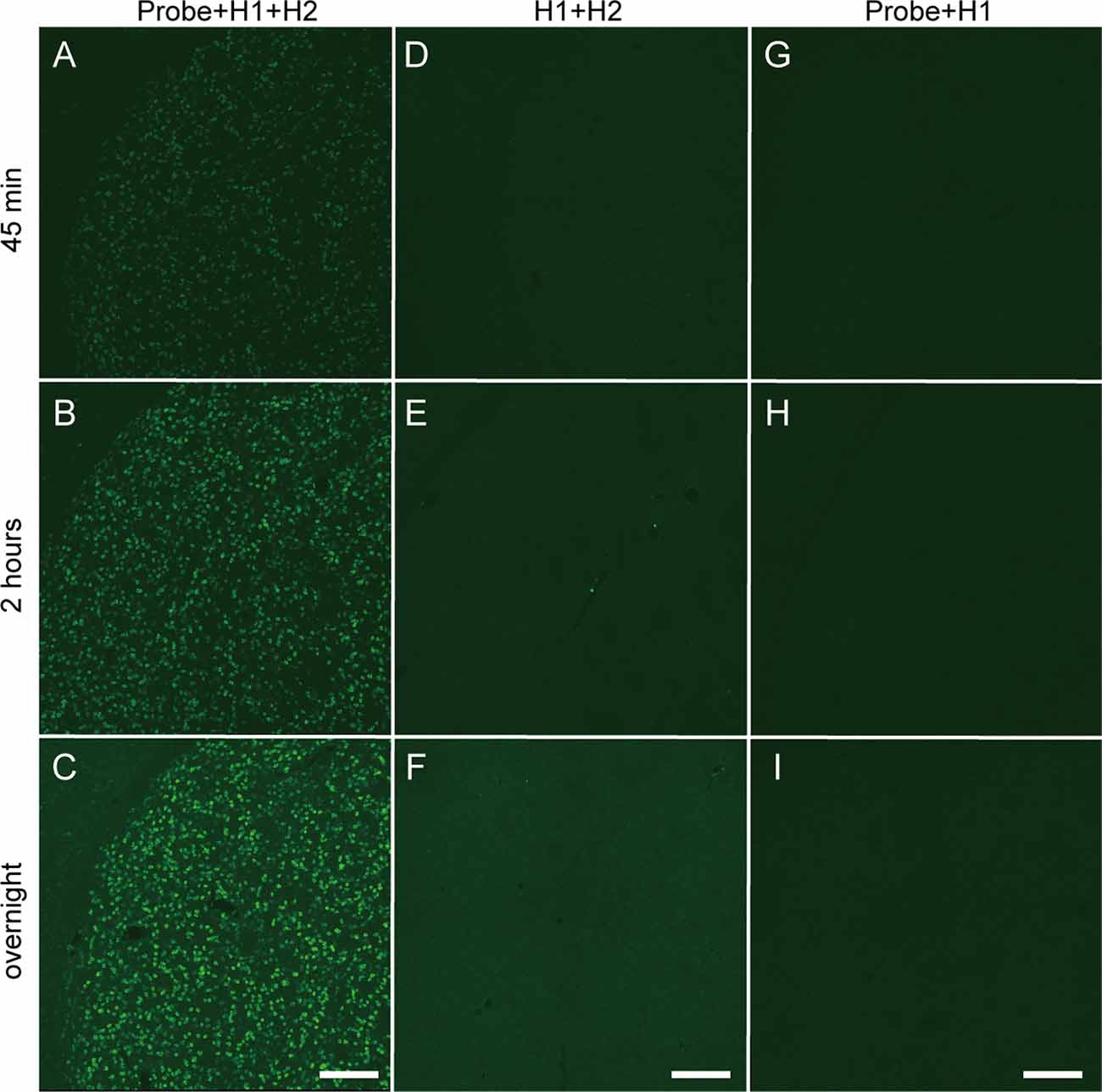
Figure 3. Time-dependent increase in fluorescence of in situ HCR using short hairpin DNAs. Penk mRNA expression in the mouse striatum was detected by Alexa647-conjugated hairpin DNA (#S41) and corresponding probe sets. Background signals were not subtracted. (A–C) Sections were hybridized with split-initiator probes followed by HCR amplification. (D–F) Sections were treated without probes, which was followed by HCR amplification. (G–I) Sections were hybridized with probes followed by only H1 hairpin DNA hybridization. (A,D,G) Incubation for 45 min with hairpin DNAs. (B,E,H) Incubation for 2 h with hairpin DNAs. (C,F,I) Overnight incubation with hairpin DNAs. Scale bars: 200 μm.
Since conjugated fluorophores and linkers affected the HCR efficiency in microtubes, we examined the effect of fluorophores and linkers on in situ HCR for Penk mRNA using the same five probe sets. Although the autofluorescence and detection conditions differed among the color spectra, Penk-positive neurons were observed using hairpins labeled with ATTO390, Alexa488, ATTO488, ATTO550, ATTO565, and Alexa647, but the signal intensity of ATTO390, ATTO488 and ATTO565 was weak (Figure 4). We obtained the best and brightest signals with the conjugation of ATTO550, and Alexa647, which is consistent with HCR efficiencies observed in microtubes (Figures 2B,C).
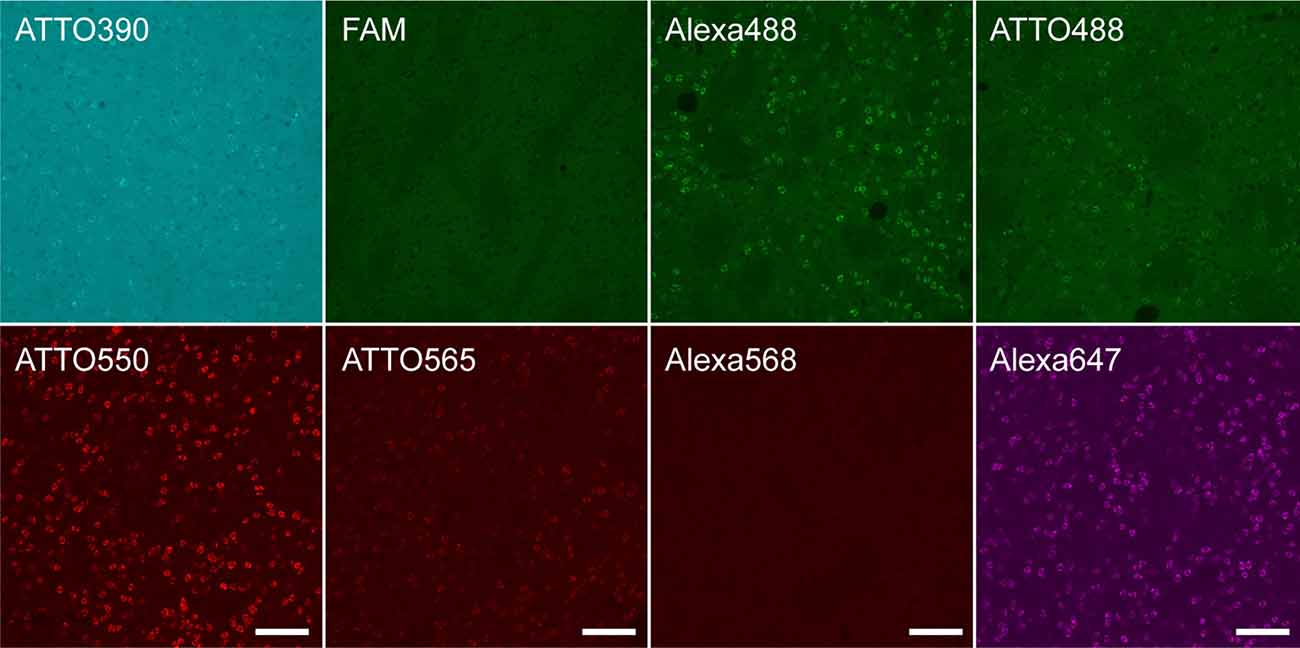
Figure 4. Fluorophores conjugated to hairpin DNAs affect in situ HCR signal intensity. in situ HCR for Penk mRNAs in the mouse striatum using #S41 hairpin DNA. Various fluorophores were conjugated to the hairpin DNAs. All tissue sections were prepared from the same mouse. Background signals were not subtracted. Scale bars: 100 μm.
Consistent with in vitro HCR (Supplementary Figure S2), ssH-linked ATTO550 produced higher signal intensity for Penk-positive neurons than C6-linked ATTO550 (Supplementary Figure S4). The use of the ssH linker for Alexa488 or Alexa647 resulted in slightly stronger signals for Penk mRNA than that of the C6 linker (Supplementary Figure S4). PAGE purification of split-initiator probes for Penk mRNA increased fluorescent intensity compared with that of unpurified probes (data not shown).
We compared the sensitivity of in situ HCR using short hairpins with other ISH methods by detecting Oxtr mRNA, which we selected as an example of very low abundance mRNAs. After hybridization of 10 split-initiator probe sets for Oxtr mRNA, in situ HCR detected Oxtr mRNA signals in the rhomboid nucleus of the bed nucleus of the stria terminalis (BNST) and the magnocellular nucleus (MN) and weak signals in the principal nucleus of the BNST and the medial preoptic area (MPOA; Figures 5A,D; Young et al., 1997; Okabe et al., 2017). ISH with tyramide signal amplification failed to visualize Oxtr-positive cells in the BNST (Figures 5B,E). ISH using BCIP/NBT as chromogens demonstrated the distribution of Oxtr mRNA-positive cells consistent with that of in situ HCR, but there were much weaker and imprecise signals (Figures 5C,F,I). Almost all the HCR signals were observed near cell nuclei (Figure 5G). ISH with tyramide signal amplification produced a low signal-to-noise ratio results in cells at the BNST (Figure 5H).
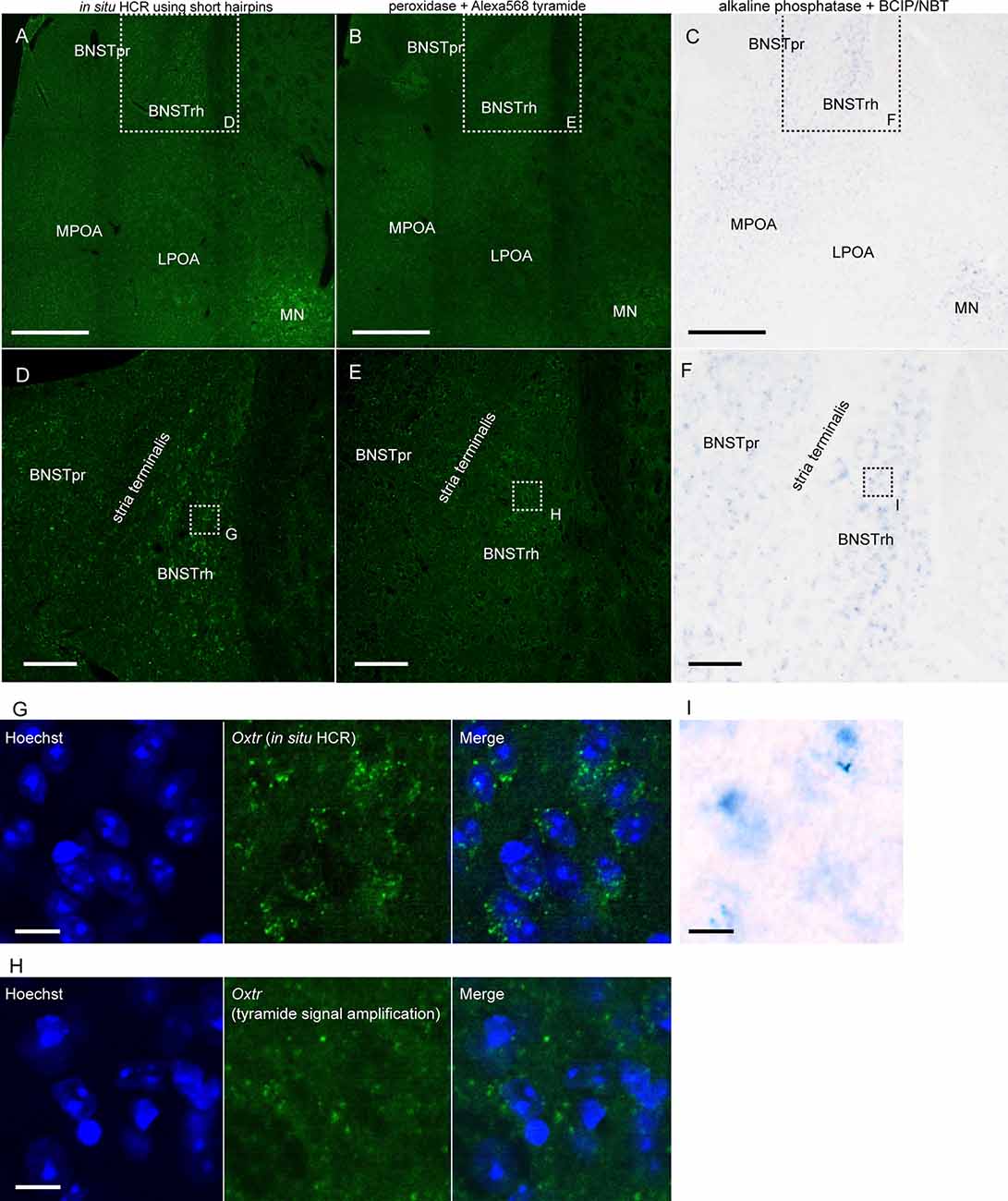
Figure 5. Detection of low abundance mRNA using in situ HCR. (A,D) In situ HCR using ATTO550-conjugated hairpin DNA (#S23) detected Oxtr mRNA in mouse brain. Amplification was performed overnight. (B,E) Fluorescent ISH with tyramide signal amplification for Oxtr mRNA. Oxtr mRNA was visualized by a combination of DIG-labeled probe, peroxidase-conjugated anti-DIG antibody, and tyramide Alexa568. (C,F) Chromogenic ISH for Oxtr mRNA by a combination of DIG-labeled probe, alkaline phosphatase-conjugated anti-DIG antibody, and BCIP/NBT. Color development was performed for 3 days. Panels (D–F) corresponds to dashed squares in panels (A–C), respectively. Panels (G–I) corresponds to dashed squares in (D–F). Scale bars: 400 μm (A–C), 100 μm (D–F) and 10 μm (G–I). BNST, bed nucleus of stria terminalis; BNSTpr, principal nucleus of BNST; BNSTrh, rhomboid nucleus of BNST; LPOA, lateral preoptic area; MN, magnocellular nucleus; MPOA, medial preoptic area.
Proteinase K treatment has been used for ISH and in situ HCR to improve the penetration of RNA/DNA probes and fluorophore-labeled hairpins by digesting proteins. However, proteinase K treatment damages endogenous proteins such as c-Fos, which is a commonly used marker of neural activity, which limits the usefulness of ISH combined with immunohistochemistry. Since our modified in situ HCR uses shorter probes than what has been previously used, proteinase K treatment may not be necessary. As predicted, in situ HCR using 42-nt hairpins and five sets of 39- and 36-nt split-initiator probes without proteinase K treatment detected vesicular glutamate transporter (Vglut) 2 in the MPOA (Figure 6C) and the paraventricular thalamus (PVT; Figure 6D) with the same or slightly decreased signal levels as tissues that did undergo proteinase K treatment.
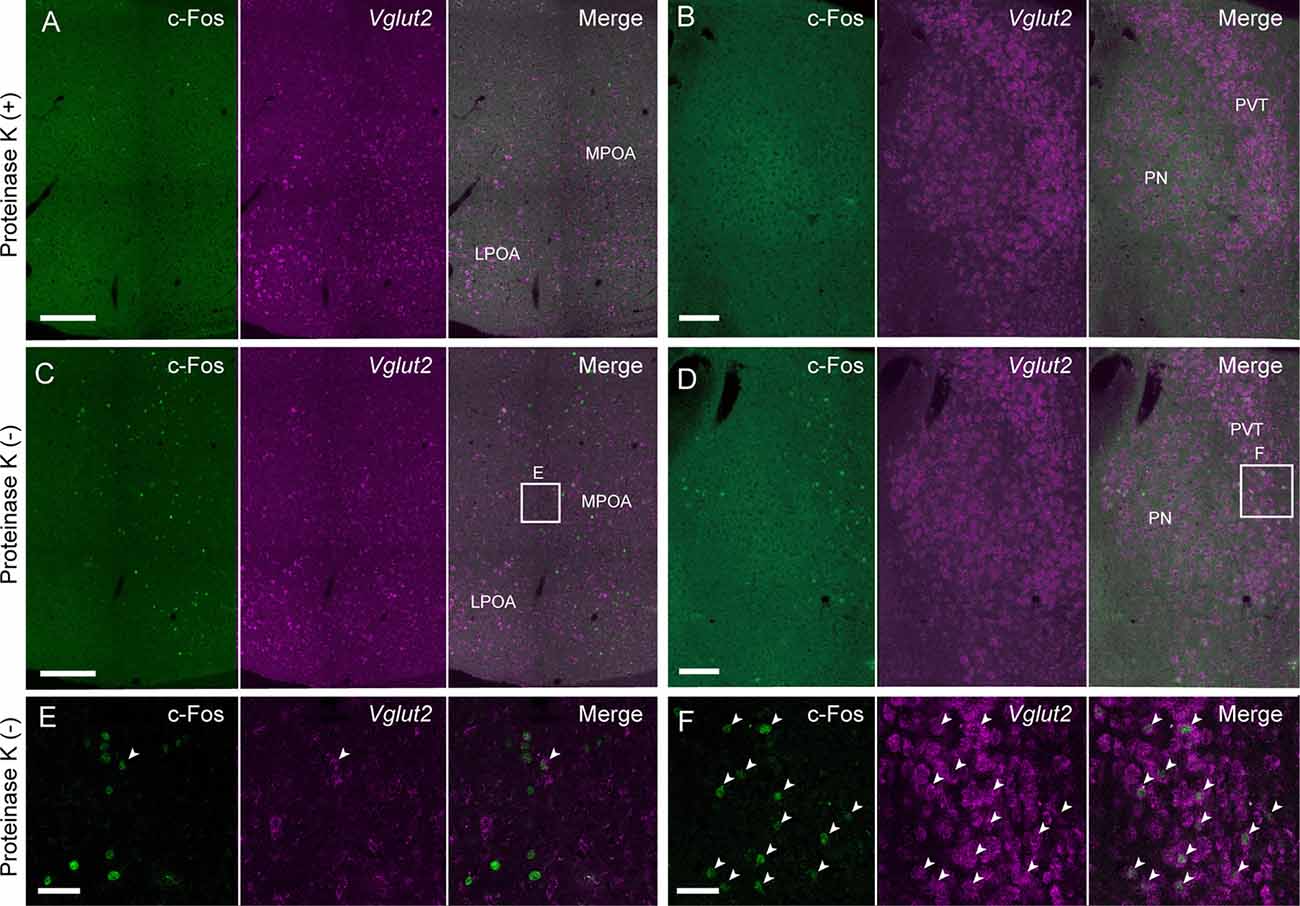
Figure 6. Modified in situ HCR without proteinase K treatment. Vglut2 mRNAs were detected in the mouse medial preoptic area (A,C,E) and PVT (B,D,F) using Alexa647-conjugated hairpin DNA (#S41). Amplification was performed for 2 h. (A,B) Sections were pretreated with proteinase K. (C,D) Sections were not treated with proteinase K. Maternal behavior-induced c-Fos immunoreactivity that was diminished by proteinase K treatment before hybridization. Panels (E,F) correspond to dashed areas in panels (C,D). Arrowheads indicate double-positive cells of c-Fos protein and Vglut2 mRNA. LPOA, lateral preoptic area; MPOA, medial preoptic area; PN, parataenial nucleus; PVT, paraventricular thalamus. Scale bars: 200 μm (A,C), 100 μm (B,D) and 40 μm (E,F).
Female mice induced c-Fos expression in the MPOA after the retrieval of pups (Tsuneoka et al., 2013, 2017b; Moffitt et al., 2018). In situ HCR without proteinase K treatment successfully detected abundant c-Fos-positive cells in the MPOA and PVT (Figures 6C,D), whereas the number of c-Fos-positive cells and the intensity of c-Fos immunoreactivity were drastically decreased after proteinase K treatment (Figures 6A,B). Vglut2-positive neurons were scattered in the MPOA, but most of them were not c-Fos positive (Figures 6E,F).
Multiplex in situ HCR
To test whether the hairpin DNAs separately detect different mRNAs in a single tissue section, we performed multiplexed in situ HCR. After 2 h of HCR amplification, dopamine receptor d1 (Drd1), Drd2 and Penk mRNAs were simultaneously detected in the mouse striatum using the corresponding probes (five probe sets for each mRNA) and hairpin DNA pairs conjugated to different fluorophores (Figure 7). Although Drd1 and Drd2 mRNA were abundantly expressed in the striatum, double-positive cells for Drd1 and Drd2 mRNAs were rarely observed (Figure 7). In contrast, most of the Drd2-positive cells were also Penk-positive. When observed at higher magnification, the subcellular distribution and abundance of fluorescence for Drd2 mRNA were distinct from those for Penk mRNA.
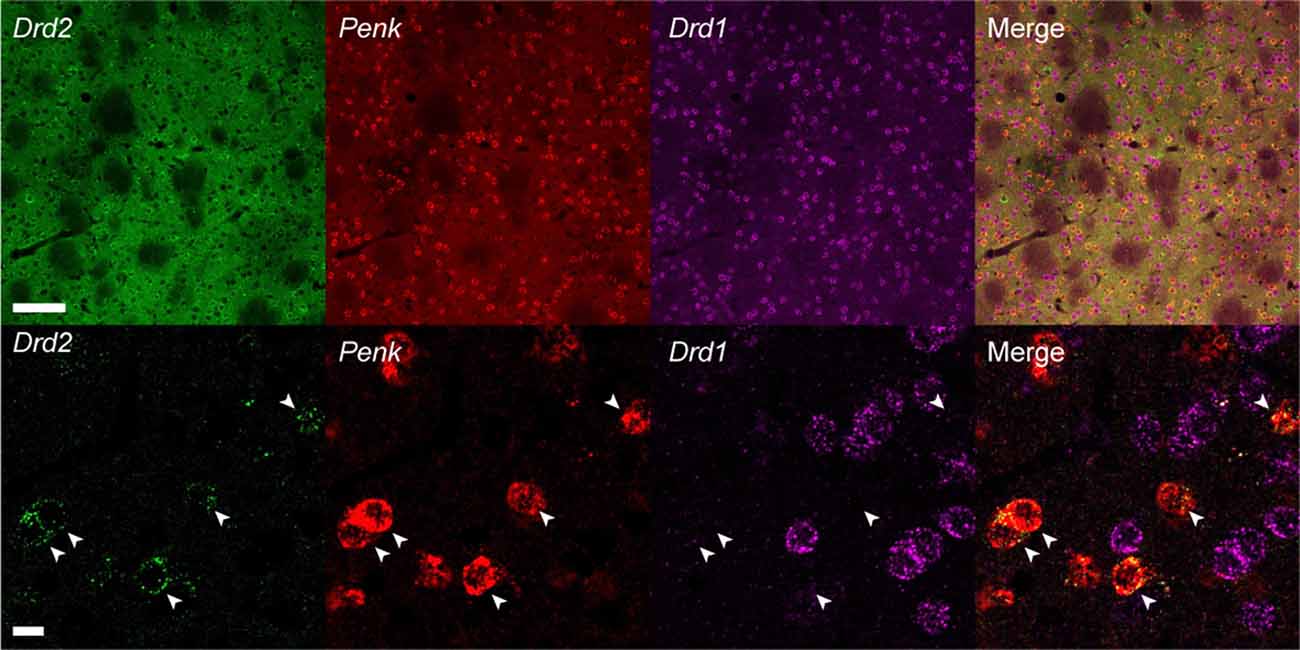
Figure 7. Multiplex in situ HCR in the mouse striatum. Drd2, Penk and Drd1 mRNAs were detected by 2-h HCR that simultaneously used different probe-hairpin pairs: Alexa488-conjugated hairpin DNA (#S23) for Drd2, ATTO565-conjugated hairpin DNA (#S10) for Penk, and Alexa647-conjugated hairpin DNA (#S25) for Drd1. In the upper panels, background signals were not subtracted. Drd2-positive cells were also Penk-positive (arrowheads). Scale bars: 100 μm (Upper panel) and 10 μm (Lower panel).
We next examined the sensitivity and resolution of modified in situ HCR using two different probe sets for Moxd1 mRNA (Supplementary Table S2), which is a marker of sexually dimorphic nuclei (Tsuneoka et al., 2017a). One of them was five split-initiator probes containing #S41 initiator sequence and detected by ATTO550-conjugated hairpin pairs (#S41) and another was five split-initiator probes containing #S25 initiator sequence and detected by Alexa647-conjugated hairpin pairs (#S25). Both probe-hairpin combinations successfully detected Moxd1-positive cells in the BNST by 45-min HCR amplification (Figure 8A), which is consistent with the previous report (Tsuneoka et al., 2017a). At the highest magnification, almost all the ATTO550 and Alexa647 signals were observed as granules that colocalized within the cell (Figure 8B). Whereas 76.9% of ATTO550-positive granules were Alexa647-positive, 74.8% of Alexa647-positive granules were ATTO550-positive (247 and 254 granules in the total count, respectively).
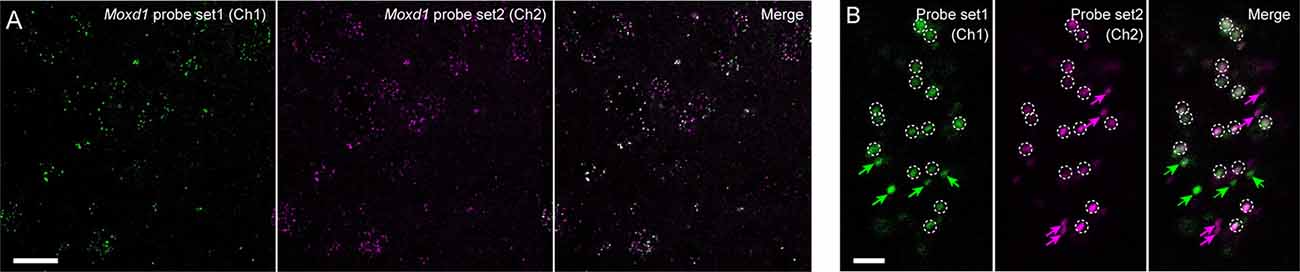
Figure 8. Redundant detection of Moxd1 mRNA using different probes. (A) In situ HCR using different probe-hairpin sets designed for different regions of the Moxd1 mRNAs. HCR for 45 min detected Moxd1 mRNAs in the bed nucleus of the stria terminalis. Five probe pairs were used for each channel. Fluorescent signals in Ch1 and Ch2 were detected using ATTO550-conjugated hairpin DNA (#S41) and Alexa647-conjugated hairpin DNA (#S25), respectively. (B) Representative photomicrograph showing subcellular resolution using the same probe-hairpin sets as in panel (A). Dotted circles indicate granules detected in both channels, and arrows indicate granules detected in one of the two channels. Scale bars: 20 μm (A) and 2 μm (B).
Discussion
In this study, we designed new short hairpin DNAs and corresponding split initiator probe sets to achieve sensitive detection of various mRNAs. This study proposed short hairpin design rules as well as linker types and fluorophores suitable for in situ HCR. These hairpin sets successfully performed HCR both in vitro and in situ and detected multiple mRNAs simultaneously with virtually the same procedures for a single mRNA. Short probes and hairpin DNAs can penetrate tissues without proteinase K treatment as well as they can tissues with proteinase K treatment. Also, our modified in situ HCR technique provided highly sensitive mRNA detection for the visualization of less abundant mRNAs such as Oxtr.
Design of Short Hairpin DNA
To achieve efficient and optimal HCR, the lengths of the total hairpin and each domain are crucial. HCR progresses using the difference in free energy between a long double-strand formed by hairpin pairs and a short double strand of the stem domain of each monomer hairpin. Therefore, longer toehold/loop domains enhance HCR. The HCR efficiency was increased by the extension of the toehold sequence and was independent of the stem sequences shown in Figure 2A. The difference in free energy also depended on the GC contents in the toehold/loop domain. However, increased GC content in the toehold/loop and longer toehold/loop may lead to initiator-independent HCR (Ang and Yung, 2016).
The stability of each hairpin largely depends on the stem length and its sequence. In general, increased stem length and GC content enhance hairpin stability due to an increased number of hydrogen bonds. In this respect, we extended the stem length from 12-nt to 13- or 14-nt by shifting the transition between the stem and loop by one or two nucleotides [e.g., S23-H1: toehold (9 nt) -stem (12 nt)—A—loop (7 nt)—T—stem (12-nt); A and T are transition bases], which often decreased initiator-independent HCR but did not exhibit a decrease in HCR efficiency. Many designed hairpins showed a certain level of initiator-independent HCR, polymerization without an initiator. Many factors cause initiator-independent HCR (Chen et al., 2013). Practically, the purification of synthesized oligos is effective for reducing initiator-independent HCR, although the HPLC purification of DNA hairpins sometimes led to a low HCR efficiency compared with that of PAGE purification (data not shown). We also found that a biased abundance of G or C on one side of the stem domain decreased the stability of the hairpin monomer (Table 1). Although further investigation of the hairpin stability is needed, these data provide useful information regarding the design of new hairpin-based nonenzymatic circuits.
We also found that fluorophores conjugated with hairpin DNAs affect HCR, and the linker length between hairpin DNAs and fluorophores and the structure of fluorophores may be involved in this effect. When a C6-amino linker (6 carbon atom-length) was used to link hairpin DNAs with ATTO550 succinimidyl ester, HCR did not form long products (Supplementary Figure S2). In contrast, the ssH-amino linker (11 atom-length) enabled the HCR using the same hairpin DNA conjugated with ATTO550 (Figure 2 and Supplementary Figure S2), indicating that the use of a longer linker enhances the HCR. Similarly, the signal intensity of ATTO550-labeled hairpin DNAs via the ssH linker was brighter than via the C6 linker in situ (Supplementary Figure S4). Also, the ssH-amino linker achieves a high conjugation efficiency with succinimidyl esters and easy purification (Komatsu et al., 2008). We found efficient HCR amplification with fluorophore-labeled hairpin DNAs conjugated through an ssH-amino linker.
The presence of a linker-like linear structure in fluorophores is an additional factor to be considered for higher signal gain. Among the fluorophores examined, Alexa647, ATTO550, and ATTO488 have a linker-like linear structure (Figure 2). Hairpin DNAs conjugated with Alexa647 using the C6-amino linker led to over 10 kb HCR products (Supplementary Figure S2). Many fluorophores, such as the Alexa and ATTO series, have a comparable molecular structure and steric size to a single nucleotide molecule. Therefore, the use of a long linker and fluorophore with a linker-like linear structure is preferred because it is unlikely to cause steric hindrance against forming double-strands between hairpin DNA pairs. We found efficient HCR amplification with fluorophore-labeled hairpin DNAs conjugated through an ssH-amino linker, except ATTO565 and Alexa568 (Figure 2B), which have a very large molecular structure without any linker-like structure, although Alexa568 is widely used bright fluorophore in the immunohistochemistry and ISH. Such steric hindrance was further suggested in performing in situ HCR (Figure 4). The molecular structures of ATTO550 and Alexa647 succinimidyl esters are also very large, but they have a linker-like structure near the binding site, enabling efficient HCR amplification.
In situ HCR Using Short Hairpin DNA
The signal gain of HCR amplification by short hairpin DNAs was also confirmed in the mouse brain. In situ HCR showed that Penk mRNA was uniformly expressed in the mouse striatum (Figure 3), which was consistent with the results from previous ISH studies (Turchan et al., 1997; Lobo et al., 2006; Labouesse et al., 2018). The signal for Penk developed intensely after an overnight incubation as HCR amplification proceeded, although the overnight incubation also resulted in slight background staining. Such Penk-positive cells were never observed in the reaction buffer without H2 hairpin DNA, suggesting that simple hybridization of H1 with a probe for Penk mRNA was not enough to visualize Penk mRNA-positive cells.
Although modification and adoption of the short hairpin in situ HCR was reported to detect very abundant neuropeptide mRNAs (Sui et al., 2016), the amplification reached a plateau within an hour and the polymerized products were much smaller (approximately 1 kb) than those observed in the current study.
The gel electrophoresis of our short hairpin DNAs showed at least 400-fold amplification to develop more than 10 kb double-strand products from 42 nt single-strand hairpin DNAs (Figure 2). Compared with 42-nt hairpin DNAs, 36-nt hairpin DNAs exhibited lower HCR efficiency in vitro (Figure 2A) and in situ (Supplementary Figure S3), which may be due to a short toehold/loop length that renders hairpin monomer stable.
Such strong amplification by HCR using our short hairpin DNAs enabled us to visualize low abundance mRNAs such as Oxtr mRNA. Although G-protein-coupled receptors are often difficult to visualize by enzyme-based fluorescent ISH because of their low abundance, in situ HCR successfully detected weak Oxtr mRNA expression in the MPOA and BNST, consistent with previous studies (Young et al., 1997; Okabe et al., 2017) and Allen brain atlas3. Oxtr mRNA was detected using chromogenic ISH (CISH) using alkaline phosphatase and BCIP/NBT after a long chromogenic reaction which is thought to be most sensitive among enzyme-based conventional ISH (Bonn et al., 2012). We also confirmed that the sensitivity of CISH was superior to that of ISH with tyramide signal amplification for low abundance mRNA. Importantly, the distribution of Oxtr mRNA detected by in situ HCR was largely matched with the result of CISH, suggesting that the sensitivity of in situ HCR was comparable to that of CISH. Furthermore, in situ HCR has several advantages over CISH such as simultaneous multiplex imaging and an elevated ability for the probes to penetrate.
Probe Design
In this study, we detected six genes using five or ten sets of split probes per gene. In the original report of split probes (Choi et al., 2018), the authors recommended more than 20 sets of probes to increase signal/noise ratio and precision, because the number of probes theoretically correlates signal intensity. As shown in Figure 8, five sets of probes can visualize a single mRNA, although the imaging of a single-molecule requires a high N.A. lens (e.g., >1.3). Ten sets of probes allowed us to visualize low abundance mRNA such as Oxtr at a relatively low magnification using a 20× objective lens with 0.75 N.A. (Figure 5). Additional probe sets may increase signal/noise ratio and precision, while it costs more. The minimum number of probes for appropriate imaging needs to be determined by users based on many factors including the imaging environment, the intensity of autofluorescence, conjugated fluorophore, required resolution, and precision.
We designed split probes according to Choi et al. (2018). The sequence for probes were chosen in consideration of GC contents and homology without further optimization and evaluation, because the split probes were very robust for non-specific hybridization (Choi et al., 2018). Gene expressions shown in this study were consistent with previous studies. Therefore, the design of new probes for target genes is as easy and fast as probes for conventional ISH.
Probe penetration is a key factor for the sensitivity of ISH. To enhance penetration, proteinase K treatment has been widely used in ISH in addition to the fragmentation of long probes, although some endogenous proteins are degraded by proteinase K treatment. Some reports proposed organic solvents as an alternative to proteinase K treatment in the Drosophila embryo (Nagaso et al., 2001; Jaeger et al., 2004), but these techniques were not as effective as the proteinase K treatment in mouse neural tissue (data not shown). Therefore, in the case of ISH with immunohistochemistry, there was a trade-off in the use of proteinase K between better ISH probe penetration and better preservation of antigens, such as c-Fos, for immunohistochemistry of mouse neural tissue. There was a report on the combined use of in situ HCR with immunohistochemistry to detect both mRNA and proteins (Zhuang et al., 2020). However, the authors adopted proteinase K treatment, which requires additional optimization of immunohistochemistry such as adjusting the concentration of antibodies. In situ HCR using short hairpins overcomes these issues. In situ HCR without proteinase K treatment showed Vglut2 mRNA signals comparable to those observed following proteinase K treatment (Figure 6).
Multiplex in situ HCR
Using three hairpin DNA sets, in situ HCR demonstrated that Penk-positive cells in the mouse striatum were also Drd2-positive, but Drd1-negative (Figure 7). Almost all striatal neurons in mice express either Drd1 or Drd2 mRNA, and Drd2-positive neurons express Penk mRNA, as demonstrated by ISH, cell-assembly microarray using transgenic reporter mice and single-cell RNA-seq (Lobo et al., 2006; Heiman et al., 2008; Gokce et al., 2016; Labouesse et al., 2018). In situ HCR in this study exactly reproduced cell data revealed by the previous studies. Thus, multiplex in situ HCR works to visualize different sets of mRNAs simultaneously with a high signal-to-noise ratio. Tyramide signal amplification has been a popular approach for performing multiplexed fluorescent ISH (Zaidi et al., 2000; Lauter et al., 2011a,b; Bonn et al., 2012; Tsuneoka et al., 2015, 2017b). It has been used with different hapten-labeled probes, peroxidase-conjugated anti-hapten antibodies, and fluorophore-conjugated tyramides. ISH using tyramide signal amplification requires optimization for each step, requiring substantial labor from researchers. Also, antibody reactions and subsequent amplification steps should be performed separately for each target mRNA after the deactivation of the enzyme. In contrast, the in situ HCR system can simultaneously develop multiple colors and show autonomous suppression of noise without further optimization per target mRNA (Choi et al., 2014, 2018). Such advantages were also observed with the short hairpin DNAs designed in this study. Moreover, the current modification for the short hairpin DNA design implies further signal amplification by modifying the hairpin sequence, such as is used in the branched HCR system (Xu and Zheng, 2016; Bi et al., 2017; Liu et al., 2018; Wu et al., 2019).
Single-molecule fluorescent ISH has become a powerful technique not only for analyzing subcellular localization of specific mRNAs (Chen et al., 2016; Samacoits et al., 2018) but also for demonstrating the presence of low abundance mutant mRNAs (Haimovich and Gerst, 2018; Marras et al., 2019), long noncoding RNAs (Chen et al., 2016), ribosome-mRNA interactions (Burke et al., 2017) and comprehensive transcriptional analyses in situ (Shah et al., 2016; Moffitt et al., 2018). The signals from two different probes designed for detection of Moxd1 mRNA were largely matched to each other at subcellular resolution (Figure 8). This suggests that in situ HCR using short hairpin DNAs is also applicable for single-molecule fluorescent ISH with a specific imaging device, similar to the original hairpin in situ HCR methods (Choi et al., 2014, 2018). Because, we used a small number of probe sets (five probe sets) and the thresholds were determined to minimize false-positive signals derived from tissue autofluorescence, not all the signal from one channel coincide with those from another channel. Increasing the number of probe sets will gain a higher signal/noise ratio and precision.
Summary and Advantage of Short Hairpin DNA
In summary, our short hairpin in situ HCR enables the visualization of low abundance mRNA and multiple mRNAs and the simultaneous detection of mRNA and protein without proteinase K treatment. When designing short hairpins, the following points should be taken into account; length of toehold/loop, end bases of toehold/loop, the number of G and C on one strand of the stem domain, structures of fluorophores and linker length. The advantages of short hairpin DNAs are low cost and high permeability without proteinase K treatment. Given that the length of DNA decreases from 72 nt to 42 nt, the cost for oligo DNA synthesis per mole decreases by 66% (1–422/722). Also, as the oligos get longer, mis-synthesized oligos increase exponentially, which further makes short hairpins cost-effective.
Data Availability Statement
All data are available from the corresponding authors upon reasonable request.
Ethics Statement
The animal study was reviewed and approved by Institutional Animal Care and Use Committee of Toho University.
Author Contributions
YT: conceptualization, methodology and formal analysis. YT and HF: writing, funding acquisition and supervision.
Funding
This work was supported by Takeda Science Foundation (to YT), Grant for Basic Science Research Projects from the Sumitomo Foundation (to YT), Uehara Memorial Foundation (to HF) and Japan Society for the Promotion of Science (JSPS) Kakenhi (18K06509 to YT and 17H05583 to HF).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to Hiroko Arai, Akane Iijima, Naomi Ohno, Moana Ohnishi and Haruka Arai for excellent technical assistance for this study, and Sachine Yoshida and Mayuko Oka for helpful discussions.
Footnotes
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnmol.2020.00075/full#supplementary-material.
References
Ang, Y. S., and Yung, L. L. (2016). Rational design of hybridization chain reaction monomers for robust signal amplification. Chem. Commun. 52, 4219–4222. doi: 10.1039/c5cc08907g
Bi, S., Yue, S., and Zhang, S. (2017). Hybridization chain reaction: a versatile molecular tool for biosensing, bioimaging, and biomedicine. Chem. Soc. Rev. 46, 4281–4298. doi: 10.1039/c7cs00055c
Bonn, M., Schmitt, A., and Asan, E. (2012). Double and triple in situ hybridization for coexpression studies: combined fluorescent and chromogenic detection of neuropeptide Y (NPY) and serotonin receptor subtype mRNAs expressed at different abundance levels. Histochem. Cell Biol. 137, 11–24. doi: 10.1007/s00418-011-0882-3
Burke, K. S., Antilla, K. A., and Tirrell, D. A. (2017). A fluorescence in situ hybridization method to quantify mRNA translation by visualizing ribosome-mRNA interactions in single cells. ACS Cent. Sci. 3, 425–433. doi: 10.1021/acscentsci.7b00048
Chen, X., Briggs, N., McLain, J. R., and Ellington, A. D. (2013). Stacking nonenzymatic circuits for high signal gain. Proc. Natl. Acad. Sci. U S A 110, 5386–5391. doi: 10.1073/pnas.1222807110
Chen, F., Wassie, A. T., Cote, A. J., Sinha, A., Alon, S., Asano, S., et al. (2016). Nanoscale imaging of RNA with expansion microscopy. Nat. Methods 13, 679–684. doi: 10.1038/nmeth.3899
Choi, H. M. T., Beck, V. A., and Pierce, N. A. (2014). Next-generation in situ hybridization chain reaction: higher gain, lower cost, greater durability. ACS Nano 8, 4284–4294. doi: 10.1021/nn405717p
Choi, H. M. T., Calvert, C. R., Husain, N., Huss, D., Barsi, J. C., Deverman, B. E., et al. (2016). Mapping a multiplexed zoo of mRNA expression. Development 143, 3632–3637. doi: 10.1242/dev.140137
Choi, H. M. T., Chang, J. Y., Trinh, L. A., Padilla, J. E., Fraser, S. E., and Pierce, N. A. (2010). Programmable in situ amplification for multiplexed imaging of mRNA expression. Nat. Biotechnol. 28, 1208–1212. doi: 10.1038/nbt.1692
Choi, H. M. T., Schwarzkopf, M., Fornace, M. E., Acharya, A., Artavanis, G., Stegmaier, J., et al. (2018). Third-generation in situ hybridization chain reaction: multiplexed, quantitative, sensitive, versatile, robust. Development 145:dev165753. doi: 10.1242/dev.165753
Dirks, R. M., and Pierce, N. A. (2004). Triggered amplification by hybridization chain reaction. Proc. Natl. Acad. Sci. U S A 101, 15275–15278. doi: 10.1073/pnas.0407024101
Funato, H., Saito-Nakazato, Y., and Takahashi, H. (2000). Axonal growth from the habenular nucleus along the neuromere boundary region of the diencephalon is regulated by semaphorin 3F and netrin-1. Mol. Cell. Neurosci. 16, 206–220. doi: 10.1006/mcne.2000.0870
Gokce, O., Stanley, G. M., Treutlein, B., Neff, N. F., Camp, J. G., Malenka, R. C., et al. (2016). Cellular taxonomy of the mouse striatum as revealed by single-Cell RNA-seq. Cell Rep. 16, 1126–1137. doi: 10.1016/j.celrep.2016.06.059
Haimovich, G., and Gerst, J. E. (2018). Single-molecule fluorescence in situ hybridization (smFISH) for RNA detection in adherent animal cells. Bioprotocol 8:21. doi: 10.21769/bioprotoc.3070
Heiman, M., Schaefer, A., Gong, S., Peterson, J. D., Day, M., Ramsey, K. E., et al. (2008). A translational profiling approach for the molecular characterization of CNS cell types. Cell 135, 738–748. doi: 10.1016/j.cell.2008.10.028
Jaeger, J., Surkova, S., Blagov, M., Janssens, H., Kosman, D., Kozlov, K. N., et al. (2004). Dynamic control of positional information in the early Drosophila embryo. Nature 430, 368–371. doi: 10.1038/nature02678
Jensen, E. (2014). Technical review: in situ hybridization. Anat. Rec. 297, 1349–1353. doi: 10.1002/ar.22944
Komatsu, Y., Kojima, N., Sugino, M., Mikami, A., Nonaka, K., Fujinawa, Y., et al. (2008). Novel amino linkers enabling efficient labeling and convenient purification of amino-modified oligonucleotides. Bioorg. Med. Chem. 16, 941–949. doi: 10.1016/j.bmc.2007.10.011
Krumlauf, R., Holland, P. W. H., McVey, J. H., and Hogan, B. L. M. (1987). Developmental and spatial patterns of expression of the mouse homeobox gene, Hox 2.1. Development 99, 603–617.
Labouesse, M. A., Sartori, A. M., Weinmann, O., Simpson, E. H., Kellendonk, C., and Weber-Stadlbauer, U. (2018). Striatal dopamine 2 receptor upregulation during development predisposes to diet-induced obesity by reducing energy output in mice. Proc. Natl. Acad. Sci. U S A 115, 10493–10498. doi: 10.1073/pnas.1800171115
Larsson, C., Grundberg, I., Söderberg, O., and Nilsson, M. (2010). in situ detection and genotyping of individual mRNA molecules. Nat. Methods 7, 395–397. doi: 10.1038/nmeth.1448
Lauter, G., Soll, I., and Hauptmann, G. (2011a). Multicolor fluorescent in situ hybridization to define abutting and overlapping gene expression in the embryonic zebrafish brain. Neural Dev. 6:10. doi: 10.1186/1749-8104-6-10
Lauter, G., Söll, I., and Hauptmann, G. (2011b). Two-color fluorescent in situ hybridization in the embryonic zebrafish brain using differential detection systems. BMC Dev. Biol. 11:43. doi: 10.1186/1471-213x-11-43
Liu, L., Liu, J.-W., Wu, H., Wang, X.-N., Yu, R.-Q., and Jiang, J.-H. (2018). Branched hybridization chain reaction circuit for ultrasensitive localizable imaging of mRNA in living cells. Anal. Chem. 90, 1502–1505. doi: 10.1021/acs.analchem.7b04848
Lobo, M. K., Karsten, S. L., Gray, M., Geschwind, D. H., and Yang, X. W. (2006). FACS-array profiling of striatal projection neuron subtypes in juvenile and adult mouse brains. Nat. Neurosci. 9, 443–452. doi: 10.1038/nn1654
Marcus, J. N., Aschkenasi, C. J., Lee, C. E., Chemelli, R. M., Saper, C. B., Yanagisawa, M., et al. (2001). Differential expression of orexin receptors 1 and 2 in the rat brain. J. Comp. Neurol. 435, 6–25. doi: 10.1002/cne.1190
Marras, S. A. E., Bushkin, Y., and Tyagi, S. (2019). High-fidelity amplified FISH for the detection and allelic discrimination of single mRNA molecules. Proc. Natl. Acad. Sci. U S A 116, 13921–13926. doi: 10.1073/pnas.1814463116
Moffitt, J. R., Bambah-Mukku, D., Eichhorn, S. W., Vaughn, E., Shekhar, K., Perez, J. D., et al. (2018). Molecular, spatial, and functional single-cell profiling of the hypothalamic preoptic region. Science 5324:eaau5324. doi: 10.1126/science.aau5324
Moorman, A. F. M., Houweling, A. C., de Boer, P. A. J., and Christoffels, V. M. (2001). Sensitive nonradioactive detection of mRNA in tissue sections: novel application of the whole-mount in situ hybridization protocol. J. Histochem. Cytochem. 49, 1–8. doi: 10.1177/002215540104900101
Nagaso, H., Murata, T., Day, N., and Yokoyama, K. K. (2001). Simultaneous detection of RNA and protein by in situ hybridization and immunological staining. J. Histochem. Cytochem. 49, 1177–1182. doi: 10.1177/002215540104900911
Okabe, S., Tsuneoka, Y., Takahashi, A., Ooyama, R., Watarai, A., Maeda, S., et al. (2017). Pup exposure facilitates retrieving behavior via the oxytocin neural system in female mice. Psychoneuroendocrinology 79, 20–30. doi: 10.1016/j.psyneuen.2017.01.036
Samacoits, A., Chouaib, R., Safieddine, A., Traboulsi, A.-M., Ouyang, W., Zimmer, C., et al. (2018). A computational framework to study sub-cellular RNA localization. Nat. Commun. 9:4584. doi: 10.1038/s41467-018-06868-w
Shah, S., Lubeck, E., Schwarzkopf, M., He, T. F., Greenbaum, A., Sohn, C. H., et al. (2016). Single-molecule RNA detection at depth by hybridization chain reaction and tissue hydrogel embedding and clearing. Development 143, 2862–2867. doi: 10.1242/dev.138560
Sui, Q.-Q., Zhu, J., Li, X., Knight, G. E., He, C., Burnstock, G., et al. (2016). A modified protocol for the detection of three different mRNAs with a new-generation in situ hybridization chain reaction on frozen sections. J. Mol. Histol. 47, 511–529. doi: 10.1007/s10735-016-9696-x
Tsuneoka, Y., Maruyama, T., Yoshida, S., Nishimori, K., Kato, T., Kuroda, K. O., et al. (2013). Functional, anatomical, and neurochemical differentiation of medial preoptic area subregions in relation to maternal behavior in the mouse. J. Comp. Neurol. 521, 1633–1663. doi: 10.1002/cne.23251
Tsuneoka, Y., Tokita, K., Yoshihara, C., Amano, T., Esposito, G., Huang, A. J., et al. (2015). Distinct preoptic-BST nuclei dissociate paternal and infanticidal behavior in mice. EMBO J. 34, 2652–2670. doi: 10.15252/embj.201591942
Tsuneoka, Y., Tsukahara, S., Yoshida, S., Takase, K., Oda, S., Kuroda, M., et al. (2017a). Moxd1 is a marker for sexual dimorphism in the medial preoptic area, bed nucleus of the stria terminalis and medial amygdala. Front. Neuroanat. 11:26. doi: 10.3389/fnana.2017.00026
Tsuneoka, Y., Yoshida, S., Takase, K., Oda, S., Kuroda, M., and Funato, H. (2017b). Neurotransmitters and neuropeptides in gonadal steroid receptor-expressing cells in medial preoptic area subregions of the male mouse. Sci. Rep. 7:9809. doi: 10.1038/s41598-017-10213-4
Turchan, J., Lasoń, W., Budziszewska, B., and Przewłocka, B. (1997). Effects of single and repeated morphine administration on the prodynorphin, proenkephalin and dopamine D2 receptor gene expression in the mouse brain. Neuropeptides 31, 24–28. doi: 10.1016/s0143-4179(97)90015-9
Urbanek, M. O., Nawrocka, A. U., and Krzyzosiak, W. J. (2015). Small RNA detection by in situ hybridization methods. Int. J. Mol. Sci. 16, 13259–13286. doi: 10.3390/ijms160613259
Wu, Q., Wang, H., Gong, K., Shang, J., Liu, X., and Wang, F. (2019). Construction of an autonomous nonlinear hybridization chain reaction for extracellular vesicles-associated MicroRNAs discrimination. Anal. Chem. 91, 10172–10179. doi: 10.1021/acs.analchem.9b02181
Xu, Y., and Zheng, Z. (2016). Direct RNA detection without nucleic acid purification and PCR: combining sandwich hybridization with signal amplification based on branched hybridization chain reaction. Biosens. Bioelectron. 79, 593–599. doi: 10.1016/j.bios.2015.12.057
Yamaguchi, T., Kawakami, S., Hatamoto, M., Imachi, H., Takahashi, M., Araki, N., et al. (2015). In situ DNA-hybridization chain reaction (HCR): a facilitated in situ HCR system for the detection of environmental microorganisms. Environ. Microbiol. 17, 2532–2541. doi: 10.1111/1462-2920.12745
Young, L. J., Muns, S., Wang, Z., and Insel, T. R. (1997). Changes in oxytocin receptor mRNA in rat brain during pregnancy and the effects of estrogen and interleukin-6. J. Neuroendocrinol. 9, 859–865. doi: 10.1046/j.1365-2826.1997.00654.x
Zaidi, A. U., Enomoto, H., Milbrandt, J., and Roth, K. A. (2000). Dual fluorescent in situ hybridization and immunohistochemical detection with tyramide signal amplification. J. Histochem. Cytochem. 48, 1369–1375. doi: 10.1177/002215540004801007
Keywords: short hairpin DNA, hybridization chain reaction, in situ hybridization, fluorophore, mouse brain, striatum, medial preoptic area
Citation: Tsuneoka Y and Funato H (2020) Modified in situ Hybridization Chain Reaction Using Short Hairpin DNAs. Front. Mol. Neurosci. 13:75. doi: 10.3389/fnmol.2020.00075
Received: 28 January 2020; Accepted: 16 April 2020;
Published: 12 May 2020.
Edited by:
Jean-Marc Taymans, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceReviewed by:
Dagmar Galter, Karolinska Institutet (KI), SwedenFrançoise Gofflot, Catholic University of Louvain, Belgium
Copyright © 2020 Tsuneoka and Funato. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yousuke Tsuneoka, eW91c3VrZS50c3VuZW9rYUBtZWQudG9oby11LmFjLmpw; Hiromasa Funato, aGlyb21hc2EuZnVuYXRvQG1lZC50b2hvLXUuYWMuanA=