 Masaharu Takamori
Masaharu Takamori- Neurological Center, Kanazawa-Nishi Hospital, Kanazawa, Japan
Myasthenia gravis (MG) is a disease of the postsynaptic neuromuscular junction (NMJ) where nicotinic acetylcholine (ACh) receptors (AChRs) are targeted by autoantibodies. Search for other pathogenic antigens has detected the antibodies against muscle-specific tyrosine kinase (MuSK) and low-density lipoprotein-related protein 4 (Lrp4), both causing pre- and post-synaptic impairments. Agrin is also suspected as a fourth pathogen. In a complex NMJ organization centering on MuSK: (1) the Wnt non-canonical pathway through the Wnt-Lrp4-MuSK cysteine-rich domain (CRD)-Dishevelled (Dvl, scaffold protein) signaling acts to form AChR prepatterning with axonal guidance; (2) the neural agrin-Lrp4-MuSK (Ig1/2 domains) signaling acts to form rapsyn-anchored AChR clusters at the innervated stage of muscle; (3) adaptor protein Dok-7 acts on MuSK activation for AChR clustering from “inside” and also on cytoskeleton to stabilize AChR clusters by the downstream effector Sorbs1/2; (4) the trans-synaptic retrograde signaling contributes to the presynaptic organization via: (i) Wnt-MuSK CRD-Dvl-β catenin-Slit 2 pathway; (ii) Lrp4; and (iii) laminins. The presynaptic Ca2+ homeostasis conditioning ACh release is modified by autoreceptors such as M1-type muscarinic AChR and A2A adenosine receptors. The post-synaptic structure is stabilized by: (i) laminin-network including the muscle-derived agrin; (ii) the extracellular matrix proteins (including collagen Q/perlecan and biglycan which link to MuSK Ig1 domain and CRD); and (iii) the dystrophin-associated glycoprotein complex. The study on MuSK ectodomains (Ig1/2 domains and CRD) recognized by antibodies suggested that the MuSK antibodies were pathologically heterogeneous due to their binding to multiple functional domains. Focussing one of the matrix proteins, biglycan which functions in the manner similar to collagen Q, our antibody assay showed the negative result in MG patients. However, the synaptic stability may be impaired by antibodies against MuSK ectodomains because of the linkage of biglycan with MuSK Ig1 domain and CRD. The pathogenic diversity of MG is discussed based on NMJ signaling molecules.
Introduction
The neuromuscular junction (NMJ) functions as acetylcholine (ACh)-mediated synapse (Gray frame, Figure 1); its signal transmission depends on coordinated interaction between the pre-synaptic active zone (Südhof, 2012) and the post-synaptic nicotinic acetylcholine receptor (AChR; Kummer et al., 2006). In confirming the synaptic architecture, the trans-synaptic communications mediated by nerve-secreted or muscle membrane-bound proteins such as agrin (heparin sulfate proteoglycan) and Wnts (belonging to the Wingless-type integration site family of glycoproteins) play the pivotal roles via interactions with the muscle-specific tyrosine kinase (MuSK) and the low-density lipoprotein-related protein 4 (Lrp4; Wu et al., 2010; Shi et al., 2012; Takamori, 2013, 2017a). The review focusses on: (1) the AChR cluster formation by MuSK activation from inside (Dok-7 and Wnts) and outside (agrin and Wnts) of the muscle; (2) the synaptic compensatory mechanisms based on retrograde signals from muscle to nerve and presynaptic Ca2+ homeostasis by auto-receptors; and (3) the synaptic stabilization based on cytoskeletal dynamics by extracellular matrix proteins and dystrophin-associated glycoprotein complex. These have paved the way to search for the mechanisms underlying myasthenia gravis (MG) weakness (Burden et al., 2018; Sudres et al., 2018; Koneczny and Herbst, 2019). MG, an autoimmune NMJ disease characterized by fatigable weakness of voluntary muscles, are generally reviewed from the viewpoints of clinical subgroups and antibody characteristics (Vincent et al., 2018; Gilhus et al., 2019).
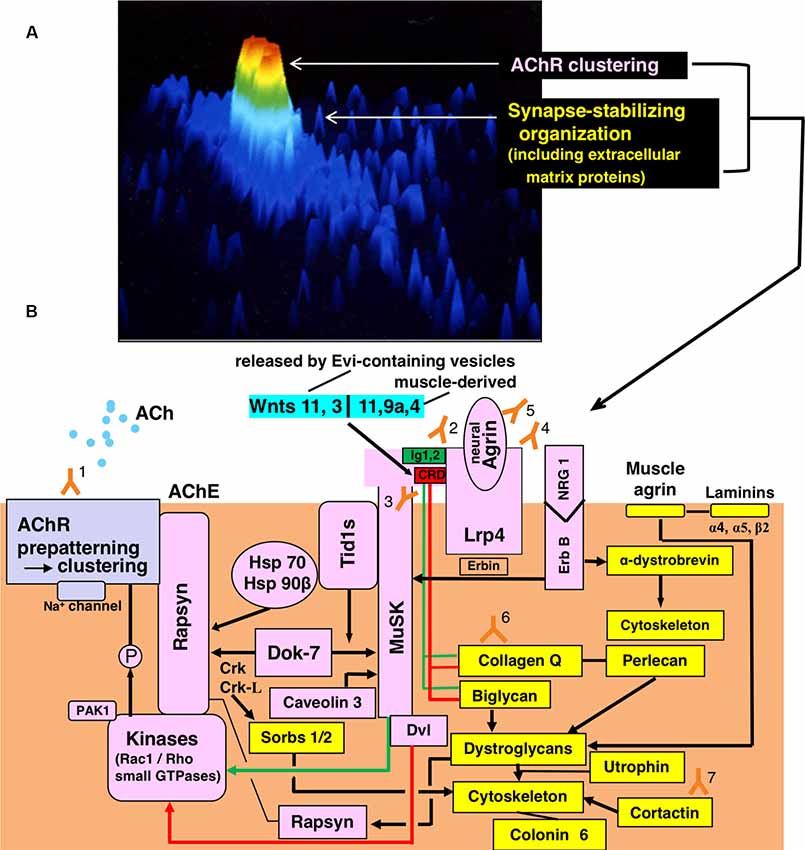
Figure 1. Functional organization for synaptic transmission in neuromuscular junction (NMJ) and antibody-targets. (A) Presentation by staining of cultured rat myotube with fluorescence-labeled α-bungarotoxin and by the image analyzing using a laser cytometer, indicating acetylcholine receptor (AChR) cluster (red), a synaptic stabilizing organization including extracellular matrix proteins (gree and light blue). The image is constructed on ACAS 570 (Meridian Instruments Inc., Okemos, MI, USA) which provides a graded pseudocolor image on the computer display. (B) Schematic presentation of the post-synaptic structures. Y marks attached with numbers indicate the antibodies to recognize respective targets of the functional structures. Gray frame indicates the acetylcholine receptor (AChR) cluster formation. Pink frames indicate AChR clustering by way of two signaling pathways mediated via the muscle-specific tyrosine kinase (MuSK) 1/2 domains (green-limit in the pink MuSK ectodomain and green line with arrowhead) and MuSK cysteine-rich domain (CRD; red-limit in the pink MuSK ectodomain and red-line with arrowhead), the signals of which are mediated by Dishevelled (Dvl, adaptor protein). The low-density lipoprotein receptor-related protein 4 (Lrp4) is the receptor for agrin (partly for Wnts as described in the text). The small GTPases (shown in the pink frame of Kinases) effector PAK1 (p21-activated kinase 1) acts as a bridging molecule between the Wnt- and agrin-signaling pathways. From “inside” the muscle cell, MuSK is activated by Dok7 (downstream kinase); Dok7 recruits two adaptor protein, Crk and Crk-L (CT10 regulators of kinase) for rapsyn-anchored AChR cluster formation. The formed AChR clusters are anchored at the endplate membrane by rapsyn and immobilized by MuSK-linking heat-shock proteins (HSPs): tumorous imaginal disc 1 short form (Tid1s), HSP 70 and HSP 90β. Tid1s is required for the MuSK-Dok7 signaling during the MuSK activation. The interaction of neuregulin 1 (NRG 1) with ErbB receptor (receptor tyrosine kinase of epidermal growth factor receptor family) increases the MuSK tyrosine phosphorylation (via Erbin) and thereby modulates the MuSK-dependent AChR clustering. Caveolin 3 binds with the MuSK kinase domain and thereby driving AChR clustering. Yellow frames indicate the organizations for synaptic stability and maintenance. The synaptic stability of NMJ including AChR clusters (gray frame), MuSK (pink frame), Lrp4 (pink frame) and acetylcholinesterase (AChE) is modulated by extracellular matrix proteins (collagen Q, perlecan, biglycan, laminin-network including muscle agrin and laminins and dystroglycan) worked in cooperation with the cytoskeleton. The interaction of NRG 1 (neuregulin 1) with ErbB receptor (pink frames) contributes to the cytoskeletal organization through α-dystrobrevin phosphorylation on one hand (yellow frame) and the MuSK activation via Erbin on the other hand (pink frame). The downstream effector of Dok7-recruited Crk-L (Sorbs1/2) acts on the cytoskeleton for synaptic stability. Collagen Q-Perlecan and Biglycan act on Dystroglycans in cooperation with cytoskeleton for synaptic stability on one hand (yellow frame) and implicate in AChR cluster formation via their interaction with pink-MuSK ectodomains (Ig1 shown by green limit with a green line and CRD shown by red limit with red line) on the other hand. The former is cooperated by linking to dystrophin/utrophin-associated protein complex; the latter is cooperated by linking with rapsyn to firmly anchor AChR clusters at the post-synaptic membrane. Cortactin (yellow frame) has the function of phosphorylation-dependent signaling downstream from Agrin/Lrp4/MuSK (pink frame) in promoting actin polymerization and also stabilizing AChR clusters at the postsynaptic membrane. Coronin 6 is the actin-binding protein contributive to synaptic stability. Targets recognized by antibodies: Y1, Acetylcholine receptor (AChR); Y2, Muscle-specific tyrosine kinase (MuSK) 1/2 domains; Y3, MuSK cysteine-rich domain (CRD); Y4, Low-density lipoprotein receptor-related protein 4 (Lrp4); Y5, neural Agrin; Y6, Collagen Q; Y7, Cortactin.
Myasthenia Gravis (MG) Caused by Antibodies Targeting Key Molecules in Neuromuscular Junction
Anti-AChR Antibodies
MG is the B cell-mediated, T cell-dependent autoimmune disease including thymic pathologies (Marx et al., 2013; Berrih-Aknin and Le Panse, 2014; Nemazee, 2017; Behin and Le Panse, 2018; Villegas et al., 2018; Yi et al., 2018; Koneczny and Herbst, 2019). In typical MG, most (~85%) patients have AChR (Gray frame in Figure 1) antibodies (Y1 in Figure 1), the pathogenic mechanisms of which are classified into (1) removal of AChRs due to cross-linking and subsequent internalization (antigenic modulation; Drachman et al., 1978), (2) functional AChR block (Drachman et al., 1982; Takamori et al., 1988) and (3) activation of complement with the formation of membrane attack complexes that cause focal lysis. The main immunogenic region targeted by MG antibodies is the AChR α-subunit 67–76 (Tzartos et al., 1982, 1988). The AChR antibodies are predominantly IgG1 and IgG3 subtypes that bind the complement causing the post-synaptic disorder in NMJ (Sahashi et al., 1980; Vincent and Newsom-Davis, 1980; Tüzün and Christadoss, 2013; Cetin and Vincent, 2018; Howard, 2018); this mechanism is most pathogenic in patients with refractory MG, who have efficaciously been treated with C5 inhibitor (eculizumab and Zilucoplan; Muppidi et al., 2019; Howard et al., 2020). Experimentally, the investigational RNAi targeting C5 (Kusner et al., 2019) and the single-chain AChR antibody coupled to complement decay-accelerating factor (Kusner et al., 2014) are shown to be efficacious in the models of MG. Promising therapeutic strategies include the B cell-related therapies (Rituximab; Tandan et al., 2017; Beecher et al., 2019; Di Stefano et al., 2020; Huda, 2020; Litchman et al., 2020) and the neonatal Fc receptor antagonists (Efgartigimod and Rozanolixizumab; Ulrichts et al., 2018; Howard et al., 2019; Huijbers et al., 2019a; Zuercher et al., 2019; Gable and Guptill, 2020). Taking into consideration of the reduced suppressive activity of T regulatory cells (Treg) in MG (reflecting on elevated inflammatory cytokines in MG; Thiruppathi et al., 2012; Ha and Richman, 2015; Wen et al., 2016; Molin et al., 2017), the Treg-based therapy achieved with the use of IL-2/anti-IL-2 monoclonal antibody complexes could be useful for treating MG as shown by the experiment in experimental autoimmune MG (Liu et al., 2010; Danikowski et al., 2017). Thymic abnormalities (hyperplasia tending to be potential in MG patients before the age of 50) have long been paid attention as an underlying pathology of MG and the target for therapy. We have participated in the MGTX (enrollment at 18–65 years of age; the disease severity of America classification class II–IV) randomized trial of thymectomy (with alternative-day prednisone), resulting in the benefit in non-thymomatous MG patients (Wolfe et al., 2019).
Anti-striational Antigen Antibodies
The suggestive biomarkers for the presence of thymoma indicative for thymectomy are the antibodies against muscle striational antigens such as ryanodine receptor (a foot protein in the junctional gap between the dihydropyridine receptor containing T-tubules and the sarcoplasmic reticulum, functioning as Ca2+ release channel for E-C coupling) and titin (a protein to keep the thick filaments centered in the sarcomere, allowing optimum active force development; Mygland et al., 1992; Skeie et al., 2002; Somnier and Engel, 2002; Takamori et al., 2004; Romi et al., 2007; Romi, 2011; Choi Decroos et al., 2014; Stergiou et al., 2016). We electrophysiologically confirmed the muscular contractile (twitch and tetanus) weakness caused by anti-ryanodine receptor antibodies in the spontaneous thymoma rat (Buffalo/Mna strain suppled by Charles River Japan, Inc.; Iwasa et al., 1998).
Immune Checkpoint Inhibitors for Cancer Therapy and MG
The therapies by use of immune checkpoint inhibitors (anti-programmed cell death protein-1 (PD1), anti-programmed death ligand-1 (PD-L1) and anti-cytotoxic T lymphocyte associated-4) to manage various cancers are occasionally associated with immune-related adverse events (due to loss of Treg homeostasis; Danikowski et al., 2017; Dalakas, 2018; Kumar et al., 2018, 2019; Möhn et al., 2019; Dubey et al., 2020) including myasthenia gravis (Becquart et al., 2019; Johansen et al., 2019; Sato et al., 2019; Choi and Lee, 2020) Reportedly, PD-L1 mRNA is highly expressed in MG muscle cells (Iwasa et al., 2018); this may suggest the participation of the local immune-mediated damage in MG muscle by the checkpoint inhibitor effect which leads to abnormal T-regulatory (Treg) cell function and altered Treg/Th17 cell axis (Dalakas, 2018). Also, PD-1 and PD-L1 are expressed in thymic epithelial tumors, suggesting a poor prognostic factor in thymomas (Bagir et al., 2018; Song et al., 2019).
Anti-MuSK Antibodies
MuSK (Pink frame, Figure 1), a member of the receptor tyrosine kinase superfamily, is composed of four ligand-binding ectodomains (linking with neural agrin <Pink frame, Figure 1> via first and second immunoglobulin-like domains, Ig1/2 <green -limit in pink- MuSK, Figure 1>, and with Wnts <Blue frames, Figure 1> via cysteine-rich domain (CRD), <red- limit in pink-MuSK, Figure 1>, through Lrp4 <Pink frame, Figure 1>), a single transmembrane-spanning domain and an intracellular kinase domain; this receptor tyrosine kinase acts as the key organizer to form AChR clusters in the post-synaptic membrane of NMJ (Ghazanfari et al., 2011; Burden et al., 2013; Takamori, 2013; Zong and Jin, 2013; Koneczny et al., 2014; Herbst, 2019; green and red lines via Dishevelled (Dvl, Pink frame, Figure 1). Mesdec2 plays a key role in cell-surface expression of Lrp4 and postsynaptic specialization (Hoshi et al., 2013).
Hoch and colleagues (Hoch et al., 2001) detected the antibodies against MuSK (as discussed later, two types of antibodies are determined: Y2 targeting the Ig1/2 domains and Y3 targeting CRD in Figure 1) in the serum samples of MG patients without AChR antibodies. In general, the patients show predominant involvement in the facial, pharyngeal, tongue (often with muscle atrophy), neck, and shoulder, but extraocular muscles are rarely involved at onset in contrast to AChR antibody-positive MG; they are occasionally seized with a respiratory crisis. The patients respond to immunosuppressant therapy (Evoli et al., 2008, 2018; Pasnoor et al., 2010; Guptill et al., 2011; Evoli and Padura, 2013; Koneczny et al., 2014; Morren and Li, 2018; Li et al., 2019). The B-cell depletion therapy (Rituximab) provides more benefit to MuSK-MG patients than AChR-MG patients (Evoli et al., 2008, 2018; Díaz-Manera et al., 2012; Evoli and Padura, 2013; Tandan et al., 2017; Morren and Li, 2018; Beecher et al., 2019; Di Stefano et al., 2020; Huda, 2020; Litchman et al., 2020). In the experimental model of MuSK MG, the fetal Fc receptor antibodies (Gable and Guptill, 2020), antigen-specific immunoadsorption (Skriapa et al., 2014; Lazaridis et al., 2020) and the inhibition of tyrosine phosphatase inhibitor (SHP2; Huda et al., 2020) alleviate the MuSK antibody-induced AChR cluster deformation. One must be cautious that the hypersensitivity and worsening of symptoms to cholinesterase inhibitors tend to occur (Punga et al., 2006; Evoli and Padura, 2013). In general, thymic abnormalities are not detected and thus no apparent clinical benefit of thymectomy is obtained (Leite et al., 2005; Evoli et al., 2008; Evoli and Padura, 2013; Nakata et al., 2013; Clifford et al., 2019).
Immunologically, MuSK antibodies are mainly IgG4 subclass (Koneczny, 2018) and known to cause MG by inhibiting binding between MuSK and Lrp4 independently of complement activation (Huijbers et al., 2013), but IgG1-3 MuSK antibodies can disperse preformed agrin-independent AChR clusters (Koneczny et al., 2013).
Anti-Lrp4 Antibodies
Lrp4 (Pink frame, Figure 1) which plays a pivotal role in mediating agrin (via green-limit, MuSK lg1/2, Figure 1)—and Wnt (via red-limit, MuSK CRD, Figure 1)—signalings for AChR clustering at the postsynaptic membrane and also serves the retrograde signaling from muscle to nerve (Barik et al., 2014a; Guarino et al., 2020). The molecular structure of Lrp4 consists of a large extracellular domain that contains eight low-density lipoprotein receptor domains class A (LDLa), EGF-like domains and four β-propeller domains; its N-terminal region (including the last few LDLa repeats and the first β-propeller domain) interacts with agrin (Zhang et al., 2011; Zong et al., 2012); in its third β-propeller domain, the edge part mediates the MuSK signaling and the central part mediates the Wnt signaling (Ohkawara et al., 2014). Autoantibodies (complement-activating IgG1 and IgG2; Y4 in Figure 1) are detected in a proportion of MG with or without antibodies to AChR and MuSK (Higuchi et al., 2011; Pevzner et al., 2011; Cossins et al., 2012; Zhang et al., 2012b; Shen et al., 2013; Zouvelou et al., 2013; Zisimopoulou et al., 2014; Marino et al., 2015; Li et al., 2017; Rivner et al., 2018; Yan et al., 2018b; Aoki et al., 2020). The severity of disease in most patients is mild to moderate, but some patients have severe symptoms causing the myasthenic crisis. Most patients respond to anti-acetylcholinesterases, steroids, and immunosuppressant medications. Thymic pathology is usually not present but underlies in some patients including thymomas (Zisimopoulou et al., 2014; Marino et al., 2015; Aoki et al., 2020). The active immunization of mice with Lrp4 was proved to induce myasthenia gravis; the pathogenicity of Lrp4 antibodies was confirmed by the study in animal models that showed both pre- and post-synaptic impairments and active complements (Mori et al., 2012, 2017; Shen et al., 2013; Ulusoy et al., 2017).
Anti-agrin Antibodies
Agrin, a proteoglycan expressed in both motor neuron (Pink frame, Figure 1) and skeletal muscle (Yellow frame, Figure 1), is essential to form NMJ formation (Burden et al., 2018). The neural agrin-dependent pathways include the two muscle-bound transmembrane co-receptors Lrp4 and MuSK (Pink frames, Figure 1). In the functional structure of Agrin (Hoch et al., 1994; Gesemann et al., 1995; Burgess et al., 1999; Bezakova and Ruegg, 2003; Stetefeld et al., 2004; Scotton et al., 2006; Zong et al., 2012; Tezuka et al., 2014; Zhang et al., 2015), the N-terminus is required for agrin-immobilization at the NMJ; the C-terminus segment contains two of the three laminin-like G domains (G2 and G3). G2 is essential for binding of muscle agrin (Yellow frame, Figure 1) to α-dystroglycan which contributes, via β-dystroglycan, to the stabilization of the post-synaptic apparatus at the NMJ through the organization of the intracellular cytoskeleton of muscle (Yellow frames with black linking lines, Figure 1; muscle agrin: this is >5,000 less active compared with neural agrin as far as AChR aggregation is concerned). G3 consists of four amino acid insertion at the A/y splicing site and 8, 11 or 19 amino acid insertion at the B/z splicing site (neural agrin: Pink frame, Figure 1) and is essential for the AChR clustering activity of agrin through Lrp4-MuSK signaling (green and red lines via pink-framed Dvl).
Anti-agrin antibodies (Y5 in Figure 1) have been detected in some of MG patients with or without antibodies against AChR, MuSK, and Lrp4 (Gasperi et al., 2014; Zhang et al., 2014; Cordts et al., 2017; Yan et al., 2018b). Induction of anti-agrin antibodies was proved to cause MG in mice by an impairment of agrin-Lrp4 interaction (Yan et al., 2018a). We must be cautious of the antigen used for agrin-antibody assay because of the above-mentioned difference of functional structure between neural agrin and muscle agrin.
The myasthenic weakness in anti-agrin antibody-positive patients is mild to severe but tends to be moderate to the treatment, suggesting that the early detection of anti-agrin antibodies could help disease management (Cordts et al., 2017). It is worthy of note as a therapeutic device in future that a fragment of neural agrin (NT-1654) has the potential to counteract the symptoms of the diseased NMJ in the sarcopenia-like phenotype in neurotrypsin-overexpressing mice (Hettwer et al., 2014; Rudolf et al., 2014). The engineered agrin was reported to attenuate the severity of experimental autoimmune myasthenia gravis (Li Z. et al., 2018). The active form of vitamin D (1.25D) upregulates the agrin-induced AChR clustering in association with the upregulated expression of rapsyn in C2C12 myotubes (Arakawa and Wagatsuma, 2020); this pharmacologic effect could apply to a group of MG, the antibodies of which are maleficient more to rapsyn-clustered AChR than non-clustered AChR (Cetin et al., 2020).
Matrix metalloproteinases (MMPs) are membrane-anchored extracellular proteinases capable of degrading a variety of proteins including agrin; the function of agrin is regulated by the site-specific cleavage by MMPs (Patel et al., 2012; Chan et al., 2020). Additionally shown was that MMP-3 removes agrin from synaptic basal lamina (Werle and VanSaun, 2003). Reportedly, the MMP-3 level is high in the serum samples from a proportion of MG patients (Helgeland et al., 2011; Romi et al., 2012), suggesting that the NMJ might be affected by the pathogenic mechanisms as the result of the dysfunction of agrin that is brought about non-immunologically by high MMP-3 level in addition to the immune response to agrin (Luckman et al., 2011; Chan et al., 2020).
Agrin/Wnt-Lrp4-Musk Signalings in Neuromuscular Junction
Agrin/Dok7 Signaling via Lrp4/MuSK Contributes to AChR Clustering
In the ligand-binding ectodomains of MuSK, the first and second immunoglobulin-like domains (Ig1 and Ig2: Ig1/2; green limit in the pink-MuSK frame, Figure 1) mediate the agrin-Lrp4 signaling (Stiegler et al., 2006) via binding of the Ig1 domain (green limit in the pink-MuSK frame, Figure 1) with Lrp4 to form a tetrameric complex (Zhang et al., 2011; Zong et al., 2012). The MuSK (Pink frame, Figure 1) is stimulated from “inside” the muscle cell by an adaptor protein Dok7 (Pink frame, Figure 1) which forms a dimeric unit to dimerize and activate MuSK (Pink frame, Figure 1; Okada et al., 2006; Bergamin et al., 2010) and also recruits two adaptor proteins, CT10 regulators of the kinase (Crk and Crk-L) contributive to rapsyn (Pink frame, Figure 1)—anchored AChR cluster formation (Gray frame, Figure 1; Hallock et al., 2010). The results from the experiments to modify the Dok7 functions are such as follows: in muscles lacking Dok7, agrin failed to stimulate MuSK tyrosine phosphorylation (Inoue et al., 2009); silencing of Dok7 in rat muscle increases susceptibility to passive transfer MG (Gomez et al., 2016); overexpression of Dok7 in rat muscle enhances neuromuscular transmission with structural alterations of NMJ (Eguchi et al., 2020).
From “outside” the muscle cell, MuSK is stimulated by neural agrin (Pink frame, Figure 1) via Lrp4 (Pink frame, Figure 1) which is the agrin receptor and also activates MuSK without agrin (Weatherbee et al., 2006; Kim et al., 2008; Zhang et al., 2008, 2011, 2012a; Barik et al., 2014b; Stamatakou and Salinas, 2014).
Rapsyn and Tid1s Stabilize AChR Clusters at the Membrane
The AChR clusters formed by the agrin-Lrp4-MuSK signaling are anchored at the end-plate membrane by rapsyn (Pink frame, Figure 1; Lee et al., 2009; Zuber and Unwin, 2013; Xing et al., 2020) which is immobilized by the short-form of tumorous imaginal discs-1 (Tid1s: MuSK- and Dok7-binding protein belongs to a family of heat-shock protein 40, HSP40) in cooperation with HSP70 and HSP90β (Pink frames, Figure) and thereby maintains AChR clusters (Gray frame, Figure 1) at the end-plate membrane (Linnoila et al., 2008; Luo et al., 2008; Zong and Jin, 2013). Tid1s (Pink frame, Figure 1) is also contributive to the MuSK-Dok7 signaling during the activation of MuSK (Linnoila et al., 2008). The RING domain of rapsyn contains E3 ligase activity to induce AChR clustering and NMJ formation, possibly by regulation of AChR neddylation (Li et al., 2016).
Wnt-Mediating Signal via MuSK Contributes to AChR Cluster Prepatterning
In addition to the above-mentioned role as the mediator in agrin-Lrp4-MuSK signaling, MuSK acts as the mediator for Wnts (Blue frames, Figure 1) which belong to a family of secreted glycoproteins and released from motor neurons or derived from muscle (Wu et al., 2010; Gordon et al., 2012; Koles and Budnik, 2012; Zhang et al., 2012a; Barik et al., 2014b; Stamatakou and Salinas, 2014). Of 19 different Wnt molecules existing in mice and humans (Wu et al., 2010), Wnt 11 (Jing et al., 2009; Wu et al., 2010; Zhang et al., 2012a) and Wnt 3 (Henriquez et al., 2008) are released from the motor neurons; the neuron-released Wnts are transferred across the synapse by exosome-like Evi-containing vesicles (Koles et al., 2012). Wnt 11 (Wu et al., 2010; Zhang et al., 2012a), Wnt 9a (Zhang et al., 2012a), and Wnt4 (Strochlic et al., 2012; Zhang et al., 2012a) are derived from muscles. The binding of the Ig4 domain (CRD) of MuSK (red-limit in pink MuSK, Figure 1) with Wnts contributes to AChR clustering through the non-canonical pathway mediated by the Dishevelled protein (Dvl; Pink frame, Figure 1), leading to the formation of AChR microclusters at the early non-innervated stage, termed as the AChR prepatterning (Kummer et al., 2006) which forms the clusters at the central part of the muscle and guides the incoming axon (Gray frame, Figure 1; Luo et al., 2002; Jing et al., 2009; Stiegler et al., 2009; Wu et al., 2010; Gordon et al., 2012; Koles and Budnik, 2012; Barik et al., 2014b; Burden et al., 2018; Guarino et al., 2020). The motoneuron-derived Wnt7a and Wnt7b serve nerve terminal development (Shen et al., 2018).
In the clustering of AChR, Wnt 11 and Wnt 9a require Lrp4 but not agrin (Zhang et al., 2012a); Wnt 4 requires neither agrin nor Lrp4 (Strochlic et al., 2012). Wnt 3 promotes the agrin-Lrp4-MuSK (Ig1/2 domains)-mediated clustering of AChR (Henriquez et al., 2008; Wnts 11, 9a, 4 and 3: Blue frames in Figure 1). In contrast to the neuron-released Wnt 3, the muscle-derived Wnt 3a reduces the agrin-mediated AChR clustering via the canonical Dvl pathway to β-catenin causing the reduced rapsyn expression (Wang et al., 2008); this negative regulation maintains balance with the positive regulator (neuron-released Wnt 3) and thereby helps to sculpt the mature synaptic architecture (Henriquez and Salinas, 2012; Koles and Budnik, 2012).
Linking to the intracellular kinase domain of MuSK, the small GTPases (Rac1 and Rho) effector p21-activated kinase 1 (PAK1; Pink frame, Figure 1) acts as a bridging molecule between the Wnt- and agrin-signaling pathways required for AChR clustering (Luo et al., 2002; Wu et al., 2010; Koles and Budnik, 2012). Wnt activates Rac1 in a more efficient usage than agrin which preferentially increases Rho activity (Henriquez and Salinas, 2012).
In the innervated stage of muscle when the agrin-Lrp4-MuSK (Ig1/2 domains) signaling plays a principal role in AChR cluster formation, the cyclin-dependent kinase 5 (Cdk5) concentrated at the postsynaptic NMJ is activated and required for ACh-induced removal of prepatterning AChR clusters which are not stabilized by agrin; this is mediated through two molecular mechanisms (the upregulation of calpain activity and the interaction of Cdk5 with nestin; Lin et al., 2005; Chen et al., 2007; Mohseni et al., 2011; Yang et al., 2011; Shi et al., 2012).
Neuregulin 1-ErbB Signaling to MuSK Activation
The ErbB receptors (receptor tyrosine kinase of EGF, epidermal growth factor, receptor family) for neuregulin 1 (NRG 1; Pink frames, Figure 1) link to the MuSK through the adaptor protein, Erbin (Simeone et al., 2010), suggesting that this signaling cascade increases the tyrosine phosphorylation of MuSK and thereby modulates MuSK-dependent AChR clustering (Ngo et al., 2012). Anti-ErbB antibodies were not detected in the sera of MG patients; however, the thymic expression of ErbB receptors was reduced in these patients (Vrolix et al., 2011).
A Variety of Immunogenicity of MuSK
By immunoblotting against recombinant proteins (expressed in HEK 293F cells) of MuSK ectodomains, we showed that the MuSK Ig1/2 antibodies (Y2 in Figure 1) were positive in the serum samples from 33 of 43 AChR antibody-negative MG patients; 10 of them (30%) were additionally positive for MuSK CRD antibodies (Y3 in Figure 1), suggesting an impairment of both the agrin-Lrp4-MuSK (Ig1/2 domains) signaling pathway (the green line from the green limit in pink-MuSK to Kinases via Dvl, Figure 1) and Wnt-Lrp4-MuSK(CRD) signaling pathway (the red line from the red limit in pink-MuSK to Kinases via Dvl, Figure 1) in the neuromuscular transmission; however, none was positive for anti-MuSK CRD alone in our study (Takamori et al., 2013). Reportedly, the experiments by the inhibition assay in the presence of unlabelled competitive proteins (McConville et al., 2004) and the immunoprecipitation assay using MuSK Ig1 domain and CRD (Otsuka et al., 2015) suggested the co-existence of antibodies against MuSK Ig1/2 domains and MuSK CRD in MuSK antibody-positive MG. The longitudinal epitope mapping study in 53 MuSK antibody-positive MG patients by the European group showed that 22.6% of the patients were positive for MuSK CRD antibodies, although they emphasized the MuSK Ig 1 domain as the main immunogenic region (Huijbers et al., 2016). It seems likely that the MuSK antibodies have pathological heterogeneity based on their binding to functionally different domains.
To determine the functional significance of MuSK CRD in neuromuscular transmission, Messéant and associates showed that the CRD deletion of MuSK in mice caused exuberant axonal growth bypassing AChR clusters and decreased synaptic vesicle density (presynaptic impairment), and a drastic deficit in AChR clustering (postsynaptic impairment; Messéant et al., 2015). They also suggested an implication of MuSK CRD in the Wnt-canonical signaling by demonstrating that this pre- and post-synaptic impairments were rescued by lithium chloride which acts as an inhibitor of the glycogen synthese kinase-3β (Gsk3β) and an activator of the Wnt/β-catenin signaling (Messéant et al., 2015). This study was followed by the demonstration that MuSK ΔCRD is malfunctioning since MuSKΔCRD/− embryos present defective pre- and post-synaptic differentiation (Boëx et al., 2018). In addition to the above-mentioned Wnt-canonical signaling pathway, they proposed the β catenin-dependent and vangl 2 (coimmunoprecipitated with Wnts 4 and 11 in muscle)-dependent Wnt non-canonical signaling pathway (Messéant et al., 2017). The experiment in mice suggested that the NMJ pre- and post-synaptic organizations by β-catenin-dependent or Wnt-dependent signaling is regulated by Yes-associated protein (Yap) in the muscle (Zhao et al., 2017). Reportedly, the inhibition of the MuSK-Dvl signaling pathway (Wu et al., 2010; Koles and Budnik, 2012) decreased not only the amplitude of spontaneous synaptic currents but also their frequency (Luo et al., 2002), suggesting a defect in ACh-release upregulation to compensate for the post-synaptic dysfunction (Takamori et al., 1984; Plomp et al., 1995; Plomp, 2017; Takamori, 2019). This, in turn, may reflect an impairment in the retrograde Wnt-MuSK (CRD)-Dvl-Gsk3β inhibition-β catenin-slit 2 signaling at the nerve terminal (Luo et al., 2002; Salinas, 2005; Li et al., 2008; Budnik and Salinas, 2011; Koles and Budnik, 2012; Wu et al., 2012a, 2015; Barik et al., 2014b). Postsynaptically shown in the experiment is that β catenin regulates agrin-induced AChR clustering through interaction with rapsyn in a manner dependent on α catenin-associated cytoskeleton (Zhang et al., 2007). These can help explain the reports showing both pre- and post-synaptic defects in MuSK antibody-positive MG patients (Niks et al., 2010) and animal models (Cole et al., 2008; Klooster et al., 2012; Mori et al., 2012; Richman et al., 2012; Viegas et al., 2012). In the study of congenital myasthenic syndrome due to MuSK mutation, the changes in the presynaptic morphology were assumed to reflect a disturbance in muscle-derived retrograde signals resulting from the impairment of agrin- and Dok7-induced MuSK phosphorylation (Maselli et al., 2010; Ben Ammar et al., 2013).
We noted the debate by Remédio and associates who proposed that dependently on species, the prepatterning of AChR clusters is influenced by an additional presence of Kringle domain in the MuSK extracellular region (Remédio et al., 2016; Burden et al., 2018); this is likely to augue against the participation of MuSK CRD in signaling variety in MuSK antibody-positive MG patients. In this regard, Legay and Mei (2017) suggested that the controversial result may reflect the genotype-phenotype relationships because opposite phenotypes can be found depending on the genetic background (Sittig et al., 2016).
In the MuSK antibody-positive MG patients, the recent research reported that the monovalent MuSK antibodies abolished agrin-induced MuSK phosphorylation and AChR clustering, while the divalent monospecific MuSK antibodies had the opposing effect which leads to partially induced AChR clustering; this is independent of agrin (Huijbers et al., 2019b). It is also noticeable that Takata and his associates generated human MuSK monoclonal autoantibodies (2 IgG4 and 1 IgG3 subclasses obtained using isolation of MuSK autoantibody-expressing B cells from MuSK antibody-positive MG patients) and showed that these antibodies inhibited AChR clustering, but enhanced MuSK phosphorylation; they suggested an alternative mechanism for inhibiting AChR clustering (Takata et al., 2019).
Bone-Morphogenic Proteins (BMPs) and MuSK Activation
The BMPs, which are secreted ligands belonging to the transforming growth factor β (TGFβ) superfamily of signaling proteins, have been focused in highlighting insight into the possibility that BMPs work in vertebrate organisms as diffusible signals to ensure proper communication between motor neurons and skeletal muscles (Osses and Henriquez, 2015). In a variety of function and localization of BMPs, it is an interest to note the reports that the Wnt-MuSK Ig3 domain interaction acts as a BMP co-receptor which shapes BMP transcriptional output and cholinergic signaling to communicate motor neurons and skeletal muscles in the vertebrate (Henríquez et al., 2011; Yilmaz et al., 2016). The BMP-4 has been confirmed to be expressed in the skeletal muscle and localized in close vicinity to postsynaptic densities at the NMJ (Chou et al., 2013). Further work may provide a clue to the understanding of BMPs-mediated pathology in MuSK antibody-positive MG patients.
Synaptic Retrograde Signalings from Muscle to Nerve
Wnt-MuSK, Lrp4, and Laminins
As previously discussed in a variety of immunogenicity of MuSK, the interaction of Wnt with MuSK CRD leading to Dvl-GSK3β inhibition-β catenin-Slit 2 signaling (Luo et al., 2002; Salinas, 2005; Li et al., 2008; Budnik and Salinas, 2011; Koles and Budnik, 2012; Wu et al., 2012a; Barik et al., 2014b; Messéant et al., 2015) and the muscle Lrp4 (extracellular eight low-density lipoproteins repeats; Wu et al., 2012b; Yumoto et al., 2012) contribute to the retrograde signaling and thereby modulate the pre-synaptic differentiation including synaptic vesicles and active zone in the nerve terminal. Additional retrograde signaling in the synapse is mediated by the muscle-derived laminins α4, α5, and β2 which contribute to the presynaptic organization acting for ACh release; this includes the action of laminin β2 which tethers P/Q- and N-types voltage-gated Ca2+ channels (VGCCs) to the active zone proteins (Nishimune et al., 2008; Carson et al., 2010; Chen et al., 2011; Nishimune, 2012; Samuel et al., 2012; Yamada and Sekiguchi, 2015; Zhang et al., 2015; Rogers and Nishimune, 2017).
Presynaptic Ca2+ Homeostasis and Autoreceptors
From the standpoint of clinical electrophysiology, the AChR antibody-positive MG and animal models showed that the compensatory ACh release upregulation cannot be sustained at the high frequency of nerve stimulation (Plomp et al., 2015). This may reflect the impairment of the presynaptic Ca2+ homeostasis for ACh release (Takamori, 2012) to compensate for postsynaptic dysfunction. The presynaptic Ca2+ homeostasis is promoted by the activation of G protein-coupled receptors such as M1-type muscarinic AChR (M1 mAChR) and A2A adenosine receptor along with the interaction of brain-derived neurotrophic factor (BDNF) with receptor tyrosine kinase B (TrkB); these biological mechanisms lead to P/Q-type VGCC activation by the signaling mediated by phospholipase C (PLC)-phosphatidylinositol 4,5-bisphosphate (PIP2)-diacylglycerol (DAG)-protein kinase C (PKC; Amaral and Pozzo-Miller, 2012; Santafé et al., 2015; Nadal et al., 2016; Hurtado et al., 2017; Simö et al., 2018; Tomàs et al., 2018). Also, PLC-generated DAG regulates the presynaptic vesicle priming protein (Munc13-1) to recruit ACh-containing vesicles for the immediately releasable pool (Bauer et al., 2007). Taking these into consideration, the presynaptic impairments to compensate postsynaptic dysfunction could, at least in part, be compatible with the result of our immunological study that the M1 mAChR antibodies were positive in 7 (28%) of our 25 post-synaptic nicotinic AChR antibody-positive MG patients (Takamori et al., 2007; Takamori, 2019); additionally, the anti-M1 mAChR antibodies were often (76%) positive in our 25 patients with Lambert-Eaton myasthenic syndrome (an autoimmune presynaptic disease caused by antibodies against mainly P/Q-type VGCC and partly synaptotagmin 1; Takamori et al., 2007; Takamori, 2008a). Of note, the M1 mAChR is physiologically associated with the non-voltage-gated Ca2+-dominant influx channel (Transient receptor potential canonical; Kim and Saffen, 2005), the antibodies against which were positive in 7 (28%) of our 25 MG patients (Takamori, 2008b). An alternative theory postulated is that the impaired compensatory mechanism could be referred to the reduction in homeostatic small reserve pool of synaptic vesicles that depends on presynaptic adenosine A2A receptor operating Ca2+ influx via L-type VGCC (Oliverira et al., 2004; Ma et al., 2015; Wang et al., 2016). In a long-tetanic load in muscle, the complex compensatory mechanisms including the presynaptic autoreceptors and Ca2+ influx channel(s) may underlie to compensatory postsynaptic dysfunction.
Therapeutic Option of β2-Adrenoceptor Agonists in Myasthenias
Besides the biological signal (compensatory presynaptic Ca2+ homeostasis) triggered by G protein-coupled receptor, it is worthy to note that BDNF in the synapse is activated by β2-adrenoceptor agonists and thereby contributes to the maintenance of the structural and functional integrity of motor end-plate; this signaling pathway may include the cyclic adenosine monophosphate (cAMP)/protein kinase A/cAMP-responsive element-binding protein; Bartus et al., 2015). The therapeutic pre- and post-synaptic benefits of β2-adrenoceptor agonists (salbutamol and ephedrine) have been reported in MuSK MG animal model (Ghazanfari et al., 2014), autoimmune MG patients (Rodríquez Cruz et al., 2015b; Lipka et al., 2017; Vanhaesebrouck et al., 2019) and various types of congenital myasthenic syndromes (Engel et al., 2015; Beeson, 2016) including the patients with mutations in MuSK (Gallenmller et al., 2014; Rodríquez Cruz et al., 2020; Vanhaesebrouck and Beeson, 2019), Lrp4 (Selcen et al., 2015; Beeson, 2016), Dok7 (Lashley et al., 2010; Liewluck et al., 2011; Burke et al., 2013; Lorenzoni et al., 2013), agrin (Nicole et al., 2014; Beeson, 2016; Vanhaesebrouck and Beeson, 2019), collagen Q (McMacken et al., 2019) and collagen XIII (Beeson, 2016; Rodríguez Cruz et al., 2018, 2019; Dusl et al., 2019). In pharmacotherapeutic options, the β2-agonist stimulation could be chosen as a novel therapeutic strategy in autoimmune and genetic myasthenias.
Candidates as The Molecules Implicating in Pathogenicities of Myasthenias
The studies including clinical researches may suggest the following molecules as candidates to implicate in the pathogenic mechanisms of myasthenic diseases: (1) caveolin 3 (Pink frame, Figure 1; MuSK kinase domain-binding protein which participates in agrin-induced phosphorylation and activation of MuSK, thereby driving AChR clustering; Hezel et al., 2010; Iwasa et al., 2016); (2) doublecortin (expressed in motor neurons and skeletal muscles to orderly form the pre- and post-synaptic morphology; Bourgeois et al., 2015); (3) R-spondin 2 (expressed in motor neurons and reactive with leucine-rich repeat-containing G-protein-coupled receptor which is expressed in skeletal muscles and enriched in the NMJ so as to regulate Wnt-mediated AChR clustering and also acting on an effect for synaptic vesicle recycling and number of active zones in the nerve terminal; Vieira et al., 2015; Nakashima et al., 2016); (4) amyloid precursor protein (APP)/APP-like protein (contributive to AChR clustering via the Lrp4-MuSK signal in cooperation with agrin-mediated signal and also to the presynaptic differentiation of the NMJ, and additionally contributive to the density of ACh-containing synaptic vesicles which is modulated by glia cell-derived neurotrophic factor, GDNF, expressed in muscle cells; Akaaboune et al., 2000; Wang et al., 2009; Caldwell et al., 2013; Choi et al., 2013; Stanga et al., 2016), and (5) α-neurexins (contributive to neurotransmission and differentiation of the synapse by functioning as the trans-synaptic complex with postsynaptic proteins such as neuroligins (Sons et al., 2006; Reissner et al., 2013; Plomp, 2017).
Synaptic Stability and Maintenance by Extracellular Matrix and Dystrophin-Associated Glycoprotein Complex
To be precisely opposed to the nerve terminal including the active zone artitecture (Nishimune, 2012; Südhof, 2012; Harris and Littleton, 2015; Körber and Kuner, 2016), the postsynaptic structure is stabilized by the extracellular matrix proteins (including synaptic collagens such as Q, IV and XIII, perlecan, biglycan, laminins, muscle agrin, nidogens and α-dystroglycan; Yellow frames, Figure 1), dystrophin-associated glycoprotein complex (including β-dystroglycan which links to rapsyn and utrophin; Yellow frames, Figure 1), cortactin (Yellow frame, Figure 1; cortactin-directing antibodies: Y7 in Figure 1; acting as not only a synaptic kinase substrate but also a regulator of actin polymerization), coronin 6 (Yellow frame, Figure 1; actin-binding protein which plays a role in synaptic stability), α-dystrobrevin (Yellow frame, Figure 1; contributive to cytoskeletal organization due to its phosphorylation by neuregulin 1(NRG 1)-ErbB receptor interaction: Pink frames in Figure 1) and Sorbs 1/2 (Yellow frame, Figure 1; contributive to synaptic stabilization by its interaction with Dok7-recruited Crk-L Nishimune et al., 2008; Madhavan et al., 2009; Hallock et al., 2010, 2015; Pilgram et al., 2010; Schmidt et al., 2011; Singhal and Martin, 2011; Yurchenco, 2011; Amenta et al., 2012; Maselli et al., 2012; Nastase et al., 2012; Shi et al., 2012; Hochenester and Yurchenco, 2013; Karmouch et al., 2013; Chen et al., 2014; Constantin, 2014; Takamori, 2017a,b; Belhasan and Akaaboune, 2020). The following is the synaptic partners organizing molecules that regulate synaptic stability.
Collagen Q (Col Q) and Perlecan
Collagen Q (Yellow frame, Figure 1) is one of the muscle basal lamina proteins (junctional collagens; Singhal and Martin, 2011). Its N-terminus interacts with acetylcholinesterase (AChE); the two heparin-binding sites of its central collagenic domain mediate the interaction with perlecan (Yellow frame, Figure 1) which binds α-dystroglycan (Yellow frame, Figure 1) for linking the extracellular matrix to the cytoskeleton (Yellow frame, Figure 1). Its C-terminus binds the Ig1 domain (green limit) and CRD (red limit) in MuSK (Pink frame, Figure 1); these links make ColQ (via green and red lines, Figure 1) to modulate the action of the other ligands of the MuSK/Lrp4 complex for NMJ formation (Karmouch et al., 2013). The ColQ anchors AChE in the synaptic basal lamina, and thereby the association between Col Q and MuSK conforms Col Q/AChE to the synaptic localization (Cartaud et al., 2004). The MuSK antibodies were found to block the Col Q-MuSK interaction (Kawakami et al., 2011). Reportedly, the Col Q and MuSK antibodies competitively suppress agrin/Lrp4/MuSK signaling (Otsuka et al., 2015). Col Q antibodies (Y6 in Figure 1) were detected in some MG patients positive for MuSK and AChR antibodies (Cossins et al., 2012; Zoltowska Katarzyna et al., 2015). These findings are compatible with the electrophysiological finding characterized by slow miniature end-plate potential kinetics and hypersensitivity to AChE inhibitors in MuSK antibody-positive MG (Shin et al., 2014) and its animal model (Mori et al., 2012).
Collagen XIII
Collagen XIII is encoded as an extracellular matrix protein and is known to be crucial for the formation and function of pre- and post-synaptic organization of the NMJ, including the synaptic integrity through binding to the Col Q tail of AChE, AChR clustering, synaptic vesicle accumulation and axonal neurofilament (Latvanlehto et al., 2010; Logan et al., 2015; Härönen et al., 2017, 2019; Beeson et al., 2018; Heikkinen et al., 2019; Rodríquez Cruz et al., 2019). Autoantibodies to the collagen XIII have recently been reported in MG (Tu et al., 2018), suggesting the test as to if the Collagen XIII could be a pathogenic antigen in MG patients.
Biglycan
Biglycan (Yellow frame, Figure 1) is one of the proteoglycans and is enriched in the postsynaptic membrane and is a ubiquitous structural component of the extracellular matrix, and also acts as signaling molecules (Amenta et al., 2012; Nastase et al., 2012). The glycosaminoglycan-binding form of biglycan mediates its binding to extracellular α-dystroglycan (Yellow frame, Figure 1); the transmembrane β-dystroglycan (Yellow frame, Figure 1) binds to rapsyn (Pink frame, Figure 1) for firmly anchoring AChR clusters at the postsynaptic membrane (from Yellow frame to Pink frame, being connected by a black line, Figure 1) and also binding to the dystrophin/utrophin (Yellow frame, Figure 1)—associated protein complex contributive to the organization of cytoskeleton (Yellow frame, Figure 1) for synaptic stability. The non-glycosylated form of biglycan (lacking glycosaminoglycan side chain) directly interacts with two extracellular domains of MuSK: one (green line) is the agrin/Lrp4-mediating Ig1 domain of MuSK (green limit in pink-MuSK, Figure 1; Weatherbee et al., 2006; Kim et al., 2008; Zhang et al., 2008, 2011; Zong et al., 2012; Zong and Jin, 2013) and the other (red line) is the Wnt-mediating MuSK CRD (red limit in pink-MuSK, Figure 1; Jing et al., 2009; Koles and Budnik, 2012; Strochlic et al., 2012; Zhang et al., 2012a). The evidence that the two sites of MuSK ectodomain bind biglycan suggest possible participation of this matrix protein in reinforcing a functional bridge between the agrin-signaling (via Ig1 domain) and Wnt-signaling (via CRD; Barik et al., 2014b). Since biglycan becomes incapable of linking to MuSK by mutations of Ig1 domain and CRD (Amenta et al., 2012), the interaction between MuSK and biglycan (non-glycanated form) is potentially blocked by MuSK antibodies in the manner similar to the block of the MuSK-Col Q linkage by MuSK antibodies (Kawakami et al., 2011). In the serum samples from MG patients that we previously studied for the detection of antibodies recognizing MuSK Ig1/2 domains and CRD (Takamori et al., 2013), we tested if they harbor antibodies that directly recognize biglycan, but obtained the negative result (Takamori, 2017b). However, the MuSK antibodies may impair the synaptic stability by the disturbed MuSK-biglycan linkage. In addition to the immunological participation, biglycan stimulates multifunctional proinflammatory signaling linking the innate to adaptive immune systems when it is in a soluble form; this is based on that biglycan is capable of clustering several types of pathogen recognition receptors and orchestrating their signalings (Bavelova et al., 2009; Moreth et al., 2012; Nastase et al., 2012; Frey et al., 2013). In MG, biglycan produced in thymic myoid cells is known to play in generation and maintenance of the hyperplastic change of MG thymus (Tomoyasu et al., 1998; Takamori, 2017b).
Cortactin
Cortactin (Yellow frame, Figure 1) is a protein that is a tyrosine kinase substrate and regulates actin polymerization, and is co-enriched at AChR clusters; its tyrosine phosphorylation is enhanced by agrin, suggesting a novel function of phosphorylation-dependent cortactin signaling downstream from agrin/MuSK in promoting actin polymerization (through actin-related proteins 2/3 complex activation) and stabilizing AChR cluster at the postsynaptic membrane (Madhavan et al., 2009). Cortactin antibodies (Y7 in Figure 1) were detected in some of the MG patients (Gallardo et al., 2014; Cortés-Vicente et al., 2016; Illa et al., 2018), and also found in a patient with Lambert-Eaton myasthenic syndrome (a presynaptic autoimmune disease; Gallardo et al., 2014); this may reflect the fact that cortactin expresses in the presynaptic side (Peng et al., 1997) and acts as the Wnt signaling-dependent presynaptic effector molecule (Alicea et al., 2017).
Conclusion and Perspective
We emphasize: (1) the AChR cluster formation based on the Wnts-Lrp4-MuSK (CRD)-Dvl signaling (pre-innervated stage) which converges upon the agrin-Lrp4-MuSK (Ig1/2 domains) pathway (innervated stage); (2) the retrograde signaling (from muscle to the nerve) for presynaptic differentiation via MuSK CRD, Lrp4 and Laminins; (3) presynaptic Ca2+ homeostasis by autoreceptors; and (4) the synaptic stability based on the laminin-network including the extracellular matrix and the cytoskeleton. The insight into the NMJ structural mechanisms discussed in this review will foster further immunological approaches to search for new antigenic targets (Li L. et al., 2018; Koneczny and Herbst, 2019) and to enhance antibody detection (Vincent et al., 2012, 2018; Rodríguez Cruz et al., 2015a; Huda et al., 2017; Lazaridis and Tzartos, 2020) in MG, and may thereby help to view clinical profiles in MG subtypes (Verschuuren et al., 2013; Gilhus et al., 2019) and to provide potential therapeutic approaches (Losen et al., 2005; Ghazanfari et al., 2014; Gomez et al., 2014; Dalakas, 2015, 2019; Behin and Le Panse, 2018; Ito and Ohno, 2018; Morren and Li, 2020).
Author Contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Akaaboune, M., Allinguant, B., Farza, H., Roy, K., Magoul, R., Fiszman, M., et al. (2000). Developmental regulation of amyloid precursor protein at the neuromuscular junction in mouse skeletal muscle. Mol. Cell Neurosci. 15, 355–367. doi: 10.1006/mcne.2000.0834
Alicea, D., Perez, M., Maodonado, C., Dominicci-Cotto, C., and Marie, B. (2017). Cortactin is a regulator of activity-dependent synaptic plasticity controlled by Wingless. J. Neurosci. 37, 2203–2215. doi: 10.1523/JNEUROSCI.1375-16.2017
Amaral, M. D., and Pozzo-Miller, L. (2012). Intracellular Ca2+ stores and Ca2+ influx are both required for BDNF to rapidly increase quantal vesicular transmitter release. Neural Plast. 2012:203536. doi: 10.1155/2012/203536
Amenta, A. R., Creely, H. E., Mercado, M. L. T., Hagiwara, H., McKechnie, B. A., Lechner, B. E., et al. (2012). Biglycan is an extracellular MuSK binding protein important for synaptic stability. J. Neurosci. 32, 2324–2334. doi: 10.1523/JNEUROSCI.4610-11.2012
Aoki, S., Nagashima, K., Furuta, M., Makioka, K., Fujita, Y., Saito, K., et al. (2020). A case of anti-Lrp4 antibody-associated myasthenia gravis with a rare complication of thymoma successfully treated by thymectomy. Intern. Med. 59, 1219–1222. doi: 10.2169/internalmedicine.3828-19
Arakawa, M., and Wagatsuma, A. (2020). 1α, 25(OH)2D3 regulates agrin-induced acetylcholine receptor clustering through upregulation of rapsyn expression in C2C12 myotubes. Biochem. Biophys. Res. Commun. 525, 80–86. doi: 10.1016/j.bbrc.2020.02.027
Bagir, E. K., Acikalin, A., Gumurdulu, D., and Paydas, S. (2018). PD-1 and PD-L1 expression in thymic epithelial tumours and non-neoplastic thymus. J. Clin. Pathol. 71, 637–641. doi: 10.1136/jclinpath-2017-204788
Barik, A., Lu, Y., Sathyamurthy, A., Bowman, A., Shen, C., Li, L., et al. (2014a). LRP4 is critical for neuromuscular junction maintenance. J. Neurosci. 34, 13892–13905. doi: 10.1523/JNEUROSCI.1733-14.2014
Barik, A., Zhang, B., Sohal, G. S., Xiong, W. C., and Mei, L. (2014b). Crosstalk between agrin and Wnt signaling in development of vertebrate neuromuscular junction. Dev. Neurobiol. 74, 828–838. doi: 10.1002/dneu.22190
Bartus, R. T., Bétourné, A., Basile, A., Peterson, B. L., Glass, J., and Boulis, N. M. (2015). β-adrenoceptor agonists as novel, safe and potentially effective therapies for amyotrophic lateral sclerosis (ALS). Neurobiol. Dis. 85, 11–24. doi: 10.1016/j.nbd.2015.10.006
Bauer, C. S., Woolley, R. J., Teschemacher, A. G., and Seward, E. P. (2007). Potentiation of exocytosis by phospholipase C-coupled G-protein-coupled receptors requires the priming protein Munc13–1. J. Neurosci. 27, 212–219. doi: 10.1523/JNEUROSCI.4201-06.2007
Bavelova, A., Moreth, K., Tsalastra-Grreul, W., Zeng-Brouwers, J., Eickelberg, O., Young, M. F., et al. (2009). Biglycan, a danger signal that activates the NLRP3 inflammasome via Toll-like and P2X receptors. J. Biol. Chem. 284, 24035–24048. doi: 10.1074/jbc.m109.014266
Becquart, O., Lacotte, J., Malissart, P., Nadal, J., Lesage, C., Guillot, B., et al. (2019). Myasthenia gravis induced by immune checkpoint inhibitors. J. Immunother. 42, 309–312. doi: 10.1097/CJI.0000000000000278
Beecher, S., Putko, B. N., Wagner, A. N., and Siddiqi, Z. A. (2019). Therapeutic directed against B-cells and downstream effects in generalized autoimmune myasthenia gravis: current status. Drugs 79, 353–364. doi: 10.1007/s40265-019-1065-0
Beeson, D. (2016). Congenital myasthenic syndromes: recent advances. Curr. Opin. Neurol. 29, 565–571. doi: 10.1097/WCO.0000000000000370
Beeson, D., Cossins, J., Rodriquez Cruz, P. M., Maxwell, S., Liu, W. W., and Palace, J. (2018). Myasthenic syndromes due to defects in COL13A1 and in the N-linked glycosylation pathway. Ann. N Y Acad. Sci. 1413, 163–169. doi: 10.1111/nyas.13576
Behin, A., and Le Panse, R. (2018). New pathway and therapeutic targets in autoimmune myasthenia gravis. J. Neuromuscul. Dis. 5, 265–277. doi: 10.3233/jnd-170294
Belhasan, D. C., and Akaaboune, M. (2020). The role of the dystrophin glycoprotein complex on the neuromuscular system. Neurosci. Lett. 722:134833. doi: 10.1016/j.neulet.2020.134833
Ben Ammar, A., Soltanzadeh, P., Bauché, S., Richard, P., Goillot, E., Herbst, R., et al. (2013). A mutation causes MuSK reduced sensitivity to agrin and congenital myasthenia. PLoS One 8:e53826. doi: 10.1371/journal.pone.0053826
Bergamin, E., Hallock, P. T., Burden, S. J., and Hubbard, S. R. (2010). The cytoplasmic adaptor protein Dok7 activates the receptor tyrosine kinase MuSK via dimerization. Mol. Cell 39, 100–109. doi: 10.1016/j.molcel.2010.06.007
Berrih-Aknin, S., and Le Panse, R. (2014). Myasthenia gravis: a complrehensive review of immune dysregulation and etiological mechanisms. J. Autoimmun. 52, 90–100. doi: 10.1016/j.jaut.2013.12.011
Bezakova, G., and Ruegg, M. A. (2003). New insight into the roles of agrin. Nat. Rev. Mol. Cell Biol. 4, 295–308. doi: 10.1038/nrm1074
Boëx, M., Messéant, J., Bauché, S., Fontaine, B., Legay, C., and Strochlic, L. (2018). Regulation of mammalian neuromuscular junction formation and maintenance by Wnt signaling. Curr. Opin. Physiol. 4, 88–95. doi: 10.1016/j.cophys.2018.07.002
Bourgeois, F., Messéant, J., Kordeli, E., Petit, J. M., Delers, P., Bahi-Buisson, N., et al. (2015). A critical and previously unsuspected role for doublecortin at the neuromuscular junction in mouse and human. Neuromuscul. Disord. 25, 461–473. doi: 10.1016/j.nmd.2015.01.012
Budnik, V., and Salinas, P. C. (2011). Wnt signaling during synaptic development and plasticity. Curr. Opin. Neurobiol. 21, 151–159. doi: 10.1016/j.conb.2010.12.002
Burden, S. J., Huijbers, M. G., and Remedio, L. (2018). Fundamental molecules and mechanisms for forming and maintaining neuromuscular synapses. Int. J. Mol. Sci. 19:E490. doi: 10.3390/ijms19020490
Burden, S. J., Yomoto, N., and Zhang, W. (2013). The role of MuSK in synapse formation and neuromuscular disease. Cold Spring Harb. Perspect. Biol. 5:e009167. doi: 10.1101/cshperspect.a009167
Burgess, R. W., Nguyen, Q. T., Son, Y. J., Lichtman, J. W., and Sanes, J. R. (1999). Alternatively spliced isoforms of nerve- and muscle-derived agrin: their roles at the neuromuscular junction. Neuron 23, 33–44. doi: 10.1016/s0896-6273(00)80751-5
Burke, G., Hiscock, A., Klein, A., Niks, E. H., Main, M., Manzur, A. Y., et al. (2013). Sulbutamol benefits children with congenital myasthenic syndrome due to DOk7 mutations. Neuromuscul. Disord. 23, 170–175. doi: 10.1016/j.nmd.2012.11.004
Caldwell, J. H., Klevanski, M., Saar, M., and Müller, U. C. (2013). Roles of the amyloid precursor protein family in the peripheral nervous system. Mech. Dev. 130, 433–446. doi: 10.1016/j.mod.2012.11.001
Carson, S. S., Valdez, G., and Sanes, J. R. (2010). Presynaptic calcium channels and α3-integrins are complexed with synaptic cleft laminins, cytoskeletal elements and active zone components. J. Neurochem. 115, 654–666. doi: 10.1111/j.1471-4159.2010.06965.x
Cartaud, A., Strochlic, L., Guerra, M., Blanchard, B., Lambergeon, M., Krejci, E., et al. (2004). MuSK is required for anchoring acetylcholinesterase at the neuromuscular junction. J. Cell Biol. 165, 505–515. doi: 10.1083/jcb.200307164
Cetin, H., and Vincent, A. (2018). Pathogenic mechanisms and clinical correlations in autoimmune myasthenic syndromes. Semin. Neurol. 38, 344–354. doi: 10.1055/s-0038-1660500
Cetin, H., Webster, R., Liu, W. W., Nagaishi, A., Koneczny, I., Zimprich, F., et al. (2020). Myasthenia gravis AChR antibodies inhibit function of rapsyn-cluster AChRs. J. Neurol. Neurosurg. Psychiatry 91, 526–532. doi: 10.1136/jnnp-2019-322640
Chen, J., Billings, S. E., and Nishimune, H. (2011). Calcium channels link the muscle-derived synapse organizer laminin β2 to Bassoon and CAST/Erc2 to organize presynaptic active zones. J. Neurosci. 31, 512–525. doi: 10.1523/JNEUROSCI.3771-10.2011
Chen, Y., Ip, F. C. F., Shi, L., Zhang, Z., Tang, H., Ng, Y. P., et al. (2014). Coronin 6 regulates acetylcholine receptor clustering through modulating receptor anchorage to actin cytoskeleton. J. Neurosci. 34, 2413–2421. doi: 10.1523/JNEUROSCI.3226-13.2014
Chen, F., Qian, L., Yang, Z. H., Huang, Y., Ngo, S. T., Ruan, N. J., et al. (2007). Rapsyn interaction with calpain stabilizes AChR clusters at the neuromuscular junction. Neuron 55, 247–260. doi: 10.1016/j.neuron.2007.06.031
Choi Decroos, E., Hobson-Webb, L. D., Juel, V. C., Massey, J. M., and Sanders, D. B. (2014). Do acetylcholine receptor and striated muscle antibodies predict the presence of thymoma in patients with myasthenia gravis? Muscle Nerve 49, 30–34. doi: 10.1002/mus.23882
Choi, J., and Lee, S. Y. (2020). Clinical characteristics and treatment of immune-related adverse events of immune checkpoint inhibitors. Immune Netw. 20:e9. doi: 10.4110/in.2020.20.e9
Choi, H. Y., Liu, Y., Tennert, C., Sugiura, Y., Karakatsani, A., Kröger, S., et al. (2013). APP interacts with Lrp4 and agrin to coordinate the development of the neuromuscular junction in mice. Elife 2:e00220. doi: 10.7554/eLife.00220
Chou, H. J., Lai, D. M., Huang, C. W., McLennan, I. S., Wang, H. D., and Wang, P. Y. (2013). BMP4 is a peripherally-derived factor for motor neurons and attenuates glutamate-induced excitotoxicity in vitro. PLoS One 8:e58441. doi: 10.1371/journal.pone.0058441
Chan, Z., Oentaryo, M. J., and Lee, C. W. (2020). MMP-mediated modulation of ECM environment during axonal growth and NMJ development. Neurosci. Lett. 724:134822. doi: 10.1016/j.neulet.2020.134822
Clifford, K. M., Hobson-Webb, L. D., Benatar, M., Burns, T. M., Barnett, C., Silvestri, N. J., et al. (2019). Thymectomy may not be associated with clinical improvement in MuSK myasthenia gravis. Muscle Nerve 59, 404–410. doi: 10.1002/mus.26404
Cole, R. N., Reddel, S. W., Gervásio, O. L., and Phillips, W. D. (2008). Anti-MuSK patient antibodies disrupt the mouse neuromuscular junction. Ann. Neurol. 63, 782–789. doi: 10.1002/ana.21371
Constantin, B. (2014). Dystrophin complex functions as a scaffold for signaling proteins. Biochim. Biophys. Acta 1838, 635–642. doi: 10.1016/j.bbamem.2013.08.023
Cordts, I., Bodart, N., Hartmann, K., Karagiorgou, K., Tzartos, J. S., Mei, L., et al. (2017). Screening for lipoprotein receptor-protein 4-, agrin- and titin-antibodies and exploring the autoimmune spectrum in myasthenia gravis. J. Neurol. 264, 1193–1203. doi: 10.1007/s00415-017-8514-z
Cortés-Vicente, E., Gallardo, E., Martínez, M. Á., Díaz-Manera, J., Querol, L., Rojas-García, R., et al. (2016). Clinical characteristics of patients with double-seronegative myasthenia gravis and antibodies to cortactin. JAMA Neurol. 73, 1099–1104. doi: 10.1001/jamaneurol.2016.2032
Cossins, J., Belaya, K., Zoltowska, K., Koneczny, I., Maxwell, S., Jacobson, L., et al. (2012). The search for new antigenic targets in myasthenia gravis. Ann. N Y Acad. Sci. 1275, 123–128. doi: 10.1111/j.1749-6632.2012.06833.x
Dalakas, M. C. (2015). Future perspectives in target-specific immunotherapies of myasthenia gravis. Ther. Adv. Neurol. Disord. 8, 316–327. doi: 10.1177/1756285615605700
Dalakas, M. C. (2018). Neurological complications of immune checkpoint inhibitors: what happens when you ‘take the brakes off’ the immune system. Ther. Adv. Neurol. Disord. 11:1756286418799864. doi: 10.1177/1756286418799864
Dalakas, M. C. (2019). Immunotherapy in myasthenia gravis in the era of biologics. Nat. Rev. Neurol 15, 113–124. doi: 10.1038/s41582-018-0110-z
Danikowski, K. M., Jayaraman, S., and Prabhaakar, B. S. (2017). Regulatory T cells in multiple sclerosis and myasthenia gravis. J. Neuroinflammation 14:117. doi: 10.1186/s12974-017-0892-8
Di Stefano, V., Lupica, A., Rispoli, M. G., Di Muzio, A., Brighina, F., and Rodolico, C. (2020). Rituximab in AChR subtype of myasthenia gravis: systematic review. J. Neurol. Neurosurg. Psychiatry 91, 392–395. doi: 10.1136/jnnp-2019-322606
Díaz-Manera, J., Martínez-Hernández, E., Querol, L., Klooster, R., Rojas-García, R., Suárez-Calvet, X., et al. (2012). Long-lasting treatment effect of rituximab in MuSK myasthenia. Neurology 78, 189–193. doi: 10.1212/WNL.0b013e3182407982
Drachman, D. B., Adams, R. N., Josifek, L. F., and Self, S. G. (1982). Functional activities of antibodies to acetylcholine receptors and clinical severity of myasthenia gravis. N. Engl. J. Med. 307, 769–775. doi: 10.1056/NEJM19820.9233071.301
Drachman, D. B., Angus, C. W., Adams, R. N., Michelson, J. D., and Hoffamn, G. J. (1978). Myasthenic antibodies cross-link acetylcholine receptors to accelerate degradation. N. Engl. J. Med. 298, 1116–1122. doi: 10.1056/nejm197805182982004
Dubey, D., David, W. S., Reynolds, K. L., Chute, D. F., Chement, N. F., Cohen, J. V., et al. (2020). Severe neurological toxicity in immune checkpoint inhibitors: growing spectrum. Ann. Neurol. 87, 659–669. doi: 10.1002/ana.25708
Dusl, M., Moreno, T., Munell, F., Macaya, A., Gratacòs, M., Abicht, A., et al. (2019). Congenital myasthenic syndrome caused by novel COL13A1 mutations. J. Neurol. 266, 1107–1112. doi: 10.1007/s00415-019-09239-7
Eguchi, T., Tezuka, T., Fukudome, T., Watanabe, Y., Sagara, H., and Yamanashi, Y. (2020). Overespression of Dok-7 in skeletal muscle enhances neuromuscular transmission with structural alterations of neuromuscular junctions: Implications in robustness of neuromuscular transmission. Biochem. Biophys. Res. Commun. 523, 214–219. doi: 10.1016/j.bbrc.2019.12.011
Engel, A. G., Shen, X. M., Selcen, D., and Sine, S. M. (2015). Congenital myasthenic syndromes: pathogenesis, diagnosis, and treatment. Lancet Neurol. 144, 420–434. doi: 10.1016/S1474-4422(14)70201-7
Evoli, A., Alboini, P. E., Damato, V., Iorio, R., Provenzano, C., Bartoccioni, E., et al. (2018). Myasthenia gravis with antibodies to MuSK: an update. Ann. N Y Acad. Sci. 1412, 82–89. doi: 10.1111/nyas.13518
Evoli, A., Bianchi, M. R., Riso, R., Minicuci, G. M., Batocchi, A. P., Servidei, S., et al. (2008). Response to therapy in myasthenia gravis with anti-MuSK antibodies. Ann. N Y Acad. Sci. 1132, 76–83. doi: 10.1196/annals.1405.012
Evoli, A., and Padura, J. (2013). Dianosis and therapy of myasthenia gravis with antibodies to muscle-specific kinase. Autoimmun. Rev. 12, 931–935. doi: 10.1016/j.autrev.2013.03.004
Frey, H., Schroeder, N., Manon-Jensen, T., Iozzo, R. V., and Schaefer, L. (2013). Biological interplay between proteoglycans and their innate immune receptors in inflammation. FEBS J. 280, 2165–2179. doi: 10.1111/febs.12145
Gable, K. L., and Guptill, J. T. (2020). Antagonism of the neonatal Fc receptor as an emerging treatment for myasthenia gravis. Front. Immunol. 10:3052. doi: 10.3389/fimmu.2019.03052
Gallardo, E., Martinez-Hernández, E., Titulaer, M. J., Huijbers, M. G., Martinez, M. A., Ramos, A., et al. (2014). Cortactin autoantibodies in myasthenia gravis. Autoimmun. Rev. 13, 1003–1007. doi: 10.1016/j.autrev.2014.08.039
Gallenmller, C., Mller-Felber, W., Dusl, M., Strucka, R., Guergueltcheva, V., Blaschek, A., et al. (2014). Salbutamol-responsive limb-girdle congenital myasthenic syndome due to a novel missense mutation and heteroallelic deletion in MuSK. Neuromuscul. Disord. 24, 31–35. doi: 10.1016/j.nmd.2013.08.002
Gasperi, C., Melms, A., Schoser, B., Zhang, Y., Meltoranta, J., Risson, V., et al. (2014). Anti-agrin autoantibodies in myasthenia gravis. Neurology 82, 1976–1983. doi: 10.1212/WNL.0000000000000478
Gesemann, M., Denzer, A. J., and Ruegg, M. A. (1995). Acetylcholine receptor-aggregating activity of agrin isoforms and mapping of the active state. J. Cell Biol. 128, 625–636. doi: 10.1083/jcb.128.4.625
Ghazanfari, N., Fernandez, K. J., Murata, Y., Morsch, M., Ngo, S. T., Reddel, S. W., et al. (2011). Muscle specific kinase: organizer of synaptic membrane domains. Int. J. Biochem. Cell Biol. 43, 295–298. doi: 10.1016/j.biocel.2010.10.008
Ghazanfari, N., Morsch, M., Tse, N., Reddel, S. W., and Phillips, W. D. (2014). Effects of the β2-adrenoceptor agonist, alubuterol, in a mouse model of anti-MuSK myasthenia gravis. PLoS One 9:e87840. doi: 10.1371/journal.pone.0087840
Gilhus, N. E., Tzartos, S., Evoli, A., Palace, J., Burns, T. M., and Verschuuren, J. J. G. M. (2019). Myasthenia gravis. Nat. Rev. Dis. Primers 5:30. doi: 10.1038/s41572-019-0079-y
Gomez, A. M., Stevens, J. A., Mané-Damas, M., Molenaar, P., Duimel, H., Verheyen, F., et al. (2016). Silencing of Dok-7 in adult rat muscle increases susceptibility to passive transfer myasthenia gravis. Am. J. Pathol. 186, 2559–2568. doi: 10.1016/j.ajpath.2016.05.025
Gomez, A. M., Willcox, N., Vrolix, K., Hummel, J., Nogales-Gadea, G., Saxena, A., et al. (2014). Proteasome inhibition with bortezomid depletes plasma cells and specific autoantibody production in primary thymic cell cultures from early-onset myasthenia gravis patients. J. Immunol. 193, 1055–1063. doi: 10.4049/jimmunol.1301555
Gordon, L. R., Gribble, K. D., Syrett, C. M., and Granato, M. (2012). Initiation of synapse formation by Wnt-induced MuSK endocytosis. Development 139, 1023–1033. doi: 10.1242/dev.071555
Guarino, S. R., Canciani, A., and Ferneris, F. (2020). Dissecting the extracellular complexity of neuromuscular junction organizers. Front. Mol. Biosci. 6:156. doi: 10.3389/fmolb.2019.00156
Guptill, J. T., Sanders, D. B., and Evoli, A. (2011). Anti-MuSK antibody myasthenia gravis: clinical findings and response to treatment in two large cohorts. Muscle Nerve 44, 36–40. doi: 10.1002/mus.22006
Ha, J. C., and Richman, D. P. (2015). Myasthenia gravis and related disorders: pathology and molecular pathogenesis. Biochem. Biophys. Acta 1852, 651–657. doi: 10.1016/j.bbadis.2014.11.022
Hallock, P. T., Chin, S., Blais, S., Neubert, T. A., and Glass, D. J. (2015). Sorbs1 and -2 interact with CrkL and are required for acetylcholine receptor cluster formation. Mol. Cell. Biol. 36, 262–270. doi: 10.1128/mcb.00775-15
Hallock, P. T., Xu, C. F., Park, T. J., Neubert, T. A., Curran, T., and Burden, S. J. (2010). Dok-7 regulates neuromuscular synapse formation by recruiting Crk and Crk-L. Genes Dev. 24, 2451–2461. doi: 10.1101/gad.1977710
Harris, K. P., and Littleton, J. T. (2015). Transmission, development, and plasticity of synapses. Genetics 201, 345–375. doi: 10.1534/genetics.115.176529
Heikkinen, A., Härönen, H., Norman, O., and Pihlajaniemi, T. (2019). Collagen XIII and other ECM components in the assembly and disease of the neuromuscular junction. Anat. Rec. doi: 10.1002/ar.24092 [Epub ahead of print].
Helgeland, G., Petzold, A., Luckman, S. P., Gilhus, N. E., Plant, G. T., and Romi, F. R. (2011). Matrix metalloproteinases in myasthenia gravis. Eur. Neurol. 65, 53–58. doi: 10.1159/000322737
Henríquez, J. P., Krull, C. E., and Osses, N. (2011). The Wnt and BMP families of signaling morphogens at the vertebrate neuromuscular junction. Int. J. Mol. Sci. 12, 8924–8946. doi: 10.3390/ijms12128924
Henriquez, J. P., and Salinas, P. C. (2012). Dual roles for Wnt signaling during the formation of the vertebrate neuromuscular junction. Acta Physiol. 204, 128–136. doi: 10.1111/j.1748-1716.2011.02295.x
Henriquez, J. P., Webb, A., Bence, M., Bildose, H., Sahores, M., Hughes, S. M., et al. (2008). Wnt signaling promotes AChR aggregation at the neuromuscular synapse in collaboration with agrin. Proc. Natl. Acad. Sci. U S A 105, 18812–18817. doi: 10.1073/pnas.0806300105
Herbst, R. (2019). MuSK function during health and disease. Neurosci. Lett. 716:134676. doi: 10.1016/j.neulet.2019.134676
Hettwer, S., Lin, S., Kucsera, S., Haubitz, M., Oliveri, F., Fariello, R. G., et al. (2014). Injection of soluble fragment of neural agrin (N-1654) considerably improves the muscle pathology caused by the disassembly of the neuromuscular junction. PLoS One 9:e88739. doi: 10.1371/journal.pone.0088739
Hezel, M., de Groat, W. C., and Galbiati, F. (2010). Caveolin-3 promotes nicotinic acetylcholine receptor clustering and regulates neuromuscular junction activity. Mol. Biol. Cell 21, 302–310. doi: 10.1091/mbc.e09-05-0381
Higuchi, O., Hamuro, J., Motomura, M., and Yamanashi, Y. (2011). Autoantibodies to low-density lipoprotein receptor-related protein 4 in myasthenia gravis. Ann. Neurol. 69, 418–422. doi: 10.1002/ana.22312
Hoch, W., Campanelli, J. T., Harrison, S., and Scheller, R. H. (1994). Structural domains of agrin required for clustering of nicotinic acetylcholine receptors. EMBO J. 13, 2814–2821. doi: 10.1002/j.1460-2075.1994.tb06575.x
Hoch, W., McConville, J., Helmes, S., Newsom-Davis, J., Melms, A., and Vincent, A. (2001). Auto-antibodies to the receptor tyrosine kinase MuSK in patients with myasthenia gravis without acetylcholine receptor antibodies. Nat. Med. 7, 365–368. doi: 10.1038/85520
Hochenester, E., and Yurchenco, P. D. (2013). Laminins in basement membrane assembly. Cell Adh. Migr. 7, 56–63. doi: 10.4161/cam.21831
Hoshi, T., Tezuka, T., Yokoyama, K., Iemura, S., Natsume, T., and Yamanashi, Y. (2013). Mesdc2 plays a key role in cell-surface expression of Lrp4 and postsynaptic specialization in myotubes. FEBS Lett. 587, 3749–3754. doi: 10.1016/j.febslet.2013.10.001
Howard, J. F. Jr. (2018). Myasthenia gravis: the role of complement at the neuromuscular junction. Ann. N Y Acad. Sci. 1412, 113–128. doi: 10.1111/nyas.13522
Howard, J. F. Jr., Bril, V., Burns, T. M., Mantegazza, R., Bilinska, M., Szczudlik, A., et al. (2019). Randomized phase 2 study of FcRn antagonist efgartigimod in generalized myasthenia gravis. Neurology 92, e2661–e2673. doi: 10.1212/wnl.0000000000007600
Howard, J. F. Jr., Nowak, R. J., Wolfe, G. I., Freimer, M. L., Vu, T. H., Hinton, J. L., et al. (2020). Clinical effects of the self-administered subcutaneous complement inhibitor Zilucoplan in patients with moderate to severe generalized myasthenia gravis: results of a phase 2 randomized double-blind, placebo-controlled, multicenter clinical trial. JAMA Neurol. doi: 10.1001/jamaneurol.2019.5125 [Epub ahead of print].
Härönen, H., Zainul, Z., Naumenko, N., Sormunen, R., Miinalainen, I., Shakirzyanova, A., et al. (2019). Correct expression and localization of collagen XIII are crucial for the normal formation and function of the neuromuscular system. Eur. J. Neurosci. 49, 1491–1511. doi: 10.1111/ejn.14346
Härönen, H., Zainul, Z., Tu, H., Naumenko, N., Sormunen, R., Miinalainen, I., et al. (2017). Collagen XIII secures pre- and postsynaptic integrity of the neuromuscular synapse. Hum. Mol. Genet. 26, 2076–2090. doi: 10.1093/hmg/ddx101
Huda, R. (2020). New approaches to targeting B cells for myasthenia gravis. Front. Immunol. 11:240. doi: 10.3389/fimmu.2020.00240
Huda, S., Cao, M., De Rosa, A., Woodhall, M., Rodriquez Cruz, P. M., Cossins, J., et al. (2020). SHP2 inhibitor protects AChRs from effects of myasthenia gravis MuSK antibody. Neurol. Neuroimmunol. Neuroinflamm. 7:e645. doi: 10.1212/nxi.0000000000000645
Huda, S., Waters, P., Woodhall, M., Leite, M. I., Jacobson, L., De Rosa, A., et al. (2017). IgG-specific cell-based assay detected potentially pathogenic MuSK-abs in seronegative MG. Neurol. Neuroimmunol. Neuroinflamm. 4:e357. doi: 10.1212/nxi.0000000000000357
Huijbers, M. G., Plomp, J. J., van Es, I. E., Fillié-Grijpma, Y. E., Kamar-Al Majidi, S., Ulrichts, P., et al. (2019a). Efgartigimod improves muscle weakness in a mouse model for muscle-specific kinase myasthenia gravis. Exp. Neurol. 317, 133–143. doi: 10.1016/j.expneurol.2019.03.001
Huijbers, M. G., Vergoossen, D. L., Fillié-Grijpma, Y. E., van Es, I. E., Koning, M. T., Slot, L. M., et al. (2019b). MuSK myasthenia gravis monoclonal antibodies: valency dictates pathogenicity. Neurol. Neuroimmunol. Neuroinflamm. 6:e547. doi: 10.1212/nxi.0000000000000547
Huijbers, M. G., Vink, A. F., Niks, E. H., Westhuis, R. H., van Zwet, E. W., de Meel, R. H., et al. (2016). Longitudinal epitope mapping in MuSK myasthenia gravis: implications for disease severity. J. Neuroimmunol. 291, 82–88. doi: 10.1016/j.jneuroim.2015.12.016
Huijbers, M. G., Zhang, W., Klooster, R., Niks, E. H., Friese, M. B., Staasheijm, K. R., et al. (2013). MuSK IgG4 autoantibodies cause myasthenia gravis by inhibiting binding between MuSK and Lrp4. Proc. Natl. Acad. Sci. U S A 110, 20783–20788. doi: 10.1073/pnas.1313944110
Hurtado, E., Cilleros, V., Nadal, L., Simö, A., Obis, T., Gracia, N., et al. (2017). Muscle contraction regulates BDNF/TrkB signaling to modulate synaptic function through presynaptic cPKCα and cPKCβI. Front. Mol. Neurosci. 10:147. doi: 10.3389/fnmol.2017.00147
Illa, I., Cortés-Vicente, E., Martinez, M. Á., and Gallardo, E. (2018). Dioagnostic utility of cortactin antibodies in myasthenia gravis. Ann. N Y Acad. Sci. 1412, 90–94. doi: 10.1111/nyas.13502
Inoue, A., Setoguchi, K., Matsubara, Y., Okada, K., Sato, N., Iwakura, Y., et al. (2009). Dok-7 activates the muscle receptor kinase MuSK and shapes synaptic formation. Sci. Signal. 2:ra7. doi: 10.1126/scisignal.2000113
Ito, M., and Ohno, K. (2018). Protein-anchoring therapy to target extracellular matrix proteins to their physiological destinations. Matrix Biol. 68–69, 628–636. doi: 10.1016/j.matbio.2018.02.014
Iwasa, K., Furukawa, Y., Yoshikawa, H., and Yamada, M. (2016). Caveolin-3 is aberrantly expressed in skeletal muscle cells in myasthenia gravis. J. Neuroimmunol. 301, 30–34. doi: 10.1016/j.jneuroim.2016.10.011
Iwasa, K., Komai, K., and Takamori, M. (1998). Spontaneous thymoma rat as a model for myasthenic weakness caused by anti-ryanodine receptor antibodies. Muscle Nerve 21, 1655–1660. doi: 10.1002/(sici)1097-4598(199812)21:12<1655::aid-mus5>3.0.co;2-f
Iwasa, K., Yoshikawa, H., Furukawa, Y., and Yamada, M. (2018). Programmed cell death ligand 1 expression is upregulated in the skeletal muscle of patients with myasthenia gravis. J. Neuroimmunol. 325, 74–78. doi: 10.1016/j.jneuroim.2018.09.012
Jing, L., Lefebvre, J. L., Gordon, L. R., and Granato, M. (2009). Wnt signals organize synaptic prepattern and axon guidance through the Zebrafish unplugged/MuSK receptor. Neuron 61, 721–733. doi: 10.1016/j.neuron.2008.12.025
Johansen, A., Christensen, S. J., Scheie, D., Højgaard, J. L. S., and Kondziella, D. (2019). Neuromuscular adverse events associated with anti-PD-1 monoclonal antibodies: systemic review. Neuerology 92, 663–674. doi: 10.1212/wnl.0000000000007235
Karmouch, J., Dobbertin, A., Sigoillot, S., and Legay, C. (2013). Developmental consequences of the ColQ/MuSK interactions. Chem. Biol. Interact. 203, 287–291. doi: 10.1016/j.cbi.2012.10.006
Kawakami, Y., Ito, M., Hirayama, M., Sahashi, K., Ohkawara, B., Masuda, A., et al. (2011). Anti-MuSK autoantibodies block binding of collagen Q to MuSK. Neurology 77, 1819–1826. doi: 10.1212/WNL.0b013e318237f660
Kim, Y., and Saffen, D. (2005). Activation of M1 muscarinic acetylcholine receptors stimulates the formation of multiprotein complex centered on TRPC6 channels. J. Biol. Chem. 280, 32035–32047. doi: 10.1074/jbc.m500429200
Kim, N., Stiegler, A. L., Cameron, T. O., Hallock, P. T., Gomez, A. M., Huang, J. H., et al. (2008). Lrp4 is a receptor for agrin and forms a complex with MuSK. Cell 135, 334–342. doi: 10.1016/j.cell.2008.10.002
Klooster, R., Plomp, J. J., Huijbers, M. G., Niks, E. H., Straasheijm, K. R., Detmers, F. J., et al. (2012). Muscle-specific kinase myasthenia gravis IgG4 autoantibodies cause severe neuromuscular junction dysfunction in mice. Brain 135, 1081–1101. doi: 10.1093/brain/aws025
Koles, K., and Budnik, V. (2012). Wnt signaling in neuromuscular junction development. Cold Spring Harb. Perspect. Biol. 4:a008045. doi: 10.1101/cshperspect.a008045
Koles, K., Nunnari, J., Korkut, C., Barria, R., Brewer, C., Li, Y., et al. (2012). Mechanism of evenness interrupted (Evi)-exosome release at synaptic boutons. J. Biol. Chem. 287, 16820–16834. doi: 10.1074/jbc.m112.342667
Koneczny, I. (2018). A new classification systemfor IgG4 autoantibodies. Front. Immunol. 9:97. doi: 10.3389/fimmu.2018.00097
Koneczny, I., Cossins, J., and Vincent, A. (2014). The role of muscle-specific tyrosine kinase (MuSK) and mystery of MuSK myasthenia gravis. J. Anat. 224, 29–35. doi: 10.1111/joa.12034
Koneczny, I., Cossins, J., Waters, P., Beeson, D., and Vincent, A. (2013). MuSK myasthenia gravis IgG4 disrupts the interaction of Lrp4 with MuSK but both IgG4 and IgG1–3 can disperse preformed agrin-independent AChR clusters. PLoS One 8:e80695. doi: 10.1371/journal.pone.0080695
Koneczny, I., and Herbst, R. (2019). Myasthenia gravis: pathogenic effects of autoantibodies on neuromuscular architecture. Cells 8, 671–705. doi: 10.3390/cells8070671
Körber, C., and Kuner, T. (2016). Molecular machines regulating the release probability of synaptic vesicles at the active zone. Front. Synaptic Neurosci. 8:5. doi: 10.3389/fnsyn.2016.00005
Kumar, P., Bhattacharya, P., and Prabhakar, B. S. (2018). A comprehensive review on the role of co-signalling receptors and Treg homeostasis in autoimmunity and tumor immunity. J. Autoimmun. 95, 77–99. doi: 10.1016/j.jaut.2018.08.007
Kumar, P., Saini, S., and Prabhakar, B. S. (2019). Cancer immunotherapy with ckeck point inhibitor can cause autoimmune adverse events due to loss of Treg. Semin. Cancer Biol. doi: 10.1016/j.semcancer.2019.01.006 [Epub ahead of print].
Kummer, T. T., Misgeld, T., and Sanes, J. R. (2006). Assembly of the postsynaptic membrane at the neuromuscular junction: paradigm lost. Curr. Opin. Neurobiol. 16, 74–82. doi: 10.1016/j.conb.2005.12.003
Kusner, L. L., Satija, N., Cheng, G., and Kaminski, H. J. (2014). Targeting therapy to the neuromuscular junction: proof of concept. Muscle Nerve 49, 749–756. doi: 10.1002/mus.24057
Kusner, L. L., Yucius, K., Sengupta, M., Sprague, A. G., Desai, D., Nguyen, T., et al. (2019). Investigational RNAi therapeutic targeting C5 is efficacious in pre-clinical models of myasthenia gravis. Mol. Ther. Methods Clin. Dev. 13, 484–492. doi: 10.1016/j.omtm.2019.04.009
Lashley, D., Palace, J., Jayawant, S., Robb, S., and Beeson, D. (2010). Ephedrine treatment in congenital myasthenic syndrome due to mutations in Dok7. Neurology 74, 1517–1523. doi: 10.1212/wnl.0b013e3181dd43bf
Latvanlehto, A., Fox, M. A., Sormunen, R., Tu, H., Oikarainen, T., Koski, A., et al. (2010). Muscle-derived collagen XIII regulates maturation of the skeletal neuromuscular junction. J. Neurosci. 30, 12230–12241. doi: 10.1523/JNEUROSCI.5518-09.2010
Lazaridis, K., Baltatzidour, V., Tektonidis, N., and Tzartos, S. J. (2020). Antigen-specific immunoadsorption of MuSK autoantibodies as a treatment of MuSK-induced experimental autoimmune myasthenia gravis. J. Neuroimmunol. 339:577136. doi: 10.1016/j.jneuroim.2019.577136
Lazaridis, K., and Tzartos, S. J. (2020). Antibody specificities in myasthenia gravis; implications for improved diagnostics and therapeutics. Front. Immunol. 11:212. doi: 10.3389/fimmu.2020.00212
Lee, Y., Rudell, J., and Ferns, M. (2009). Rapsyn interacts with the muscle acetylcholine receptor via α-helical domains in the α, β, and ε subunit intracellular loops. Neuroscience 163, 222–232. doi: 10.1016/j.neuroscience.2009.05.057
Legay, C., and Mei, L. (2017). Moving forward with the neuromuscular junction. J. Neurochem. 142, 59–63. doi: 10.1111/jnc.14028
Leite, M. I., Ströbel, P., Jones, M., Micklem, K., Moritz, R., Gold, R., et al. (2005). Fewer thymic changes in MuSK antibody-positive than in MuSK antibody-negative MG. Ann. Neurol. 57, 444–448. doi: 10.1002/ana.20386
Li, L., Cao, Y., Wu, H., Ye, X., Zhu, Z., Xing, G., et al. (2016). Enzymatic activity of the scaffold protein rapsyn for synapse formation. Neuron 92, 1007–1019. doi: 10.1016/j.neuron.2016.10.023
Li, X. M., Dong, X. P., Luo, S. W., Zhang, B., Lee, D. H., Ting, A. K., et al. (2008). Retrograde regulation of motoneuron differentiation by muscle-β-catenin. Nat. Neurosci. 11, 262–268. doi: 10.1038/nn2053
Li, Y., Guptill, J. T., Russo, M. A., Massey, J. M., Jud, V. C., Hobson-Webb, L. D., et al. (2019). Tacrolimus inhibits Th1 and Th12 responses in MuSK-antibody positive myasthenia gravis patients. Exp. Neurol. 312, 43–50. doi: 10.1016/j.expneurol.2018.11.006
Li, Z., Li, M., Wood, K., Hettwer, S., Muley, S. A., Shi, F. D., et al. (2018). Engineered agrin attenuates the severity of experimental autoimmune myasthenia gravis. Muscle Nerve 57, 814–820. doi: 10.1002/mus.26025
Li, L., Xiong, W. C., and Mei, L. (2018). Neuromuscular junction formation, aging, and disorders. Annu. Rev. Physiol. 80, 159–188. doi: 10.1146/annurev-physiol-022516-034255
Li, Y., Zhang, Y., Cai, G., He, D., Dai, Q., Xu, Z., et al. (2017). Anti-Lrp4 autoantibodies in chinese patients with myasthenia gravis. Muscle Nerve 56, 938–942. doi: 10.1002/mus.25591
Liewluck, T., Selecen, D., and Engel, A. G. (2011). Beneficial effects of Albuterol in congenital endplate acetylcholinesterase deficiency and DOK-7 myasthenia. Muscle Nerve 44, 789–794. doi: 10.1002/mus.22176
Lin, W., Dominguez, B., Yang, J., Aryal, P., Brandon, E. P., Gage, F. H., et al. (2005). Neurotransmitter acetylcholine negatively regulates neuromuscular synapse formation by a Cdk5-dependent mechanism. Neuron 46, 569–579. doi: 10.1016/j.neuron.2005.04.002
Linnoila, J., Wang, Y., Yao, Y., and Wang, Z. Z. (2008). A mammalian homolog of Drosophila tumorous imaginal discs, Tid1, mediates agrin signaling at the neuromuscular junction. Neuron 60, 625–641. doi: 10.1016/j.neuron.2008.09.025
Lipka, A. F., Vrinten, C., van Zwet, E. W., Schirmmel, K. J., Cornel, M. C., Kuijpers, M. R., et al. (2017). Ephedrine treatment for autoimmune myasthenia gravis. Neuromuscul. Disord. 27, 259–265. doi: 10.1016/j.nmd.2016.11.009
Litchman, T., Roy, B., Kumar, A., Sharma, A., Nike, V., and Nowak, R. J. (2020). Differential response to rituximab in anti-AChR and anti-MuSK positive myasthenia gravis patients: a single-center retrospective study. J. Neurol. Sci. 411:116690. doi: 10.1016/j.jns.2020.116690
Liu, R., Zhou, Q., La Cava, A., Campagnolo, D. I., Van Kaer, L., and Shi, F. D. (2010). Expansion of regulatory T cells via IL-2/anti-IL-2 mAb complexes suppresses experimental myasthenia. Eur. J. Immunol. 40, 1577–1589. doi: 10.1002/eji.200939792
Logan, C. V., Cossins, J., Rodríguez Cruz, P. M., Parry, D. A., Maxwell, S., Martínez-Martínez, P., et al. (2015). Congenital myasthenic syndrome type 19 is caused by mutations in COL13A1, encoding the atypical non-fibrillar collagen type XIII α1 chain. Am. J. Hum. Genet. 97, 878–885. doi: 10.1016/j.ajhg.2015.10.017
Lorenzoni, P. J., Scola, R. H., Kay, C. S. K., Filla, L., Miranda, A. P. P., Pinherio, J. M. R., et al. (2013). Salbutamol therapy in congenital myasthenic syndrome due to DOK7 mutation. J. Neurol. Sci. 331, 155–157. doi: 10.1016/j.jns.2013.05.017
Losen, M., Stassen, M. H., Martínez-Martínez, P., Machiels, B. M., Duimel, P., Frederik, P., et al. (2005). Increased expression of rapsyn in muscle prevents acetylcholine receptor loss in experimental autoimmune myasthenia gravis. Brain 128, 2327–2337. doi: 10.1093/brain/awh612
Luckman, S. P., Gilhus, N. E., and Romi, F. (2011). Matrix metalloproteinase-3 in myasthenia gravis compared to other neurological disorders and healthy controls. Autoimmune Dis. 2011, 151258–151261. doi: 10.4061/2011/151258
Luo, Z. G., Wang, Q., Zhou, J. Z., Wang, J., Luo, Z., Liu, M., et al. (2002). Regulation of AChR clustering by Dishevelled interacting with MuSK and PAK1. Neuron 35, 489–505. doi: 10.1016/s0896-6273(02)00783-3
Luo, S., Zhang, B., Dong, X. P., Tao, Y., Ting, A., Zhou, Z., et al. (2008). HSP90 β regulates rapsyn turnover and subsequent AChR cluster formation and maintenance. Neuron 60, 97–110. doi: 10.1016/j.neuron.2008.08.013
Ma, J., Kelly, L., Ingram, J., Price, T. J., Meriney, S. D., and Dittrich, M. (2015). New insights into short-term synaptic facilitation at the frog neuromuscular junction. J. Neurophysiol. 113, 71–87. doi: 10.1152/jn.00198.2014
Madhavan, R., Gong, Z. L., Ma, J. J., Chan, A. W. S., and Peng, H. B. (2009). The function of cortactin in the clustering of acetylcholine receptors at the vertebrate neuromuscular junction. PLoS One 4:e8478. doi: 10.1371/journal.pone.0008478
Marino, M., Scuderi, F., Samengo, D., Saltelli, G., Maiuri, M. T., Shen, C., et al. (2015). Flow cytofluorimetric analysis of anti-Lrp4 (LDL receptor-related protein 4) autoantibodies in Italian patients with myasthenia gravis. PLoS One 10:e0135378. doi: 10.1371/journal.pone.0135378
Marx, A., Pfister, F., Schalke, B., Saruhan-Direskeneli, G., Melms, A., and Strobel, P. (2013). The different roles of the thymus in the pathogenesis of the various myasthenia gravis subtypes. Autoimmun. Rev. 12, 875–884. doi: 10.1016/j.autrev.2013.03.007
Maselli, R. A., Arredondo, J., Cagney, O., Ng, J. J., Anderson, J. A., Williams, C., et al. (2010). Mutations in MUSK causing congenital myasthenic syndrome impair MuSK-Dok-7 interaction. Hum. Mol. Genet. 19, 2370–2379. doi: 10.1093/hmg/ddq110
Maselli, R. A., Arredondo, J., Ferns, M. J., and Wollmann, R. L. (2012). Synaptic basal lamina-associated congenital myasthenic syndrome. Ann. N Y Acad. Sci. 1275, 36–48. doi: 10.1111/j.1749-6632.2012.06807.x
McConville, J., Farrugia, M. E., Beeson, D., Kishore, U., Metcalfe, R., Newsom-Davis, J., et al. (2004). Detection and characterization of MuSK antibodies in seronegative myasthenia gravis. Ann. Neurol. 55, 580–584. doi: 10.1002/ana.20061
McMacken, G. M., Spendiff, S., Whittaker, R. G., O’Connor, E., Howarth, B. M., Boczonadi, V., et al. (2019). Salbutamol modifies the neuromuscular junction in a mouse model of ColQ myasthenic syndrome. Hum. Mol. Genet. 28, 2339–2351. doi: 10.1093/hmg/ddz059
Messéant, J., Dobbertin, A., Girard, E., Delers, P., Manuel, M., Mangione, F., et al. (2015). MuSK Frizzled-like domain is critical for mammalian neuromuscular junction formation and maintenance. J. Neurosci. 35, 4926–4941. doi: 10.1523/JNEUROSCI.3381-14.2015
Messéant, J., Ezan, J., Delers, P., Glebov, K., Marchiol, C., Lager, F., et al. (2017). Wnt proteins contribute to neuromuscular junction formation through distinct signaling pathways. Development 144, 1712–1724. doi: 10.1242/dev.146167
Möhn, N., Beutel, G., Gutzmer, R., Ivanyi, P., Satzger, I., and Skripu, T. (2019). Neurological immune related adverse events associated with Nivolumab, Ipilimumab, and pembrolizumab therapy-review of the literature and future outlook. J. Clin. Med. 8:E1777. doi: 10.3390/jcm8111777
Mohseni, P., Sung, H. K., Murphy, A. J., Laliberte, C. L., Pallari, H. M., Henkelman, M., et al. (2011). Nestin is not essential for development of the CNS but required for dispersion of acetylcholine receptor clusters at the area of neuromuscular junctions. J. Neurosci. 31, 11547–11552. doi: 10.1523/JNEUROSCI.4396-10.2011
Molin, C. J., Westerberg, E., and Pung, A. R. (2017). Profile of upregulated inflammatory proteins in sera of myasthenia gravis patients. Sci. Rep. 7:39716. doi: 10.1038/srep39716
Moreth, K., Iozzo, R. V., and Schaefer, L. (2012). Small leucine-rich proteoglycans orchestrate receptor crosstalk during inflammation. Cell Cycle 11, 2084–2901. doi: 10.1038/srep39716
Mori, S., Kubo, S., Akiyoshi, T., Yamada, S., Miyazaki, T., Hotta, H., et al. (2012). Antibodies against muscle-specific kinase impair both presynaptic and postsynaptic functions in a murine model of myasthenia gravis. Am. J. Pathol. 180, 798–810. doi: 10.1016/j.ajpath.2011.10.031
Mori, S., Motohashi, N., Takashima, R., Kishi, M., Nishimune, H., and Shigemoto, K. (2017). Immunization of mice with Lrp4 induces myasthenia similar to MuSK-associated myasthenia gravis. Exp. Neurol. 297, 158–167. doi: 10.1016/j.expneurol.2017.08.006
Morren, J., and Li, Y. (2018). Myasthenia gravis with musk-specific tyrosine kinase antibodies: a narrative review. Muscle Nerve 58, 344–358. doi: 10.1002/mus.26107
Morren, J., and Li, Y. (2020). Maintenance immunosuppression in myasthenia gravis, an update. J. Neurol. Sci. 410:116648. doi: 10.1016/j.jns.2019.116648
Muppidi, S., Utsugisawa, K., Benatar, M., Murai, H., Barohn, R. J., Illa, I., et al. (2019). Long-term safety and efficacy of eculizumab in generalized myasthenia gravis. Muscle Nerve 60, 14–24. doi: 10.1002/mus.26447
Mygland, A., Tysnes, O. B., Matre, R., Volpe, P., Aarli, J. A., and Gilhus, N. E. (1992). Ryanodine receptor autoantibodies in myasthenia gravis patients with a thymoma. Ann. Neurol. 32, 589–591. doi: 10.1002/ana.410320419
Nadal, L., Garcia, N., Hurtado, E., Simó, A., Tomàs, M., Lanuza, M. A., et al. (2016). Presynaptic muscarinic acetylcholine autoreceptors (M1, M2 and M4 subtypes), adenosine receptors (A1 and A2A) and tropomyosin-related kinase B receptor (TrkB) modulate the developmental synapse elimination process at the neuromuscular junction. Mol. Brain 9:67. doi: 10.1186/s13041-016-0248-9
Nakashima, H., Ohkawara, B., Ishigaki, S., Fukudome, T., Ito, K., Tsushima, M., et al. (2016). R-spondin 2 promotes acetylcholine receptor clustering at the neuromuscular junction via Lgr 5. Sci. Rep. 6:28512. doi: 10.1038/srep28512
Nakata, R., Motomura, M., Masuda, T., Shiraishi, H., Tokuda, M., Fukuda, T., et al. (2013). Thymus histology and concomitant autoimmune diseases in Japanese patients with muscle-specific receptor tyrosine kinase-antibody-positive myasthenia gravis. Eur. J. Neurol. 20, 1272–1276. doi: 10.1111/ene.12169
Nastase, M. V., Young, M. F., and Schaefer, L. (2012). Biglycan: a multivalent proteoglycan providing structure and signals. J. Histochem. Cytochem. 60, 963–975. doi: 10.1369/0022155412456380
Nemazee, D. (2017). Mechanisms of central tolerance for B cells. Nat. Rev. Immunol. 17, 281–294. doi: 10.1038/nri.2017.19
Ngo, S. T., Cole, R. N., Sunn, N., Phillips, W. D., and Noakes, P. G. (2012). Neuregulin-1 potentiates agrin-induced acetylcholine receptor clustering through muscle-specific kinase phosphorylation. J. Cell Sci. 125, 1531–1543. doi: 10.1242/jcs.095109
Nicole, S., Chaouch, A., Torbergsen, T., Bauché, S., de Bruyckere, E., Fontenille, M. J., et al. (2014). Agrin mutations lead to a congenital myasthenic syndrome with distal muscle weakness and atrophy. Brain 137, 2429–2443. doi: 10.1093/brain/awu160
Niks, E. H., Kuks, J. B., Wokke, J. H., Veldman, H., Bakker, E., Verschuuren, J. J., et al. (2010). Pre- and postsynaptic neuromuscular junction abnormalities in musk myasthenia. Muscle Nerve 42, 283–288. doi: 10.1002/mus.21642
Nishimune, H. (2012). Molecular mechanism of active zone organization at vertebrate neuromuscular junctions. Mol. Neurobiol. 45, 1–16. doi: 10.1007/s12035-011-8216-y
Nishimune, H., Valdez, G., Jarad, G., Moulson, C. L., Müller, J. H., Miner, J. H., et al. (2008). Laminins promote postsynaptic maturation by an autocrine mechanism at the neuromuscular junction. J. Cell Biol. 182, 1201–1215. doi: 10.1083/jcb.200805095
Ohkawara, B., Cabrera-Serrano, M., Nakata, T., Milone, M., Asai, N., Ito, K., et al. (2014). Lrp4 third β-propeller domain mutations cause novel congenital myasthenia by compromising agrin-mediated MuSK signaling in a position-specific manner. Hum. Mol. Genet. 23, 1856–1868. doi: 10.1093/hmg/ddt578
Okada, K., Inoue, A., Okada, M., Murata, Y., Kakuta, S., Jigami, T., et al. (2006). The muscle protein Dok-7 is essential for neuromuscular synaptogenesis. Science 312, 1802–1805. doi: 10.1126/science.1127142
Oliverira, L., Timóteo, M. A., and Correira-de-Sá, P. (2004). Tetanic depression is overcome by tonic adenosine A2A receptor facilitation of L-type Ca2+ influx into rat motor nerve terminals. J. Physiol. 560, 157–168. doi: 10.1113/jphysiol.2004.067595
Osses, N., and Henriquez, J. P. (2015). Bone morphogenetic protein signaling in vertebrate motor neurons and neuromuscular communication. Front. Cell. Neurosci. 8:453. doi: 10.3389/fncel.2014.00453
Otsuka, K., Ito, M., Ohkawara, B., Masuda, A., Kawakami, Y., Sahashi, K., et al. (2015). Collagen Q and anti-MuSK autoantibody competitively suppress agrin/Lrp4/MuSK signaling. Sci. Rep. 5:13928. doi: 10.1038/srep13928
Pasnoor, M., Wolfe, G. I., Nations, S., Trivedi, J., Barohn, R. J., Herbelin, L., et al. (2010). Clinical findings in MuSK-antibody positive myasthenia gravis: a US experience. Muscle Nerve 41, 370–374. doi: 10.1002/mus.21533
Patel, T. R., Butler, G., McFarlane, A., Xie, I., Overall, C. M., and Stetefeld, J. (2012). Site specific cleavage mediated by MMPs regulates function of agrin. PLoS One 7:e43669. doi: 10.1371/journal.pone.0043669
Peng, H. B., Xie, H., and Dai, Z. (1997). Association of cortactin with developing neuromuscular specializations. J. Neurocytol. 26, 637–650. doi: 10.1023/a:1018545827369
Pevzner, A., Schoser, B., Peters, K., Cosma, N. C., Karakatsani, A., Schalke, B., et al. (2011). Anti-LRP4 autoantibodies in AChR- and MuSK-antibody-negative myasthenia gravis. J. Neurol. 259, 427–435. doi: 10.1007/s00415-011-6194-7
Pilgram, G. S. K., Potikanond, S., Baines, R. A., Fradkin, L. G., and Noordermeer, J. N. (2010). The roles of the dystrophin-associated glycoprotein complex at the synapse. Mol. Neurobiol. 41, 1–21. doi: 10.1007/s12035-009-8089-5
Plomp, J. J. (2017). Trans-synaptic homeostasis at the myasthenic neuromuscular junction. Front. Biosci. 22, 1033–1051. doi: 10.2741/4532
Plomp, J. J., Morsch, M., Phillips, W. D., and Verschuuren, J. J. (2015). Electrophysiological analysis of neuromuscular synaptic function in myasthenia gravis patients and animal models. Exp. Neurol. 270, 41–54. doi: 10.1016/j.expneurol.2015.01.007
Plomp, J. J., Van Kempen, G. T., De Baets, M. B., Graus, Y. M., Kuks, J. B., and Molenaar, P. C. (1995). Acetylcholine release in myasthenia gravis: regulation at single end-plate level. Ann. Neurol. 37, 627–636. doi: 10.1002/ana.410370513
Punga, A. R., Flink, R., Askmark, H., and Stalberg, E. V. (2006). Cholinergic neuromuscular hypersensitivity in patients with myasthenia gravis seropositive for MuSK antibody. Muscle Nerve 34, 111–115. doi: 10.1002/mus.20515
Reissner, C., Runkel, F., and Missler, M. (2013). Neurexins. Genome Biol. 14:213. doi: 10.1186/gb-2013-14-9-213
Remédio, L., Gribble, K. D., Lee, J. K., Kim, N., Hallock, P. T., Delestrée, N., et al. (2016). Diverging roles for Lrp4 and Wnt signaling in neuromuscular synapse development during evolution. Genes Dev. 30, 1058–1069. doi: 10.1101/gad.279745.116
Richman, D. P., Nishi, K., Ferns, M. J., Schnier, J., Pytel, P., Maselli, R. A., et al. (2012). Animal models of antimuscle-specific kinase myasthenia. Ann. N Y Acad. Sci. 1274, 140–147. doi: 10.1111/j.1749-6632.2012.06782.x
Rivner, M. H., Pasnoor, M., Dimachkie, M. M., Barohn, R. J., and Mei, L. (2018). Muscle-specific tyrosine kinase and myasthenia gravis owing to other antibodies. Neurol. Clin. 36, 293–310. doi: 10.1016/j.ncl.2018.01.004
Rodríguez Cruz, P. M., Al-Hajjar, M., Huda, S., Jacobson, L., Woodhall, M., Jayawant, S., et al. (2015a). Clinical features and diagnostic usefulness of antibodies to clustered acetylcholine receptors in the diagnosis of seronegative myasthenia gravis. JAMA Neurol. 72, 642–649. doi: 10.1001/jamaneurol.2015.0203
Rodríquez Cruz, P. M., Palace, J., Ramjattan, H., Jayawand, S., Eoff, S. A., and Beeson, D. (2015b). Salbutamol and ephedrine in the treatment of severe AChR deficiency syndromes. Neurology 85, 1043–1047. doi: 10.1212/wnl.0000000000001952
Rodríquez Cruz, P. M., Cossin, J., Estephan, E. P., Munell, F., Selby, K., Hirano, M., et al. (2019). The clinical spectrum of the congenital myasthenic syndrome resulting from COL13A mutations. Brain 142, 1547–1560. doi: 10.1093/brain/awz107
Rodríquez Cruz, P. M., Cossins, J., Cheung, J., Maxwell, S., Jayawant, S., Herbst, R., et al. (2020). Congenital myasthenic syndrome due to mutations in MuSK suggests that the level of MuSK phosphorylation is crucial for govering synaptic structure. Hum. Mutat. 41, 619–631. doi: 10.1002/humu.23949
Rodríguez Cruz, P. M., Palace, J., and Beeson, D. (2018). The neuromuscular junction and wide heterogeneity of congenital myasthenic syndromes. Int. J. Mol. Sci. 19:1677. doi: 10.3390/ijms19061677
Rogers, R. S., and Nishimune, H. (2017). The role of laminins in the organization and function of neuromuscular junctions. Matrix Biol. 57, 86–105. doi: 10.1016/j.matbio.2016.08.008
Romi, F. (2011). Thymoma in myasthenia gravis: from diagnosis to treatment. Autoimmune Dis. 2011:474512. doi: 10.4061/2011/474512
Romi, F., Aarli, J. A., and Gilhus, N. E. (2007). Myasthenia gravis patients with ryanodine receptor antibodies have distinctive clinical features. Eur. J. Neurol. 14, 617–620. doi: 10.1111/j.1468-1331.2007.01785.x
Romi, F., Helgeland, G., and Gilhus, N. E. (2012). Serum levels of matrix metalloproteinases: implications in clinical neurology. Eur. Neurol. 67, 121–128. doi: 10.1159/000334862
Rudolf, R., Khan, M. M., Labeit, S., and Deschenes, M. R. (2014). Degeneration of neuromuscular junction in age and dystrophy. Front. Aging Neursci. 6:99. doi: 10.3389/fnagi.2014.00099
Sahashi, K., Engel, A. G., Lambert, E. H., and Howard, F. M. Jr. (1980). Ultrastructural localization of the terminal and lytic ninth complement (C9) at the motor end-plate in myasthenia gravis. J. Neuropathol. Exp. Neurol. 39, 160–172. doi: 10.1097/00005072-198003000-00005
Salinas, P. C. (2005). Retrograde signalling at the synapse: a role for Wnt proteins. Biochem. Soc. Trans. 33, 1295–1298. doi: 10.1042/bst20051295
Samuel, M. A., Valdez, G., Tapia, J. C., Lichtman, J. W., and Sanes, J. R. (2012). Agrin and synaptic laminin are required to maintain adult neuromuscular junctions. PLoS One 7:e46663. doi: 10.1371/journal.pone.0046663
Santafé, M. M., Priego, M., Obis, T., Garcia, N., Tomàs, M., Lanuza, M. A., et al. (2015). Adenosine receptors and muscarinic receptors cooperate in acetylcholine release modulation in the neuromuscular synapse. Eur. J. Neurosci. 42, 1775–1787. doi: 10.1111/ejn.12922
Sato, K., Mano, T., Iwata, A., and Toda, T. (2019). Neurological and related adverse events in immune checkpoint inhibitors: a pharmacovigilance study from the Japanese adverse drug event report database. J. Neurooncol. 145, 1–9. doi: 10.1007/s11060-019-03273-1
Schmidt, N., Akaaboune, M., Gajendran, N., Martinez-Pena y Valenzuela, I., Wakefield, S., Thurnheer, R., et al. (2011). Neuregulin/ErbB regulate neuromuscular junction development by phosphorylation of α-dystrobrevin. J. Cell Biol. 195, 1171–1184. doi: 10.1083/jcb.201107083
Scotton, P., Bleckmann, D., Stebler, M., Sciandra, F., Brancaccio, A., Meier, T., et al. (2006). Activation of muscle-specific receptor tyrosine kinase and binding to dystroglycan are regulated by alternative mRNA splicing of agrin. J. Biol. Chem. 281, 36835–36845. doi: 10.1074/jbc.m607887200
Selcen, D., Ohkawara, B., Shen, X. M., McEvoy, K., Ohno, K., and Engel, A. G. (2015). Impaired synaptic development, maintenance, and neuromuscular transmission in Lrp4-related myasthenia. JAMA Neurol. 72, 889–896. doi: 10.1001/jamaneurol.2015.0853
Shen, C., Li, L., Zhao, K., Bai, L., Wang, A., Shu, X., et al. (2018). Motoneuron Wnts regulate neuromuscular junction development. Elife 7:e34625. doi: 10.7554/elife.34625
Shen, C., Lu, Y., Zhang, B., Figueiredo, D., Bean, J., Jung, J., et al. (2013). Antibodies against low-density lipoprotein receptor-related protein 4 induce myasthenia gravis. J. Clin. Investig. 123, 5190–5202. doi: 10.1172/JCI66039
Shi, L., Fu, A. K., and Ip, N. Y. (2012). Molecular mechanisms underlying maturation and maintenance of the vertebrate neuromuscular junction. Trends Neurosci. 35, 441–453. doi: 10.1016/j.tins.2012.04.005
Shin, H. Y., Park, H. J., Lee, H. E., Choi, Y. C., and Kim, S. M. (2014). Clinical and electrophysiologic responses to acetylcholinesterase inhibitors in MuSK-antibody-positive myasthenia gravis: Evidence for cholinergic neuromuscular hypersensitivity. J. Clin. Neurol. 10, 119–124. doi: 10.3988/jcn.2014.10.2.119
Simeone, L., Straubinger, M., Khan, M. A., Nalleweg, N., Cheusova, T., and Hashemolhosseini, S. (2010). Identification of Erbin interlinking MuSK and ErbB2 and its impact on acetylcholine receptor aggregation at the neuromuscular junction. J. Neurosci. 30, 6620–6634. doi: 10.1523/jneurosci.5778-09.2010
Simö, A., Just-Borràs, L., Cilleros-Mañé, V., Hurtado, E., Nadal, L., Tomàs, M., et al. (2018). BDNF-TrkB signaling coupled to nPKCε and cPKCβI modulate the phosphorylation of the exocytotic protein Munc18–1 during synaptic activity at the neuromuscular junction. Front. Mol. Neurosci. 11:207. doi: 10.3389/fnmol.2018.00207
Singhal, N., and Martin, P. T. (2011). Role of extracellular matrix proteins and their receptors in the development of the vertebrate neuromuscular junction. Dev. Neurobiol. 71, 982–1005. doi: 10.1002/dneu.20953
Sittig, L. J., Carbonetto, P., Engel, K. A., Krauss, K. S., Barrios-Camacho, C. M., and Palmer, A. A. (2016). Genetic background limits generalizability genotype-phenotype relationships. Neuron 91, 1253–1259. doi: 10.1016/j.neuron.2016.08.013
Skeie, G. O., Mygland, A., Treves, S., Gilhus, N. E., Aarli, J. A., and Zorzato, F. (2002). Ryanodine receptor antibodies in myasthenia gravis: epitope mapping and effect on calcium release in vitro. Muscle Nerve 27, 81–89. doi: 10.1002/mus.10294
Skriapa, L., Zisimopoulou, P., Trakas, N., Grapas, E., and Tzartos, S. J. (2014). Expression of extracellular domains of muscle specific kinase (MuSK) and use as immunoadsorbents for the development of an antigen-specific therapy. J. Neuroimmunol. 276, 150–158. doi: 10.1016/j.jneuroim.2014.09.013
Somnier, F. E., and Engel, P. J. (2002). The occurrence of anti-titin antibodies and thymomas: a population survey of MG 1970–1999. Neurology 59, 92–98. doi: 10.1212/wnl.59.1.92
Song, J. S., Kim, D., Kwon, J. H., Kim, H. R., Choi, C. M., and Jang, S. J. (2019). Clinicopathologic significance and immunogenomic analysis of programmed death-ligand 1 (PD-L1) and programmed death 1 (PD-1) expression in thymic epithelial tumors. Front. Oncol. 9:1055. doi: 10.3389/fonc.2019.01055
Sons, M. S., Busche, N., Strenzke, N., Moser, T., Ernsberger, U., Mooren, F. C., et al. (2006). α-Neurexions are required for efficient transmitter release and synaptic homeostasis at the mouse neuromuscular junction. Neuroscience 138, 433–446. doi: 10.1016/j.neuroscience.2005.11.040
Stamatakou, E., and Salinas, P. C. (2014). Postsynaptic assembly: a role for Wnt signaling. Dev. Neurobiol. 74, 818–827. doi: 10.1002/dneu.22138
Stanga, S., Zanou, N., Audouard, E. T., Tasiaux, B., Contino, S., Vandermeulen, G., et al. (2016). APTT-dependent glial cell line-derived neurotrophic gactor gene expression drives neuromuscular junction. FASEB J. 30, 1696–1711. doi: 10.1096/fj.15-278739
Stergiou, C., Lazaridis, K., Zouvelou, V., Tzartos, J., Mantegazza, R., Antozzi, C., et al. (2016). Titin antibodies in “seronegative” myasthenia gravis-A new role for an old antigen. J. Neuroimmunol. 292, 108–115. doi: 10.1016/j.jneuroim.2016.01.018
Stetefeld, J., Alexandrescu, A. T., Maciejewski, M. W., Jenny, M., Rathgeb-szabo, K., Schulthess, T., et al. (2004). Modulation of agrin function by alternative splicing and Ca2+ binding. Structure 12, 503–515. doi: 10.1016/j.str.2004.02.001
Stiegler, A. L., Burden, S. J., and Hubbard, S. R. (2006). Crystal structure of the agrin-responsive immunoglobulin-like domains 1 and 2 of the receptor tyrosine kinase MuSK. J. Mol. Biol. 364, 424–433. doi: 10.1016/j.jmb.2006.09.019
Stiegler, A. L., Burden, S. J., and Hubbard, S. R. (2009). Crystal structure of the Frizzled-like cycteine-rich domain of the receptor tyrosine kinase MuSK. J. Mol. Biol. 393, 1–9. doi: 10.1016/j.jmb.2009.07.091
Strochlic, L., Falk, J., Goillot, E., Sigoillot, S., Bourgeois, F., Delers, P., et al. (2012). Wnt4 participates in the formation of vertebrate neuromuscular junction. PLoS One 7:e29976. doi: 10.1371/journal.pone.0029976
Südhof, T. C. (2012). The presynaptic active zone. Neuron 75, 11–25. doi: 10.1016/j.neuron.2012.06.012
Sudres, M., Verdier, J., Truffault, F., Le Panse, R., and Berrih-Aknin, S. (2018). Pathophyusiological mechanisms of autoimmunity. Ann. N Y Acad. Sci. 1413, 59–68. doi: 10.1111/nyas.13560
Takamori, M. (2008a). Lambert-Eaton myasthenic syndrome: search for alternative autoimmune targets and possible compensatory mechanisms based on presynaptic calcium homeostasis. J. Neuroimmunol. 201–202, 145–152. doi: 10.1016/j.jneuroim.2008.04.040
Takamori, M. (2008b). Autoantibodies against TRPC3 and ryanodine receptor in myasthenia gravis. J. Neuroimmunol. 200, 142–144. doi: 10.1016/j.jneuroim.2008.06.001
Takamori, M. (2012). Structure of the neuromuscular junction: function and cooperative mechanisms in the synapse. Ann. N Y Acad. Sci. 1274, 14–23. doi: 10.1111/j.1749-6632.2012.06784.x
Takamori, M. (2013). Structural and pathogenic mechanisms centered on muscle-specific tyrosine kinase in autoimmune neuromuscular junction disorders. Curr. Immunol. Rev. 9, 207–213. doi: 10.2174/1573395510666140318003732
Takamori, M. (2017a). Synaptic homeostasis and its immunological disturbance in neuromuscular junction disorders. Int. J. Mol. Sci. 18:E896. doi: 10.3390/ijms18040896
Takamori, M. (2017b). Pathogenic participation of MuSK-biglycan linkage contributive to synaptic stability and signaling in myasthenia gravis. J. Autoimmun. Disord. 3, 41–48. doi: 10.21767/2471-8513
Takamori, M. (2019). Synaptic compensatory mechanism and its impairment in autoimmune myasthenic diseases. J. Immunol. Sci. 3, 6–13. doi: 10.29245/2578-3009/2019/3.1173
Takamori, M., Motomura, M., Fukudome, T., and Yoshikawa, H. (2007). Autoantibodies against M1 muscarinic acetylcholine receptor in myasthenic disorders. Eur. J. Neurol. 14, 1230–1235. doi: 10.1111/j.1468-1331.2007.01931.x
Takamori, M., Motomura, M., Kawaguchi, N., Nemoto, Y., Hattori, T., Yoshikawa, H., et al. (2004). Anti-ryanodine receptor antibodies and FK506 in myasthenia gravis. Neurology 62, 1894–1896. doi: 10.1212/01.wnl.0000125254.99397.68
Takamori, M., Nakamura, T., and Motomura, M. (2013). Antibodies against Wnt receptor of muscle-specific tyrosine kinase in myasthenia gravis. J. Neuroimmunol. 254, 183–186. doi: 10.1016/j.jneuroim.2012.09.001
Takamori, M., Okumura, S., Nagata, M., and Yoshikawa, H. (1988). Myasthenogenic significance of synthetic α-subunit peptide 183–200 of Torpedo californica and human acetylcholine receptor. J. Neurol. Sci. 85, 121–129. doi: 10.1016/0022-510x(88)90150-5
Takamori, M., Sakato, S., and Okumura, S. (1984). Presynaptic function modified by acetylcholine-receptor interaction in experimental autoimmune myasthenia gravis. J. Neurol. Sci. 66, 245–253. doi: 10.1016/0022-510x(84)90013-3
Takata, K., Stathopoulos, P., Cao, M., Mané-Damas, M., Fichtner, M. L., Benotti, E. S., et al. (2019). Characterization of pathogenic monoclonal autoantibodies derived from muscle-specific kinase myasthenia gravis patients. JCI Insight 4:e127167. doi: 10.1172/jci.insight.127167
Tandan, R., Hehir, M. K. II., Waheed, W., and Howard, D. B. (2017). Rituximab treatment of myasthenia gravis: a systematic review. Muscle Nerve 56, 185–196. doi: 10.1002/mus.25597
Tezuka, T., Inoue, A., Hoshi, T., Wheatherbee, S. D., Burgees, R. W., Ueta, R., et al. (2014). The MuSK activator agrin has a separate role essential for postnatal maintenance of neuromuscular synapses. Proc. Natl. Acad. Sci. U S A 111, 16556–16561. doi: 10.1073/pnas.1408409111
Thiruppathi, M., Rowin, J., Ganesh, B., Sheng, J. R., Prabbakar, B. S., and Meriggioli, M. N. (2012). Impaired regulatory function in circulating CD4+CD25highCD127low/- T cells in patients with myasthenia gravis. Clin. Immunol. 145, 209–223. doi: 10.1016/j.clim.2012.09.012
Tomàs, J., Gracia, N., Lanuza, M. A., Santafé, M. M., Tomàs, M., Nodal, L., et al. (2018). Adenosine receptors in developing and adult mouse neuromuscular junctions and functional links with other metabotrophic receptor pathways. Front. Pharmacol. 9:397. doi: 10.3389/fphar.2018.00397
Tomoyasu, H., Kikuchi, A., and Kamo, I. (1998). Identification of haemopoietic biglycan in hyperplastic thymus associated with myasthenia gravis. J. Neuroimmunol. 89, 59–63. doi: 10.1016/s0165-5728(98)00092-7
Tu, H., Pirskanen-Matell, R., Heikkinen, A., Oikarainen, T., Risteli, J., and Pihlajaniemi, T. (2018). Autoimmune antibodies to collagen XIII in myasthenia gravis patients. Muscle Nerve 57, 506–510. doi: 10.1002/mus.25969
Tüzün, E., and Christadoss, P. (2013). Complement associated pathogenic mechanisms in myasthenia gravis. Autoimmun. Res. 12, 904–911. doi: 10.1016/j.autrev.2013.03.003
Tzartos, S. J., Kokla, A., Walgrave, S. L., and Conti-Tronconi, B. M. (1988). Localization of the main immunogenic region of human muscle acetylcholine receptor to residues 67–76 of the α subunit. Proc. Natl. Acad. Sci. U S A 85, 2899–2903. doi: 10.1073/pnas.85.9.2899
Tzartos, S. J., Seybold, M. E., and Lindstrom, J. M. (1982). Specificities of antibodies to acetylcholine receptors in sera from myasthenia gravis patients measured by monoclonal antibodies. Proc. Natl. Acad. Sci. U S A 79, 188–192. doi: 10.1073/pnas.79.1.188
Ulrichts, P., Guglietta, A., Dreier, T., van Bragt, T., Hanssens, V., Hofman, E., et al. (2018). Neonatal Fc receptor antagonist Efgartigimod safely and sustainably reduces IgGs in humans. J. Clin. Invest. 128, 4372–4386. doi: 10.1172/JCI97911
Ulusoy, C., Cavus, F., Yilmaz, V., and Tuzűn, E. (2017). Immunization with recombinantly expressed LRP4 induces experimental autoimmune myasthenia gravis in C57BL/6 mice. Immunol. Invest. 46, 490–499. doi: 10.1080/08820139.2017.1299754
Vanhaesebrouck, A. E., and Beeson, D. (2019). The congenital myasthenic syndromes: expanding genetic and phenotypic spectrums and refining treatment strategies. Curr. Opin. Neurol. 32, 696–703. doi: 10.1097/wco.0000000000000736
Vanhaesebrouck, A. E., Webster, R., Maxwell, S., Rodriquez Cruz, P. M., Cossins, J., Wickens, J., et al. (2019). β2-Adrenergic receptor agonists ameliorate the adverse effect of long-term pyridostigmine on neuromuscular junction structure. Brain 142, 3713–3727. doi: 10.1093/brain/awz322
Verschuuren, J. J. G. M., Huijbers, M. G., Plomp, J. J., Niks, E. H., Molenaar, P. C., Martinez-Martinez, P., et al. (2013). Pathophysiology of myasthenia gravis with antibodies to the acetylcholine receptor, muscle-specific kinase and low-density lipoprotein receptor-related protein 4. Autoimmun. Rev. 12, 918–923. doi: 10.1016/j.autrev.2013.03.001
Viegas, S., Jacobson, L., Waters, P., Cossins, J., Jacob, S., Leite, M. I., et al. (2012). Passive and active immunization models of MuSK-ab positive myasthenia: electrophysiological evidence for pre and postsynaptic defects. Exp. Neurol. 234, 506–512. doi: 10.1016/j.expneurol.2012.01.025
Vieira, G. C., Chockalingam, S., Melegh, Z., Greenhough, A., Malik, S., Szemes, M., et al. (2015). LGR5 regulates pro-survival MEK/ERK and proliferative Wnt/β-catenin signaling in neuroblastoma. Oncotarget 6, 40053–40067. doi: 10.18632/oncotarget.5548
Villegas, J. A., Van Wassenhove, J., Le Panse, R., Berrih-Aknin, S., and Dragin, N. (2018). An imbalance between regulatory T cells and T helper 17 cells in acetylcholine receptor-positive myasthenia gravis pateints. Ann. N Y Acad. Sci. 1413, 154–162. doi: 10.1111/nyas.13591
Vincent, A., Huda, S., Cao, M., Cetin, H., Koneczny, I., Rodriguez Cruz, P. M., et al. (2018). Serological and experimental studies in different forms of myasthenia gravis. Ann. N Y Acad. Sci. 1413, 143–153. doi: 10.1111/nyas.13592
Vincent, A., and Newsom-Davis, J. (1980). Anti-acetylcholine receptor antibodies. J. Neurol. Neurosurg. Psychiatry 43, 590–600. doi: 10.1136/jnnp.43.7.590
Vincent, A., Waters, P., Leite, M. I., Jacobson, L., Koneczny, I., Cossins, J., et al. (2012). Antibodies identified by cell-based assays in myasthenia gravis and associated diseases. Ann. N Y Acad. Sci. 1274, 92–98. doi: 10.1111/j.1749-6632.2012.06789.x
Vrolix, K., Niks, E. H., Le Panse, R., van Ostaijen-Ten Dam, M. M., Murin, A. H., Jol-van der Zijde, C. M., et al. (2011). Reduced thymic expression of ErbB receptors without auto-antibodies against synaptic ErbB in myasthenia gravis. J. Neuroimmunol. 232, 158–165. doi: 10.1016/j.jneuroim.2010.10.024
Wang, J., Ruan, N. J., Qian, L., Lei, W. L., Chen, F., and Luo, Z. G. (2008). Wnt/β-catenin signaling suppresses rapsyn expression and inhibits acetylcholine receptor clustering at the neuromuscular junction. J. Biol. Chem. 283, 21668–21675. doi: 10.1074/jbc.m709939200
Wang, X., Pinter, M. J., and Rich, M. M. (2016). Reversible recruitment of a homeostatic reserve pool of synaptic vesicles underlies rapid homeostatic plasticity of quantal content. J. Neurosci. 36, 828–836. doi: 10.1523/jneurosci.3786-15.2016
Wang, Z., Wang, B., Yang, L., Guo, Q., Aithmitti, N., Songyang, Z., et al. (2009). Presynaptic and postsynaptic interaction of the amyloid precursor protein promotes peripheral and central synaptogenesis. J. Neurosci. 29, 10788–10801. doi: 10.1523/jneurosci.2132-09.2009
Weatherbee, S. D., Anderson, K. V., and Niswander, L. A. (2006). LDL-receptor-related protein 4 is critical for formation of the neuromuscular junction. Development 133, 4993–5000. doi: 10.1242/dev.02696
Wen, Y., Yang, B., Lu, J., Zhang, J., Yang, H., and Li, J. (2016). Imbalance of circulating CD+CXCR5+FOXP3+ Tfr-like cells and CD+CXCR5+FOXP3− Tfh-like cells in myasthenia gravis. Neurosci. Lett. 630, 176–182. doi: 10.1016/j.neulet.2016.07.049
Werle, M. J., and VanSaun, M. (2003). Activity dependent removal of agrin from synaptic basal lamina by matrix metalloproteinase 3. J. Neurocytol. 32, 905–915. doi: 10.1023/b:neur.0000020631.69804.f5
Wolfe, G. I., Kaminski, H. J., Aban, I. B., Minisman, G., Kuo, H. C., Marx, A., et al. (2019). Long-term effect of thymectomy plus prednisone versus prednisone alone in patients with non-thymomatous myasthenia gravis: 2-year extension of MGTX randmised trial. Lancet Neurol. 18, 259–268. doi: 10.1016/S1474-4422(18)30392-2
Wu, H., Barik, A., Lu, Y., Shen, C., Bowman, A., Li, L., et al. (2015). Slit2 as a β-catenin/Ctnnb1-dependent retrograde signal for presynaptic differentiation. Elife 4:e07266. doi: 10.7554/elife.07266
Wu, H., Lu, Y., Barik, A., Joseph, A., Taketo, M. M., Xiong, W. C., et al. (2012a). β-catenin gain of function in muscles impairs neuromuscular junction formation. Development 139, 2392–2404. doi: 10.1242/dev.080705
Wu, H., Lu, Y., Shen, C., Patel, N., Gan, L., Xiong, W. C., et al. (2012b). Distict roles of muscle and motoneuron Lrp4 in neuromuscular junction formation. Neuron 75, 94–107. doi: 10.1016/j.neuron.2012.04.033
Wu, H., Xiong, W. C., and Mei, L. (2010). To bild a synapse: signaling pathways in neuromuscular junction assembly. Development 137, 1017–1033. doi: 10.1242/dev.038711
Xing, G., Xiong, W. C., and Mei, L. (2020). Rapsyn as a signaling and scaffolding molecule in NMJ formation and maintenance. Neurosci. Lett. 135013. doi: 10.1016/j.neulet.2020.135013
Yamada, M., and Sekiguchi, K. (2015). Molecular basis of laminin-integrin interactions. Curr. Top. Membr. 76, 197–229. doi: 10.1016/bs.ctm.2015.07.002
Yan, M., Liu, Z., Fei, E., Chen, W., Lai, X., Luo, B., et al. (2018a). Induction of anti-agrin antibodies causes myasthenia gravis in mice. Neuroscience 373, 113–121. doi: 10.1016/j.neuroscience.2018.01.015
Yan, M., Xin, G. L., Xiong, W. C., and Mei, L. (2018b). Agrin and Lrp4 antibodies as new biomarkers of myasthenia gravis. Ann. N Y Acad. Sci. 1413, 126–135. doi: 10.1111/nyas.13573
Yang, J., Dominguez, B., de Winter, F., Gould, T. W., Eriksson, J. E., and Lee, K. F. (2011). Nestin negatively regulates postsynaptic differentiation of the neuromuscular synapse. Nat. Neurosci. 14, 324–330. doi: 10.1038/nn.2747
Yi, J. S., Guptill, J. T., Stathopoulos, P., Nowak, R. J., and O’Connor, K. C. (2018). B cells in the pathophysiology of myasthenia gravis. Muscle Nerve 57, 172–184. doi: 10.1002/mus.25973
Yilmaz, A., Kattamuri, C., Ozdeslik, R. N., Schmiedel, C., Mentzer, S., Schorl, C., et al. (2016). MuSK is a BMP co-receptor that shapes BMP responses and calcium signaling in muscle cells. Sci. Signal. 9:ra87. doi: 10.1126/scisignal.aaf0890
Yumoto, N., Kim, N., and Burden, S. J. (2012). Lrp4 is a retrograde signal for presynaptic differentiation at neuromuscular synapses. Nature 489, 438–442. doi: 10.1038/nature11348
Yurchenco, P. D. (2011). Basement membranes: Cell scaffoldings and signaling platforms. Cold Spring Harb. Perspect. Biol. 3:e004911. doi: 10.1101/cshperspect.a004911
Zhang, W., Coldefy, A. S., Hubbard, S. R., and Burden, S. J. (2011). Agrin binds to the N-terminal region of Lrp4 protein and stimulates association between Lrp4 and the first immunoglobulin-like domain in muscle-specific kinase (MuSK). J. Biol. Chem. 286, 40624–40630. doi: 10.1074/jbc.m111.279307
Zhang, B., Liang, C., Bates, R., Yin, Y., Xiong, W. C., and Mei, L. (2012a). Wnt proteins regulate acetylcholine receptor clustering in muscle cells. Mol. Brain 5:7. doi: 10.1186/1756-6606-5-7
Zhang, B., Tzartos, J. S., Belimezi, M., Ragheb, S., Bealmear, B., Lewis, R. A., et al. (2012b). Autoantibodies to lipoprotein-related protein 4 in patients with double-seronegative myasthenia gravis. Arch. Neurol. 69, 445–451. doi: 10.1001/archneurol.2011.2393
Zhang, B., Luo, S., Dong, X. P., Zhang, X., Liu, C., Luo, Z., et al. (2007). β-catenin regulates acetylcholine receptor clustering in muscle cells through interaction with rapsyn. J. Neurosci. 27, 3968–3973. doi: 10.1523/JNEUROSCI.4691-06.2007
Zhang, B., Luo, S., Wang, Q., Suzuki, T., Xiong, W. C., and Mei, L. (2008). Lrp4 serves as a coreceptor of agrin. Neuron 60, 285–297. doi: 10.1016/j.neuron.2008.10.006
Zhang, B., Shen, C., Bealmear, B., Ragheb, S., Xiong, W. C., Lewis, R. A., et al. (2014). Autoantibodies to agrin in myasthenia gravis patients. PLoS One 9:e91816. doi: 10.1371/journal.pone.0091816
Zhang, B. G., Quigley, A. F., Bourke, J. L., Nowell, C. J., Myers, D. E., Choong, P. F., et al. (2015). Combination of agrin and laminin increase acetylcholine receptor clustering and enhance functional neuromuscular junction formation in vitro. Dev. Neurobiol. 76, 551–565. doi: 10.1002/dneu.22331
Zhao, K., Shen, C., Lu, Y., Huang, Z., Li, L., Rand, C. D., et al. (2017). Muscle Yap is a regulator of neuromuscular junction formation and regeneration. J. Neurosci. 37, 3465–3477. doi: 10.1523/NEUROSCI.2934-16.2017
Zisimopoulou, P., Evangelakou, P., Tzartos, J., Lazaridis, K., Zouvelou, V., Mantegazza, R., et al. (2014). A comprehensive analysis of the epidemiology and clinical characteristics of anti-Lrp4 in myasthenia gravis. J. Autoimmun. 52, 139–145. doi: 10.1016/j.jaut.2013.12.004
Zoltowska Katarzyna, M., Belaya, K., Leite, M., Patrick, W., Vincent, A., and Beeson, D. (2015). Collagen Q-a potential target for autoantibodies in myasthenia gravis. J. Neurol. Sci. 348, 241–244. doi: 10.1016/j.jns.2014.12.015
Zong, Y., and Jin, R. (2013). Structural mechanisms of the agrin-Lrp4-MuSK signaling pathway in neuromuscular junction differentiation. Cell. Mol. Life Sci. 70, 3077–3088. doi: 10.1007/s00018-012-1209-9
Zong, Y., Zhang, B., Gu, S., Lee, K., Zhou, J., Yao, G., et al. (2012). Structural basis of agrin-Lrp4-MuSK signaling. Genes Dev. 26, 247–258. doi: 10.1101/gad.180885.111
Zouvelou, V., Zisimopoulou, P., Rentzos, M., Karandreas, N., Evangelakou, P., Stramboulis, E., et al. (2013). Double seronegative myasthenia gravis with anti-LRP4 antibodies. Neuromuscul. Disord. 23, 568–570. doi: 10.1016/j.nmd.2013.03.013
Zuber, B., and Unwin, N. (2013). Structure and superorganization of acetylcholine receptor-rapsyn complexes. Proc. Natl. Acad. Sci. U S A 110, 10622–10627. doi: 10.1073/pnas.1301277110
Keywords: neuromuscular junction, myasthenia gravis, acetylcholine receptor, agrin, wnts, MuSK, Lrp4, matrix proteins
Citation: Takamori M (2020) Myasthenia Gravis: From the Viewpoint of Pathogenicity Focusing on Acetylcholine Receptor Clustering, Trans-Synaptic Homeostasis and Synaptic Stability. Front. Mol. Neurosci. 13:86. doi: 10.3389/fnmol.2020.00086
Received: 15 January 2020; Accepted: 28 April 2020;
Published: 28 May 2020.
Edited by:
Hanns Lochmuller, University of Ottawa, CanadaReviewed by:
Lin Mei, Case Western Reserve University, United StatesJoern R. Steinert, University of Leicester, United Kingdom
Copyright © 2020 Takamori. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Masaharu Takamori, bS10YWthbW9yaUB2YW5pbGxhLm9jbi5uZS5qcA==